Hydrous Ferric Oxides in Sediment Catalyze Formation of Reactive Oxygen Species during Sulfide Oxidation
 Sarah A. Murphy1
Sarah A. Murphy1  Shengnan Meng1 Benson M. Solomon1 Dewamunnage M. C. Dias1 Timothy J. Shaw1,2
Shengnan Meng1 Benson M. Solomon1 Dewamunnage M. C. Dias1 Timothy J. Shaw1,2  John L. Ferry1,2*
John L. Ferry1,2*- 1Department of Chemistry and Biochemistry, University of South Carolina, Columbia, SC, USA
- 2Nanocenter at the University of South Carolina, Columbia, SC, USA
This article describes the formation of reactive oxygen species as a result of the oxidation of dissolved sulfide by Fe(III)-containing sediments suspended in oxygenated seawater over the pH range 7.00 and 8.25. Sediment samples were obtained from across the coastal littoral zone in South Carolina, US, at locations from the beach edge to the forested edge of a Spartina dominated estuarine salt marsh and suspended in aerated seawater. Reactive oxygen species (superoxide and hydrogen peroxide) production was initiated in sediment suspensions by the addition of sodium bisulfide. The subsequent loss of HS−, formation of Fe(II) (as indicated by Ferrozine), and superoxide and hydrogen peroxide were monitored over time. The concentration of superoxide rose from the baseline and then persisted at an apparent steady state concentration of ~500 nM at pH 8.25 and 200 nM at pH 7.00, respectively, until >97% hydrogen sulfide was consumed. Measured superoxide was used to predict hydrogen peroxide yield based on superoxide dismutation. Dismutation alone quantitatively predicted hydrogen peroxide formation at pH 8.25 but over predicted hydrogen peroxide formation at pH 7 by a factor of approximately 102. Experiments conducted with episodic spikes of added hydrogen peroxide indicated rapid hydrogen peroxide consumption could account for its apparent low instantaneous yield, presumably the result of its reaction with Fe(II) species, polysulfides or bisulfite. All sediment samples were characterized for total Fe, Cu, Mn, Ni, Co, and hydrous ferric oxide by acid extraction followed by mass spectrometric or spectroscopic characterization. Sediments with the highest loadings of hydrous ferric oxide were the only sediments that produced significant dissolved Fe(II) species or ROS as a result of sulfide exposure.
Introduction
Reactive oxygen species (ROS, including superoxide, hydrogen peroxide, and hydroxyl radical) are critical for enabling abiotic reaction paths between organic carbon and atmospheric oxygen in surface waters. Abiotic ROS production in seawater is usually attributed to photoprocesses involving the direct reduction of oxygen by photoexcited natural organic matter or by Fe(II) generated by photoinduced ligand to metal charge transfer (Zepp et al., 1998; Powers and Miller, 2014). In the latter case, oxidizable ligands can include a wide variety of organic molecules and some ligands that are not ordinarily considered reductants, including water and chloride (Sulzberger and Laubscher, 1995; Sima and Makanova, 1997; Voelker et al., 1997). However, there are other abiotic sources of reductive equivalents that can reduce Fe(III) to Fe(II) without the need for sunlight; including hydrogen sulfide, polysulfides, some forms of organic carbon (e.g., polyhydroxylated phenols, organothiols etc.), and superoxide (Poulton et al., 2004; Carey and Taillefert, 2005; Larsen et al., 2006; Ma et al., 2006; Konovalov et al., 2007; Rickard and Luther, 2007; Gartman et al., 2011; Johnston et al., 2011; Chirita and Schlegel, 2012; Wan et al., 2014; Chirita and Schlegel, 2015; Havig et al., 2015; Peiffer et al., 2015; Duinea et al., 2016). Our own interest in Fe(II) is associated with the tidally driven efflux of anoxic porewater and this has led us to investigate the potential for ROS formation as a consequence of the non-photochemical reduction of Fe(III) by sulfide and other reduced sulfur species (Figure 1).
