 Peter Lamprecht1*
Peter Lamprecht1* Anja Kerstein1
Anja Kerstein1 Sebastian Klapa1Susanne Schinke1
Sebastian Klapa1Susanne Schinke1 Christian M. Karsten2
Christian M. Karsten2 Xinhua Yu3,4
Xinhua Yu3,4 Marc Ehlers5Jörg T. Epplen6,7Konstanze Holl-Ulrich8
Marc Ehlers5Jörg T. Epplen6,7Konstanze Holl-Ulrich8 Thorsten Wiech9
Thorsten Wiech9 Kathrin Kalies10Tanja Lange1Martin Laudien11
Kathrin Kalies10Tanja Lange1Martin Laudien11 Tamas Laskay12Timo Gemoll13Udo Schumacher14Sebastian Ullrich14,15
Tamas Laskay12Timo Gemoll13Udo Schumacher14Sebastian Ullrich14,15 Hauke Busch16
Hauke Busch16 Saleh Ibrahim16
Saleh Ibrahim16 Nicole Fischer17Katrin Hasselbacher18Ralph Pries18Frank Petersen4Gesche Weppner1
Nicole Fischer17Katrin Hasselbacher18Ralph Pries18Frank Petersen4Gesche Weppner1 Rudolf Manz2Jens Y. Humrich1Relana Nieberding1
Rudolf Manz2Jens Y. Humrich1Relana Nieberding1 Gabriela Riemekasten1
Gabriela Riemekasten1 Antje Müller1
Antje Müller1
- 1Department of Rheumatology and Clinical Immunology, University of Lübeck, Lübeck, Germany
- 2Institute for Systemic Inflammation Research, University of Lübeck, Lübeck, Germany
- 3Xiamen-Borstel Joint Laboratory of Autoimmunity, Medical College of Xiamen University, Xiamen, China
- 4Priority Area Asthma and Allergy, Research Center Borstel, Airway Research Center North (ARCN), German Center for Lung Research (DZL), Borstel, Germany
- 5Laboratories of Immunology and Antibody Glycan Analysis, Institute for Nutrition Medicine, University of Lübeck and University Medical Center Schleswig Holstein, Lübeck, Germany
- 6Department of Human Genetics, Ruhr-University, Bochum, Germany
- 7University of Witten/Herdecke, ZBAF, Witten, Germany
- 8Institute of Pathology, Cath. Mary’s Hospital, Hamburg, Germany
- 9Institute of Pathology, University Hospital Hamburg-Eppendorf, Hamburg, Germany
- 10Institute of Anatomy, University of Lübeck, Lübeck, Germany
- 11Department of Otorhinolaryngology, Head and Neck Surgery, University of Kiel, Kiel, Germany
- 12Department for Infectious Diseases and Microbiology, University of Lübeck, Lübeck, Germany
- 13Department of Surgery, Section for Translational Surgical Oncology and Biobanking, University of Lübeck, University Medical Center Schleswig-Holstein, Lübeck, Germany
- 14Institute of Anatomy and Experimental Morphology, Center for Experimental Medicine, University Cancer Center, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 15Medical Department 3, Gastroenterology/Rheumatology, Municipal Hospital Kiel, Kiel, Germany
- 16Lübeck Institute of Experimental Dermatology, University of Lübeck, Lübeck, Germany
- 17Institute of Medical Microbiology, Virology and Hygiene, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 18Department of Otorhinolaryngology, University of Lübeck, Lübeck, Germany
Anti-neutrophil cytoplasmic autoantibodies (ANCA) targeting proteinase 3 (PR3) and myeloperoxidase expressed by innate immune cells (neutrophils and monocytes) are salient diagnostic and pathogenic features of small vessel vasculitis, comprising granulomatosis with polyangiitis (GPA), microscopic polyangiitis, and eosinophilic GPA. Genetic studies suggest that ANCA-associated vasculitides (AAV) constitute separate diseases, which share common immunological and pathological features, but are otherwise heterogeneous. The successful therapeutic use of anti-CD20 antibodies emphasizes the prominent role of ANCA and possibly other autoantibodies in the pathogenesis of AAV. However, to elucidate causal effects in AAV, a better understanding of the complex interplay leading to the emergence of B lymphocytes that produce pathogenic ANCA remains a challenge. Different scenarios seem possible; e.g., the break of tolerance induced by a shift from non-pathogenic toward pathogenic autoantigen epitopes in inflamed tissue. This review gives a brief overview on current knowledge about genetic and epigenetic factors, barrier dysfunction and chronic non-resolving inflammation, necro-inflammatory auto-amplification of cellular death and inflammation, altered autoantigen presentation, alternative complement pathway activation, alterations within peripheral and inflamed tissue-residing T- and B-cell populations, ectopic lymphoid tissue neoformation, the characterization of PR3-specific T-cells, properties of ANCA, links between autoimmune disease and infection-triggered pathology, and animal models in AAV.
Introduction
Anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitides (AAV) are classified into three distinct diseases based on clinical and pathological features: granulomatosis with polyangiitis (GPA, formerly Wegener’s granulomatosis), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA, Churg–Strauss syndrome). Etiology and pathogenesis of AAV are multifactorial (1, 2). The pathogenesis appears to be initiated by a combination of predisposing genetic and environmental factors, altered autoantigen presentation, ectopic lymphoid tissue neoformation, and imbalance of effector and regulatory B- and T-cells. Consequently, production of pathogenic autoantibodies originating from precursor natural autoantibodies results in ANCA-induced activation of neutrophils and monocytes with subsequent activation of the alternative complement pathway, vascular damage, and self-perpetuating non-resolving chronic inflammation (2–4). Herein, we briefly summarize current ideas, observations, and evidence on the pathogenesis of AAV.
Clinical Manifestations
While each of the three AAV retains a unique clinical phenotype, many manifestations are shared among them owing to the systemic nature of the underlying small-vessel vasculitis, and, in GPA and EGPA, granulomatous inflammation. Thus, pulmonary–renal syndrome is the dominating clinical feature in GPA and MPA (5, 6). In EGPA, renal involvement is associated with positive ANCA-status (7, 8). Prodromes such as malaise, arthralgias, myalgias—and rhinitis and/or sinusitis in GPA and EGPA—often precede manifestations of the pulmonary–renal syndrome by weeks or months (5–7). Fulminant AAV is rare (9). EGPA patients typically have a long-standing history of asthma and allergic rhinitis (7, 10). Other organs frequently affected are peripheral and central nervous system, skin, gut, and heart (5–7). Laboratory findings show elevated markers of inflammation (3, 11). GPA is highly associated with proteinase 3 (PR3)-specific ANCA, whereas MPA and—less commonly—EGPA are associated with myeloperoxidase (MPO)-specific ANCA (12–14). The majority of AAV patients (approximately 80–90%) present with renal or other organ-threatening manifestations, i.e., generalized disease. Relapse is more common in GPA (15). Renal-limited AAV is less common. Fewer than 10% of patients have a localized phenotype restricted to the upper and/or lower respiratory tract or early-systemic phenotype without imminent organ failure with less frequently detected ANCA (5, 11, 12, 16, 17). Progression from localized to generalized GPA is rare (16).
Treatment is guided by severity of organ involvement and disease activity. Various cytotoxic immunosuppressants and the monoclonal anti-CD20 antibody rituximab are recommended for the induction and maintenance of remission (18, 19). While treatment options have improved the prognosis of AAV, therapy de-escalation still carries an immanent risk of relapse (20). Major causes of death are vasculitis and infections (21). Optimizing treatment strategies according to prognostic subsets, autoantibody- and autoantigen-targeted therapies, and therapeutic interference with chronic inflammation and break of tolerance will set the stage for individualized precision medicine, further improvement of outcomes, and eventually cure of AAV (20, 22, 23).
