 Gillian L. Dornan
Gillian L. Dornan John E. Burke
John E. Burke- Department of Biochemistry and Microbiology, University of Victoria, Victoria, BC, Canada
The signaling lipid phosphatidylinositol 3,4,5, trisphosphate (PIP3) is an essential mediator of many vital cellular processes, including growth, survival, and metabolism. PIP3 is generated through the action of the class I phosphoinositide 3-kinases (PI3K), and their activity is tightly controlled through interactions with regulatory proteins and activating stimuli. The class IA PI3Ks are composed of three distinct p110 catalytic subunits (p110α, p110β, and p110δ), and they play different roles in specific tissues due to disparities in both expression and engagement downstream of cell-surface receptors. Disruption of PI3K regulation is a frequent driver of numerous human diseases. Activating mutations in the PIK3CA gene encoding the p110α catalytic subunit of class IA PI3K are frequently mutated in several cancer types, and mutations in the PIK3CD gene encoding the p110δ catalytic subunit have been identified in primary immunodeficiency patients. All class IA p110 subunits interact with p85 regulatory subunits, and mutations/deletions in different p85 regulatory subunits have been identified in both cancer and primary immunodeficiencies. In this review, we will summarize our current understanding for the molecular basis of how class IA PI3K catalytic activity is regulated by p85 regulatory subunits, and how activating mutations in the PI3K catalytic subunits PIK3CA and PIK3CD (p110α, p110δ) and regulatory subunits PIK3R1 (p85α) mediate PI3K activation and human disease.
Introduction
Phosphoinositide 3-kinases (PI3Ks) are essential mediators of signaling downstream of cell-surface receptors and play essential roles in numerous cellular processes, including growth, metabolism, and differentiation (1). PI3Ks generate the lipid phosphatidylinositol 3,4,5, trisphosphate (PIP3), which recruits signaling proteins containing PIP3 binding domains. Many signaling proteins are activated by PIP3, including AGC family Ser/Thr kinases, TEK family tyrosine kinases, and modulators of Ras superfamily GTPases, specifically Guanine nucleotide exchange factors (GEFs), and GTPase activating proteins (2). One of the most well studied PIP3 effectors is the AGC protein kinase Akt, which plays key roles in regulating growth and metabolism (3).
The class IA PI3Ks are composed of three p110 catalytic subunits (p110α, p110β, and p110δ), which form an obligate constitutive heterodimer (4) with one of five p85-like regulatory subunits (p85α, p85β p55α, p50α, and p55γ). Class IA PI3Ks are activated downstream of receptor tyrosine kinases (RTKs) and other tyrosine phosphorylated receptors/adaptors, G-protein-coupled receptors (GPCRs), and Ras superfamily GTPases. The p110α and p110β catalytic subunits are ubiquitously expressed, while the p110δ and p110γ subunits share a more restricted immune cell-specific expression profile (5). Knockin genetic models and isoform-selective inhibitors have revealed the essential roles of specific PI3K isoforms, including p110α in insulin and growth factor signaling (6, 7), and p110δ and p110γ in mediating immune cell development and function (8–11).
Due to this fundamental role in a plethora of vital functions, the misregulation of PI3K signaling occurs in various human diseases (2). Underlying the importance of maintaining regulated levels of PI3K activity, disease can be caused by overactive and inactive PI3K signaling. The first clinically approved therapeutic Idelalisib specifically targeting p110δ was FDA approved in 2014 and has shown efficacy in the treatment of B cell-related malignancies (12–16). Even though p110δ inhibitors have shown promise as therapeutics, careful consideration of unexpected complications is critical, as long-term inhibition of p110δ signaling can lead to B cell genomic instability through an Activation-induced cytidine deaminase (AID)-dependent mechanism (17).
Mutations in both catalytic and regulatory subunits frequently activate lipid kinase activity through modification/disruption of inhibitory interfaces between the two subunits. Fundamental to understanding how mutations in different catalytic and regulatory subunits modify PI3K signaling in different cells/tissues is understanding how class IA PI3Ks are regulated by their p85 regulatory subunits, and how they are activated downstream of different activating stimuli. This review will specifically focus on the molecular mechanisms of how class IA PI3Ks are regulated, and how both oncogenic and primary immunodeficiency mutations/deletions in catalytic and regulatory subunits lead to disease.
Class IA PI3K Regulation
The class IA regulatory subunits have three key roles: they stabilize the p110 catalytic subunit, they inhibit p110 catalytic activity, and they allow for the activation of activity downstream of proteins containing phosphorylated YXXM motifs through engagement of p85 SH2 domains (18, 19). While class IA catalytic subunits require a regulatory subunit for stability, the p85 subunits have been postulated to exist alone and can mediate cellular functions free of p110 (20, 21).
Both the class IA PI3K p110 catalytic subunit and p85 regulatory subunit are large, dynamic multi-domain proteins (Figures 1A–C). p110 is composed of an adaptor-binding domain (ABD), which interacts with p85, a Ras-binding domain (RBD), which mediates interaction with Ras superfamily GTPases, a C2 domain, a helical domain, and a bi-lobed kinase domain, composed of an N-lobe and a C-lobe. All class IA regulatory subunits contain two Src homology 2 domains [referred to as nSH2 and cSH2 to denote N-terminal and C-terminal] connected by a coiled-coil domain known as the inter SH2 (iSH2). The main interface holding the PI3K heterodimer together is the very tight interaction of the ABD of p110 with the iSH2 domain of p85 (22). Both p85α and p85β subunits also contain a Src homology 3 domain (SH3) and a bar cluster region homology domain (BH). A comparison of class IA PI3K domain organization compared with other SH2 containing protein kinases including Src family and Syk family kinases reveals the large size and complexity of the p110/p85 complex relative to other signaling kinases (Figures 1C,D).