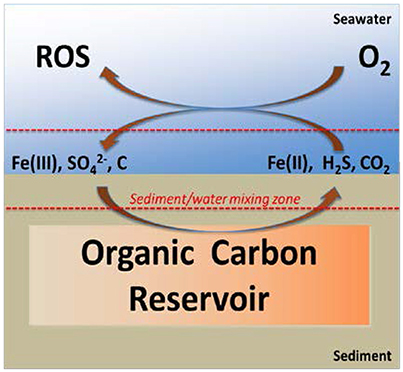
Figure 1. Microbial oxidation of buried carbon results in the production of Fe(II), HS−, and ROS. Anaerobic microbial respiration is often based on the use of Fe(III) or as electron acceptors. When resulting Fe(II) and HS− in porewater (present as ions or FeS) mix with the oxic portion of the water column their abiotic oxidation leads to the formation of ROS along with the regeneration of Fe(III) and .
The oxic portion of the biosphere is a metastable mixture of different oxidation states of carbon, sulfur and oxygen energetically poised from equilibrium by the net rate differentials between photosynthetic carbon fixation and its metabolic or abiotic oxidation (Watson et al., 1978). Sedimentary carbon burial widens this gap by imposing a significant mass transfer limitation on the rate of carbon transport between the lithosphere, atmosphere and hydrosphere. It also restricts oxygen transport, forcing microbial metabolism of buried material to rely on alternative electron acceptors such as sulfate or carbon dioxide. The anaerobic microbial metabolism of buried carbon results in the reduction of ~11.3–75 × 1012 moles of sulfate to sulfide per year in marine sediments and coastal marshes (Bottrell and Newton, 2006; Luther et al., 2011; Bowles et al., 2014). This range is compiled from recent efforts to reconcile older sulfate reduction estimates based on spatial averaging to more recent measurements correlating the global carbon flux to sediment with sulfate reduction (see the recent work by Bowles et al., 2014 and references therein). Based on the more conservative estimate of 11.3 × 1012 moles of microbially produced sulfide/yr, and applying accepted percentage outcomes for the fate of sulfur in the sulfur cycle, approximately 2.3 × 1012 moles of this is immobilized annually in the process of pyrite burial. The remaining 9.0 × 1012 moles sulfide is reoxidized and returned to the water column (primarily as sulfate). The direct oxidation of sulfide by dioxygen is thermodynamically favorable but kinetically unfavorable and requires the intercession of a catalyst such as sedimentary Fe(III) or Mn(IV) (Equations 1–9; Vazquez et al., 1989; Roden et al., 2004; Carey and Taillefert, 2005; Bottrell and Newton, 2006; Aller R. C. et al., 2010; Johnston et al., 2011; Kubo et al., 2011; Luther et al., 2011; Lin et al., 2012; Rickard, 2012; Bowles et al., 2014; Murphy et al., 2014).
Here we focus on the impact of sulfide oxidation on the ferric/ferrous iron system given its relative geographical importance, kinetic facility, and potential for generating ROS. The non-photochemical interaction between the carbon, oxygen and sulfur cycles as ROS sources is interesting because the potential ROS generation capacity is so large, as indicated by the number of moles of Fe(II) produced/yr globally by the reoxidation of microbially produced hydrogen sulfide (vide supra; Bottrell and Newton, 2006; Luther et al., 2011; Bowles et al., 2014). The initial oxidation of sulfide and bisulfide by Fe(III)aq or hydrous ferric oxides [represented collectively Fe(III)OHx in the following equations] results in a mixture of Fe(II)-containing species as summarized in Equations (1–6) (Rickard, 1975; Afonso and Stumm, 1992; Rickard et al., 1995; Luther et al., 1996; Rickard and Luther, 1997, 2007; Poulton et al., 2004). These may undergo rapid oxidation by dissolved O2, generating the reactive oxygen species superoxide () and hydrogen peroxide (H2O2) while regenerating Fe(III)OHx to continue sulfide oxidation (Equations 5, 6). The fate of HS· is unknown, although dimerization to produce H2S2 or reaction with excess HS− to produce polysulfides are possible (Rickard et al., 1995; Rickard and Luther, 2007; Wan et al., 2014). The self-reaction of superoxide with its conjugate acid HOO·(Equation 8) is the kinetically favored outcome for superoxide at typical seawater pH unless there is a significant quantity of Fe(II) present, in which case superoxide may be reduced directly by Fe(II) to yield Fe(III) and hydrogen peroxide (Equation 9; Bielski, 1978; Bielski et al., 1985).
