 Miriam G. Jasiulionis
Miriam G. Jasiulionis- Laboratory of Ontogeny and Epigenetics, Pharmacology Department, Universidade Federal de São Paulo, São Paulo, Brazil
Epigenetics refers to the study of mechanisms controlling the chromatin structure, which has fundamental role in the regulation of gene expression and genome stability. Epigenetic marks, such as DNA methylation and histone modifications, are established during embryonic development and epigenetic profiles are stably inherited during mitosis, ensuring cell differentiation and fate. Under the effect of intrinsic and extrinsic factors, such as metabolic profile, hormones, nutrition, drugs, smoke, and stress, epigenetic marks are actively modulated. In this sense, the lifestyle may affect significantly the epigenome, and as a result, the gene expression profile and cell function. Epigenetic alterations are a hallmark of aging and diseases, such as cancer. Among biological systems compromised with aging is the decline of immune response. Different regulators of immune response have their promoters and enhancers susceptible to the modulation by epigenetic marks, which is fundamental to the differentiation and function of immune cells. Consistent evidence has showed the regulation of innate immune cells, and T and B lymphocytes by epigenetic mechanisms. Therefore, age-dependent alterations in epigenetic marks may result in the decline of immune function and this might contribute to the increased incidence of diseases in old people. In order to maintain health, we need to better understand how to avoid epigenetic alterations related to immune aging. In this review, the contribution of epigenetic mechanisms to the loss of immune function during aging will be discussed, and the promise of new means of disease prevention and management will be pointed.
Epigenetics: How the Genome Talks with the Environment
Epigenetics (epi = beyond) refers to the study of the heritable information on the chromatin beyond that given by the DNA sequence. Epigenetic marks, represented by different chemical groups added on both the DNA molecule and histone proteins, play key roles in the control of chromatin structure and function (1, 2). Among the most studied epigenetic mechanisms are DNA methylation and posttranslational histone modifications. The chromatin remodeling, the presence of structural and functional variants of histones, and the regulation by noncoding RNAs are additional epigenetic mechanisms working together with DNA methylation and histone modifications to maintain genome stability and control gene expression.
DNA Methylation
In mammals, each cytosine in the context of a CpG dinucleotide is a potential site for the covalent addition of a methyl group, yielding 5-methylcytosine (5mC) (3). This reaction is catalyzed by DNA methyltransferases (DNMTs)—DNMT1, 3A, and 3B, which transfer the methyl group from S-adenosylmethionine (SAM) to the 5-carbon of the cytosine (5mC) (4). SAM, a component of methionine cycle, is considered the universal donor of methyl groups to various biomolecules, including DNA and histones (5). Different components coming from the diet, such as B6 and B12 vitamin, choline, and betaine act as cofactors in reactions taking part in methionine cycle, and the folate cycle is coupled to the methionine cycle, bringing up how cellular nutrient status may modulate epigenetic marks (6). In normal cells, most CpG-rich (CpG islands) gene promoters are unmethylated, making these genes permissive to transcription. Hypermethylated CpG-rich promoters are typically associated with gene silencing, since methylated CpGs can both impair the binding of transcriptional factors and recruit repressive complexes (7, 8). Regions, such as repetitive DNA sequences and transposons, present in high number in our genome, have CpGs heavily methylated in normal cells. The loss of methylation in these regions favors homologous recombination and the expression of undesirable elements, contributing to chromosomal instability (9). The technological advances in genome-wide chromatin profiling have revealed that in fact the role of DNA methylation in gene regulation depends on its position and context. While in promoters it is associated with transcriptional silencing, in other regions it can modulate enhancer activity and splicing (10). The methylation at gene bodies is frequent in ubiquitously expressed genes and correlated with transcriptional activation (11). The tissue-specific DNA methylation seems to occur not at CpG islands, but at regions of lower CpG density about 2 kb distant from CpG islands, named CpG island shores (12). In addition, other non-CpG sites were more recently described as sites for DNA methylation in humans, such as CHG and CHH sites (where H is A, C, or T) (13), but the mechanisms involved in this process are still unknown. The complexity of the covalent modification of DNA has additionally increased with the recent identification of a new family of enzymes known as ten-eleven translocation 1-3 (TET1-3), which are able to oxidize 5mC to 5-hydroxymethylcytosine (5hmC) in a reaction that generates other intermediates (5-methylformylcytosine and 5-carboxylcytosine) (14, 15). These modified bases may be excised by thymine DNA glycosylases (TDG) and, through the base excision repair (BER) process, yield demethylated cytosines (16), which makes this process a mechanism of active DNA demethylation. The DNA can be also actively demethylated by the deamination of 5mC and 5hmC by the activation-induced deaminase/apolipoprotein B editing complex enzymes, followed by BER/TDG activity (17). Alternatively, 5hmC can be passively demethylated during DNA replication, since it is not recognized by DNMT1, yielding in this position non-methylated cytosine in the newly synthesized DNA strand.
Histone Modifications
In the nucleosomes, not only the DNA molecule but also histone proteins carry chemical modifications, which are fundamental for chromatin-dependent gene regulation (18). Several posttranslational histone modifications (PTMs) regulate the chromatin structure, by affecting inter-nucleosomal interactions, and recruit proteins and complexes that influence not only the gene transcription but also mediate processes, such as DNA replication, DNA repair, alternative splicing, and recombination (19). A large number of proteins acting as writers, erasers, and readers have been described as components of histone modifier machinery, targeting all core histones H2A, H2B, H3, and H4, and the linker H1 histone, which have their amino acids subjected to covalent modifications mainly in the N-terminal tails. Among these modifications are acetylation, phosphorylation, methylation, ubiquitylation, sumoylation, and ADP ribosylation (19). By decreasing the positive charge of histones, acetylation and phosphorylation weaken interactions between histones and DNA, facilitating transcription machinery to access the DNA. Histone methylation occurs mainly on lysines and arginines, which can be, respectively, mono-, di- or trimethylated, and mono- and di-methylated, resulting in a high level of complexity regarding their effects. For example, high levels of trimethylated H3K4, H3K36, and H3K79 are associated with actively transcribed chromatin, while methylated H3K9, H3K27, and H4K20 are associated with transcriptionally inactive chromatin (20). The covalent attachment of the large ubiquitin molecule changes the nucleosome conformation, affecting both intra-nucleosomal interactions and interactions with effector proteins. Sumoylation involves the addition of small ubitiquin-like molecules to histones, and has been associated with repressive functions. Histone mono- and poly-ADP ribosylation has been correlated with a relaxed chromatin state. The number of possible histone modifications has increased with the continuous identification of novel histone PTMs, such as lysine propionylation (21, 22), butyrylation (21), crotonylation (23), succinylation, and malonylation (24), coupling cell metabolism with chromatin structure and function. It is important to keep in mind that a single histone mark is not responsible for the final effect on chromatin, but rather the combination of all marks in a chromatin region defines the biological outcome (25, 26). Besides that, there is interplay of DNA methylation, histone modifications, and nucleosome positioning, and the outcome is a result of the sum of these interactions.
The Plasticity of the Epigenetic Marks
Although presenting the same genome, each cell type in the same individual has a specific group of epigenetic marks, named epigenome. Epigenetic marks are established during embryonic development and transmitted through mitosis, stabilizing gene expression programs, and defining cell-type identities and function (27). In addition to their role in cell differentiation, these marks are fundamental to X chromosome inactivation in females and genomic imprinting during development (28, 29). Although being relatively stable over time, epigenetic marks can change dynamically in response to cellular conditions and environmental cues (2, 30, 31). Recent studies have shown that the methylation patterns determined by the binding of factors on DNA motifs are less responsive to environment within the lifetime of an individual, and that these patterns would persist across generations (32, 33). However, other DNA regions would have their epigenetic marks more susceptible to internal and external environment, such as stress, smoke, drugs, hormones, circadian rhythms, and metabolic variations caused by diet. In this way, the environment can modulate the epigenotype, and consequently the phenotype, being decisive to direct to health or disease states. In fact, many studies have shown the relation between epigenetic alterations and a wide variety of diseases, including aging-related diseases (34–36).
Changes in Epigenetic Marks During Aging
During the aging of an organism, there is a gradual decline of normal physiological functions. In humans, these include decreased immune function, chronic inflammation, sarcopenia, and most importantly increased susceptibility to diseases, such as cancer, cardiovascular disorders, and metabolic and neurodegenerative diseases. Although systemic, these phenotypes are a result of alterations in different cellular processes, such as DNA damage response, mitochondrial and proteasome function, and cell death regulation (37–40). At the molecular level, transcriptional dysregulation is observed with aging, resulting in gene expression changes (41, 42). Epigenetic alterations are important contributors to these changes in the aged transcriptome, and are known as “epigenetic drift” (43–45).
DNA Methylation
Regarding DNA methylation, a progressive global hypomethylation occurs invariably with advanced age (46). Repetitive DNA sequences normally silenced by epigenetic marks become expressed, being at least partly responsible for the well-characterized loss of heterochromatin observed during aging (47, 48). An age-dependent hypomethylation of specific gene promoters, such as IL17RC, occurs and induces their transcription (49). At the same time, some gene promoters become hypermethylated and abnormally silenced (50–52). Regarding 5hmC, it was shown that although the global level of 5hmC in the brain increases with aging both in mice (53) and humans (54), it decreases in other tissues, such as blood (55).
Besides the epigenetic drift, which is stochastic non-site-specific changes in DNA methylation that contributes to variability during aging, DNA methylation signatures in specific CpGs, both tissue-specific and present in several tissue types, were identified as associated with chronological age (56). Although age-related DNA methylation alterations are more frequent in CpG islands, tissue-specific changes occur in other genomic regions (57). In a comprehensive study of DNA methylation, Yuan and colleagues (58) showed that besides hypermethylated CpG islands, a great number of age-related differentially methylated regions fell into open sea (regions of megabase extension characterized by low CG content) or shore/shelf regions, which were found hypomethylated with age. These authors have also identified large age-associated hypomethylated blocks, similar to those described associated with cancer (59). Based on the genome-wide methylation profile of whole blood from 656 individuals spanning a wide age range, a quantitative model was built to determine the rate at which an individual’s methylome ages, and was shown to represent a strong and reproducible mean to discriminate relevant factors in aging (60).