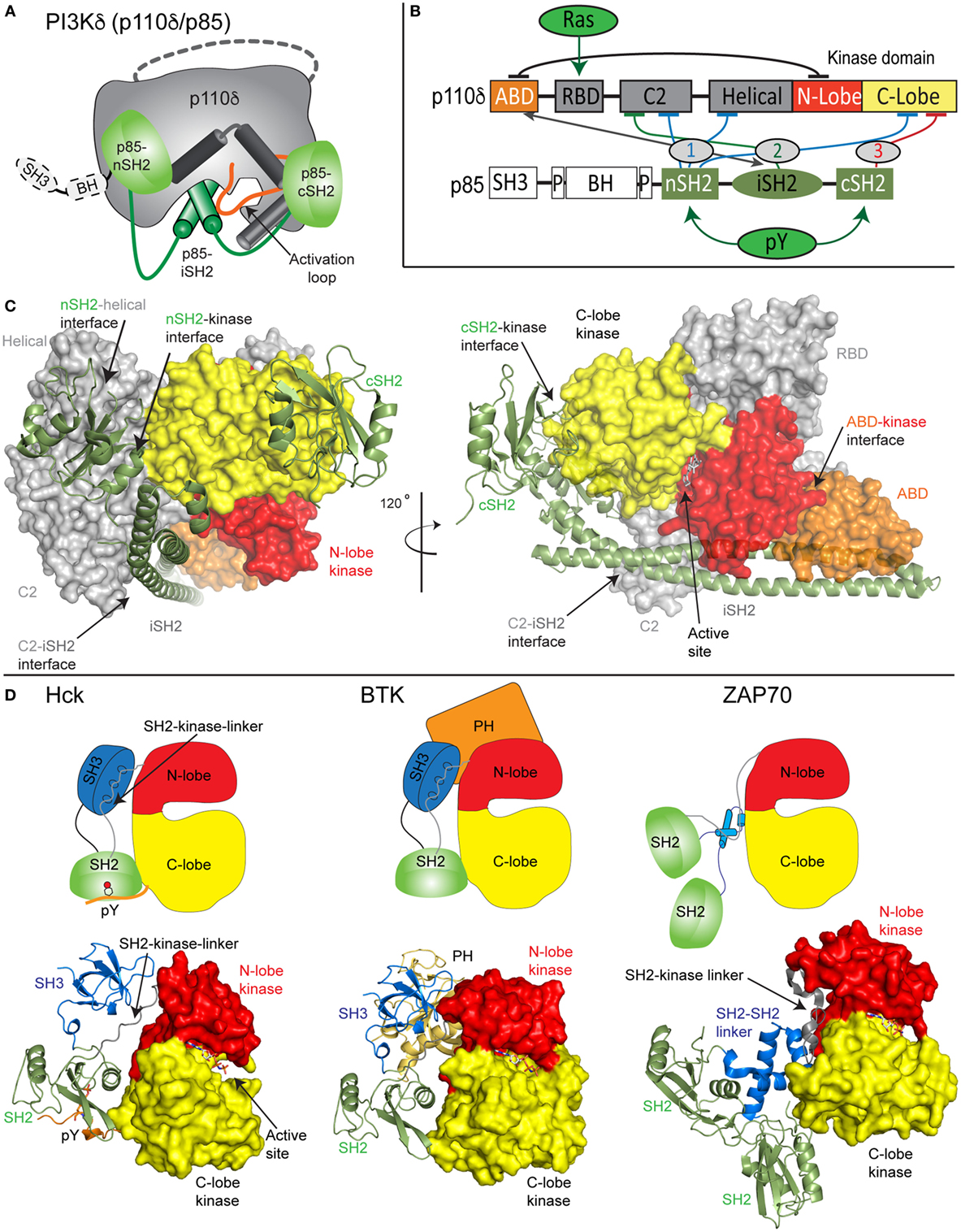
Figure 1. Model of class IA phosphoinositide 3-kinase (PI3K) complex of p110δ/p85α and comparison with other SH2-regulated kinases. (A) Cartoon model of the complex of p110δ/p85α, with key features annotated. (B) Domain schematic of p110δ and p85α with binding interfaces indicated by the double-sided arrow and inhibitory interfaces indicated by the numbered lines. Activators of PI3K [Ras and phosphorylated receptors (pY)], and their sites of interaction are indicated. The cSH2 domain of p85 only inhibits the p110β and p110δ isoforms and does not inhibit p110α. (C) Structural model of p110δ/p85α generated from multiple structures (23–25). The domains are colored according to the scheme shown in panel (B). The p110 catalytic subunit is shown as a surface, and the p85 regulatory subunit shown in cartoon representation. Inhibitory intra- and inter-domain interfaces are annotated, and an inhibitor bound in the active site is shown in sticks. (D) Structures of the inhibited forms of SH2-regulated protein kinases involved in immune signaling, Hck (26), BTK (27), and ZAP70 (28) are shown along with cartoon representations indicating how SH2 domains inhibit kinase activity. This shows the various mechanisms of how SH2 domains can inhibit kinase activity, and the key differences in how p85 SH2 domains inhibit PI3K lipid kinase activity.
Biochemical/biophysical studies have informed the molecular mechanism of how regulatory subunits bind and inhibit the different p110 catalytic subunits (18, 19, 22, 24, 25, 29–35). A number of inter- and intra-subunit interactions mediate inhibition of each of the class IA catalytic subunits (annotated on the domain schematic in Figure 1B). In all class IA PI3Ks, the ABD domain forms an intra-subunit inhibitory contact with the N-lobe of the kinase domain (32). How the ABD interacts with kinase domain is mediated by the ABD–RBD linker, which packs against the ABD. The C2 domain of p110 forms an inhibitory contact with the iSH2 domain of p85 regulatory subunits. Intriguingly, different p110 subunits have diverse capabilities to be inhibited by this interaction, with p110β being less inhibited by the C2–iSH2 interaction (36), compared with p110α and p110δ.
The nSH2 forms inhibitory interactions with the C2, helical, and C-lobe of all class IA p110 catalytic subunits (22, 24, 29, 30). The C-terminal SH2 domain, which interacts with the C-lobe of the kinase domain, only inhibits p110β (25) and p110δ (30). This interaction cannot occur in p110α due to a loop extension that sterically prevents this inhibitory interaction. Intriguingly, the nSH2 and cSH2 domains have different inhibitory interfaces with p110, with the nSH2 interacting with p110 through its pY binding site, and the cSH2-p110 interface not directly involving the pY binding site. Upon interaction with pYXXM motifs in phosphorylated receptors and their adaptors, the nSH2 and cSH2 interfaces with p110 are disrupted. Different regulation of class IA PI3Ks by their regulatory subunits has important functional implications for how they can be activated by different activating stimuli.