If one conservatively assumes a 1:1 stoichiometry between Fe(III) and HS− this implies a 1:1 conversion of the reductive equivalents in sulfide to superoxide. Given the most significant loss of superoxide is through formation of hydrogen peroxide (Equations 7–9) this implies an annual global sulfide-driven ROS formation potential of 9.0–4.5 × 1012 moles, depending on whether the terminal ROS is superoxide or hydrogen peroxide and based on the assumption that 75–90% HS− oxidation is abiotic (Bottrell and Newton, 2006, and references therein). There are relatively few estimates of annual photochemical ROS production in surface waters to compare this to, but recent work by Powers and Miller suggests the marine average is between 2.9 and 10.9 × 1012 moles ROS in the top meter of the oceans/yr (Powers and Miller, 2014). There are other sources of sulfide not considered in this estimate such as volcanic and hydrothermal vents so the overlap between potential sulfide-driven ROS production and the estimated photochemical ROS production demonstrated here is not quantitative (Field and Sherrell, 2000; Moore et al., 2009a; Yucel et al., 2009; Gartman et al., 2011). There are also other sources of Fe(II), such as the direct reduction of Fe(III) by facultative anaerobes (Perry et al., 1993; Coates et al., 1998; Dollhopf et al., 2000; Sekar and DiChristina, 2014). Nonetheless, the estimated values are intriguingly close, certainly within the same order of magnitude, and that serves as justification for studying potential mechanisms for sulfide-driven ROS generation.
Here we report an investigation of the sources and mechanisms of ROS formation in sediment suspensions containing dissolved hydrogen sulfide (and bisulfide) and oxygen. These conditions are rarely observed in the open water column but are often encountered at the sediment/water interface. Specific examples include conditions associated with bioturbation, undersea mudflow, dredging, wave-driven mixing, and the trailing edges of the tidal prism (Luther et al., 1991; Precht et al., 2004; Aller and Blair, 2006; Rickard and Luther, 2007; Moore et al., 2009b; Aller J. Y. et al., 2010; Aller R. C. et al., 2010; Michaud et al., 2010; Santos et al., 2012). This work is a continuation of an investigation of ROS generation associated with the oxidation of reduced transition metals at sediment surfaces (Burns et al., 2010, 2011a,b; Murphy et al., 2014). It reports a test of the hypothesis that the conditions of frequent episodic anoxia set the stage for pulsed ROS production in marine littoral zones, focusing on the roles of hydrogen sulfide, ferric oxides, and pH on superoxide and hydrogen peroxide production (Figure 1). The oxidation of reduced sulfur species by dissolved Fe(III) and hydrous ferric oxides is much more rapid than by more crystalline iron oxides such as goethite, lepidocrite, or magnetite, and a central hypothesis tested by this work was that hydrous ferric oxides would play a correspondingly more important role in ROS formation than other ferric iron sources (Kostka and Luther, 1995; Poulton et al., 2004; Ma et al., 2006; Rickard and Luther, 2007; Cai et al., 2010; Luther, 2010; Luther et al., 2011). Sediments were collected from across the marine littoral zone in South Carolina, from the beach face to the forested inland edge of a saline Spartina alterniflora-dominated estuary. The addition of pulses of HS− to aerated suspensions of collected sediments resulted in rapid Fe(II) production with concomitant superoxide and hydrogen peroxide formation. After a brief initiation phase superoxide essentially reached a steady state in the tested systems while hydrogen peroxide was more dynamic and sensitive to instantaneous concentration of Fe(II). This pump-and-probe experimental strategy of interrogating sediments for ROS production capacity was applied to all sediments tested and marsh sediments were additionally exposed to multiple sequential pulses of HS− and hydrogen peroxide. All sediments tested consumed HS- but only marsh sediments produced significant ROS.
