 Sophie Jung
Sophie Jung Laura Gámez-Díaz
Laura Gámez-Díaz Michele Proietti
Michele Proietti Bodo Grimbacher
Bodo Grimbacher- 1CNRS, UPR 3572 (I2CT), Institut de Biologie Moléculaire et Cellulaire (IBMC), Strasbourg, France
- 2Hôpitaux Universitaires de Strasbourg, Pôle de Médecine et de Chirurgie Bucco-Dentaires, Strasbourg - Université de Strasbourg, Faculté de Chirurgie Dentaire, Strasbourg, France
- 3Center for Chronic Immunodeficiency (CCI), Medical Center – Faculty of Medicine, University of Freiburg, Freiburg, Germany
Primary immunodeficiencies (PIDs) represent a group of mostly monogenic disorders caused by loss- or gain-of-function mutations in over 340 known genes that lead to abnormalities in the development and/or the function of the immune system. However, mutations in different genes can affect the same cell-signaling pathway and result in overlapping clinical phenotypes. In particular, mutations in the genes encoding for members of the phosphoinositide3-kinase (PI3K)/AKT/mTOR/S6 kinase (S6K) signaling cascade or for molecules interacting with this pathway have been associated with different PIDs that are often characterized by the coexistence of both immune deficiency and autoimmunity. The serine/threonine kinase mechanistic/mammalian target of rapamycin (mTOR), which acts downstream of PI3K and AKT, is emerging as a key regulator of immune responses. It integrates a variety of signals from the microenvironment to control cell growth, proliferation, and metabolism. mTOR plays therefore a central role in the regulation of immune cells’ differentiation and functions. Here, we review the different PIDs that share an impairment of the PI3K/AKT/mTOR/S6K pathway and we propose to name them “immune TOR-opathies” by analogy with a group of neurological disorders that has been originally defined by PB Crino and that are due to aberrant mTOR signaling (1). A better understanding of the role played by this complex intracellular cascade in the pathophysiology of “immune TOR-opathies” is crucial to develop targeted therapies.
Introduction
Primary immunodeficiencies (PIDs) comprise more than 350 inherited disorders that affect the development and/or the functions of the components of the immune system (2, 3). They are individually rare but collectively, they are “more common than thought” (4), particularly due to the rapid increase in the number of newly described disorders and of causative genes that have been identified. In fact, the study of PIDs has frequently contributed to the discovery of new genes that are pivotal in immune cell development, effector functions, or in the maintenance of immune homeostasis (5). Susceptibility to severe and recurrent infections is a constant clinical manifestation in PID patients. However, an overlap between immune deficiency (infections and/or malignancies) and immune dysregulation (autoimmunity, autoinflammation, and/or allergy) is often observed in certain types of PIDs (2, 3, 6). Although PIDs are mostly inherited as monogenic disorders, disease penetrance, as well as disease expressivity, may result from interactions between genetic, epigenetic, and/or environmental factors. This contributes to the wide phenotypic diversity, even between individuals with an identical mutation in the same gene (2, 3, 7). The International Union of Immunological Societies (IUIS) PID expert committee regularly publishes a classification based on shared pathogenesis and/or clinical phenotypes with the latest update in 2017 (2, 3).
The serine/threonine kinase mechanistic/mammalian target of rapamycin (mTOR) plays a central role within the phosphoinositide3-kinase (PI3K)/AKT/mTOR/S6 kinase (S6K) signaling pathway. It acts as a downstream effector of AKT in two structural and functional distinct protein complexes named mTOR complex 1 and 2 (mTORC1 and mTORC2, respectively) (8). mTOR integrates the different cues from the microenvironment to control cell growth, proliferation, and metabolism, thereby exerting crucial functions in the regulation of immune homeostasis (8, 9).
Defects in the genes encoding for the different members of the PI3K/AKT/mTOR/S6K cascade or for molecules interacting with this pathway are frequently associated with immune dysfunction. We therefore propose here to cluster the different PIDs that share an impairment of the PI3K/AKT/mTOR/S6K pathway. Considering the central role of mTOR in the signaling cascade, this subgroup of PIDs will be referred hereafter as “immune TOR-opathies.” The term “mTOR-opathies” was initially coined in 2007 by PB Crino to define a wide spectrum of neurological disorders due to abnormal mTOR signaling that are characterized by focal malformations of cortical development, epilepsy, and neurobehavioral disabilities (1, 10).
In this review, we describe the PI3K/AKT/mTOR/S6K signaling cascade, focusing on the genetic and molecular defects of the different “immune TOR-opathies,” and on the impact of this pivotal pathway in the development of immune deficiency and immune dysregulation, a hallmark of “immune TOR-opathies.”
PI3K/AKT/mTOR/S6K Signaling Pathway Plays a Crucial Role in Immune Homeostasis
S6 kinase activation involves a complex signaling cascade that connects a number of critical kinases, including PI3Ks, AKT (also called PKB for protein kinase B), and mTOR (11) (Figure 1). The PI3K/AKT/mTOR/S6K pathway plays a major role in the control of cell proliferation (increase in number), cell growth (increase in size), survival, and metabolism (12). It is therefore crucial in the regulation of immune responses, as well as in the promotion of B cells, T cells, and myeloid cells differentiation, activation, and function (9).
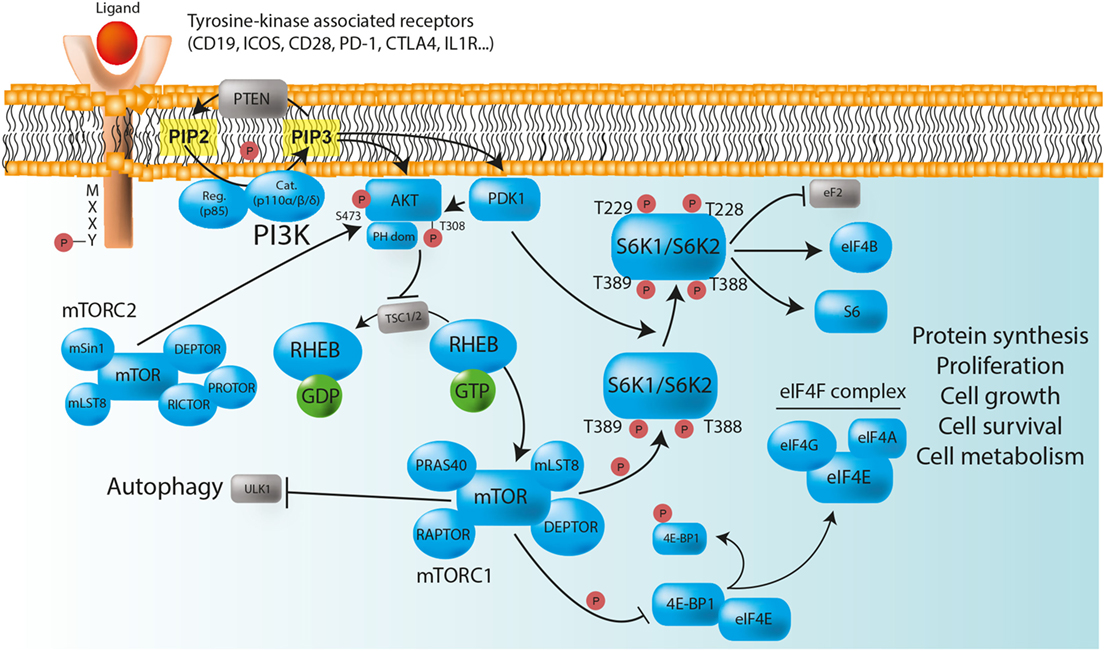
Figure 1. The PI3K/AKT/mTOR/S6K pathway plays a major role in the control of immune cell homeostasis. Class IA PI3Ks are heterodimeric molecules composed of a p110 catalytic subunit (p110α, p110β, or p110δ) and a p85 regulatory subunit. In immune cells, class IA PI3Ks can be activated via multiple surface tyrosine kinase-associated receptors [e.g., BCR, TCR, TLR, CD19, ICOS, PD-1, and CTLA-4] that bear YXXM motifs in their cytoplasmic domain. In the absence of ligand binding, the TSC1/TSC2 complex negatively regulates mTORC1, and therefore protein synthesis, by converting RHEB into its inactive GDP-bound state. After receptor activation, phosphorylated YXXM motifs provide binding sites for the p85 regulatory subunit that brings the p110 catalytic subunit to the membrane, where it converts PIP2 to PIP3. PIP3 serves as plasma membrane docking sites for PH-domain containing proteins, such as AKT, and its upstream activator PDK1. The activity of AKT is also positively regulated by mTORC2. Once phosphorylated, AKT inhibits the TSC1/TSC2 complex, and allows the release of GTP-bound RHEB, thereby enabling mTORC1 activation. Activated mTORC1 triggers biosynthetic pathways (protein synthesis) essential for cell proliferation, survival, and metabolism through S6Ks and 4E-BP1 phosphorylation, while inhibiting ULK1, and therefore autophagy. S6K phosphorylate numerous substrate, including ribosomal protein S6, eukaryotic translation initiation factor eIF4B, and eukaryotic elongation factor 2 (eEF2) kinase. The phosphorylation of 4E-BP1 prevents its binding to the cap-binding protein eIF4E, allowing it to participate in the formation of the eIF4F complex, which is composed of the DEAD-box RNA helicase eIF4A, the cap-binding protein eIF4E, and the large “scaffold” protein eIF4G, and which is required for the initiation of cap-dependent translation. PTEN is a negative regulator of PI3K/AKT/mTOR/S6K signaling pathway that dephosphorylates PIP3 back to PIP2. Red circles: phosphorylation; normal arrows: activation; blunt arrows: inhibition.
