Comparative Metagenomics of Viral Assemblages Inhabiting Four Phyla of Marine Invertebrates
 Brent M. Gudenkauf
Brent M. Gudenkauf Ian Hewson
Ian Hewson- Department of Microbiology, Cornell University, Ithaca, NY, USA
Viruses are the most abundant biological entities on Earth, killing 10–20% of oceanic biomass each day. However, despite their ecological importance, viruses inhabiting many echinoderms, cnidarians, urochordates, and marine arthropods have not been investigated with significant breadth. We conducted a broad survey of the viral assemblages inhabiting these hosts through viral metagenomics and phylogenetic analysis. Results indicate that different invertebrate groups harbor distinct viral assemblages. Interestingly, however, no significant difference is observed between the viral assemblages of echinoderms and arthropods. These similarities and differences may be due to cellular, immunological, geographical, and ecological differences amongst host phyla, although mechanistic determination is beyond the purview of this work. Additionally, we present evidence of the detection of several viral families that have not yet been observed in these hosts. Finally, we confirm the result of previous investigation that method of library construction significantly biases metagenomic results by altering the representation of ssDNA and dsDNA viral genomes.
Introduction
Viruses are the most abundant biological entities on Earth, with an estimated 1030 viruses in the oceans (Suttle, 2007). Surface seawater typically harbors 107 viral particles mL−1, with fewer viruses at depth (105 mL−1), and greater abundance in algal blooms (Wommack and Colwell, 2000). These viruses cause mortality in 10–20% of oceanic biomass each day (Munn, 2006; Suttle, 2007). Viruses associated with metazoan host microbiomes include viruses that infect hosts (i.e., pathogens, neutral-effect viruses, and endogenous viruses), and also viruses that infect non-metazoan components, including bacterial, archaeal, and eukaryotic microbial constituents.
Despite their significance as agents of mortality, investigation of viruses associated with marine invertebrates has historically been limited. Viruses of many ecologically important marine invertebrate faunae have not yet been investigated; Entire phyla lack described viruses (Suttle, 2005). This is due to several obstacles that make conventional investigation difficult. For instance, cell lines that model marine invertebrate metazoan tissues are unavailable, and cell culturing techniques are incompatible with microbes that have unknown growth requirements or require symbioses. Metagenomics allows investigators to overcome some of these obstacles via next generation shotgun-sequencing, targeting nucleic acids to observe entire microbial communities. Recent viral metagenomic studies have exposed novel families of invertebrate viruses (e.g., Ng, 2010; Rosario et al., 2011, 2012; Dunlap et al., 2013). This technique may be used as viral surveillance (Ng, 2010), or as a starting point for further investigation into microbe–microbe interactions, microbe–host interactions, pathogenesis, and disease diagnosis. The lack of representative viral genomes in public databases may decrease confidence in metaviromes annotation (Weynberg et al., 2014). However, metaviromics provides a strong starting point for future study of viral–host relationships.
Echinoderms, cnidarians, urochordates, and arthropods are ecologically important benthic marine invertebrates. Many are keystone species, disproportionately affecting ecological structure through predation and consumption (Paine, 1969). Others are filter-feeders, clarifying water by altering turbidity and light penetration by controlling local populations of free-floating microbes and particulate matter (Berke, 2010). Still others are decomposers and participate in bioturbation, oxygenating anoxic sediments, and releasing stored nutrients into the water column. Because benthic invertebrates play important roles in marine habitat ecology and biogeochemistry, investigation into the viral diversity of their microbiomes is relevant to understanding how viruses influence marine ecosystems. There have been few reports of viruses of echinoderms (Gudenkauf et al., 2014; Hewson et al., 2014), very few of non-commercially important (but ecologically important) marine arthropods (e.g., Dunlap et al., 2013), only one of viruses infecting non-scleractinian corals (Hewson et al., 2011a), and no previous reports of viruses inhabiting urochordates.
While most populations of echinoderms are long-lived in the absence of pathogenic agents (Bluhm et al., 1998), bacteria and protozoan parasites can cause mortality (Jangoux, 1987). However, there are some instances of mass mortality that remain unexplained by bacteria or protozoan influences alone and for which viruses are the most likely factor. For instance, a mass mortality event of the echinoid Diadema antillarum occurred starting in 1983. Death of 97% of the population occurred rapidly throughout the Caribbean, with infection radiating outward from a locus at the Panama Canal. While these characteristics alone resemble classical elements of viral epidemiology, the most striking resemblance lies in the fact that infection was transmissible by material < 0.2 μm in size, which is smaller than the smallest known bacterial genus, Mycoplasma (Lessios et al., 1984; Lessios, 1988). The loss of D. antillarum profoundly affected the ecology of reef ecosystems within the Caribbean as macroalgae became overgrown, inhibiting coral growth and contributing to further decline of scleractinian corals (Carpenter, 1990).
Asteroids have also been decimated by several still-unexplained mass mortality events off the coast of California since 1978 (Bates et al., 2009). Recently, a densovirus (the sea star associated densovirus or SSaDV) was identified as the most likely cause of the most recent sea star “wasting disease” that occurred in 2013 and 2014 along the western coast of North America (Hewson et al., 2014). Gorgonian sea fans and tunicates also underwent a mass-mortality event in the northwest Mediterranean Sea in the summer of 1999, with this event correlating with higher than average oceanic temperatures (Cerrano et al., 2000). This correlation may support a switch from commensal to pathogenic microorganisms. However, this does not explain those events in which oceanic temperatures were not above normal, like the sea star wasting event. While the sudden decline of echinoderm populations may be related to environmental shifts (e.g., restricted water flow, anoxia, or slight changes in temperature), collapse in the absence of great physical change is consistent with viral or other pathogen attack.
With this work we seek to characterize the viruses found in the tissues of echinoderms, marine arthropods, cnidarians, and urochordates and compare the assemblages to identify common constituents within different host phyla. We attempt to answer the question: Do similar viruses inhabit broad host taxa, or do different taxa host distinct viruses?
Methods
Sampling
All samples were adults (i.e., no juveniles or larvae) and were collected by snorkel (Peramphithoe femorata), SCUBA diving (Gorgonia ventalina), from an artisanal fish market (Pyura chilensis), or by hand in the intertidal zone (arthropoda species) (Table 1). Data from asteroids and echinoids were collected as part of two previous studies (Gudenkauf et al., 2014; Hewson et al., 2014). In all cases, samples were immediately placed into Whirlpak© bags and frozen.