Materials and Methods
Reagents
Iron(III) chloride hexahydrate (99+%) and sodium sulfide nonahydrate (99.99+% trace metal free), potassium superoxide (98%), and chromatographic sand were purchased from Aldrich and used without further purification. Hydrochloric acid (ACS grade) was obtained from BDH. Horseradish peroxidase and 2-methyl-6-(p-methoxyphenyl)-3,7-dihydroimidazo[1,2-a]pyrazine-3-one (MCLA) were obtained from Sigma-Aldrich. 10-Acetyl-3,7-dihydroxyphenoxazine (Amplex Red, 97%) was purchased from American Advanced Scientific and the latter three reagents were stored in a desiccator at −5°C. Diethylenetriaminepentaacetic acid (98+%) and iron(II) chloride anhydrous (99.5+%) were purchased from Alfa Aesar. Iron(II) chloride was stored in a desiccator and all solutions were kept under nitrogen in a glove box. Ferrozine iron reagent (98%) was purchased from VWR. All other salts used as purchased from (Fisher, 99%).
Sediment Characterization
Sediment samples were obtained from the top 2 cm of material at five locations (Figure 2) across the marine littoral zone of coastal South Carolina. The total organic carbon content (TOC, in %) for each sample was determined by loss of mass on ashing. Sample locations included (progressing toward the ocean) the landward forested edge of a S. alterniflora dominated salt marsh (33°20′24.36″N; 79°12′9.23″W, TOC 1.47%), the bank of a dredged canal between the marsh and a barrier island (33°42′51.22″N; 78°55′17.79″W, TOC 0.79%), the sand dunes on the same barrier island (33°42′0.74″N; 78°52′10.77″W, TOC 0.16%), the swash zone at the surf's edge (33°42′0.69″N; 78°52′8.54″W, TOC 0.01%), and at a depth of 1 m below the surf edge at low tide (33°42′0.11″N; 78°52′7.28″W. TOC 0.13%). A control sample was prepared from commercially available sand (Sigma Aldrich) that was triple washed with aqua regia (60 min exposure/wash) followed by a triple rinse with 18 MΩ deionized water to purify the sample of solution accessible acid soluble metals (TOC 0.00%). Samples were sieved, dried and analyzed for metal content using inductively coupled plasma mass spectrometry. Fe was the dominant transition metal in all samples. Samples were digested in alternately concentrated HCl or ascorbic acid in accordance with the procedures detailed in Kostka and Luther (1994) to determine total Fe (crystalline and amorphous) and the hydrous ferric oxide fraction respectively (Kostka and Luther, 1994). Approximately 25% of the total Fe in sample A from the forested marsh edge was hydrous ferric oxides, all other samples were less than 10% hydrous ferric oxides (Figure 2).
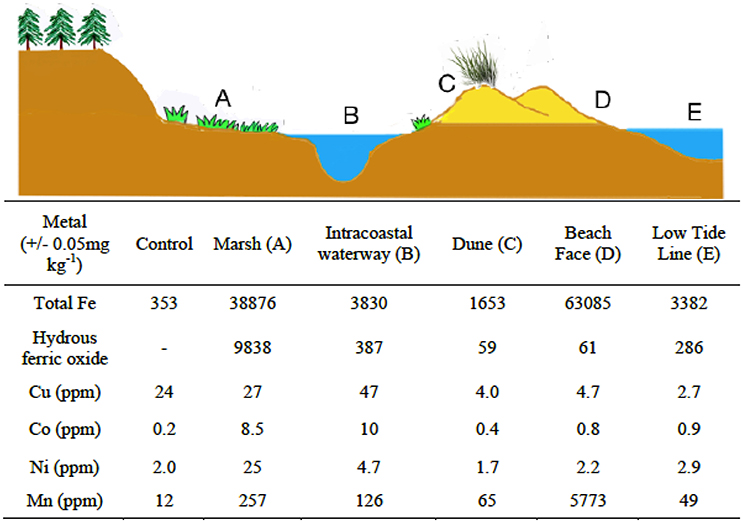
Figure 2. Sediment collection points and metal content (dry weight). Sediment surface samples were collected from several points across the South Carolina marine littoral zone, including a coastal marsh (A), manmade intracoastal water way (B), a barrier island dune crest (C), the beach face or swash zone (D), and at a depth of 1 m below the low tide line (E). Samples were sieved (4 mm) and air dried before analysis for metals or use in experiments. The control sample was chromatographic sand treated with aqua regia and washed. Characterization for transition metals in all samples was achieved by acid extraction followed by inductively coupled plasma mass spectrometry or ascorbate/HCl extraction followed by Ferrozine (amorphous Fe only).
