 Bahar Miraghazadeh1,2
Bahar Miraghazadeh1,2 Matthew C. Cook1,2,3*
Matthew C. Cook1,2,3*
- 1Centre for Personalised Immunology, John Curtin School of Medical Research, Australian National University, Acton, ACT, Australia
- 2Translational Research Unit, Canberra Hospital, Acton, ACT, Australia
- 3Department of Immunology, Canberra Hospital, Acton, ACT, Australia
NF-κB (nuclear factor-kappa B) is a transcription complex crucial for host defense mediated by innate and adaptive immunity, where canonical NF-κB signaling, mediated by nuclear translocation of RelA, c-Rel, and p50, is important for immune cell activation, differentiation, and survival. Non-canonical signaling mediated by nuclear translocation of p52 and RelB contributes to lymphocyte maturation and survival and is also crucial for lymphoid organogenesis. We outline NF-κB signaling and regulation, then summarize important molecular contributions of NF-κB to mechanisms of self-tolerance. We relate these mechanisms to autoimmune phenotypes described in what is now a substantial catalog of immune defects conferred by mutations in NF-κB pathways in mouse models. Finally, we describe Mendelian autoimmune syndromes arising from human NF-κB mutations, and speculate on implications for understanding sporadic autoimmune disease.
Introduction
NF-κB is family of proteins that mediate transcriptional regulation crucial to many biological functions (1). NF-κB was discovered and named for its action in regulating κ light chain expression in B cells. Remarkably, however, as elucidation of Drosophila immunity has demonstrated, NF-κB and its orthologs are highly conserved regulators of signaling and transcription crucial for cellular and humoral host defense even in organisms that lack adaptive immunity (2). In mammals, the actions of NF-κB are complex and extensive, and encompass activation, proliferation in cells of the innate and adaptive immune system, and organogenesis of lymphoid tissue and its microarchitecture (3–5).
Under normal circumstances, NF-κB proteins are latent in the cytoplasm, poised for rapid responses after their inhibition is temporarily removed. Uninhibited NF-κB molecules then shuttle between nucleus and cytoplasm as transcriptionally active homo- and heterodimers (Figure 1). In addition to this fundamental inhibitory constraint, many other negative regulatory loops exist to either prevent, dampen, or terminate NF-κB signaling, including sequestration in multi-molecular complexes, posttranscriptional regulation, and posttranslational modifications of proteins by phosphorylation and ubiquitination (of various forms). Furthermore, many components of NF-κB, including both positive and negative regulators, are under transcriptional regulation by NF-κB itself.
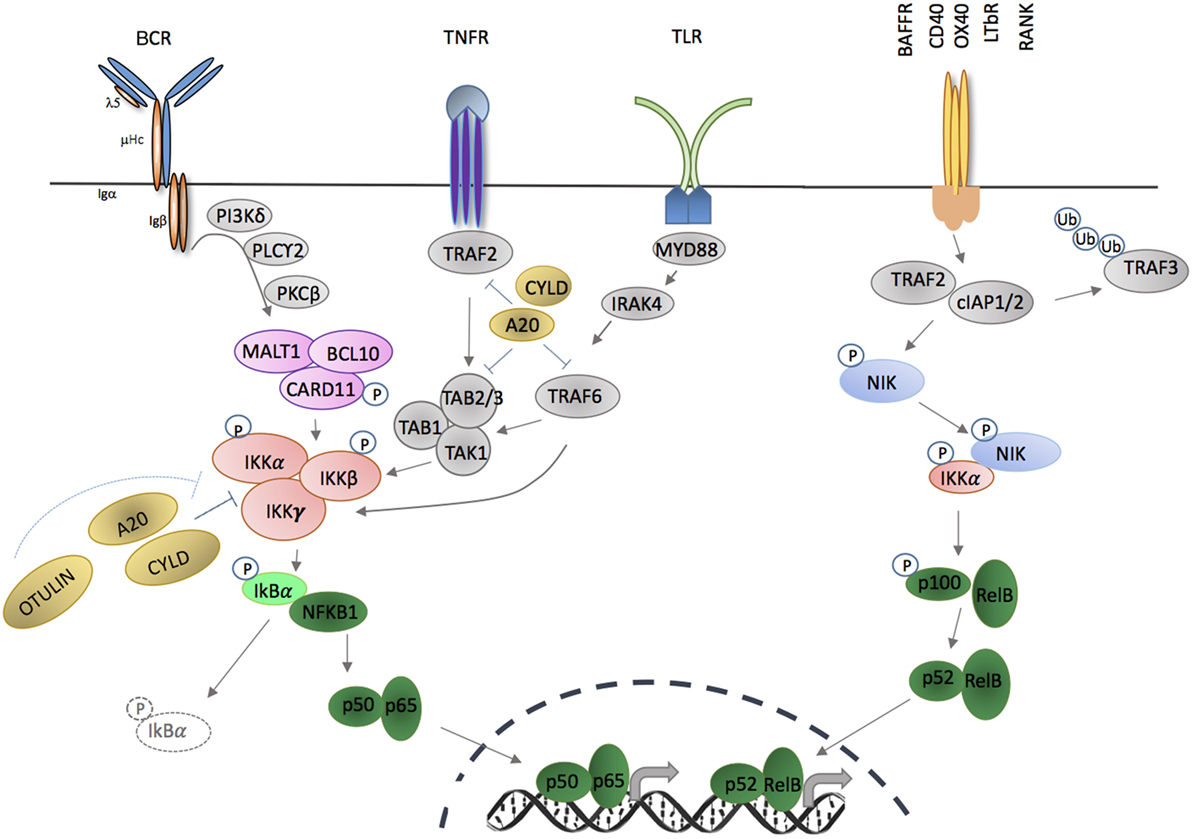
Figure 1. Activation of canonical and non-canonical NF-κB signaling pathways through membrane-bound extracellular ligands. TNFR and toll-like receptor (TLR) family members, as well as antigen receptors activate the canonical pathway; and regulation of B cell activating factor receptor (BAFFR), CD40, OX40, LTβR, and receptor activator of nuclear factor kappa-B (RANK) activate the non-canonical pathway. Triggering of canonical pathway results in activation of p50/p65 (RelA), while the non-canonical pathway signaling leads to activation of p52/RelB complexes. Both pathways require phosphorylation and activation of inhibitor of kappa B kinase (IKK) subunit(s) in order to release NF-κB molecules that are sequestered by an inhibitor, e.g., IκBα or p100. Phosphorylation and ubiquitination of the inhibitors by IKKs release NF-κB that translocate into nucleus in the forms of homodimers or heterodimers complexes and bind to the κB site of their target genes.
Despite this complex regulatory network, specific defects in individual molecules within the NF-κB pathway have been shown to disrupt cellular homeostasis, and immune pathology is an important consequence (1, 6). In this review, we will concentrate on how NF-κB contributes to immunological self-tolerance, and how defects in NF-κB contribute to autoimmune disease. Defects in NF-κB have also been shown to cause immune deficiency and autoinflammatory diseases, and somatic mutations are frequent drivers of lymphoid malignancy, for which authoritative reviews are available (7, 8). As will be discussed here, however, it is notable that in some cases, a single mutation confers both autoimmunity and immune deficiency, reflecting the complex regulatory actions of NF-κB.
Outline of Normal NF-κB Signaling
The NF-κB family of transcription factors form hetero- and homodimers that regulate transcription by binding to a palindromic DNA sequence, κB (1), located within promoters and enhancers of a large number of genes (9, 10). In vertebrates, there are five NF-κB family members, RelA, c-rel, RelB, NF-κB1, and NF-κB2. N-terminal Rel-homology domains (RHD, from v-Rel, reticuloendotheliosis viral oncogene homolog) are common to all and mediate κB binding and interactions with other proteins, including inhibitor of kappa B (IκB) (see below) (10–12).
NF-κB proteins are classified in two groups according to structure and function. p105 (NF-κB1) and p100 (NF-κB2) are precursor proteins that undergo partial proteolysis to remove their C-terminal ankyrin repeats, yielding p50 and p52, respectively. p50 and p52 lack transactivation domains (TAD) unless heterodimerized with Rel or coactivator non-Rel proteins (13). By contrast, RelA (p65), RelB, and c-Rel are active in the absence of proteolysis because they contain TAD that positively regulate expression of target genes (14, 15).
Differences in transcriptional activity of NF-κB dimers helps explain the plasticity of responses to both quantitative and qualitative variation in cell stimulation (16). p50/65 heterodimers are near ubiquitous, and positively regulate NF-κB target genes (10). By contrast p50 homodimers repress TNF-α transcription in response to lipopolysaccharide (LPS) (17–19). Homodimers of p50 are abundant in resting T cells, but their expression is reduced after antigenic receptor ligation (20), when p50/p65 become abundant in cell nuclei, reversing the NF-κB-dependent suppression of the target genes, i.e., IL-2 or IL-6 and iNOS in response to LPS (21). RelB does not homodimerise, but confers transcriptional activity when complexed with p52 or p50 (22). RelB constitutively localizes to the nucleus, but binding may be inhibited by association with p100 (23–25). Under some circumstances, RelB represses NF-κB activity by forming RelA/RelB heterodimers that fail to bind DNA and sequesters RelA (9, 26, 27). Similarly to RelB, c-Rel is expressed in lymphoid tissues, and both c-Rel homodimers and c-Rel/p50 heterodimers are detected predominantly in hematopoietic cells. c-Rel homodimers and c-Rel/p65 are essential for B and T cells survival and effector cell function (28–30).
Activation of NF-κB in Immunity
In the absence of specific signals, NF-κB is maintained in latent form bound to IκB in the cytoplasm. The IκB family includes IκBα, IκBβ, IκBε, BCL-3, IκBζ, IκBNS, as well as unprocessed p100 and p105, which are all characterized by multiple ankyrin repeats (31). In addition to their IκB function, so-called atypical IκBs (IκBζ, IκB-NS, and Bcl-3) have intrinsic nuclear localization propensity where they bind preferentially to p50 (Bcl-3, IκBζ) and p52 (Bcl-3), which under different conditions can either promote or terminate NF-κB binding to DNA (32, 33). This mechanism has been well characterized for Bcl-3, which has a TAD and can act as a coactivator when associated with p50 or p52 homodimers (34, 35), or by facilitating replacement of transcriptionally inactive p50 homodimers with transcriptionally active heterodimers (36, 37). On the other hand, Bcl-3 can also function as a repressor by stabilizing p50 homodimers on κB site of the target genes. For example, Bcl-3 has been reported to mediate LPS tolerance in macrophages by stabilizing p50 homodimer on the TNF promoter (38–40).
NF-κB members are liberated by the action of the IκB kinase complex [IKKα, IKKβ and NF-κB essential modulator (NEMO), encoded by IKBKA, IKBKB and IKBKG], which leads to temporary degradation of IκBs (Figure 1). IKKγ (NEMO) is a regulatory subunit that does not have intrinsic catalytic activity but is important for kinase activation of IKKα and IKKβ (41). IKKα and IKKβ are serine/threonine kinases that share an N-terminal kinase domain and are responsible for phosphorylating several members of the IκB family (42, 43). Serine phosphorylation of IKKα and IKKβ [serines 177 and 181 for IKKβ; serines 176 and 180 for IKKα (44, 45)] results in conformational change and kinase activation. Activated IKKβ operates within the inhibitor of kappa B kinase (IKK) complex to phosphorylate IκBα, leading to K48-linked ubiquitination and proteosomal degradation, which releases NF-κB factors for nuclear translocation (46, 47). Loss of IKKβ results in significant reduction of NF-κB activity in response to TNF-α and IL-1α, a defect that cannot be completely compensated for by IKKα. As result, Ikbkb deficiency confers a mouse phenotype similar to Rela deficiency (48, 49) (Table 1).