Pathology
ANCA-associated vasculitidies are systemic necrotizing small-vessel vasculitides, predominantly affecting intraparenchymal small arteries, arterioles, capillaries, venules, and less often medium-sized arteries and veins. In addition, patients with GPA and EGPA display extravascular inflammatory lesions with predilection for the upper and/or lower respiratory tract. Immunohistology discloses few or no immunoglobulin and C3 deposits at inflammatory sites. AAV are hence designated pauci-immune vasculitides (1, 24, 25). Swelling, necrosis, and detachment of endothelial cells are the earliest histomorphological alterations of necrotizing vasculitis. At the vessel wall, both marginating and transmigrating neutrophils undergo apoptosis and karyorrhexis (leukocytoclasia). In the kidney, degranulation of neutrophils induces rupture of glomerular basement membranes and necrosis of adjacent cells, followed by fibrin precipitation. Necrotic debris, fibrin, and proinflammatory factors spill into Bowman’s space. Subsequently, monocytes accumulate and parietal (Bowman’s) epithelial cells proliferate forming crescents. Neutrophils within glomerular lesions also display NETosis, i.e., cellular death characterized by the formation of neutrophil extracellular traps (NETs) (26). Proinflammatory cytokines, chemokines, and complement factors of the alternative pathway locate within inflammatory glomerular lesions. Later stages are characterized by growing influx of monocytes, macrophages, and T- and B-cells. Finally, fibrocellular accumulations progress to fibrotic (sclerotic) lesions. Chronic injury occur more frequently in MPA (27). Notably, renal outcome and risk of relapse correlate with the proportion of sclerotic glomeruli (28, 29) and inversely with the proportion of normal glomeruli (30). In GPA, early extravascular lesions are characterized by the accumulation of neutrophils forming microabscesses. In advanced lesions, geographic patterns of necrosis with peripheral accumulation of macrophages and giant cells are found. Concomitant cellular infiltrates eventually also contain dendritic cells, T- and B-cells, and plasma cells (24, 25). Histopathological classification for extravascular granulomatous lesions in GPA has been proposed (31). Ectopic lymphatic structures have been found in both extravascular granulomatous lesions and glomerulonephritis (32, 33). In EGPA, extravascular and vascular lesions are characterized by eosinophilic infiltration (24, 25).
Epidemiology and Genetic Background
A yearly incidence between 10 and 20 per million inhabitants and a prevalence of 120 and 140 per million inhabitants has been reported for AAV in Europe and the USA. Prevalence doubling during the last decade has been attributed to improved outcomes (34, 35). Recently, higher incidence of GPA than previously reported has been identified in a UK population (36). A cyclical pattern of occurrence is observed in GPA, but not MPA (37). By contrast, MPA has a higher prevalence than GPA in Japan and China (35). Familial cases are rare in AAV (38). While individual disease-associated alleles carry modest degrees of risk, multiple genetic factors of relatively small effect combine to convey susceptibility to chronic inflammation and autoimmunity (39). Notably, the HLA polymorphism shapes self-epitope specific regulatory T-cell (Treg) responses, thereby mediating protection or causation of autoimmunity (40). Among AAV, GPA displays a remarkable HLA-DP association (p = 6.2 × 10−89) (41). The HLA-DPB1*0401 allele is closely linked to PR3-ANCA+ GPA (p = 1.2 × 10−22) (42, 43). Reduced HLA-DP protein expression is observed in GPA patients with the associated HLA-DP allele (44). By contrast, MPO-ANCA+ MPA is HLA-DQ-associated (p = 2.1 × 10−8) (41). Furthermore, GPA is associated with polymorphisms of α1-anti-trypsin and PR3 encoding genes (41). In EGPA, association with HLA-DRB1*04 was reported, whereas HLA-DR*13 is underrepresented (45). The IL10.2 haplotype is associated with ANCA-negative EGPA and increased interleukin (IL)-10 production (46). By contrast, disease-associated PTPN22 R620W allele correlates with reduced IL-10 transcription in GPA and MPA (47, 48). Aberrant microRNA expression with dysregulation of genes involved in inflammation and autoimmunity has been reported for various autoimmune diseases (49). In AAV, miRNA expression patterns correlate with renal involvement and steroid doses (50, 51). Upregulated expression of miR-634 induces a proinflammatory phenotype of monocytes in PR3-ANCA+ AVV (52). Furthermore, a GPA-specific miRNA expression pattern has been found in nasal tissue differentiating GPA from healthy and disease controls (53). These findings underscore a multifactorial process in which genetic and epigenetic factors play important roles in the genesis of AAV (54–57).
Barrier Dysfunction in GPA and EGPA
Granulomatosis with polyangiitis is known for its propensity toward necrotizing neutrophilic granulomatous inflammation of the respiratory tract (31). Accordingly, respiratory tract manifestations and flu-like symptoms represent the most frequent initial features and affect virtually all patients with GPA during follow-up in large cohorts (5, 58, 59). Moreover, the so-called “grumbling disease” related to persistent ENT disease activity is observed in many patients being otherwise in clinical remission and despite immunosuppressive treatment (17, 60). Asthma, ENT manifestations, and eosinophilic pulmonary infiltrates also represent the most frequent manifestations in EGPA (7, 61). The nasal mucosa displays a unique gene expression signature with differentially expressed transcripts of antimicrobial peptides, cytokines, extracellular matrix proteins, and molecules important for epithelial barrier integrity in GPA (62, 63). Ciliary motility is severely reduced (64). Chronic nasal carriage of Staphylococcus aureus is associated with endonasal activity and relapse in GPA and facilitated by decreased production of human beta-defensin-3 and anomalous cytokine expression pattern of nasal epithelial cells (65–68). Virulence genes, e.g., genes for pore-forming toxins such as leukocidins, may contribute to disease progression and/or relapse in PR3-ANCA+ GPA (69). Sensitization to fungi resulting in allergic bronchopulmonary aspergillosis may play a role in the development of EGPA (70). Moreover, exposure to silica is associated with increased risk for AAV (71). Altogether, these findings suggest a link between respiratory barrier dysfunction, infection, and chronic inflammation in GPA and in EGPA (31).
Cell Death and Chronic Inflammation
Anti-neutrophil cytoplasmic autoantibody-induced pathogenesis is linked to neutrophil activation leading to vascular injury, generation of NETs, apoptosis, and necrosis (25). However, defects in the cell death machinery itself can trigger inflammation and autoimmunity in AAV in extravascular tissues. Dysregulation in neutrophil apoptosis and clearance of apoptotic cells was demonstrated in vitro and in vivo for GPA (72–74). Apoptotic cells displaying membrane-located PR3 contribute to non-resolving inflammation by disturbing proper clearance (efferocytosis) and subsequent proinflammatory polarization of macrophages (72). Prolonged survival of neutrophils lead to accumulation within inflamed tissue, demonstrated recently in vivo in a human transgenic PR3 mouse model (75). Without proper clearing, apoptotic neutrophils undergo secondary necrosis with subsequent release of proinflammatory cytokines, damage-associated molecular patterns (DAMPs), and potential autoantigens (76). Within necrotizing granulomatous inflammation of GPA, release of DAMPs such as high-mobility-group-protein B1 (HMGB1) and IL-33 perpetuate receptor-dependent local auto-amplificatory loops (77). HMGB1, also described as endogenous adjuvant (78), is expressed together with the autoantigen PR3 on the surface of apoptotic neutrophils, potentially contributing to the induction of a pathogenic autoimmune response (79). NETosis is linked to the pathogenesis of AAV by contributing to endothelial damage, stimulation of lymphocytes, and activation of the alternative complement cascade (80, 81). Furthermore, ANCA-induced NET formation has recently been shown to be controlled by caspase-independent form of regulated necrosis, namely necroptosis, in an MPA mouse model (82). Altogether, dysregulation in the cell death machinery can lead to a necro-inflammatory auto-amplification loop (83), a relevant pathogenic feature in AAV and potential target for future treatment strategies.
Alternative Complement Pathway Activation
Evidence for involvement of the complement system in AAV comes from murine models of MPO-ANCA-induced glomerulonephritis and vasculitis, demonstrating activation of the alternative complement pathway and specifically C5 being essential for disease induction (84, 85). This has been confirmed by proteomic analysis of kidney biopsies (86). Moreover, in the murine model of MPO-ANCA vasculitis, liver-derived C5a is a key mediator (87). MPO- and PR3-ANCA-activated neutrophils elicit C5a release. Subsequent interaction of C5a with the C5a receptor 1 (C5aR1) may represent a proinflammatory amplification loop in AAV (88, 89). Consequently, elevated plasma and serum concentrations of C5a and C3a have been reported in active AAV, especially MPO-ANCA+ AAV (90–92). Deposition of complement components is detected in human renal biopsies in AAV (93). Similarly, C5aR1 is expressed scarcely, whereas expression of C5aR2 is upregulated in MPO-ANCA+ glomerulonephritis (92). Interestingly, the exact role of C5aR2 has not been clearly defined yet. In the context of AAV, human neutrophil-expressed C5aR2 interacting with C5a seem to play a proinflammatory role (94). However, in the murine model of MPO-ANCA-induced glomerulonephritis, blocking of C5aR1 with a small molecule antagonist (avacopan) is protective, whereas lack of C5aR2 lead to aggravated disease (95). The oral C5aR1-antagonist avacopan yields promising results as glucocorticoid-sparing agent in two randomized phase II clinical trials (NCT01363388 and NCT02222155) (96). In general, tissue expression and function of the anaphylatoxin receptors (C5aR1/2) are still poorly understood, specifically in humans (97). Therefore, it remains to be examined why results regarding tissue expression of C5aR1/2 in MPO-AAV and the function of C5aR2 differ between murine and human studies. Altogether, C5a can be regarded as one of the central players in the pathogenesis of AAV (98). Contribution of the corresponding C5aRs, however, needs to be further investigated.