Experimental Procedure
Sediment samples (1.00 wt%) were suspended in 500 mL pH-adjusted seawater (adjusted by dropwise addition of HCl; Luther, 2010; Murphy et al., 2014). Reactions took place in 1 L beakers open to the atmosphere with suspension and aeration achieved simultaneously with rapid vortex mixing, using procedures published in earlier work (Murphy et al., 2014). All reactions and sample handling took place in darkened laboratories. Minimal lighting was provided from sources with 500 nm cutoff filters. All samples and stock solutions were stored in light-tight, blackened drawers until needed. After a 30 min equilibration period sufficient aqueous hydrogen sulfide was added to each suspension to yield a concentration of 300 μM. This level was chosen because it represented a conservative midpoint between the concentrations for HS− often reported at dynamic oxic/anoxic interfaces in estuaries or bays (0–30 μM) and anoxic porewater (600–1000 μM; Rozan et al., 2002; Taillefert et al., 2002; Snyder et al., 2004; Carey and Taillefert, 2005; Lewis et al., 2007). Aliquots of known volume were periodically withdrawn for analysis of sulfide accessible Fe, HS−, and H2O2 over time (Stookey, 1970; Zhou et al., 1997; Simpson, 2001). Aqueous sediment loading was 10.00 g L−1 of air dried, sieved sediment, consistent with the low range of solid/liquid ratio (99% porosity) observed in the top layers of many coastal surface sediments (Aller J. Y. et al., 2010). Samples were removed from the reactors and centrifuged on a Baxter Dade Immufuge II centrifuge at 3225 rpm for 30 s to remove suspended solids before subsequent spectroscopic assays. This basic experimental design was varied by adding replicates that included episodically spiked “refreshers” of hydrogen sulfide or hydrogen peroxide to determine the effect of rapidly resupplying or depleting Fe(II) on the system. Samples were withdrawn during the 30 min equilibration period prior to HS− addition to serve as controls and quantify background Fe(II), superoxide or hydrogen peroxide. Ferrozine-responsive Fe(II) was detected prior to HS− addition in salt marsh suspensions (sterile and nonsterile) at a concentration of ~20 μM and this was stable on the timescale of the equilibration period. Fe(II) was below the method detection limit in all other sediment suspensions during the equilibration phase. Superoxide and hydrogen peroxide were observed at concentrations of ~10 nM or 500–800 nM, respectively, during the equilibrium phase in all sediment suspensions and these concentrations were stable. The latter measurements included the aqua regia washed sand control sediment so these measurements were indicative of background. The stability of the background hydrogen peroxide and superoxide in marsh sediment during the equilibration phase indicated detected Fe(II) was stable toward oxidation, presumably as a result of complexation by humics (Anastacio et al., 2008; Catrouillet et al., 2014; Veverica et al., 2016). Previously published work has demonstrated that HS− is kinetically stable in sterile, trace-metal free solutions over the time scale of these experiments and does not evolve detectable levels of hydrogen peroxide (Luther, 2010; Murphy et al., 2014). Experimental data reports t0 as equivalent to the time of hydrogen sulfide addition for the sake of graphical clarity.
Analytical
Iron(II) and sulfide measurement. Fe(II)aq and hydrogen sulfide were monitored colorimetrically using the Ferrozine and methylene blue methods respectively as previously reported (Cline, 1969; Stookey, 1970; Burns et al., 2011a,b). Samples were withdrawn from the reactors and added directly to developing solutions (varied by analyte). Particulates were removed by immediate centrifugation (3225 rpm; Dade Immufuge II). Supernatant was removed by pipetting directly into a 96-well glass microplate. Absorption spectra were recorded on a Spectramax M5 plate reader.