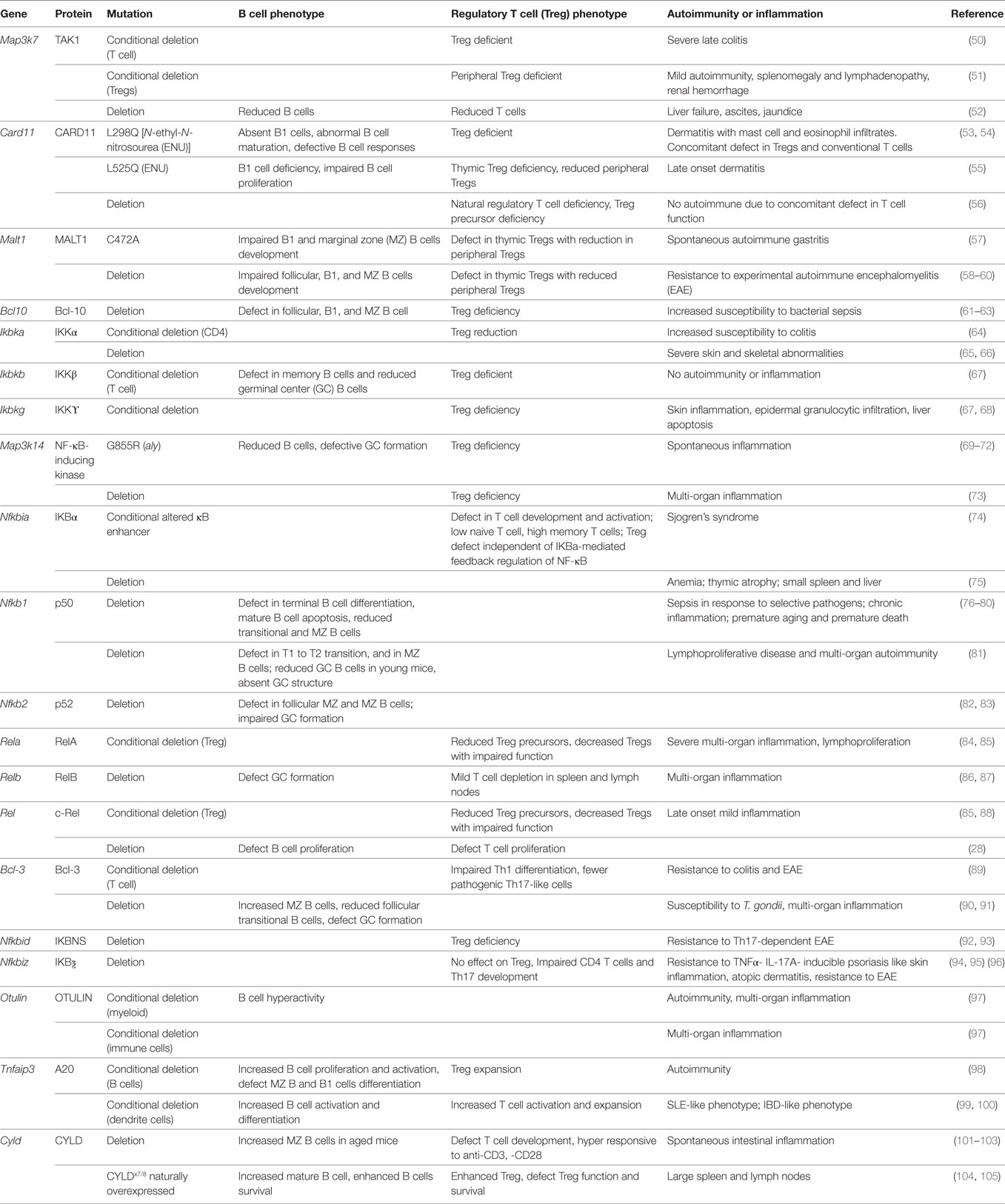
Table 1. Summary of autoimmune phenotypes in mice with genetic manipulation of NF-κB.
Canonical Pathway Stimuli
The canonical NF-κB pathway includes NFκB1 (p105), RelA (p65), and c-Rel and is activated by many ligands, including those that engage members of the tumor necrosis factor receptor superfamily, toll-like receptors (TLR), interleukin 1 receptor, and T and B cell antigen receptors (Figure 1). Different receptor-proximal signaling cascades connect these receptors to the IKK complex. TLR ligation activates a complex composed of TAK1, TAB 1, TAB 2, which phosphorylates IKKβ (106, 107). Activation of TNF receptor leads to interaction of series of adaptor proteins that contains TRAF-binding domains (108, 109). TRAF2 recruits the E3 ubiquitin ligases, cIAP1/2, which are necessary for IKK activation via recruitment of linear ubiquitination assembly complex (LUBAC) (directly) and TAK1 (indirectly) (110, 111). Ligation of B and T cell antigen receptors leads to phosphorylation of CARD11 by protein kinase C and assembly of the CARD/BCL-10/MALT1 (CBM) complex.
Modification of Canonical Signaling
Posttranslational regulation by ubiquitination is crucial for NF-κB regulation. A series of ubiquitinating and deubiquitinating enzymes are known that both activate and modify NF-κB transcriptional regulation (112). The best characterized negative regulators of NF-κB are deubiquitinase (DUB) A20 (encoded by TNFAIP3), CYLD, and OTULIN (113–116).
TNFAIP3 is upregulated in response to TNF receptor and TLR ligation, and A20 negatively controls NF-κB-dependent gene expression (Figure 1) (117). A20 removes or modifies K63 polyubiquitin from several substrates within NF-κB signaling, including TRAF6, NEMO, MALT1, and TNFR1 (118–120). In addition to its deubiquitin domain in the N-terminus, A20 also contains seven zinc finger domains in the C-terminus with E3 ligase functions, enabling ubiquitin-editing. Thus, A20 replaces K63- with K48-polyubiquitin chains of RIPK1, flagging it for proteasomal degradation (99, 113). The importance of A20 in modifying NF-κB activity and immune responses was confirmed in A20-deficient mice that developed profound inflammation and cachexia, and died prematurely (117) (Table 1), and cell-specific deletion of A20 in myeloid cells, dendritic cells (DCs) or B cells, results in autoimmune phenotypes including polyarthritis and enteritis (Table 1) (121–124). Loss of catalytic activity might not account for this pro-inflammatory action, however, as specific introduction of a deubiquitination domain inactivating mutation resulted in a much less prominent inflammatory phenotype (125).
NF-κB is also modified by addition of methionine (M)1-linked linear ubiquitin chains by the LUBAC that consists of HOIL-1L (RBCK1), HOIP (RNF31), and SHARPIN (112). LUBAC promotes canonical signaling in part through direct binding of the E3 ligase activity of the ring domains of HOIP to drive linear ubiquitination of NEMO and RIP1 (126), which promotes IKKβ phosphorylation and activation through TAK1 (107, 127, 128). CYLD modifies canonical signaling by removing K63 and M1-polyubiquitin chains from NF-κB signaling proteins, including TRAF2, TRADD, NEMO, and TAK1 (Figure 1). CYLD appears to be particularly important in promoting necroptosis via RIPK1. CYLD deficiency results in several non-immunological phenotypes, but also inflammatory bowel disease, defects in thymic medullary epithelial cells (see below), and increased B cell activity (Table 1) (105, 129). OTULIN specifically hydrolyzes M1-polyubiquitins. In mice, germline deficiency of Otulin is lethal, but conditional myeloid deficiency results in profound sterile inflammation (Table 1) (97). Interestingly, after caspase-mediated cleavage during apoptosis, the N-terminal fragment of HOIP also binds to the DUBs OTULIN and CYLD, which are down-regulators of LUBAC-mediated ubiquitination, providing a further regulatory feedback loop (130).
Non-Canonical Signaling
NFκB2 (p100) and RelB participate in the non-canonical NF-κB pathway (Figure 1). Unlike the canonical pathway, which is poised for rapid response, the non-canonical pathway depends on constant protein synthesis (131). This might explain the preferential action of the non-canonical pathway in cellular homeostasis and organogenesis, whereas the canonical pathway mediates acute inflammatory responses and immune activation.
Non-canonical NF-κB activation is stimulated by a relatively small number of ligands, including lymphotoxin, CD40, B cell activating factor (BAFF), and receptor activator of NF-κB (RANK). In resting cells, TRAF2 mediates ubiquitination of NF-κB-inducing kinase (NIK) in association with TRAF3 and cIAP1/2. NIK is maintained in limiting concentrations by rapid degradation after phosphorylation by TRAF6, but after receptor ligation, TRAF2-induced proteolysis and degradation of TRAF3 and TRAF6 leads to NIK accumulation, which phosphorylates IKKα (132, 133). pIKKα is crucial for phosphorylation of p100, leading to proteosomal processing to p52 (134–140).
Non-canonical signaling activates complex regulatory loops because p100 exerts IκB activity by associating with p52, p65 (RelA), c-Rel, and RelB. Interestingly, and by contrast with the IκB activity of the canonical counterpart p105, p52 is formed from recently synthesized p100. Elegant experiments have revealed that the balance between p100 activation and NIK degradation is maintained within a kappaBsome, which is a complex of NIK-IKKα-p100 and RelB. Almost all p100 is maintained within the kappaBsome, since free cytoplasmic p100 undergoes complete degradation, whereas processing from within the kappaBsome yields p52 (141). RelB promotes formation of the kappaBsome, and competes with IKKα–NIK complexes to inhibit p100 activation and processing. In addition to promoting p100 processing, IKKα serves to limit activity and abundance of NIK within the complex, and p100 inhibits NIK degradation. RelB dimerizes with both p52 and p100 in the complex to permit the proper processing of p100. In the absence of RelB, p100 undergoes complete degradation (141, 142).
Transcriptional Regulation by NF-κB
NF-κB lacks the enzymatic activity to directly regulate transcriptional responses, and this is achieved through binding to transcriptional co-regulators including histone acetyltransferases (HATs) and histone deacetylases (HDACs). For instance, acetylation of p65 at different lysines by HATs (and the associated binding proteins) promotes its activity, while the co-repressor, HDACs, can deacetylate important sites on p65 and reverse the effect (143). NF-κB recruit these co-regulators to the enhancer region of the target genes, leading to conformational changes in chromatin to make genes accessible for transcriptional machinery (143–145).
Binding to HATs is also controlled partially by posttranscriptional modifications (PTMs). For example, kinases including PKA, MSK1, and MSK2 phosphorylate p65 at Serine 276, which is crucial for the interaction of p65 with HATs (21, 26, 146, 147). Interestingly, this amino acid substitution only affects expression of a subset of the genes, which suggests other PTMs might contribute to p65-dependent chromatin remodeling (148). One example is Akirin2, which binds to HATs and IκBζ and facilitates recruitment of other co-regulators to p50 (149).
NF-κB and Self-Tolerance
Canonical and non-canonical NF-κB pathways are crucial for T and B cell activation, regulation of inflammatory effector responses, antigen presentation and regulation of tissue-specific cellular targets of immunity. We will concentrate our attention on mechanisms that regulate lymphocyte self-tolerance, since these are most relevant to autoimmunity. These actions can be divided into those that act intrinsically within lymphocytes, and those that are mediated by lymphocyte extrinsic actions, particularly for thymic selection.