Alterations of Effector and Tregs
Disruption of the balance between regulatory and effector T-cells and accumulation of effector T-cells in tissues with disease propagation favor chronic inflammation and autoimmunity (99). Alterations of the peripheral T-cell compartment have been reported in GPA such as the expansion of circulating effector memory T-cells (TEM) including Th1-type CD4+ and CD8+ T-cell populations lacking costimulatory CD28 expression (CD28−), Th17 and Th22 cells, IL-21-producing cells, and CD4+CD8+ double-positive cells. Conversely, the percentage of circulating naïve T-cells is decreased (4, 100–105). Depletion of circulating Vδ2 T-cells, mucosal-associated invariant T-cells, and innate lymphoid T-cell subsets have been observed (106, 107). Remission is associated with a shift toward circulating total Th2, Th9, and Treg populations in GPA (105). Notably, the cytokine profile of PR3-specific T-cells is skewed toward Th2-type, Th17, and Th22 cells independent of disease activity (102, 108, 109). PR3-specific T-cells are CCR7−CD45RA− TEM with either “intermediate” CD28+CD27− or “early” CD28+CD27+ phenotype (4, 108, 109). They may express the costimulatory c-type lectin CD161 (Figure 1).
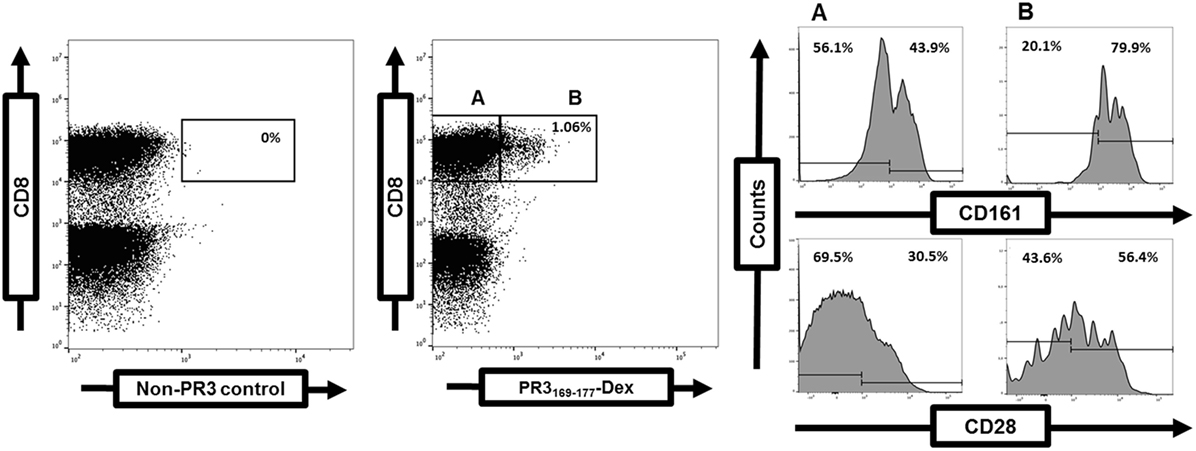
Figure 1. Frequency and phenotype of proteinase 3 (PR3)-specific T-cells determined by flow cytometry. Detection of PR3-specific T-cells within the gated CD3+CD8+ T-cell population in a GPA-patient (right dot plot). A negative control (left dot plot). Costimulatory CD28 and CD161 expression on gated PR3-specific CD8+ T-cells (A) in comparison to PR3-negative CD8+ T-cells (B) (4).
Percentages of total circulating TEM decrease during active disease, suggestive of TEM migration toward inflamed sites (101). Accordingly, CD28− TEM are abundant in bronchoalveolar fluid, inflammatory lesions, and, during renal activity, in urine (100, 110, 111). In EGPA, the frequency of circulating total Th2 and Th17 cell populations is increased, whereas lower numbers of Treg are detected (112–114). Phenotype and function of Treg are altered and impaired, respectively, in AAV (115, 116). Expansion of circulating and tissue-resident CD28− TEM lacking IL-2 production could favor Treg dysfunction in GPA (100). In line with an antigen-driven pathogenesis, oligoclonal T-cell proliferation has been reported in AAV previously (117, 118). Expansion of circulating CD28− T-cells has been attributed to latent cytomegalovirus (CMV) infection in GPA (119). However, concomitant cellular CMV- and Epstein–Barr virus (EBV)-infection rather than sole CMV or EBV infection is associated with expansion of CD28− TEM (4). Transcriptome analysis suggests pathogen (S. aureus, EBV, and others) and inflammation (cytokine and chemokine)-driven alterations of the peripheral T-cell compartment linking autoimmune disease and infection-triggered pathology in GPA (4).
ANCA Immunoglobulins
Myeloperoxidase- and PR3-ANCA are hallmarks of autoimmune vasculitis in AAV (Table S1 in Supplementary Material) (23, 120, 121). Functionally, numerous in vitro studies demonstrate IgG MPO- and PR3-ANCA interaction with neutrophils and monocytes. Interaction between circulating ANCA and neutrophils and monocytes causing their activation, PR3- and MPO-translocation to the cellular surface, microvascular adherence, vascular inflammation, and cell death is known as ANCA-cytokine-sequence theory (122). Notably, PR3- and MPO-ANCA display differences in neutrophil activation, e.g., respiratory burst (123, 124). In known or suspected AAV, the revised consensus on the use of ANCA testing now recommends high-quality immunoassays as primary screening method (13). Regarding IgG MPO-ANCA, epitope specificity determines pathogenicity, mainly by identifying an epitope of MPO (anti-MPO447-459) that is exclusively linked to active MPA (125). IgG MPO-ANCA can be masked by a fragment of ceruloplasmin (125). Thus, true ANCA-negative vasculitis is considered as being less than 10% using contemporary ANCA tests (13, 121). In EGPA, IgA MPO-ANCA can occur together with IgG MPO-ANCA, but seem of less value as a biomarker (126). Of note, IgG, IgA, and IgM MPO-ANCA representing low-titer natural autoantibodies are found in healthy donors (126–128). With respect to PR3-ANCA, especially IgG isotypes (122), but IgA and IgM as well, have been linked to GPA. IgA PR3-ANCA are observed in about a quarter of GPA patients, being less prevalent in severe renal disease (129). Transient and recurring presence of IgM PR3-ANCA is reported in proportions of two large cohorts of patients with GPA or MPA and associated with a higher rate of alveolar hemorrhage (130). Similar to MPO-ANCA, IgG, IgA, and IgM PR3-ANCA are detected in healthy donors. However, the epitope specificity of natural ANCA in healthy donors differs from that of pathogenic ANCA in AAV (125, 127–129). Several studies indicate that ANCA and other autoantibodies present in AAV recognize targets apart from MPO and PR3 (Table S1 in Supplementary Material). Some of these studies though still lack confirmatory data or functional relevance.
ANCA Glycosylation
Altered Fc N-glycosylation patterns of IgG, differing between proinflammatory (agalactosylated) and antiinflammatory (galactosylated and sialylated) IgG antibodies (131), may contribute to the pathogenicity of IgG-ANCA and be a useful biomarker. Agalactosylation of total IgG has been associated with MPA, GPA, and EGPA (132). Instead, sialylation of IgG PR3-ANCA inversely correlates with disease activity and ROS production by neutrophils in GPA (133). Employing mass spectroscopy, reduced galactosylation and sialylation of IgG1 PR3-ANCA or total IgG was confirmed (134, 135). IgG-ANCA bind via their Fc part to FcγRIIa and FcγRIIIb, thereby co-operating with the antigen-binding site in neutrophil activation (136). Alteration of IgG-ANCA glycosylation attenuates experimental ANCA-induced glomerulonephritis (137).
Animal Models of ANCA-Induced Vasculitis and Extravascular Granulomatosis
While murine models demonstrated convincing evidence for MPO-ANCA-induced glomerulonephritis and vasculitis (138), it has proven extremely difficult to show pathogenicity of PR3-ANCA in vivo (139). Transfer of human hematopoietic stem cells and anti-PR3 antibodies in NOD-SCID-IL-2Rγ−/− mice induced disease manifestations partially resembling systemic vasculitis (140), giving a hint of PR3-ANCA pathogenicity. A transgenic mouse model expressing human PR3 displays enhanced neutrophil survival and accumulation reflecting aspects of granulomatous inflammation in GPA (75). Besides active immunization and passive transfer of autoimmunity (141), transplantation of GPA tissue into immunodeficient mice provides evidence that fibroblasts induce nasal cartilage and bone destruction seen in GPA (142).