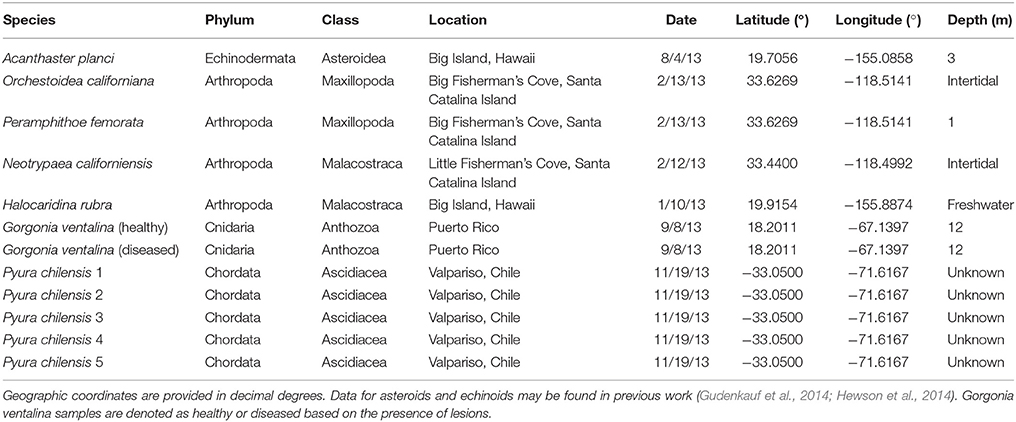
Table 1. Sample details for Arthropoda, Echinodermata, Chordata, and Cnidaria samples used in metagenomic analyses.
Metagenomic Library Preparation
We prepared 41 viromes from four phyla of marine invertebrates following the approach of Ng et al. (2009). Invertebrate tissue samples were removed from storage at −80°C and thawed. Once at room temperature, we amended tissues with 20 mL of 0.02 μm-filtered phosphate buffered saline and ground them in a sterile mortar and pestle. Homogenates were centrifuged for 10 min at 5000 × g to remove large particulate material. Next, we syringe-filtered the supernatant (0.2 μm Polyethersulfone) to remove cell debris and bacteria-size particles. Then, we amended filtrates with polyethylene glycol 8000 (10% weight-to-volume), mixed by inversion for 1 min, and precipitated them overnight at 4°C. Following precipitation, samples were centrifuged for 45 min at 12,000 × g. Next we removed supernatants by pipette and suspended viral pellets in 1 mL nuclease-free deionized water. Resuspended pellets were extracted with 200 μL CHCl3, inverted to mix, and allowed to settle for 10 min at room temperature. Subsequently we centrifuged samples for 1 min at 15,000 × g and transferred the aqueous layer to a new tube. Invitrogen TURBO DNase (14 U), Promega RNase One (20 U), 1 μL of Benzonase Nuclease, and 11.5 μL of nuclease-free deionized water were added to each sample, which were then incubated in a water bath at 37°C for 3 h. After incubation, we added 20 μmol EDTA to each sample, which we then stored at −80°C.
We confirmed the absence of bacterial and eukaryotic genomic DNA by PCR targeting 16S and 18S genes (Thurber et al., 2009). Next, we extracted DNA from purified viruses using Zymo Research Viral DNA Kit and amplified viral DNA using the GenomePlex Whole Genome Amplification (WGA2) kit (Sigma-Aldrich), or for the case of P. chilensis, we used both WGA2 and GenomiPhi ϕ29 HY DNA Amplification kits (Illustra) to compare the effects of amplification strategy on library composition. Next, we used the Zymo Research DNA Clean and Concentrator kit to purify amplicons. Whole genome amplicons were sequenced with Illumina MiSeq 2 × 250 bp paired-end chemistry at Cornell University Core Sequencing Center, where each sample was multiplexed with other samples. Sequence libraries have been deposited at GenBank SRA under accession numbers SAMN03653376, SAMN03653669, SAMN03653670, and SAMN03653673–SAMN03653676. Data from 28 asteroid samples (Hewson et al., 2014) and data from three echinoid samples (Gudenkauf et al., 2014) were included as part of this analysis.
Metagenomic Analyses
We imported resulting forward and reverse Illumina read sequences into CLC Genomics Workbench 4.0 and conducted de novo assembly on sequence reads using an assembly algorithm (length fraction 0.5, similarity 0.8, global alignment, vote on uncertain nucleotides, non-specific-matches randomized, and minimum contiguous sequence length of 500 nucleotides), generating contiguous fragments (contigs). Duplicate reads made up only a small percentage of reads within each library. Next, we identified open reading frames on contiguous sequences using the GetORF algorithm (EMBOSS) and subjected the resulting sequences to BLASTx analysis against the NCBI non-redundant (nr) protein database at the Cyberinfrastructure for Advanced Microbial Ecology Research and Analysis (CAMERA2). The strongest ORF similarity (i.e., lowest E-value, with an E-value cutoff of 0.001) per contig was used in further analyses.
Statistical Analysis
Two analyses of metagenomic composition were performed. First, we noted the number of assembled contigs per identified organism for quantitative statistical analyses on virome composition. The relative proportion of each viral genotype within a particular library was calculated as a percentage of the total number of viral genotypes identified in the library. Next, we binned viral genotypes at the family level of phylogeny. Then, we calculated a Bray–Curtis pairwise similarity matrix between libraries using XLSTAT (Addinsoft SARL) and heat mapping on the similarity matrix to permit visualization of the similarity of viral metagenomic compositions. Next, we performed two-dimensional scaling analysis (absolute model, Kruskal's stress, random initial configuration, five repetitions, stop condition at 0.00001 convergence/500 iterations) and agglomerative hierarchical clustering (unweighted pair-group average, automatically truncated) on non-bacteriophage annotations (also not including unclassified viruses). We also calculated mean similarity (by averaging Bray–Curtis similarity index values) and standard error between and across the host phyla studied both including and removing bacteriophage sequences.
Phylogenetic Analysis
Three groups of eukaryotic viruses that were observed in viromes from multiple host phyla were chosen for phylogenetic analysis. These groups consisted of viruses from families Parvoviridae, Circoviridae, and Phycodnaviridae. The phylogeny of the viral sequence with the greatest representation in each library was determined by comparison with the closest BLASTx match against the RefSeq Database at NCBI. Metagenomic sequences and their closest matches in GenBank were aligned using CLUSTALW (Thompson et al., 1994) and bootstrapped phylogenetic trees (1000 iterations) were drawn by the Neighbor Joining algorithm (complete linkage) and visualized using TREEVIEW (Page, 1996). Trees were rooted with an evolutionarily distant out-group.
Results and Discussion
Metagenomic Annotation
Illumina sequence reads varied from about 180 to 600 bp and sequencing of viromes prepared from echinoderms, arthropods, cnidarians, and urochordates yielded between 33,673 and 1,677,426 contiguous sequence per library (Table 2). Of these, between 0.06 and 88.03% matched viral genomes, with the remaining sequences corresponding to bacterial, archaeal, or eukaryotic genome fragments. This is similar to the results of previous studies that utilized similar viral genome isolation protocols, and likely results from DNA released from host, bacterial, and/or archaeal cells during the tissue homogenization process (Hewson et al., 2011a, 2013; Gudenkauf et al., 2014). The low number of viral annotations may be a result of lack of representative viral genomes in public databases (Weynberg et al., 2014). The wide range of contig sequences could have resulted from differences in host tissue mass, amounts of mucosal epithelia from which viruses were extracted, or from differences in viral load.