Histone Modifications
The global DNA hypomethylation observed during aging was shown to be associated with changes in histone modification patterns (61, 62). Changes in the activity, function, and abundance of enzymes of the epigenetic machinery are present with aging (63, 64). Genes identified as hypermethylated in blood cells during aging were associated with the presence of bivalent chromatin domains in embryonic stem cells and with the repressive histone marks H3K27me3 and H3K9me3 in differentiated cells (65–67). A global loss of histones, as well as an imbalance of activating and repressive histone marks, occurs with age (68, 69). For example, a diminished content of acetylated H3K9 (70) and trimethylated H3K27 (71) was described in aged cells. Reduced levels of H3K9me3, which can be a result of the downregulation of SUV39H1/2 (72), were found with age in human and murine tissues and cells and seem to contribute to the loss of heterochromatin (72, 73). An age-decrease in the expression of HP1 (74) and DNMTs (75) could favor DNA demethylation in the heterochromatin. Another alteration that could contribute to a more opened chromatin state is the increased level of H4K16Ac with replicative age, as described in human fibroblasts in culture (76). H4K16 is among the targets of the NAD+-dependent histone deacetylase SIRT1, which is associated with aging extent and genome maintenance in different organisms (77).
While the levels of the canonical histones decrease during aging, alterations in the replication-independent incorporation of histone variants occur during aging. The replication-independent histone variant H3.3 becomes more abundant with age in general, not just in non-replicating cells, such as neurons (78). Again, this could favor a chromatin state more accessible to the transcription machinery. Another replication-independent histone variant that seems to be linked to aging is the H2A.Z, since H2A.Z knockdown fibroblasts were shown to develop premature senescence (79). The H2A variant macroH2A is characteristic of senescence-associated heterochromatin foci, heterochromatin regions over proliferation-promoting genes in senescent cells (80). An age-dependent increase in the macroH2A level was described both during replicative senescence in cultured human fibroblasts and in many tissues of aged mammals (81).
Effects of the Environmental and Lifestyle Factors on Epigenetic Changes and Aging
A classical study by Esteller’s group (82) showed significant differences in epigenetic marks in old monozygotic twin pairs compared to very young twin pairs, which presented these marks indistinguishable. More interestingly, those old twin pairs that had spent less of their lifetime together and/or had a more different natural health-medical history were those presenting the greatest differences in epigenetic marks. Studies with human population have shown that genetic factors cause no more than 20–30% of the differences observed in the lifespan of identical twins, the epigenetic drift being the main responsible for variation during the lifetime (83, 84). These and other studies (85–88) illustrate how epigenetic marks change with aging and are under the effect of environment. As a whole, these alterations change the chromatin accessibility, resulting in abnormal gene transcription and genomic instability, and have been proposed to be key regulators of the aging process, contributors to the development of age-related diseases and even predictors of the chronological age (52, 89–93). Age-related changes in multiple CpG sites across the genome were shown to accurately predict the biological age of an individual. This epigenetic clock has been shown a potential biomarker of aging in humans and associated with several aging-related disease phenotypes (60, 90, 94, 95). Epigenetic age assessed in blood was able to predict, independently of chronological age, all-cause mortality in different cohorts, including different racial/ethnic groups (93, 95–97).
It is important to emphasize that the epigenome acts as a molecular interface between the genome and the environment. In this way, the lifestyle, including diet habits, exercises, life stressors, smoke, substance abuse, chemical exposition, among others, could alter the epigenetic landscape, affecting the chromatin structure and function, and, consequently, favoring the development of aging-related disease phenotypes. Exercise and nutritional habits remodel epigenetic marks in human skeletal muscle and adipose tissue (98–100). The effect of exercise on the improved cardiorespiratory fitness and running performance, as well as on the decreased low-density lipoprotein levels, was accompanied by a widespread demethylation of CpG islands, opposed of the methylation changes observed during aging (101, 102). Several studies have demonstrated the adverse effect of smoke associated with changes in epigenetic marks. Prenatal smoke exposure affects DNA methylation of blood cells from children of smoking mothers (103). Epigenetic alterations caused by chronic cigarette smoke sensitize bronchial epithelial cells to malignant transformation (104). Tobacco smoking may induce DNA methylation alterations in cell types of both the innate and adaptive immune system (105). Offspring DNA methylation alterations were associated with maternal alcohol consumption (106). The turnover of histones and histone variants was shown to be affected by the alcohol exposure in rats (107). Many of these effects of the environment on aging involve oxidative stress, both in humans and animal models. Although severe acute or chronic stress exposure accelerates aging by favoring error accumulation due to exhausting defense mechanisms, moderate stress has shown to delay aging process by activating defense mechanisms to prevent and/or eliminate errors (108). In the last years, several studies have demonstrated the relation between cellular stress and epigenetic alterations (104, 109–114). Reactive oxygen species (ROS) lead to oxidized DNA lesions that can contribute to DNA methylation alterations. One of the major DNA oxidative damage products is 8-hydroxy-2′-deoxy-guanosine that impairs binding of DNMTs and methyl-CpG binding proteins to DNA (115). In addition, ROS may interfere with TET-mediated DNA demethylation (116). SAM availability can also be decreased by the depletion of glutathione (GSH) because of redox status, inhibiting all methylation reactions (117). Sirtuins play important role in response to a variety of stresses, such as oxidative or genotoxic stress and are crucial for cell metabolism. ROS can both induce DNA damage and SIRT1 relocation to these damage sites, for where SIRT1 recruits other epigenetic machinery components, such as DNMTs and polycomb proteins in order to silence these regions. O’Hagan and coworkers (110, 112) showed that this process could result in stable aberrant epigenetic and gene transcription changes, similarly to alterations observed in cancer. In murine embryonic mesenchymal fibroblasts, increased levels of hydrogen peroxide induce SIRT1 to relocate from repressed DNA sequences to DNA breaks to promote repair, resulting in transcriptional changes that parallel those in the aging mouse brain (118). By responding to environmental stress, sirtuins promote cell survival and, as a result, increase replicative and chronological lifespan. Although not clearly established in mammals, the association of sirtuins with aging and lifespan is suggested by the overexpression of SIRT1 in murine tissues during caloric restriction (CR) (119), the requirement of Sirt1 to the increased physical activity and extended lifespan during caloric restriction (120), and the improved health and survival of mice submitted to a high-calorie diet after resveratrol treatment, which activates Sirt1 (121). Several studies in different model organisms show the role of sirtuins in lifespan extension by CR (122–125), and evidences indicate that epigenetic mechanisms have crucial roles in this process (126, 127). In this context, new and known compounds have been tested as “CR mimetics,” including sirtuin-activating compounds, such as resveratrol (128). Compounds inhibiting histone acetylation, such as spermidine, also extend lifespan (129).
As mentioned before, ROS may modify the TET-mediated DNA demethylation (116). Both the increase in endogenous antioxidants and caloric restriction were shown to impair the increase in 5hmC levels in murine aged brains (130). The demethylase activity of TET enzymes can be stimulated by nutrients, such as ascorbic acid (131, 132). Since the activity of many epigenetic enzymes depend on intracellular levels of essential metabolites (methionine, iron, ketoglutarate, NAD+, acetyl-CoA, SAM), the cellular metabolism controls epigenetic modifications and may regulate longevity (133, 134).
In another aspect, studies in human cohorts have shown that life stressors, in special during early development, can induce lasting epigenome alterations (135–139). Stress and glucocorticoids may induce long-lasting changes in DNA methylation both at the genome-wide level and within selective gene loci, as observed both in humans and rodent models (140–142).
Epigenetic Regulation of the Immune System
An important characteristic of the immune system is its adaptive capacity to recognize self from non-self to protect the organism in response to environmental signals of different types and duration, such as potentially pathogenic agents and substances. Several immune cell populations act against potentially hazard environment by both innate and adaptive mechanisms, and their functions depend on highly controlled regulation of hematopoietic cell differentiation. Increasing number of studies have demonstrated the crucial role of epigenetic mechanisms in the development and differentiation of immune system, as well as in related pathologies (143–145). With age, the immunocompetence becomes compromised and this has been linked to the repression of immune cell differentiation genes along with the activation of autoimmunity genes because of DNA methylation alterations (49, 146–148).
Innate Immune Cells
The innate immune system, consisting of macrophages, neutrophils, dendritic cells (DCs), and natural killer (NK) cells, is the first response to pathogenic agents. Macrophages and DCs are professional antigen-presenting cells (APCs) able to capture antigens for processing and presentation to lymphocytes. When activated, resident macrophages can act directly by destroying their targets or indirectly via initiating an acute inflammatory response by producing cytokines, chemoattractants, and inflammatory mediators, and recruiting neutrophils, monocytes, and DCs (149). Activated macrophages release different factors in response to the extracellular environment, being able to acquire functionally distinct phenotypes, classic M1 and alternative M2. Activated M1 macrophages are induced by the cytokine interferon-gamma (IFN-γ) and bacterial products and have a pro-inflammatory profile, playing an important role in host defense. Differently, M2 macrophages are induced by interleukin-4 and -10 (IL-4 and IL-10) and helminthic products and have an anti-inflammatory profile, promoting tissue repair. Since mature cells of the immune system have to rapidly respond to pathogens, the contribution of epigenetic mechanisms to the regulation of genes involved in these responses has been substantially described. In this context, epigenetic mechanisms were shown to be involved in the modulation of macrophage polarization, mainly by histone marks present in enhancers of specific genes (150). The first study showing the epigenetic regulation of inflammation was that by Saccani and Natoli (151). They demonstrated the induction of inflammatory cytokines, such as IL-8 and macrophage inflammatory protein 1-alpha (MIP-1α), by the loss of H3K9 methylation at the promoter regions after exposing cultured human monocyte-derived DCs to bacterial endotoxin lipopolysaccharide (LPS). Innate immune cells have a degree of specificity by presenting pattern recognition receptors (PRRs) to recognize damage- or pathogen-associated molecular patterns in non-infectious substances or microbes, respectively (152). Recent evidences indicate that, different from previously believed, cells of innate immune system may keep a memory of past stimulations, named “trained immunity,” changing the response upon new stimuli and becoming able to respond to a larger number of microbes than the initial agent (153, 154). This immunological memory involves changes in transcriptional programs by reprogramming epigenetic marks. For example, metabolic changes in monocytes activated by β-glucan from Candida are associated with increased levels of the active histone marks, H3K4 trimethylation, and H3K27 acetylation, leading to increased production of IL-6 and TNF cytokines, inflammation, and “trained immunity” (155). Macrophages restimulated with LPS induce an attenuated inflammatory response, although maintaining an intact antimicrobial response. Foster and colleagues (156) showed that genes involved in LPS-tolerance lose the active histone marks H3K4me3 and H4Ac in their promoters during restimulation with LPS, while non-tolerizeable genes maintain these active marks after a secondary challenge with LPS, correlated with a permissive gene transcription. Epigenetic mechanisms also regulate the differentiation of human monocytes into DCs under specific stimuli. For example, the observed increased expression of CD209 during differentiation was shown to be a result of the acquisition of H3K9Ac and loss of H3K9me3, H4K20me3, and DNA methylation in its promoter (157).