Autoreactive Circulating and Lesional B Lymphocytes
Increased proportion of circulating matured B-cells recognizing PR3 in PR3-ANCA+ AAV (143), presence of ectopic lymphoid structures and autoreactive B-cells in inflamed tissue of GPA (144, 145) and glomerulonephritis (32) provide a rationale for targeting B-cells. B-cell depletion proves to be an efficient therapy in AAV (146–148). Although IL-10-producing regulatory B-cells (Bregs) and CD24high CD38high Bregs, respectively, are reduced in AAV (149–151), their role in AAV remains to be further defined as such circulating cells are probably removed by B-cell-depleting therapies. Altogether, as possible scenario for ANCA-mediated pathogenesis, one could envision that B-cells producing natural autoantibodies which recognize PR3, MPO, or other antigens are selected in an inflamed microenvironment and mature with the help of autoantigen-specific T-cells. Subsequently, this could lead to the emergence and survival of pathogenic plasma cells (152) producing proinflammatory ANCA.
Conclusion
Much progress has been achieved in elucidating pathogenetic mechanisms in AAV, especially regarding the discovery of links between genetic predispositions and ANCA as well as deciphering mostly deleterious functions of PR3- and MPO-ANCA by in vitro and in case of the latter by in vivo studies. Future research projects should, for instance, focus on investigating the role of microbes (bacteria and viruses) in the etiology of AAV. Altogether, a better understanding of the complex interplay between endogenous and exogenous factors contributing to pathogenic PR3- and MPO-ANCA may support the development of individualized therapies.
Author Contributions
PL, AM, and AK substantially contributed to this review with regard to content and structure of the manuscript. All other authors listed have made direct and/or intellectual contribution to sections according to their area of expertise. All authors approved the manuscript for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by German Research Foundation grants EXC306 (HB and GR) and RTG1727 (AM, PL, and GR).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.00680/full#supplementary-material.
References
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum (2013) 65:1–11. doi:10.1002/art.37715
2. Jennette JC, Falk RJ, Gasim AH. Pathogenesis of antineutrophil cytoplasmic autoantibody vasculitis. Curr Opin Nephrol Hypertens (2011) 20:263–70. doi:10.1097/MNH.0b013e3283456731
3. Kerstein A, Holl-Ulrich K, Müller A, Riemekasten G, Lamprecht P. Granulomatose mit polyangiitis. Dtsch Medizinische Wochenschrift (2017) 142:24–31. doi:10.1055/s-0042-111610
4. Kerstein A, Schüler S, Cabral-Marques O, Fazio J, Häsler R, Müller A, et al. Environmental factor and inflammation-driven alteration of the total peripheral T-cell compartment in granulomatosis with polyangiitis. J Autoimmun (2017) 78:79–91. doi:10.1016/j.jaut.2016.12.004
5. Holle JU, Gross WL, Latza U, Nölle B, Ambrosch P, Heller M, et al. Improved outcome in 445 patients with Wegener’s granulomatosis in a German vasculitis center over four decades. Arthritis Rheum (2011) 63:257–66. doi:10.1002/art.27763
6. Schirmer JH, Wright MN, Vonthein R, Herrmann K, Nölle B, Both M, et al. Clinical presentation and long-term outcome of 144 patients with microscopic polyangiitis in a monocentric German cohort. Rheumatology (Oxford) (2016) 55:71–9. doi:10.1093/rheumatology/kev286
7. Comarmond C, Pagnoux C, Khellaf M, Cordier J-F, Hamidou M, Viallard J-F, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum (2013) 65:270–81. doi:10.1002/art.37721
8. Cottin V, Bel E, Bottero P, Dalhoff K, Humbert M, Lazor R, et al. Revisiting the systemic vasculitis in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): a study of 157 patients by the Groupe d’Etudes et de Recherche sur les Maladies Orphelines Pulmonaires and the European respiratory society taskforce on eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Autoimmun Rev (2017) 16:1–9. doi:10.1016/j.autrev.2016.09.018
9. Demiselle J, Auchabie J, Beloncle F, Gatault P, Grangé S, Du Cheyron D, et al. Patients with ANCA-associated vasculitis admitted to the intensive care unit with acute vasculitis manifestations: a retrospective and comparative multicentric study. Ann Intensive Care (2017) 7:39. doi:10.1186/s13613-017-0262-9
10. Gioffredi A, Maritati F, Oliva E, Buzio C. Eosinophilic granulomatosis with polyangiitis: an overview. Front Immunol (2014) 5:549. doi:10.3389/fimmu.2014.00549
11. Kallenberg CG. Antineutrophil cytoplasmic autoantibody-associated small-vessel vasculitis. Curr Opin Rheumatol (2007) 19:17–24. doi:10.1097/BOR.0b013e3280119842
12. Csernok E, Lamprecht P, Gross WL. Diagnostic significance of ANCA in vasculitis. Nat Clin Pract Rheumatol (2006) 2:174–5. doi:10.1038/ncprheum0159
13. Bossuyt X, Cohen Tervaert J-W, Arimura Y, Blockmans D, Flores-Suárez LF, Guillevin L, et al. Position paper: revised 2017 international consensus on testing of ANCAs in granulomatosis with polyangiitis and microscopic polyangiitis. Nat Rev Rheumatol (2017) 13(11):683–92. doi:10.1038/nrrheum.2017.140
14. Damoiseaux J, Csernok E, Rasmussen N, Moosig F, van Paassen P, Baslund B, et al. Detection of antineutrophil cytoplasmic antibodies (ANCAs): a multicentre European Vasculitis Study Group (EUVAS) evaluation of the value of indirect immunofluorescence (IIF) versus antigen-specific immunoassays. Ann Rheum Dis (2017) 76:647–53. doi:10.1136/annrheumdis-2016-209507
15. Specks U, Merkel PA, Seo P, Spiera R, Langford CA, Hoffman GS, et al. Efficacy of remission-induction regimens for ANCA-associated vasculitis. N Engl J Med (2013) 369:417–27. doi:10.1056/NEJMoa1213277
16. Holle JU, Gross WL, Holl-Ulrich K, Ambrosch P, Noelle B, Both M, et al. Prospective long-term follow-up of patients with localised Wegener’s granulomatosis: does it occur as persistent disease stage? Ann Rheum Dis (2010) 69:1934–9. doi:10.1136/ard.2010.130203
17. Kallenberg CGM. Key advances in the clinical approach to ANCA-associated vasculitis. Nat Rev Rheumatol (2014) 10:484–93. doi:10.1038/nrrheum.2014.104
18. Yates M, Watts RA, Bajema IM, Cid MC, Crestani B, Hauser T, et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann Rheum Dis (2016) 75:1583–94. doi:10.1136/annrheumdis-2016-209133
19. Schirmer JH, Aries PM, de Groot K, Hellmich B, Holle JU, Kneitz C, et al. S1-Leitlinie Diagnostik und Therapie der ANCA-assoziierten Vaskulitiden. Z Rheumatol (2017) 76:77–104. doi:10.1007/s00393-017-0394-1
20. Schinke S, Riemekasten G, Lamprecht P. [De-escalation of therapy in ANCA-associated vasculitides]. Z Rheumatol (2017) 76:15–20. doi:10.1007/s00393-016-0241-9
21. Flossmann O, Berden A, de Groot K, Hagen C, Harper L, Heijl C, et al. Long-term patient survival in ANCA-associated vasculitis. Ann Rheum Dis (2011) 70:488–94. doi:10.1136/ard.2010.137778
22. Guarino C, Hamon Y, Croix C, Lamort AS, Dallet-Choisy S, Marchand-Adam S, et al. Prolonged pharmacological inhibition of cathepsin C results in elimination of neutrophil serine proteases. Biochem Pharmacol (2017) 131:52–67. doi:10.1016/j.bcp.2017.02.009
23. van der Geest KSM, Brouwer E, Sanders J-S, Sandovici M, Bos NA, Boots AMH, et al. Towards precision medicine in ANCA-associated vasculitis. Rheumatology (2017):1–8. doi:10.1093/rheumatology/kex367
24. Holl-Ulrich K. [Histopathology of systemic vasculitis]. Pathologe (2010) 31:67–76. doi:10.1007/s00292-009-1156-x
25. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol (2014) 10:463–73. doi:10.1038/nrrheum.2014.103
26. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15:623–5. doi:10.1038/nm.1959
27. Hauer HA, Bajema IM, Van Houwelingen HC, Ferrario F, Noël L-H, Waldherr R, et al. Renal histology in ANCA-associated vasculitis: differences between diagnostic and serologic subgroups. Kidney Int (2002) 61:80–9. doi:10.1046/j.1523-1755.2002.00089.x
28. Berden AE, Ferrario F, Hagen EC, Jayne DR, Jennette JC, Joh K, et al. Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol (2010) 21:1628–36. doi:10.1681/ASN.2010050477
29. Göçeroğlu A, Berden AE, Fiocco M, Floßmann O, Westman KW, Ferrario F, et al. ANCA-associated glomerulonephritis: risk factors for renal relapse. PLoS One (2016) 11:e0165402. doi:10.1371/journal.pone.0165402
30. Hilhorst M, Wilde B, van Breda Vriesman P, van Paassen P, Cohen Tervaert JW, Limburg Renal Registry. Estimating renal survival using the ANCA-associated GN classification. J Am Soc Nephrol (2013) 24:1371–5. doi:10.1681/ASN.2012090912
31. Lamprecht P, Holl-Ulrich K, Gross WL. Granulomatosis with polyangiitis (Wegener’s granulomatosis): pathogenesis. In: Ball GV, Fessler BJ, Bridges SL Jr, editors. Oxford Textbook of Vasculitis. 3rd ed. Oxford: Oxford University Press (2014). p. 385–400.