Thymic Development
The thymus is derived from the endoderm of the third pharyngeal pouch. Mature thymic epithelium is made up of cortical thymic epithelial cells (cTEC) and medullary thymic epithelial cells (mTEC). Unlike other epithelia, thymic epithelium lacks apical polarity, and cells are dispersed in a three-dimensional reticular meshwork (150). The two best understood mechanisms of central T cell tolerance are negative selection of self-reactive conventional T cells (Tconv), and selection of regulatory T cells (Tregs). Both mechanisms depend on maturation of mTECs, and expression of autoimmune regulator (Aire), which in turn regulates expression of tissue specific antigens (TSA) (151–153).
Both mTECs and cTECs have been postulated to arise from CD205+ β5t+ and CD45− bipotential precursors (154). SSEA-1+ Claudin3/4+ expression identifies a self-renewing population of CD80lo MHCIIlo mTEC precursors (155), which exhibit some epithelial stem cells characteristics. cTECs are characterized by epithelial cellular adhesion molecule (Ep-CAM) β5t, CD205, and Ly51 expression (156). Mature mTECs are UAE-1+ Ly51−, MHCIIhi, CD80hi, CD40+, and express Aire. mTECs are responsible for maintenance of T cell tolerance via negative selection of αβ T cells (157, 158), and positive selection of FOXP3+ Tregs, by virtue of promiscuous expression of tissue specific antigens (TSA) (154, 159, 160). Terminally differentiated mTECs (which form Hassall’s corpuscles) are marked by loss of AIRE, MHCII, and CD80, acquisition of desmogleins 1 and 3, and of involucrin, a global marker of epithelial terminal differentiation (161).
NF-κB and Development of Thymic Epithelium
Thymus development depends on an unusual hematopoietic–epithelial interplay between epithelial cell precursors and nascent lymphocytes (162). Interestingly, lymphocytes exert a similar influence on development of the specialized epithelium overlaying Peyer’s patches (M cells) (163). In both thymus and Peyer’s patches, epithelial development is at least partially dependent on NF-κB activation by RANKL and other ligands, delivered by lymphocytes (164–166). Selection of developing T cells depends on lymphocyte–epithelial interactions within the thymus, and interactions between immature T cells and thymic epithelium regulates T cell development by negative selection of self-reactive Tconv, and positive selection of regulatory Tregs (157, 161, 167).
Signaling via the non-canonical NF-κB pathway appears to regulate mTEC maturation and the size of the mTEC compartment, but not necessarily specification of the mTEC lineage. This action hinges on NF-κB activation by ligation of TNF receptor superfamily members RANK, CD40, and LTβR (86, 166, 168). Single positive thymocytes, γδ T cells, invariant NK cells, and lymphoid tissue inducer cells are all sources of RANKL and CD40L (165, 166). Fate-mapping studies have provided more information on the sequential actions of non-canonical stimuli for development and maintenance of mTECs. Thus, RANK operates after mTEC precursors have differentiated and coincides with onset of RelB expression (169). RelB appears to act cell-intrinsically to determine mTEC numbers (86, 168), while RelB expression mediates the non-canonical pathway, it is also modified by RelA and c-Rel, and therefore, mTEC development can be influenced by canonical NF-κB signaling (170). Late mTEC differentiation is also disrupted by altered CYLD function, suggesting that regulation of RANK signaling via TRAF6 deubiqutination might be important during late mTEC development (171), possibly by regulating RelB induction by the canonical NF-κB pathway.
As well as RelB deficiency, deficiencies of NIK, IKKα, LTβR, NF-κB2 (p100), and Bcl-3 result in mTEC deficiencies of varying severity (172–174). Interestingly, the thymic defect observed with Nfkb2 deficiency is less marked than that observed with Relb deficiency (175). mTEC-specific deletion of LTβR results in disordered thymic architecture, but not altered negative selection, although negative selection is defective when LTβR is deleted from thymic DCs (159). Indeed, distinguishing the actions of defects in mTECs from those in DCs requires construction of chimeras in which the mutation is confined to either hematopoietic or stromal compartments, which has not yet been performed for all mutations.
Bcl-3 action appears to be redundant with NF-κB2 for mTEC development (176). Thus, mice doubly deficient for Bcl3 and Nfkb2 lack mTECs, thymic Aire expression, and some thymic DCs, and they develop severe organ-specific autoimmunity. NIK is crucial for non-canonical NF-κB activation, but the mechanism of Bcl-3 is less obvious. While Bcl-3 is an atypical IκB, it does not regulate RelA or c-Rel. Uncertainties regarding the mechanism of Bcl-3 notwithstanding, there is solid empirical evidence for a crucial cell-intrinsic action of non-canonical signaling for maintaining mTECs, and for central T cell tolerance. In addition, several findings suggest that these actions are modified by regulation of RelB expression and activation via the canonical pathway.
NF-κB Defects Confer Susceptibility to Tissue-Specific Autoimmunity
Deficiency of Aire leads to widespread tissue-specific inflammation in man and mouse as a result of impaired expression of TSA by mTECs (158, 177, 178). This discovery represented a landmark in autoimmunity research, providing empirical evidence that negative selection mediated by antigen expression in mTECs is a bulwark against autoimmune disease (160). NF-κB signaling is important for Aire expression independently of mTEC development and homeostasis. The conserved non-coding sequence 1 (CNS1) located 3′ to the Aire transcription start site is an Aire enhancer, and contains two NF-κB-binding motifs, one with a preference for RelA and c-Rel, and the other for RelB (179, 180). There is compelling evidence for the interaction of RelA with CNS1, implicating the canonical NF-κB pathway in Aire expression. CD40 and RANK ligation appears to be important for Aire induction by NF-κB, whereas Aire expression is independent of LTβR (181).
More recently, FEZF2 was also identified as a transcriptional regulator of TSA expression in mTECs. NF-κB has been implicated in Fezf2 expression, although the relative importance of LTβR and RANKL remains controversial (159, 182). Fezf2 deficiency results in a pattern of organ-specific autoimmunity targeted at lung, kidney, liver, and intestine, but not retina or pancreas, which is observed in Aire deficiency (182). PRDM1 is the most recent addition to transcriptional regulators of TSA in mTECs, and while the regulation of expression PRDM1 in mTECs is unclear, it is known to be induced by RANKL-mediated NF-κB signaling in other tissues (183, 184).
As might be predicted from its crucial action on mTECs, Relb deficiency results in multi-organ inflammation, indicating that non-canonical signaling acting to regulate mTEC development is crucial for maintaining self-tolerance and prevention of organ-specific autoimmunity (86, 168). The autoimmune phenotypes of Relb mutants and related strains are summarized in Table 1. The severity of the autoimmune phenotype conferred by Relb deficiency may reflect its importance not only for mTEC development but also on Aire expression. Mice deficient in NIK (Map3k14−/−) also develop autoimmunity, providing additional evidence that the non-canonical pathway regulates self-tolerance (70, 71). Similarly, autoimmune phenotypes have been induced by deletion of CD40L, RANKL, or LTβR, the ligands crucial for inducing non-canonical signaling for mTEC maturation and maintenance (166, 185, 186).
Mice rendered deficient in Nfkb2 also display modest defects in central tolerance and an autoimmune phenotype of inflammatory infiltrates in lungs, liver, kidneys, and pancreas, together with autoantibodies, thus resembling the phenotype of Aire deficiency (175). In this case, reciprocal bone marrow chimeras confirmed the defect was in the radio-resistant stroma, and further analysis revealed a numerical defect in Ep-Cam+ (G8.8) and Ulex europaeus agglutinin 1+ mTECs, a cell-for-cell defect in LTβR-induction of Aire, and a reduction in expression of Aire-dependent TSA. No numerical or functional Treg defect was observed. Interestingly, and by contrast with complete NF-κB2 deficiency, specific deficiency of p52 conferred only a T-cell dependent antibody response with no overt autoimmune phenotype (83, 187, 188).
NF-κB and Treg Selection
NF-κB appears to be important for maintaining Treg function via mTEC development, thymic dendritic cell development, and T-cell-intrinsic actions (Table 1). TCR ligation and co-stimulation, particularly via CD28, activates the canonical NF-κB pathway, which appears to be important for FoxP3 induction (189). Engagement of TCR and CD28 activates the CBM complex, via the upstream and mediators, TAK1 and PKCθ. Deficiency in each of these signaling components alone also results in Treg deficiency (54–56, 190, 191). Activation of the canonical pathway induces c-Rel, which binds the conserved non-coding sequence (CNS)-3 enhancer of FoxP3 to promote its expression (192, 193). Thus, c-Rel deficiency results in a similar Treg deficiency, while Tregs are preserved in the face of p50 deficiency (88). Furthermore, constitutive activation of IKKβ appears to be sufficient to drive Treg development even in the absence of TCR ligation (194).
NF-κB, Treg Homeostasis and Susceptibility to Autoimmunity
Once selected, ongoing NF-κB signals appear to be necessary to maintain Tregs and prevent end-organ inflammation. Comparison with Tconv reveals that there is greater accumulation of canonical NF-κB components in nuclei of Tregs (84). CD28 signals are important for Treg homeostasis in the periphery. Disruption of CD80/86 with CTLA4Ig results in a loss of Tregs (195, 196). T cell-intrinsic defects in the canonical pathway do not, however, always result in autoimmunity or organ-specific inflammation, as Tconv also exhibit a defect in activation.
Resting Tregs are predominantly located in secondary lymphoid organs due to specific chemokine receptor expression, while activated Tregs migrate into sites of inflammation and are characterized by a greater array of suppressive functions, including IL-10, Lag-3, TIGIT, CD73, and PD-1 expression (197). NF-κB also regulates Treg activation. Acquisition of the effector phenotype depends on TCR ligation (198). RelA appears to play a dominant role in maintaining effector Treg function in the periphery. Rela (p65) deletion is embryonic lethal, but recent evidence from conditional deficiency indicates that p65 is crucial for maintaining Treg lineage stability and activity (Table 1) (84, 85). Treg-specific p65 deficiency confers a multi-organ inflammatory phenotype. c-Rel deficiency confers a less prominent inflammatory phenotype (199). In addition to Treg homeostasis, canonical signaling appears to specify whether Tregs adopt either resting (CD62Lhi CD44lo) or activated/effector (CD62Llo CD44hi) phenotypes (200).
NF-κB Contributes to B Cell Homeostasis and Self-Tolerance
In mouse models, B cell survival depends on signals via BCR and BAFF (201–203), implicating signaling though canonical and non-canonical NF-κB pathways. Several mechanisms normally operate to eliminate autoreactive B cells (204). In the bone marrow, high affinity ligation by antigen results in receptor internalization, attenuation of NF-κB-mediated induction of Bcl-2, increased Bcl-2-interacting mediator (BIM), and reduced expression of regulation of B cell activating factor receptor (BAFFR). V(D)J recombination continues by virtue of ongoing RAG1/2 expression resulting in receptor editing but if immature B cells remain autoreactive they undergo clonal deletion. Receptor editing is marked by enhanced canonical signaling (205).
Clonal anergy represents a third mechanism by which autoreactive B cells enter the periphery with diminished capacity for activation. B cell anergy is characterized by reduced BCR expression, which attenuates canonical NF-κB survival signals and BAFFR expression is reduced, while pro-apoptotic signals such as BIM continue (206). BAFFR signals predominantly through the non-canonical pathway, and enhances B cell survival via induction of Bcl-2, PIM2, and PKCδ (207, 208). These actions reduce the risk of activation and survival of autoreactive B cells. BAFF is normally available in limiting amounts, which regulates the size of the transitional B cell compartment, but also regulates survival of autoreactive anergic B cells. Experimental manipulations demonstrate that excessive BAFF production results in improved survival of anergic B cells (206, 209).
