Draft Genomes Shed Light on the Dual Bacterial Symbiosis that Dominates the Microbiome of the Coral Reef Sponge Amphimedon queenslandica
 Marie-Emilie A. Gauthier
Marie-Emilie A. Gauthier Jabin R. Watson
Jabin R. Watson  Sandie M. Degnan
Sandie M. Degnan- Marine Genomics Lab, School of Biological Sciences, University of Queensland, Brisbane, QLD, Australia
Amphimedon queenslandica is a coral reef demosponge that houses a low complexity and low abundance microbiota dominated by a proteobacterial duo for which draft genomes are presented here. The most prevalent symbiont, AqS1, is a sulfur-oxidizing gammaproteobacterium closely related to other demosponge symbionts and to free-living Ectothiorhodospiraceae (Chromatiales). The predicted gene repertoire of AqS1 indicates that it is capable of sulfur oxidation, carbon monoxide oxidation and inorganic phosphate assimilation, and that some of its metabolic capabilities may have been acquired via horizontal gene transfer from alphaproteobacteria. The second most prevalent symbiont, AqS2, is a betaproteobacterium whose closest known relatives are other demosponge symbionts. AqS1 has characteristic sponge symbiont features, including a versatile nutrient use with large number of transporters, ankyrin-repeat-containing proteins, and a CRISPR system. Based on the size of its genome assembly, AqS2 is predicted to have a much smaller genome with many fewer symbiotic features than AqS1. The smaller is reflected in its more limited metabolic capabilities that include carbohydrate metabolism, but not sulfur oxidation or phosphorus metabolism. Within-pathway complementation and resource partitioning potentially occur between the two bacteria. The addition of these symbiont genomes to extensive genome and transcriptome resources already available for the sponge host now permits the development of mixed-species genome-scale metabolic models as a foundation for experimental investigations of resource partitioning between symbionts and host.
Introduction
Marine sponges (Phylum Porifera) are abundant sessile, benthic filter feeders that have a major influence on biogeochemical fluxes in coral reef and coastal environments (Bell, 2008; Southwell et al., 2008b; de Goeij et al., 2013; Colman, 2015). Their ecosystem engineering roles are enabled by diverse communities of symbiotic prokaryotes that can account for up to 40% of the sponge body volume (Taylor et al., 2007a,b; Webster et al., 2010; Hentschel et al., 2012). The symbionts extend the metabolic capabilities of the host by mediating processes, such as photosynthesis (Wilkinson, 1983; Arillo et al., 1993; Steindler et al., 2002), carbon (De Goeij et al., 2008a,b; de Goeij et al., 2013), nitrogen (Wilkinson and Fay, 2004; Mohamed et al., 2008; Southwell et al., 2008a; Hoffmann et al., 2009; Schläppy et al., 2010; Fiore et al., 2013), methane (Vacelet et al., 1995), sulfur (Hoffmann et al., 2005), and phosphorus (Sabarathnam et al., 2010; Zhang et al., 2015b) cycling. Metagenomic and transcriptomic studies of sponge microbiomes have provided independent molecular evidence of the contribution of symbiotic bacteria, in particular, to biogeochemical and nutrient cycling (Hentschel et al., 2012; Fiore et al., 2015; Rua et al., 2015), underscoring the significance of the holobiont perspective in understanding ecological roles of animal hosts in marine ecosystems.
More broadly, recent metatranscriptomic, and metaproteomic analyses of sponge-associated microbial consortia have begun to unveil their global expressed gene repertoire (Liu et al., 2012; Radax et al., 2012; Moitinho-Silva et al., 2013, 2014; Fiore et al., 2015; Rua et al., 2015). Single-cell genomic and metagenomics studies have contributed to correlating particular traits with specific symbiont lineages (Thomas et al., 2010; Siegl et al., 2011; Kamke et al., 2013; Gao et al., 2014; Tian et al., 2014; Wilson et al., 2014; Burgsdorf et al., 2015; Britstein et al., 2016), revealing functional gene convergence in phylogenetically-distinct microbial communities with core features that likely reflect adaptation of microorganisms to the sponge host environment (Fan et al., 2012; Hentschel et al., 2012). Traits that have repeatedly been reported in prokaryote symbionts of poriferans include versatile nutrient utilization, protection from environmental and host-specific stress, and eukaryotic-like proteins (Fan et al., 2012; Hentschel et al., 2012). Unsurprisingly, however, other traits are specific to particular symbionts, including variable carbon fixation and secondary metabolism processes, motility, and reduced genome size. A more complete picture will emerge only as more sponge and associated symbiont genomes become available.
The persistent presence of dominant bacterial phylotypes in several species of sponge indicates that many specific host-microbe associations are stable over time, likely facilitated by vertical inheritance (Taylor et al., 2007b; Erwin et al., 2012; Hentschel et al., 2012; Thomas et al., 2016). In non-sponge host-symbiont systems, the temporal stability of associations involving two or more unrelated bacterial symbionts (Takiya et al., 2006; Wu et al., 2006; Dubilier et al., 2008; Zimmermann et al., 2014) has been postulated to involve the use of different energy sources to reduce competition between the bacterial species (Duperron et al., 2006; Kleiner et al., 2012b). It has also been suggested that some symbionts might rely on one another for their metabolism, in a mutual dependency resulting from loss of metabolic redundancies that originally existed within symbiotic partners at the time of their acquisition by the host (Wu et al., 2006; Rao et al., 2015). The extent to which these phenomena occur in sponge symbionts is currently largely unknown.
Here we report draft genomes of the dominant bacterial symbionts of the genome- and transcriptome-enabled coral reef demosponge Amphimedon queenslandica (Srivastava et al., 2010; Conaco et al., 2012; Anavy et al., 2014; Fernandez-Valverde et al., 2015; Levin et al., 2016). In their native habitat on the Great Barrier Reef of Australia, adult A. queenslandica support a relatively low diversity bacterial symbiont community characterized by a core group that are inherited vertically from the maternal parent during early embryonic development (Fieth et al., in review). The metagenomics approach that we adopt herein reveals that the adult microbiome of this sponge is dominated by just two phylotypes that we name AqS1 (A. queenslandica Symbiont 1) and AqS2, corresponding to OTU1 and OTU4, respectively, in Fieth et al. (in review). We successfully assemble draft genomes for both of these, which allows us to present the gene repertoire of the two species. We discuss potential metabolic interactions that might take place between the symbionts, and between the symbionts and their sponge host. We also report phylogenetic affinities of a third, lower abundance bacterial symbiont (AqS3, corresponding to OTU5 in Fieth et al., in review) for which we retrieve a draft genome of lesser quality.
Materials and Methods
Acquisition of Bacterial gDNA
Six adult sponges (Amphimedon queenslandica) were collected from Shark Bay, Heron Island, Great Barrier Reef, QLD (23° 27′S, 151° 55′ E), three in September 2012 (HSep1-3), and three in December 2012 (HDec1-3). Each sponge was removed from the substrate with a sterile scalpel and pieces were stored in tubes of 0.22 μm filter sterilized seawater (FSW) half submerged in iced water and quickly transported back to the Heron Island Research Station for immediate processing. Enrichment of sponge-associated bacteria from sponge cells followed the protocol of Thomas et al. (2010) with slight modifications, and total genomic DNA was extracted from these cell preps using a standard phenol-chloroform method. Details are provided in text of the Supplementary Material.
Genome Sequencing and Assembly
Each of the six samples was multiplexed and sequenced by Macrogen (http://www.macrogen.com) in one lane using the HiSeq Illumina platform. Pair-end reads were 100 bp long and had an insert size ranging between 150 and 250 bp. Reads were filtered with Trimmomatic (Bolger et al., 2014), removing the first five bases from the start of the read, performing a sliding window trimming with a window of four bases and an average quality threshold of 15, and eliminating resulting reads that were less than 50 bp long. We also did not retain ambiguous reads with Ns, nor reads that aligned to contigs that unambiguously belonged to the genome of the host sponge, Amphimedon queenslandica, using Bowtie2 (Langmead and Salzberg, 2012). Metabinning and genome construction were performed following the protocol of Albertsen et al. (2013) with slight modifications (Supplementary Material text; Figure S1).
Small Subunit 16S rRNA Gene Reconstruction from Metagenomic Shotgun Data
Small subunit (i.e., 16S) rRNA gene-containing reads were identified from the filtered FASTQ files using Metaxa v. 2.0.1 (Bengtsson et al., 2011). Extracted reads were assembled with IDBA-UD (Peng et al., 2012) using a 99% overlap identity. We first performed assemblies on these reads keeping samples separate, and then another assembly pooling reads from all samples. We used CAP3 (Huang and Madan, 1999) on all assemblies to merge overlapping contigs, and checked manually the resulting singletons and collapsed contigs. These derived 16S rRNA sequences were broadly classified using BLASTN and the NR database, excluding uncultured and environmental samples. After confirming that we recovered most reads that had been initially extracted by Metaxa within our final list of assembled contigs, we then used the original FASTQ files and our derived 16S rRNA sequences as reference, and applied the closed-reference OTU picking strategy of the QIIME program (Caporaso et al., 2010b) to derive 16S rRNA gene abundance estimates for each sample.
Gene Annotation
We obtained gene annotation for the two bacterial genomes that we deemed had adequate coverage (AqS1 and AqS2). Contigs for each genome were uploaded on the RAST server (Aziz et al., 2008; Overbeek et al., 2014) and annotated using the Classic annotation scheme, the RAST gene caller, and FIGfams v. 70. We opted for errors and frame shifts to be fixed automatically, and for the pipeline to BLAST large gaps for missing genes. We compared the gene content of AqS1, AqS2, Thioalkalivibrio sp. HK1, three of AqS1 closest relatives and other known sulfur-oxidizing symbionts using the function based comparative tool available on the SEED viewer within the RAST platform. For each genome, we either used predicted protein-coding sequences that were publically available or obtained these ourselves using the RAST server. Although a gene annotation for T. sp. HK1 is available on NCBI, we derived a new one via the RAST server for ease of comparison with the AqS1 genome. This annotation can be made available upon request.
We searched for the presence of domains associated with proteins postulated to facilitate host-symbiont interactions in the predicted protein-coding sequences of AqS1, AqS2, other known sulfur-oxidizing symbionts and the three closest free-living relatives to AqS1. To do so, we scanned for specific PFAM domains using hmmsearch from the program hmmer 3.1 (hmmer.org), using the trusted cutoff bit score thresholding option (–cut_tc). We used Interpro (http://www.ebi.ac.uk/interpro/), NCBI conserved domains (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/) to predict domains and transmembrane regions of the high-affinity branched-chain amino acid ABC transporter proteins. An example of putative 2D topology for one of the transmembrane protein complex was derived using TOPO2 (http://www.sacs.ucsf.edu/TOPO2/topo2-help.html). Finally, we used the CRISPRfinder program (http://crispr.u-psud.fr/Server/) to deduce the organization of the CRISPR in AqS1. The 16S rRNA gene sequences of AqS1, AqS2, and AqS3 are available from the NCBI GenBank database under accession numbers KX772780-2. The draft genomes of AqS1 and AqS2 have been deposited as Whole Genome Shotgun projects at DDBJ/ENA/GenBank under the accession numbers MEID00000000 and MEIE00000000, respectively. The versions described in this paper are MEID01000000 and MEIE01000000, respectively. The genome browsers of AqS1 and AqS2 are hosted on the RAST server (http://rast.nmpdr.org/seedviewer.cgi) under genome IDs 6666666.207661 and 6666666.208689, respectively, and are accessible via username 'guest' with password 'guest').