32. Steinmetz OM, Velden J, Kneissler U, Marx M, Klein A, Helmchen U, et al. Analysis and classification of B-cell infiltrates in lupus and ANCA-associated nephritis. Kidney Int (2008) 74:448–57. doi:10.1038/ki.2008.191
33. Mueller A, Brieske C, Schinke S, Csernok E, Gross WL, Hasselbacher K, et al. Plasma cells within granulomatous inflammation display signs pointing towards autoreactivity and destruction in granulomatosis with polyangiitis. Arthritis Res Ther (2014) 16:R55. doi:10.1186/ar4490
34. Herlyn K, Buckert F, Gross WL, Reinhold-Keller E. Doubled prevalence rates of ANCA-associated vasculitides and giant cell arteritis between 1994 and 2006 in northern Germany. Rheumatology (Oxford) (2014) 53:882–9. doi:10.1093/rheumatology/ket440
35. Pearce FA, Craven A, Merkel PA, Luqmani RA, Watts RA. Global ethnic and geographic differences in the clinical presentations of anti-neutrophil cytoplasm antibody-associated vasculitis. Rheumatology (Oxford) (2017) 56:1962–9. doi:10.1093/rheumatology/kex293
36. Pearce FA, Grainge MJ, Lanyon PC, Watts RA, Hubbard RB. The incidence, prevalence and mortality of granulomatosis with polyangiitis in the UK Clinical Practice Research Datalink. Rheumatology (Oxford) (2017) 56:589–96. doi:10.1093/rheumatology/kew413
37. Watts RA, Mooney J, Skinner J, Scott DGI, Macgregor AJ. The contrasting epidemiology of granulomatosis with polyangiitis (Wegener’s) and microscopic polyangiitis. Rheumatology (Oxford) (2012) 51:926–31. doi:10.1093/rheumatology/ker454
38. Knudsen BB, Joergensen T, Munch-Jensen B. Wegener’s granulomatosis in a family. A short report. Scand J Rheumatol (1988) 17:225–7. doi:10.3109/03009748809098787
39. Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med (2011) 365:1612–23. doi:10.1056/NEJMra1100030
40. Ooi JD, Petersen J, Tan YH, Huynh M, Willett ZJ, Ramarathinam SH, et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature (2017) 545:243–7. doi:10.1038/nature22329
41. Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DRW, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med (2012) 367:214–23. doi:10.1056/NEJMoa1108735
42. Heckmann M, Holle JU, Arning L, Knaup S, Hellmich B, Nothnagel M, et al. The Wegener’s granulomatosis quantitative trait locus on chromosome 6p21.3 as characterised by tagSNP genotyping. Ann Rheum Dis (2008) 67:972–9. doi:10.1136/ard.2007.077693
43. Hilhorst M, Arndt F, Joseph Kemna M, Wieczorek S, Donner Y, Wilde B, et al. HLA-DPB1 as a risk factor for relapse in antineutrophil cytoplasmic antibody-associated vasculitis: a cohort study. Arthritis Rheumatol (2016) 68:1721–30. doi:10.1002/art.39620
44. Merkel PA, Xie G, Monach PA, Ji X, Ciavatta DJ, Byun J, et al. Identification of functional and expression polymorphisms associated with risk for antineutrophil cytoplasmic autoantibody-associated vasculitis. Arthritis Rheumatol (2017) 69:1054–66. doi:10.1002/art.40034
45. Wieczorek S, Hellmich B, Gross WL, Epplen JT. Associations of Churg-Strauss syndrome with the HLA-DRB1 locus, and relationship to the genetics of antineutrophil cytoplasmic antibody-associated vasculitides: comment on the article by Vaglio et al. Arthritis Rheum (2008) 58:329–30. doi:10.1002/art.23209
46. Wieczorek S, Hellmich B, Arning L, Moosig F, Lamprecht P, Gross WL, et al. Functionally relevant variations of the interleukin-10 gene associated with antineutrophil cytoplasmic antibody-negative Churg-Strauss syndrome, but not with Wegener’s granulomatosis. Arthritis Rheum (2008) 58:1839–48. doi:10.1002/art.23496
47. Jagiello P, Aries P, Arning L, Wagenleiter SEN, Csernok E, Hellmich B, et al. The PTPN22 620W allele is a risk factor for Wegener’s granulomatosis. Arthritis Rheum (2005) 52:4039–43. doi:10.1002/art.21487
48. Cao Y, Yang J, Colby K, Hogan SL, Hu Y, Jennette CE, et al. High basal activity of the PTPN22 gain-of-function variant blunts leukocyte responsiveness negatively affecting IL-10 production in ANCA vasculitis. PLoS One (2012) 7:e42783. doi:10.1371/journal.pone.0042783
49. O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol (2010) 10:111–22. doi:10.1038/nri2708
50. Kurz T, Weiner M, Skoglund C, Basnet S, Eriksson P, Segelmark M. A myelopoiesis gene signature during remission in anti-neutrophil cytoplasm antibody-associated vasculitis does not predict relapses but seems to reflect ongoing prednisolone therapy. Clin Exp Immunol (2014) 175:215–26. doi:10.1111/cei.12236
51. Skoglund C, Carlsen AL, Weiner M, Kurz T, Hellmark T, Eriksson P, et al. Circulating microRNA expression pattern separates patients with anti-neutrophil cytoplasmic antibody associated vasculitis from healthy controls. Clin Exp Rheumatol (2015) 33:S–64–71.
52. Bertram A, Lovric S, Engel A, Beese M, Wyss K, Hertel B, et al. Circulating ADAM17 level reflects disease activity in proteinase-3 ANCA-associated vasculitis. J Am Soc Nephrol (2015) 26:2860–70. doi:10.1681/ASN.2014050477
53. Schinke S, Laudien M, Müller A, Gross WL, Häsler R. Disease-associated micro-RNA profiles in granulomatosis with polyangiitis nasal tissue indicate a regulatory network targeting pathophysiological processes. Ann Rheum Dis (2013) 71:477. doi:10.1136/annrheumdis-2012-eular.2967
54. Arning L, Holle JU, Harper L, Millar DS, Gross WL, Epplen JT, et al. Are there specific genetic risk factors for the different forms of ANCA-associated vasculitis? Ann Rheum Dis (2011) 70:707–8. doi:10.1136/ard.2010.130971
55. Wieczorek S, Holle JU, Cohen Tervaert JW, Harper L, Moosig F, Gross WL, et al. The SEM6A6 locus is not associated with granulomatosis with polyangiitis or other forms of antineutrophil cytoplasmic antibody-associated vasculitides in Europeans: comment on the article by Xie et al. Arthritis Rheumatol (2014) 66:1400–1. doi:10.1002/art.38367
56. Husmann CA, Holle JU, Moosig F, Mueller S, Wilde B, Cohen Tervaert JW, et al. Genetics of toll like receptor 9 in ANCA associated vasculitides. Ann Rheum Dis (2014) 73:890–6. doi:10.1136/annrheumdis-2012-202803
57. Ciavatta DJ, Yang J, Preston GA, Badhwar AK, Xiao H, Hewins P, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest (2010) 120:3209–19. doi:10.1172/JCI40034DS1
58. Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med (1992) 116:488–98. doi:10.7326/0003-4819-116-6-488
59. Reinhold-Keller E, Beuge N, Latza U, de Groot K, Rudert H, Nölle B, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: long-term outcome in 155 patients. Arthritis Rheum (2000) 43:1021–32. doi:10.1002/1529-0131(200005)43:5<1021:AID-ANR10>3.0.CO;2-J
60. Garske U, Haack A, Beltrán O, Flores-Suárez LF, Bremer JP, Lamprecht P, et al. Intra- and inter-rater reliability of endonasal activity estimation in granulomatosis with polyangiitis (Wegener’s). Clin Exp Rheumatol (2012) 30:S22–8.