T Lymphocytes
The age-dependent deterioration of the immune system, named immunesenescence, is accompanied by alterations in epigenetic marks. Kuwahara and colleagues (158) showed that CD4 T-cell senescence and cytokine homeostasis is controlled by the maintenance of histone acetylation on the Bach2 locus promoted by the binding of menin. In addition, the increased genomic instability in the thymus with age is associated with a loss of heterochromatin marks, including H3K9me3 with corresponding reduction in SUV39H1 expression (159). The senescence seems to be also activated by DNA hypomethylation since the hypomethylation is observed in senescing but not in immortalized cells (160), and the DNA methylation inhibition leads immortal cells to cell arrest (161).
Cells from the innate immune system present antigens to both B and T lymphocytes, activating them to proliferate and differentiate into effector cells. APCs activate T cell receptor and costimulatory molecules of naïve T cells, initiating T cell differentiation by the activation of the nuclear factor of activated T cells and production of interleukin-2 (IL-2). IL-2 orchestrates the molecular switch of transcriptional programs of immune-responsive genes in response to T cell activation. Naïve and resting CD4+ T cells do not express IL-2, but this cytokine is expressed in T cells under antigen stimulation. Murayama and colleagues (162) showed that demethylation of a single specific CpG site in an enhancer region is a prerequisite for IL-2 transcription and, more interestingly, that this epigenetic change constitutes a memory that CD4+ T cells encountered the antigen.
Peptide antigens are presented by APCs to T cells in the context of the major histocompatibility complex (MHC) molecules. Cytotoxic T cells, expressing CD8, recognize antigens presented by normal cells in the context of MHC class I molecules, being able to directly destroy the infected cells. Activated CD8+ T cells have increased levels of H3Ac at the IFN-γ promoter and enhancer, modification that is maintained through memory CD8+ T cells, and permits a quicker and stronger cytotoxic response to additional antigen stimulation (163). MHC class II are the MHC molecules involved in the antigen presentation to CD4+ helper T cells. The class II transactivator (CIITA) is a key factor controlling the expression of MHC-II, and both CIITA expression and CIITA-dependent MHC-II expression are epigenetically regulated (164). Analysis of chromatin accessibility in PBMCs identified memory CD8+ T cells as the subpopulation with the most profound chromatin remodeling with aging (165, 166).
After antigen recognition, depending on cytokine environment, naïve T lymphocytes differentiate into effector T “helper” (Th1, Th2, and Th17) or regulatory (Treg) CD4+ T cells, and coordinate specific immune responses by producing distinct sets of cytokines (167). The differentiation toward a Th1 profile is induced by IFN-γ, IL-12, or IL-15, whereas differentiation toward the Th2 profile—by IL-4, IL-10, or IL-13; both pathways involve the regulated expression of multiple effector genes. Transforming growth factor beta and IL-6 are responsible for inducing naïve T cell differentiation into Th17 cells. The CD4+ T cells differentiation into these different profiles is tightly regulated to assure specific cytokine signatures and changes in the epigenetic marks are fundamental to complete this process. The IFNG promoter, hypermethylated in human naïve T cells, becomes demethylated during the differentiation into Th1 profile (168). Specific histone marks were identified across the IFNG locus, where H4Ac and H3K4me3 are present in Th1 cells and H3K27me2 and H3K27me3 in Th2 cells (169). Naïve and Th1 cells present IL-4 promoter highly methylated, while Th2 cells have the intron 2 of IL-4 partly demethylated (170). Th17 cells are characterized by the expression of IL-17 cytokine and RAR-related orphan receptor C (RORC) transcription factor. The demethylation of both IL-17A and RORC loci correlates with gene expression in human Th17 cells (171), and the active histone marks H3Ac and H3K4me3 were found in the IL-17 locus (172). The demethylation of Foxp3 locus, as well the hyperacetylation of histones, was shown to be important to maintain the stable expression of forkhead box P3 (FOXP3) and stabilize the regulatory phenotype in Treg cells (173, 174).
B Lymphocytes
After binding to an antigen and be induced by T helper cells, B cells differentiate into antibody-secreting plasma cells. Antibodies bind to the specific antigen, leading to a better recognition and destruction of the pathogen (such as bacteria, virus, and tumor cells) by activating complement and/or interacting with lytic cells. During B cell differentiation, lineage-specific genes are expressed, whereas genes related to multipotent progenitors and alternative lineages are repressed. Complex epigenetic regulatory mechanisms coordinate B cell differentiation and function, including monoallelic V(D)J rearrangement and antibody diversity (175–178). A key transcriptional factor involved in B cell commitment is paired box 5 (Pax5) that, besides having its expression regulated by epigenetic mechanisms (179, 180), recruits chromatin-modifying proteins to regulate the expression of its targets. For example, CD79a gene promoter, hypermethylated in the progenitor stage, becomes demethylated during early stages of B cell differentiation, followed by the action of histone acetyltransferases recruited by Pax5, allowing gene expression (181). Pax5 can also interact with chromatin-modifying enzymes to repress genes specific for other lineages (182). V(D)J rearrangement and antibody diversity are necessary for the production of effective antibodies and require the activation-induced cytidine deaminase (AID), expressed by B cells at specific stages of differentiation. In naïve B cells, AID gene promoter is hypermethylated and the gene is not expressed. Upon B cells activation, AID gene becomes demethylated and acquires increased levels of the active histone mark H3Ac (183). The acquisition of this histone mark in active promoters and distal enhancers is also crucial for gene expression changes occurring during the differentiation of B cells to plasma cells (184). Blimp-1, a transcriptional repressor that maintains plasma cell identity, has its expression epigenetically induced and epigenetically suppresses the expression of mature B cell genes by recruiting histone modifiers (185, 186). After V(D)J rearrangement and antibody diversity processes, B cells can differentiate into memory B cells, which acquire additional epigenetic marks beyond those acquired during B cell activation (187). Different epigenetic modifications, as well as epigenetic enzymes, such as enhancer of zeste homolog 2 (188), histone acetyltransferase monocytic leukemia zinc finger protein (189), and DNMT3a (190), are observed in resting and activated B cells, and indicate that the memory B cell epigenome could favor a faster and more efficient activation than that of naïve cells.
Contribution of Epigenetic Alterations to Immune Aging
Age-associated defects are observed in all cells from the immune system, affecting their activation and cytokines production.
Innate Immune Cells
Regarding the innate immune system, many immune responses decrease during aging, but at the same time hyperreactivity of some responses are also observed (191). Epigenetic alterations seem to affect the monocyte differentiation with age, since older hematopoietic progenitor cells (HPCs) present hypomethylation of differentiation-related genes compared to progenitor cells from umbilical cord blood (192). It could be related to the reduced pluripotency and decreased potential of differentiation of HPCs from older donors (193). At the same time, older HPCs presented de novo methylation of a subset of genes associated with the Polycomb repressive complex that could contribute to the reduced phenotypic plasticity of aged stem cells (192). Indeed, epigenetic dysfunction could be a precursor to hematologic disease in elderly individuals (194). In macrophages, epigenetic mechanisms contribute to the decreased expression of MHC-II observed with age (195). Although the number of NK cells increases in older individuals, their cytotoxic activity decrease, and DNA methylation regulation of IFN-γ and IL-2 seems to contribute to this defected function of NK cells (196). Aging is well characterized by an imbalance between inflammatory and anti-inflammatory responses, where increased levels of inflammatory mediators, such as IL-6 and tumor necrosis factor-alpha (TNF-α), are observed even in the absence of acute infection or other physiologic stress (process known as “inflammaging”) (197). TNF-α has its expression increased during aging linked to its promoter demethylation (198). This epigenetic alteration contributes to the increased levels of TNF-α and also IL-1α (199, 200), which initiate the low-grade inflammation associated with resting neutrophils from aged donors. A major cause of worldwide morbidity in the elderly is the age-associated inflammatory lung disease (201). In this context, promoter hypomethylation of inflammatory genes, such as toll-like receptor 2, carnitine O-acetyltransferase, and coagulation factor III, were associated with decreased lung function (202). Zinc is a micronutrient crucial for the development and function of immune system, and its deficiency, frequently observed during aging, contributes to a wide range of immune defects (203), including an enhanced inflammatory response by inducing IL-6 promoter demethylation (204). Using the C-reactive protein (CRP) as an inflammatory biomarker, Ligthart and coworkers (205) performed a meta-analysis of epigenome wide association studies of DNA methylation on chronic low-grade inflammation. In this study, the authors demonstrated that several inflammation-related CpG sites were associated with the expression of nearby genes, and that many of these CpGs presented association with cardiometabolic phenotypes and incident coronary heart disease. Among these genes is AIM2, important in innate immune response since it takes part of host defense mechanisms against bacterial and viral pathogens, and that was found hypermethylated and expressed at low levels in samples with low levels of CRP.