Rare Genetic Variants of NF-κB and Human Autoimmune Disease
Rare single gene disorders provide important evidence to support mechanistic pathways of human disease. Mutations affecting NF-κB activation, and its proximal signaling pathways, have so far been implicated overwhelmingly in susceptibility to infection (i.e., primary immune deficiency diseases). For the most part, these are loss-of-function mutations arising from homozygous or biallelic mutations. An exception is gain-of-function (GoF) mutations in NFKBIA (IκBα), but since this is the fundamental negative regulator of canonical signaling, GoF reinforces lack of activation and, therefore, results in a clinical and cellular phenotype that resembles LoF mutations in IKKγ and IKKβ (210, 211).
Several Mendelian human diseases nevertheless provide evidence that NF-κB protects against autoimmunity (Table 2). In other cases, NF-κB defects have been shown to cause auto-inflammatory diseases, which also result in tissue-specific inflammatory responses, but are distinguished by the absence of evidence for adaptive immune responses to autoantigens.
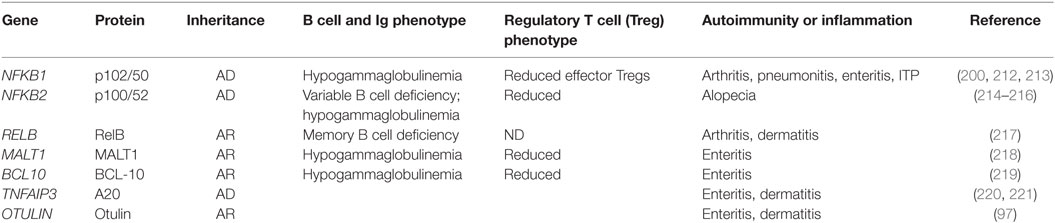
Table 2. Mendelian defects in NF-κB pathway genes that confer syndromes including autoimmune manifestations.
Syndromes arising from heterozygous mutations in NFKB1 include autoimmune manifestations of arthritis, lung inflammation, gut inflammation, and immune-mediated thrombocytopenic purpura (ITP). There appears to be considerable clinical and cellular heterogeneity and postulated incomplete penetrance (212). The same mutation can also lead to antibody deficiency (213). Interestingly, as noted above, recent evidence from mouse studies showed that canonical signaling via RelA is important for maintaining Tregs in the periphery. Consistent with this, patients with NFKB1 haploinsufficiency also show a defect in Tregs, with a diminution in effector Tregs (as judged by ICOS expression) (200).
NFKB2 mutations result in a syndrome of antibody deficiency with variable B cell deficiencies (214–216). Patients with NFKB2 mutations exhibit other clinical features as well, including defects of the anterior pituitary and alopecia. In at least some cases, the alopecia is reversible, consistent with alopecia areata. The fundamental defect in self-tolerance conferred by NFKB2 mutations is yet to be resolved. Mouse models (outlined above) point to the possibility of a defect in central tolerance, and human NFKB2 defects have been shown to confer a reduction in circulating Tregs (214). Whether there is a causal association between this observation and end-organ pathology remains to be resolved.
RelB deficiency conferred by a rare homozygous nonsense mutation has been reported in a single patient with severe polyarthritis and inflammatory skin disease, as well as immune deficiency (217). Interestingly, this phenotype was associated with enhanced nuclear p65 translocation, but a defect in T cell activation and thymus biopsy was reported to show a defect in T cell production.
MALT1 and BCL-10 deficiencies both appear to result in an IPEX (immune dysregulation polyendocrinopathy, enteropathy, X-linked; OMIM 304790)-like syndrome, which was originally identified in young boys with FOXP3 deficiency and subsequently also observed with heterozygous mutations in CTLA4 and IL2RA (CD25) (222–225). Homozygous MALT1 mutations result in a phenotype of primary antibody deficiency together with enteropathy and a reduction in Tregs (218). A similar phenotype of enteropathy and Treg deficiency has been observed with BCL10 deficiency (219). Again, the precise mechanism of action remains to be determined, although as noted above, CBM defects in mice are typically associated with Treg deficiency.
Several patients have been reported with a Mendelian syndromes arising from haploinsufficiency of TNFAIP3 (220, 221). This autoinflammatory syndrome is reminiscent of Behcet’s disease, with manifestations, including aphthous ulceration, inflammatory bowel disease, and neutrophilic dermatoses. Biochemical analysis confirmed increased canonical pathway activation, prolonged nuclear translocation of p65, and enhanced transcription of NF-κB-dependent genes, including IL6 and TNF. A similar autoinflammatory syndrome of panniculitis and dermatitis has been reported to arise from homozygous loss-of-function OTULIN mutations affecting the catalytic OTU domain. These mutations result in increased ubiquitination of several NF-κB proteins, including NEMO and RIPK1 (97).
Polygenic Autoimmunity
The mechanisms explaining sporadic autoimmune disease remain poorly understood. Rare monogenic cases provide clues to pathogenic pathways that become dysregulated in more common versions of the disease, even if in any given individual these cellular and biochemical defects arise from more than one gene defect. Human AIRE mutations provide evidence for the importance of promiscuous thymic TSA expression in protecting against organ-specific autoimmune disease (160, 177, 226). Interestingly, autosomal-dominant defects in AIRE have now been reported, raising the possibility of more prevalent single gene causes of autoimmunity (227).
Thymic hyperplasia and thymoma have been associated with some forms of organ-specific autoimmunity, most notably, myasthenia gravis (MG), an autoimmune disease of the muscle end-plate. MG is characterized by muscle fatigability and weakness, is often associated with thymic hyperplasia or thymoma, together with defects in CD4+ T cell selection and intrathymic cytokine production. Thymectomy is sometimes curative (228). Certain common AIRE polymorphisms segregate with MG, particularly in early-onset cases (229).
While plausible, there is no empirical evidence that genetic variants affecting non-canonical NF-κB signaling segregate with organ-specific autoimmune disease. The most consistent NF-κB genetic association with autoimmunity is TNFAIP3. Numerous coding and non-coding polymorphisms have been shown to segregate with autoimmune diseases, including lupus and organ-specific autoimmune diseases (230–232). Polymorphisms of REL (c-Rel) have been shown to segregate with both psoriatic arthritis and rheumatoid arthritis, and altered expression has been noted in other autoimmune diseases (233–236). Common NFKB1 polymorphisms have been shown to segregate with several autoimmune and inflammatory diseases (237–239). More recently, a polymorphism within the NFKB1 locus was shown to segregate with multiple sclerosis. Interestingly, MS patients were found to have increased levels of p65 (RelA) activation, and the disease associated polymorphism was found to be associated with increased expression of BCL-3, TNFAIP3, and CIAP1 (240). This represents important progress toward understanding how common variants in NF-κB might contribute to sporadic and more common autoimmune diseases, via pathways now well understood from investigations of animal models and patients with rare monogenic disease.
Conclusion
NF-κB is well established as a regulator of innate and adaptive mechanisms of host defense, and specific genetic defects that confer immune deficiency confirm the importance of these mechanisms. As described here, there is also abundant evidence that NF-κB is crucial for maintaining immunological tolerance, as a result of its actions during thymic selection, both for negative selection of autoreactive T cells, and selection and maintenance of Tregs. The non-canonical NF-κB pathway appears to be particularly important for normal mTec function; nevertheless, evidence has also emerged for a significant lymphocyte-intrinsic action of the canonical pathway for maintaining T cell tolerance. Further work is required to accomplish the challenging task of separating the actions of canonical and non-canonical components, cell-intrinsic versus cell extrinsic actions, actions on negative selection versus Treg defects. Finally, it is important to note that since NF-κB is crucial for Tconv function in the periphery, it is possible that defects in selection may be offset by defects in the Tconv compartment.
Author Contributions
Both the authors contributed to conceptualizing and writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by NHMRC program grant 1113577, NHMRC project grant 1079648, and NHMRC Centre of Research Excellence grant 1107464.
Abbreviations
Aire, autoimmune regulator; BAFFR, regulation of B cell activating factor receptor; BIM, Bcl-2-interacting mediator; CBM, CARD/BCL-10/MALT1 complex; cTEC, cortical thymic epithelial cell; DC, dendritic cell; DUB, deubiquitinase; EAE, experimental autoimmune encephalomyelitis; ENU, N-ethyl-N-nitrosourea; Ep-CAM, epithelial cellular adhesion molecule; GC, germinal center; IκB, inhibitor of kappa B; IKK, inhibitor of kappa B kinase; LPS, lipopolysaccharide; mTEC, medullary thymic epithelial cell; MZ, marginal zone; NEMO, NF-κB essential modulator; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NIK, NF-κB-inducing kinase; nTreg, natural regulatory T cell; RANK, receptor activator of nuclear factor kappa-B; TAD, transactivation domain; Tconv, Conventional T cell; Treg, regulatory T cell; TSA, tissue specific antigens; UEA-1, Ulex europaeus agglutinin 1.
References
1. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell (2017) 168:37–57. doi:10.1016/j.cell.2016.12.012
2. Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu Rev Immunol (2007) 25:697–743. doi:10.1146/annurev.immunol.25.022106.141615
3. Oh H, Ghosh S. NF-κB: roles and regulation in different CD4(+) T-cell subsets. Immunol Rev (2013) 252:41–51. doi:10.1111/imr.12033
4. Kaileh M, Sen R. NF-κB function in B lymphocytes. Immunol Rev (2012) 246:254–71. doi:10.1111/j.1600-065X.2012.01106.x
5. Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol (2009) 27:693–733. doi:10.1146/annurev.immunol.021908.132641
6. Courtois G, Gilmore TD. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene (2006) 25:6831–43. doi:10.1038/sj.onc.1209939
7. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation—the negative regulation of NF-κB and the NLRP3 inflammasome. Nat Immunol (2017) 18:861–9. doi:10.1038/ni.3772
8. Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol (2010) 2:a000109. doi:10.1101/cshperspect.a000109
9. Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene (1999) 18:6853–66. doi:10.1038/sj.onc.1203239
10. Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev (2004) 18:2195–224. doi:10.1101/gad.1228704
11. Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell (1996) 87:13–20. doi:10.1016/S0092-8674(00)81318-5
12. Rothwarf DM, Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE (1999) 1999:RE1–1. doi:10.1126/stke.1999.5.re1
13. Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol (1994) 10:405–55. doi:10.1146/annurev.cb.10.110194.002201
14. Sun S-C, Ley SC. New insights into NF-kappaB regulation and function. Trends Immunol (2008) 29:469–78. doi:10.1016/j.it.2008.07.003
15. Heissmeyer V, Krappmann D, Hatada EN, Scheidereit C. Shared pathways of IkappaB kinase-induced SCF(betaTrCP)-mediated ubiquitination and degradation for the NF-kappaB precursor p105 and IkappaBalpha. Mol Cell Biol (2001) 21:1024–35. doi:10.1128/MCB.21.4.1024-1035.2001
16. Saccani S, Pantano S, Natoli G. Modulation of NF-kappaB activity by exchange of dimers. Mol Cell (2003) 11:1563–74. doi:10.1016/S1097-2765(03)00227-2
17. Plaksin D, Baeuerle PA, Eisenbach L. KBF1 (p50 NF-kappa B homodimer) acts as a repressor of H-2Kb gene expression in metastatic tumor cells. J Exp Med (1993) 177:1651–62. doi:10.1084/jem.177.6.1651
18. Bohuslav J, Kravchenko VV, Parry GC, Erlich JH, Gerondakis S, Mackman N, et al. Regulation of an essential innate immune response by the p50 subunit of NF-kappaB. J Clin Invest (1998) 102:1645–52. doi:10.1172/JCI3877
19. Ledebur HC, Parks TP. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-kappa B site and p65 homodimers. J Biol Chem (1995) 270:933–43. doi:10.1074/jbc.270.2.933
20. Kang SM, Tran AC, Grilli M, Lenardo MJ. NF-kappa B subunit regulation in nontransformed CD4+ T lymphocytes. Science (1992) 256:1452–6. doi:10.1126/science.1604322
21. Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell (2002) 9:625–36. doi:10.1016/S1097-2765(02)00477-X
22. Ryseck RP, Bull P, Takamiya M, Bours V, Siebenlist U, Dobrzanski P, et al. RelB, a new Rel family transcription activator that can interact with p50-NF-kappa B. Mol Cell Biol (1992) 12:674–84. doi:10.1128/MCB.12.2.674
23. Dobrzanski P, Ryseck RP, Bravo R. Both N- and C-terminal domains of RelB are required for full transactivation: role of the N-terminal leucine zipper-like motif. Mol Cell Biol (1993) 13:1572–82. doi:10.1128/MCB.13.3.1572
24. Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer’s patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J (2003) 22:121–30. doi:10.1093/emboj/cdg004
25. Derudder E, Dejardin E, Pritchard LL, Green DR, Korner M, Baud V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation: critical roles for p100. J Biol Chem (2003) 278:23278–84. doi:10.1074/jbc.M300106200
26. Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev (2012) 26:203–34. doi:10.1101/gad.183434.111
27. Marienfeld R, May MJ, Berberich I, Serfling E, Ghosh S, Neumann M. RelB forms transcriptionally inactive complexes with RelA/p65. J Biol Chem (2003) 278:19852–60. doi:10.1074/jbc.M301945200
28. Köntgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev (1995) 9:1965–77. doi:10.1101/gad.9.16.1965
29. Liou HC, Sha WC, Scott ML, Baltimore D. Sequential induction of NF-kappa B/Rel family proteins during B-cell terminal differentiation. Mol Cell Biol (1994) 14:5349–59. doi:10.1128/MCB.14.8.5349
30. Weih F, Carrasco D, Bravo R. Constitutive and inducible Rel/NF-kappa B activities in mouse thymus and spleen. Oncogene (1994) 9:3289–97.
31. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell (2008) 132:344–62. doi:10.1016/j.cell.2008.01.020
32. Yamamoto M, Yamazaki S, Uematsu S, Sato S, Hemmi H, Hoshino K, et al. Regulation of toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature (2004) 430:218–22. doi:10.1038/nature02738
33. Motoyama M, Yamazaki S, Eto-Kimura A, Takeshige K, Muta T. Positive and negative regulation of nuclear factor-kappaB-mediated transcription by IkappaB-zeta, an inducible nuclear protein. J Biol Chem (2005) 280:7444–51. doi:10.1074/jbc.M412738200
34. Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, et al. The oncoprotein Bcl-3 directly transactivates through kappa B motifs via association with DNA-binding p50B homodimers. Cell (1993) 72:729–39. doi:10.1016/0092-8674(93)90401-B
35. Nolan GP, Fujita T, Bhatia K, Huppi C, Liou HC, Scott ML, et al. The bcl-3 proto-oncogene encodes a nuclear I kappa B-like molecule that preferentially interacts with NF-kappa B p50 and p52 in a phosphorylation-dependent manner. Mol Cell Biol (1993) 13:3557–66. doi:10.1128/MCB.13.6.3557
36. Franzoso G, Bours V, Azarenko V, Park S, Tomita-Yamaguchi M, Kanno T, et al. The oncoprotein Bcl-3 can facilitate NF-kappa B-mediated transactivation by removing inhibiting p50 homodimers from select kappa B sites. EMBO J (1993) 12:3893–901.
37. Inoue J, Kerr LD, Kakizuka A, Verma IM. I kappa B gamma, a 70 kd protein identical to the C-terminal half of p110 NF-kappa B: a new member of the I kappa B family. Cell (1992) 68:1109–20. doi:10.1016/0092-8674(92)90082-N
38. Wessells J, Baer M, Young HA, Claudio E, Brown K, Siebenlist U, et al. BCL-3 and NF-kappaB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J Biol Chem (2004) 279:49995–50003. doi:10.1074/jbc.M404246200
39. Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative regulation of toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science (2007) 317:675–8. doi:10.1126/science.1142953
40. Hatada EN, Nieters A, Wulczyn FG, Naumann M, Meyer R, Nucifora G, et al. The ankyrin repeat domains of the NF-kappa B precursor p105 and the protooncogene bcl-3 act as specific inhibitors of NF-kappa B DNA binding. Proc Natl Acad Sci U S A (1992) 89:2489–93. doi:10.1073/pnas.89.6.2489
41. Israël A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol (2010) 2:a000158. doi:10.1101/cshperspect.a000158
42. Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell (1997) 91:243–52. doi:10.1016/S0092-8674(00)80406-7
43. Zandi E, Chen Y, Karin M. Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: discrimination between free and NF-kappaB-bound substrate. Science (1998) 281:1360–3. doi:10.1126/science.281.5381.1360
44. Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science (1999) 284:309–13. doi:10.1126/science.284.5412.309
45. Mercurio F, Murray BW, Shevchenko A, Bennett BL, Young DB, Li JW, et al. IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol (1999) 19:1526–38. doi:10.1128/MCB.19.2.1526
46. Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene (2006) 25:6685–705. doi:10.1038/sj.onc.1209934
47. Häcker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE (2006) 2006:re13. doi:10.1126/stke.3572006re13
48. Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, et al. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity (1999) 10:421–9. doi:10.1016/S1074-7613(00)80042-4
49. Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science (1999) 284:321–5. doi:10.1126/science.284.5412.321
50. Sato S, Sanjo H, Tsujimura T, Ninomiya-Tsuji J, Yamamoto M, Kawai T, et al. TAK1 is indispensable for development of T cells and prevention of colitis by the generation of regulatory T cells. Int Immunol (2006) 18:1405–11. doi:10.1093/intimm/dxl082
51. Chang J-H, Hu H, Sun S-C. Survival and maintenance of regulatory T cells require the kinase TAK1. Cell Mol Immunol (2015) 12:572–9. doi:10.1038/cmi.2015.27
52. Tang M, Wei X, Guo Y, Breslin P, Zhang S, Zhang S, et al. TAK1 is required for the survival of hematopoietic cells and hepatocytes in mice. J Exp Med (2008) 205:1611–9. doi:10.1084/jem.20080297
53. Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, et al. Identifying the MAGUK protein carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity (2003) 18:751–62. doi:10.1016/S1074-7613(03)00141-9
54. Altin JA, Tian L, Liston A, Bertram EM, Goodnow CC, Cook MC. Decreased T-cell receptor signaling through CARD11 differentially compromises forkhead box protein 3-positive regulatory versus T(H)2 effector cells to cause allergy. J Allergy Clin Immunol (2011) 127:1277–85.e5. doi:10.1016/j.jaci.2010.12.1081
55. Barnes MJ, Krebs P, Eidenschenk C, Arnold CN, Crozat K, Sovath S, et al. Commitment to the regulatory T cell lineage requires CARMA1 in the thymus but not in the periphery. PLoS Biol (2009) 7:e51. doi:10.1371/journal.pbio.1000051
56. Molinero LL, Yang J, Gajewski T, Abraham C, Farrar MA, Alegre M-L. CARMA1 controls an early checkpoint in the thymic development of FoxP3+ regulatory T cells. J Immunol (2009) 182:6736–43. doi:10.4049/jimmunol.0900498
57. Jaworski M, Marsland BJ, Gehrig J, Held W, Favre S, Luther SA, et al. Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J (2014) 33:2765–81. doi:10.15252/embj.201488987
58. Ruefli-Brasse AA, French DM, Dixit VM. Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science (2003) 302:1581–4. doi:10.1126/science.1090769
59. Ruland J, Duncan GS, Wakeham A, Mak TW. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity (2003) 19:749–58. doi:10.1016/S1074-7613(03)00293-0
60. Brüstle A, Brenner D, Knobbe-Thomsen CB, Cox M, Lang PA, Lang KS, et al. MALT1 is an intrinsic regulator of regulatory T cells. Cell Death Differ (2017) 24:1214–23. doi:10.1038/cdd.2015.104
61. Ruland J, Duncan GS, Elia A, del Barco Barrantes I, Nguyen L, Plyte S, et al. Bcl10 is a positive regulator of antigen receptor-induced activation of NF-κ B and neural tube closure. Cell (2001) 104:33–42. doi:10.1016/S0092-8674(01)00189-1
62. Xue L, Morris SW, Orihuela C, Tuomanen E, Cui X, Wen R, et al. Defective development and function of Bcl10-deficient follicular, marginal zone and B1 B cells. Nat Immunol (2003) 4:857. doi:10.1038/ni963
63. Schmidt-Supprian M, Tian J, Grant EP, Pasparakis M, Maehr R, Ovaa H, et al. Differential dependence of CD4(+)CD25(+) regulatory and natural killer-like T cells on signals leading to NF-κB activation. Proc Natl Acad Sci U S A (2004) 101:4566–71. doi:10.1073/pnas.0400885101
64. Chen X, Willette-Brown J, Wu X, Hu Y, Howard OMZ, Hu Y, et al. IKKα is required for the homeostasis of regulatory T cells and for the expansion of both regulatory and effector CD4 T cells. FASEB J (2015) 29:443–54. doi:10.1096/fj.14-259564
65. Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, et al. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science (1999) 284:316–20. doi:10.1126/science.284.5412.316
66. Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, et al. Limb and skin abnormalities in mice lacking IKKalpha. Science (1999) 284:313–6. doi:10.1126/science.284.5412.313
67. Schmidt-Supprian M, Courtois G, Tian J, Coyle AJ, Israël A, Rajewsky K, et al. Mature T cells depend on signaling through the IKK complex. Immunity (2003) 19:377–89. doi:10.1016/S1074-7613(03)00237-1
68. Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israël A, Rajewsky K, et al. NEMO/IKKγ-deficient mice model incontinentia pigmenti. Mol Cell (2000) 5:981–92. doi:10.1016/S1097-2765(00)80263-4
69. Miyawaki S, Nakamura Y, Suzuka H, Koba M, Yasumizu R, Ikehara S, et al. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur J Immunol (1994) 24:429–34. doi:10.1002/eji.1830240224
70. Shinkura R, Kitada K, Matsuda F, Tashiro K, Ikuta K, Suzuki M, et al. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat Genet (1999) 22:74–7. doi:10.1038/8780
71. Tsubata R, Tsubata T, Hiai H, Shinkura R, Matsumura R, Sumida T, et al. Autoimmune disease of exocrine organs in immunodeficient alymphoplasia mice: a spontaneous model for Sjögren’s syndrome. Eur J Immunol (1996) 26:2742–8. doi:10.1002/eji.1830261129
72. Yamada T, Mitani T, Yorita K, Uchida D, Matsushima A, Iwamasa K, et al. Abnormal immune function of hemopoietic cells from alymphoplasia (<em>aly</em>) mice, a natural strain with mutant NF-κB-inducing kinase. J Immunol (2000) 165:804–12. doi:10.4049/jimmunol.165.2.804
73. Häcker H, Chi L, Rehg JE, Redecke V. NIK prevents the development of hypereosinophilic syndrome-like disease in mice independent of IKKα activation. J Immunol (2012) 188:4602–10. doi:10.4049/jimmunol.1200021
74. Peng B, Ling J, Lee AJ, Wang Z, Chang Z, Jin W, et al. Defective feedback regulation of NF-κB underlies Sjögren’s syndrome in mice with mutated κB enhancers of the IκBα promoter. Proc Natl Acad Sci U S A (2010) 107:15193–8. doi:10.1073/pnas.1005533107
75. Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev (1995) 9:2736–46. doi:10.1101/gad.9.22.2736
76. Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell (1995) 80:321–30. doi:10.1016/0092-8674(95)90415-8
77. Grumont RJ, Rourke IJ, O’Reilly LA, Strasser A, Miyake K, Sha W, et al. B lymphocytes differentially use the rel and nuclear factor κB1 (NF-κB1) transcription factors to regulate cell cycle progression and apoptosis in quiescent and mitogen-activated cells. J Exp Med (1998) 187:663–74. doi:10.1084/jem.187.5.663
78. Cariappa A, Liou H-C, Horwitz BH, Pillai S. Nuclear factor κb is required for the development of marginal zone B lymphocytes. J Exp Med (2000) 192:1175–82. doi:10.1084/jem.192.8.1175
79. Oakley F, Mann J, Nailard S, Smart DE, Mungalsingh N, Constandinou C, et al. Nuclear factor-κB1 (p50) limits the inflammatory and fibrogenic responses to chronic injury. Am J Pathol (2005) 166:695–708. doi:10.1016/S0002-9440(10)62291-2
80. Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun (2014) 2:4172. doi:10.1038/ncomms5172