Among the different classes of PI3Ks, class IA molecules have the most important function in immune cells (13). Those heterodimeric proteins are formed by the association of a catalytic subunit of approximately 110 kDa (p110α, p110β, or p110δ encoded by PIK3CA, PIK3CB, and PIK3CD respectively), and a Src-homology 2 (SH2) domain-containing regulatory subunit (p85, p50, and p55α encoded by PIK3R1; p85β encoded by PIK3R2; and p55γ encoded by PIK3R3). The catalytic subunits p110α and p110β are widely expressed, whereas the expression of p110δ is restricted to leukocytes (13, 14). The regulatory subunit controls the cellular location and the activity of the enzyme by recruiting the catalytic subunit to membrane-associated proteins that have been phosphorylated on YXXM motifs by tyrosine kinases (12, 13). In immune cells, class IA PI3Ks can be activated via multiple surface tyrosine-kinase-associated receptors, including the T- and B-cell receptors (TCR and BCR, respectively), toll-like receptors (TLRs), as well as various co-receptors [CD19, inducible T-cell costimulator (ICOS), CD28, PD-1, and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)], and cytokine receptors (IL-1, IL-2, IL-4, IL-12, and IFN-γ) that contain YXXM motifs in their cytoplasmic domain (12). After activation, class I PI3Ks catalyze the conversion of phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2 or PIP2] to phosphatidylinositol-(3,4,5)-trisphosphate [PI(3,4,5)P3 or PIP3] (12). PIP3 acts as binding sites for various intracellular enzymes harboring pleckstrin-homology (PH) domains, in particular for the serine/threonine kinase AKT, which is then recruited at the inner leaflet of the cell membrane to be phosphorylated. The activity of AKT is positively regulated by the binding of PIP3 to its PH domain, but also by the phosphorylation at position Thr308 by phosphoinositide-dependent kinase-1 (PDK1) and at position Ser473 by mTORC2 (15) (Figure 1). Once AKT is activated, it inhibits the tuberous sclerosis heterodimeric complex (TSC1/TSC2 complex), inducing the release of the GTP-binding protein Ras homolog enriched in brain (RHEB) from the inhibition by TSC2, therefore enabling the activation of mTORC1 (16) (Figure 1).
The serine/threonine kinase mTOR was identified while investigating the mechanism of action of rapamycin (also known as sirolimus), an immunosuppressive drug inhibiting mTOR enzymatic activity that is currently used to prevent organ transplant rejection and to treat lymphoproliferative diseases (17, 18). mTOR associates with distinct sets of proteins to form the intracellular signaling complexes mTORC1 and mTORC2 (8). Both complexes contain mammalian lethal with SEC13 protein 8/G protein β subunit-like (mLST8/GβL) and DEP domain-containing mTOR interacting protein (DEPTOR). In contrast, the partners regulatory-associated protein of mTOR (RAPTOR) and proline-rich AKT substrate 40 kDa (PRAS40) define the mTORC1 network, whereas rapamycin-insensitive companion of mTOR (RICTOR), stress-activated map kinase-interacting protein 1 (mSIN1), and protein observed with Rictor (PROTOR) are specific to the mTORC2 complex (8, 19, 20) (Figure 1). The major function of mTORC1 is to sense nutrients and mitogenic signals (8, 19, 20). Thus, when conditions are favorable, mTORC1 triggers biosynthetic pathways essential for cell growth and proliferation, mainly through direct phosphorylation of ribosomal S6K and eukaryotic translation initiation factor 4E (eIF4E) binding protein 1 (4E-BP1) (8, 19, 20). mTORC1 also inhibits the serine/threonine kinase ULK1, thereby suppressing autophagy, a conserved catabolic process by which double-membrane vesicles (autophagosomes) engulf cytoplasmic contents for lysosomal degradation. Autophagy allows the recycling of cellular components and the generation of nutrients under metabolic stress, promoting cell survival (8, 21, 22). It is also implicated in more complex functions and participates in the regulation of immunity (23). Overall, the phosphorylation of S6Ks and 4E-BP1, along with the suppression of autophagy by active mTORC1, are essential for cell growth (24). Conversely, in case of starvation, AMP-activated protein kinase (AMPK) inactivates mTORC1 and phosphorylates the active sites of ULK1, therefore, enabling autophagy initiation (8, 21, 22). mTORC2 plays various roles in cell survival, metabolism, proliferation, and cytoskeleton organization via the phosphorylation of AKT on Ser473 (mTORC2-dependent), leading to the phosphorylation, sequestration, and further inhibition of Forkhead box protein O (FOXO) (9). Negative regulators controlling PI3K/AKT/mTOR/S6K pathway include the phosphatase and tensin homolog (PTEN) that dephosphorylates PIP3 back to PIP2, thereby downregulating AKT signaling (25) (Figure 1).
Together with 4E-BP1, ribosomal S6Ks represent the best characterized substrates of mTORC1 (11, 26, 27). Like AKT, S6K1 (isoforms p70- and p85-S6K1), and S6K2 (isoforms p54- and p60-S6K2) belong to the AGC serine/threonine kinases family (26). The S6K activation begins with the phosphorylation of serine residues in the C-terminal domain that expose the internal region of the protein, allowing mTOR to phosphorylate Thr389 in S6K1 and Thr388 in S6K2. Indeed, S6K activation absolutely requires mTORC1-mediated phosphorylation (28). The subsequent phosphorylation by PDK1 at Thr229 in S6K1 and at Thr228 in S6K2 leads to their full activation (26) (Figure 1). S6K proteins originally gained their name due to their ability to phosphorylate ribosomal protein S6, a component of the 40S ribosome subunit, and their preferred phosphorylation motif has been characterized as RXRXXS/T (26). S6K1 and S6K2 have many functional similarities. They regulate several cellular and molecular processes, including transcription, protein synthesis, metabolism, cell proliferation, and survival (11, 26, 28). Although S6K1 has been more extensively studied, some distinct functions of S6K2 have been described (29). For instance, it has been shown that S6K2 plays a role in Th17 differentiation through the regulation of the transcription factor RORγ (30) despite a more recent study suggesting that this function may be context-specific (31). Ribosomal protein S6 was the first discovered substrate of S6Ks. It promotes biosynthetic pathways that are important for cell growth (27, 28), but the functional significance of its phosphorylation still remains not fully understood (28). However, the analysis of the phosphorylation status of p70-S6K1 (at Thr 389) and its substrate ribosomal protein S6 (at Ser240/244; S6K dependent) is widely and routinely used as a readout of mTORC1 activity (32, 33), in particular in lymphocytes populations, where other mTOR signaling markers are more difficult to monitor. A number of other S6K1 substrates have been involved in the regulation of protein synthesis at levels of initiation (eIF4B: eukaryotic translation initiation factor 4B), and elongation (eEF2: eukaryotic elongation factor 2), but also in RNA splicing (CBC: cap binding complex; SKAR: S6K1 Aly/REF-like target) (Figure 1). In addition, S6K1 plays a role in cell survival by blocking apoptosis through phosphorylation of the pro-apoptotic protein Bcl-2-associated death promoter (BAD), thereby preventing its interaction with BCL-X or BCL-2 (11, 26, 28). Some evidences also indicate that S6K1 may participate in cytoskeleton dynamics, in particular in F-actin reorganization (34).
Studies in animal models have suggested that reduced PI3K/AKT/mTOR/S6K signaling (hypoactivation) can lead to immune deficiency, whereas uncontrolled PI3K/AKT/mTOR/S6K signaling (hyperactivation) is associated with autoimmunity and hematological malignancies (12). Nevertheless, this simplistic dichotomous model does not reflect the highly complex regulation of this pathway. Indeed, several human PIDs that are associated with a hyperactivation of the PI3K/AKT/mTOR/S6K pathway have features of both immunodeficiency and immune dysregulation, suggesting a tight and dynamic modulation of the signaling cascade for optimal immune cell function.
mTOR plays a central role in the regulation of immune responses evidenced in numerous studies showing that mTOR or mTORC1 inhibition can have both positive and negative effects on lymphocytes, in particular on T-cell development and functions [reviewed in Ref. (9)]. The mTOR hypomorphic mouse, which is a model of mTORC1/mTORC2 inhibition [murine Mtor knockout (KO) is lethal and there are no reported cases of human loss-of-function (LOF) mutations in MTOR] is characterized by an immunodeficient phenotype with impaired development, proliferation, and migration of lymphocytes, as well as abnormal antibody production (35). Reduced mTOR expression results in decreased phosphorylation of the mTORC1 target p70-S6K1 and of the mTORC2 target AKT (phosphorylation at Ser473) in fibroblasts and TCR stimulated T cells. However, despite reduction of p70-S6K1 phosphorylation in murine B cells activates with lipopolysaccharide (LPS), mTORC2 activity is increased, suggesting that AKT regulation may be cell-type specific (35). In addition, PI3K/AKT/mTOR pathway seems to play differing roles during the differentiation and function of regulatory T cells (Tregs). Tissue tolerance is associated with the upregulation of enzymes that consume many of the essential amino acids (36). These starvation conditions lead to mTOR inhibition, promoting the expression of FoxP3 in naïve T cells, and therefore the generation of CD4+ FoxP3+ Tregs (37). In fact, continued TCR signaling and constitutive PI3K/AKT/mTOR activity antagonizes Foxp3 induction (9, 37, 38). However, under mTOR inhibitory conditions, Tregs are not optimally functional, requiring mTOR re-activation or inflammatory conditions to acquire their full suppressive potential. Alternate cycles of mTOR activity may therefore be needed for optimal functional induction of Tregs (37, 39). The mTOR downstream effectors S6Ks are essential in controlling the cell size and proliferation of certain cell types such as hepatocytes (40, 41). However, in contrast to mTOR, the functions of S6K1 and S6K2 in lymphocytes still remain controversial (33). Simultaneous deletion of S6K1 and S6K2 genes in a murine model was associated with a severe reduction in viability due to perinatal lethality, but single S6K1 or S6K2 KO mice did not exhibit obvious immune defects (although no detailed immunological study was performed) (41, 42). In addition, it has been shown in vitro using S6K1/S6K2 double KO T and B cells that S6K activity is dispensable for lymphocytes growth and proliferation after antigen receptor engagement (33). Germline deletion of Rps6 that encodes for ribosomal protein S6 is embryonically lethal (43) and T cell-specific deletion of Rps6 abolishes thymic T-cell development (44). By contrast, the role of S6 phosphorylation is not well understood. Knockin mice in which all serine residues of S6 protein have been mutated to alanine to prevent phosphorylation by S6Ks are viable (45) and show normal T-cell activation and differentiation (46). All these data clearly demonstrate the complexity of PI3K/AKT/mTOR/S6K pathway regulation.