T Lymphocytes
The involution of thymic structure and function, characterized by a reduced number and functional defects of thymic naïve T cells, is other process contributing to the immune aging (206). By analyzing the methylome of CD4+ T cells from newborn and centenarian individuals, Heyn and coworkers (207) showed these immune cells present the same DNA methylation changes that are observed in other tissues during aging, a global DNA hypomethylation and a higher variability of DNA methylation. Later, by an integrated analysis of transcriptome, methylome, and miRNAome in the same CD4+ T cells, Zhao and colleagues (148) found a potential relationship between gene transcription and DNA methylation for age- or immune-related genes, indicating the involvement of DNA methylation in the transcription regulation related to the development and functions of T cells in aging. Mice with a heterozygous Dnmt1 null mutation have hypomethylated DNA and showed to be phenotypically normal, but presented immune senescence and developed early autoimmunity compared with normal mice of the same age (208). By analyzing naïve CD4+ T cells from 74 healthy 19- to 66-year-old individuals, Dozmorov and colleagues (209) identified sites hypomethylated with age presenting T cell-specific enrichment in active enhancers marked with H3K27Ac and H3K4me1, suggesting a progressive age-associated shift in T-cell epigenomes toward pro-inflammatory and T cell activating phenotype that could contribute to increased autoimmunity with age. It was also shown that aged individuals, who have higher levels of autoantibodies, have T cells presenting demethylation and overexpression in the same genes demethylated and overexpressed in T cells from lupus patients (146). The progressive loss of the costimulatory molecule CD28 in CD4+ T lymphocytes during aging is associated with impaired immune response. Recently, a unique DNA methylation landscape was described in CD28null T cells, leading to the expression of inflammasome-related genes (210). Other recent study found two CpG sites present in the promoter region of KLF14, involved in CD4+ T cell differentiation via suppression of FOXP3, that exhibit stable methylation early in life and a rapid increase late in life in peripheral whole blood, monocytes, and isolated CD4+ T cells (211). Dysfunctional Treg cells have been considered to be contributors to immune senescence and increased susceptibility to age-associated diseases by suppressing T cell responses. Garg and colleagues (212) showed that the high number of Treg cells observed in aged mice is associated with hypomethylation of the upstream FoxP3 enhancer, resulting in its increased expression. They also demonstrated that Treg cells from aged mice release more IL-10, are more efficient in downregulating the costimulatory molecule CD86 on DCs, and modulate the extracellular redox environment, suppressing T cells proliferation.
Immune senescence is also characterized by a loss of naïve and central memory cells and an expansion of effector memory cells within the CD8+ T cell compartment. A shift toward more differentiated state of chromatin openness was observed in naïve and central memory cells from older individuals, as well a loss of chromatin accessibility at gene promoters mediated in part by the loss of nuclear respiratory factor 1 (NRF1) binding in aged naïve cells (213). By analyzing PBMCs methylation data set in an Italian population, Horvath and colleagues (214) showed that the centenarians are younger than expected based on their chronological age. McEwen and colleagues (215), by examining one of the highest old-age life expectancies populations from Costa Rica (Nicoyans), found this population to possess a significant higher abundance of predicted CD8+ T naïve cells and a lower abundance of estimated CD8+ T memory cells compared with non-Nicoyans, suggesting a younger immune cell profile. In addition, they showed a lower variability in the DNA methylation in Nicoyans compared with non-Nicoyans as an epigenetic characteristic of the longevity in this population.
B Lymphocytes
Considering the role of epigenetic mechanisms in B cell differentiation and function, age-associated epigenetic changes could be the responsible for the decline of humoral immunity in elderly individuals. Loss of function of B cells and their progenitors, reduction in the immunoglobulin diversity and affinity, and shifts in the proportion of naïve and antigen-experienced peripheral B cells subpopulation are characteristics of immune aging (216, 217). Hematopoietic stem cells (HSCs) lose their capacity to differentiate with age, and epigenetic alterations are important contributors to this change. Aged mice present HSCs with an aberrant gene expression profile because of epigenetic deregulation (218). Defects on both B-lineage commitment and transit through early development stage are observed during aging (219).
Conclusion and Perspectives
Considerable progress has been made in understanding epigenetic alterations involved in aging over the recent years. Most of recent knowledge about epigenetics and aging is almost lack regarding immune aging. Many gaps and questions are still open and should be deeply investigated in this area. However, there are also important challenges and limitations in this study. For example, several analyses in this field use samples contained mixed cell types, instead isolated cells, which may bring confusion in the data interpretation. The dynamic nature of the immune system per se becomes challenging to study plastic molecular alterations involved in immune responses. It remains to be determined which epigenetic changes are causally related to aging process, and how they cause immune aging. Future studies are needed to determine the overlapping epigenetic signatures between immune aging and age-related immune diseases.
Independently of these challenges, and taking into account that: (1) epigenetic mechanisms modulate chromatin states, defining gene expression profiles, (2) epigenetic mechanisms play crucial roles in the development and function of immune system, (3) a tightly regulated functioning of immune system is necessary to maintain a healthy state, (4) the environment modifies epigenetic marks throughout lifetime, and (5) epigenetic marks are potentially reversible, the knowledge about how the environment modulates the immune system by epigenetic mechanisms contributing to age-related diseases may lead to the design of novel strategies for prevention and therapeutics. Since some age-related epigenetic alterations are similar across a range of tissues (52, 220), these alterations could be also potentially used as biomarkers for aging-related disease phenotypes in biological samples, such as blood or saliva. But most importantly, considering that both intrinsic and external factors modify epigenetic marks throughout life, it is important to have in mind that healthier lifestyle may be still the most effective way to prevent diseases later in life (Figure 1) (221). In conclusion, huge efforts should be undertaken to better understand the relation among epigenetics, immune aging and age-related diseases, in order to define interventions in the lifestyle able to modulate our epigenome for a healthy aging.
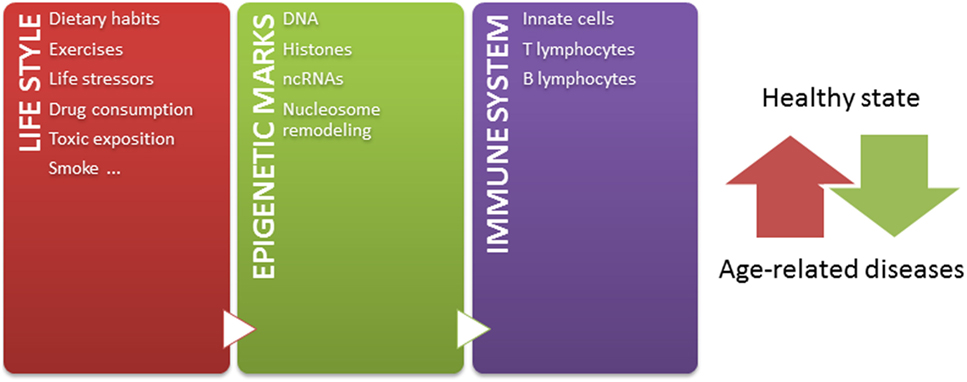
Figure 1. The epigenetic landscape, modulated by intrinsic and extrinsic factors, regulates immune system and contributes to define a healthy or pathological state. Our life style, including nutritional habit, exercises, life stressor, drugs, toxics exposition, and smoke, dynamically sculpts our epigenome along lifetime. The immune system is one of the systems in which functions are importantly regulated by epigenetic mechanisms. In this way, the accumulation of abnormal epigenetic marks in immune cells might contribute to the development of age-related diseases.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grant 2014/13663-0) and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, grant 400036/2016-9).
Abbreviations
ncRNA, noncoding RNA; 5mC, 5-methylcytosine; DNMT, DNA methyltransferase; SAM, S-adenosylmethionine; TET, ten-eleven translocation; 5hmC, 5-hydroxymethylcytosine; 5fC, 5-methylformylcytosine; 5caC, 5-caboxylcytosine; PTM, posttranslational modification; 8-OHdG, 8-hydroxy-2′-deoxy-guanosine; DC, dendritic cell; NK, natural killer; APC, antigen-presenting cell; IFN-γ, interferon-gamma; IL, interleukin; MIP-1α, macrophage inflammatory protein 1-alpha; LPS, lipopolysaccharide; PRR, pattern recognition receptor; DAMP, damage-associated molecular pattern; PAMP, pathogen-associated molecular pattern; TCR, T cell receptor; NFAT, nuclear factor of activated T cells; MHC, major histocompatibility complex; CIITA, class II transactivator; Treg cell, regulatory T cell; TGFβ, transforming growth factor β; RORC, RAR-related orphan receptor C; FOXP3, forkhead box P3; Pax5, paired box 5; AID, activation-induced cytidine deaminase; Ezh2, enhancer of zeste homolog 2; MOZ, monocytic leukemia zinc finger protein; HPC, hematopoietic progenitor cell; TNF-α, tumor necrosis factor-alpha; TLR-2, toll-like receptor 2; CRAT, carnitine O-acetyltransferase; F3, coagulation factor III; CRP, C-reactive protein; EWAS, epigenome wide association studies; HSC, hematopoietic stem cell.