Signaling Inputs
The ability of PI3K isoforms to mediate signaling in different tissues is a balance between differential PI3K expression and their unique ability to be activated by GPCRs, Ras superfamily GTPases, and phosphorylated receptors/adaptors. All class IA isoforms can be activated by proteins containing phosphorylated YXXM motifs, as this leads to SH2-mediated recruitment of regulatory subunits, and disruption of SH2 inhibitory contacts (22, 29, 30, 35) with the p110 catalytic subunits. p110α is more sensitive to activation downstream of a phosphopeptide derived from platelet-derived growth factor receptor than either p110β or p110δ in vitro (29), and this is likely due to the absence of the cSH2 inhibitory interface, which makes the cSH2 more accessible to interact with pYXXM motifs. In vivo evidence in support of free SH2 domains being more available to pYXXM motifs is that the E545K mutant of p110α, which disrupts the nSH2–helical interface (described in the following section), is more readily recruited to phosphorylated insulin receptor substrate proteins (37).
Class IA PI3Ks are activated downstream of the Ras superfamily of GTPases through interactions with the RBD domain present in p110 catalytic subunits (38, 39). The Ras superfamily is large and diverse, composed of five main families (Ras, Rho, Rab, Ran, and Arf) (40). The PI3K isoforms are differentially activated downstream of Ras superfamily members (39, 41), with p110α and p110δ being activated downstream of Ras family GTPases, and p110β being activated downstream of Rho family GTPases. Ras activates PI3K through enhanced membrane interaction, with Ras activation being strongly synergistic with activation downstream of phosphorylated receptors (42, 43). Mutant p110α deficient in its ability to be activated by Ras leads to decreased oncogenic transformation, tumor maintenance, and angiogenesis downstream of mutant Ras (44–46).
Class IA PI3Ks can synergize direct and indirect inputs downstream of specific upstream stimuli. p110β is unique in being activated downstream of phosphorylated receptors/adaptors, GPCRs, and Rho family GTPases (47). The ability of p110β to integrate signals from RTKs and GPCRs is critical in its signaling role in myeloid cells (48). p110α is sensitive to activation downstream of insulin receptors due to it being both directly and indirectly activated through RTK-mediated activation of Ras. The ability of different isoforms to be activated downstream of different upstream stimuli plays a key role in determining the capability for activating somatic point mutations to mediate human disease.
Mutations of PIK3CA, PIK3CD, and PIK3R1 in Cancer, Developmental Disorders, and Primary Immunodeficiencies
Class IA PI3Ks in Cancer and Developmental Disorders
The importance of PI3K activity being properly regulated in human health is underscored by a vast array of human diseases caused by mutations in class IA PI3Ks (mutations in class I PI3Ks in immune disorders and developmental disorders are summarized in Table S1 in Supplementary Material). Mutations can arise in the germline de novo or be inherited in an autosomal dominant or recessive manner, and can also arise somatically in specific tissues. Somatic point mutation frequency in cancer in both PIK3CA (49) and PIK3R1 (20, 50) is indicated in Figures 2C,D. Intriguingly, de novo germline and postzygotic, somatic mosaic mutations in similar locations in PIK3CA and PIK3R2 (p85β) also lead to overgrowth and developmental disorder syndromes (51–56), revealing that the same mutant can lead to cancer and/or developmental disorders. There are two hotspot regions in PIK3CA located at the nSH2–helical interface (E542K and E545K) and the C-terminus of the kinase domain (H1047R) involved in membrane binding (Figures 2B,C). However, in addition, there are numerous rare mutations distributed throughout the primary sequence, primarily localized at the ABD–kinase interface, ABD–RBD linker, C2–iSH2 interface, and the regulatory arch of the kinase domain which is situated over the active site (Figures 2A,B). Rare mutations activate lipid kinase activity, induce oncogenic transformation (31, 57, 58), and are found in endometrial cancers (59).
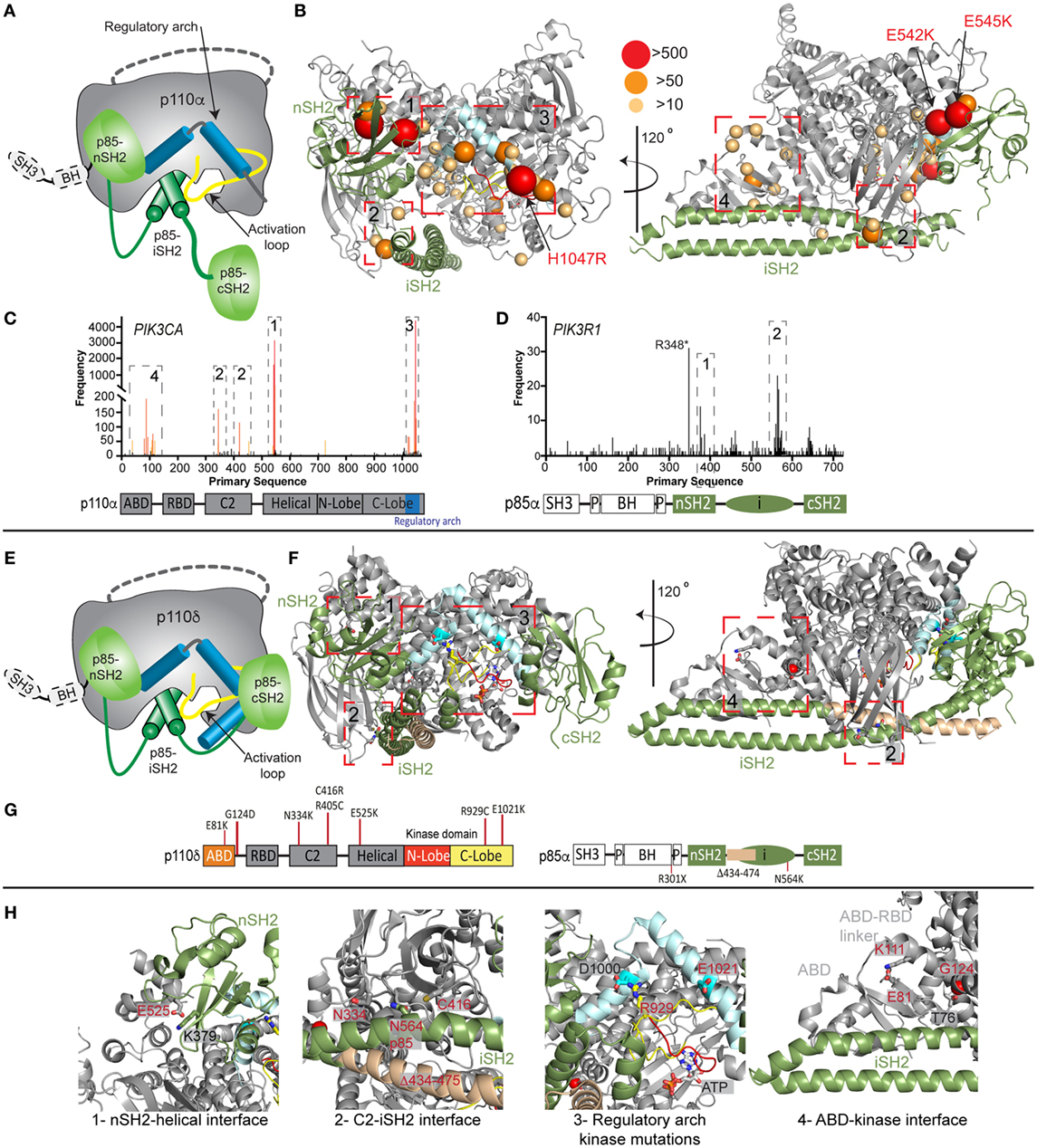
Figure 2. Oncogenic and primary immunodeficiency mutations in PIK3CA, PIK3CD, and PIK3R1. (A) Cartoon schematic of the complex between p110α and p85α with key regulatory features annotated. (B) The locations of oncogenic mutations in PIK3CA are shown on a structural model of p110α and p85α (24), with the frequency of mutations annotated according to the legend [frequency derived from the Catalogue of Somatic Mutations in Cancer (COSMIC), http://cancer.sanger.ac.uk/cosmic]. The proteins are colored according to the cartoon in panel (A). Regulatory interfaces [N-terminal SH2 domain (nSH2)–helical, C2–inter SH2 (iSH2), regulatory arch, and adaptor binding domain (ABD)–kinase] are boxed and numbered. Boxed regions 1–4 represent mutation hotspots in key regulatory regions. These are enlarged in panel (H) in the context of patient mutations in p110δ and p85α. (C,D) Frequency of somatic mutations in PIK3CA and PIK3R1 shown on the primary sequence, with the domain schematic