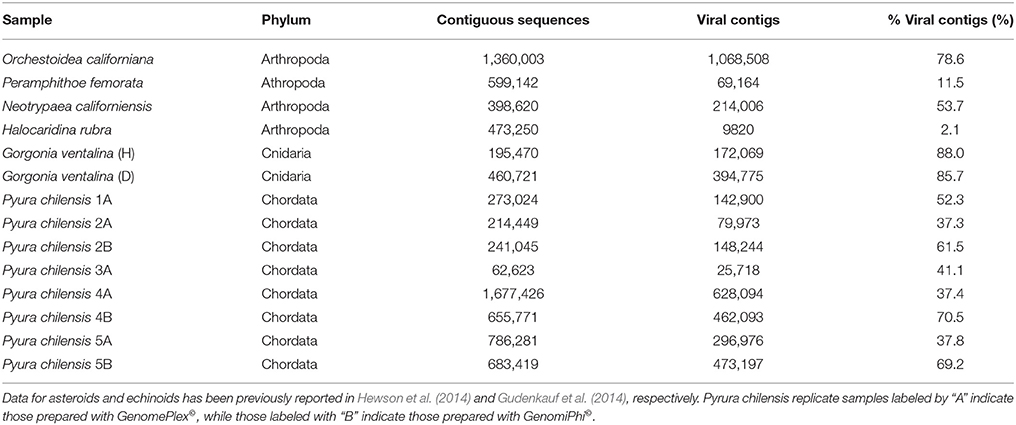
Table 2. The number of contiguous sequences (viral, bacterial, archaeal, eukaryotic), annotated viral contigs, and percentage of viral contigs resulting from the sequencing of viromes from samples listed in Table 1.
Overview of Viral Metagenomic Composition
Thirty-four viral families were represented within 41 viromes. Pisaster ochraceus (diseased 2, Santa Cruz) and Pycnopodia helianthoides (diseased 2, Seattle Aquarium), members of the Echinodermata phylum, harbored the most diverse viral assemblages, with 15 viral families represented in each. Evasterias troscheli (healthy, Friday Harbor Lab) and P. helianthoides (healthy, Friday Harbor Lab), also echinoderms, had the least diverse viral assemblages, with only two families represented in each virome.
Bacteriophage comprised the vast majority of viral annotations in all samples (Figure 1). This is not surprising, as bacteria exist within healthy epithelial tissues of echinoderms (Becker et al., 2007, 2009), on healthy cnidarian sea fan tissues (Gil-Agudelo et al., 2006), within epithelial and hepatopancreatic tissue of decapoda (Goffredi et al., 2008; Guri et al., 2012; Nunan et al., 2013), within healthy epithelial tissues of amphipoda (Mengoni et al., 2013), and tunicates (Pérez-Matos et al., 2007). Because the surfaces of the metazoans were not cleaned or scrubbed prior to tissue amendment, some of these observations likely derive from viruses that resided upon the organisms' exterior surface, caught in epithelial mucus or infecting bacteria there.
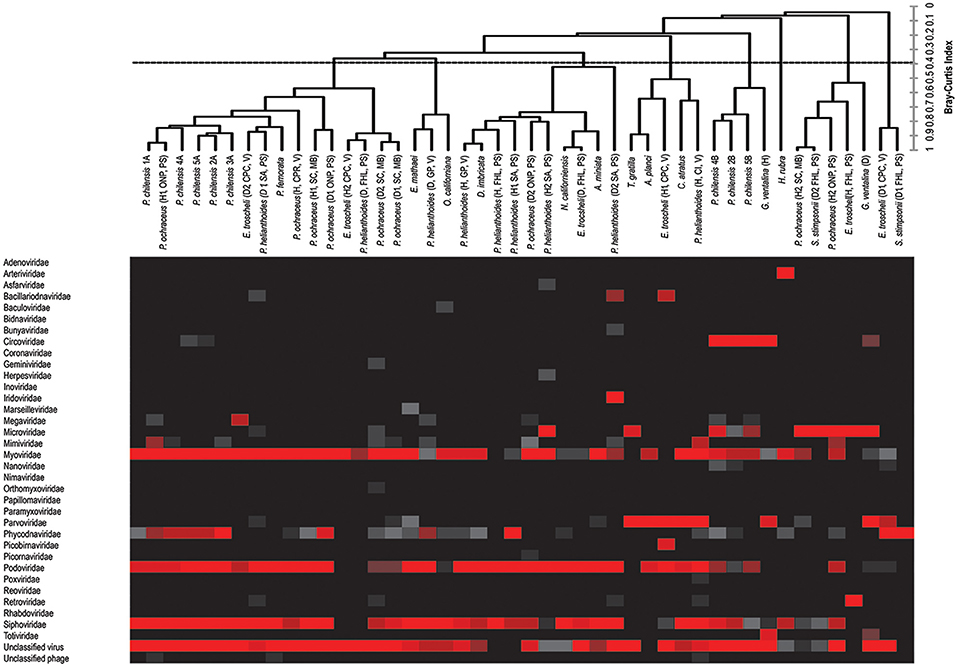
Figure 1. Combined heat map of viral family abundance and agglomerative hierarchical clustering (AHC) dendrogram visualizing relatedness between viral communities of sampled hosts at the viral family level. The dendrogram was created by unweighted-pair-group-mean average (UPGMA) and was based on the Bray–Curtis Similarity Index, and includes bacteriophage (see Figure 2 for patterns of clustering in the absence of bacteriophage).
The viral group comprising the greatest proportion of sequences in libraries—the phycodnaviruses—was significantly higher (p < 0.0001, Student's t-test, df = 38) compared to the next most well-represented group, the parvoviruses, which in turn was significantly higher (p < 0.000001, Student's t-test, df = 38) than the next most abundant group, the retroviruses. Our observation of bidnaviruses and asfiviruses in Echinodermata, circoviruses, and densoviruses in Chordata, iridoviruses in Echinodermata and Chordata, and arteriviruses and nucleopolyhedroviruses in Arthropoda extend the potential host ranges of these viral families. Of viral groups, only the totiviruses (in Cnidaria), nimaviruses (in Urochordata), and arteriviruses (in Arthropods) were unique to the phyla sampled, with the remaining viral annotations shared between at least two phyla.
Similarities and Differences between Viromes of Different Marine Invertebrate Phyla
The viral assemblages of marine invertebrates are generally disparate both within and between host phyla (Tables 3, 4). Chordate-derived viromes are the most similar, at 69.4% mean similarity (Bray–Curtis similarity index), while arthropod-derived viromes are the least similar, at 18.1%. These differences could have resulted from differences in feeding patterns, as P. chilensis is a filter-feeder, while the arthropods studied are herbivores or detritivores. These observations could also result from differences in motility, as P. chilensis is stationary throughout its lifespan, encountering the same types of environmental viruses, while the arthropods are motile and could be exposed to different viruses as they traverse novel environments.

Table 3. Pair-wise matrix of mean similarity and standard error (in brackets) of total viromes (i.e., including bacteriophage) within and between host phyla, between asteroids and echinoids, and between GenomePlex and GenomiPhi prepared libraries (from Pyura chilensis urochordate samples).