Gain-of-Function (GOF) Mutations in the Genes Encoding Class I PI3K Cause Activated PI3Kδ Syndrome (APDS)
Hyperactivation of the PI3K/AKT/mTOR/S6K signaling pathway in immune cells can be the consequence of heterozygous GOF mutations in the genes encoding for PI3Kδ that cause an immune dysregulation disorder called activated PI3Kδ syndrome [APDS; also known as “p110δ activating mutation causing senescent T cells, lymphadenopathy, and immunodeficiency” (PASLI)] (47). Molecularly, APDS encompasses two different disorders: APDS1 and APDS2. APDS1 (or PASLI-CD) is the consequence of mutations in the PIK3CD gene encoding for p110δ, the catalytic subunit of PI3Kδ that result in single-amino-acid substitutions leading to p110δ overactivation. APDS2 (or PASLI-R1) results from mutations in the PIK3R1 gene encoding for p85α, the regulatory subunit of PI3Kδ. These mutations impair the binding of p85α to its cognate partner p110δ that is, therefore, inefficiently inhibited (47–51). Up to date, more than 150 APDS patients have been reported (48–68). They display features of both immune deficiency and immune dysregulation, and all of them present with early-onset, as well as severe and recurrent sino-pulmonary infections, mostly by encapsulated bacteria (47, 58). Benign lymphoproliferation (hepatosplenomegaly, lymphadenopathy, focal nodular lymphoid hyperplasia), various autoimmune manifestations, and B cell lymphomas are also frequently observed (47, 54, 55, 58, 61). Growth retardation is, however, commonly associated with APDS2, but not APDS1 (58, 66).
Most APDS patients have elevated transitional B cells, reduced class-switched memory B cells, variable immunoglobulin levels (mainly reduced IgG and increased IgM levels, hypogammaglobulinemia, or in some cases agammaglobulinemia) associated with a poor vaccine response, and an impaired in vitro B cell isotype switching (47, 51, 64, 69). Abnormalities in B lymphocytes from APDS patients recapitulate the defects of class-switch recombination that are observed in B lymphocytes from PTEN-deficient mice (70). Although APDS was initially described as a common variable immunodeficiency (CVID)-like disease, affected patients also suffer from recurrent herpes virus infections (i.e., EBV, CMV, and VZV), indicating an impaired T cell function (47, 52, 54, 56–58, 65). In addition, the majority of APDS patients show a progressive CD4+ T cell lymphopenia with a decreased frequency of CD4+ naïve T cells [in contrast to the lethal CD4+ T cell hyperplasia that is described in mice with a T cell-specific deletion of PTEN (71)], but an excessive accumulation of terminally differentiated, senescent CD8+ effector T cells (64). Considering the T cell abnormalities, APDS may be classified as combined immunodeficiency (CID) rather than as CVID-like disease.
In T cells, PI3Kδ is activated downstream of CD28, leading to enhanced AKT and mTOR signaling, which blocks autophagy but stimulates T cell proliferation and terminal differentiation through the phosphorylation of S6K (12). Activated AKT also mediates the phosphorylation and subsequent degradation of FOXO transcription factors that regulate T cell expansion and memory T cell differentiation (72). The analysis of PI3K signaling in T cells from APDS patients showed a constitutive hyperphosphorylation of both AKT (on Thr308: PI3K/PDK1 dependent and on Ser473: mTORC2 dependent) and S6 (on Ser235/236 and Ser240/244: mTORC1 dependent) (50–52, 64, 65, 67). The general overactivation of the PI3K/mTOR/S6K signaling pathway promotes the switch to an anabolic cellular state with increased aerobic glycolysis that is required for the expansion of effector T cells (73). Downregulation of mTOR signaling and reversion to a catabolic cellular state by autophagy induction, are, however, crucial for memory T cell formation and prolonged survival (73). In APDS patients, the constant maintenance of aerobic glycosis restrains the function and survival of memory CD8+ T cells, leading to an abundance of senescent effector and short-lived effector memory CD8+ T cells that exhibit a poor recall response in vitro and could account for the defective antiviral immunity in vivo (64, 65, 74). Similarly, high AKT and S6 phosphorylation levels were observed in transformed EBV-B cells, peripheral blood mononuclear cells, and isolated B cells (total B cells and isolated B cell subsets) from APDS patients at basal state and after B cell stimulation (48, 51, 52, 65). However, the link between the increased PI3K/mTOR/S6K signaling in B cells and the observed B cell phenotype is still a focus of research.
The insights into the pathophysiology of APDS allowed refining the therapeutic approaches. Indeed, it has been shown that in vitro treatment of unstimulated T cell blasts with the mTOR inhibitor rapamycin (sirolimus) leads to a decrease of S6 hyperphosphorylation (64). More notably, the administration of rapamycin was found to improve the clinical and immunological phenotype of two APDS patients with a reduction of hepatosplenomegaly and lymphadenopathy, as well as a normalization of T cell subpopulations (64, 67). However, PI3Kδ regulates additional pathways to mTOR (such as FOXO for example) and mTOR is also controlled by PI3K-independent pathways (13). Therefore, selective inhibitors of the PI3Kδ subunit, which have already shown remarkable success in certain hematologic malignancies, should be considered as future therapeutic options in APDS patients. Both in vitro and in vivo data support the specific inhibition of PI3Kδ as a promising therapy. Indeed, the selective p110δ inhibitor IC87114 is able to dampen the activity of the mutated PI3Kδ in vitro in APDS1 patients’ T cells (52), and both p110δ (APDS1) and p85α (APDS2) are strongly inhibited in vitro by the PI3Kδ-specific inhibitor idelalisib (GS-1101 or CAL-101), which is currently approved by the US-Food and Drug Administration for the treatment of chronic lymphocytic leukemia (50, 75). In addition, the first clinical trial (#NCT02435173) that has been conducted by Novartis with the PI3Kδ-specific inhibitor leniolisib (CDZ173) in six APDS patients produced encouraging results (76). Oral administration of leniolisib during 12 weeks was well tolerated and was associated with an improvement of both laboratory and clinical parameters (reduction of peripheral transitional B cells, naive B cells, and senescent T cells; decrease of IgM and inflammatory cytokines levels; reduction of splenomegaly and lymphadenopathy) (76). Another clinical trial for an inhaled PI3Kδ inhibitor, sponsored by GlaxoSmithKline, is currently ongoing in patients with APDS (#NCT02593539) (47).
LOF Mutations in PTEN Lead to an Activated PIK3δ Syndrome-Like Deficiency (APDS-Like)
PTEN encodes a lipid and protein phosphatase that dephosphorylates PIP3 back to PIP2 (77), thereby inhibiting the PI3K/mTOR/AKT/S6K signaling cascade (25). Impairment of PTEN activity is associated with an overabundance of PIP3 and a constitutive downstream activation of AKT, leading to cellular proliferation and overgrowth (78).
A complete disruption of Pten in mouse results in early embryonic death (79), whereas Pten heterozygous mutant mice display hyperplastic-dysplastic features, develop spontaneously tumors (80), and present a lethal polyclonal autoimmune disorder with a phenotype that is reminiscent of Fas-deficient mice (81). Mice carrying a B cell-specific deletion of Pten show abnormal B cell differentiation and function, with increased numbers of marginal zone and B1-a B cells in the spleen, a production of serum autoantibodies, an impaired response to T-dependent and T-independent immunizations, as well as a defect in immunoglobulin class-switch recombination (70, 82, 83).
In humans, heterozygous germline mutations in PTEN may cause different autosomal dominant disorders including Cowden syndrome (CWS; OMIM 158350), Bannayan–Riley–Ruvalcaba syndrome (OMIM 153480), and Proteus syndrome (OMIM 176920), which are characterized by the development of multiple benign hamartoma and malignant tumors (84–86). The term PTEN hamartoma tumor syndrome (PHTS) is therefore used to describe any patient with a germline PTEN mutation regardless of the phenotype (78). Browning et al. reported a case of CWS associated with CID (87). In line with this observation, recent studies indicated that heterozygous LOF mutations in PTEN lead to immunodeficiency and immune dysregulation, with a clinical and immunological presentation that resembles APDS phenotype, including recurrent infections, organomegaly, and CD4+ T cell lymphopenia (68, 88). However, immunodeficiency seems to occur only in some, but not all, patients with PTEN LOF mutations (68). Similarly to patients with heterozygous GOF mutations in PIK3CD, PTEN mutations are associated with an aberrant hyperactivation of the PI3K/AKT/mTOR/S6K pathway with increased phosphorylation of AKT, mTOR, and S6 in T cells (68, 87). Driessen et al. further studied, in a cohort of nine PHTS patients, the impact of germline PTEN mutations on the peripheral B cell development and the humoral immune response (89). They observed decreased counts of switched memory B cells associated with a dysregulated T-dependent B cell response, abnormalities in class-switch recombination, and decreased somatic hypermutation, resulting in hypogammaglobulinemia in about one-third of the patients (89). In mice, it has been shown that the level of activation-induced cytidine deaminase, the main regulator of somatic hypermutation and class-switch recombination, is regulated by the PI3K/AKT signaling cascade (70, 83, 90). This could explain, at least in part, the dysregulated humoral immune response observed in human PTEN deficiency (89).
Surprisingly, despite PTEN dysfunction, PHTS patients display a normal frequency and phenotype of CD4+ FoxP3+ Tregs, as well as a normal activation of the downstream signaling pathway with similar percentages of S6-phosphorylated Tregs in PHTS patients and controls subjects (88). In this cell subset, the enzyme PH domain leucine-rich repeat protein phosphatase (PHLPP), located downstream of PTEN and highly expressed in normal Tregs, provides a complementary phosphatase activity that is important for limiting PI3K hyperactivation (88). PTEN haploinsufficiency leads to APDS-like immune dysregulation, but the compensatory activity of the phosphatase PHLPP may help to maintain checkpoint control at the immunological synapse in human Tregs (88), possibly preventing the development of autoimmune manifestations.