indicated below. The locations boxed on the structure in panel (B) are also indicated on primary sequence. (E) Cartoon schematic of the complex between p110δ and p85α with key regulatory features annotated. (F) The locations of primary immunodeficiency mutations in PIK3R1 are shown on a structural model of p110δ and p85α (23). Boxed regions 1–4 represent mutation hotspots in key regulatory regions. These are enlarged in panel (H) in the context of patient mutations in p110δ and p85α. (G) Domain schematic of p110δ and p85α with locations of immune-linked mutations in PIK3CD and PIK3R1 indicated. (H) Zoom in on molecular details of activating phosphoinositide 3-kinase (PI3K) delta syndrome mutations in p110δ and p85α, focused on the regulatory interfaces boxed in panel (F), with all mutated residues and their interacting residues shown as sticks.
Mutants located at the ABD–kinase, C2–iSH2, and nSH2–helical interfaces activate lipid kinase activity through disruption of these inhibitory contacts. Intriguingly, there appears to be allosteric long range coupling between these sites, as disruption of the C2–iSH2 interface also leads to disruption of the ABD–kinase interface (31). Mutations within the regulatory arch (a region composed of the two most C-terminal helices, kα10 and kα11, residues 1017–1049) appear to work through a separate mechanism, where conformational changes induced by these mutations drive increased membrane recruitment (31, 60). The regulatory arch lies directly over the active site of the enzyme (Figure 2A). Different mutations induce oncogenic transformation through different mechanisms, with the H1047R mutant requiring p85-mediated recruitment to RTKs, and no longer requiring Ras for transformation, while the E545K mutation still requires input from Ras, and no longer requires p85-mediated RTK activation (58). This is consistent with the putative mechanism of Ras activation, where Ras drives membrane recruitment, and H1047R evades this requirement due to enhanced membrane binding (42, 43).
Somatic cancer-associated point mutations in PIK3R1 are similarly localized at regulatory interfaces (Figures 2B–D), with the most frequent mutation occurring at the C2–iSH2 interface (N564K/D). These mutants primarily activate PI3K signaling through p110α activation (50, 61, 62). Loss of p85α is also a driver of cancer as it acts as a tumor suppressor, and oncogenic transformation due to loss of p85α is also driven by p110α (63). Several deletions/truncations identified in PIK3R1 also can mediate oncogenic transformation through different mechanisms. Truncations at the C-terminus of the iSH2 domain can still interact with p110 subunits and disrupt inhibitory contacts (62), leading to increased PI3K activity. Intriguingly, oncogenic truncations also occur N-terminal to the iSH2 domain, and they are unable to bind p110 subunits. These truncations are proposed to function through modification of free p85 interactions with binding partners (20, 21, 64), including the antagonist of PI3K signaling, the phosphatase PTEN.
Mutations in PIK3R1 leading to decreased PI3K signaling are also found in patients with developmental disorders, with autosomal-dominant or de novo mutations in the cSH2 (R649W, K653*, and Y657*) leading to insulin resistance, and dramatically decreased PI3K signaling (65–71). This condition is defined as SHORT syndrome (Short stature, hyperextensibility of joints and/or inguinal hernia, ocular depression, Rieger anomaly, and teething delay) and is caused by the inability of the cSH2 domain to interact with phosphorylated RTKs, as mutation of R649 disrupts the FLVR motif critical for SH2 binding to phosphorylated pYXXM motifs.
Class IA PI3Ks in Primary Immunodeficiencies
Activating, autosomal-dominant and de novo mutations in PIK3CD and PIK3R1 have been discovered in patients with primary immunodeficiencies, and this condition is called activating PI3K delta syndrome (APDS), which is also referred to as PASLI (p110 delta activating mutation causing senescent T cells, lymphadenopathy, and immunodeficiency). Mutations in PIK3CD, referred to as APDS1, are found in similar locations to oncogenic mutations in p110α, with mutations discovered at the ABD (E81K), ABD–RBD linker (G124D), C2–iSH2 interface (N334K, R405C, and C416R), nSH2–helical interface (E525K and E525A), and at the C-terminus of the kinase domain (R929C, E1021K, and E1025G) (Figures 2E–H) (72–86). Biochemical experiments have revealed, similar to p110α mutations, that activation occurs due to disruption of p85-mediated regulatory inputs and conformational changes that promote membrane binding (83, 87). The most prevalent mutation in APDS1 is E1021K (similar location to H1047R in p110α); however, APDS mutations in p110δ are more frequently found distributed throughout the primary sequence compared with p110α (Figures 2C,D,G). In line with this observation, E1021K leads to a smaller increase in p110δ lipid kinase activity compared with H1047R p110α. It is likely that additional mutations in PIK3CD will be discovered that mimic previously discovered oncogenic mutations in PIK3CA, highlighting the need to sequence the entire PIK3CD gene in patients presenting with complex immunodeficiencies.