Table 4. Pair-wise matrix of mean similarity and standard error (in brackets) of non-bacteriophage viromes within and between host phyla, between asteroids and echinoids, and between GenomePlex and GenomiPhi prepared libraries (from Pyura chilensis urochordate samples).
While total viral assemblages of marine invertebrates are disparate in general, the non-bacteriophage echinoidea-derived viromes (i.e., including only the virus families which infect eukaryotes) are more similar to each other than to any other group, and are clearly separate from urochordate-derived viromes (Figure 2). Also, the individual asteroidea viromes are highly dissimilar to one another, a difference which could have resulted from the wide geographic range of sample collection and concomitant exposure to different sources of viruses.
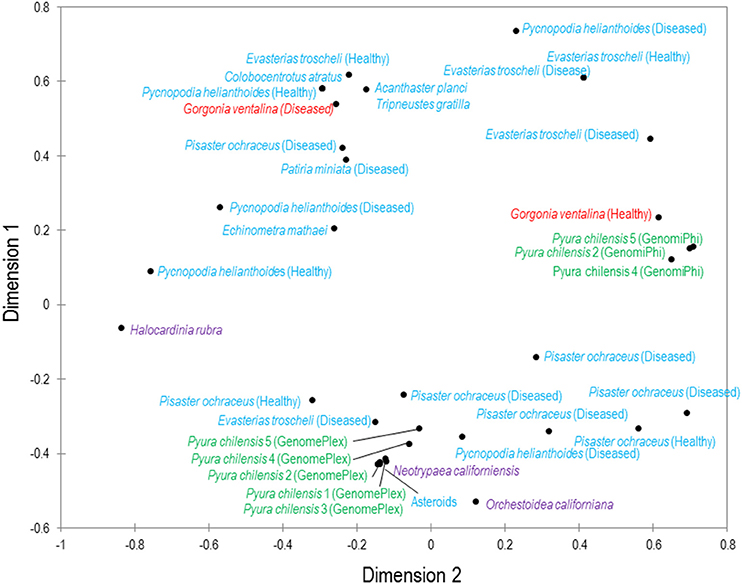
Figure 2. Multiple dimensional scaling plot of viral metagenomes prepared from four phyla. MDS plot represents two-dimensional scaling analysis (absolute model, Kruskal's stress, random initial configuration, five repetitions, stop condition at 0.00001 convergence/500 iterations). The phyla are color-coded: blue are echinoderms, red are gorgonian octocorals, green are urochordates and purple are crustaceans. Bacteriophage and unclassified viruses are not included in this analysis.
The similarities and differences observed both within and between host phyla could have resulted from differences in location of sample collection, feeding patterns, host physiology, immune response, viral entry receptors, or active infection, although pathogenesis and immune activity is not the focus of this work. Further metagenomic studies should incorporate greater numbers and phyla of these organisms from various locations for greater statistical power and discrimination between these different variables.
Trends in Viral Detection across Host Phyla
Circoviruses were found in high proportion in diseased G. ventalina (cnidaria) and were found in low proportion in echinoderms (Figure 1). Interestingly, these viruses were completely absent from arthropod-derived viromes. Viruses from the Parvoviridae family are particularly widespread in marine invertebrate viromes, comprising a large proportion of those from echinoderms (both asteroidea and echinoidea), but a relatively low proportion in viromes of urochordates. Again, these viruses were completely absent from arthropod-derived viromes (Figure 1). Further investigation should clarify why circoviruses and parvoviruses were not detected in this host phylum. Phycodnaviruses were present in high proportions in arthropod, echinoderm, and urochordate-derived viromes, but were present in low proportion in viromes of cnidaria (Figure 1). These differences may suggest differential exposure of these hosts to infected algae in the surrounding water and these algae-associated viruses. Mimiviruses were detected in high proportion in four echinoderm samples (echinoidea), suggesting that amoeba species are present in/on these host species (Figure 1). Lastly, retroviruses were found in high proportions in two echinoderm samples (echinoidea), perhaps indicating active infection in these hosts (Figure 1). Future investigation should clarify the epidemiological importance of these observations in marine invertebrates as well as their physiological basis.
Unexpected Viruses Found within Marine Invertebrates
Single-Stranded RNA Viruses
It is surprising that RNA viruses were detected, as only DNA viruses were targeted using our approach. These could have been intermediate DNA stages after conversion from viral RNA to DNA in the host cell by reverse transcriptase enzymes. Arteriviruses were detected in Halocaridina rubra, with observations representing strong homology to arterivirus proteins since the match was stringent (E-value = 0.00). Arteriviruses have pleomorphic (usually icosahedral) enveloped capsids and linear, positive sense genomes that are 12–16 kb in length, encoding eight to twelve genes (Snijder et al., 2013). Isolated arteriviruses have only been found in mammals, although viruses from the order Nidovirales (to which arteriviruses belong) have also been found infecting shrimp, fish, and insects (Snijder et al., 2013). Arterivirus infection can result in a range of effects from total asymptomatic infection to fatal disease (Snijder et al., 2013).
Single-Stranded DNA Viruses
Previously characterized bacilladnaviruses have an enveloped icosahedral capsid and a closed circular genome of approximately 6.7 kb (Tomaru et al., 2011). These viruses infect diatoms from the phylum Bacillariophyta and their presence within metazoans (two P. helianthoides from Seattle Aquarium and three E. troscheli from Cape Roger Curtis) suggests they ingested or came into contact with infected diatoms. Sequence homology to bacilladnaviral proteins was strong, with e-values ranging from 1.57 × 10−6 to 1.90 × 10−34.
Bidnaviruses were detected in two asteroids, P. ochraceus from Cates Park Reef and E. troscheli from Cape Roger Curtis. They consist of non-enveloped spherical capsids enclosing two sets of complimentary linear genomes of 6.5 and 6 kb that encode four genes (Hu et al., 2013). Hits within these asteroids represent homologous proteins within the bidnaviral genomes, since they had low e-values (2.24 × 10−21 and 1.08 × 10−8). Bidnaviruses have only been detected previously in the insect Bombyx mori, where they are highly pathogenic.
Circoviruses were particularly widespread amongst the viromes and detected in cnidarians, urochordates, and echinoderms; until recently they were only known to exist in mammals, birds, arthropods and amphipods (Dunlap et al., 2013). They are non-enveloped, icosahedral viruses with a circular genome of about 2 kb, and infection ranges from asymptomatic to fatal (Delwart and Li, 2012). An echinoderm circovirus from E. troscheli appears very similar to a circovirus from the crustacean Labidocera aestiva. G. ventalina circoviruses appear to be the most distantly related to crustacean, echinoderm, and tunicate circoviruses (Figure 3). All of these circoviruses were distantly related to mammalian and avian circoviruses yet distinct from cycloviruses.

Figure 3. (A, Left panel) Phylogenetic representation based on a 96 aa alignment of Circovirus replication initiator protein (rep), including closest relatives based on BLASTX comparison against the non-redundant database at the NCBI. (B, Right panel) Phylogenetic representation based on a 126 aa alignment of Densovirus non-structural protein (NS1), including closest relatives based on BLASTX comparison against the non-redundant database at the NCBI. The alignment was performed with CLUSTAL W and the tree drawn using neighbor joining. Bootstrap values of 1000 tree iterations that are >50% are indicated at branch nodes. Scale bar represents 0.1 substitutions per site. The phyla are color-coded: blue are echinoderms, red are gorgonian octocorals, green are urochordates.