Phylogenetic Analyses
Phylogenetic analyses were performed for the three most abundant 16S rRNA sequences in our metagenome data. Nearest related and described sequences were retrieved from the 2013 Greengene and NCBI NR databases. The partial sequence from T. sp. HK1 reported in Tian et al. (2014) was kindly provided by Dr Ren-Mao Tian. A sequence alignment was derived with PyNAST (Caporaso et al., 2010a) using the template file “core_set_aligned.fasta.imputed” from QIIME v. 1.8.0 (Caporaso et al., 2010b) and manually checked using AliView (Larsson, 2014). A maximum-likelihood tree was constructed with RAxML v. 8 (Stamatakis, 2014) with 1000 bootstrap resamples, and visualized using FigTree v. 1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/). For trees with low bootstrap support at given nodes, we checked whether some taxa had ambiguous or insufficient phylogenetic signal by deriving their leaf stability indices using the program RogueNaRok (Aberer et al., 2013).
Since a large proportion of genes that endow the AqS1 symbiont with heterotrophic capabilities are most closely related to genes of several alphaproteobacteria, we wanted to test the hypothesis that some of these have been acquired horizontal gene transfers (HGT). To do so, we performed a phylogenetic analysis on the concatenated alignment of high-affinity branched-chain amino acid ABC transporter proteins. Nearest related and described sequences were retrieved from the NCBI NR database. We also included two species from the order Chromatiales as an outgroup. We derived alignments using AliView and checked for the presence of ambiguously aligned positions using Aliscore and Alicut (Kück et al., 2010). We used prottest3 (Darriba et al., 2011) to choose a model of evolution for each gene. We concatenated our sequence alignment into one supermatrix using FASconCAT (Kuck and Meusemann, 2010).
Results
Genome Sequencing and Genome Assembly
We sequenced six bacterial-enriched samples from the sponge Amphimedon queenslandica and recovered between 23,906,452 and 27,938,004 pairs of Illumina reads from each library. Filtering out low quality reads and contaminating sponge host DNA resulted in 14,563,169 to 16,046,247 read pairs contributing to bacterial genome assemblies (Table S1). To isolate the symbiont genomes of interest—AqS1, AqS2, and AqS3—we derived scaffold coverage plots with GC content and taxonomic information overlayed, using a slight modification of the protocol developed by Albertsen et al. (2013) (Supplementary Material; Figures S1, S2). The AqS1 genome was assembled into 127 contigs (median length 13 Kbp, total length 4, 2 Mbp, with 59% average GC content) and the AqS2 genome into 239 contigs (median length 3 Kbp, total length 1,6 Mbp, with 69% average GC content). We also recovered a third genome (AqS3) that we were able to assemble into 233 contigs (median length 5 Kbp, total length 3,17 Mbp, with 62% average GC content) (Table 1). To evaluate the quality of the assemblies, we checked for the presence of 105 conserved single-copy genes in each genome (Supplementary Material; Table S2). In general, we found that the total number of genes recovered in each of the three symbionts was on par with the unique essential gene list. Specifically, we estimate that the reconstructed AqS1 genome is 99% complete (we recovered 104 out of 105 genes) and that of AqS2 and AqS3 are 85% and 94% complete, respectively. However, since the coverage of AqS3 was low (<10X read coverage) and a principal component analysis on the tetranucleotide frequencies of its genome scaffolds indicated the presence of possible contamination that could not be removed, we elected to omit this genome from our downstream gene annotation, which is thus restricted to the two most abundant symbionts, AqS1 and AqS2.
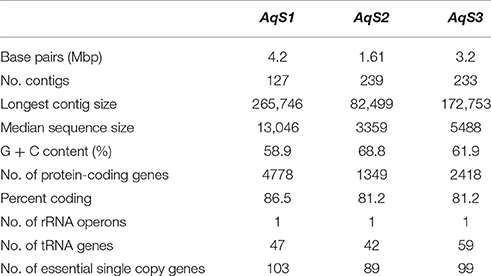
Table 1. Summary of genomic features for the three main symbionts present in Amphimedon queenslandica.
Phylogeny of the Three Main Phylotypes Present in Amphimedon queenslandica
Based on maximum-likelihood phylogenetic analysis of the 16S rRNA gene, the AqS1 symbiont clusters admist symbionts of various other sponge species (identity range 95–99%); sister to these is a small clade of marine environmental samples (Figure 1; Figure S3). These clades fall at the base of the family Ectothiorhodospiraceae, order Chromatiales (commonly known as sulfur-oxidizing bacteria). The closest cultured bacterial relatives (identity range 90–92%) are from Ectothiorhodospira and Thioalkalivibrio, both in the family Ectothiorhodospiraceae; the top BLAST NR hit was to the free-living Thioalkalivibrio paradoxus ARh 1. The AqS1 symbiont falls into the same sponge-specific clade as bacteria derived from the demosponges Callyspongia vaginalis and Gelliodes carnosa; also in this clade is a symbiont recently—but, we suggest, perhaps prematurely—named Thioalkalivibrio sp. HK1, whose genome was assembled from a metagenome, obtained from the sponge Haliclona cymaeformis (Tian et al., 2014). The other sponge-specific clade includes symbionts from a homoscleromorph (Plakortis halichondrioides) and several Demospongiae. For AqS1, we also performed phylogenetic analysis based on 30 concatenated essential marker genes, and including the genome of the close relative T. sp. HK1 plus a selection of 11 other publically-available genomes from free-living Chromatiales bacteria. In agreement with the 16S rRNA phylogenetic analysis, both the AqS1 and the HK1 sponge symbionts were recovered in a cluster of bacterial species belonging mostly to the family Ectothiorhodospiraceae (data not shown).
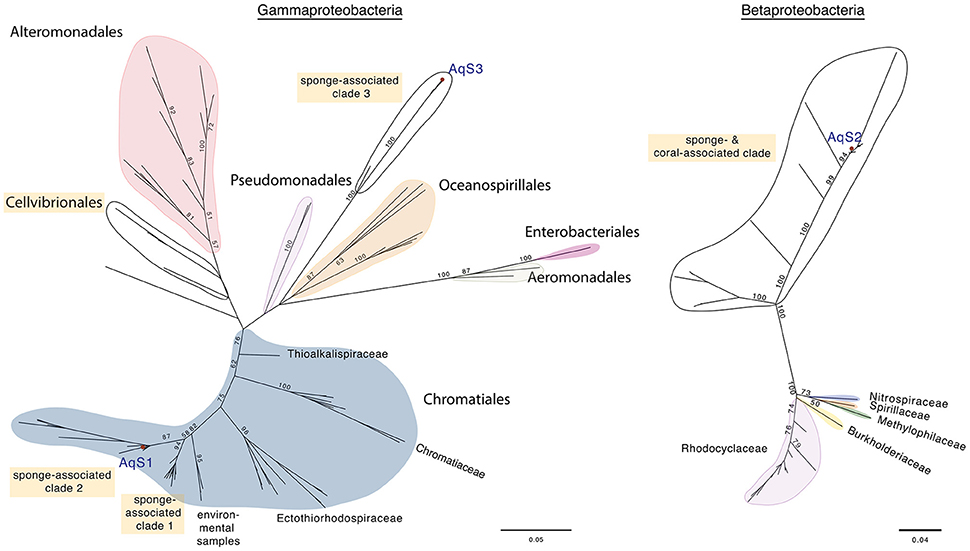
Figure 1. Maximum-likelihood phylogenetic trees for AqS1, AqS2, and AqS3. (A) The symbionts AqS1 and AqS3 are affiliated with the class Gammaproteobacteria. The AqS1 symbiont is recovered within the order Chromatiales in a sponge-specific clade; the nearest described species belong to the family Ectothiorhodospiraceae. The AqS3 symbiont clusters separately with bacterial clones derived from other demosponges in a clade located between the orders Pseudomonadales and Alteromonodales, but its phylogenetic placement is not well resolved. The detailed tree is presented in Figure S3. (B) The AqS2 symbiont sits within a large, well-supported sponge-specific cluster that is quite divergent from described families of the class Betaproteobacteria. The detailed tree is presented in Figure S4. The symbionts identified in A. queenslandica are indicated by a red dot and labeled as AqS1-3. Bootstrap values >50 are specified at major nodes.
The 16S rRNA sequence of the AqS2 symbiont forms part of a large, well-supported cluster of demosponge and one coral-derived bacteria (identity range 84–99%), that is quite divergent from recognized families within the class Betaproteobacteria. Similar to AqS1, the AqS2 sequence clades with bacteria whose genome was assembled from metagenomic data derived from C. vaginalis and G. carnosa, as well as another Amphimedon sp. from the Red Sea (Figure 1; Figure S4). The closest cultivated relatives of the 16S rRNA of AqS2 are from the genera Azoarcus and Thauera (identity range 82–84%), both in the family Rhodocyclaceae; the top BLAST NR free-living hit was to the Azoarcus sp. CIB, but only with 84% identity.
The 16S rRNA gene sequence of symbiont AqS3 shows highest percent sequence similarity to bacterial clones originating from several demosponges (identity range 84–99%). The closest described relative is the Chromatiales Thioprofundum hispidum strain gps61, but the identity score is low (86%). In our phylogenetic analysis, AqS3 clustered separately with gammaproteobacteria derived from other demosponges, but this apparently novel clade has low bootstrap support, and thus its placement within the gammaproteobacteria is not yet resolved (Figure 1, Figure S3).
The Microbial Community of Amphimedon queenslandica Is Stable and Clearly Dominated by AqS1 and AqS2
Since bacterial genomes can contain more than one copy of the 16S rRNA gene, the relative abundance of detected phylotypes from metagenomic DNA is potentially biased. We were interested in validating the binning strategy of our assembly by establishing whether the three assembled genomes would be also recovered as the most prevalent in the 16S rRNA pool, and whether any other prominent genome had not been accounted for. We also wanted to assess whether the overall composition of the microbial population found in the sponge A. queenslandica was broadly consistent across the six individual sponges that we sampled, and between the two different sampling seasons. It is also worth noting that we recovered the 16S rRNA gene in our single-copy gene list derived from our binned contigs for each of the three AqS genomes (Table S2).
Our 16S rRNA sequence reconstruction from the metagenomic Illumina data confirmed that the three symbionts for which we retrieved a genome assembly were also the most highly represented in the small subunit rRNA (ssrRNA) sequence pool derived from both separate and combined sponge metagenomic DNA samples (Table S3). In all of the individuals tested, and regardless of season, the microbiome was dominated either by the AqS1 or the AqS2 phylotype, which together represented as much as 90% of the overall microbial 16S rRNA pool. Reads matching to the ssrRNA sequence of the AqS3 symbiont were detected across all samples but recovered on average 20-fold less than those matching to the AqS1 phylotype. The remaining phylotypes that we detected all occurred at much lower abundance, with most displaying a relative abundance of <1% (Figure S5; Table S3), and could represent either rarer members of the A. queenslandica symbiont community or of the seawater, or contaminants introduced during sampling or processing. Because we could not discriminate between these scenarios, we do not discuss these further. Although we also recovered 16S rRNA sequences matching to members of the domain Archaea in three sponge individuals, these were from single pair-ended reads that could not be assembled into contigs.
We observed very little variation between the microbiome composition of individual sponges within a season; the two dominant phylotypes represented a similar proportion of the microbial community across sponge individuals. The AqS1 phylotype was more prevalent than the AqS2 phylotype in the samples collected in September, while the relative abundance of the two symbionts was more equivalent in those sponges collected in December, suggesting a slight seasonal variation (Figure S5; Table S3). Altogether, these data indicate that A. queenslandica individuals support a stable microbiome, the bulk of which comprises a coexisting gammaproteobacterium and betaproteobacterium. These observations are consistent with that reported in a companion paper, Fieth et al. (in review) in which OTU1 and OTU4 correspond to AqS1 and AqS2, respectively. The genome contents of both of these symbionts are described below.