Mutations in PIK3R1, referred to as APDS2, have also been identified in a number of immunodeficiency patients, with the most frequent mutation resulting in a splice variant that removes exon 11 (resulting in a p85α with region 434–475 deleted, located at the N-terminus of the iSH2 domain) (88–92). In vitro, this deletion leads to increased activation of p110δ compared with p110α, and this is mediated through disruption of all p85 regulatory inputs for p110δ, and only partial disruption of p85 regulatory inputs for p110α (87). This mutant may decrease protein stability of p110 subunits, and there have been reports of these patients having symptoms consistent with both SHORT syndrome and APDS (92, 93). This may be due to increased p110δ signaling, and decreased p110α signaling caused by decreased stability of p110α. Activating point mutations in the iSH2 domain of PIK3R1 at the C2–iSH2 interface (N564K) also cause APDS2 symptoms (86). This mutant is also found in solid tumors, and it appears in certain situations it can drive p110α-mediated oncogenesis or drive p110δ-mediated immunodeficiency. Loss of function mutations in PIK3R1 also occur in immune disorders, with patients identified with autosomal recessive nonsense mutations in PIK3R1 (W298*, R301*) leading to agammaglobulinemia, and severe defects in B-cell development (94, 95).
Conclusion
Tremendous advances in our understanding of PI3K structure, function, and regulation have occurred in the last decade. Detailed cellular and mice studies have revealed unexpected mechanisms of how PI3Ks are activated. The discovery of patients containing PI3K mutations in cancer, developmental disorders, and immunodeficiencies has revealed the key role of these enzymes in disease. PI3K-specific inhibitors have been developed, and the first PI3K inhibitor, selective for p110δ, has entered the clinic for treatment of blood cancers (14, 96), and other PIK3CD-specific inhibitors have showed efficacy in the treatment of APDS (97, 98). PI3K inhibitors may also be useful in targeting the tumor microenvironment (99), and in promoting tumor-specific immune responses (100). However, many PI3K inhibitors have failed in clinical trials for cancer, and there is still extensive work that needs to be done to understand PI3K signaling in human disease. For example, why do the same mutations occur in both cancer and immunodeficiencies, what are the other factors that predispose the same mutation toward a particular disease? Continued examination of PI3K signaling will be essential to fully understand its role in human disease and may reveal unexpected paths to novel therapeutic development.
Author Contributions
All authors contributed to the writing and editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
JB is supported by funding from the Canadian Institutes of Health Research (New Investigator award and an Open Operating grant CRN-142393), the Cancer Research Society (Operating grant CRS-22641), and the Natural Sciences and Engineering Research Council of Canada (Discovery Grant NSERC-05218).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.00575/full#supplementary-material.
References
1. Burke JE, Williams RL. Synergy in activating class I PI3Ks. Trends Biochem Sci (2015) 40:88–100. doi:10.1016/j.tibs.2014.12.003
2. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell (2017) 170:605–35. doi:10.1016/j.cell.2017.07.029
3. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell (2017) 169:381–405. doi:10.1016/j.cell.2017.04.001
4. Geering B, Cutillas P, Nock G, Gharbi S, Vanhaesebroeck B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci U S A (2007) 104:7809–14. doi:10.1073/pnas.0700373104
5. Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci (2009) 34:115–27. doi:10.1016/j.tibs.2009.01.003
6. Knight Z, Gonzalez B, Feldman M, Zunder E, Goldenberg D, Williams O, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell (2006) 125:733–47. doi:10.1016/j.cell.2006.03.035
7. Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikami A, et al. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A (2006) 103:16296–300. doi:10.1073/pnas.0607899103
8. Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science (2000) 287:1049–53. doi:10.1126/science.287.5455.1049
9. Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kδ and primary immunodeficiencies. Nat Rev Immunol (2016) 16:702–14. doi:10.1038/nri.2016.93
10. Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol (2013) 31:675–704. doi:10.1146/annurev-immunol-032712-095946
11. Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science (2002) 297:1031–4. doi:10.1126/science.1073560
12. Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, et al. Idelalisib, an inhibitor of phosphatidylinositol 3 kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood (2014) 123(22):3390–97. doi:10.1182/blood-2013-11-535047
13. Flinn IW, Kahl BS, Leonard JP, Furman RR, Brown JR, Byrd JC, et al. Idelalisib, a selective inhibitor of phosphatidylinositol 3-kinase-δ, as therapy for previously treated indolent non-Hodgkin lymphoma. Blood (2014) 123 (22):3406–13. doi:10.1182/blood-2013-11-538546
14. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med (2014) 370:997–1007. doi:10.1056/NEJMoa1315226
15. Kahl BS, Spurgeon SE, Furman RR, Flinn IW, Coutre SE, Brown JR, et al. Results of a phase I study of idelalisib, a PI3Kδ inhibitor, in patients with relapsed or refractory mantle cell lymphoma (MCL). Blood (2014) 123(22): 3398–405. doi:10.1182/blood-2013-11-537555
16. Yang Q, Modi P, Newcomb T, Quéva C, Gandhi V. Idelalisib: first-in-class PI3K delta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin Cancer Res (2015) 21:1537–42. doi:10.1158/1078-0432.CCR-14-2034