Densoviruses (family Parvoviridae) are non-enveloped viruses with icosahedral capsids and linear genomes of 3–5 kb that encode two or three genes (Wang et al., 2010). Parvoviridae were known, until recently, to only infect vertebrates, arthropods, and echinoderms (Gudenkauf et al., 2014; Hewson et al., 2014) but this study detected them within echinoderms, cnidarians, and urochordates. Infection may be asymptomatic (Berns and Parrish, 2007) or lead to mass mortality amongst populations like Hawaiian blue shrimp (Bell and Lightner, 1984). Phylogenetic analysis corroborates observations of virome similarity (Figure 3). Acanthaster planci and E. troscheli densoviruses appear to be closely related to each other and to insect densoviruses, again suggesting that echinoderm-associated viruses are similar to those associated with arthropods. P. chilensis densoviruses are somewhat distant from those of echinoderms or cnidarians. All densovirus-like sequences are distantly related to parvoviruses and bocaviruses. Parvoviruses (genus Parvovirus of the Parvoviridae family) contain two open reading frames while Bocaviruses (genus Bocavirus, also within Parvoviridae) contain three open reading frames, with the third located in the middle of the viral genome (Zhang et al., 2011; Cotmore et al., 2014). Densoviruses infect invertebrate hosts while parvoviruses and bocaviruses infect vertebrates (Cotmore et al., 2014).
Double-Stranded DNA Viruses
Asfiviruses were detected in two asteroids, P. ochraceus from Cates Park Reef and P. helianthoides from Seattle Aquarium (second healthy sample). They have been detected primarily in mammals (Rodríguez et al., 1996) but also in terrestrial arthropods (Labuda and Nuttall, 2004; Hubálek and Rudolf, 2012) and have enveloped icosahedral capsids enclosing a linear genome of 170–193 kb that encodes 70 genes (Dixon et al., 2013). Infection in arthropods is asymptomatic but results in severe hemorrhagic fever in mammals. These observations had relatively weak expect values (8.10 × 10−5 and 1.60 × 10−8) and hence may represent homologous proteins from asfiviruses or other organisms.
Nucleopolyhedroviruses (NPV's) along with granuloviruses comprise a suborder of Baculoviridae and were detected as a particularly strong hit in the amphipod Orchestoidea californiana (E-value of 1.24 × 10−89). They have enveloped baculo-shaped capsids enclosing a closed circular genome of 120–140 kb that encodes 100–180 proteins (Blissard and Rohrmann, 1990; Thiem and Cheng, 2009; Hewson et al., 2011b). NPV's typically infect insects but a recent study found them heavily enriched in sea foam and kelp detritus (Thiem and Cheng, 2009; Hewson et al., 2011b). By inhabiting and consuming kelp detritus O. californiana could be hosting this virus and release desiccation-resistant multivirion capsids necessary for viral transmission. This would indicate that NPV's detected in the 2011 Hewson et al. study were most likely from amphipod hosts.
Iridoviruses have been found within insects, mollusks, crustacean, and cnidarians in previous studies (Williams, 2008; Weynberg et al., 2014), but were detected in Demasterias imbricata, P. helianthoides (second diseased sample from the Seattle Aquarium), E. troscheli (second diseased sample from Cates Park Reef), and GenomePlex-prepared P. chilensis samples from Chile. They may have enveloped or non-enveloped icosahedral capsids (depending on if they resulted from budding or release via lysis) and contain a linear genome of 140–303 kb that encodes a highly variable number of genes (Darai, 1989). These hits have strong homology to iridoviral proteins, with e-values of 5.51 × 10−20, 5.17 × 10−10, 2.63 × 10−5, 4.90 × 10−15, and 5.24 × 10−16, respectively.
Phycodnaviruses are enveloped icosahedral viruses with dsDNA genomes from 160 to 560 kb in length, encoding dozens of genes (Grimsley et al., 2012). These viruses infect algae, suggesting that their presence within echinoderms and tunicates indicates that these populations consumed infected algae. Phycodnaviruses associated with tunicates appeared distinct from those associated with echinoderms and all phycodnaviruses in invertebrate viromes were distinct from distantly related phycodnavirus genotypes like Ectocarpus siliculosis virus (Figure 4).
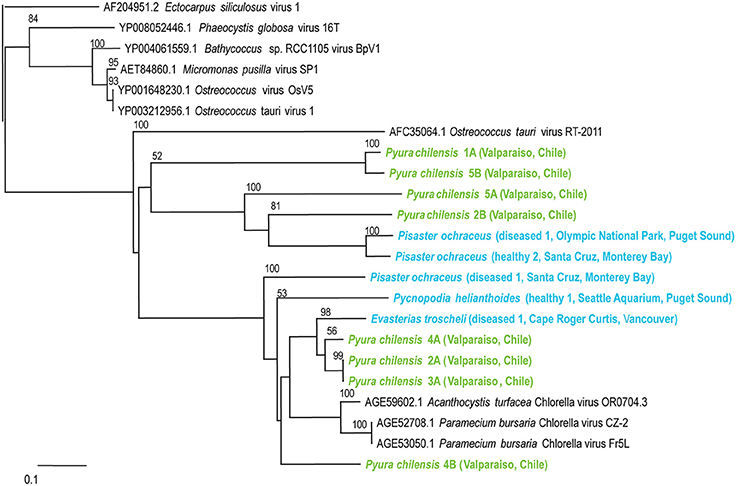
Figure 4. Phylogenetic representation based on a 76 aa alignment of Phycodnavirus adenosylcobalamin-dependent ribonucleoside-triphosphate reductase protein, including closest relatives based on BLASTX comparison against the non-redundant database at the NCBI. The alignment was performed with CLUSTAL W and the tree drawn using neighbor joining. Bootstrap values of 1000 tree iterations that are >50% are indicated at branch nodes. Scale bar represents 0.1 substitutions per site. The phyla are color-coded: blue are echinoderms, green are urochordates.
Pseudomonas bacteriophages were also particularly widespread amongst the viromes and were detected in echinoderms, crustaceans, and tunicates (Figure 1). These phages are mostly from the Myoviridae family and have non-enveloped icosahedral capsids enclosing genomes of 33–244 kb encoding 40–415 proteins (Drulis-Kawa et al., 2014). Pseudomonas phages infect the Pseudomonas genus of γ-proteobacteria and their presence reveals the existence of Pseudomonas within or associated with hosts. Those from P. ochraceus appear distinct from those associated with P. chilensis. All phage were distantly related to Pseudomonas phage PAKP1 and distinct from Vibrio phage KVP40.