Hydrogen peroxide measurements were episodic. Slurry samples were withdrawn from reactors and dispensed into precharged vials containing 0.01 M Diethylenetriaminepentaacetic acid adjusted to pH 7.4. Particles were removed by immediate centrifugation and an aliquot of the supernatant was transferred to a 96-well plate before subsequent derivatization and spectroscopic analysis using the Amplex Red technique (Zhou et al., 1997). Horseradish peroxidase was dissolved in a 0.05 M sodium phosphate buffer at pH = 7.4 and a 100 μL aliquot was added to each sample, followed by 100 μL of 10 mM 10-acetyl-3,7-dihydroxyphenoxazine prepared in dimethyl sulfoxide. The samples were incubated at room temperature for 30 min and then analyzed for development of the indicator resorufin by fluorescence and absorbance spectroscopy (Zhou et al., 1997). At least one full calibration curve was run with each plate, for a minimum of 5 replicate calibration curves per day of analysis. All glassware was cleaned in a muffle furnace and acid washed in a 10% HCl/1 M oxalic acid mixture. After rinsing with 18MΩ deionized water, glassware was handled and stored as trace metal clean glassware to prevent inadvertent oxidation of sulfide in the absence of added metals. Superoxide was continuously measured by flow injection analysis (Waterville Analytical) with the MCLA chemiluminescence technique (Rose et al., 2008; Godrant et al., 2009). All initial flow rates (sample and MCLA) were 2.5 mL/min. The flow cell volume was 2.0 mL and the PMT integration time set to 0.200 s. Calibration was performed daily against spectroscopically verified superoxide stock solutions (UV absorbance at 240 nm) made up at pH 10 (NaOH) or higher.
Quality Assurance/Quality Control
Replicate blanks (n = 3) were obtained for all reagents. Blanks were updated with preparation of fresh reagent solutions. Reference standards were interrogated for peroxide analysis at a frequency of 1 reference check/5 unknown determinations. Peroxide reference standards were externally calibrated against the optical absorbance of the concentrated stock at 254 nm. The detection limit for each method was defined by the linear dynamic range of the calibration curves. All experiments were run in triplicate. All experimental data were reported as the mean of the triplicate experiments. All error estimates were ± one standard deviation about the mean.
Results and Discussion
Hydrogen sulfide was added to separate, aerated suspensions of all sediments studied or a sediment-free control at pH 7.00, 7.50, 8.00, and 8.25. The systems were monitored for changes in sulfide, Fe(II), superoxide, and hydrogen peroxide for 120 min following sulfide addition. Sulfide consumption followed two profiles; an extremely rapid decay (95+%) in the first 30 s with a slow decay thereafter or an overall slow decay that in many cases was not statistically different than the control (Figure 3). Sediments from the marsh fell in the former category at all pHs studied, whereas the sediments from the intracoastal waterway, dune, or beach swash zone displayed the latter. Marsh sediments were also the only samples to experience significant increases in Fe(II) (as indicated by Ferrozine) over the timescale of the experiments (Figure 4), obtaining a maximum Fe(II) of 45.0 × 10−6 M very rapidly (<60 s) at pH 8.25. In contrast, intracoastal waterway sediment yielded the second highest apparent Fe(II) concentration of 7.3 × 10−6 M at ~600 s. These results justified focusing primarily on the marsh system. For marsh sediments the instantaneous concentration of Fe(II) at