The AqS1 Genome Encodes a Larger Gene Repertoire than the AqS2 Genome
A total of 4778 protein-encoding genes are predicted in the draft genome of AqS1, 54% of which are hypothetical at time of writing, while 29% are incorporated into 347 subsystems derived from the RAST server. The genome contains one copy of the 5S rRNA, the 16S rRNA, and the 23S rRNA genes – the latter bridging two scaffolds (scaffold 49 and 73), and 47 predicted transfer RNAs (tRNA). The gene repertoire of AqS1 is very similar to that of T. sp. HK1 (Figure 2; Table S4), and their contigs show high synteny (data not shown). Compared to AqS1, AqS2 displays a much smaller gene repertoire that reflects its reduced genome assembly size. A total of 1349 protein-encoding genes are predicted in the genome of AqS2, but only 22% of these are hypothetical, while 50% are incorporated into 244 subsystems. For AqS2, we recovered a single copy each of the 5S rRNA, the 16S rRNA, and the 23S rRNA genes, plus 42 putative tRNAs.

Figure 2. (A) Functional comparison between AqS1, three closest free-living and two symbiont sulfur-oxidizing strains using FIGfam-based subsystems derived from RAST annotations. THK1 Thioalkalivibrio sp. HK1 (isolated from the sponge Haliclona cymaeformis); Crm Candidatus Ruthia magnifica (isolated from the clam Calyptogena magnifica); Cvo Candidatus Vesicomyosocius okutanii (isolated from the clam Calyptogena okutanii); Tp Thioalkalivibrio paradoxus ARh 1; Tn Thioalkalivibrio nitroreducens; Ts Thioalkalivibrio sulfidophilus HL-EbGr7. Genes might contribute to more than one category. The detailed gene counts can be found in Table S4. (B) Number and functional categories of annotated genes present in AqS1 that are not recovered in T. paradoxus ARh 1, T. nitroreducens or T. sulfidophilus HL-EbGr7 (columns to the left). The FIGfam-based subcategories are further divided into subsystems (columns to the right). Numbers in brackets refer to the number of genes that had a top BLAST hit to an alphaproteobacterium.
The Symbiont AqS1 is a Putative Sulphur-Oxidising Bacterium
AqS1, along with its close relative T. sp. HK1 (Tian et al., 2014), show the highest phylogenetic similarity to Ectothiorhodospiraceae and like most bacteria belonging to this order, the two bacteria appear capable of sulfur oxidation. The genome of AqS1 encodes for a Sox enzyme complex (for periplasmic oxidation of various reduced sulfur compounds) coupled with a reverse dissimilatory sulfate reduction pathway (for sulfide oxidation in the cytoplasm) (Figure 3). The Sox pathway encoded by AqS1 is truncated (that is, it includes a soxBHXAYZ but no soxCD), as is the case for T. sp. HK1, although a point of difference is that no soxH gene was recovered in the latter. The Sox genes are spatially separated in the genome of AqS1, with soxB and soxH not occurring contiguously with soxAXYZ (Table S5). In the absence of the soxCD genes, obligatory intermediate sulfur globules are predicted to form as pathway byproducts, and these can be subsequently oxidized to sulfate by the dissimilatory sulfite reductase (dsr) pathway (Pott and Dahl, 1998). Homologues to the DsrABC proteins, as well as associated proteins needed to build the DsrMKOJP complex, are present in both the AqS1 and T. sp. HK1 genomes. In the current AqS1 assembly, the dsr genes are organized in a single polycistronic gene cassette (dsrABEFHCMKLJOP) that spans two contigs.
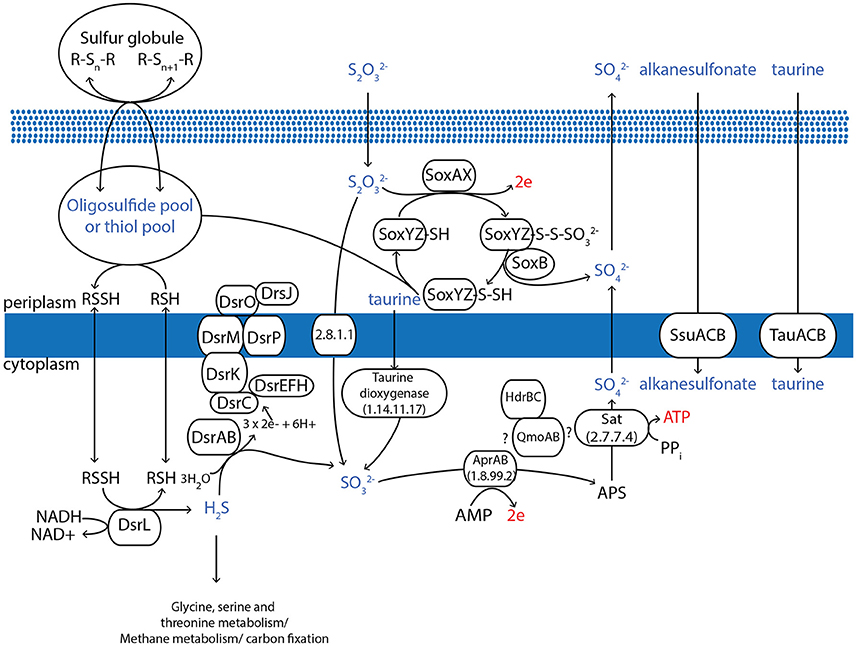
Figure 3. Overview of the proposed pathways for the oxidative sulfur metabolism of AqS1. All of the genes depicted in the figure are identifiable in the genome assembly. Specifically, AqS1 contains a Sox enzyme complex for periplasmic oxidation of reduced sulfur compounds, coupled with a reverse dissimilatory sulfate reduction pathway for sulfide oxidation in the cytoplasm. The AqS1 genome also includes other genes that can process extracellular taurine, alkanesulfonate and dimethylsulfoniopropionate, all of which can contribute to sulfide incorporation into carbon. Figure adapted from Frigaard and Dahl (2008).
AqS1 and T. sp. HK1 also encode a gene encoding for ATP sulphurylase (sat) and for adenosine-5′-phosphosulphate (APS) reductase (aprAB), both of which enable the conversion of sulfide to sulfate via APS. We did not detect the membrane-binding protein or the QmoABC complex that participate in the electron transfer during this conversion step in other bacteria (Frigaard and Bryant, 2008). However, the genomes of both these symbionts possess an operon-like gene cluster, adjacent to the sat gene, that comprises a qmoA—and a qmoB-like gene followed by a heterodisulphide reductase (hdr) C and hdrB gene (Figure 3; Table S5). This putative QmoAB-HdrCB protein complex could provide an alternative means of cytoplasmic electron transfer during sulfide oxidation, as described in another sulfur-oxidizing bacterium Thiolapillus brandeum (Nunoura et al., 2014).
Another enzyme involved in sulfur metabolism and that catalyzes the formation of sulfur globules is sulfide:quinone oxidoreductase, but we could not identify this gene in either of the AqS1 or the T. sp. HK1 genomes. However, we did detect the presence of a thiosulphate sulphurtransferase (rhodanese) gene and two rhodanese-related sulphurtransferases in both symbionts, with one copy located near the soxH gene in AqS1. The proteins encoded by these genes could participate in thiosulphate import to the cytoplasm (Aussignargues et al., 2012). Finally, both genomes include genes whose products can process extracellular taurine (i.e. tauA, B and C), alkanesulfonate (ssuB) and dimethylsulfoniopropionates (dmdA and dmdB), all of which can contribute to sulfide metabolism (Figure 3; Table S5).
AqS1 Has a Versatile, Heterotrophic Metabolism
Our RAST-based gene annotation analyses indicate that components of the Calvin-Benson-Bassham (CBB) pathway are present (Table S5), but both the AqS1 and the T. sp. HK1 genomes are missing homologs to key enzymes: (a) ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO) form I and II, and (b) others known to be essential in alternative carbonfixation routes (Hügler and Sievert, 2011).
To establish the heterotrophic capabilities of AqS1, we focussed on annotated genes found in the AqS1 symbiont but absent in its three closest free-living relatives, all of which are known to be obligate autotrophs (Figure 2B). From this list of 285 genes (86% of which are also present in the genome of T. sp. HK1), we recovered several genes capable of utilizing a wide range of sugars (monosaccharides, polysaccharides, organic acids and sugar alcohol subcategories) as well as amino acids and derivatives (e.g., polyamine, aromatic compound and creatine metabolism) as energy sources. We also found multiple genes encoding membrane transporters. These include four tripartite adenosine triphosphate (ATP)-independent periplasmic (TRAP) type transporters that are required for dicarboxylic acid import, as well as 29 tripartite adenosine triphosphate (ATP) binding cassette (ABC) transporter genes responsible for carbohydrate, amino acid and peptide uptake (Table S6A). Focusing on the high-affinity branched-chain amino acid ABC transporters, the AqS1 genome encodes multiple copies of the polycistronic LivFGHMK gene cassette, five of which are complete, and one of which is missing a livM homolog. Each cassette displays a different gene organization. Nonetheless, based on the transmembrane domain and protein domain predictions for each of the five complete cassettes, they all have the capacity to form functional transmembrane complexes (Figures 4A,B; Table S5). Finally, both D-octopine dehydrogenase and opine oxidases A, B and C are present in the genome of AqS1 (Table S5; Table S6A), which could enable AqS1 to use methyl-group donors like opines for carbon and nitrogen source. Therefore, as was observed for the symbiont T. sp. HK1 (Tian et al., 2014), the symbiont bacterium AqS1 harbors a large repertoire of genes that play a role in heterotrophic metabolism. This suggests that it might not need to rely on carbon fixation, as it is able to acquire and oxidize organic compounds.
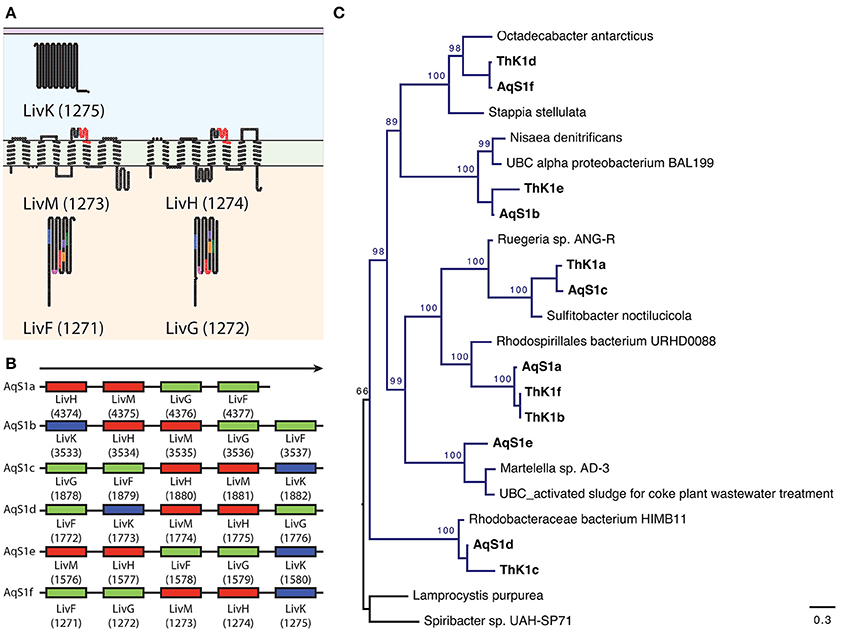
Figure 4. Repertoire of high-affinity branched-chain amino acid ATP binding cassette (A,B,C) transporters present in AqS1. The symbiont encodes multiple copies of the polycistronic LivFGHMK gene cassettes that are ATP-dependent high-affinity branched-chain amino acid transport systems involved in leucine metabolism and transport. (A) An example of putative 2D topology for one of the LivFGHMK transmembrane protein complex, derived using the program TOPO2. Conserved motifs are represented in different colors on the diagram. For LivF and LivG: blue AP-loop, pink Q-loop, red ATP transporter signature motif, orange Walker B-loop, purple D-loop, green, H-loop/switch region; For LivM and LivH: red TM-ABC transporter signature motif. The number in brackets corresponds to the RAST gene model id. (B) The AqS1 genome encodes six complete LivFGHMK gene cassettes; five of these cassettes are complete, while one – AqS1a – is missing a LivK homolog. Each cassette displays a different gene arrangement. (C) Maximum-likelihood phylogenetic tree for concatenated genes LivF, LivG, LivH, LivM, and LivK present within a polycistronic cassette for both AqS1 and T. sp. HK1. We used two Chromatiales representatives as outgroup. All cassettes nested with different representatives of the class Alphaproteobacteria, within the orders Rhodobacterales and Rhodospirilales. Bootstrapping percentages greater than 50% are shown.