Persicivirga bacteriophages are recently characterized as part of the Siphoviridae family, with non-enveloped icosahedral capsids containing genomes of 35 kb, encoding seven proteins (Kang et al., 2012). These phages infect the Persicivirga genus of the Flavobacteriaceae family of the Bacteroidetes phylum and their presence within echinoderms, crustaceans, and tunicates demonstrates the existence of Persicivirga within or associated with these organisms. Persicivirga can degrade complex polysaccharides produced by algae, suggesting that their presence is closely linked to marine algae (Kang et al., 2012). Phylogenetic analysis corresponds with observations of virome similarity (Figure 5) as a clear separation exists between those associated with P. chilensis and E. troscheli. Other Persicivirga phage-like sequences are spurious hits; those from P. ochraceus (Friday Harbor Lab, diseased 2 Santa Cruz, healthy 1 Santa Cruz, and Olympic National Park), P. helianthoides, P. chilensis (1A, 2A, 4A, 4B), and E. troscheli (diseased 1 Cape Roger Curtis and Friday Harbor Lab) appeared most related to Pseudoalteromonas phage RIO-1, indicating that these phages are most likely phage of γ-proteobacteria like Pseudoalteromonas. P. chilensis 5B, and P. ochraceus (diseased 1, Santa Cruz) phages appeared related to Synechococcus phage S-CBS4 and are most likely not bacteriophages of Persicivirga or Pseudoalteromonas.
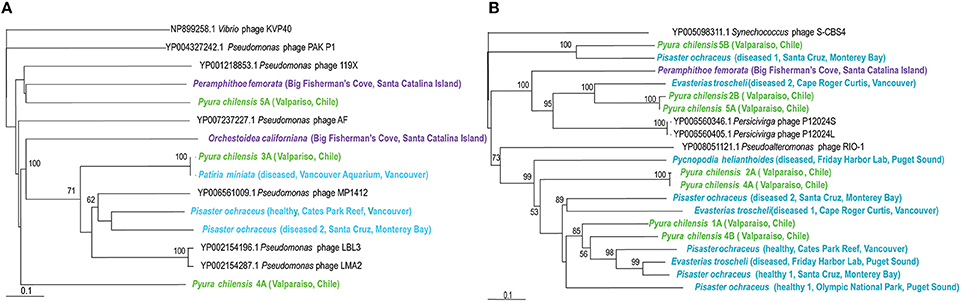
Figure 5. (A, Left panel) Phylogenetic representation based on a 146 aa alignment of Persicivirga phage portal protein, including closest relatives based on BLASTX comparison against the non-redundant database at the NCBI. (B, Right panel) Phylogenetic representation based on a 156 aa alignment of Pseudomonas phage helicase protein, including closest relatives based on BLASTX comparison against the non-redundant database at the NCBI. The alignment was performed with CLUSTAL W and the tree drawn using neighbor joining. Bootstrap values of 1000 tree iterations that are >50% are indicated at branch nodes. Scale bar represents 0.1 substitutions per site. The phyla are color-coded: blue are echinoderms, green are urochordates and purple are crustaceans.
Effect of Library Preparation Method on Metagenomic Composition
Viromes prepared using two different library preparation methods are highly dissimilar (Figures 1, 2). The mean between-library method similarity was significantly different (p = 10−23, Student's t-test, df = 15) to within-library method similarity. GenomiPhi (which utilizes multiple displacement amplification) appears to bias results in favor of circular single stranded viruses, confirming the results of previous work (Ng et al., 2011). Circoviruses were detected in abundances several orders of magnitude larger within GenomiPhi-prepared samples than in those prepared with GenomePlex, comprising 4.09% of the P. chilensis sample 4 virome prepared with GenomiPhi and only 0.01% of the virome prepared with GenomePlex. In contrast, phycodnaviruses were under-represented in the GenomiPhi library, representing only 0.02% of reads (compared to 0.93% of reads in the GenomePlex-prepared library). Differences also resulted from GenomiPhi's failure to amplify several types of linear dsDNA viruses which were detected in libraries prepared by GenomePlex. For instance, the virome derived from GenomiPhi-prepared P. chilensis sample 4 lacked all hits to Iridoviridae, Marseilleviridae, and Nimaviridae linear dsDNA virus families that were present within the GenomePlex-prepared sample. Overall, there were fewer circular ssDNA viruses, and more strains of linear dsDNA viruses in viromes from P. chilensis that were prepared with GenomePlex.
Our result corroborates previous studies that found that GenomiPhi biases viromes in favor of circular single stranded DNA viruses, resulting in variable coverage of viral communities, leaving entire families unamplified (Kim and Bae, 2011; Marine et al., 2014). These results reflect the fact that the phi29 DNA polymerase preferentially amplifies genomic regions associated with circular ssDNA viruses during initial priming events (Dichosa et al., 2012). We agree with previous work and suggest that researchers choose an amplification strategy that will minimally bias results if viral community proportions are to be studied. If possible, PCR-free approaches will result in the least bias. However, if viral detection is the only goal of a study, it would be wise to use both GenomePlex and GenomiPhi to ensure all linear ssDNA and circular ssDNA viruses are detected.
Conclusions
This study indicates that viromes of marine invertebrates are generally disparate both within and between host phyla. Urochordates have the most similar viromes, while arthropods have the least. Non-bacteriophage echinoidea viromes appear most similar by multidimensional scaling analysis and distinct from those derived from urochordates. Asteroidea, cnidaria, and arthropod-derived non-bacteriophage viromes are highly disparate. Several virus families were observed in certain host phyla but not others, and viruses of Parvoviridae and Phycodnaviridae were found in all phyla except arthropoda. The viral communities of many marine invertebrates have not been investigated but may be influencing host ecology, and this study may provide some starting points for further investigation into similarities and differences observed. These may be due to cellular, immunological, geographical, and ecological niche differences amongst these host phyla. Additionally, several viral families were observed within viromes that have not been detected in these host phyla in previous investigation. Finally, this paper confirms previous results indicating substantial differences between viromes built by multiple displacement amplification and other library preparation approaches by describing artifactual differences in community composition and representation introduced by the two methods.
Author Contributions
IH, and BG designed and performed laboratory analyses, analyzed data, and helped to write the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Ernesto Weil (University of Puerto Rico), David Needham, and Wiebke Ziebis (University of Southern California), Senifa Annandale (University of Hawaii, Hilo), David Tapia Espinoza, Yoanna Eissler, and Juan Kuznar (University of Valparaiso), and Nelson Hairston (Cornell University) for sample collection. The authors are also grateful to James Eaglesham (Cornell University) for assistance with virome preparation. This work was supported by NSF grants OCE-1356964, OCE-1537111, DEB-1028898, and OCE-1049670 awarded to IH and a grant from the Mario Einaudi Center for International Studies at Cornell University.