References
1. Putiri EL, Robertson KD. Epigenetic mechanisms and genome stability. Clin Epigenetics (2011) 2(2):299–314. doi:10.1007/s13148-010-0017-z
2. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet (2003) 33:245–54. doi:10.1038/ng1089
3. Bestor TH. DNA methylation: evolution of a bacterial immune function into a regulator of gene expression and genome structure in higher eukaryotes. Philos Trans R Soc Lond B Biol Sci (1990) 326:179–87. doi:10.1098/rstb.1990.0002
4. Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem (2005) 74:481–514. doi:10.1146/annurev.biochem.74.010904.153721
5. Struck AW, Thompson ML, Wong LS, Micklefield J. S-adenosyl-methionine-dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. Chembiochem (2012) 13:2642–55. doi:10.1002/cbic.201200556
6. Anderson OS, Sant KE, Dolinoy DC. Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism, and DNA methylation. J Nutr Biochem (2012) 23(8):853–9. doi:10.1016/j.jnutbio.2012.03.003
7. Bird A, Wolffe AP. Methylation-induced repression – belts, braces, and chromatin. Cell (1999) 99:451–4. doi:10.1016/S0092-8674(00)81532-9
8. Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science (2001) 293:1068–70. doi:10.1126/science.1063852
9. Jones PA, Gonzalgo ML. Altered DNA methylation and genome instability: a new pathway to cancer? Proc Natl Acad Sci U S A (1997) 94(6):2103–5. doi:10.1073/pnas.94.6.2103
10. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet (2012) 13(7):484–92. doi:10.1038/nrg3230
11. Lee SM, Choi WY, Lee J. The regulatory mechanisms of intragenic DNA methylation. Epigenomics (2015) 7(4):527–31. doi:10.2217/epi.15.38
12. Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet (2009) 41:1350–3. doi:10.1038/ng.471
13. Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, et al. Dynamic changes in the human methylome during differentiation. Genome Res (2010) 20:320–31. doi:10.1101/gr.101907.109
14. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (2009) 324(5929):930–5. doi:10.1126/science.1170116
15. Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature (2010) 466(7310):1129–33. doi:10.1038/nature09303
16. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science (2011) 333(6047):1303–7. doi:10.1126/science.1210944
17. Delatte B, Deplus R, Fuks F. Playing TETris with DNA modifications. EMBO J (2014) 33:1198–211. doi:10.15252/embj.201488290
18. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet (2016) 17:487–500. doi:10.1038/nrg.2016.59
19. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res (2011) 21:381–95. doi:10.1038/cr.2011.22
20. Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell (2007) 128:707–19. doi:10.1016/j.cell.2007.01.015
21. Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, et al. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics (2007) 6:812–9. doi:10.1074/mcp.M700021-MCP200
22. Kebede AF, Nieborak A, Sharidian LZ, Gras SL, Richter F, Gómez DA, et al. Histone propionylation is a mark of active chromatin. Nat Struct Mol Biol (2017) 24(12):1048–56. doi:10.1038/nsmb.3490
23. Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell (2011) 146:1016–28. doi:10.1016/j.cell.2011.08.008
24. Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, et al. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics (2012) 11:100–7. doi:10.1074/mcp.M111.015875
25. Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet (2008) 40(7):897–903. doi:10.1038/ng.154
26. Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol (2010) 28:817–25. doi:10.1038/nbt.1662
27. Barrero MJ, Boué S, Izpisúa Belmonte JC. Epigenetic mechanisms that regulate cell identity. Cell Stem Cell (2010) 7(5):565–70. doi:10.1016/j.stem.2010.10.009
28. Kacem S, Feil R. Chromatin mechanisms in genomic imprinting. Mamm Genome (2009) 20:544–56. doi:10.1007/s00335-009-9223-4
29. Reik W, Lewis A. Co-evolution of X-chromosome inactivation and imprinting in mammals. Nat Rev Genet (2005) 6:403–10. doi:10.1038/nrg1602
30. Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet (2009) 18(21):4046–53. doi:10.1093/hmg/ddp353
31. Aguilera O, Fernández AF, Muñoz A, Fraga MF. Epigenetics and environment: a complex relationship. J Appl Physiol (2010) 10(1):243–51. doi:10.1152/japplphysiol.00068.2010
32. Teh AL, Pan H, Chen L, Ong ML, Dogra S, Wong J, et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res (2014) 24(7):1064–74. doi:10.1101/gr.171439.113
33. Whitaker JW, Chen Z, Wang W. Predicting the human epigenome from DNA motifs. Nat Methods (2015) 12(3):265–72. doi:10.1038/nmeth.3065
34. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature (2004) 429(6990):457–63. doi:10.1038/nature02625
35. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol (2010) 28(10):1057–68. doi:10.1038/nbt.1685
36. Brunet A, Berger SL. Epigenetics of aging and aging-related disease. J Gerontol (2014) 69(1):S1–20. doi:10.1093/gerona/glu042
37. Rajawat YS, Hilioti Z, Bossis J. Aging: central role for autophagy and the lysosomal degradative system. Aging Res Rev (2009) 8:199–213. doi:10.1016/j.arr.2009.05.001
38. Freitas AA, de Magalhães JP. A review and appraisal of the DNA damage theory of aging. Mutat Res (2011) 728:12–22. doi:10.1016/j.mrrev.2011.05.001
39. Saez I, Vilchez D. The mechanistic links between proteasome activity, aging and age-related diseases. Curr Genomics (2014) 15(1):38–51. doi:10.2174/138920291501140306113344
40. Payne BAI, Chinnery PF. Mitochondrial dysfunction in aging: much progress but many unresolved questions. Biochim Biophys Acta (2015) 1847(11):1347–53. doi:10.1016/j.bbabio.2015.05.022
41. Tan Q, Christensen K, Christiansen L, Frederiksen H, Bathum L, Dahlgaard J, et al. Genetic dissection of gene expression observed in whole blood samples of elderly Danish twins. Hum Genet (2005) 117(2–3):267–74. doi:10.1007/s00439-005-1308-x
42. Zahn JM, Sonu R, Vogel H, Crane E, Mazan-Mamczarz K, Rabkin R, et al. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet (2006) 2(7):e115. doi:10.1371/journal.pgen.0020115
43. Feser J, Tyler J. Chromatin structure as a mediator of aging. FEBS Lett (2011) 585:2041–8. doi:10.1016/j.febslet.2010.11.016
44. Tsurumi A, Li WX. Global heterochromatin loss: a unifying theory of aging? Epigenetics (2012) 7:680–8. doi:10.4161/epi.20540
45. Moskalev AA, Aliper AM, Smit-McBride Z, Buzdin A, Zhavoronkov A. Genetics and epigenetics of aging and longevity. Cell Cycle (2014) 13:1063–77. doi:10.4161/cc.28433
46. Wilson VL, Smith RA, Ma S, Cutler RG. Genomic 5-methyldeoxycytidine decreases with age. J Biol Chem (1987) 262:9948–51.
47. Bolati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, et al. Decline in genomic DNA methylation through aging in a cohort of eldery subjects. Mech Ageing Dev (2009) 130:234–9. doi:10.1016/j.mad.2008.12.003
48. Barbot W, Dupressoir A, Lazar V, Heidmann T. Epigenetic regulation of an IAP retrotransposon in the aging mouse: progressive demethylation and de-silencing of the element by its repetitive induction. Nucleic Acids Res (2012) 30:2365–73. doi:10.1093/nar/30.11.2365
49. Wei L, Liu B, Tuo J, Shen D, Chen P, Li Z, et al. Hypomethylation of the IL17RC promoter associates with age-related macular degeneration. Cell Rep (2012) 2(5):1151–8. doi:10.1016/j.celrep.2012.10.013
50. Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell (2003) 113:703–16. doi:10.1016/S0092-8674(03)00401-X
51. Salpea P, Russanova VR, Hirai TH, Sourlingas TG, Sekeri-Pataryas KE, Romero R, et al. Postnatal development- and age-related changes in DNA-methylation patterns in the human genome. Nucleic Acids Res (2012) 40:6477–94. doi:10.1093/nar/gks312
52. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol (2013) 14:R115. doi:10.1186/gb-2013-14-10-r115
53. Chen H, Dzitoyeva S, Manev H. Effect of aging on 5-hydroxymethylcytosine in the mouse hippocampus. Restorat Neurol Neurosci (2012) 30:237–45. doi:10.3233/RNN-2012-110223
54. Wagner M, Steinbacher J, Kraus TF, Michalakis S, Hackner B, Pfaffeneder T, et al. Age-dependent levels of 5-methyl-, 5-hydroxymethyl-, and 5-formylcytosine in human and mouse brain tissues. Angew Chem Int Ed Engl (2015) 54:12511–4. doi:10.1002/anie.201502722
55. Xiong J, Jiang HP, Peng CY, Deng QY, Lan MD, Zeng H, et al. DNA hydroxymethylation age of human blood determined by capillary hydrophilic-interaction liquid chromatography/mass spectrometry. Clin Epigenetics (2015) 7:72. doi:10.1186/s13148-015-0109-x
56. Farré P, Jones MJ, Meaney MJ, Emberly E, Turecki G, Kobor MS. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin (2015) 8:19. doi:10.1186/s13072-015-0011-y
57. Weidner CI, Wagner W. The epigenetics tracks of aging. Biol Chem (2014) 395:1307–14. doi:10.1515/hsz-2014-0180
58. Yuan T, Jiao Y, Jong S, Ophoff RA, Beck S, Teschendorff AE. An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLoS Genet (2015) 11(2):e1004996. doi:10.1371/journal.pgen.1004996
59. Timp W, Bravo HC, McDonald OG, Goggins M, Umbricht C, Zeiger M, et al. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med (2014) 6(8):61. doi:10.1186/s13073-014-0061-y
60. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell (2013) 49(2):359–67. doi:10.1016/j.molcel.2012.10.016
61. Xu Z, Taylor JA. Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer. Carcinogenesis (2014) 35:356–64. doi:10.1093/carcin/bgt391
62. Sidler C, Kovalchuk O, Kovalchuk I. Epigenetic regulation of cellular senescence and aging. Front Genet (2017) 8:138. doi:10.3389/fgene.2017.00138
63. Pogribny IP, Vanyushin BF. Age-related genomic hypomethylation. In: Tollefsbol TO, editor. Epigenetics of Aging. New York: Springer (2010). p. 11–26.