Lipopolysaccharide-Responsive Beige-Like Anchor Protein (LRBA) Deficiency is Associated with Impaired mTOR/S6K Signaling in T Cells
Lipopolysaccharide-responsive beige-like anchor protein (LRBA) belongs to the Beige and Chediak-Higashi (BEACH) domain-containing protein (BDCP) family together with eight other human proteins (91, 92). Although the exact functions of BDCPs remain unclear, they are considered to act as scaffolding molecules forming multiprotein complexes involved in vesicle trafficking and receptor signaling (92). Biallelic mutations in LRBA cause a PID and immune dysregulation disorder known as LRBA deficiency (93). LRBA-deficient patients show an early-onset broad spectrum of clinical and immunological manifestations, including recurrent infections, organomegaly, inflammatory bowel-like disease, hypogammaglobulinemia, and autoimmunity (94, 95). Several LRBA-deficient patients present with an immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX)-like syndrome, indicating Treg cells impairment, that might contribute to the development of the various autoimmune manifestations (96). In fact, nearly two-thirds of LRBA-deficient patients have reduced Tregs frequency (95) with decreased expression of the canonical Treg markers (FOXP3, CD25, Helios, CTLA-4) and impaired Treg cell-mediated suppression (96). Additional perturbations observed in the T cell compartment such as increased proportion of circulating follicular helper T cells (TFH) and decreased proportion of circulating follicular Tregs suggest an ineffective regulation of autoantibodies’ production (96). Although the frequency of recent thymic emigrants seems to be normal, conventional T cells and Tregs from LRBA-deficient patients exhibit an increased apoptosis (96). In mice, Treg-specific disruption of mTORC1 (through the deletion of Raptor) leads to a profound loss of Treg suppressive activity with early development of a lethal autoimmunity and lymphoproliferation (39). Mechanistically, mTORC1 signaling promotes the cholesterol/lipid metabolism that is crucial for cell proliferation and for CTLA-4 upregulation, thereby establishing functional Treg competency (39). CTLA-4 belongs to the T cell co-stimulatory molecule family, including CD28, ICOS, and PD1. It is a critical negative regulator of T cell proliferation that serves as a “checkpoint” of immune responses (97). Interestingly, the role of LRBA in CTLA-4 post-transcriptional regulation is currently the only proven cellular function for LRBA (98). Specifically, LRBA binds through its BEACH domain to the cytoplasmic tail of CTLA-4, allowing its vesicular transport to the plasma membrane of Tregs, and activated conventional T cells (98). CTLA-4 is then able to remove, via transendocytosis, the CD80 and CD86 co-stimulatory molecules from the cell surface of antigen-presenting cells, thereby controlling T cell activation (99). However, when LRBA is absent, the adaptor protein AP-1 binds to CTLA-4, leading to its lysosomal degradation (98). Decreased CTLA-4 expression might therefore contribute to the high frequency of autoimmune manifestations observed in patients with LRBA deficiency (94, 95). Indeed, patients with heterozygous LOF mutations in CTLA-4 develop an immune dysregulation syndrome with an LRBA-deficiency-like clinical phenotype (100–102) known since 2014 as CTLA-4 deficiency. Surprisingly, CTLA-4 was assessed to bind to PI3K with the same avidity as CD28, possibly leading to the activation of PDK1 that phosphorylates AKT at position Thr308 (103, 104), thereby activating mTORC1 signaling cascade. Moreover, in T cells, CTLA-4 dependent activation of PI3K and AKT was shown to sustain T cell anergy without cell death (105). However, the intracellular signaling capacity of CTLA-4 was recently questioned (106). In contrast, it has been reported that activated LRBA-deficient CD4+ and CD8+ T cell subsets show an impaired mTORC1 and mTORC2 activity with a reduced phosphorylation of downstream mTORC1 (S6 and 4E-BP1) and mTORC2 (AKT at position Ser473) substrates (96). Therefore, the PI3K/mTOR/S6K signaling pathway should also be investigated in patients with CTLA-4 deficiency.
Besides Tregs dysfunction, patients with LRBA deficiency present defects in the B cell compartment with reduced numbers of switched memory B cells and plasmablasts, impaired immunoglobulin secretion, low proliferative responses, and a high susceptibility to apoptosis (95, 96). In addition, LRBA-deficient B cells show an impairment of the autophagic flux with an abnormal accumulation of autophagosomes (93). Pengo et al. have shown that autophagy is required for plasma cell homeostasis and long-lived humoral immunity by limiting endoplasmic reticulum stress and immunoglobulin synthesis, while sustaining energy metabolism and plasma cell viability (107). The impaired B cell differentiation and hypogammaglobulinemia observed in LRBA-deficient individuals may therefore be attributable to an increased B cell apoptosis and a reduced plasma cell survival due to defective autophagy. In fact, autophagy is also essential for the survival of memory B cells, and for the maintenance of protective antibody responses required to control viral infections in mice (108). In addition, the accumulation of apoptotic cells may trigger as well the development of autoimmunity (109). mTOR plays a key role at the interface of the pathways controlling cell growth and autophagy. Under nutrient starvation, reduced growth factor signaling, or stress conditions, mTOR is inhibited, and autophagy is therefore promoted. Conversely, the activation of the PI3K/AKT pathway negatively regulates autophagy induction (22). It has been previously suggested that LRBA might act as a scaffold protein, coordinating the assembly and activation of mTOR complexes or of protein networks involved in the autophagic process, as well as the recruitment of downstream molecules (96). Future studies addressing mTOR/S6K signaling in the B cell compartment of LRBA-deficient patients may help to further clarify the links between LRBA, autophagy, and B cell homeostasis.
Mutations Affecting the CARD11-BCL10-MALT1 (CBM) Signalosome Complex are Responsible for Novel PID Phenotypes with an Abnormal Activation of the mTOR/S6K Signaling Pathway
Upon TCR and CD28 activation, the adapter protein caspase recruitment domain-containing protein 11 (CARD11, also called CARMA1), which is specifically expressed in hematopoietic cells, becomes phosphorylated by protein kinase C and other kinases including AKT (110). Phosphorylated CARD11 recruits B-cell lymphoma/leukemia 10 (BCL10) and mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) to form a scaffold called the CBM (CARD11-BCL10-MALT1) signalosome complex that is necessary for optimal activation of the canonical nuclear factorp-κB (NF-κB) pathway (111). Recently, it has been shown that CARD11 and the paracaspase MALT1, but not BCL10, are also required for an optimal activation of the mTOR/S6K pathway in T cells in response to TCR and CD28 co-receptor stimulation (112).
LOF autosomal recessive mutations in CARD11, MALT1, and BCL10 are the cause of a new group of CIDs characterized by recurrent sinopulmonary infections, dysregulated B cell development, and abnormal T cell proliferation despite normal lymphocytes counts, due to a defective canonical NF-κB activation after antigen receptor stimulation (113–118). However, these recently described disorders have a distinct phenotype from other known PIDs affecting the NF-κB axis (113). In addition, there are notable differences between the clinical presentation of CARD11, MALT1, and BCL10 deficiencies (113). For instance, CARD11-deficient patients display variable immunoglobulin levels and Tregs numbers, a predominance of Pneumocystis jirovecii infections, but no gastrointestinal inflammation, whereas BCL10 deficiency has an impact on lymphocytes (low memory T cells) and fibroblasts but not on myeloid cells. CARD11 and BCL10 deficiencies are both characterized by the lack of autoimmune manifestations despite reduced Treg numbers, possibly reflecting the individual nuanced and independent functions of the CBM proteins (113). CARD11-deficient and MALT1-knockdown cells are characterized by a reduced phosphorylation of S6K and S6, emphasizing the role of CARD11 and MALT1 in the mTOR/S6K signaling pathway (112). In addition, the metabolic reprogramming and the proliferation of CD4+ T cells that are also mTORC1 dependent are impaired after MALT1 inhibition (112).
Very recently, Ma et al. have described rare heterozygous hypomorphic CARD11 mutations in eight individuals from four unrelated families with severe atopic dermatitis (119). The phenotype also included variable cutaneous and respiratory infections (88%), eosinophilia (86%), B cell lymphopenia (29%) with low IgM, but normal or elevated IgA (43%), and hyper-IgE (71%) (119). Transfection of mutant CARD11 constructs into T cell lines demonstrated both LOF and a dominant-negative effect on mTORC1 (indicated by reduced S6 phosphorylation), but also on NF-κB signaling, at basal state and after antigen-receptor-induced stimulation. Similarly, mTORC1 activity was also attenuated in T cells, and to a lesser extent in B cells, from patients with heterozygous hypomorphic mutations in CARD11, whereas AKT phosphorylation on Ser473 (mTORC2-dependent) was normal (119). mTOR activity is known to be crucial for T helper (TH) cell differentiation (120). Patients’ T cells were characterized by an impaired TH1 cytokine production (low IFN-γ) and a TH2-skewed phenotype, consistent with their atopic predisposition. The reduced CARD11-dependent mTORC1 activation could contribute to impaired TH1 differentiation in these patients, allowing mTORC2-dependent TH2 response to dominate (119). The role of CARD11 in the regulation of mTORC1 activation depends on its ability to facilitate TCR-induced upregulation, but also on its capacity to activate sodium-dependent neutral amino acid transporter type 2 (ASCT2, also known as SLC1A5), an essential amino acid transporter required for extracellular glutamine import during T cell activation (121). Indeed, T cells from patients with germline hypomorphic CARD11 mutations showed reduced ASCT2 upregulation after TCR activation (119). However, the addition of exogenous glutamine in T cell culture medium was able to boost mTORC1 activation with increased S6 phosphorylation, and to partially correct the TH1 cell defect including proliferation and IFN-γ production (119). Further studies are required to evaluate whether glutamine supplementation, a very simple therapeutic intervention, could ameliorate atopic dermatitis in patients with CARD11 mutations (119). This clearly illustrates that a fine comprehension of the mechanisms regulating the mTOR/S6K signaling pathway is an essential prerequisite for a proper improvement of the patients’ therapeutic management.
Germline heterozygous GOF mutations in CARD11 have been linked to a novel congenital B cell lymphoproliferative disorder called BENTA for “B cell Expansion with NF-κB and T cell Anergy” (122, 123). Five different GOF CARD11 mutations in 16 patients have been described so far (74, 122–124). They abrogate the requirement for antigen receptor engagement in CARD11 activation, resulting in spontaneous CBM signalosome formation, and constitutive NF-κB activation that is responsible for an excessive accumulation of both immature transitional B cells, and polyclonal mature naive B cells (122, 125). BENTA patients develop massive B cell lymphocytosis early in life accompanied by splenomegaly and lymphadenopathy, but without obvious signs of autoimmunity (122, 123). Moreover, GOF CARD11 mutations can potentially predispose to B cell malignancies (74, 122, 126). Despite excessive B cell accumulation, BENTA disease is associated with an underlying immunodeficiency characterized by low frequencies of circulating memory and class-switched B cells, poor humoral response to T cell-independent polysaccharide-based vaccines, impaired plasma cell differentiation, and low IgM as well as variable IgA/IgG secretion. Recurrent sinopulmonary infections are common, and opportunistic viral infections have been noted in some patients (74). Although circulating T cells are present at normal numbers, they are hyporesponsive upon in vitro stimulation, suggesting that they may be anergic (74, 113, 122–124). GOF mutations in CARD11 affect B and T cells differently, promoting proliferation and survival of B lymphocytes versus anergy in T lymphocytes, but the underlying mechanisms remain poorly understood (74). Similarly to LOF CARD11 mutations, BENTA-associated mutations may therefore perturb other CARD11-dependent downstream signaling cascades including the mTOR/S6K pathway (74). However, to our knowledge, there are currently no published data on mTOR and S6K phosphorylation in the context of BENTA disease.