References
Bates, A. E., Hilton, B. J., and Harley, C. D. (2009). Effects of temperature, season and locality on wasting disease in the keystone predatory sea star Pisaster ochraceus. Dis. Aquat. Organ. 86, 245–251. doi: 10.3354/dao02125
Becker, P., Gillan, D. C., and Eeckhaut, I. (2007). Microbiological study of the body wall lesions of the echinoid Tripneustes gratilla. Dis. Aquat. Organ. 77, 73–82. doi: 10.3354/dao01821
Becker, P. T., Gillan, D. C., and Eeckhaut, I. (2009). Characterization of the bacterial community associated with body wall lesions of Tripneustes gratilla (Echinoidea) using culture-independent methods. J. Invertebr. Pathol. 100, 127–130. doi: 10.1016/j.jip.2008.11.002
Bell, T. A., and Lightner, D. V. (1984). IHHN virus - infectivity and pathogenicity studies in Penaeus stylirostris and Penaeus vannamei. Aquaculture 38, 185–194. doi: 10.1016/0044-8486(84)90142-X
Berke, S. K. (2010). Functional groups of ecosystem engineers: a proposed classification with comments on current issues. Integr. Comp. Biol. 50, 147–157. doi: 10.1093/icb/icq077
Berns, K., and Parrish, C. R. (2007). “Parvoviridae,” in Field's Virology, eds D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (Philadelphia, PA: Lippincott Williams and Wilkins), 2437–2477.
Blissard, G. W., and Rohrmann, G. F. (1990). Baculovirus diversity and molecular ecology. Annu. Rev. Entomol. 35, 127–155. doi: 10.1146/annurev.en.35.010190.001015
Bluhm, B. A., Piepenburg, D., and von Juterzenka, K. (1998). Distribution, standing stock, growth, mortality and production of Strongylocentrotus pallidus (Echinodermata: Echinodea) in the northern Barents Sea. Polar Biol. 20, 325–334. doi: 10.1007/s003000050310
Carpenter, R. C. (1990). Mass mortality of Diadema antillarum. I. Long-term effects on sea urchin population-dynamics and coral reef algal communities. Mar. Biol. 104, 67–77. doi: 10.1007/BF01313159
Cerrano, C. B., Bavastrello, G., Nike Bianchi, C., Cattaneo-vietti, R., Bava, S., Morganti, C., et al. (2000). A catastrophic mass-mortality episode of gorgonia and other organisms in the Ligurian Sea (North-western Mediterranean), summer 1999. Ecol. Lett. 3, 284–293. doi: 10.1046/j.1461-0248.2000.00152.x
Cotmore, S. F., Agbandje-McKenna, M., Chiorini, J. A., Mukha, D. V., Pintel, D. J., Qiu, J., et al. (2014). The family Parvoviridae. Arch. Virol. 159, 1239–1247. doi: 10.1007/s00705-013-1914-1
Delwart, E., and Li, L. (2012). Rapidly expanding genetic diversity and host range of the Circoviridae viral family and other Rep encoding small circular ssDNA genomes. Virus Res. 164, 114–121. doi: 10.1016/j.virusres.2011.11.021
Dichosa, A. E., Fitzsimons, M. S., Lo, C. C., Weston, L. L., Preteska, L. G., Snook, J. P., et al. (2012). Artificial polyploidy improves bacterial single cell genome recovery. PLoS ONE 7:e37387. doi: 10.1371/journal.pone.0037387
Dixon, L. K., Chapman, D. A., Netherton, C. L., and Upton, C. (2013). African swine fever virus replication and genomics. Virus Res. 173, 3–14. doi: 10.1016/j.virusres.2012.10.020
Drulis-Kawa, Z., Olszak, T., Danis, K., Majkowska-Skrobek, G., and Ackermann, H. W. (2014). A giant Pseudomonas phage from Poland. Arch. Virol. 159, 567–572. doi: 10.1007/s00705-013-1844-y
Dunlap, D. S., Ng, T. F., Rosario, K., Barbosa, J. G., Greco, A. M., Breitbart, M., et al. (2013). Molecular and microscopic evidence of viruses in marine copepods. Proc. Natl. Acad. Sci. U.S.A. 110, 1375–1380. doi: 10.1073/pnas.1216595110
Gil-Agudelo, D. L., Myers, C., Smith, G. W., and Kim, K. (2006). Changes in the microbial communities associated with Gorgonia ventalina during aspergillosis infection. Dis. Aquat. Organ. 69, 89–94. doi: 10.3354/dao069089
Goffredi, S. K., Jones, W. J., Ehrlich, H., Springer, A., and Vrijenhoek, B. C. (2008). Epibiotic bacteria associated with the recently discovered Yeti crab, Kiwi hirsutai. Environ. Microbiol. 10, 2623–2634. doi: 10.1111/j.1462-2920.2008.01684.x
Grimsley, N. H., Thomas, R., Kegel, J. U., Jacquet, S., Moreau, H., and Desdevises, Y. (2012). Genomics of algal host–virus interactions. Adv. Bot. Res. 64, 343–381. doi: 10.1016/B978-0-12-391499-6.00009-8
Gudenkauf, B. M., Eaglesham, J. B., Aragundi, W. M., and Hewson, I. (2014). Discovery of urchin-associated densoviruses (family Parvoviridae) in coastal waters of the Big Island, Hawaii. J. Gen. Virol. 95, 652–658. doi: 10.1099/vir.0.060780-0
Guri, M., Durand, L., Cueff-Gauchard, V., Zbinden, M., Crassous, P., Shillito, B., et al. (2012). Acquisition of epibiotic bacteria along the life cycle of the hydrothermal shrimp Rimicaris exoculata. ISME J. 6, 597–609. doi: 10.1038/ismej.2011.133
Hewson, I., Brown, J. M., Burge, C. A., Couch, C. S., LaBarre, B. A., Mouchka, M. E., et al. (2011a). Description of viral assemblages associated with the Gorgonia ventalina holobiont. Coral Reefs 31, 487–491. doi: 10.1007/s00338-011-0864-x
Hewson, I., Brown, J. M., Gitlin, S. A., and Doud, D. F. (2011b). Nucleopolyhedrovirus detection and distribution in terrestrial, freshwater, and marine habitats of Appledore Island, Gulf of Maine. Microb. Ecol. 62, 48–57. doi: 10.1007/s00248-011-9856-1
Hewson, I., Button, J. B., Gudenkauf, B. M., Miner, B., Newton, A. L., Gaydos, J. K., et al. (2014). Densovirus associated with sea-star wasting disease and mass mortality. Proc. Natl. Acad. Sci. U.S.A. 111, 17278–17283. doi: 10.1073/pnas.1416625111
Hewson, I., Eaglesham, J. B., Hook, T. O., LaBarre, B. A., Sepulveda, M. S., Thompson, P. D., et al. (2013). Investigation of viruses in Diporeia spp. from the Laurentian Great Lakes and Owasco Lake as potential stressors of declining populations. J. Great Lakes Res. 39, 499–506. doi: 10.1016/j.jglr.2013.06.006
Hu, Z. Y., Li, G. H., Li, G. T., Yao, Q., and Chen, K. P. (2013). Bombyx mori bidensovirus: the type species of the new genus Bidensovirus in the new family Bidnaviridae. Chin. Sci. Bull. 58, 4528–4532. doi: 10.1007/s11434-013-5876-1
Hubálek, Z., and Rudolf, I. (2012). Tick-borne viruses in Europe. Parasitol. Res. 111, 9–36. doi: 10.1007/s00436-012-2910-1
Jangoux, M. (1987). Diseases of Echinodermata. I. Agents microorganisms and protistans. Dis. Aquat. Organ. 2, 147–162. doi: 10.3354/dao002147
Kang, I., Jang, H., and Cho, J. C. (2012). Complete genome sequences of two Persicivirga bacteriophages, P12024S and P12024L. J. Virol. 86, 8907–8908. doi: 10.1128/JVI.01327-12
Kim, K. H., and Bae, J. W. (2011). Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 77, 7663–7668. doi: 10.1128/AEM.00289-11
Labuda, M., and Nuttall, P. A. (2004). Tick-borne viruses. Parasitology 129(Suppl.), S221–S245. doi: 10.1017/S0031182004005220
Lessios, H. A. (1988). Mass mortality of Diadema antillarum in the Caribbean: what have we learned? Annu. Rev. Ecol. Syst. 19, 371–393. doi: 10.1146/annurev.es.19.110188.002103
Lessios, H. A., Robertson, D. R., and Cubit, J. D. (1984). Spread of Diadema mass mortality through the Caribbean. Science 226, 335–337. doi: 10.1126/science.226.4672.335
Marine, R., McCarren, C., Vorrasane, V., Nasko, D., Crowgey, E., Polson, S. W., et al. (2014). Caught in the middle with multiple displacement amplification: the myth of pooling for avoiding multiple displacement amplification bias in a metagenome. Microbiome 2:3. doi: 10.1186/2049-2618-2-3
Mengoni, A., Focardi, A., Bacci, G., and Ugolini, A. (2013). High genetic diversity and variability of bacterial communities associated with the sandhopper Talitrus saltator (Montagu) (Crustacea, Amphipoda). Est. Coast. Shelf Sci. 131, 75–82. doi: 10.1016/j.ecss.2013.08.011
Munn, C. B. (2006). Viruses as pathogens of marine organisms - from bacteria to whales. J. Mar. Biol. Assoc. UK 86, 453–467. doi: 10.1017/S002531540601335X
Ng, T. F. (2010). Discovery of Novel Viruses from Animals, Plants, and Insect Vectors using Viral Metagenomics. Graduate School Theses and Dissertations. University of South Florida.