61. Petersen H, Götz P, Both M, Hey M, Ambrosch P, Bremer JP, et al. Manifestation of eosinophilic granulomatosis with polyangiitis in head and neck. Rhinology (2015) 53:277–85. doi:10.4193/Rhin14.074
62. Laudien M, Häsler R, Wohlers J, Böck J, Lipinski S, Bremer L, et al. Molecular signatures of a disturbed nasal barrier function in the primary tissue of Wegener’s granulomatosis. Mucosal Immunol (2011) 4:564–73. doi:10.1038/mi.2011.9
63. Grayson PC, Steiling K, Platt M, Berman JS, Zhang X, Xiao J, et al. Defining the nasal transcriptome in granulomatosis with polyangiitis (Wegener’s). Arthritis Rheumatol (2015) 67:2233–9. doi:10.1002/art.39185
64. Ullrich S, Gustke H, Lamprecht P, Gross WL, Schumacher U, Ambrosch P, et al. Severe impaired respiratory ciliary function in Wegener granulomatosis. Ann Rheum Dis (2009) 68:1067–71. doi:10.1136/ard.2008.096974
65. Stegeman CA, Tervaert JW, Sluiter WJ, Manson WL, de Jong PE, Kallenberg CG. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med (1994) 120:12–7. doi:10.7326/0003-4819-120-1-199401010-00003
66. Laudien M, Gadola SD, Podschun R, Hedderich J, Paulsen J, Reinhold-Keller E, et al. Nasal carriage of Staphylococcus aureus and endonasal activity in Wegener’s granulomatosis as compared to rheumatoid arthritis and chronic rhinosinusitis with nasal polyps. Clin Exp Rheumatol (2010) 28:51–5.
67. Hui Y, Wohlers J, Podschun R, Hedderich J, Lamprecht P, Ambrosch P, et al. Antimicrobial peptides in nasal secretion and mucosa with respect to S. aureus colonisation in Wegener’s granulomatosis. Clin Exp Rheumatol (2011) 29:S49–56.
68. Wohlers J, Breucker K, Podschun R, Hedderich J, Lamprecht P, Ambrosch P, et al. Aberrant cytokine pattern of the nasal mucosa in granulomatosis with polyangiitis. Arthritis Res Ther (2012) 14:R203. doi:10.1186/ar4041
69. Glasner C, de Goffau MC, van Timmeren MM, Schulze ML, Jansen B, Tavakol M, et al. Genetic loci of Staphylococcus aureus associated with anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitides. Sci Rep (2017) 7:12211. doi:10.1038/s41598-017-12450-z
70. Ishiguro T, Takayanagi N, Takaku Y, Kagiyama N, Kurashima K, Sugita Y. Combined allergic bronchopulmonary aspergillosis and eosinophilic granulomatosis with polyangiitis: three cases and a review of the literature. Intern Med (2016) 55:793–7. doi:10.2169/internalmedicine.55.5431
71. Hogan SL, Cooper GS, Savitz DA, Nylander-French LA, Parks CG, Chin H, et al. Association of silica exposure with anti-neutrophil cytoplasmic autoantibody small-vessel vasculitis: a population-based, case-control study. Clin J Am Soc Nephrol (2007) 2:290–9. doi:10.2215/CJN.03501006
72. Millet A, Martin KR, Bonnefoy F, Saas P, Mocek J, Alkan M, et al. Proteinase 3 on apoptotic cells disrupts immune silencing in autoimmune vasculitis. J Clin Invest (2015) 125:4107–21. doi:10.1172/JCI78182
73. Abdgawad M, Pettersson Å, Gunnarsson L, Bengtsson AA, Geborek P, Nilsson L, et al. Decreased neutrophil apoptosis in quiescent ANCA-associated systemic vasculitis. PLoS One (2012) 7:e32439. doi:10.1371/journal.pone.0032439
74. Gabillet J, Millet A, Pederzoli-Ribeil M, Tacnet-Delorme P, Guillevin L, Mouthon L, et al. Proteinase 3, the autoantigen in granulomatosis with polyangiitis, associates with calreticulin on apoptotic neutrophils, impairs macrophage phagocytosis, and promotes inflammation. J Immunol (2012) 189:2574–83. doi:10.4049/jimmunol.1200600
75. Martin KR, Pederzoli-Ribeil M, Pacreau E, Burgener SS, Dahdah A, Candalh C, et al. Transgenic mice expressing human proteinase 3 exhibit sustained neutrophil-associated peritonitis. J Immunol (2017) 199:3914–24. doi:10.4049/jimmunol.1601522
76. Muñoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol (2010) 6:280–9. doi:10.1038/nrrheum.2010.46
77. Kerstein A, Erschig A, Müller A, Riemekasten G, Lamprecht P. Dangerous signaling in granulomatosis with polyangiitis – how alarmins perpetuate the granulomatous inflammation. gms | 43. Kongress der Deutschen Gesellschaft für Rheumatologie, 29. Jahrestagung der Deutschen Gesellschaft für Orthopädische Rheumatologie, 25. Wissenschaftliche Jahrestagung der Gesellschaft für Kinder- und Jugendrheumatologie. German Medical Science GMS Publishing House. ER.21 (2015). Available from: http://www.egms.de/static/de/meetings/dgrh2015/15dgrh075.shtml (Accessed: September 1, 2015).
78. Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep (2004) 5:825–30. doi:10.1038/sj.embor.7400205
79. Kerstein A, Müller A, Holl-Ulrich K, Lamprecht P. High-mobility group box 1 (HMGB1) as a link between the granulomatous inflammation and the induction of autoreactivity in granulomatosis with polyangiitis. Nephron (2015) 129(Suppl):205.
80. Lange C, Csernok E, Moosig F, Holle JU. Immune stimulatory effects of neutrophil extracellular traps in granulomatosis with polyangiitis. Clin Exp Rheumatol (2017) 35Suppl 103(1):33–9.
81. Söderberg D, Segelmark M. Neutrophil extracellular traps in ANCA-associated vasculitis. Front Immunol (2016) 7:256. doi:10.3389/fimmu.2016.00256
82. Schreiber A, Rousselle A, Becker JU, von Mässenhausen A, Linkermann A, Kettritz R. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc Natl Acad Sci U S A (2017) 114:E9618–25. doi:10.1073/pnas.1708247114
83. Linkermann A, Stockwell BR, Krautwald S, Anders H-J. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol (2014) 14:759–67. doi:10.1038/nri3743
84. Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol (2007) 170:52–64. doi:10.2353/ajpath.2007.060573
85. Huugen D, van Esch A, Xiao H, Peutz-Kootstra CJ, Buurman WA, Tervaert JWC, et al. Inhibition of complement factor C5 protects against anti-myeloperoxidase antibody-mediated glomerulonephritis in mice. Kidney Int (2007) 71:646–54. doi:10.1038/sj.ki.5002103
86. Sethi S, Zand L, De Vriese AS, Specks U, Vrana JA, Kanwar S, et al. Complement activation in pauci-immune necrotizing and crescentic glomerulonephritis: results of a proteomic analysis. Nephrol Dial Transplant (2017) 32:i139–45. doi:10.1093/ndt/gfw299
87. Freeley SJ, Popat RJ, Parmar K, Kolev M, Hunt BJ, Stover CM, et al. Experimentally-induced anti-myeloperoxidase vasculitis does not require properdin, MASP-2 or bone marrow-derived C5. J Pathol (2016) 240:61–71. doi:10.1002/path.4754
88. Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol (2009) 20:289–98. doi:10.1681/ASN.2008050497
89. Camous L, Roumenina L, Bigot S, Brachemi S, Frémeaux-Bacchi V, Lesavre P, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood (2011) 117:1340–9. doi:10.1182/blood-2010-05-283564
90. Gou S-J, Yuan J, Chen M, Yu F, Zhao M-H. Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int (2012) 83:129–37. doi:10.1038/ki.2012.313
91. Le Roux S, Pepper R, Dufay A, Néel M, Meffray E, Lamandé N, et al. Elevated soluble Flt1 inhibits endothelial repair in PR3-ANCA-associated vasculitis. J Am Soc Nephrol (2012) 23:155–64. doi:10.1681/ASN.2010080858
92. Yuan J, Gou S-J, Huang J, Hao J, Chen M, Zhao M-H. C5a and its receptors in human anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Arthritis Res Ther (2012) 14:R140. doi:10.1186/ar3873