Future Studies Should Explore mTOR/S6K Signaling Pathway in T Cells from CARMIL2-Deficient Patients
Biallelic LOF mutations in the gene encoding for the cell membrane-cytoskeleton-associated protein RLTPR (RGD, leucine-rich repeat, tropomodulin and proline-rich-containing protein), also known as CARMIL2 (capping protein regulator and myosin 1 linker 2), have been shown to be responsible for a novel PID disorder characterized by cutaneous and pulmonary allergy, by various bacterial, fungal, and mycobacterial infections, as well as by EBV lymphoproliferation (EBV+ smooth muscle tumors) (127, 128). In addition to its involvement in cell polarity and migration (129), CARMIL2 plays an important role in T cells by acting as a scaffold protein, bridging CD28 to CARD11 and therefore to the NF-κB signaling axis (130). Mutations in CARMIL2 prevent the association of CARMIL2 with CARD11 (130). CARMIL2-deficient T cells have a perturbed cytoskeletal organization leading to abnormalities in T cell polarity and migration, but also an impaired CD28-mediated co-signaling with a defective activation of the canonical NF-κB pathway (127, 128, 130). CARMIL2-deficient patients have a normal production of TH2 cytokines, but a reduced secretion of TH1, as well as TH17 effector cytokines, and therefore the strong decrease in Treg numbers does not result in the development of autoimmunity (127, 130). This phenotype is reminiscent of CARD11-deficient patients (119). Considering the newly described role of CARD11 in the mTOR/S6K pathway activation following TCR and CD28 stimulation, future studies should also address this signaling cascade in T cells from CARMIL2-deficient patients.
Mutations in Genes Encoding for the CD19-Complex Could be Associated with a Disturbed PI3K/mTOR/S6K Signaling
CD19 is a B cell lineage-specific transmembrane protein expressed from the pro B cell stage until plasma cell differentiation (131). It forms the CD19-complex together with CD21, CD81, and CD225 on the membrane of mature B cells. This complex is recruited to the BCR after ligation by complement (C3d) opsonized antigen via the complement receptor 2 (CR2, also known as CD21). This increases the BCR-mediated signal into B cells, as the BCR itself only delivers a weak tonic signal. CD19, with its many tyrosine residues, amplifies this signal to properly activate B cells (131–133). Biallelic mutations in CD19, leading to loss of CD19 membrane expression, to concomitant reduction of CD21 levels, and hence B cell activation, have been described in CVID patients (134–137). Affected patients have recurrent bacterial infections, hypogammaglobulinemia, decreased memory B cell numbers, defective antibody response after vaccination, as well as impaired somatic hypermutation, class-switch recombination, and immunoglobulin repertoire selection (134–138). As expected, they show neither T cell defects nor signs of lymphoproliferation (134–137). However, autoimmune manifestations (thrombocytopenia, glomerulonephritis) and autoantibody production have been reported (134, 135, 137, 139). Since CD81 is required for CD19 expression on the plasma membrane, patients with CD81 deficiency display a phenotype that is highly similar to CD19-deficient patients (140, 141). Upon BCR ligation, CD19 is rapidly phosphorylated at multiple tyrosine residues, leading to the recruitment of various downstream signaling intermediates. A prominent feature of CD19 signaling is the binding of the p85α regulatory subunit and the subsequent activation of class IA PI3K, thereby promoting AKT phosphorylation (132). In the absence of CD19, AKT activity is reduced in B cells (142). However, CD19 amplifies not only BCR signaling, but also plays a crucial role in the regulation of TLR9 responses in human B cells (143). It recruits PI3K and mediates AKT as well as Bruton’s tyrosine kinase (BTK) phosphorylation after ligation of nucleic acids, controlling both early B cell activation and proliferation (143). In fact, although AKT phosphorylation at position Ser473 is still induced after BCR triggering in CD19-deficient B cells, it is strongly reduced after CpG stimulation. In addition, inhibition of PI3K and AKT results in TLR9-induced B cell activation defects that are similar to those observed in CD19-deficient B cells (143). Therefore, CD19 deficiency may also be associated with abnormal mTOR/S6K signaling in B cells, but no data are currently available in the literature. However, since the phenotype of p85α-deficient mice is much more severe than the one of CD19-deficient mice, other signaling components might compensate for the loss of CD19 (142, 144–146).
PI3K/mTOR/S6K Signaling Should be Investigated in ICOS-Deficient Patients
Inducible T-cell costimulator (ICOS, also known as CD278) is another member of the CD28 T cell co-stimulatory molecules family (147). CD28 is expressed in resting and activated T cells, whereas ICOS expression is induced only upon T cell activation. Like CD28, ICOS delivers a positive signal that sustains T cell responses, and it is crucial for cell proliferation as well as cytokine production (148). CD28 and ICOS share a common signaling pathway, including PI3K recruitment (149, 150). In addition, ICOS plays an essential role in TFH differentiation as well as in germinal center formation, and hence in isotype switching and in the development of memory B cells (151, 152). ICOS deficiency was the first monogenic defect reported to cause CVID (153). To date, homozygous mutations (deletions) in ICOS have been identified in 16 patients, resulting in the absence of ICOS protein on T cells (153–158). ICOS deficiency was initially considered as a “predominantly antibody deficiency” by the IUIS PID expert committee (159), but following published patients with more complex phenotypes [reviewed by Ref. (154)], allowed a reclassification of the disease as a CID (2, 3). Besides hypogammaglobulinemia (93% of the cases) associated with an increased susceptibility to bacterial infections, more than two-thirds of the patients presented with autoimmunity and immune dysregulation (mainly enteropathy and psoriasis). Viral and opportunistic infections were frequently observed, and two patients developed malignancies (154). ICOS deficiency is associated with several immunological abnormalities including decreased numbers of switched memory B cells and circulating CXCR5+ TFH that coincide with an impaired germinal center formation (151, 154). B cell counts seem to decline progressively during the course of the disease, possibly as a consequence of a bone marrow production failure. IL-17 levels are markedly decreased in all patients who have been assessed for cytokine production, but without being associated with an increased susceptibility to Candida infection (154). ICOS is responsible for a greater PI3K activity than CD28, leading to a strong subsequent phosphorylation of AKT (150, 160). It bears a unique YMFM motif in its cytoplasmic tail that binds to the p85α regulatory subunit of PI3K (149).
In addition, ICOS interaction with its ligand ICOSL induces the recruitment of the PI3K regulatory subunit p50α at the synapse of T cell/antigen-presenting cells conjugates (160). ICOS deficiency should therefore be associated with impaired PI3K signaling. The activity of PI3K, as well as of downstream effector signaling molecules including mTOR and S6K, should be explored in T cells from ICOS-deficient individuals.
Regarding CD28, no PID has been associated so far with mutations in the gene encoding for this other T cell co-stimulatory receptor.
Conclusion
There are several lines of evidence that link the PI3K/AKT/mTOR/S6K signaling pathway to PIDs. Further studies are nevertheless required to characterize more deeply the crosstalk between the PI3K/AKT/mTOR/S6K cascade and other signaling molecules, as well as the disease-specific defects. Understanding the genetics and mechanisms behind the “immune TOR-opathies” is crucial to improve the management of the patients. The use of inhibitors such as mTOR and PI3K inhibitors that specifically target this signaling pathway and could restore properly the immune function represent very promising therapeutic approaches. Selective PI3K inhibitors should be considered as future treatment options, in particular in APDS patients, with encouraging preliminary results in ongoing clinical trials.
Author Contributions
SJ, LG-D, and BG wrote the review. MP prepared the figure. All authors concur with the submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. F. Gros for proofreading of the manuscript.
Funding
SJ is supported by grants from Hôpitaux Universitaires de Strasbourg (HUS), Université de Strasbourg (UdS), Centre National de la Recherche Scientifique (CNRS), and by EU-funded (ERDF, European Regional Development Fund) project INTERREG V Upper Rhine programma “RARENET.” BG, LG-D, and MP are supported by the German Ministry for Education and Research (BMBF, Bundesministerium für Bildung und Forschung) with the grants IFB/CCI:01EO1303 and E:med/SysInflame:01ZX1306F, by the German Center for Infection Research (DZIF, Deutsches Zentrum für Infektionsforschung): 8000805-3, and by the German Research Society (DFG, Deutsche Forschungsgemeinschaft) with the grants GR 1617/8-1 and SFB1160-IMPATH.
Abbreviations
AMPK, AMP-activated protein kinase; APDS, activated PI3Kδ syndrome; ASCT2, sodium-dependent neutral amino acid transporter type 2; BAD, Bcl-2-associated death promoter; BCL10, B-cell lymphoma/leukemia 10; BCR, B-cell receptor; BDCP, BEACH domain-containing protein; BEACH, Beige and Chediak-Higashi; BENTA, B cell expansion with NF-κB and T cell anergy; BTK, Bruton’s tyrosine kinase; CARD11, caspase recruitment domain-containing protein 11; CARMIL2, capping protein regulator and myosin 1 linker 2; CBM, CARD11-BCL10-MALT1; CID, combined immunodeficiency; CMV, cytomegalovirus; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; CR2, complement receptor 2; CVID, common variable immunodeficiency; CWS, Cowden syndrome; DEPTOR, DEP domain-containing mTOR interacting protein; EBV, Epstein-Barr virus; 4E-BP1, eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1; FOXO, Forkhead box protein O; GOF, gain-of-function; ICOS, inducible T-cell costimulator; IPEX, immune dysregulation, polyendocrinopathy, enteropathy, X-linked; IUIS, International Union of Immunological Societies; KO, knockout; LOF, loss-of-function; LPS, lipopolysaccharide; LRBA, lipopolysaccharide-responsive beige-like anchor protein; MALT1, mucosa-associated lymphoid tissue lymphoma translocation protein 1; mLST8/GβL, mammalian lethal with SEC13 protein 8/G protein β subunit-like; mSIN, stress-activated map kinase-interacting protein 1; mTOR, mechanistic/mammalian target of rapamycin; mTORC, mTOR complex; NF-κB, nuclear factor-kappa B; PASLI, p110δ activating mutation causing senescent T cells, lymphadenopathy, and immunodeficiency; PDK1, phosphoinositide-dependent kinase-1; PH, pleckstrin-homology; PHTS, PTEN hamartoma tumor syndrome; PHLPP, PH domain leucine-rich repeat protein phosphatase; PID, primary immunodeficiency; PIP2, phosphatidylinositol-(4,5)-bisphosphate; PIP3, phosphatidylinositol-(3,4,5)-trisphosphate; PI3K, phosphoinositide 3-kinase; PKB, protein kinase B; PRAS40, proline-rich AKT substrate 40 kDa; PROTOR, protein observed with Rictor; PTEN, phosphatase and tensin homolog; RAPTOR, regulatory-associated protein of mTOR; RHEB, Ras homolog enriched in brain; RICTOR, rapamycin-insensitive companion of mTOR; RLTPR, RGD, leucine-rich repeat, tropomodulin and proline-rich-containing protein; S6K, S6 kinase; SH2, Src-homology 2; TCR, T-cell receptor; TFH, follicular helper T cells; TH, T helper; TLR, toll-like receptor; Tregs, regulatory T cells; VZV, varicella zoster virus.