As further evidence of the versatile metabolism of AqS1, we also found genes encoding small, medium and large subunits of the aerobic-type carbon monoxide (CO) dehydrogenase, a CO dehydrogenase F and a CO oxidation accessory protein (Table S5). These imply that the symbiont can generate reductive energy from the oxidation not only of sulfide, as described above, but also of CO. The RAST-based gene model predictions suggest that for catabolism, AqS1 is equipped with a complete Embden-Meyerhof-Parnas (glycolysis) pathway for the metabolism of hexose sugar, and a near-complete TCA cycle for the production of intermediates in cofactors and amino acid biosynthesis. A near complete pentose phosphate pathway is also present, providing a mechanism for production of NADPH and ribose sugars for nucleotide biosynthesis.
AqS1 is well equipped for phosphorus metabolism, including mechanisms to regulate the assimilation of inorganic phosphate (Pi). Specifically, the AqS1 genome encodes for components of the Phosphate (Pho) regulon, namely a Phosphate regulon sensor protein (PhoR) that detects levels of Pi in the environment, and downstream response genes (i.e., Phosphate regulon transcriptional regulatory protein [PhoB], Phosphate-starvation-inducible protein, predicted ATPase [PhoH], Phosphate transport system regulatory protein [PhoU], and two genes encoding response regulators in a two-component regulatory system) (Table S5). Also present is a gene encoding a polyphosphate kinase (EC 2.7.4.1) that catalyzes the formation of long-chain polyphosphate (polyP) (Table S5).
Finally, this symbiont has retained either partial or complete biosynthesis pathways for most essential amino acids (Figure 5A) and, like most bacteria, is capable of assimilating ammonia (genes encoding EC 1.7.1.4, EC 1.4.1.13, and EC 1.4.1.2 all are present) (Table S5). On the other hand, the absence of ureases and their associated proteins suggests that AqS1 is unable to use urea to cover its nitrogen needs. AqS1 also appears to lack a denitrification pathway; we could not detect genes encoding the intermediate enzymes of the anaerobic respiratory chain, nitrite reductase (NO forming) (EC 1.7.2.1) or nitric oxide reductase (EC 1.7.99.7).
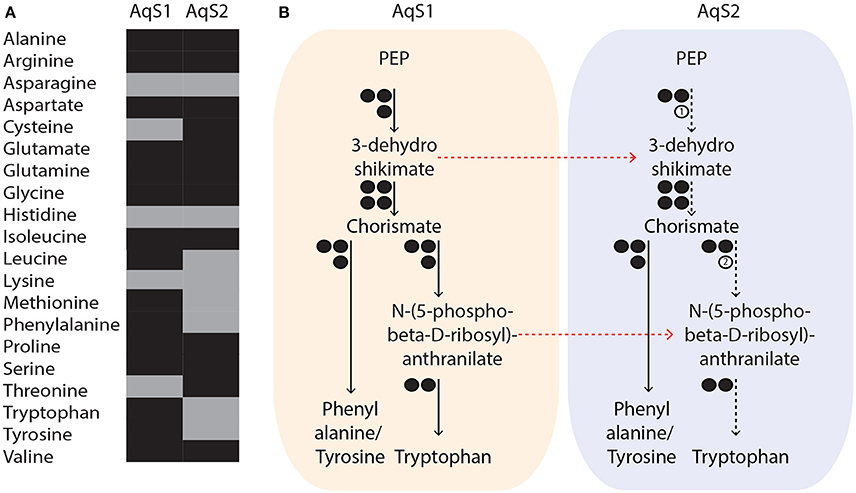
Figure 5. (A) Putative metabolic capacity of amino acid biosynthesis in AqS1 and AqS2. Amino acids are represented in different colors, depending on whether their biosynthesis pathway was predicted to be complete (black) or partially complete (gray) by the RAST server. (B) Example of possible metabolic complementation occurring between AqS1 and AqS2. AqS2 could rely on the metabolism of AqS1 to supply intermediate substrates that are required for the synthesis of aromatic amino acids. The number of genes involved in each pathway is shown as circles. Presence or absence of enzymes is indicated by black or white circles, respectively. (1) 3-dehydroshikimate and (2) N-(5-phospho-beta-D-ribosyl)-anthranilate. All paths that are partial are represented by dashed instead of solid arrows and red arrows indicate the possible movement of intermediary metabolites between the two symbionts. PEP: phosphoenolpyruvate.
There Is Evidence of Extensive Past Horizontal Gene Transfer in the AqS1 Genome
The identification of 285 genes found in the AqS1 symbiont, but not in its closest free-living relatives, led us to examine the origin of these genes. Interestingly, 136 of the 285 genes showed highest BLASTP similarity to various species in the class Alphaproteobacteria, with the order Rhodobacterales being the most represented (please refer to numbers that are reported in brackets in Figure 2B; Tables S7A,B). This suggests that the versatile metabolic capacity of the AqS1 symbiont could be, at least in part, a result of horizontal gene transfer (HGT) from one or more alphaproteobacteria. To further explore this possibility, we performed a phylogenetic analysis focusing on the multiple high-affinity branched amino acid ABC transporter protein cassettes found in both AqS1 and T. sp. HK1 (see above). These all clade with different representatives of Alphaproteobacteria, and specifically within the orders Rhodobacterales and the Rhodospirilales. This same association was also discussed by Tian et al. (2014) for T. sp. HK1 (Figure 4C).
Further support for an HGT origin, albeit not alone compelling, is that the AqS1 genome encodes genes implicated in HGT mechanisms, including four copies of the ISSpo3 transposase, mobile element proteins and phage-associated proteins (i.e., adenine DNA methyltransferase, phage-associated; putative prophage protein ps3). Genetic systems for conjugation and transformation were not detected, but DNA recombination and repair enzymes are present (i.e., Rec F, N, O and R), as well as protein families involved in general DNA-modifying activities (i.e., HNH endonuclease), and restriction-modification (R-M) and toxin–antitoxin systems (Type I, Type II, and Type III R-M systems, proteins from the Doc/Phd family, and VapI of the HigAB system) (Table S5).
Interestingly, mechanisms to control excessive genetic exchange and to minimize the introduction of foreign DNA are also encoded by the AqS1 genome. Single clustered, regularly interspaced, short, palindromic repeats (CRISPRs) and their associated proteins form adaptive immunity systems that occur in most archaea and many bacteria and act against invading genetic elements, such as viruses and plasmids (Makarova et al., 2011). A single CRISPR is found in the genome of AqS1. It is composed of at least fifteen direct repeat regions that are each 36 nucleotides in length, separated by at least thirteen spacers that are each 30 nucleotides in length. A gap present in our assembly, which bridges the first recovered spacer at the 5′ end of the predicted CRISPR, precludes the recovery of its leader sequence. The AqS1 CRISPR system has a canonical genomic arrangement (Makarova et al., 2011), with CRISPR-associated (CAS) genes (Csn1 and Cas1) located upstream of and directly adjacent to the CRISPR array (Figure S6; Table S5).
AqS2 Also Has a Versatile Metabolism
Our RAST-based gene annotation analyses recovered complete or near-complete key functional pathways in AqS2, despite its smaller genome assembly size. These included glycolysis, the TCA cycle and the pentose phosphate pathway. This symbiont does not seem to have nitrogen reduction, carbon fixation or sulfur oxidation capacity and, in stark contrast to the AqS1 genome, there are no genes associated with phosphorus metabolism. However, AqS2 does possess a gene operon that includes the three subunits of carbon monoxide (CO) dehydrogenase form I, D and F, and CO oxidation accessory protein (CoxG), suggesting that, like AqS1, it can generate reductive energy from the oxidation of CO. AqS2 might also be capable of assimilating ammonia, as it encodes NADP-specific glutamate dehydrogenase (EC 1.4.1.4) (Table S5). It also (like AqS1) has retained partial or complete biosynthesis pathways for most essential amino acids (Figure 5A), and genes involved in carbohydrate metabolism and membrane transport are also present, indicating AqS2 has heterotrophic abilities (Table S7). On the other hand, the genome of this symbiont does not show any evidence of past horizontal gene transfer (including no transposable insertion elements, or CRISPR-Cas resistance systems). We did recover genes involved in protection against foreign DNA (e.g., R-M systems) and cellular stress response (e.g., chaperones, membrane proteases) (Table S5).
Evidence of Resource Partitioning and Gene Repertoire Complementarity between AqS1 and AqS2
It has been postulated that co-occurring symbionts avoid competition by utilizing different carbon and energy sources (Duperron et al., 2006; Kleiner et al., 2012a). To establish preliminary evidence of resource partitioning between AqS1 and AqS2, we scanned for differences in their overall repertoire of predicted proteins involved in carbohydrate metabolism and transport. AqS1 encodes a wider range of membrane transporters than AqS2 (e.g., ABC transporter branched-chain amino acid, ABC transporter dipeptide, ABC transporter tungstate, copper transport system) (Table S7), and a genome-wide comparison clearly shows that each symbiont is equipped with a unique suite of proteins that would permit them to import or export different substrates (Figure 6). These include sulfur-related compounds, ammonium, vitamin B12, putrescine, spermidine, glutamate, aspartate, L-proline, glycine betaine, benzoate and chromate in AqS1; and biotin, methionine and manganese in AqS2.

Figure 6. Counts of putative substrates derived from predicted transporters present in the genome of AqS1 only (blue), AqS2 only (red), or both (black).
Differences also exist in the means by which the two bacteria exploit carbohydrates (Table S7). AqS2 can utilize N-acetylglucosamine and chitin, both of which are important sources of carbon and nitrogen for marine organisms (Driscoll et al., 2007; Souza et al., 2011). It is also predicted to metabolize and catabolise a larger number of monosaccharides than AqS1, with subcategories, such as D-galactonate catabolism, D-gluconate and ketogluconates metabolism, deoxyribose and deoxynucleoside catabolism, mannose metabolism and xylose utilization being only recovered in AqS2. On the other hand, AqS1 is able to use the organic acid tricarballylate, which is a citrate analog and a potential source of carbon and energy (Gutnick et al., 1969) (Table S7).