93. Hilhorst M, Van Paassen P, Van Rie H, Bijnens N, Heerings-Rewinkel P, Van Breda Vriesman P, et al. Complement in ANCA-associated glomerulonephritis. Nephrol Dial Transplant (2017) 32:1302–13. doi:10.1093/ndt/gfv288
94. Hao J, Wang C, Yuan J, Chen M, Zhao MH. A pro-inflammatory role of C5L2 in C5a-primed neutrophils for ANCA-induced activation. PLoS One (2013) 8:e66305. doi:10.1371/journal.pone.0066305
95. Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J Am Soc Nephrol (2014) 25:225–31. doi:10.1681/ASN.2013020143
96. Jayne DRW, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol (2017) 28(9):2756–67. doi:10.1681/ASN.2016111179
97. Laumonnier Y, Karsten CM, Köhl J. Novel insights into the expression pattern of anaphylatoxin receptors in mice and men. Mol Immunol (2017) 89:44–58. doi:10.1016/j.molimm.2017.05.019
98. Chen M, Jayne DRW, Zhao M-H. Complement in ANCA-associated vasculitis: mechanisms and implications for management. Nat Rev Nephrol (2017) 13:359–67. doi:10.1038/nrneph.2017.37
99. Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest (2015) 125:1–6. doi:10.1172/JCI78088
100. Komocsi A, Lamprecht P, Csernok E, Mueller A, Holl-Ulrich K, Seitzer U, et al. Peripheral blood and granuloma CD4+CD28− T cells are a major source of interferon-γ and tumor necrosis factor-α in Wegener’s granulomatosis. Am J Pathol (2002) 160:1717–24. doi:10.1016/S0002-9440(10)61118-2
101. Abdulahad WH, van der Geld YM, Stegeman CA, Kallenberg CGM. Persistent expansion of CD4+ effector memory T cells in Wegener’s granulomatosis. Kidney Int (2006) 70:938–47. doi:10.1038/sj.ki.5001670
102. Abdulahad WH, Stegeman CA, Limburg PC, Kallenberg CGM. Skewed distribution of Th17 lymphocytes in patients with Wegener’s granulomatosis in remission. Arthritis Rheum (2008) 58:2196–205. doi:10.1002/art.23557
103. Wilde B, Thewissen M, Damoiseaux J, Hilhorst M, van Paassen P, Witzke O, et al. Th17 expansion in granulomatosis with polyangiitis (Wegener’s): the role of disease activity, immune regulation and therapy. Arthritis Res Ther (2012) 14:R227. doi:10.1186/ar4066
104. Abdulahad WH, Lepse N, Stegeman CA, Huitema MG, Doornbos-van der Meer B, Tadema H, et al. Increased frequency of circulating IL-21 producing Th-cells in patients with granulomatosis with polyangiitis (GPA). Arthritis Res Ther (2013) 15:R70. doi:10.1186/ar4247
105. Szczeklik W, Jakieła B, Wawrzycka-Adamczyk K, Sanak M, Hubalewska-Mazgaj M, Padjas A, et al. Skewing toward Treg and Th2 responses is a characteristic feature of sustained remission in ANCA-positive granulomatosis with polyangiitis. Eur J Immunol (2017) 47:724–33. doi:10.1002/eji.201646810
106. Fazio J, Quabius ES, Müller A, Adam-Klages S, Wesch D, Sebens S, et al. Vδ2 T cell deficiency in granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Immunol (2013) 149:65–72. doi:10.1016/j.clim.2013.06.003
107. Braudeau C, Amouriaux K, Néel A, Herbreteau G, Salabert N, Rimbert M, et al. Persistent deficiency of circulating mucosal-associated invariant T (MAIT) cells in ANCA-associated vasculitis. J Autoimmun (2016) 70:73–9. doi:10.1016/j.jaut.2016.03.015
108. Fagin U, Csernok E, Müller A, Pitann S, Fazio J, Krause K, et al. Distinct proteinase 3-induced cytokine patterns in Wegener’s granulomatosis, Churg-Strauss syndrome, and healthy controls. Clin Exp Rheumatol (2011) 29:S57–62.
109. Fagin U, Pitann S, Gross WL, Lamprecht P. Flow cytometric characterization of “early” and “late differentiated” T-cells including PR3-specific cells in granulomatosis with polyangiitis (Wegener’s). Cytometry B Clin Cytom (2012) 82:173–5. doi:10.1002/cyto.b.21006
110. Lamprecht P. CD28 negative T cells are enriched in granulomatous lesions of the respiratory tract in Wegener’s granulomatosis. Thorax (2001) 56:751–7. doi:10.1136/thorax.56.10.751
111. Abdulahad WH, Kallenberg CGM, Limburg PC, Stegeman CA. Urinary CD4+ effector memory T cells reflect renal disease activity in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum (2009) 60:2830–8. doi:10.1002/art.24747
112. Kiene M, Csernok E, Müller A, Metzler C, Trabandt A, Gross WL. Elevated interleukin-4 and interleukin-13 production by T cell lines from patients with Churg-Strauss syndrome. Arthritis Rheum (2001) 44:469–73. doi:10.1002/1529-0131(200102)44:2<469:AID-ANR66>3.0.CO;2-0
113. Tsurikisawa N, Saito H, Tsuburai T, Oshikata C, Ono E, Mitomi H, et al. Differences in regulatory T cells between Churg-Strauss syndrome and chronic eosinophilic pneumonia with asthma. J Allergy Clin Immunol (2008) 122:610–6. doi:10.1016/j.jaci.2008.05.040
114. Saito H, Tsurikisawa N, Tsuburai T, Oshikata C, Akiyama K. Cytokine production profile of CD4+ T cells from patients with active Churg-Strauss syndrome tends toward Th17. Int Arch Allergy Immunol (2009) 149(Suppl 1):61–5. doi:10.1159/000210656
115. Klapa S, Mueller A, Csernok E, Fagin U, Klenerman P, Holl-Ulrich K, et al. Lower numbers of FoxP3 and CCR4 co-expressing cells in an elevated subpopulation of CD4+CD25highregulatory T cells from Wegener’s granulomatosis. Clin Exp Rheumatol (2010) 28:72–80.
116. Free ME, Bunch DO, McGregor JA, Jones BE, Berg EA, Hogan SL, et al. Patients with antineutrophil cytoplasmic antibody-associated vasculitis have defective Treg cell function exacerbated by the presence of a suppression-resistant effector cell population. Arthritis Rheum (2013) 65:1922–33. doi:10.1002/art.37959
117. Grunewald J, Halapi E, Wahlström J, Giscombe R, Nityanand S, Sanjeevi C, et al. T-cell expansions with conserved T-cell receptor beta chain motifs in the peripheral blood of HLA-DRB1*0401 positive patients with necrotizing vasculitis. Blood (1998) 92:3737–44.
118. Boita M, Guida G, Circosta P, Elia AR, Stella S, Heffler E, et al. The molecular and functional characterization of clonally expanded CD8+ TCR BV T cells in eosinophilic granulomatosis with polyangiitis (EGPA). Clin Immunol (2014) 152:152–63. doi:10.1016/j.clim.2014.03.001
119. Morgan MD, Pachnio A, Begum J, Roberts D, Rasmussen N, Neil DAH, et al. CD4+CD28− T cell expansion in granulomatosis with polyangiitis (Wegener’s) is driven by latent cytomegalovirus infection and is associated with an increased risk of infection and mortality. Arthritis Rheum (2011) 63:2127–37. doi:10.1002/art.30366
120. Cornec D, Gall EC-L, Fervenza FC, Specks U. ANCA-associated vasculitis—clinical utility of using ANCA specificity to classify patients. Nat Rev Rheumatol (2016) 12:570–9. doi:10.1038/nrrheum.2016.123
121. Jennette JC, Nachman PH. ANCA glomerulonephritis and vasculitis. Clin J Am Soc Nephrol (2017) 12:1680–91. doi:10.2215/CJN.02500317
122. Csernok E. Anti-neutrophil cytoplasmic antibodies and pathogenesis of small vessel vasculitides. Autoimmun Rev (2003) 2:158–64. doi:10.1016/S1568-9972(03)00010-7
123. Franssen CF, Huitema MG, Muller Kobold AC, Oost-Kort WW, Limburg PC, Tiebosch A, et al. In vitro neutrophil activation by antibodies to proteinase 3 and myeloperoxidase from patients with crescentic glomerulonephritis. J Am Soc Nephrol (1999) 10:1506–15.