81. de Valle E, Grigoriadis G, O’Reilly LA, Willis SN, Maxwell MJ, Corcoran LM, et al. NFκB1 is essential to prevent the development of multiorgan autoimmunity by limiting IL-6 production in follicular B cells. J Exp Med (2016) 213:621–41. doi:10.1084/jem.20151182
82. Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, et al. Mice deficient in nuclear factor (NF)-κB/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med (1998) 187:147–59. doi:10.1084/jem.187.2.147
83. Caamaño JH, Rizzo CA, Durham SK, Barton DS, Raventós-Suárez C, Snapper CM, et al. Nuclear factor (NF)-κB2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med (1998) 187:185–96. doi:10.1084/jem.187.2.185
84. Messina N, Fulford T, O’Reilly L, Loh WX, Motyer JM, Ellis D, et al. The NF-κB transcription factor RelA is required for the tolerogenic function of Foxp3(+) regulatory T cells. J Autoimmun (2016) 70:52–62. doi:10.1016/j.jaut.2016.03.017
85. Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, et al. An NF-κB transcription-factor-dependent lineage-specific transcriptional program promotes regulatory T cell identity and function. Immunity (2017) 47:450–65.e5. doi:10.1016/j.immuni.2017.08.010
86. Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, et al. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell (1995) 80:331–40. doi:10.1016/0092-8674(95)90416-6
87. Weih DS, Yilmaz ZB, Weih F. Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J Immunol (2001) 167:1909–19. doi:10.4049/jimmunol.167.4.1909
88. Deenick EK, Elford AR, Pellegrini M, Hall H, Mak TW, Ohashi PS. c-Rel but not NF-kappaB1 is important for T regulatory cell development. Eur J Immunol (2010) 40:677–81. doi:10.1002/eji.201040298
89. Tang W, Wang H, Claudio E, Tassi I, Ha H-L, Saret S, et al. The oncoprotein and transcriptional regulator Bcl-3 governs plasticity and pathogenicity of auto-immune T cells. Immunity (2014) 41:555–66. doi:10.1016/j.immuni.2014.09.017
90. Franzoso G, Carlson L, Scharton-Kersten T, Shores EW, Epstein S, Grinberg A, et al. Critical roles for the Bcl-3 oncoprotein in T cell-mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity (1997) 6:479–90. doi:10.1016/S1074-7613(00)80291-5
91. Zhang X, Paun A, Claudio E, Wang H, Siebenlist U. The tumor promoter and NF-κB modulator Bcl-3 regulates splenic B cell development. J Immunol (2013) 191:5984–92. doi:10.4049/jimmunol.1300611
92. Schuster M, Glauben R, Plaza-Sirvent C, Schreiber L, Annemann M, Floess S, et al. IκBNS protein mediates regulatory T cell development via induction of the Foxp3 transcription factor. Immunity (2012) 37:998–1008. doi:10.1016/j.immuni.2012.08.023
93. Kobayashi S, Hara A, Isagawa T, Manabe I, Takeda K, MaruYama T. The nuclear IκB family protein IκB(NS) influences the susceptibility to experimental autoimmune encephalomyelitis in a murine model. PLoS One (2014) 9:e110838. doi:10.1371/journal.pone.0110838
94. Shiina T, Konno A, Oonuma T, Kitamura H, Imaoka K, Takeda N, et al. Targeted disruption of MAIL, a nuclear IkappaB protein, leads to severe atopic dermatitis-like disease. J Biol Chem (2004) 279:55493–8. doi:10.1074/jbc.M409770200
95. Okamoto K, Iwai Y, Oh-Hora M, Yamamoto M, Morio T, Aoki K, et al. IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature (2010) 464:1381–5. doi:10.1038/nature08922
96. Johansen C, Mose M, Ommen P, Bertelsen T, Vinter H, Hailfinger S, et al. IκBζ is a key driver in the development of psoriasis. Proc Natl Acad Sci U S A (2015) 112:E5825–33. doi:10.1073/pnas.1509971112
97. Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, et al. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell (2016) 166:1215–30.e20. doi:10.1016/j.cell.2016.07.019
98. Chu Y, Soberon V, Glockner L, Beyaert R, Massoumi R, van Loo G, et al. A20 and CYLD do not share significant overlapping functions during B cell development and activation. J Immunol (2012) 189:4437–43. doi:10.4049/jimmunol.1200396
99. Evans PC, Ovaa H, Hamon M, Kilshaw PJ, Hamm S, Bauer S, et al. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem J (2004) 378:727–34. doi:10.1042/BJ20031377
100. Kool M, van Loo G, Waelput W, De Prijck S, Muskens F, Sze M, et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity (2011) 35:82–96. doi:10.1016/j.immuni.2011.05.013
101. Reiley WW, Jin W, Lee AJ, Wright A, Wu X, Tewalt EF, et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med (2007) 204:1475–85. doi:10.1084/jem.20062694
102. Reiley WW, Zhang M, Jin W, Losiewicz M, Donohue KB, Norbury CC, et al. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat Immunol (2006) 7:411–7. doi:10.1038/ni1315
103. Jin W, Reiley WR, Lee AJ, Wright A, Wu X, Zhang M, et al. Deubiquitinating enzyme CYLD regulates the peripheral development and naive phenotype maintenance of B cells. J Biol Chem (2007) 282:15884–93. doi:10.1074/jbc.M609952200
104. Reissig S, Hövelmeyer N, Weigmann B, Nikolaev A, Kalt B, Wunderlich TF, et al. The tumor suppressor CYLD controls the function of murine regulatory T cells. J Immunol (2012) 189:4770–6. doi:10.4049/jimmunol.1201993
105. Hövelmeyer N, Wunderlich FT, Massoumi R, Jakobsen CG, Song J, Wörns MA, et al. Regulation of B cell homeostasis and activation by the tumor suppressor gene CYLD. J Exp Med (2007) 204:2615–27. doi:10.1084/jem.20070318
106. Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature (1999) 398:252–6. doi:10.1038/18465
107. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature (2001) 412:346–51. doi:10.1038/35085597
108. Ermolaeva MA, Michallet M-C, Papadopoulou N, Utermöhlen O, Kranidioti K, Kollias G, et al. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat Immunol (2008) 9:1037–46. doi:10.1038/ni.1638
109. Chen N-J, Chio IIC, Lin W-J, Duncan G, Chau H, Katz D, et al. Beyond tumor necrosis factor receptor: TRADD signaling in toll-like receptors. Proc Natl Acad Sci U S A (2008) 105:12429–34. doi:10.1073/pnas.0806585105
110. Vince JE, Wong WW-L, Khan N, Feltham R, Chau D, Ahmed AU, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell (2007) 131:682–93. doi:10.1016/j.cell.2007.10.037
111. Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci U S A (2008) 105:11778–83. doi:10.1073/pnas.0711122105
112. Iwai K, Tokunaga F. Linear polyubiquitination: a new regulator of NF-kappaB activation. EMBO Rep (2009) 10:706–13. doi:10.1038/embor.2009.144
113. Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature (2004) 430:694–9. doi:10.1038/nature02794
114. Tokunaga F, Nishimasu H, Ishitani R, Goto E, Noguchi T, Mio K, et al. Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-κB regulation. EMBO J (2012) 31:3856–70. doi:10.1038/emboj.2012.241
115. Verhelst K, Verstrepen L, Carpentier I, Beyaert R. Linear ubiquitination in NF-κB signaling and inflammation: what we do understand and what we do not. Biochem Pharmacol (2011) 82:1057–65. doi:10.1016/j.bcp.2011.07.066
116. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer (2013) 12:86. doi:10.1186/1476-4598-12-86
117. Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science (2000) 289:2350–4. doi:10.1126/science.289.5488.2350
118. Coornaert B, Baens M, Haegman M, Sun L, Marynen P. T cell antigen receptor stimulation induces MALT1 paracaspase–mediated cleavage of the NF-κB inhibitor A20. Nat Immunol (2008) 9:263–71. doi:10.1038/ni1561
119. Heyninck K, Beyaert R. The cytokine-inducible zinc finger protein A20 inhibits IL-1-induced NF-kappaB activation at the level of TRAF6. FEBS Lett (1999) 442:147–50. doi:10.1016/S0014-5793(98)01645-7
120. Song HY, Rothe M, Goeddel DV. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc Natl Acad Sci U S A (1996) 93:6721–5. doi:10.1073/pnas.93.13.6721
121. Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wójtowicz E, et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood (2011) 117:2227–36. doi:10.1182/blood-2010-09-306019
122. Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S, et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol (2011) 12:1184–93. doi:10.1038/ni.2135
123. Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature (2014) 512:69–73. doi:10.1038/nature13322
124. Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol (2014) 35:22–31. doi:10.1016/j.it.2013.10.005
125. De A, Dainichi T, Rathinam CV, Ghosh S. The deubiquitinase activity of A20 is dispensable for NF-κB signaling. EMBO Rep (2014) 15:775–83. doi:10.15252/embr.201338305
126. Fujita H, Rahighi S, Akita M, Kato R, Sasaki Y, Wakatsuki S, et al. Mechanism underlying IκB kinase activation mediated by the linear ubiquitin chain assembly complex. Mol Cell Biol (2014) 34:1322–35. doi:10.1128/MCB.01538-13
127. Ea C-K, Deng L, Xia Z-P, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell (2006) 22:245–57. doi:10.1016/j.molcel.2006.03.026
128. Kanayama A, Seth RB, Sun L, Ea C-K, Hong M, Shaito A, et al. TAB 2 and TAB 3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell (2004) 15:535–48. doi:10.1016/j.molcel.2004.08.008
129. Lork M, Verhelst K, Beyaert R. CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: so similar, yet so different. Cell Death Differ (2017) 24:1172–83. doi:10.1038/cdd.2017.46
130. Goto E, Tokunaga F. Decreased linear ubiquitination of NEMO and FADD on apoptosis with caspase-mediated cleavage of HOIP. Biochem Biophys Res Commun (2017) 485:152–9. doi:10.1016/j.bbrc.2017.02.040
131. Sun S-C. The noncanonical NF-κB pathway. Immunol Rev (2012) 246:125–40. doi:10.1111/j.1600-065X.2011.01088.x
132. Liao G, Zhang M, Harhaj EW, Sun S-C. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem (2004) 279:26243–50. doi:10.1074/jbc.M403286200
133. He JQ, Zarnegar B, Oganesyan G, Saha SK, Yamazaki S, Doyle SE, et al. Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J Exp Med (2006) 203:2413–8. doi:10.1084/jem.20061166
134. Park SW, Huq MDM, Hu X, Wei L-N. Tyrosine nitration on p65: a novel mechanism to rapidly inactivate nuclear factor-kappaB. Mol Cell Proteomics (2005) 4:300–9. doi:10.1074/mcp.M400195-MCP200
135. Senftleben U, Li ZW, Baud V, Karin M. IKKbeta is essential for protecting T cells from TNFalpha-induced apoptosis. Immunity (2001) 14:217–30. doi:10.1016/S1074-7613(01)00104-2
136. Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IkappaB kinase. Cell (1997) 90:373–83. doi:10.1016/S0092-8674(00)80344-X
137. Xiao G, Cvijic ME, Fong A, Harhaj EW, Uhlik MT, Waterfield M, et al. Retroviral oncoprotein tax induces processing of NF-kappaB2/p100 in T cells: evidence for the involvement of IKKalpha. EMBO J (2001) 20:6805–15. doi:10.1093/emboj/20.23.6805
138. Uhlik M, Good L, Xiao G, Harhaj EW, Zandi E, Karin M, et al. NF-kappaB-inducing kinase and IkappaB kinase participate in human T-cell leukemia virus I tax-mediated NF-kappaB activation. J Biol Chem (1998) 273:21132–6. doi:10.1074/jbc.273.33.21132
139. Lin L, DeMartino GN, Greene WC. Cotranslational biogenesis of NF-kappaB p50 by the 26S proteasome. Cell (1998) 92:819–28. doi:10.1016/S0092-8674(00)81409-9
140. Liu J, Sudom A, Min X, Cao Z, Gao X, Ayres M, et al. Structure of the nuclear factor κB-inducing kinase (NIK) kinase domain reveals a constitutively active conformation. J Biol Chem (2012) 287:27326–34. doi:10.1074/jbc.M112.366658
141. Tao Z, Fusco A, Huang D-B, Gupta K, Kim DY, Ware CF, et al. p100/IκBδ sequesters and inhibits NF-κB through kappaBsome formation. Proc Natl Acad Sci U S A (2014) 111:15946–51. doi:10.1073/pnas.1408552111
142. Fusco AJ, Mazumder A, Wang VY-F, Tao Z, Ware C, Ghosh G. The NF-κB subunit RelB controls p100 processing by competing with the kinases NIK and IKK1 for binding to p100. Sci Signal (2016) 9:ra96–96. doi:10.1126/scisignal.aad9413
143. Chen L-F, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J (2002) 21:6539–48. doi:10.1093/emboj/cdf660
144. Ashburner BP, Westerheide SD, Baldwin AS. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol (2001) 21:7065–77. doi:10.1128/MCB.21.20.7065-7077.2001
145. Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science (2001) 293:1653–7. doi:10.1126/science.1062374
146. Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev (2006) 210:171–86. doi:10.1111/j.0105-2896.2006.00375.x
147. Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell (1998) 1:661–71. doi:10.1016/S1097-2765(00)80066-0
148. Natoli G, Saccani S, Bosisio D, Marazzi I. Interactions of NF-kappaB with chromatin: the art of being at the right place at the right time. Nat Immunol (2005) 6:439–45. doi:10.1038/ni1196
149. Bonnay F, Nguyen X-H, Cohen-Berros E, Troxler L, Batsche E, Camonis J, et al. Akirin specifies NF-κB selectivity of drosophila innate immune response via chromatin remodeling. EMBO J (2014) 33:2349–62. doi:10.15252/embj.201488456
150. van Ewijk W, Wang B, Hollander G, Kawamoto H, Spanopoulou E, Itoi M, et al. Thymic microenvironments, 3-D versus 2-D? Semin Immunol (1999) 11:57–64. doi:10.1006/smim.1998.0158
151. Meredith M, Zemmour D, Mathis D, Benoist C. Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat Immunol (2015) 16:942–9. doi:10.1038/ni.3247
152. Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science (2015) 348:589–94. doi:10.1126/science.aaa7017
153. Abramson J, Husebye ES. Autoimmune regulator and self-tolerance – molecular and clinical aspects. Immunol Rev (2016) 271:127–40. doi:10.1111/imr.12419
154. Baik S, Jenkinson EJ, Lane PJL, Anderson G, Jenkinson WE. Generation of both cortical and Aire(+) medullary thymic epithelial compartments from CD205(+) progenitors. Eur J Immunol (2013) 43:589–94. doi:10.1002/eji.201243209
155. Sekai M, Hamazaki Y, Minato N. Medullary thymic epithelial stem cells maintain a functional thymus to ensure lifelong central T cell tolerance. Immunity (2014) 41:753–61. doi:10.1016/j.immuni.2014.10.011
156. Rossi SW, Jenkinson WE, Anderson G, Jenkinson EJ. Clonal analysis reveals a common progenitor for thymic cortical and medullary epithelium. Nature (2006) 441:988–91. doi:10.1038/nature04813
157. Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol (2001) 2:1032–9. doi:10.1038/ni723
158. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the Aire protein. Science (2002) 298:1395–401. doi:10.1126/science.1075958
159. Cosway EJ, Lucas B, James KD, Parnell SM, Carvalho-Gaspar M, White AJ, et al. Redefining thymus medulla specialization for central tolerance. J Exp Med (2017) 214:3183–95. doi:10.1084/jem.20171000
160. Mathis D, Benoist C. Aire. Annu Rev Immunol (2009) 27:287–312. doi:10.1146/annurev.immunol.25.022106.141532
161. Hamazaki Y, Sekai M, Minato N. Medullary thymic epithelial stem cells: role in thymic epithelial cell maintenance and thymic involution. Immunol Rev (2016) 271:38–55. doi:10.1111/imr.12412
162. Irla M, Hugues S, Gill J, Nitta T, Hikosaka Y, Williams IR, et al. Autoantigen-specific interactions with CD4+ thymocytes control mature medullary thymic epithelial cell cellularity. Immunity (2008) 29:451–63. doi:10.1016/j.immuni.2008.08.007
163. Wood MB, Rios D, Williams IR. TNF-α augments RANKL-dependent intestinal M cell differentiation in enteroid cultures. Am J Physiol Cell Physiol (2016) 311:C498–507. doi:10.1152/ajpcell.00108.2016
164. Anderson G, Jenkinson WE. Border control: anatomical origins of the thymus medulla. Eur J Immunol (2015) 45:2203–7. doi:10.1002/eji.201545829
165. Hikosaka Y, Nitta T, Ohigashi I, Yano K, Ishimaru N, Hayashi Y, et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity (2008) 29:438–50. doi:10.1016/j.immuni.2008.06.018
166. Akiyama T, Shimo Y, Yanai H, Qin J, Ohshima D, Maruyama Y, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity (2008) 29:423–37. doi:10.1016/j.immuni.2008.06.015
167. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol (2014) 14:377–91. doi:10.1038/nri3667
168. Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature (1995) 373:531–6. doi:10.1038/373531a0
169. Baik S, Sekai M, Hamazaki Y, Jenkinson WE, Anderson G. Relb acts downstream of medullary thymic epithelial stem cells and is essential for the emergence of RANK(+) medullary epithelial progenitors. Eur J Immunol (2016) 46:857–62. doi:10.1002/eji.201546253
170. Riemann M, Andreas N, Fedoseeva M, Meier E, Weih D, Freytag H, et al. Central immune tolerance depends on crosstalk between the classical and alternative NF-κB pathways in medullary thymic epithelial cells. J Autoimmun (2017) 81:56–67. doi:10.1016/j.jaut.2017.03.007
171. Reissig S, Hövelmeyer N, Tang Y, Weih D, Nikolaev A, Riemann M, et al. The deubiquitinating enzyme CYLD regulates the differentiation and maturation of thymic medullary epithelial cells. Immunol Cell Biol (2015) 93:558–66. doi:10.1038/icb.2014.122
172. Onder L, Nindl V, Scandella E, Chai Q, Cheng H-W, Caviezel-Firner S, et al. Alternative NF-κB signaling regulates mTEC differentiation from podoplanin-expressing precursors in the cortico-medullary junction. Eur J Immunol (2015) 45:2218–31. doi:10.1002/eji.201545677
173. Kinoshita D, Hirota F, Kaisho T, Kasai M, Izumi K, Bando Y, et al. Essential role of IkappaB kinase alpha in thymic organogenesis required for the establishment of self-tolerance. J Immunol (2006) 176:3995–4002. doi:10.4049/jimmunol.176.7.3995
174. Kajiura F, Sun S, Nomura T, Izumi K, Ueno T, Bando Y, et al. NF-kappa B-inducing kinase establishes self-tolerance in a thymic stroma-dependent manner. J Immunol (2004) 172:2067–75. doi:10.4049/jimmunol.172.4.2067
175. Zhu M, Chin RK, Christiansen PA, Lo JC, Liu X, Ware C, et al. NF-kappaB2 is required for the establishment of central tolerance through an Aire-dependent pathway. J Clin Invest (2006) 116:2964–71. doi:10.1172/JCI28326
176. Zhang Q, Didonato JA, Karin M, McKeithan TW. BCL3 encodes a nuclear protein which can alter the subcellular location of NF-kappa B proteins. Mol Cell Biol (1994) 14:3915–26. doi:10.1128/MCB.14.6.3915
177. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional cloning of the APECED gene. Nat Genet (1997) 17:393–8. doi:10.1038/ng1297-393
178. Finnish-German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet (1997) 17:399–403. doi:10.1038/ng1297-399
179. Haljasorg U, Bichele R, Saare M, Guha M, Maslovskaja J, Kõnd K, et al. A highly conserved NF-κB-responsive enhancer is critical for thymic expression of Aire in mice. Eur J Immunol (2015) 45:3246–56. doi:10.1002/eji.201545928
180. LaFlam TN, Seumois G, Miller CN, Lwin W, Fasano KJ, Waterfield M, et al. Identification of a novel cis-regulatory element essential for immune tolerance. J Exp Med (2015) 212:1993–2002. doi:10.1084/jem.20151069
181. Seach N, Ueno T, Fletcher AL, Lowen T, Mattesich M, Engwerda CR, et al. The lymphotoxin pathway regulates Aire-independent expression of ectopic genes and chemokines in thymic stromal cells. J Immunol (2008) 180:5384–92. doi:10.4049/jimmunol.180.8.5384
182. Takaba H, Morishita Y, Tomofuji Y, Danks L, Nitta T. Fezf2 Orchestrates a thymic program of self-antigen expression for immune tolerance. Cell (2015) 163:975–87. doi:10.1016/j.cell.2015.10.013
183. Nishikawa K, Nakashima T, Hayashi M, Fukunaga T, Kato S, Kodama T, et al. Blimp1-mediated repression of negative regulators is required for osteoclast differentiation. Proc Natl Acad Sci U S A (2010) 107:3117–22. doi:10.1073/pnas.0912779107
184. Wang X, Belguise K, O’Neill CF, Sánchez-Morgan N, Romagnoli M, Eddy SF, et al. RelB NF-kappaB represses estrogen receptor alpha expression via induction of the zinc finger protein Blimp1. Mol Cell Biol (2009) 29:3832–44. doi:10.1128/MCB.00032-09
185. Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature (1999) 397:315–23. doi:10.1038/16852
186. Mouri Y, Yano M, Shinzawa M, Shimo Y, Hirota F, Nishikawa Y, et al. Lymphotoxin signal promotes thymic organogenesis by eliciting RANK expression in the embryonic thymic stroma. J Immunol (2011) 186:5047–57. doi:10.4049/jimmunol.1003533
187. Tucker E, O’Donnell K, Fuchsberger M, Hilton AA, Metcalf D, Greig K, et al. A novel mutation in the Nfkb2 gene generates an NF-kappa B2 “super repressor”. J Immunol (2007) 179:7514–22. doi:10.4049/jimmunol.179.11.7514
188. Miosge LA, Blasioli J, Blery M, Goodnow CC. Analysis of an ethylnitrosourea-generated mouse mutation defines a cell intrinsic role of nuclear factor kappaB2 in regulating circulating B cell numbers. J Exp Med (2002) 196:1113–9. doi:10.1084/jem.20020959
189. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity (2000) 12:431–40. doi:10.1016/S1074-7613(00)80195-8
190. Gupta S, Manicassamy S, Vasu C, Kumar A, Shang W, Sun Z. Differential requirement of PKC-theta in the development and function of natural regulatory T cells. Mol Immunol (2008) 46:213–24. doi:10.1016/j.molimm.2008.08.275
191. Schmidt-Supprian M, Tian J, Ji H, Terhorst C, Bhan AK, Grant EP, et al. I kappa B kinase 2 deficiency in T cells leads to defects in priming, B cell help, germinal center reactions, and homeostatic expansion. J Immunol (2004) 173:1612–9. doi:10.4049/jimmunol.173.3.1612
192. Vang KB, Yang J, Pagán AJ, Li L-X, Wang J, Green JM, et al. Cutting edge: CD28 and c-Rel-dependent pathways initiate regulatory T cell development. J Immunol (2010) 184:4074–7. doi:10.4049/jimmunol.0903933
193. Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med (2009) 206:3001–14. doi:10.1084/jem.20091411
194. Long M, Park S-G, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity (2009) 31:921–31. doi:10.1016/j.immuni.2009.09.022
195. Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, et al. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol (2003) 171:3348–52. doi:10.4049/jimmunol.171.7.3348
196. Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol (2001) 19:225–52. doi:10.1146/annurev.immunol.19.1.225
197. Cretney E, Kallies A, Nutt SL. Differentiation and function of Foxp3(+) effector regulatory T cells. Trends Immunol (2013) 34:74–80. doi:10.1016/j.it.2012.11.002
198. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol (2014) 15:1070–8. doi:10.1038/ni.3004
199. Grinberg-Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA, et al. κB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell (2017) 170:1096–108.e13. doi:10.1016/j.cell.2017.08.004
200. Vasanthakumar A, Liao Y, Teh P, Pascutti MF, Oja AE, Garnham AL, et al. The TNF receptor superfamily-NF-κB axis is critical to maintain effector regulatory T cells in lymphoid and non-lymphoid tissues. Cell Rep (2017) 20:2906–20. doi:10.1016/j.celrep.2017.08.068
201. Khan WN. B cell receptor and BAFF receptor signaling regulation of B cell homeostasis. J Immunol (2009) 183:3561–7. doi:10.4049/jimmunol.0800933
202. Kitamura D, Roes J, Kühn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature (1991) 350:423–6. doi:10.1038/350423a0
203. Lam KP, Kühn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell (1997) 90:1073–83. doi:10.1016/S0092-8674(00)80373-6
204. Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature (2005) 435:590–7. doi:10.1038/nature03724
205. Cadera EJ, Wan F, Amin RH, Nolla H, Lenardo MJ, Schlissel MS. NF-kappaB activity marks cells engaged in receptor editing. J Exp Med (2009) 206:1803–16. doi:10.1084/jem.20082815
206. Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu H-B, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity (2004) 20:441–53. doi:10.1016/S1074-7613(04)00079-2
207. Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol (2002) 3:958–65. doi:10.1038/ni842
208. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol (2003) 21:231–64. doi:10.1146/annurev.immunol.21.120601.141152
209. Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity (2004) 20:785–98. doi:10.1016/j.immuni.2004.05.010
210. Courtois G, Smahi A, Reichenbach J, Doffinger R, Cancrini C, Bonnet M, et al. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest (2003) 112:1108–15. doi:10.1172/JCI18714
211. Schimke LF, Rieber N, Rylaarsdam S, Cabral-Marques O, Hubbard N, Puel A, et al. A novel gain-of-function IKBA mutation underlies ectodermal dysplasia with immunodeficiency and polyendocrinopathy. J Clin Immunol (2013) 33:1088–99. doi:10.1007/s10875-013-9906-1
212. Kaustio M, Haapaniemi E, Göös H, Hautala T, Park G, Syrjänen J, et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J Allergy Clin Immunol (2017) 140:782–96. doi:10.1016/j.jaci.2016.10.054
213. Fliegauf M, Bryant VL, Frede N, Slade C, Woon S-T, Lehnert K, et al. Haploinsufficiency of the NF-κB1 subunit p50 in common variable immunodeficiency. Am J Hum Genet (2015) 97:389–403. doi:10.1016/j.ajhg.2015.07.008
214. Lee CE, Fulcher DA, Whittle B, Chand R, Fewings N, Field M, et al. Autosomal-dominant B-cell deficiency with alopecia due to a mutation in NFKB2 that results in nonprocessable p100. Blood (2014) 124:2964–72. doi:10.1182/blood-2014-06-578542
215. Chen K, Coonrod EM, Kumánovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am J Hum Genet (2013) 93:812–24. doi:10.1016/j.ajhg.2013.09.009
216. Brue T, Quentien M-H, Khetchoumian K, Bensa M, Capo-Chichi J-M, Delemer B, et al. Mutations in NFKB2 and potential genetic heterogeneity in patients with DAVID syndrome, having variable endocrine and immune deficiencies. BMC Med Genet (2014) 15:139. doi:10.1186/s12881-014-0139-9
217. Sharfe N, Merico D, Karanxha A, Macdonald C, Dadi H, Ngan B, et al. The effects of RelB deficiency on lymphocyte development and function. J Autoimmun (2015) 65:90–100. doi:10.1016/j.jaut.2015.09.001
218. Charbit-Henrion F, Jeverica AK, Begue B, Markelj G, Parlato M, Avčin SL, et al. Deficiency in mucosa-associated lymphoid tissue lymphoma translocation 1: a novel cause of IPEX-like syndrome. Nutrition (2017) 64:378–84. doi:10.1097/MPG.0000000000001262
219. Torres JM, Martinez-Barricarte R, García-Gómez S, Mazariegos MS, Itan Y, Boisson B, et al. Inherited BCL10 deficiency impairs hematopoietic and nonhematopoietic immunity. J Clin Invest (2014) 124:5239–48. doi:10.1172/JCI77493
220. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet (2016) 48:67–73. doi:10.1038/ng.3459
221. Duncan CJA, Dinnigan E, Theobald R, Grainger A, Skelton AJ, Hussain R, et al. Early-onset autoimmune disease due to a heterozygous loss-of-function mutation in TNFAIP3 (A20). Ann Rheum Dis (2017). doi:10.1136/annrheumdis-2016-210944
222. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27:20–1. doi:10.1038/83713
223. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27:18–20. doi:10.1038/83707
224. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (2014) 345:1623–7. doi:10.1126/science.1255904
225. Sharfe N, Dadi HK, Shahar M, Roifman CM. Human immune disorder arising from mutation of the α chain of the interleukin-2 receptor. Proc Natl Acad Sci U S A (1997) 94:3168–71. doi:10.1073/pnas.94.7.3168
226. Aaltonen J, Björses P. Cloning of the APECED gene provides new insight into human autoimmunity. Ann Med (1999) 31:111–6. doi:10.3109/07853899708998786
227. Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, et al. Dominant mutations in the autoimmune regulator AIRE are associated with common organ-specific autoimmune diseases. Immunity (2015) 42:1185–96. doi:10.1016/j.immuni.2015.04.021
228. Sun L, Li H, Luo H, Zhao Y. Thymic epithelial cell development and its dysfunction in human diseases. Biomed Res Int (2014) 2014:206929. doi:10.1155/2014/206929
229. Hong Y, Skeie GO, Zisimopoulou P, Karagiorgou K, Tzartos SJ, Gao X, et al. Juvenile-onset myasthenia gravis: autoantibody status, clinical characteristics and genetic polymorphisms. J Neurol (2017) 264:955–62. doi:10.1007/s00415-017-8478-z
230. Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet (2011) 43:253–8. doi:10.1038/ng.766
231. Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, Ortmann W, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet (2008) 40:1062–4. doi:10.1038/ng.202
232. Musone SL, Taylor KE, Nititham J, Chu C, Poon A, Liao W, et al. Sequencing of TNFAIP3 and association of variants with multiple autoimmune diseases. Genes Immun (2011) 12:176–82. doi:10.1038/gene.2010.64
233. Eyre S, Hinks A, Flynn E, Martin P, Wilson AG, Maxwell JR, et al. Confirmation of association of the REL locus with rheumatoid arthritis susceptibility in the UK population. Ann Rheum Dis (2010) 69:1572–3. doi:10.1136/ard.2009.122887
234. Bowes J, Ho P, Flynn E, Ali F, Marzo-Ortega H, Coates LC, et al. Comprehensive assessment of rheumatoid arthritis susceptibility loci in a large psoriatic arthritis cohort. Ann Rheum Dis (2012) 71:1350–4. doi:10.1136/annrheumdis-2011-200802
235. Garcia-Etxebarria K, Jauregi-Miguel A, Romero-Garmendia I, Plaza-Izurieta L, Legarda M, Irastorza I, et al. Ancestry-based stratified analysis of immunochip data identifies novel associations with celiac disease. Eur J Hum Genet (2016) 24:1831–4. doi:10.1038/ejhg.2016.120
236. Chen F, Xu L, Zhao T, Xiao X, Pan Y, Hou S. Genetic variation in the REL gene increases risk of Behcet’s disease in a Chinese han population but that of PRKCQ does not. PLoS One (2016) 11:e0147350. doi:10.1371/journal.pone.0147350
237. Karban AS, Okazaki T, Panhuysen CIM, Gallegos T, Potter JJ, Bailey-Wilson JE, et al. Functional annotation of a novel NFKB1 promoter polymorphism that increases risk for ulcerative colitis. Hum Mol Genet (2004) 13:35–45. doi:10.1093/hmg/ddh008
238. Kurylowicz A, Hiromatsu Y, Jurecka-Lubieniecka B, Kula D, Kowalska M, Ichimura M, et al. Association of NFKB1 -94ins/del ATTG promoter polymorphism with susceptibility to and phenotype of Graves’ disease. Genes Immun (2007) 8:532–8. doi:10.1038/sj.gene.6364418
239. Yenmis G, Oner T, Cam C, Koc A, Kucuk OS, Yakicier MC, et al. Association of NFKB1 and NFKBIA polymorphisms in relation to susceptibility of Behçet’s disease. Scand J Immunol (2015) 81:81–6. doi:10.1111/sji.12251
Keywords: NF-κB, autoimmunity, self-tolerance, thymic development, mutation
Citation: Miraghazadeh B and Cook MC (2018) Nuclear Factor-kappaB in Autoimmunity: Man and Mouse. Front. Immunol. 9:613. doi: 10.3389/fimmu.2018.00613
Received: 15 December 2017; Accepted: 12 March 2018;
Published: 09 April 2018
Edited by:
Raffi Gugasyan, Burnet Institute, AustraliaReviewed by:
Andrew S. Day, University of Otago, New ZealandLorraine Ann O’Reilly, Walter and Eliza Hall Institute of Medical Research, Australia
Copyright: © 2018 Miraghazadeh and Cook. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew C. Cook, matthew.cook@anu.edu.au