While both the AqS1 and AqS2 genomes have the capacity to independently synthesize de novo most amino acids, they also lack key enzymes required for the production of others (Figure 5A). For instance, 3-dehydroquinate dehydratase (EC 4.2.1.10), an enzyme-encoding gene that participates in the production of aromatic acids (phenylalanine, tyrosine and tryptophan), is not present in the AqS2 genome. AqS2 also lacks a homolog for phosphoribosylanthranilate isomerase (EC 5.3.1.24) that catalyzes the third step of tryptophan synthesis. Interestingly, however, these missing genes are present in the genome of AqS1, suggesting that AqS2 could synthezise all three aromatic acids provided that AqS1 metabolism supplies the intermediate substrates, 3-dehydroshikimate and N-(5-phospho-beta-D-ribosyl)-anthranilate (Table S5; Figure 5B). The alternative—that AqS2 could could directly take up the synthesized amino acid provided by AqS1—is not supported, because we are unable to detect transporters for phenalanine, tryptophan or tyrosine amongst the predicted proteins of AqS2. These observations suggest that both essential and non-essential amino acid synthesis could depend on within-pathway complementation between the two symbionts.
The Gene Repertoire Reflects a Symbiotic Lifestyle for Both AqS1 and AqS2
Sponge-associated bacteria are predicted to use characteristic eukaryote-like domain-containing proteins to facilitate host-microbe interactions; these include ankyrin-repeats, tetratricopeptide-repeats, leucine-rich repeats, fibronectin domain IIIs, and cadherins (Thomas et al., 2010; Fan et al., 2012; Hentschel et al., 2012). Our comparison of AqS1 with its free-living relatives provided support for this prediction in that the genome of AqS1 is enriched in ankyrin-repeat containing proteins (Figure 7); a similar trend was reported for T. sp. HK1 (Tian et al., 2014). On the other hand, the AqS2 genome does not show enrichment for any of these domains (Figure 7). There is little evidence of virulence factors in either of the symbiont genomes; they share just a small number of predicted protein-coding genes conferring resistance to copper, cobalt-zinc-cadmium, and/or virulence. In AqS1, the predicted proteins belonging to the virulence resistance category are involved in the production of bacteriocin Colicin V, universal stress proteins, such as heat shock protein UspA, as well as PotD, PotA and PotH. Resistance to chromium is predicted only in AqS1 (Table S5).
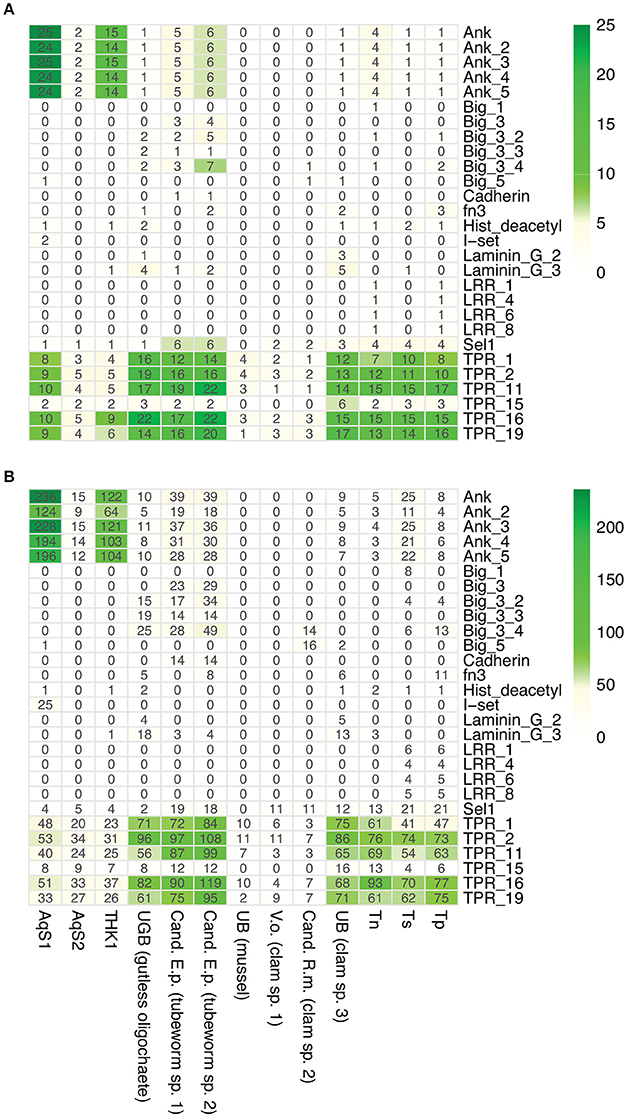
Figure 7. Occurrence of eukaryotic-like containing proteins in AqS1 and AqS2. (A) Counts of eukaryotic-like containing proteins. (B) Counts of eukaryotic domains. THK1 Thioalkalivibrio sp. HK1; UGB (gutless oligochaete) Gamma3 isolated from Olavius algarvensis JGI genome accession number 2004178004; Cand. E.p. (tubeworm sp. 1) Candidatus Endoriftia persephone isolated from Riftia pachyptila, accession number AFOC00000000; Cand. E.p. (tubeworm sp. 2) Candidatus E. persephone isolated from Tevnia jerichonana, accession number AFZB00000000; UB (mussel) Unknown thiotroph isolated from Bathymodiolus sp., accession number CAEB01000001; V.o. (clam sp. 1) Candidatus Vesicomyosocius okutanii isolated from Calyptogena okutanii, accession number NC_009465.1; Cand. R.m. (clam sp. 2) Candidatus Ruthia magnifica isolated from Calyptogena magnifica, accession number NC_008610.1; UB (clam sp. 3) Unknown symbiont isolated from Solemya velum, accession number NZ_JRAA00000000.1; Tn Thioalkalivibrio nitroreducens; Ts Thioalkalivibrio sulfidophilus HL-EbGr7; Tp Thioalkalivibrio paradoxus ARh 1.
Finally, AqS1 and AqS2 have the genomic capacity to produce products that could be beneficial to the host, although it must be noted that many non-symbiotic bacteria share the same capacity. For instance, the AqS1 genome encodes genes associated with the synthesis of coenzyme A, as well as the essential pyrophosphate (vitamin B1), which animals must obtain from their diet (Begley et al., 2008) (Table S5). The ability to synthezise vitamin B1 has also been observed in other sponge associated bacteria and archaea (Fan et al., 2012). The two Aq symbionts also contain genes involved in pyridoxine (a form of vitamin B6), menaquinone/phylloquinone and folate biosynthesis. AqS2 is additionally equipped with genes that are involved in biotin (vitamin B7) synthesis (Table S5).
Discussion
Here we show that the coral reef demosponge Amphimedon queenslandica supports a microbiota dominated by two species of proteobacteria, and present the first draft genomes of both. These “core” bacterial symbionts are stable across individuals and across seasons, and represent the bulk of the microbiome, in sponge hosts sampled from Heron Island reef in the southern Great Barrier Reef of Australia (see Fieth et al., in review). The most prevalent symbiont, AqS1, is a putative sulfur-oxidizing gammaproteobacterium of the order Chromatiales, and the second most prevalent symbiont, AqS2, is an unknown betaproteobacterium whose closest relatives also include other sponge symbionts.
The co-occurrence of bacteria genetically related to AqS1, AqS2, and AqS3 in Calyspongia vaginalis (Giles et al., 2012), and to AqS1 and AqS2 in Haliclona simulans (Kennedy et al., 2008), suggests that they could have undergone long-term codiversification with their hosts, as has been demonstrated in other systems involving multiple stable symbionts within a specific host (Dubilier et al., 2001, 2008; Takiya et al., 2006; Zimmermann et al., 2014). Because of limitations with our AqS3 genome assembly, we choose not to discuss its gene repertoire; thus the remainder of this discussion focuses solely on AqS1 and AqS2.
Sulfur-oxidizing symbioses are found in various marine habitats ranging from shallow water sediments, organic rich mud and mangrove peat, to deep-sea hydrothermal vents and cold seeps. These associations rely on sulfide produced either geothermally or biologically, and have evolved independently numerous times in phylogenetically diverse hosts (Dubilier et al., 2008). Several lines of evidence point to their widespread occurrence in marine sponges. Our own phylogenetic analysis confirm that bacteria belonging to the family Ectothiorhodospiraceae within the order Chromatiales, and closely related to AqS1 and T. sp. HK1, have been reported in taxonomically distant sponges that live at varying latitudes and depths (Taylor et al., 2007b; Jensen et al., 2008; Kennedy et al., 2008; Giles et al., 2012; Radax et al., 2012; Karlinska-Batres and Wörheide, 2013; Pita et al., 2013; Olson et al., 2014; Della Sala et al., 2014). Furthermore, other studies on sponge symbionts have recorded the presence of both known and novel Chromatiales representatives belonging to either the Ectothiorhodospiraceae or Chromatiaceae family (Imhoff and Trüper, 1976; White et al., 2012), as well as sulfur-oxidizing bacteria from the Alphaproteobacteria class (Rhodospirillaceae, Rhodobacteraceae) and the phylum Chlorobi (Imhoff and Trüper, 1976; Bondarev et al., 2013).
The Chromatiales are generally lithotrophs, capable of synthesizing carbohydrates from the fixation of carbon dioxide using sulfur oxidation as an energy source (Grimm et al., 2008; Frigaard and Dahl, 2008; Ghosh and Dam, 2009). The presence of a putative Sox enzyme complex and a reverse dissimilatory sulfate reduction pathway in the genome of AqS1 suggests that this symbiont, like its close relative T. sp. HK1, does indeed have the ability to oxidize sulfur. Further evidence of sulfur oxidation has been obtained via transmission electron microscopy, with the detection of pseudo-spherical, membrane-bound vacuoles in AqS1 cells (Fieth et al., in review); these vacuoles resemble commonly-found bacterial organelles that contain sulfur globules (Lawry et al., 1981; Robertson and Kuenen, 2006; Docampo et al., 2010).
The origin of the reduced sulfur compounds that would be required by AqS1 for oxidation is unknown at this stage. Sulphur metabolism has been detected previously in several marine sponge species, and sulfate-reducing bacteria—sometimes co-occurring with sulfur oxidisers—have been isolated from sponge tissue (Imhoff and Trüper, 1976; Manz et al., 2000; Hoffmann et al., 2005; Taylor et al., 2007b; Zhang et al., 2015a). An endosymbiotic sulfur cycle, coupling sulfate-reducing with sulfide-oxidizing bacteria, has been reported in a marine oligochaete (Dubilier et al., 2001) and Hoffmann et al. (2005) proposed that a similar process might take place in some sponges. Sulphides can be toxic at high concentrations, but sulfide-oxidizing bacteria, such as AqS1 could detoxify sulfides generated by sulfate-reducing bacteria in the sponge host body. Anoxic zones can form in sponge tissue, particularly during times of pumping inactivity, and would provide the right environment for sulfate-reducing bacteria (Hoffmann et al., 2005, 2008). Since A. queenslandica adults have the ability to close their oscula and thus stop pumping water for long period of times, both in natural and in laboratory environments, hypoxic pockets are also expected to form in its tissue; indeed, sites of reduced oxygen have been detected in A. queenslandica even while it continues to pump (J. Watson and J. Kroemer, personal observations). Reduced sulfur compounds might not be only symbiotic in origin, but could also be environmentally derived. The habitat of A. queenslandica on Heron Island reef is characterized by fine sand patches and often heavy siltation (Degnan et al., 2008). During times of high sand movement or high silt deposition, adult sponges can easily be partially buried to the extent that they may be subject to anoxic conditions with high sulfide concentrations. Together, these observations indicate a functional role for AqS1 in mediating sulfur nutrient cycling between host and symbiont.