124. Harper L, Radford D, Plant T, Drayson M, Adu D, Savage COS. IgG from myeloperoxidase-antineutrophil cytoplasmic antibody-positive patients stimulates greater activation of primed neutrophils than IgG from proteinase 3-antineutrophil cytoplasmic antibody-positive patients. Arthritis Rheum (2001) 44:921–30. doi:10.1002/1529-0131(200104)44:4<921:AID-ANR149>3.0.CO;2-4
125. Roth AJ, Ooi JD, Hess JJ, van Timmeren MM, Berg EA, Poulton CE, et al. Epitope specificity determines pathogenicity and detectability in ANCA-associated vasculitis. J Clin Invest (2013) 123:1773–83. doi:10.1172/JCI65292
126. Oommen E, Hummel A, Allmannsberger L, Cuthbertson D, Carette S, Pagnoux C, et al. IgA antibodies to myeloperoxidase in patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Clin Exp Rheumatol (2017) 35:98–101. doi:10.1007/128
127. Finnern R, Bye JM, Dolman KM, Zhao MH, Short A, Marks JD, et al. Molecular characteristics of anti-self antibody fragments against neutrophil cytoplasmic antigens from human V gene phage display libraries. Clin Exp Immunol (1995) 102:566–74. doi:10.1111/j.1365-2249.1995.tb03854.x
128. Cui Z, Zhao M, Segelmark M, Hellmark T. Natural autoantibodies to myeloperoxidase, proteinase 3, and the glomerular basement membrane are present in normal individuals. Kidney Int (2010) 78:590–7. doi:10.1038/ki.2010.198
129. Kelley JM, Monach PA, Ji C, Zhou Y, Wu J, Tanaka S, et al. IgA and IgG antineutrophil cytoplasmic antibody engagement of Fc receptor genetic variants influences granulomatosis with polyangiitis. Proc Natl Acad Sci U S A (2011) 108:20736–41. doi:10.1073/pnas.1109227109
130. Clain JM, Hummel AM, Stone JH, Fervenza FC, Hoffman GS, Kallenberg CGM, et al. Immunoglobulin (Ig)M antibodies to proteinase 3 in granulomatosis with polyangiitis and microscopic polyangiitis. Clin Exp Immunol (2017) 188:174–81. doi:10.1111/cei.12925
131. Collin M, Ehlers M. The carbohydrate switch between pathogenic and immunosuppressive antigen-specific antibodies. Exp Dermatol (2013) 22:511–4. doi:10.1111/exd.12171
132. Holland M, Takada K, Okumoto T, Takahashi N, Kato K, Adu D, et al. Hypogalactosylation of serum IgG in patients with ANCA-associated systemic vasculitis. Clin Exp Immunol (2002) 129:183–90. doi:10.1046/j.1365-2249.2002.01864.x
133. Espy C, Morelle W, Kavian N, Grange P, Goulvestre C, Viallon V, et al. Sialylation levels of anti-proteinase 3 antibodies are associated with the activity of granulomatosis with polyangiitis (Wegener’s). Arthritis Rheum (2011) 63:2105–15. doi:10.1002/art.30362
134. Wuhrer M, Stavenhagen K, Koeleman CAM, Selman MHJ, Harper L, Jacobs BC, et al. Skewed Fc glycosylation profiles of anti-proteinase 3 immunoglobulin G1 autoantibodies from granulomatosis with polyangiitis patients show low levels of bisection, galactosylation, and sialylation. J Proteome Res (2015) 14:1657–65. doi:10.1021/pr500780a
135. Kemna MJ, Plomp R, van Paassen P, Koeleman CAM, Jansen BC, Damoiseaux JGMC, et al. Galactosylation and sialylation levels of IgG predict relapse in patients with PR3-ANCA associated vasculitis. EBioMedicine (2017) 17:108–18. doi:10.1016/j.ebiom.2017.01.033
136. Kettritz R. How anti-neutrophil cytoplasmic autoantibodies activate neutrophils. Clin Exp Immunol (2012) 169:220–8. doi:10.1111/j.1365-2249.2012.04615.x
137. van Timmeren MM, van der Veen BS, Stegeman CA, Petersen AH, Hellmark T, Collin M, et al. IgG glycan hydrolysis attenuates ANCA-mediated glomerulonephritis. J Am Soc Nephrol (2010) 21:1103–14. doi:10.1681/ASN.2009090984
138. Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest (2002) 110:955–63. doi:10.1172/JCI15918
139. Schreiber A, Eulenberg-Gustavus C, Bergmann A, Jerke U, Kettritz R. Lessons from a double-transgenic neutrophil approach to induce antiproteinase 3 antibody-mediated vasculitis in mice. J Leukoc Biol (2016) 100:1443–52. doi:10.1189/jlb.5A0116-037R
140. Little MA, Al-Ani B, Ren S, Al-Nuaimi H, Leite M, Alpers CE, et al. Anti-proteinase 3 anti-neutrophil cytoplasm autoantibodies recapitulate systemic vasculitis in mice with a humanized immune system. PLoS One (2012) 7:e28626. doi:10.1371/journal.pone.0028626
141. Petersen F, Yue X, Riemekasten G, Yu X. Dysregulated homeostasis of target tissues or autoantigens – a novel principle in autoimmunity. Autoimmun Rev (2017) 16:602–11. doi:10.1016/j.autrev.2017.04.006
142. Kesel N, Köhler D, Herich L, Laudien M, Holl-Ulrich K, Jüngel A, et al. Cartilage destruction in granulomatosis with polyangiitis (Wegener’s granulomatosis) is mediated by human fibroblasts after transplantation into immunodeficient mice. Am J Pathol (2012) 180:2144–55. doi:10.1016/j.ajpath.2012.01.021
143. Cornec D, Berti A, Hummel A, Peikert T, Pers J-O, Specks U. Identification and phenotyping of circulating autoreactive proteinase 3-specific B cells in patients with PR3-ANCA associated vasculitis and healthy controls. J Autoimmun (2017) 84:122–31. doi:10.1016/j.jaut.2017.08.006
144. Voswinkel J, Mueller A, Kraemer JA, Lamprecht P, Herlyn K, Holl-Ulrich K, et al. B lymphocyte maturation in Wegener’s granulomatosis: a comparative analysis of VH genes from endonasal lesions. Ann Rheum Dis (2006) 65:859–64. doi:10.1136/ard.2005.044909
145. Thurner L, Müller A, Cérutti M, Martin T, Pasquali J-L, Gross WL, et al. Wegener’s granuloma harbors B lymphocytes with specificities against a proinflammatory transmembrane protein and a tetraspanin. J Autoimmun (2011) 36:87–90. doi:10.1016/j.jaut.2010.09.002
146. Unizony S, Villarreal M, Miloslavsky EM, Lu N, Merkel PA, Spiera R, et al. Clinical outcomes of treatment of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis based on ANCA type. Ann Rheum Dis (2016) 75:1166–9. doi:10.1136/annrheumdis-2015-208073
147. Koster MJ, Warrington KJ. Recent advances in understanding and treating vasculitis. F1000Res (2016) 5:1436. doi:10.12688/f1000research.8403.1
148. Cortazar FB, Pendergraft WF, Wenger J, Owens CT, Laliberte K, Niles JL. Effect of continuous B cell depletion with rituximab on pathogenic autoantibodies and total IgG levels in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol (2017) 69:1045–53. doi:10.1002/art.40032
149. Wilde B, Thewissen M, Damoiseaux J, Knippenberg S, Hilhorst M, van Paassen P, et al. Regulatory B cells in ANCA-associated vasculitis. Ann Rheum Dis (2013) 72:1416–9. doi:10.1136/annrheumdis-2012-202986
150. Lepse N, Abdulahad WH, Rutgers A, Kallenberg CGM, Stegeman CA, Heeringa P. Altered B cell balance, but unaffected B cell capacity to limit monocyte activation in anti-neutrophil cytoplasmic antibody-associated vasculitis in remission. Rheumatology (Oxford) (2014) 53(9):1683–92. doi:10.1093/rheumatology/keu149
151. Todd SK, Pepper RJ, Draibe J, Tanna A, Pusey CD, Mauri C, et al. Regulatory B cells are numerically but not functionally deficient in anti-neutrophil cytoplasm antibody-associated vasculitis. Rheumatology (Oxford) (2014) 53(9):1693–703. doi:10.1093/rheumatology/keu136
Keywords: anti-neutrophil cytoplasmic autoantibodies, anti-neutrophil cytoplasmic autoantibody vasculitides, microscopic polyangiitis, granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis
Citation: Lamprecht P, Kerstein A, Klapa S, Schinke S, Karsten CM, Yu X, Ehlers M, Epplen JT, Holl-Ulrich K, Wiech T, Kalies K, Lange T, Laudien M, Laskay T, Gemoll T, Schumacher U, Ullrich S, Busch H, Ibrahim S, Fischer N, Hasselbacher K, Pries R, Petersen F, Weppner G, Manz R, Humrich JY, Nieberding R, Riemekasten G and Müller A (2018) Pathogenetic and Clinical Aspects of Anti-Neutrophil Cytoplasmic Autoantibody-Associated Vasculitides. Front. Immunol. 9:680. doi: 10.3389/fimmu.2018.00680
Received: 30 November 2017; Accepted: 20 March 2018;
Published: 09 April 2018
Edited by:
Falk Nimmerjahn, Friedrich-Alexander-Universität Erlangen-Nürnberg, GermanyReviewed by:
Thomas Hellmark, Lund University, SwedenMatthew Cook, Australian National University, Australia
Copyright: © 2018 Lamprecht, Kerstein, Klapa, Schinke, Karsten, Yu, Ehlers, Epplen, Holl-Ulrich, Wiech, Kalies, Lange, Laudien, Laskay, Gemoll, Schumacher, Ullrich, Busch, Ibrahim, Fischer, Hasselbacher, Pries, Petersen, Weppner, Manz, Humrich, Nieberding, Riemekasten and Müller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Lamprecht, peter.lamprecht@uksh.de