17. Compagno M, Wang Q, Pighi C, Cheong T-C, Meng F-L, Poggio T, et al. Phosphatidylinositol 3-kinase δ blockade increases genomic instability in B cells. Nature (2017) 542:489–93. doi:10.1038/nature21406
18. Vadas O, Burke JE, Zhang X, Berndt A, Williams RL. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal (2011) 4:1–13. doi:10.1126/scisignal.2002165
19. Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3’-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol (1998) 18:1379–87. doi:10.1128/MCB.18.3.1379
20. Cheung L, Hennessy B, Li J, Yu S, Myers A, Djordjevic B, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov (2011) 1:170–85. doi:10.1158/2159-8290.CD-11-0039
21. Cheung LWT, Walkiewicz KW, Besong TMD, Guo H, Hawke DH, Arold ST, et al. Regulation of the PI3K pathway through a p85α monomer-homodimer equilibrium. Elife (2015) 4:e06866. doi:10.7554/eLife.06866
22. Miled N, Yan Y, Hon W-C, Perisic O, Zvelebil M, Inbar Y, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science (2007) 317:239–42. doi:10.1126/science.1135394
23. Heffron TP, Heald RA, Ndubaku C, Wei B, Augistin M, Do S, et al. The rational design of selective benzoxazepin inhibitors of the α-isoform of phosphoinositide 3-kinase culminating in the identification of (S)-2-((2-(1-isopropyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)oxy)propanamide (GDC-0326). J Med Chem (2016) 59:985–1002. doi:10.1021/acs.jmedchem.5b01483
24. Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang C-H, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A (2009) 106:16996–7001. doi:10.1073/pnas.0908444106
25. Zhang X, Vadas O, Perisic O, Anderson KE, Clark J, Hawkins PT, et al. Structure of lipid kinase p110b/p85b elucidatesan unusual SH2-domain-mediated inhibitory mechanism. Mol Cell (2011) 41:567–78. doi:10.1016/j.molcel.2011.01.026
26. Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature (1997) 385:602–9. doi:10.1038/385602a0
27. Wang Q, Vogan EM, Nocka LM, Rosen CE, Zorn JA, Harrison SC, et al. Autoinhibition of Bruton’s tyrosine kinase (Btk) and activation by soluble inositol hexakisphosphate. Elife (2015) 4:1948. doi:10.7554/eLife.06074
28. Deindl S, Kadlecek TA, Brdicka T, Cao X, Weiss A, Kuriyan J. Structural basis for the inhibition of tyrosine kinase activity of ZAP-70. Cell (2007) 129:735–46. doi:10.1016/j.cell.2007.03.039
29. Burke JE, Williams RL. Dynamic steps in receptor tyrosine kinase mediated activation of class IA phosphoinositide 3-kinases (PI3K) captured by H/D exchange (HDX-MS). Adv Biol Regul (2013) 53:97–110. doi:10.1016/j.jbior.2012.09.005
30. Burke JE, Vadas O, Berndt A, Finegan T, Perisic O, Williams RL. Dynamics of the phosphoinositide 3-kinase p110δ interaction with p85α and membranes reveals aspects of regulation distinct from p110α. Structure (2011) 19:1127–37. doi:10.1016/j.str.2011.06.003
31. Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA). Proc Natl Acad Sci U S A (2012) 109:15259–64. doi:10.1073/pnas.1205508109
32. Huang C, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu V, Kinzler K, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science (2007) 318:1744–8. doi:10.1126/science.1150799
33. Vadas O, Burke JE. Probing the dynamic regulation of peripheral membrane proteins using hydrogen deuterium exchange-MS (HDX-MS). Biochem Soc Trans (2015) 43:773–86. doi:10.1042/BST20150065
34. Vadas O, Jenkins ML, Dornan GL, Burke JE. Using hydrogen-deuterium exchange mass spectrometry to examine protein-membrane interactions. Methods Enzymol (2017) 583:143–72. doi:10.1016/bs.mie.2016.09.008
35. Yu J, Wjasow C, Backer JM. Regulation of the p85/p110alpha phosphatidylinositol 3’-kinase. Distinct roles for the n-terminal and c-terminal SH2 domains. J Biol Chem (1998) 273:30199–203. doi:10.1074/jbc.273.46.30199
36. Dbouk HA, Pang H, Fiser A, Backer JM. A biochemical mechanism for the oncogenic potential of the p110{beta} catalytic subunit of phosphoinositide 3-kinase. Proc Natl Acad Sci U S A (2010) 107:19897–902. doi:10.1073/pnas.1008739107/-/DCSupplemental
37. Yang X, Turke AB, Qi J, Song Y, Rexer BN, Miller TW, et al. Using tandem mass spectrometry in targeted mode to identify activators of class IA PI3K in cancer. Cancer Res (2011) 71:5965–75. doi:10.1158/0008-5472.CAN-11-0445
38. Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell (2000) 103:931–43. doi:10.1016/S0092-8674(00)00196-3
39. Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature (1994) 370:527–32. doi:10.1038/370527a0
40. Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev (2013) 93:269–309. doi:10.1152/physrev.00003.2012
41. Fritsch R, de Krijger I, Fritsch K, George R, Reason B, Kumar MS, et al. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell (2013) 153:1050–63. doi:10.1016/j.cell.2013.04.031
42. Buckles TC, Ziemba BP, Masson GR, Williams RL, Falke JJ. Single-molecule study reveals how receptor and ras synergistically activate PI3Kα and PIP3 signaling. Biophys J (2017) 113:2396–405. doi:10.1016/j.bpj.2017.09.018
43. Siempelkamp BD, Rathinaswamy MK, Jenkins ML, Burke JE. Molecular mechanism of activation of class IA phosphoinositide 3-kinases (PI3Ks) by membrane-localized HRas. J Biol Chem (2017) 292:12256–66. doi:10.1074/jbc.M117.789263
44. Castellano E, Sheridan C, Thin MZ, Nye E, Spencer-Dene B, Diefenbacher ME, et al. Requirement for interaction of PI3-kinase p110α with RAS in lung tumor maintenance. Cancer Cell (2013) 24:617–30. doi:10.1016/j.ccr.2013.09.012
45. Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell (2007) 129:957–68. doi:10.1016/j.cell.2007.03.051