Sulfur-oxidizing symbionts have varied metabolic capabilities, with some displaying obligate autotrophy while others can grow heterotrophically or mixotrophically (Kleiner et al., 2012a; Tian et al., 2014). We find no evidence for a complete carbon fixation mechanism in AqS1, although we acknowledge that this could be due to limitations of our current draft genome assembly. Nonetheless, it is also possible that the symbiont either fixes carbon using an unknown mechanism, or is a lithoheterotroph. Unlike its closest free-living relatives (Thioalkalivibrio paradoxus ARh 1, T. nitratireducens and T. sulfidophilus) that are obligate autotrophs, AqS1 harbors a large repertoire of genes involved in heterotrophic metabolism (e.g., carbohydrate uptake and metabolism, membrane transport), which would allow the symbiont to import and utilize various substrates. Thus, AqS1 might not need to rely on carbon fixation to acquire and oxidize organic compounds. For instance, the AqS1 genome is particularly rich in amino acid and oligopeptide transporters and, in combination with peptidases and proteases, these could permit the acquisition and hydrolysis of complex organic compounds for nutrition, as has been proposed by Bondarev et al. (2013). Another example is the presence of both D-octopine dehydrogenase and opine oxidases A, B and C, which could enable AqS1 to use methyl-group donors like opines for carbon and nitrogen source. The bacterium could produce this compound when maintaining redox balance under anaerobic conditions, and oxidize it when requiring energy (Strahl et al., 2011; van Os et al., 2012).
In oligotrophic marine habitats, such as coral reefs, phosphorous (P) can be one of the more limiting elements, particularly over longer time scales, and its biogeochemical cycle is intimately associated with the microbial community (Karl, 2014). Zhang et al. (2015b) recently reported a potentially important mode of P sequestration mediated by bacterial symbionts of marine sponges that likely use polyphosphate kinase (ppk) to catalyze the formation of long-chain polyphosphate (polyP). We find a ppk gene in the genome of AqS1, suggesting that it could have the same capability, and supporting the hypothesis of Zhang et al. (2015b) that oscillation between anoxic and oxic states inside sponges could select for polyP-accumulating organisms within the symbiotic community. In fact, AqS1 is well equipped for phosphorus metabolism, with the ability to detect environmental levels of inorganic phosphate and accordingly regulate its assimilation as necessary, using a phosphate regulon system (Santos-Beneit, 2015).
Heterotrophy in general has been reported in several other sponge symbionts (Thomas et al., 2010; Siegl et al., 2011; Fan et al., 2012; Bondarev et al., 2013; Tian et al., 2014). Interestingly, a large proportion of genes that endow the AqS1 symbiont with heterotrophic capabilities are most closely related to genes of several alphaproteobacteria. Our phylogenetic analysis of the high-affinity branched amino acid ABC transporter protein family present in both AqS1 and T. sp. HK1, for example, strongly supports a hypothesis that these genes have been acquired via one or multiple horizontal gene transfers (HGT). Kleiner et al. (2012a) highlighted the metabolic similarities and differences between several sulfur oxidizing gammaproteobacterial symbionts and also speculated that HGT events might play an important role in the metabolic evolution of these microbes. However, it is unknown whether these events occurred before or after the establishment of the symbioses, and thus if the acquisition of certain gene sets by the microorganism was critical for the establishment of these associations and for their subsequent evolutionary success.
Based on its genome assembly, the betaproteobacterium AqS2 is predicted to have a smaller genome compared to AqS1, and, as a result, a reduced functional toolkit. Genome streamlining reported in cyanobacterial symbionts of other sponges has been postulated to reflect adaptation to the mild intercellular environment present in the host (Gao et al., 2014; Burgsdorf et al., 2015). Despite its predicted reduced genome size, AqS2 encodes genes required for heterotrophic metabolism, for transport of various substrates and for amino acid and vitamin biosynthesis. Thus, it seems to have retained genes that could be considered to play a major nutritional role for the sponge host, a feature that might be fundamental for its maintenance as a symbiont over evolutionary time.
Previous global metagenomic analyses of sponge microbiomes have provided evidence for functional genomic signatures that separate them from bacterial communities in the surrounding seawater (Thomas et al., 2010; Fan et al., 2012; Hentschel et al., 2012; Liu et al., 2012). Some of the signatures recovered in both AqS1 and AqS2 genomes are versatile nutrient utilization (e.g., carbon and nitrogen metabolism, amino acid biosynthesis, transporters), protection against foreign DNA (e.g., CRISPR in AqS1, restriction-modification systems in both microorganisms), response to cellular stress (e.g., chaperones, membrane proteases) and a deficiency in virulence factors. Other features recovered in AqS1, but not in AqS2, include evidence of past genetic transfer (e.g., transposases; mobile elements) and possible mechanisms for evading host detection (e.g., ankyrin-repeat proteins). While the predicted reduced size of its genome might be one of the reasons why these features were not detected in AqS2, this is inconsistent with these same features being present in other small-sized cyanobacterial genomes (Burgsdorf et al., 2015), and thus other factors are likely to play a role.
The existence of stable associations involving two or more symbionts has been reported in several non-sponge animal hosts (Takiya et al., 2006; Wu et al., 2006; Dubilier et al., 2008; Zimmermann et al., 2014). In such associations, the microbes are expected to use different carbon and energy sources to minimize competition (Duperron et al., 2006; Kleiner et al., 2012b). In A. queenslandica, the bacterium AqS1 has a larger gene repertoire than AqS2, as well as a more versatile metabolism, and is therefore predicted to employ feeding strategies that differ from AqS2 (e.g., sulfide oxidation, wider range of transporters). We also established that AqS1 and AqS2 differed in the ways they use carbohydrates. For example, AqS2 can utilize N-acetylglucosamine or chitin, as well as a larger number of monosaccharide substrates than AqS1, while AqS1 is able to use the organic acid tricarballylate. As yet another contrast, AqS1 has extensive capabilities for phosphorus metabolism, while AqS2 has none at all. Together, these data strongly suggest that resource partitioning does indeed occur between the two most prevalent bacterial symbionts of A. queenslandica.
Finally, within-pathway complementation might exist between AqS1 and AqS2, as has also been reported in other (non-sponge) symbionts (Wu et al., 2006; Rao et al., 2015). For example, enzyme-encoding genes that participate in the production of aromatic amino acids are present in the AqS1, but not in the AqS2, genome, suggesting that each symbiont could synthesize particular amino acids provided that the metabolism of the other supplies the intermediate substrates. It has been postulated that metabolic redundancies existing within symbiotic partners upon host initial acquisition might be lost over time, resulting in mutual dependence to produce compounds essential to their host (Wu et al., 2006; Rao et al., 2015). Alternatively, the function of the missing genes could be performed by other genes encoded in their respective genomes, either by a novel enzyme, or by an already-known enzyme that would have changed or extended its prior function by co-option (Charles et al., 2011; Sloan and Moran, 2012; Rao et al., 2015).
Conclusions and Future Directions
Here we present draft genomes for the two dominant bacterial symbionts found in the coral reef demosponge A. queenslandica, and discuss evolutionary and functional insights based on predicted gene repertoires. These symbiont genomes complement extensive genomic and transcriptomic resources already existing for the sponge host. With these tools in hand, investigations are now underway to (i) construct genome-scale models to detail the metabolic interplay between sponge and symbionts, and (ii) characterize mechanisms of molecular crosstalk that regulate establishment and maintenance of the host-symbiont relationships (Hentschel et al., 2012, Fieth et al., in review). Thus, the A. queenslandica holobiont is poised to become a fruitful model for understanding the nature and consequences of interactions between animal hosts and their bacterial symbionts.
Author Contributions
SD and MG designed the research. JW prepared the biological material and assisted in early stages of analysis. MG analyzed the data. MG and SD wrote the paper. All authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the University of Queensland's Heron Island Research Station for providing facilities for sample collection. We thank Selene Fernandez-Valverde, Kevin Kocot, Mads Albertsen, and Yuki Sekiguchi for helpful bioinformatics advice, Rebecca Fieth for her insights on Amphimedon bacterial symbionts, and the Research Computing Centre and QCIF for provision of computing facilities. This research was supported by grant DP110104601 from the Australian Research Council to SD.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fmars.2016.00196
References
Aberer, A. J., Krompass, D., and Stamatakis, A. (2013). Pruning rogue taxa improves phylogenetic accuracy: an efficient algorithm and webservice. Syst. Biol. 62, 162–166. doi: 10.1093/sysbio/sys078
Albertsen, M., Hugenholtz, P., Skarshewski, A., Nielsen, K. L., Tyson, G. W., and Nielsen, P. H. (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31, 533–538. doi: 10.1038/nbt.2579
Anavy, L., Levin, M., Khair, S., Nakanishi, N., Fernandez-Valverde, S. L., Deqnan, B. M., et al. (2014). BLIND ordering of large-scale transcriptomic developmental timecourses. Development 141, 1161–1166. doi: 10.1242/dev.105288
Arillo, A., Bavestrello, G., Burlando, B., and Sara, M. (1993). Metabolic integration between symbiotic cyanobacteria and sponges: a possible mechanism. Mar. Biol. 117, 159–162.