References
1. Crino PB. Focal brain malformations: a spectrum of disorders along the mTOR cascade. Novartis Found Symp (2007) 288:260–72; discussion 272–81.
2. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova J-L, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol (2018) 38:96–128. doi:10.1007/s10875-017-0464-9
3. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol (2018) 38:129–43. doi:10.1007/s10875-017-0465-8
4. Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova J-L, et al. Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol (2013) 33:1–7. doi:10.1007/s10875-012-9751-7
5. Fischer A. Human primary immunodeficiency diseases: a perspective. Nat Immunol (2004) 5:23–30. doi:10.1038/ni1023
6. Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol (2010) 125:S182–94. doi:10.1016/j.jaci.2009.07.053
7. Maródi L, Notarangelo LD. Immunological and genetic bases of new primary immunodeficiencies. Nat Rev Immunol (2007) 7:851–61. doi:10.1038/nri2195
8. Huang K, Fingar DC. Growing knowledge of the mTOR signaling network. Semin Cell Dev Biol (2014) 36:79–90. doi:10.1016/j.semcdb.2014.09.011
9. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol (2012) 30:39–68. doi:10.1146/annurev-immunol-020711-075024
10. Crino PB. mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol Med (2011) 17:734–42. doi:10.1016/j.molmed.2011.07.008
11. Ismail HMS. Downstream the mTOR: S6 kinases between divergence and redundancy. J Biochem Pharmacol Res (2013) 1(2):94–105.
12. Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol (2003) 3:317–30. doi:10.1038/nri1056
13. Okkenhaug K. Signalling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol (2013) 31:675–704. doi:10.1146/annurev-immunol-032712-095946
14. Chantry D, Vojtek A, Kashishian A, Holtzman DA, Wood C, Gray PW, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem (1997) 272:19236–41. doi:10.1074/jbc.272.31.19236
15. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell (2007) 129:1261–74. doi:10.1016/j.cell.2007.06.009
16. Inoki K, Li Y, Xu T, Guan K-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev (2003) 17:1829–34. doi:10.1101/gad.1110003
17. Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell (2002) 110:177–89. doi:10.1016/S0092-8674(02)00833-4
18. Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science (1991) 253:905–9. doi:10.1126/science.1715094
19. Kim D-H, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell (2002) 110:163–75. doi:10.1016/S0092-8674(02)00808-5
20. Sarbassov DD, Ali SM, Kim D-H, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol (2004) 14:1296–302. doi:10.1016/j.cub.2004.06.054
21. Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13:132–41. doi:10.1038/ncb2152
22. Jung CH, Ro S-H, Cao J, Otto NM, Kim D-H. mTOR regulation of autophagy. FEBS Lett (2010) 584:1287–95. doi:10.1016/j.febslet.2010.01.017
23. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469:323–35. doi:10.1038/nature09782
24. Limon JJ, Fruman DA. Akt and mTOR in B cell activation and differentiation. Front Immunol (2012) 3:228. doi:10.3389/fimmu.2012.00228
25. Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell (1998) 95:29–39. doi:10.1016/S0092-8674(00)81780-8
26. Tavares MR, Pavan ICB, Amaral CL, Meneguello L, Luchessi AD, Simabuco FM. The S6K protein family in health and disease. Life Sci (2015) 131:1–10. doi:10.1016/j.lfs.2015.03.001
27. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149:274–93. doi:10.1016/j.cell.2012.03.017
28. Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J (2012) 441:1–21. doi:10.1042/BJ20110892
29. Pardo OE, Seckl MJ. S6K2: the neglected S6 kinase family member. Front Oncol (2013) 3:191. doi:10.3389/fonc.2013.00191
30. Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y, et al. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORγ. Cell Rep (2012) 1:360–73. doi:10.1016/j.celrep.2012.02.007
31. Pai C, Walsh CM, Fruman DA. Context-specific function of S6K2 in Th cell differentiation. J Immunol (2016) 197:3049–58. doi:10.4049/jimmunol.1600167
32. Ikenoue T, Hong S, Inoki K. Monitoring mammalian target of rapamycin (mTOR) activity. Methods Enzymol (2009) 452:165–80. doi:10.1016/S0076-6879(08)03611-2
33. So L, Lee J, Palafox M, Mallya S, Woxland CG, Arguello M, et al. The 4E-BP-eIF4E axis promotes rapamycin-sensitive growth and proliferation in lymphocytes. Sci Signal (2016) 9:ra57. doi:10.1126/scisignal.aad8463
34. Ip CKM, Cheung ANY, Ngan HYS, Wong AST. p70 S6 kinase in the control of actin cytoskeleton dynamics and directed migration of ovarian cancer cells. Oncogene (2011) 30:2420–32. doi:10.1038/onc.2010.615
35. Zhang S, Readinger JA, DuBois W, Janka-Junttila M, Robinson R, Pruitt M, et al. Constitutive reductions in mTOR alter cell size, immune cell development, and antibody production. Blood (2011) 117:1228–38. doi:10.1182/blood-2010-05-287821
36. Cobbold SP, Adams E, Farquhar CA, Nolan KF, Howie D, Lui KO, et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc Natl Acad Sci U S A (2009) 106:12055–60. doi:10.1073/pnas.0903919106
37. Cobbold SP. The mTOR pathway and integrating immune regulation. Immunology (2013) 140:391–8. doi:10.1111/imm.12162
38. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A (2008) 105:7797–802. doi:10.1073/pnas.0800928105
39. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish Treg cell function. Nature (2013) 499:485–90. doi:10.1038/nature12297
40. Espeillac C, Mitchell C, Celton-Morizur S, Chauvin C, Koka V, Gillet C, et al. S6 kinase 1 is required for rapamycin-sensitive liver proliferation after mouse hepatectomy. J Clin Invest (2011) 121:2821–32. doi:10.1172/JCI44203
41. Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J (1998) 17:6649–59. doi:10.1093/emboj/17.22.6649
42. Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, et al. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5’-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol (2004) 24:3112–24. doi:10.1128/MCB.24.8.3112-3124.2004
43. Panić L, Tamarut S, Sticker-Jantscheff M, Barkić M, Solter D, Uzelac M, et al. Ribosomal protein S6 gene haploinsufficiency is associated with activation of a p53-dependent checkpoint during gastrulation. Mol Cell Biol (2006) 26:8880–91. doi:10.1128/MCB.00751-06
44. Sulic S, Panic L, Barkic M, Mercep M, Uzelac M, Volarevic S. Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev (2005) 19:3070–82. doi:10.1101/gad.359305
45. Ruvinsky I, Katz M, Dreazen A, Gielchinsky Y, Saada A, Freedman N, et al. Mice deficient in ribosomal protein S6 phosphorylation suffer from muscle weakness that reflects a growth defect and energy deficit. PLoS One (2009) 4:e5618. doi:10.1371/journal.pone.0005618
46. Salmond RJ, Brownlie RJ, Meyuhas O, Zamoyska R. Mechanistic target of rapamycin complex 1/S6 kinase 1 signals influence T cell activation independently of ribosomal protein S6 phosphorylation. J Immunol (2015) 195:4615–22. doi:10.4049/jimmunol.1501473
47. Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kδ and primary immunodeficiencies. Nat Rev Immunol (2016) 16:702–14. doi:10.1038/nri.2016.93
48. Petrovski S, Parrott RE, Roberts JL, Huang H, Yang J, Gorentla B, et al. Dominant splice site mutations in PIK3R1 cause hyper IgM syndrome, lymphadenopathy and short stature. J Clin Immunol (2016) 36(5):462–71. doi:10.1007/s10875-016-0281-6
49. Rae W, Gao Y, Ward D, Mattocks CJ, Eren E, Williams AP. A novel germline gain-of-function variant in PIK3CD. Clin Immunol (2017) 181:29–31. doi:10.1016/j.clim.2017.05.020
50. Takeda AJ, Zhang Y, Dornan GL, Siempelkamp BD, Jenkins ML, Matthews HF, et al. Novel PIK3CD mutations affecting N-terminal residues of p110δ cause activated PI3Kδ syndrome (APDS) in humans. J Allergy Clin Immunol (2017) 140(4):1152–6.e10. doi:10.1016/j.jaci.2017.03.026
51. Wentink M, Dalm V, Lankester AC, van Schouwenburg PA, Schölvinck L, Kalina T, et al. Genetic defects in PI3Kδ affect B-cell differentiation and maturation leading to hypogammaglobulineamia and recurrent infections. Clin Immunol (2017) 176:77–86. doi:10.1016/j.clim.2017.01.004
52. Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science (2013) 342:866–71. doi:10.1126/science.1243292
53. Chiriaco M, Brigida I, Ariganello P, Di Cesare S, Di Matteo G, Taus F, et al. A case of APDS patient: defects in maturation and function and decreased in vitro anti-mycobacterial activity in the myeloid compartment. Clin Immunol (2015) 178:20–8. doi:10.1016/j.clim.2015.12.008
54. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: a large patient cohort study. J Allergy Clin Immunol (2017) 139:597–606.e4. doi:10.1016/j.jaci.2016.06.021
55. Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol (2014) 34:272–6. doi:10.1007/s10875-014-0012-9
56. Deau M-C, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest (2014) 124:3923–8. doi:10.1172/JCI75746
57. Elgizouli M, Lowe DM, Speckmann C, Schubert D, Hülsdünker J, Eskandarian Z, et al. Activating PI3Kδ mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol (2016) 183:221–9. doi:10.1111/cei.12706
58. Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, Heurtier L, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase δ syndrome 2: a cohort study. J Allergy Clin Immunol (2016) 138:210–8.e9. doi:10.1016/j.jaci.2016.03.022
59. Hartman HN, Niemela J, Hintermeyer MK, Garofalo M, Stoddard J, Verbsky JW, et al. Gain of function mutations of PIK3CD as a cause of primary sclerosing cholangitis. J Clin Immunol (2015) 35:11–4. doi:10.1007/s10875-014-0109-1