46. Murillo MM, Zelenay S, Nye E, Castellano E, Lassailly F, Stamp G, et al. RAS interaction with PI3K p110α is required for tumor-induced angiogenesis. J Clin Invest (2014) 124:3601–11. doi:10.1172/JCI74134
47. Dbouk HA, Vadas O, Shymanets A, Burke JE, Salamon RS, Khalil BD, et al. G protein-coupled receptor-mediated activation of p110β by Gβγ is required for cellular transformation and invasiveness. Sci Signal (2012) 5:ra89. doi:10.1126/scisignal.2003264
48. Houslay DM, Anderson KE, Chessa T, Kulkarni S, Fritsch R, Downward J, et al. Coincident signals from GPCRs and receptor tyrosine kinases are uniquely transduced by PI3Kβ in myeloid cells. Sci Signal (2016) 9:ra82–82. doi:10.1126/scisignal.aae0453
49. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science (2004) 304:554. doi:10.1126/science.1096502
50. Urick ME, Rudd ML, Godwin AK, Sgroi D, Merino M, Bell DW. PIK3R1 (p85α) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res (2011) 71:4061–7. doi:10.1158/0008-5472.CAN-11-0549
51. Lindhurst MJ, Parker VER, Payne F, Sapp JC, Rudge S, Harris J, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet (2012) 44:928–33. doi:10.1038/ng.2332
52. Mirzaa GM, Conti V, Timms AE, Smyser CD, Ahmed S, Carter M, et al. Characterisation of mutations of the phosphoinositide-3-kinase regulatory subunit, PIK3R2, in perisylvian polymicrogyria: a next-generation sequencing study. Lancet Neurol (2015) 14:1182–95. doi:10.1016/S1474-4422(15)00278-1
53. Nakamura K, Kato M, Tohyama J, Shiohama T, Hayasaka K, Nishiyama K, et al. AKT3 and PIK3R2 mutations in two patients with megalencephaly-related syndromes: MCAP and MPPH. Clin Genet (2014) 85:396–8. doi:10.1111/cge.12188
54. Orloff MS, He X, Peterson C, Chen F, Chen J-L, Mester JL, et al. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am J Hum Genet (2013) 92:76–80. doi:10.1016/j.ajhg.2012.10.021
55. Rivière J-B, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet (2012) 44:934–40. doi:10.1038/ng.2331
56. Terrone G, Voisin N, Abdullah Alfaiz A, Cappuccio G, Vitiello G, Guex N, et al. De novo PIK3R2 variant causes polymicrogyria, corpus callosum hyperplasia and focal cortical dysplasia. Eur J Hum Genet (2016) 24:1359–62. doi:10.1038/ejhg.2016.7
57. Gymnopoulos M, Elsliger M-A, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A (2007) 104:5569–74. doi:10.1073/pnas.0701005104
58. Zhao L, Vogt P. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A (2008) 105:2652–7. doi:10.1073/pnas.0712169105
59. Rudd ML, Price JC, Fogoros S, Godwin AK, Sgroi DC, Merino MJ, et al. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin Cancer Res (2011) 17:1331–40. doi:10.1158/1078-0432.CCR-10-0540
60. Hon WC, Berndt A, Williams RL. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene (2012) 31:3655–66. doi:10.1038/onc.2011.532
61. Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W, et al. Somatic mutations in p85a promote tumorigenesis through class IA PI3K activation. Cancer Cell (2009) 16:463–74. doi:10.1016/j.ccr.2009.10.016
62. Jimenez C, Jones DR, Rodriguez-Viciana P, Gonzalez-Garcia A, Leonardo E, Wennström S, et al. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. EMBO J (1998) 17:743–53. doi:10.1093/emboj/17.3.743
63. Thorpe LM, Spangle JM, Ohlson CE, Cheng H, Roberts TM, Cantley LC, et al. PI3K-p110α mediates the oncogenic activity induced by loss of the novel tumor suppressor PI3K-p85α. Proc Natl Acad Sci U S A (2017) 114:7095–100. doi:10.1073/pnas.1704706114
64. Cheung LWT, Yu S, Zhang D, Li J, Ng PKS, Panupinthu N, et al. Naturally occurring neomorphic PIK3R1 mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathway inhibitors. Cancer Cell (2014) 26:479–94. doi:10.1016/j.ccell.2014.08.017
65. Bárcena C, Quesada V, De Sandre-Giovannoli A, Puente DA, Fernández-Toral J, Sigaudy S, et al. Exome sequencing identifies a novel mutation in PIK3R1 as the cause of SHORT syndrome. BMC Med Genet (2014) 15:51. doi:10.1186/1471-2350-15-51
66. Chudasama KK, Winnay J, Johansson S, Claudi T, König R, Haldorsen I, et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet (2013) 93:150–7. doi:10.1016/j.ajhg.2013.05.023
67. Dyment DA, Smith AC, Alcantara D, Schwartzentruber JA, Basel-Vanagaite L, Curry CJ, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet (2013) 93:158–66. doi:10.1016/j.ajhg.2013.06.005
68. Huang-Doran I, Tomlinson P, Payne F, Gast A, Sleigh A, Bottomley W, et al. Insulin resistance uncoupled from dyslipidemia due to C-terminal PIK3R1 mutations. JCI Insight (2016) 1:e88766. doi:10.1172/jci.insight.88766
69. Klatka M, Rysz I, Kozyra K, Polak A, Kołłataj W. SHORT syndrome in a two-year-old girl – case report. Ital J Pediatr (2017) 43:44. doi:10.1186/s13052-017-0362-z
70. Schroeder C, Riess A, Bonin M, Bauer P, Riess O, Döbler-Neumann M, et al. PIK3R1 mutations in SHORT syndrome. Clin Genet (2014) 86:292–4. doi:10.1111/cge.12263
71. Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet (2013) 93:141–9. doi:10.1016/j.ajhg.2013.05.019
72. Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science (2013) 342:866–71. doi:10.1126/science.1243292
73. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: a large patient cohort study. J Allergy Clin Immunol (2016) 139:597–606.e4. doi:10.1016/j.jaci.2016.06.021
74. Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol (2014) 34:272–6. doi:10.1007/s10875-014-0012-9
75. Dulau Florea AE, Braylan RC, Schafernak KT, Williams KW, Daub J, Goyal RK, et al. Abnormal B-cell maturation in the bone marrow of patients with germline mutations in PIK3CD. J Allergy Clin Immunol (2017) 139:1032–5.e6. doi:10.1016/j.jaci.2016.08.028
76. Elgizouli M, Lowe DM, Speckmann C, Schubert D, Hülsdünker J, Eskandarian Z, et al. Activating PI3Kδ mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol (2016) 183:221–9. doi:10.1111/cei.12706