Aussignargues, C., Giuliani, M. C., Infossi, P., Lojou, E., Guiral, M., Giudici-Orticoni, M. T., et al. (2012). Rhodanese functions as sulfur supplier for key enzymes in sulfur energy metabolism. J. Biol. Chem. 287, 19936–19948. doi: 10.1074/jbc.M111.324863
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Begley, T. P., Chatterjee, A., Hanes, J. W., Hazra, A., and Ealick, S. E. (2008). Cofactor biosynthesis–still yielding fascinating new biological chemistry. Curr. Opin. Chem. Biol. 12, 118–125. doi: 10.1016/j.cbpa.2008.02.006
Bell, J. J. (2008). The functional roles of marine sponges. Estuar. Coast. Shelf Sci. 79, 341–353. doi: 10.1016/j.ecss.2008.05.002
Bengtsson, J., Eriksson, K. M., Hartmann, M., Wang, Z., Shenoy, B. D., Grelet, G. A., et al. (2011). Metaxa: a software tool for automated detection and discrimination among ribosomal small subunit (12S/16S/18S) sequences of archaea, bacteria, eukaryotes, mitochondria, and chloroplasts in metagenomes and environmental sequencing datasets. Antonie Van Leeuwenhoek 100, 471–475. doi: 10.1007/s10482-011-9598-6
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bondarev, V., Richter, M., Romano, S., Piel, J., Schwedt, A., and Schulz-Vogt, H. N. (2013). The genus Pseudovibriocontains metabolically versatile bacteria adapted for symbiosis. Environ. Microbiol. 15, 2095–2113. doi: 10.1111/1462-2920.12123
Britstein, M., Devescovi, G., Handley, K. M., Malik, A., Haber, M., Saurav, K., et al. (2016). A new N-Acyl homoserine lactone synthase in an uncultured symbiont of the Red Sea sponge Theonella swinhoei. Appl. Environ. Microbiol. 82, 1274–1285. doi: 10.1128/AEM.03111-15
Burgsdorf, I., Slaby, B. M., Handley, K. M., Haber, M., Blom, J., Marshall, C. W., et al. (2015). Lifestyle evolution in cyanobacterial symbionts of sponges. MBio 6, e00391–e00315. doi: 10.1128/mBio.00391-15
Caporaso, J. G., Bittinger, K., Bushman, F. D., DeSantis, T. Z., Andersen, G. L., and Knight, R. (2010a). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267. doi: 10.1093/bioinformatics/btp636
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7, 335–336. doi: 10.1038/nmeth.f.303
Charles, H., Balmand, S., Lamelas, A., Cottret, L., Pérez-Brocal, V., Burdin, B., et al. (2011). A genomic reappraisal of symbiotic function in the aphid/Buchnera symbiosis: reduced transporter sets and variable membrane organisations. PLoS ONE 6:e29096. doi: 10.1371/journal.pone.0029096
Colman, A. S. (2015). Sponge symbionts and the marine P cycle. Proc. Natl. Acad. Sci. U.S.A. 112, 4191–4192. doi: 10.1073/pnas.1502763112
Conaco, C., Neveu, P., Zhou, H., Arcila, M. L., Degnan, S. M., Degnan, B. M., et al. (2012). Transcriptome profiling of the demosponge Amphimedon queenslandica reveals genome-wide events that accompany major life cycle transitions. BMC Genomics 13:209. doi: 10.1186/1471-2164-13-209
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
Degnan, B. M., Adamska, M., Craigie, A., Degnan, S. M., Fahey, B., Gauthier, M., et al. (2008). The demosponge Amphimedon queenslandica: reconstructing the ancestral metazoan genome and deciphering the origin of animal multicellularity. CSH Protoc. 2008:pdb.emo108. doi: 10.1101/pdb.emo108
De Goeij, J. M., Moodley, L., Houtekamer, M., Carballeira, N., and Van Duyl, F. C. (2008a). Tracing 13C-enriched dissolved and particulate organic carbon in the bacteria-containing coral reef sponge Halisarca caerulea: evidence for DOM-feeding. Limnol. Oceangr. 53, 1376–1386. doi: 10.4319/lo.2008.53.4.1376
De Goeij, J. M., van den Berg, H., van Oostveen, M. M., Epping, E., and Van Duyl, F. C. (2008b). Major bulk dissolved organic carbon (DOC) removal by encrusting coral reef cavity sponges. Mar. Ecol. Prog. Ser. 357, 139–151. doi: 10.3354/meps07403
de Goeij, J. M., van Oevelen, D., Vermeij, M. J. A., Osinga, R., Middelburg, J. J., de Goeij, A. F. P. M., et al. (2013). Surviving in a marine desert: the sponge loop retains resources within coral reefs. Science 342, 108–110. doi: 10.1126/science.1241981
Della Sala, G., Hochmuth, T., Teta, R., Costantino, V., and Mangoni, A. (2014). Polyketide synthases in the microbiome of the marine sponge Plakortis halichondrioides: a metagenomic update. Mar. Drugs 12, 5425–5440. doi: 10.3390/md12115425
Docampo, R., Ulrich, P., and Moreno, S. N. J. (2010). Evolution of acidocalcisomes and their role in polyphosphate storage and osmoregulation in eukaryotic microbes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 365, 775–784. doi: 10.1098/rstb.2009.0179
Driscoll, M. E., Romine, M. F., Juhn, F. S., Serres, M. H., McCue, L. A., Beliaev, A. S., et al. (2007). Identification of diverse carbon utilization pathways in shewanella oneidensis MR-1 via expression profiling. Genome Inform. 18, 287–298.
Dubilier, N., Bergin, C., and Lott, C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Micro. 6, 725–740. doi: 10.1038/nrmicro1992
Dubilier, N., Mülders, C., Ferdelman, T., de Beer, D., Pernthaler, A., Klein, M., et al. (2001). Endosymbiotic sulphate-reducing and sulphide-oxidizing bacteria in an oligochaete worm. Nature 411, 298–302. doi: 10.1038/35077067
Duperron, S., Bergin, C., Zielinski, F., Blazejak, A., Pernthaler, A., McKiness, Z. P., et al. (2006). A dual symbiosis shared by two mussel species, Bathymodiolus azoricus and Bathymodiolus puteoserpentis (Bivalvia: Mytilidae), from hydrothermal vents along the northern Mid-Atlantic Ridge. Environ. Microbiol. 8, 1441–1447. doi: 10.1111/j.1462-2920.2006.01038.x
Erwin, P. M., Pita, L., López-Legentil, S., and Turon, X. (2012). Stability of sponge-associated bacteria over large seasonal shifts in temperature and irradiance. Appl. Environ. Microbiol. 78, 7358–7368. doi: 10.1128/aem.02035-12
Fan, L., Reynolds, D., Liu, M., Stark, M., Kjelleberg, S., Webster, N. S., et al. (2012). Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. U.S.A. 109, E1878–E1887. doi: 10.1073/pnas.1203287109
Fernandez-Valverde, S. L., Calcino, A. D., and Degnan, B. M. (2015). Deep developmental transcriptome sequencing uncovers numerous new genes and enhances gene annotation in the sponge Amphimedon queenslandica. BMC Genomics 16:387. doi: 10.1186/s12864-015-1588-z
Fiore, C. L., Baker, D. M., and Lesser, M. P. (2013). Nitrogen biogeochemistry in the Caribbean sponge, Xestospongia muta: a source or sink of dissolved inorganic nitrogen? PLoS ONE 8:e72961. doi: 10.1371/journal.pone.0072961
Fiore, C. L., Labrie, M., Jarett, J. K., and Lesser, M. P. (2015). Transcriptional activity of the giant barrel sponge, Xestospongia muta Holobiont: molecular evidence for metabolic interchange. Front. Microbiol. 6:364. doi: 10.3389/fmicb.2015.00364
Frigaard, N.-U., and Bryant, D. A. (2008). “Genomic and evolutionary perspectives on sulfur metabolism in green sulfur bacteria,” in Microbial Sulfur Metabolism, eds C. Dahl and C. G. Friedrich (Berlin; Heidelberg: Springer), 60–76.
Frigaard, N.-U., and Dahl, C. (2008). Sulfur metabolism in phototrophic sulfur bacteria. Adv. Microb. Physiol. 54, 103–200. doi: 10.1016/S0065-2911(08)00002-7
Gao, Z. M., Wang, Y., Tian, R. M., Wong, Y. H., Batang, Z. B., Al-Suwailem, A. M., et al. (2014). Symbiotic adaptation drives genome streamlining of the cyanobacterial sponge symbiont “Candidatus Synechococcus spongiarum.” mBio 5, e00079-14. doi: 10.1128/mBio.00079-14
Ghosh, W., and Dam, B. (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 33, 999–1043. doi: 10.1111/j.1574-6976.2009.00187.x
Giles, E. C., Kamke, J., Moitinho-Silva, L., Taylor, M. W., Hentschel, U., Ravasi, T., et al. (2012). Bacterial community profiles in low microbial abundance sponges. FEMS Microbiol. Ecol. 83, 232–241. doi: 10.1111/j.1574-6941.2012.01467.x
Grimm, F., Franz, B., and Dahl, C. (2008). “Thiosulfate and sulfur oxidation in purple sulfur bacteria,” in Microbial Sulfur Metabolism, eds C. Dahl and C. G. Friedrich (Berlin; Heidelberg: Springer), 101–116.
Gutnick, D., Calvo, J. M., Klopotowski, T., and Ames, B. N. (1969). Compounds which serve as the sole source of carbon or nitrogen fro Salmonella typhimurium LT-2. J. Bacteriol. 100, 215–219.
Hentschel, U., Piel, J., Degnan, S. M., and Taylor, M. W. (2012). Genomic insights into the marine sponge microbiome. Nat. Rev. Micro. 10, 641–654. doi: 10.1038/nrmicro2839
Hoffmann, F., Larsen, O., Thiel, V., Rapp, H. T., Pape, T., Michaelis, W., et al. (2005). An anaerobic world in sponges. Geomicrobiology 22, 1–10. doi: 10.1080/01490450590922505
Hoffmann, F., Radax, R., Woebken, D., Holtappels, M., Lavik, G., Rapp, H. T., et al. (2009). Complex nitrogen cycling in the sponge Geodia barretti. Environ. Microbiol. 11, 2228–2243. doi: 10.1111/j.1462-2920.2009.01944.x
Hoffmann, F., Roy, H., Bayer, K., Hentschel, U., Pfannkuchen, M., Brümmer, F., et al. (2008). Oxygen dynamics and transport in the Mediterranean sponge Aplysina aerophoba. Mar. Biol. 153, 1257–1264. doi: 10.1007/s00227-008-0905-3
Huang, X., and Madan, A. (1999). CAP3: a DNA sequence assembly program. Genome Res. 9, 868–877. doi: 10.1101/gr.9.9.868
Hügler, M., and Sievert, S. M. (2011). Beyond the Calvin cycle: autotrophic carbon fixation in the ocean. Annu. Rev. Marine. Sci. 3, 261–289. doi: 10.1146/annurev-marine-120709-142712
Imhoff, J. F., and Trüper, H. G. (1976). Marine sponges as habitats of anaerobic phototrophic bacteria. Microb. Ecol. 3, 1–9. doi: 10.1007/BF02011449
Jensen, S., Neufeld, J. D., Birkeland, N.-K., Hovland, M., and Murrell, J. C. (2008). Insight into the microbial community structure of a Norwegian deep-water coral reef environment. Deep Ses Res. I 55, 1554–1563. doi: 10.1016/j.dsr.2008.06.008
Kamke, J., Sczyrba, A., Ivanova, N., Schwientek, P., Rinke, C., Mavromatis, K., et al. (2013). Single-cell genomics reveals complex carbohydrate degradation patterns in poribacterial symbionts of marine sponges. ISME J. 7, 2287–2300. doi: 10.1038/ismej.2013.111
Karl, D. M. (2014). Microbially mediated transformations of phosphorus in the sea: new views of an old cycle. Annu. Rev. Marine. Sci. 6, 279–337. doi: 10.1146/annurev-marine-010213-135046
Karlinska-Batres, K., and Wörheide, G. (2013). Phylogenetic diversity and community structure of the symbionts associated with the coralline sponge Astrosclera willeyana of the Great Barrier Reef. Microb. Ecol. 65, 740–752. doi: 10.1007/s00248-013-0212-5
Kennedy, J., Codling, C. E., Jones, B. V., Dobson, A. D. W., and Marchesi, J. R. (2008). Diversity of microbes associated with the marine sponge, Haliclona simulans, isolated from Irish waters and identification of polyketide synthase genes from the sponge metagenome. Environ. Microbiol. 10, 1888–1902. doi: 10.1111/j.1462-2920.2008.01614.x
Kleiner, M., Petersen, J. M., and Dubilier, N. (2012a). Convergent and divergent evolution of metabolism in sulfur-oxidizing symbionts and the role of horizontal gene transfer. Curr. Opin. Microbiol. 15, 621–631. doi: 10.1016/j.mib.2012.09.003
Kleiner, M., Wentrup, C., Lott, C., Teeling, H., Wetzel, S., Young, J., et al. (2012b). Metaproteomics of a gutless marine worm and its symbiotic microbial community reveal unusual pathways for carbon and energy use. Proc. Natl. Acad. Sci. U.S.A. 109, E1173–E1182. doi: 10.1073/pnas.1121198109
Kuck, P., and Meusemann, K. (2010). FASconCAT, Version 1.0, Zool. Forschungsmuseum A. Koenig, Germany.