60. Jou S-T, Chien Y-H, Yang Y-H, Wang T-C, Shyur S-D, Chou C-C, et al. Identification of variations in the human phosphoinositide 3-kinase p110delta gene in children with primary B-cell immunodeficiency of unknown aetiology. Int J Immunogenet (2006) 33:361–9. doi:10.1111/j.1744-313X.2006.00627.x
61. Kracker S, Curtis J, Ibrahim MAA, Sediva A, Salisbury J, Campr V, et al. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase δ syndrome. J Allergy Clin Immunol (2014) 134:233–6. doi:10.1016/j.jaci.2014.02.020
62. Kuhlen M, Hönscheid A, Loizou L, Nabhani S, Fischer U, Stepensky P, et al. De novo PIK3R1 gain-of-function with recurrent sinopulmonary infections, long-lasting chronic CMV-lymphadenitis and microcephaly. Clin Immunol (2016) 162:27–30. doi:10.1016/j.clim.2015.10.008
63. Lougaris V, Faletra F, Lanzi G, Vozzi D, Marcuzzi A, Valencic E, et al. Altered germinal center reaction and abnormal B cell peripheral maturation in PI3KR1-mutated patients presenting with HIGM-like phenotype. Clin Immunol (2015) 159:33–6. doi:10.1016/j.clim.2015.04.014
64. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15:88–97. doi:10.1038/ni.2771
65. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med (2014) 211:2537–47. doi:10.1084/jem.20141759
66. Olbrich P, Lorenz M, Cura Daball P, Lucena JM, Rensing-Ehl A, Sanchez B, et al. Activated PI3Kδ syndrome type 2: two patients, a novel mutation, and review of the literature. Pediatr Allergy Immunol (2016) 27:640–4. doi:10.1111/pai.12585
67. Rae W, Ramakrishnan KA, Gao Y, Ashton-Key M, Pengelly RJ, Patel SV, et al. Precision treatment with sirolimus in a case of activated phosphoinositide 3-kinase δ syndrome. Clin Immunol (2016) 171:38–40. doi:10.1016/j.clim.2016.07.017
68. Tsujita Y, Mitsui-Sekinaka K, Imai K, Yeh T-W, Mitsuiki N, Asano T, et al. Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase δ syndrome-like immunodeficiency. J Allergy Clin Immunol (2016) 138:1672–80.e10. doi:10.1016/j.jaci.2016.03.055
69. Conley ME, Dobbs AK, Quintana AM, Bosompem A, Wang Y-D, Coustan-Smith E, et al. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J Exp Med (2012) 209:463–70. doi:10.1084/jem.20112533
70. Suzuki A, Kaisho T, Ohishi M, Tsukio-Yamaguchi M, Tsubata T, Koni PA, et al. Critical roles of Pten in B cell homeostasis and immunoglobulin class switch recombination. J Exp Med (2003) 197:657–67. doi:10.1084/jem.20021101
71. Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity (2001) 14:523–34. doi:10.1016/S1074-7613(01)00134-0
72. Sullivan JA, Kim EH, Plisch EH, Peng SL, Suresh M. FOXO3 regulates CD8 T cell memory by T cell-intrinsic mechanisms. PLoS Pathog (2012) 8:e1002533. doi:10.1371/journal.ppat.1002533
73. Van der Windt GJW, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev (2012) 249:27–42. doi:10.1111/j.1600-065X.2012.01150.x
74. Arjunaraja S, Snow AL. Gain-of-function mutations and immunodeficiency: at a loss for proper tuning of lymphocyte signaling. Curr Opin Allergy Clin Immunol (2015) 15:533–8. doi:10.1097/ACI.0000000000000217
75. Dornan GL, Siempelkamp BD, Jenkins ML, Vadas O, Lucas CL, Burke JE. Conformational disruption of PI3Kδ regulation by immunodeficiency mutations in PIK3CD and PIK3R1. Proc Natl Acad Sci U S A (2017) 114:1982–7. doi:10.1073/pnas.1617244114
76. Rao VK, Webster S, Dalm VASH, Šedivá A, van Hagen PM, Holland S, et al. Effective “activated PI3Kδ syndrome”-targeted therapy with the PI3Kδ inhibitor leniolisib. Blood (2017) 130:2307–16. doi:10.1182/blood-2017-08-801191
77. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem (1998) 273:13375–8. doi:10.1074/jbc.273.22.13375
78. Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet (2013) 163:114–21. doi:10.1002/ajmg.c.31364
79. Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol (1998) 8:1169–78. doi:10.1016/S0960-9822(07)00488-5
80. Cristofano AD, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet (1998) 19:348–55. doi:10.1038/1235
81. Cristofano AD, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired Fas response and autoimmunity in Pten+/− mice. Science (1999) 285:2122–5. doi:10.1126/science.285.5436.2122
82. Anzelon AN, Wu H, Rickert RC. Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nat Immunol (2003) 4:287–94. doi:10.1038/ni892
83. Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, et al. Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity (2006) 25:545–57. doi:10.1016/j.immuni.2006.08.015
84. Liaw D, Marsh DJ, Li J, Dahia PLM, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet (1997) 16:64–7. doi:10.1038/ng0597-64
85. Marsh DJ, Dahia PL, Zheng Z, Liaw D, Parsons R, Gorlin RJ, et al. Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat Genet (1997) 16:333–4. doi:10.1038/ng0897-333
86. Eng C, Murday V, Seal S, Mohammed S, Hodgson SV, Chaudary MA, et al. Cowden syndrome and Lhermitte-Duclos disease in a family: a single genetic syndrome with pleiotropy? J Med Genet (1994) 31:458–61. doi:10.1136/jmg.31.6.458
87. Browning MJ, Chandra A, Carbonaro V, Okkenhaug K, Barwell J. Cowden’s syndrome with immunodeficiency. J Med Genet (2015) 52:856–9. doi:10.1136/jmedgenet-2015-103266
88. Chen HH, Händel N, Ngeow J, Muller J, Hühn M, Yang H-T, et al. Immune dysregulation in patients with PTEN hamartoma tumor syndrome: analysis of FOXP3 regulatory T cells. J Allergy Clin Immunol (2016) 139(2):607–20.e15. doi:10.1016/j.jaci.2016.03.059
89. Driessen GJ, IJspeert H, Wentink M, Yntema HG, van Hagen PM, van Strien A, et al. Increased PI3K/Akt activity and deregulated humoral immune response in human PTEN deficiency. J Allergy Clin Immunol (2016) 138:1744–7.e5. doi:10.1016/j.jaci.2016.07.010
90. Werner M, Hobeika E, Jumaa H. Role of PI3K in the generation and survival of B cells. Immunol Rev (2010) 237:55–71. doi:10.1111/j.1600-065X.2010.00934.x
91. Wang JW, Howson J, Haller E, Kerr WG. Identification of a novel lipopolysaccharide-inducible gene with key features of both A kinase anchor proteins and chs1/beige proteins. J Immunol (2001) 166:4586–95. doi:10.4049/jimmunol.166.7.4586
92. Cullinane AR, Schäffer AA, Huizing M. The BEACH is hot: a LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic (2013) 14:749–66. doi:10.1111/tra.12069
93. Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet (2012) 90:986–1001. doi:10.1016/j.ajhg.2012.04.015
94. Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol (2016) 36:33–45. doi:10.1007/s10875-015-0224-7
95. Gámez-Díaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol (2016) 137:223–30. doi:10.1016/j.jaci.2015.09.025
96. Charbonnier L-M, Janssen E, Chou J, Ohsumi TK, Keles S, Hsu JT, et al. Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J Allergy Clin Immunol (2015) 135:217–27. doi:10.1016/j.jaci.2014.10.019
97. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348:56–61. doi:10.1126/science.aaa8172
98. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (2015) 349:436–40. doi:10.1126/science.aaa1663
99. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi:10.1126/science.1202947
100. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med (2014) 20:1410–6. doi:10.1038/nm.3746
101. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (2014) 345:1623–7. doi:10.1126/science.1255904
102. Lo B, Fritz JM, Su HC, Uzel G, Jordan MB, Lenardo MJ. CHAI and LATAIE: new genetic diseases of CTLA-4 checkpoint insufficiency. Blood (2016) 128:1037–42. doi:10.1182/blood-2016-04-712612
103. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev (2009) 229:12–26. doi:10.1111/j.1600-065X.2009.00770.x
104. Schneider H, Prasad KV, Shoelson SE, Rudd CE. CTLA-4 binding to the lipid kinase phosphatidylinositol 3-kinase in T cells. J Exp Med (1995) 181:351–5. doi:10.1084/jem.181.1.351
105. Schneider H, Valk E, Leung R, Rudd CE. CTLA-4 activation of phosphatidylinositol 3-kinase (PI 3-K) and protein kinase B (PKB/AKT) sustains T-cell anergy without cell death. PLoS One (2008) 3:e3842. doi:10.1371/journal.pone.0003842
106. Walker LSK, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol (2015) 36:63–70. doi:10.1016/j.it.2014.12.001
107. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol (2013) 14:298–305. doi:10.1038/ni.2524
108. Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med (2014) 20:503–10. doi:10.1038/nm.3521
109. Muñoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol (2010) 6:280–9. doi:10.1038/nrrheum.2010.46
110. Roche MI, Ramadas RA, Medoff BD. The role of CARMA1 in T cells. Crit Rev Immunol (2013) 33:219–43. doi:10.1615/CritRevImmunol.2013007056
111. Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, et al. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity (2003) 18:751–62. doi:10.1016/S1074-7613(03)00141-9
112. Hamilton KS, Phong B, Corey C, Cheng J, Gorentla B, Zhong X, et al. A Carma1/MALT1-dependent, Bcl10-independent, pathway regulates antigen receptor-mediated mTOR signaling in T cells. Sci Signal (2014) 7:ra55. doi:10.1126/scisignal.2005169
113. Turvey SE, Durandy A, Fischer A, Fung S-Y, Geha RS, Gewies A, et al. The CARD11-BCL10-MALT1 (CBM) signalosome complex: stepping into the limelight of human primary immunodeficiency. J Allergy Clin Immunol (2014) 134:276–84. doi:10.1016/j.jaci.2014.06.015
114. Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, et al. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immunol (2013) 132:151–8. doi:10.1016/j.jaci.2013.04.047