64. McCauley BS, Dang W. Histone methylation and aging: lessons learned from model systems. Biochim Biophys Acta (2014) 1839(12):1454–62. doi:10.1016/j.bbagrm.2014.05.008
65. Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res (2010) 20:434–9. doi:10.1101/gr.103101.109
66. Fernández AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H, et al. A DNA methylation fingerprint of 1628 human samples. Genome Res (2012) 22:407–19. doi:10.1101/gr.119867.110
67. Fernández AF, Bayón GF, Urdinguio RG, Toraño EG, García MG, Carella A, et al. H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells. Genome Res (2015) 25(1):27–40. doi:10.1101/gr.169011.113
68. Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature (2009) 459:802–7. doi:10.1038/nature08085
69. Sen P, Shah PP, Nativio R, Berger SL. Epigenetic mechanisms of longevity and aging. Cell (2016) 166(4):822–39. doi:10.1016/j.cell.2016.07.050
70. Kawakami K, Nakamura A, Ishigami A, Goto S, Takahashi R. Age-related difference of site-specific histone modifications in rat liver. Biogerontology (2009) 10(4):415–21. doi:10.1007/s10522-008-9176-0
71. Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. The polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev (2007) 21(5):525–30. doi:10.1101/gad.415507
72. Djeghloul D, Kuranda K, Kuzniak I, Barbieri D, Naguibneva I, Choisy C, et al. Age-associated decrease of the histone methyltransferase SUV39H1 in HSC perturbs heterochromatin and B lymphoid differentiation. Stem Cell Reports (2016) 6:970–84. doi:10.1016/j.stemcr.2016.05.007
73. Sidler C, Woycicki R, Li D, Wang B, Kovalchuk I, Kovalchuk O. A role for SUV39H1-mediated H3K9 trimethylation in the control of genome stability and senescence in WI38 human diploid lung fibroblasts. Aging (2014) 6:545–63. doi:10.18632/aging.100678
74. Zhang W, Li J, Suzuki K, Qu J, Wang P, Zhou J, et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science (2015) 348:1160–3. doi:10.1126/science.aaa1356
75. Casillas MA Jr, Lopatina N, Andrews LG, Tollefsbol TO. Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Mol Cell Biochem (2003) 252:33–43. doi:10.1023/A:1025548623524
76. O’Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol (2010) 17(10):1218–25. doi:10.1038/nsmb.1897
77. Grabowska W, Sikora E, Bielak-Zmijewska A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology (2017) 18(4):447–76. doi:10.1007/s10522-017-9685-9
78. Urban MK, Zweidler A. Changes in nucleosomal core histone variants during chicken development and maturation. Dev Biol (1983) 95(2):421–8. doi:10.1016/0012-1606(83)90043-X
79. Gevry N, Chan HM, Laflamme L, Livingston DM, Gaudreau L. p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev (2007) 21:1869–81. doi:10.1101/gad.1545707
80. Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell (2005) 8(1):19–30. doi:10.1016/j.devcel.2004.10.019
81. Kreiling JA, Tamamori-Adachi M, Sexton AN, Jeyapalan JC, Munoz-Najar U, Peterson AL, et al. Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell (2011) 10(2):292–304. doi:10.1111/j.1474-9726.2010.00666.x
82. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A (2005) 102(30):10604–9. doi:10.1073/pnas.0500398102
83. Herskind AM, McGue M, Holm NV, Sørensen TI, Harvald B, Vaupel JW. The heritability of human longevity: a population-based study of 2872 Danish twin pairs born 1870-1900. Hum Genet (1996) 97(3):319–23. doi:10.1007/BF02185763
84. Poulsen P, Esteller M, Vaag A, Fraga MF. The epigenetic basis of twin discordance in age-related diseases. Pediatr Res (2007) 61(5 Pt 2):38R–42R. doi:10.1203/pdr.0b013e31803c7b98
85. Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet (2009) 41(2):240–5. doi:10.1038/ng.286
86. Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res (2010) 20(2):170–9. doi:10.1101/gr.100289.109
88. Bell JT, Tsai PC, Yang TP, Pidsley R, Nisber J, Glass D, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet (2012) 8:e1002629. doi:10.1371/journal.pgen.1002629
89. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194–217. doi:10.1016/j.cell.2013.05.039
90. Bocklandt S, Lin W, Sehl ME, Sanchez FJ, Sinsheimer JS, Horvath S, et al. Epigenetic predictor of age. PLoS One (2011) 6:e14821. doi:10.1371/journal.pone.0014821
91. O’Sullivan RJ, Karlseder J. The great unravelling: chromatin as a modulator of the aging process. Trends Biochem Sci (2012) 37(11):466–76. doi:10.1016/j.tibs.2012.08.001
92. Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, et al. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol (2013) 15:1495–506. doi:10.1038/ncb2879
93. Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol (2015) 16:25. doi:10.1186/s13059-015-0584-6
94. Lin Q, Weidner CI, Costa IG, Marioni RE, Ferreira MR, Deary IJ, et al. DNA methylation levels at individual age-associated CpG sites can be indicative for life expectancy. Aging (2016) 8:394–401. doi:10.18632/aging.100908
95. Chen BH, Marioni RE, Colicino E, Peters MJ, Ward-Caviness CK, Tsai PC, et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (2016) 8(9):1844–65. doi:10.18632/aging.101020
96. Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, et al. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell (2016) 15:149–54. doi:10.1111/acel.12421
97. Perna L, Zhang Y, Mons U, Holleczek B, Saum KU, Brenner H. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin Epigenetics (2016) 8:64. doi:10.1186/s13148-016-0228-z
98. Barrès R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab (2012) 15(3):405–11. doi:10.1016/j.cmet.2012.01.001
99. Barrès R, Kirchner H, Rasmussen M, Yan J, Kantor FR, Krook A, et al. Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep (2013) 3(4):1020–7. doi:10.1016/j.celrep.2013.03.018
100. Rönn T, Volkov P, Davegårdh C, Dayeh T, Hall E, Olsson AH, et al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. PLoS Genet (2013) 9(6):e1003572. doi:10.1371/journal.pgen.1003572
101. Denham J, O’Brien BJ, Harvey JT, Charchar FJ. Genome-wide sperm DNA methylation changes after 3 months of exercise training in humans. Epigenomics (2015) 7(5):717–31. doi:10.2217/epi.15.29
102. Maejima H, Kanemura N, Kokubun T, Murata K, Takayanagi K. Exercise enhances cognitive function and neurotrophin expression in the hippocampus accompanied by changes in epigenetic programming in senescence-accelerated mice. Neurosci Lett (2017) 665:67–73. doi:10.1016/j.neulet.2017.11.023
103. Meyer KF, Verkaik-Schakel RN, Timens W, Kobzik L, Plösch T, Hylkema MN. The fetal programming effect of prenatal smoking on Igf1r and Igf1 methylation is organ- and sex-specific. Epigenetics (2017) 21:1–49. doi:10.1080/15592294.2017.1403691
104. Vaz M, Hwang SY, Kagiampakis I, Phallen J, Patil A, O’Hagan HM, et al. Chronic cigarette smoke-induced epigenomic changes precede sensitization of bronchial epithelial cells to single-step transformation by KRAS mutations. Cancer Cell (2017) 32(3):360–76. doi:10.1016/j.ccell.2017.08.006
105. Bauer M, Fink B, Thürmann L, Eszlinger M, Herberth G, Lehmann I. Tobacco smoking differently influences cell types of the innate and adaptive immune system-indications from CpG site methylation. Clin Epigenetics (2016) 7:83. doi:10.1186/s13148-016-0249-7
106. Sharp GC, Arathimos R, Reese SE, Page CM, Felix J, Küpers LK, et al. Maternal alcohol consumption and offspring DNA methylation: findings from six general population-based birth cohorts. Epigenomics (2018) 10(1):27–42. doi:10.2217/epi-2017-0095
107. Rachdaoui N, Li L, Willard B, Kasumov T, Previs S, Sarkar D. Turnover of histones and histone variants in postnatal rat brain: effects of alcohol exposure. Clin Epigenetics (2017) 9:117. doi:10.1186/s13148-017-0416-5
108. Gems D, Partridge L. Stress-response hormesis and aging: “that which does not kill us makes us stronger”. Cell Metab (2008) 7:200–3. doi:10.1016/j.cmet.2008.01.001
109. Campos AC, Molognoni F, Melo FH, Galdieri LC, Carneiro CR, D’Almeida V, et al. Oxidative stress modulates DNA methylation during melanocyte anchorage blockade associated with malignant transformation. Neoplasia (2007) 9(12):1111–21. doi:10.1593/neo.07712
110. O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet (2008) 4(8):e1000155. doi:10.1371/journal.pgen.1000155
111. Molognoni F, Cruz AT, Meliso FM, Morais AS, Souza CF, Xander P, et al. Epigenetic reprogramming as a key contributor to melanocyte malignant transformation. Epigenetics (2011) 6(4):450–64. doi:10.4161/epi.6.4.14917
112. O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell (2011) 20(5):606–19. doi:10.1016/j.ccr.2011.09.012
113. Molognoni F, de Melo FH, da Silva CT, Jasiulionis MG. Ras and Rac1, frequently mutated in melanomas, are activated by superoxide anion, modulate Dnmt1 level and are causally related to melanocyte malignant transformation. PLoS One (2013) 8(12):e81937. doi:10.1371/journal.pone.0081937
114. Meliso FM, Micali D, Silva CT, Sabedot TS, Jasiulionis MG. SIRT1 regulates Mxd1 during malignant melanoma progression. Oncotarget (2017) 8(70):114540–53. doi:10.18632/oncotarget.21457
115. Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res (2007) 67(3):946–50. doi:10.1158/0008-5472.CAN-06-3123
116. Chia N, Wang L, Lu X, Senut MC, Brenner C, Ruden DM. Hypothesis: environmental regulation of 5-hydroxymethylcytosine by oxidative stress. Epigenetics (2011) 6(7):853–6. doi:10.4161/epi.6.7.16461
117. Hitchler MU, Domann FE. Redox regulation of the epigenetic landscape in cancer: a role for metabolic reprogramming in remodeling the epigenome. Free Radic Biol Med (2012) 53(11):2178–87. doi:10.1016/j.freeradbiomed.2012.09.028
118. Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell (2008) 135(5):907–18. doi:10.1016/j.cell.2008.10.025
119. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. Caloric restriction promotes mammalian cell survival by inducing the Sirt1 deacetylase. Science (2004) 305(5682):390–2. doi:10.1126/science.1099196
120. Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science (2005) 310(5754):1641. doi:10.1126/science.1118357
121. Bauer JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature (2006) 444(7117):337–42. doi:10.1038/nature05354
122. Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, et al. Caloric restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature (2002) 418(6895):344–8. doi:10.1038/nature00829
123. Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature (2003) 425(6954):191–6. doi:10.1038/nature01960
124. Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature (2004) 430(7000):686–9. doi:10.1038/nature02789
125. Lee C, Longo V. Dietary restriction with and without caloric restriction for healthy aging. F1000Res (2016) 5:F1000. doi:10.12688/f1000research.7136.1
126. Vaquero A, Reinberg D. Calorie restriction and the exercise of chromatin. Genes Dev (2009) 23(16):1849–69. doi:10.1101/gad.1807009
127. Li Y, Daniel M, Tollefsbol TO. Epigenetic regulation of caloric restriction in aging. BMC Med (2011) 25(9):98. doi:10.1186/1741-7015-9-98
128. Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci (2014) 35(3):146–54. doi:10.1016/j.tips.2013.12.004
129. Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol (2009) 11(11):1305–14. doi:10.1038/ncb1975
130. Chouliaras L, van den Hove DL, Kenis G, Keitel S, Hof PR, van Os J, et al. Age-related increase in levels of 5-hydroxymethylcytosine in mouse hippocampus is prevented by caloric restriction. Curr Alzheimer Res (2012) 9(5):536–44. doi:10.2174/156720512800618035
131. Yin R, Mao SQ, Zhao B, Chong Z, Yang Y, Zhao C, et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc (2013) 135(28):10396–403. doi:10.1021/ja4028346
132. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, Mahapatra S, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature (2013) 500(7461):222–6. doi:10.1038/nature12362
133. Salminen A, Kauppinen A, Hiltunen M, Kaarniranta K. Krebs cycle intermediates regulate DNA and histone methylation: epigenetic impact on the aging process. Ageing Res Rev (2014) 16:45–65. doi:10.1016/j.arr.2014.05.004
134. Sharma U, Rando OJ. Metabolic input into the epigenome. Cell Metab (2017) 25:544–58. doi:10.1016/j.cmet.2017.02.003
135. McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci (2009) 12:342–8. doi:10.1038/nn.2270
136. Perroud N, Paoloni-Giacobino A, Prada P, Olie E, Salzmann A, Nicastro R, et al. Increased methylation of glucocorticoid receptor gene (NR3C1) in adults with a history of childhood maltreatment: a link with the severity and type of trauma. Transl Psychiatry (2011) 1:e59. doi:10.1038/tp.2011.60
137. Klengel T, Mehta KB, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci (2013) 16:33–41. doi:10.1038/nn.3275
138. Non AL, Binder AM, Kubzansky LD, Michels KB. Genome-wide DNA methylation in neonates exposed to maternal depression, anxiety, or SSRI medication during pregnancy. Epigenetics (2014) 9:964–72. doi:10.4161/epi.28853
139. Yehuda R, Daskalakis NP, Bierer LM, Bader HN, Klengel T, Holsboer F, et al. Holocaust exposure induced intergenerational effects on FKBP5 methylation. Biol Psychiatry (2015) 80(5):372–80. doi:10.1016/j.biopsych.2015.08.005
140. Weaver IC, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A (2006) 103:3480–5. doi:10.1073/pnas.0507526103
141. Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmuhl Y, Fischer D, et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci (2009) 12:1559–66. doi:10.1038/nn.2436
142. Schmauss C, Lee-McDermott Z, Medina LR. Trans-generational effects of early life stress: the role of maternal behavior. Sci Rep (2014) 4:4873. doi:10.1038/srep04873
143. Fernández-Morera JL, Calvanese V, Rodríguez-Rodero S, Menéndez-Torre E, Fraga MF. Epigenetic regulation of the immune system in health and disease. Tissue Antigens (2010) 76:431–9. doi:10.1111/j.1399-0039.2010.01587.x
144. Rodriguez-Cortez VC, Hernando H, de la Rica L, Vento R, Ballestar E. Epigenomic deregulation in the immune system. Epigenomics (2011) 3:697–713. doi:10.2217/epi.11.99
145. Busslinger M, Tarakhovsky A. Epigenetic control of immunity. Cold Spring Harb Perspect Biol (2014) 6:a019307. doi:10.1101/cshperspect.a019307
146. Zhang Z, Deng C, Lu Q, Richardson B. Age-dependent DNA methylation changes in the ITGAL (CD11a) promoter. Mech Ageing Dev (2002) 123(9):1257–68. doi:10.1016/S0047-6374(02)00014-3
147. Marttila S, Kananen L, Hayrynen S, Jylhava J, Nevalainen T, Hervonen A, et al. Ageing-associated changes in the human DNA methylome: genomic locations and effects on gene expression. BMC Genomics (2015) 16:179. doi:10.1186/s12864-015-1381-z
148. Zhao M, Qin J, Yin H, Tan Y, Liao W, Liu Q, et al. Distinct epigenomes in CD4+ T cells of newborns, middle-ages and centenarians. Sci Rep (2016) 6:38411. doi:10.1038/srep38411
149. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11:762–74. doi:10.1038/nri3070
150. van den Bossche J, Neele AE, Hoeksema MA, Winther MP. Macropaheg polarization: the epigenetic point of view. Curr Opin Lipidol (2014) 25(5):367–73. doi:10.1097/MOL.0000000000000109
151. Saccani S, Natoli G. Dynamic changes in histone H3 Lys9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev (2002) 16:2219–24. doi:10.1101/gad.232502
152. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124(4):783–801. doi:10.1016/j.cell.2006.02.015
153. Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe (2011) 9(5):355–61. doi:10.1016/j.chom.2011.04.006
154. Crisan TO, Netea MG, Joosten LAB. Innate immune memory: implications for host responses to damage-associated molecular patterns. Eur J Immunol (2016) 46:817–28. doi:10.1002/eji.201545497
155. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A (2012) 109:17537–42. doi:10.1073/pnas.1202870109
156. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature (2007) 447:972–8. doi:10.1038/nature05836
157. Bullwinkel J, Ludermann A, Debarry J, Singh PB. Epigenotype switching at the CD14 and CD209 genes during differentiation of human monocytes to dendritic cells. Epigenetics (2011) 6:45–51. doi:10.4161/epi.6.1.13314
158. Kuwahara M, Suzuki J, Tofukuji S, Yamada T, Kanoh M, Matsumoto A, et al. The Menin-Bach2 axis is critical for regulating CD4 T-cell senescence and cytokine homeostasis. Nat Commun (2014) 5:3555. doi:10.1038/ncomms4555
159. Sidler C, Woycicki R, Ilnytskyy Y, Metz G, Kovalchuk I, Kovalchuk O. Immunosenescence is associated with altered gene expression and epigenetic regulation in primary and secondary immune organs. Front Genet (2013) 4:211. doi:10.3389/fgene.2013.00211
160. Degerman S, Landfors M, Siwicki JK, Revie J, Borssen M, Evelonn E, et al. Immortalization of T-cells is accompanied by gradual changes in CpG methylation resulting in a profile resembling a subset of T-cell leukemias. Neoplasia (2014) 16:606–15. doi:10.1016/j.neo.2014.07.001
161. Vogt M, Haggblom C, Yeargin J, Christiansen-Weber T, Haas M. Independent induction of senescence by p16INK4a and p21CIP1 in spontaneously immortalized human fibroblasts. Cell Growth Differ (1998) 9:139–46.
162. Murayama A, Sakura K, Nakama M, Yasuzawa-Tanaka K, Fujita E, Tateishi Y, et al. A specific CpG site demethylation in the human interleukin 2 gene promoter is an epigenetic memory. EMBO J (2006) 25:1081–92. doi:10.1038/sj.emboj.7601012
163. Northrop JK, Thomas RM, Wells AD, Shen H. Epigenetic remodeling of the IL-2 and IFN-γ loci in memory CD8 T cells is influenced by CD4 T cells. J Immunol (2006) 177:1062–9. doi:10.4049/jimmunol.177.2.1062
164. Wright KL, Ting JP. Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol (2006) 27:405–12. doi:10.1016/j.it.2006.07.007
165. Ucar D, Márquez EJ, Chung CH, Marches R, Rossi RJ, Uyar A, et al. The chromatin accessibility signature of human immune aging stems from CD8+ T cells. J Exp Med (2017) 214(10):3123–44. doi:10.1084/jem.20170416
166. Tserel L, Kolde R, Limbach M, Tretyakov K, Kasela S, Kisand K, et al. Age-related profiling of DNA methylation in CD8+ T cells reveals changes in immune response and transcriptional regulator genes. Sci Rep (2015) 5:13107. doi:10.1038/srep13107
167. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood (2008) 112:1557–69. doi:10.1182/blood-2008-05-078154
168. Schoenborn JR, Dorschner MO, Sekimata M, Santer DM, Shnyreva M, Fitzpatrick DR, et al. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat Immunol (2007) 8:732–42. doi:10.1038/ni0807-893b
169. Aune TM, Collins PL, Chang S. Epigenetics and T helper 1 differentiation. Immunology (2009) 126:299–305. doi:10.1111/j.1365-2567.2008.03026.x
170. Santangelo S, Cousins DJ, Winkelmann N, Triantaphyllopoulos K, Staynov DZ. Chromatin structure and DNa methylation of the IL4 gene in human T(H)2 cells. Chromosome Res (2009) 17:485–96. doi:10.1007/s10577-009-9040-3
171. Cohen CJ, Crome SQ, MacDonald KG, Dai EL, Mager DL, Levings MK. Human Th1 and Th17 exhibit stability at signature cytokine and transcription factor loci. J Immunol (2011) 187:5615–26. doi:10.4049/jimmunol.1101058
172. Akimzhanov AM, Yang XO, Dong C. Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem (2007) 282(9):5969–72. doi:10.1074/jbc.C600322200
173. Baron U, Floess S, Wieczorek G, Baumann K, Grützkau A, Dong J, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol (2007) 37(9):2378–89. doi:10.1002/eji.200737594
174. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood (2010) 115(5):965–74. doi:10.1182/blood-2009-02-207118
175. Parra M. Epigenetic events during B lymphocyte development. Epigenetics (2009) 4(7):462–8. doi:10.4161/epi.4.7.10052