Kück, P., Meusemann, K., Dambach, J., Thormann, B., Reumont, von, B. M., Wagele, J. W., et al. (2010). Parametric and non-parametric masking of randomness in sequence alignments can be improved and leads to better resolved trees. Front. Zool. 7:10. doi: 10.1186/1742-9994-7-10
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Larsson, A. (2014). AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278. doi: 10.1093/bioinformatics/btu531
Lawry, N., Jani, V., and Jensen, T. (1981). Identification of the sulfur inclusion body in Beggiatoa alba B18LD by energy-dispersive X-ray microanalysis. Curr. Microbiol. 6, 71–74. doi: 10.1007/bf01569006
Levin, M., Anavy, L., Cole, A. G., Winter, E., Mostov, N., Khair, S., et al. (2016). The mid-developmental transition and the evolution of animal body plans. Nature 531, 637–641. doi: 10.1038/nature16994
Liu, M., Fan, L., Zhong, L., Kjelleberg, S., and Thomas, T. (2012). Metaproteogenomic analysis of a community of sponge symbionts. ISME J. 6, 1515–1525. doi: 10.1038/ismej.2012.1
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J., Charpentier, E., Horvath, P., et al. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477. doi: 10.1038/nrmicro2577
Manz, W., Arp, G., Schumann-Kindel, G., Szewzyk, U., and Reitner, J. (2000). Widefield deconvolution epifluorescence microscopy combined with fluorescence in situ hybridization reveals the spatial arrangement of bacteria in sponge tissue. J. Microbiol. Methods 40, 125–134. doi: 10.1016/S0167-7012(99)00103-7
Mohamed, N. M., Colman, A. S., Tal, Y., and Hill, R. T. (2008). Diversity and expression of nitrogen fixation genes in bacterial symbionts of marine sponges. Environ. Microbiol. 10, 2910–2921. doi: 10.1111/j.1462-2920.2008.01704.x
Moitinho-Silva, L., Bayer, K., Cannistraci, C. V., Giles, E. C., Ryu, T., Seridi, L., et al. (2013). Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol. Ecol. 23, 1348–1363. doi: 10.1111/mec.12365
Moitinho-Silva, L., Seridi, L., Ryu, T., Voolstra, C. R., Ravasi, T., and Hentschel, U. (2014). Revealing microbial functional activities in the Red Sea sponge Stylissa carteriby metatranscriptomics. Environ. Microbiol. 16, 3683–3698. doi: 10.1111/1462-2920.12533
Nunoura, T., Takaki, Y., Kazama, H., Kakuta, J., Shimamura, S., Makita, H., et al. (2014). Physiological and genomic features of a novel sulfur-oxidizing gammaproteobacterium gelonging to a previously uncultivated symbiotic lineage isolated from a hydrothermal vent. PLoS ONE 9:e104959. doi: 10.1371/journal.pone.0104959
Olson, J. B., Thacker, R. W., and Gochfeld, D. J. (2014). Molecular community profiling reveals impacts of time, space, and disease status on the bacterial community associated with the Caribbean sponge Aplysina cauliformis. FEMS Microbiol. Ecol. 87, 268–279. doi: 10.1111/1574-6941.12222
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Peng, Y., Leung, H. C., Yiu, S. M., and Chin, F. Y. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
Pita, L., López-Legentil, S., and Erwin, P. M. (2013). Biogeography and host fidelity of bacterial communities in Ircinia spp. from the Bahamas. Microb. Ecol. 66, 437–447. doi: 10.1007/s00248-013-0215-2
Pott, A. S., and Dahl, C. (1998). Sirohaem sulfite reductase and other proteins encoded by genes at the dsr locus of Chromatium vinosum are involved in the oxidation of intracellular sulfur. Microbiology 144, 1881–1894.
Radax, R., Rattei, T., Lanzen, A., Bayer, C., Rapp, H. T., Urich, T., et al. (2012). Metatranscriptomics of the marine sponge Geodia barretti: tackling phylogeny and function of its microbial community. Environ. Microbiol. 14, 1308–1324. doi: 10.1111/j.1462-2920.2012.02714.x
Rao, Q., Rollat-Farnier, P. A., Zhu, D. T., Santos-Garcia, D., Silva, F. J., Moya, A., et al. (2015). Genome reduction and potential metabolic complementation of the dual endosymbionts in the whitefly Bemisia tabaci. BMC Genomics 16:226. doi: 10.1186/s12864-015-1379-6
Robertson, L., and Kuenen, J. G. (2006). “The colorless sulfur bacteria,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York, NY: Springer), 985–1011.
Rua, C. P., Gregoracci, G. B., Santos, E. O., Soares, A. C., Francini-Filho, R. B., and Thompson, F. (2015). Potential metabolic strategies of widely distributed holobionts in the oceanic archipelago of St Peter and St Paul (Brazil). FEMS Microbiol. Ecol. 91, pii: fiv043. doi: 10.1093/femsec/fiv043
Sabarathnam, B., Manilal, A., Sujith, S., Kiran, G. S., Selvin, J., Thomas, A., et al. (2010). Role of sponge associated actinomycetes in the marine phosphorous biogeochemical cycles. Am-Eurasian J. Agric. Environ. Sci. 8, 253–256.
Santos-Beneit, F. (2015). The Pho regulon: a huge regulatory network in bacteria. Front. Microbiol. 6:402. doi: 10.3389/fmicb.2015.00402
Schläppy, M.-L., Schöttner, S., Lavik, G., Kuypers, M. M., de Beer, D., and Hoffmann, F. (2010). Evidence of nitrification and denitrification in high and low microbial abundance sponges. Mar. Biol. 157, 593–602. doi: 10.1007/s00227-009-1344-5
Siegl, A., Kamke, J., Hochmuth, T., Piel, J., Richter, M., Liang, C., et al. (2011). Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 5, 61–70. doi: 10.1038/ismej.2010.95
Sloan, D. B., and Moran, N. A. (2012). Genome reduction and co-evolution between the primary and secondary bacterial symbionts of psyllids. Mol. Biol. Evol. 29, 3781–3792. doi: 10.1093/molbev/mss180
Southwell, M. W., Popp, B. N., and Martens, C. S. (2008a). Nitrification controls on fluxes and isotopic composition of nitrate from Florida Keys sponges. Mar. Chem. 108, 96–108. doi: 10.1016/j.marchem.2007.10.005
Southwell, M. W., Weisz, J. B., Martens, C. S., and Lindquist, N. (2008b). In situ fluxes of dissolved inorganic nitrogen from the sponge community on Conch Reef, Key Largo, Florida. Limnol. Oceanogr. 53, 986–996. doi: 10.4319/lo.2008.53.3.0986
Souza, C. P., Almeida, B. C., Colwell, R. R., and Rivera, I. N. (2011). The importance of chitin in the marine environment. Mar. Biotechnol. 13, 823–830. doi: 10.1007/s10126-011-9388-1
Srivastava, M., Simakov, O., Chapman, J., Fahey, B., Gauthier, M. E. A., Mitros, T., et al. (2010). The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466, 720–726. doi: 10.1038/nature09201
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Steindler, L., Beer, S., and Ilan, M. (2002). Photosymbiosis in intertidal and subtidal tropical sponges. Symbiosis 33, 263–273.
Strahl, J., Dringen, R., Schmidt, M. M., Hardenberg, S., and Abele, D. (2011). Metabolic and physiological responses in tissues of the long-lived bivalve Arctica islandica to oxygen deficiency. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 158, 513–519. doi: 10.1016/j.cbpa.2010.12.015
Takiya, D. M., Tran, P. L., Dietrich, C. H., and Moran, N. A. (2006). Co-cladogenesis spanning three phyla: leafhoppers (Insecta: Hemiptera: Cicadellidae) and their dual bacterial symbionts. Mol. Ecol. 15, 4175–4191. doi: 10.1111/j.1365-294X.2006.03071.x
Taylor, M. W., Hill, R. T., Piel, J., Thacker, R. W., and Hentschel, U. (2007a). Soaking it up: the complex lives of marine sponges and their microbial associates. ISME J. 1, 187–190. doi: 10.1038/ismej.2007.32
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007b). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347. doi: 10.1128/MMBR.00040-06
Thomas, T., Moitinho-Silva, L., Lurgi, M., Björk, J. R., Easson, C., Astudillo-García, C., et al. (2016). Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 7:11870. doi: 10.1038/ncomms11870
Thomas, T., Rusch, D., DeMaere, M. Z., Yung, P. Y., Lewis, M., Halpern, A., et al. (2010). Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 4, 1557–1567. doi: 10.1038/ismej.2010.74
Tian, R.-M., Wang, Y., Bougouffa, S., Gao, Z.-M., Cai, L., Bajic, V., et al. (2014). Genomic analysis reveals versatile heterotrophic capacity of a potentially symbiotic sulfur-oxidizing bacterium in sponge. Environ. Microbiol. 16, 3548–3561. doi: 10.1111/1462-2920.12586
Vacelet, J., Boury-Esnault, N., Fiala-Medioni, A., and Fisher, C. R. (1995). A methanotrophic carnivorous sponge. Nature 377, 296–296. doi: 10.1038/377296a0
van Os, N., Smits, S. H., Schmitt, L., and Grieshaber, M. K. (2012). Control of D-octopine formation in scallop adductor muscle as revealed through thermodynamic studies of octopine dehydrogenase. J. Exp. Biol. 215, 1515–1522. doi: 10.1242/jeb.069344
Webster, N. S., Taylor, M. W., Behnam, F., Lücker, S., Rattei, T., Whalan, S., et al. (2010). Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 12, 2070–2082. doi: 10.1111/j.1462-2920.2009.02065.x
White, J. R., Patel, J., Ottesen, A., Arce, G., Blackwelder, P., and Lopez, J. V. (2012). Pyrosequencing of bacterial symbionts within axinella corrugata sponges: diversity and seasonal variability. PLoS ONE 7:e38204. doi: 10.1371/journal.pone.0038204
Wilkinson, C. R. (1983). Net Primary Productivity in Coral Reef Sponges. Science 219, 410–412. doi: 10.1126/science.219.4583.410
Wilkinson, C. R., and Fay, P. (2004). Nitrogen fixation in coral reef sponges with symbiotic cyanobacteria. Nature 279, 527–529. doi: 10.1038/279527a0
Wilson, M. C., Mori, T., Rückert, C., Uria, A. R., Helf, M. J., Takada, K., et al. (2014). An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 506, 58–62. doi: 10.1038/nature12959
Wu, D., Daugherty, S. C., Van Aken, S. E., Pai, G. H., Watkins, K. L., Khouri, H., et al. (2006). Metabolic complementarity and genomics of the dual bacterial symbiosis of sharpshooters. PLoS Biol. 4:e188. doi: 10.1371/journal.pbio.0040188
Zhang, D., Sun, W., Feng, G., Zhang, F., Anbuchezhian, R., Li, Z., et al. (2015a). Phylogenetic diversity of sulphate-reducing Desulfovibrio associated with three South China Sea sponges. Lett. Appl. Microbiol. 60, 504–512. doi: 10.1111/lam.12400
Zhang, F., Blasiak, L. C., Karolin, J. O., Powell, R. J., Geddes, C. D., and Hill, R. T. (2015b). Phosphorus sequestration in the form of polyphosphate by microbial symbionts in marine sponges. Proc. Natl. Acad. Sci. U.S.A. 112, 4381–4386. doi: 10.1073/pnas.1423768112
Keywords: bacterial symbionts, metagenomics, coral reef symbiosis, proteobacteria, porifera, Chromatiales
Citation: Gauthier M-EA, Watson JR and Degnan SM (2016) Draft Genomes Shed Light on the Dual Bacterial Symbiosis that Dominates the Microbiome of the Coral Reef Sponge Amphimedon queenslandica. Front. Mar. Sci. 3:196. doi: 10.3389/fmars.2016.00196
Received: 26 June 2016; Accepted: 26 September 2016;
Published: 13 October 2016.
Edited by:
Ute Hentschel, GEOMAR Helmholtz Centre for Ocean Research Kiel (HZ), GermanyReviewed by:
Gwenael Piganeau, Observatoire Océanologique de Banyuls-sur-Mer (CNRS), FranceLaura Steindler, University of Haifa, Israel
Copyright © 2016 Gauthier, Watson and Degnan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandie M. Degnan, s.degnan@uq.edu.au