77. Hartman HN, Niemela J, Hintermeyer MK, Garofalo M, Stoddard J, Verbsky JW, et al. Gain of function mutations of PIK3CD as a cause of primary sclerosing cholangitis. J Clin Immunol (2015) 35:11–4. doi:10.1007/s10875-014-0109-1
78. Heurtier L, Lamrini H, Chentout L, Deau M-C, Bouafia A, Rosain J, et al. Mutations in the adaptor-binding domain and associated linker region of p110δ cause activated PI3K-δ syndrome 1 (APDS1). Haematologica (2017) 102:e278–81. doi:10.3324/haematol.2017.167601
79. Liu H, Tang XL, Liu JR, Li HM, Zhao SY. [Clinical and genetic analysis for activated PI3K-δ syndrome by PIK3CD gene mutation]. Zhonghua Er Ke Za Zhi (2016) 54:698–702. doi:10.3760/cma.j.issn.0578-1310.2016.09.013
80. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15:88–97. doi:10.1038/ni.2771
81. Rae W, Gao Y, Ward D, Mattocks CJ, Eren E, Williams AP. A novel germline gain-of-function variant in PIK3CD. Clin Immunol (2017) 181:29–31. doi:10.1016/j.clim.2017.05.020
82. Saettini F, Pelagatti MA, Sala D, Moratto D, Giliani S, Badolato R, et al. Early diagnosis of PI3Kδ syndrome in a 2 years old girl with recurrent otitis and enlarged spleen. Immunol Lett (2017) 190:279–81. doi:10.1016/j.imlet.2017.08.021
83. Takeda AJ, Zhang Y, Dornan GL, Siempelkamp BD, Jenkins ML, Matthews HF, et al. Novel PIK3CD mutations affecting N-terminal residues of p110δ cause activated PI3Kδ syndrome (APDS) in humans. J Allergy Clin Immunol (2017) 140:1152–6.e10. doi:10.1016/j.jaci.2017.03.026
84. Teranishi H, Ishimura M, Koga Y, Eguchi K, Sonoda M, Kobayashi T, et al. Activated phosphoinositide 3-kinase δ syndrome presenting with gut-associated T-cell lymphoproliferative disease. Rinsho Ketsueki (2017) 58:20–5. doi:10.11406/rinketsu.58.20
85. Tsujita Y, Mitsui-Sekinaka K, Imai K, Yeh T-W, Mitsuiki N, Asano T, et al. Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase δ syndrome-like immunodeficiency. J Allergy Clin Immunol (2016) 138:1672–80.e10. doi:10.1016/j.jaci.2016.03.055
86. Wentink M, Dalm V, Lankester AC, van Schouwenburg PA, Schölvinck L, Kalina T, et al. Genetic defects in PI3Kδ affect B-cell differentiation and maturation leading to hypogammaglobulineamia and recurrent infections. Clin Immunol (2017) 176:77–86. doi:10.1016/j.clim.2017.01.004
87. Dornan GL, Siempelkamp BD, Jenkins ML, Vadas O, Lucas CL, Burke JE. Conformational disruption of PI3Kδ regulation by immunodeficiency mutations in PIK3CD and PIK3R1. Proc Natl Acad Sci U S A (2017) 114:1982–7. doi:10.1073/pnas.1617244114
88. Deau M-C, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest (2014) 124:3923–8. doi:10.1172/JCI75746
89. Hauck F, Magg T, Krolo A, Bilic I, Hirschmugl T, Laass M, et al. Variant PIK3R1 hypermorphic mutation and clinical phenotypes in a family with short statures, mild immunodeficiency and lymphoma. Klin Padiatr (2017) 229:113–7. doi:10.1055/s-0043-104218
90. Kuhlen M, Hönscheid A, Loizou L, Nabhani S, Fischer U, Stepensky P, et al. De novo PIK3R1 gain-of-function with recurrent sinopulmonary infections, long-lasting chronic CMV-lymphadenitis and microcephaly. Clin Immunol (2016) 162:27–30. doi:10.1016/j.clim.2015.10.008
91. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med (2014) 211:2537–47. doi:10.1084/jem.20141759
92. Petrovski S, Parrott RE, Roberts JL, Huang H, Yang J, Gorentla B, et al. Dominant splice site mutations in PIK3R1 cause hyper IgM syndrome, lymphadenopathy and short stature. J Clin Immunol (2016) 36:462–71. doi:10.1007/s10875-016-0281-6
93. Bravo García-Morato M, García-Miñaúr S, Molina Garicano J, Santos Simarro F, Del Pino Molina L, López-Granados E, et al. Mutations in PIK3R1 can lead to APDS2, SHORT syndrome or a combination of the two. Clin Immunol (2017) 179:77–80. doi:10.1016/j.clim.2017.03.004
94. Conley ME, Dobbs AK, Quintana AM, Bosompem A, Wang Y-D, Coustan-Smith E, et al. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J Exp Med (2012) 209:463–70. doi:10.1084/jem.20112533
95. Tang P, Upton JEM, Barton-Forbes MA, Salvadori MI, Clynick MP, Price AK, et al. Autosomal recessive agammaglobulinemia due to a homozygous mutation in PIK3R1. J Clin Immunol (2017) 62:1034–8. doi:10.1007/s10875-017-0462-y
96. Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med (2014) 370:1008–18. doi:10.1056/NEJMoa1314583
97. Hoegenauer K, Soldermann N, Zécri F, Strang RS, Graveleau N, Wolf RM, et al. Discovery of CDZ173 (Leniolisib), representing a structurally novel class of PI3K delta-selective inhibitors. ACS Med Chem Lett (2017) 8:975–80. doi:10.1021/acsmedchemlett.7b00293
98. Rao VK, Webster S, Dalm VASH, Sediva A, van Hagen PM, Holland S, et al. Effective “activated PI3Kδ syndrome-”targeted therapy with the PI3Kδ inhibitor leniolisib. Blood (2017) 130(21):2307–16. doi:10.1182/blood-2017-08-801191
99. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature (2016) 539:437–42. doi:10.1038/nature19834
Keywords: primary immunodeficiency, oncogenic mutations, phosphoinositides, phosphoinositide 3-kinase, PIK3R2, PIK3R1, PIK3CA, PIK3CD
Citation: Dornan GL and Burke JE (2018) Molecular Mechanisms of Human Disease Mediated by Oncogenic and Primary Immunodeficiency Mutations in Class IA Phosphoinositide 3-Kinases. Front. Immunol. 9:575. doi: 10.3389/fimmu.2018.00575
Received: 16 January 2018; Accepted: 07 March 2018;
Published: 19 March 2018
Edited by:
Stuart G. Tangye, Garvan Institute of Medical Research, AustraliaReviewed by:
Satoshi Okada, Hiroshima University, JapanAnita Chandra, University of Cambridge, United Kingdom
Copyright: © 2018 Dornan and Burke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John E. Burke, jeburke@uvic.ca