Ng, T. F., Manire, C., Borrowman, K., Langer, T., Ehrhart, L., and Breitbart, M. (2009). Discovery of a novel single-stranded DNA virus from a sea turtle fibropapilloma by using viral metagenomics. J. Virol. 83, 2500–2509. doi: 10.1128/JVI.01946-08
Ng, T. F. F., Duffy, S., Polston, J. E., Bixby, E., Vallad, G. E., and Breitbart, M. (2011). Exploring the diversity of plant DNA viruses and their satellites using vector-enabled metagenomics on whiteflies. PLoS ONE 6:e19050. doi: 10.1371/journal.pone.0019050
Nunan, L. M., Pantoja, C. R., Gomez-Jimenez, S., and Lightner, D. V. (2013). “Candidatus Hepatobacter penaei,” an intracellular pathogenic enteric bacterium in the hepatopancreas of the marine shrimp xi (Crustacea: Decapoda). Appl. Environ. Microbiol. 79, 1407–1409. doi: 10.1128/AEM.02425-12
Page, R. D. (1996). TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12, 357–358.
Paine, R. T. (1969). The Pisaster-Tegula interaction: prey patches, predator food preference, and intertidal community structure. Ecology 50, 950–961. doi: 10.2307/1936888
Pérez-Matos, A. E., Rosado, W., and Govind, N. S. (2007). Bacterial diversity associated with the Caribbean tunicate Ecteinascidia turbinata. Antonie Van Leeuwenhoek. 92, 155–164. doi: 10.1007/s10482-007-9143-9
Rodríguez, F., Fernández, A., Pérez, J., Mártin de las Mulas, J., Sierra, M. A., and Jover, A. (1996). African swine fever: morphopathology of a viral hemorrhagic disease. Vet. Rec. 139, 249–254. doi: 10.1136/vr.139.11.249
Rosario, K., Dayaram, A., Marinov, M., Ware, J., Kraberger, S., Stainton, D., et al. (2012). Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J. Gen. Virol. 93, 2668–2681. doi: 10.1099/vir.0.045948-0
Rosario, K., Marinov, M., Stainton, D., Kraberger, S., Wiltshire, E. J., Collings, D. A., et al. (2011). Dragonfly cyclovirus, a novel single-stranded DNA virus discovered in dragonflies (Odonata: Anisoptera). J. Gen. Virol. 92, 1302–1308. doi: 10.1099/vir.0.030338-0
Snijder, E. J., Kikkert, M., and Fang, Y. (2013). Arterivirus molecular biology and pathogenesis. J. Gen. Virol. 94, 2141–2163. doi: 10.1099/vir.0.056341-0
Suttle, C. A. (2007). Marine viruses–major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812. doi: 10.1038/nrmicro1750
Thiem, S. M., and Cheng, X.-W. (2009). Baculovirus host-range. Virol. Sin. 24, 436–457. doi: 10.1007/s12250-009-3058-8
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). Clustal-W - Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic. Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Thurber, R. V., Haynes, M., Breitbart, M., Wegley, L., and Rohwer, F. (2009). Laboratory procedures to generate viral metagenomes. Nat. Protoc. 4, 470–483. doi: 10.1038/nprot.2009.10
Tomaru, Y., Takao, Y., Suzuki, H., Nagumo, T., Koike, K., and Nagasaki, K. (2011). Isolation and characterization of a single-stranded DNA virus infecting Chaetoceros lorenzianus Grunow. Appl. Environ. Microbiol. 77, 5285–5293. doi: 10.1128/AEM.00202-11
Wang, F., Wei, Y., Zhu, C., Huang, X., Xu, Y., Yu, L., et al. (2010). Novel parvovirus sublineage in the family of Parvoviridae. Virus Genes 41, 305–308. doi: 10.1007/s11262-010-0506-3
Weynberg, K. D., Wood-Charlson, E. M., Suttle, C. A., and van Oppen, M. J. H. (2014). Generating viral metagenomes from the coral holobiont. Front. Microbiol. 5:206. doi: 10.3389/fmicb.2014.00206
Williams, T. (2008). Natural invertebrate hosts of iridoviruses (Iridoviridae). Neotrop. Entomol. 37, 615–632. doi: 10.1590/S1519-566X2008000600001
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64, 69–114.
Keywords: virus, invertebrate, circovirus, metazoa, zooplankton
Citation: Gudenkauf BM and Hewson I (2016) Comparative Metagenomics of Viral Assemblages Inhabiting Four Phyla of Marine Invertebrates. Front. Mar. Sci. 3:23. doi: 10.3389/fmars.2016.00023
Received: 14 November 2015; Accepted: 22 February 2016;
Published: 10 March 2016.
Edited by:
Rex Malmstrom, U.S. Department of Energy Joint Genome Institute, USAReviewed by:
Karen Dawn Weynberg, Australian Institute of Marine Science, AustraliaRebecca Lisette Vega Thurber, Oregon State University, USA
Copyright © 2016 Gudenkauf and Hewson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ian Hewson, hewson@cornell.edu