115. McKinnon ML, Rozmus J, Fung S-Y, Hirschfeld AF, Del Bel KL, Thomas L, et al. Combined immunodeficiency associated with homozygous MALT1 mutations. J Allergy Clin Immunol (2014) 133:1458–62, 1462.e1–7. doi:10.1016/j.jaci.2013.10.045
116. Stepensky P, Keller B, Buchta M, Kienzler A-K, Elpeleg O, Somech R, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol (2013) 131:477–85.e1. doi:10.1016/j.jaci.2012.11.050
117. Greil J, Rausch T, Giese T, Bandapalli OR, Daniel V, Bekeredjian-Ding I, et al. Whole-exome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immunol (2013) 131:1376–83.e3. doi:10.1016/j.jaci.2013.02.012
118. Torres JM, Martinez-Barricarte R, García-Gómez S, Mazariegos MS, Itan Y, Boisson B, et al. Inherited BCL10 deficiency impairs hematopoietic and nonhematopoietic immunity. J Clin Invest (2014) 124:5239–48. doi:10.1172/JCI77493
119. Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline hypomorphic CARD11 mutations in severe atopic disease. Nat Genet (2017) 49:1192–201. doi:10.1038/ng.3898
120. Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol (2014) 14:435–46. doi:10.1038/nri3701
121. Nakaya M, Xiao Y, Zhou X, Chang J-H, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40:692–705. doi:10.1016/j.immuni.2014.04.007
122. Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delalande B, Zheng L, et al. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med (2012) 209:2247–61. doi:10.1084/jem.20120831
123. Brohl AS, Stinson J, Su HC, Badgett T, Jennings CD, Sukumar G, et al. Germline CARD11 mutation in a patient with severe congenital B cell lymphocytosis. J Clin Immunol (2015) 35:32–46. doi:10.1007/s10875-014-0106-4
124. Buchbinder D, Stinson JR, Nugent DJ, Heurtier L, Suarez F, Sukumar G, et al. Mild B-cell lymphocytosis in patients with a CARD11 C49Y mutation. J Allergy Clin Immunol (2015) 136:819–21.e1. doi:10.1016/j.jaci.2015.03.008
125. Chan W, Schaffer TB, Pomerantz JL. A quantitative signaling screen identifies CARD11 mutations in the CARD and LATCH domains that induce Bcl10 ubiquitination and human lymphoma cell survival. Mol Cell Biol (2013) 33:429–43. doi:10.1128/MCB.00850-12
126. Darte JM, McClure PD, Saunders EF, Weber JL, Donohue WL. Congenital lymphoid hyperplasia with persistent hyperlymphocytosis. N Engl J Med (1971) 284:431–2. doi:10.1056/NEJM197102252840807
127. Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, et al. Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med (2016) 213:2413–35. doi:10.1084/jem.20160576
128. Schober T, Magg T, Laschinger M, Rohlfs M, Linhares ND, Puchalka J, et al. A human immunodeficiency syndrome caused by mutations in CARMIL2. Nat Commun (2017) 8:14209. doi:10.1038/ncomms14209
129. Liang Y, Niederstrasser H, Edwards M, Jackson CE, Cooper JA. Distinct roles for CARMIL isoforms in cell migration. Mol Biol Cell (2009) 20:5290–305. doi:10.1091/mbc.E08-10-1071
130. Roncagalli R, Cucchetti M, Jarmuzynski N, Grégoire C, Bergot E, Audebert S, et al. The scaffolding function of the RLTPR protein explains its essential role for CD28 co-stimulation in mouse and human T cells. J Exp Med (2016) 213:2437–57. doi:10.1084/jem.20160579
131. Wang K, Wei G, Liu D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol (2012) 1:36. doi:10.1186/2162-3619-1-36
132. Carter RH, Doody GM, Bolen JB, Fearon DT. Membrane IgM-induced tyrosine phosphorylation of CD19 requires a CD19 domain that mediates association with components of the B cell antigen receptor complex. J Immunol (1997) 158:3062–9.
133. Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science (1992) 256:105–7. doi:10.1126/science.1373518
134. Kanegane H, Agematsu K, Futatani T, Sira MM, Suga K, Sekiguchi T, et al. Novel mutations in a Japanese patient with CD19 deficiency. Genes Immun (2007) 8:663–70. doi:10.1038/sj.gene.6364431
135. van Zelm MC, Smet J, van der Burg M, Ferster A, Le PQ, Schandené L, et al. Antibody deficiency due to a missense mutation in CD19 demonstrates the importance of the conserved tryptophan 41 in immunoglobulin superfamily domain formation. Hum Mol Genet (2011) 20:1854–63. doi:10.1093/hmg/ddr068
136. Van Zelm MC, Reisli I, van der Burg M, Castaño D, van Noesel CJM, van Tol MJD, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med (2006) 354:1901–12. doi:10.1056/NEJMoa051568
137. Vince N, Boutboul D, Mouillot G, Just N, Peralta M, Casanova J-L, et al. Defects in the CD19 complex predispose to glomerulonephritis, as well as IgG1 subclass deficiency. J Allergy Clin Immunol (2011) 127:538–41.e1–5. doi:10.1016/j.jaci.2010.10.019
138. Van Zelm MC, Bartol SJW, Driessen GJ, Mascart F, Reisli I, Franco JL, et al. Human CD19 and CD40L deficiencies impair antibody selection and differentially affect somatic hypermutation. J Allergy Clin Immunol (2014) 134:135–44.e7. doi:10.1016/j.jaci.2013.11.015
139. Artac H, Reisli I, Kara R, Pico-Knijnenburg I, Adin-Çinar S, Pekcan S, et al. B-cell maturation and antibody responses in individuals carrying a mutated CD19 allele. Genes Immun (2010) 11:523–30. doi:10.1038/gene.2010.22
140. Vences-Catalán F, Kuo C-C, Sagi Y, Chen H, Kela-Madar N, van Zelm MC, et al. A mutation in the human tetraspanin CD81 gene is expressed as a truncated protein but does not enable CD19 maturation and cell surface expression. J Clin Immunol (2015) 35:254–63. doi:10.1007/s10875-015-0148-2
141. Van Zelm MC, Smet J, Adams B, Mascart F, Schandené L, Janssen F, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest (2010) 120:1265–74. doi:10.1172/JCI39748
142. Otero DC, Omori SA, Rickert RC. Cd19-dependent activation of Akt kinase in B-lymphocytes. J Biol Chem (2001) 276:1474–8. doi:10.1074/jbc.M003918200
143. Morbach H, Schickel J-N, Cunningham-Rundles C, Conley ME, Reisli I, Franco JL, et al. CD19 controls TLR9 responses in human B cells. J Allergy Clin Immunol (2016) 137:889–98.e6. doi:10.1016/j.jaci.2015.08.040
144. Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW, et al. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science (1999) 283:393–7. doi:10.1126/science.283.5400.393
145. Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature (1995) 376:352–5. doi:10.1038/376352a0
146. Engel P, Zhou LJ, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity (1995) 3:39–50. doi:10.1016/1074-7613(95)90157-4
147. Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol (2002) 2:116–26. doi:10.1038/nri727
148. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397:263–6. doi:10.1038/16717
149. Coyle AJ, Lehar S, Lloyd C, Tian J, Delaney T, Manning S, et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity (2000) 13:95–105. doi:10.1016/S1074-7613(00)00011-X
150. Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol (2003) 171:166–74. doi:10.4049/jimmunol.171.1.166
151. Warnatz K, Bossaller L, Salzer U, Skrabl-Baumgartner A, Schwinger W, van der Burg M, et al. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood (2006) 107:3045–52. doi:10.1182/blood-2005-07-2955
152. Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol (2005) 175:2340–8. doi:10.4049/jimmunol.175.4.2340
153. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol (2003) 4:261–8. doi:10.1038/ni902
154. Schepp J, Chou J, Skrabl-Baumgartner A, Arkwright PD, Engelhardt KR, Hambleton S, et al. 14 Years after discovery: clinical follow-up on 15 patients with inducible co-stimulator deficiency. Front Immunol (2017) 8:964. doi:10.3389/fimmu.2017.00964
155. Chou J, Massaad MJ, Cangemi B, Bainter W, Platt C, Badran YR, et al. A novel mutation in ICOS presenting as hypogammaglobulinemia with susceptibility to opportunistic pathogens. J Allergy Clin Immunol (2015) 136:794–7.e1. doi:10.1016/j.jaci.2014.12.1940
156. Salzer U, Maul-Pavicic A, Cunningham-Rundles C, Urschel S, Belohradsky BH, Litzman J, et al. ICOS deficiency in patients with common variable immunodeficiency. Clin Immunol (2004) 113:234–40. doi:10.1016/j.clim.2004.07.002
157. Takahashi N, Matsumoto K, Saito H, Nanki T, Miyasaka N, Kobata T, et al. Impaired CD4 and CD8 effector function and decreased memory T cell populations in ICOS-deficient patients. J Immunol (2009) 182:5515–27. doi:10.4049/jimmunol.0803256
158. Robertson N, Engelhardt KR, Morgan NV, Barge D, Cant AJ, Hughes SM, et al. Astute Clinician Report: a novel 10 bp frameshift deletion in exon 2 of ICOS causes a combined immunodeficiency associated with an enteritis and hepatitis. J Clin Immunol (2015) 35:598–603. doi:10.1007/s10875-015-0193-x
159. Al-Herz W, Bousfiha A, Casanova J-L, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol (2014) 5:162. doi:10.3389/fimmu.2014.00162
Keywords: AKT, immune dysregulation, kinase, mTOR, PI3k, primary immunodeficiency, S6K
Citation: Jung S, Gámez-Díaz L, Proietti M and Grimbacher B (2018) “Immune TOR-opathies,” a Novel Disease Entity in Clinical Immunology. Front. Immunol. 9:966. doi: 10.3389/fimmu.2018.00966
Received: 19 January 2018; Accepted: 18 April 2018;
Published: 09 May 2018
Edited by:
Carrie L. Lucas, Yale University, United StatesReviewed by:
David A. Fruman, University of California, Irvine, United StatesDavid Andrew Fulcher, Australian National University, Australia
Copyright: © 2018 Jung, Gámez-Díaz, Proietti and Grimbacher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bodo Grimbacher, bodo.grimbacher@uniklinik-freiburg.de
†These authors have contributed equally to this work.