176. Lee ST, Xiao Y, Muench MO, Xiao J, Fomin ME, Wiencke JK, et al. A global DNA methylation and gene expression analysis of early human B-cell development reveals a demethylation signature and transcription factor network. Nucleic Acids Res (2012) 40(22):11339–51. doi:10.1093/nar/gks957
177. Zan H, Casali P. Epigenetics of peripheral B-cell differentiation and the antibody response. Front Immunol (2015) 6:631. doi:10.3389/fimmu.2015.00631
178. Bao Y, Cao X. Epigenetic control of B cell development and B-cell-related immune disorders. Clin Rev Allergy Immunol (2016) 50(3):301–11. doi:10.1007/s12016-015-8494-7
179. Oguro H, Yuan J, Ichikawa H, Ikawa T, Yamazaki S, Kawamoto H, et al. Poised lineage specification in multipotential hematopoietic stem and progenitor cells by the polycomb protein Bmi1. Cell Stem Cell (2010) 6(3):279–86. doi:10.1016/j.stem.2010.01.005
180. McManus S, Ebert A, Salvagiotto G, Medvedovic J, Sun Q, Tamir I, et al. The transcription factor Pax5 regulates its target genes by recruiting chromatin-modifying proteins in committed B cells. EMBO J (2011) 30(12):2388–404. doi:10.1038/emboj.2011.140
181. Maier H, Ostraat R, Gao H, Fields S, Shinton SA, Medina KL, et al. Early B cell factor cooperates with Runx1 and mediates epigenetic changes associated with mb-1 transcription. Nat Immunol (2004) 5(10):1069–77. doi:10.1038/ni1119
182. Linderson Y, Eberhard D, Malin S, Johansson A, Busslinger M, Pettersson S. Corecruitment of the Grg4 repressor by PU.1 is critical for Pax5-mediated repression of B-cell-specific genes. EMBO Rep (2004) 5(3):291–6. doi:10.1038/sj.embor.7400089
183. Crouch EE, Li Z, Takizawa M, Fichtner-Feigl S, Gourzi P, Montano C, et al. Regulation of AID expression in the immune response. J Exp Med (2007) 204:1145–56. doi:10.1084/jem.20061952
184. Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, et al. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol (2015) 16:663–73. doi:10.1038/ni.3154
185. Yu J, Angelin-Duclos C, Greenwood J, Liao J, Calame K. Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol Cell Biol (2000) 20:2592–603. doi:10.1128/MCB.20.7.2592-2603.2000
186. Su ST, Ying HY, Chiu YK, Lin FR, Chen MY, Lin KI. Involvement of histone demethylase LSD1 in Blimp-1-mediated gene repression during plasma cell differentiation. Mol Cell Biol (2009) 29:1421–31. doi:10.1128/MCB.01158-08
187. Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol (2015) 15:149–59. doi:10.1038/nri3802
188. Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest (2013) 123:5009–22. doi:10.1172/JCI70626
189. Good-Jacobson KL. Regulation of germinal center, B-cell memory, and plasma cell formation by histone modifiers. Front Immunol (2014) 5:596. doi:10.3389/fimmu.2014.00596
190. Luckey CJ, Bhattacharya D, Goldrath AW, Weissman IL, Benoist C, Mathis D. Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc Natl Acad Sci U S A (2006) 103:3304–9. doi:10.1073/pnas.0511137103
191. Panda A, Arjona A, Sapey E, Bai F, Fikrig E, Montgomery RR, et al. Human innate immunosenescence: causes and consequences for immunity in old age. Trends Immunol (2009) 30:325–33. doi:10.1016/j.it.2009.05.004
192. Bocker MT, Hellwig I, Breiling A, Eckstein V, Ho AD, Lyko F. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood (2011) 117:e182–9. doi:10.1182/blood-2011-01-331926
193. Beerman I, Maloney WJ, Weissmann IL, Rossi DJ. Stem cells and the aging hematopoietic system. Curr Opin Immunol (2010) 22(4):500–6. doi:10.1016/j.coi.2010.06.007
194. Kramer A, Challen GA. The epigenetic basis of hematopoietic stem cell aging. Sem Hematol (2017) 54:19–24. doi:10.1053/j.seminhematol.2016.10.006
195. Chang YC, Chen TC, Lee CT, Yang CY, Wang HW, Wang CC, et al. Epigenetic control of MHC class II expression in tumor-associated macrophages by decoy receptor 3. Blood (2008) 111(10):5054–63. doi:10.1182/blood-2007-12-130609
196. Kutza J, Murasko DM. Effects of aging on natural killer cell activity and activation by interleukin-2 and IFN-alpha. Cell Immunol (1994) 155(1):195–204. doi:10.1006/cimm.1994.1112
197. Baylis D, Bartlett DB, Patel HP, Roberts HC. Understanding how we age: insights into inflammaging. Longev Healthspan (2013) 2(1):8. doi:10.1186/2046-2395-2-8
198. Gowers IR, Walters K, Kiss-Toth E, Read RC, Duff GW, Wilson AG. Age-related loss of CpG methylation in the tumour necrosis factor promoter. Cytokine (2011) 56(3):792–7. doi:10.1016/j.cyto.2011.09.009
199. Sullivan KE, Reddy AB, Dietzmann K, Suriano AR, Kocieda VP, Stewart M, et al. Epigenetic regulation of tumor necrosis factor alpha. Mol Cell Biol (2007) 27(14):5147–60. doi:10.1128/MCB.02429-06
200. van Rietschoten JG, Gal-Yam EN, Jeong S, Cortez CC, Verweij CL, Jones PA. Epigenetic regulation and nucleosome positioning in the human TATA-less IL-1 alpha promoter. Genes Immun (2008) 9(7):582–90. doi:10.1038/gene.2008.53
201. Budinger GRS, Kohanski RA, Gan W, Kobor MS, Amaral LA, Armanios M, et al. The intersection of aging biology and the pathobiology of lung diseases: a joint NHLBI/NIA workshop. J Gerontol A Biol Sci Med Sci (2017) 72(11):1492–500. doi:10.1093/gerona/glx090
202. Lepeule J, Baccarelli A, Motta V, Cantone L, Litonjua AA, Sparrow D, et al. Gene promoter methylation is associated with lung function in the elderly: the Normative Aging study. Epigenetics (2012) 7(3):261–9. doi:10.4161/epi.7.3.19216
203. Haase H, Rink L. The immune system and the impact of zinc during aging. Immun Ageing (2009) 6:9. doi:10.1186/1742-4933-6-9
204. Wong CP, Rinaldi NA, Ho E. Zinc deficiency enahnced inflammatory response by increasing imune cell activation and inducing IL6 promoter demethylation. Mol Nutr Food Res (2015) 59(5):991–9. doi:10.1002/mnfr.201400761
205. Ligthart S, Marzi C, Aslibekyan S, Mendelson MM, Conneely KN, Tanaka T, et al. DNA methylation signatures of chronic low-grade inflammation are associated with complex diseases. Genome Biol (2016) 17(1):255. doi:10.1186/s13059-016-1119-5
206. Aspinall R, Andrew D. Thymic involution in aging. J Clin Immunol (2000) 20:250–6. doi:10.1023/A:1006611518223
207. Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, et al. Distinct DNA methylation of newborn and centenarians. Proc Natl Acad Sci U S A (2012) 109:10522–7. doi:10.1073/pnas.1120658109
208. Yung R, Ray D, Eisenbraun JK, Deng C, Attwood J, Eisenbraun MD, et al. Unexpected effects of a heterozygous dnmt1 null mutation on age-dependent DNA hypomethylation and autoimmunity. J Gerontol A Biol Sci Med Sci (2001) 56(6):B268–76. doi:10.1093/gerona/56.6.B268
209. Dozmorov MG, Coit P, Maksimowicz-McKinnon K, Sawalha AH. Age-associated DNA methylation changes in naive CD4+ T cells suggest an evolving autoimmune epigenotype in aging T cells. Epigenomics (2017) 9(4):429–45. doi:10.2217/epi-2016-0143
210. Suarez-Álvarez B, Rodríguez RM, Schlangen K, Raneros AB, Márquez-Kisinousky L, Fernández AF, et al. Phenotypic characteristics of aged CD4+ CD28null T lymphocytes are determined by changes in the whole-genome DNA methylation pattern. Aging Cell (2017) 16(2):293–303. doi:10.1111/acel.12552
211. Johnson ND, Wiener HW, Smith AK, Nishitani S, Absher DM, Arnett DK, et al. Non-linear patterns in age-related DNA methylation may reflect CD4+ T cell differentiation. Epigenetics (2017) 12(6):492–503. doi:10.1080/15592294.2017.1314419
212. Garg SK, Delaney C, Toubai T, Ghosh A, Reddy P, Banerjee R, et al. Aging is associated with increased regulatory T-cell function. Aging Cell (2014) 13(3):441–8. doi:10.1111/acel.12191
213. Moskowitz DM, Zhang DW, Hu B, Le Saux S, Yanes RE, Ye Z, et al. Epigenomics of human CD8 T cell differentiation and aging. Sci Immunol (2017) 2(8):eaag0192. doi:10.1126/sciimmunol.aag0192
214. Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delledonne M, et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (2015) 7(12):1159–66. doi:10.18632/aging.100861
215. McEwen LM, Morin AM, Edgar RD, MacIssac JL, Jones MJ, Dow WH, et al. Differential DNA methylation and lymphocyte proportions in a Costa Rican high longevity region. Epigenetics Chromatin (2017) 10:21–34. doi:10.1186/s13072-017-0128-2
216. Klinman NR, Riley RL, Stone MR, Wylie D, Zharhary D. The specificity repertoire of prereceptor and mature B cells. Ann N Y Acad Sci (1983) 418:130–9. doi:10.1111/j.1749-6632.1983.tb18061.x
217. Nicoletti C, Yang X, Cerny J. Repertoire diversity of antibody response to bacterial antigens in aged mice. III. Phosphorylcholine antibody from young and aged mice differ in structure and protective activity against infection with Streptococcus pneumoniae. J Immunol (1993) 150:543–9.
218. Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol (2007) 5:e201. doi:10.1371/journal.pbio.0050201
219. Johnson KM, Owen K, Witte PL. Aging and developmental transitions in the B cell lineage. Int Immunol (2002) 14:1313–23. doi:10.1093/intimm/dxf092
220. Farré P, Jones MJ, Meaney MJ, Emberly E, Turecki G, Kobor MS. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin (2015) 8:19. doi:10.1186/s13072-015-0011-y
Keywords: immune aging, epigenetics, DNA methylation, histones, environment, age-related diseases
Citation: Jasiulionis MG (2018) Abnormal Epigenetic Regulation of Immune System during Aging. Front. Immunol. 9:197. doi: 10.3389/fimmu.2018.00197
Received: 13 November 2017; Accepted: 23 January 2018;
Published: 12 February 2018
Edited by:
Rafael Solana, Universidad de Córdoba, SpainReviewed by:
Vasily V. Ashapkin, Moscow State University, RussiaSalman M. Tajuddin, National Institute on Aging (NIH), United States
Copyright: © 2018 Jasiulionis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miriam G. Jasiulionis, mjasiulionis@gmail.com