a given time never exceeded an amount corresponding to an Fe(II) yield above 15% of HS− consumed at the same time. Given that total iron and hydrous ferric oxides were both in stoichiometric excess to HS− in marsh sediment the low yield of Fe(II) was attributed to competitive losses of HS− through formation of polysulfides (Giggenbach, 1972; Hoffmann and Lim, 1979; Fukuto et al., 2012; Cunha et al., 2016). The apparent half-life for Fe(II) was also considerably slower than would be expected based on existing Fe(II) oxidation models (e.g., over 1000 s; King et al., 1995; Santana-Casiano et al., 2005; Trapp and Millero, 2007; Burns et al., 2011a,b; Peiffer et al., 2015). These results can be explained by two exclusive models of the system; one where Fe(II) oxidation was slowed by the presence of a stabilizing ligand or one where Fe(II) oxidation was kinetically facile and the measured concentration was actually the product of simultaneous Fe(III) reduction and Fe(II) oxidation. The two models were resolved by examination of the concentration vs. time profiles for superoxide (Figure 5) in marsh sediments. Both pH conditions experienced a sudden increase in superoxide upon the addition of hydrogen sulfide, indicating at least some of the total Fe(II) was available for oxidation by dioxygen (Equation 5). The half-lives for superoxide were calculated based on dismutation (Equation 8) using the maximum observed superoxide as initial concentration (Equation 10):
where at pH 7.00 k = 5.01 × 105 M−1s−1 and superoxide = 204 × 10−9 M and at pH 8.25 k = 1.78 × 104 M−1s−1 and superoxide = 514 × 10−9 M (conditional k-values obtained from a comprehensive review by Bielski et al., 1985). T under these conditions was 9.8 and 109.0 s at pH 7.00 and 8.25, respectively. Given the apparent stability of superoxide in the experiments (Figure 5) and the truism that dismutation sets the minimum rate of superoxide decay in aqueous systems we concluded that superoxide was continually replenished by the oxidation of Fe(II) in both cases; i.e., the kinetically facile model was correct. This meant the instantaneous concentration of Fe(II) at any point was a function of the relative rates of parallel Fe(III) reduction by reduced sulfur species (including HS-, polysulfides, bisulfite etc.) and the parallel oxidation of Fe(II) by dioxygen, superoxide, and hydrogen peroxide (Warneck and Ziajka, 1995; Lichtschlag et al., 2013; Lohmayer et al., 2014; Wan et al., 2014; Cunha et al., 2016). Since only a few of the required bimolecular rate constants are known for these reactions and the concentration of the secondary sulfide oxidation products was not measured it was not possible to convert the instantaneous Fe(II) concentration to a direct estimate of the ROS formation potential of the system.
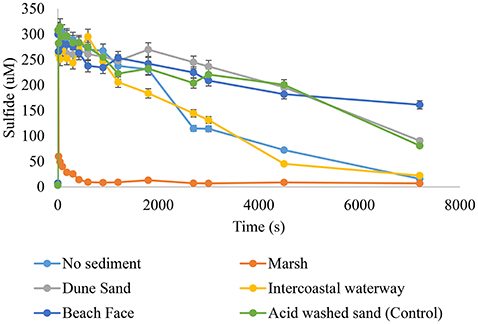
Figure 3. The effect of sediment slurries on sulfide oxidation. Sediment suspensions (1.00 wt%) were made up in pH adjusted seawater. Suspensions were maintained and aerated through rapid mixing. At time = 0 s sufficient was added to bring its solution concentration 300 μM. HS− was monitored for a minimum of 7200 s from zero. Results from pH 8.25 shown.
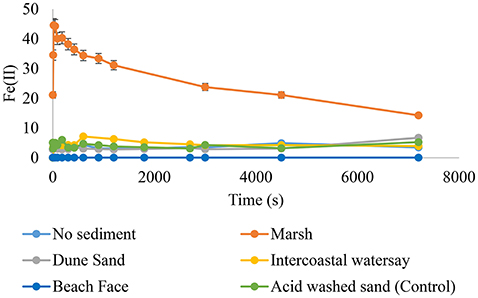
Figure 4. Fe(II) evolution after HS− addition. The introduction of to aerated sediment suspensions (1.00 wt%) seawater resulted in the formation of Ferrozine-responsive Fe(II). The highest yields of Fe(II) were obtained from sediments with high concentrations of hydrous ferric oxides. Results from pH 8.25 shown.
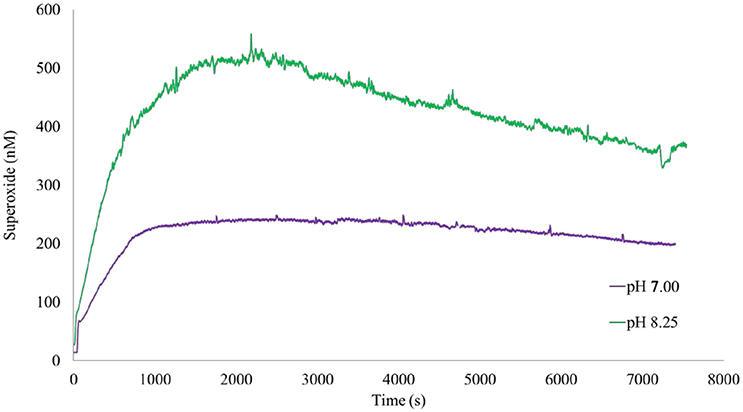
Figure 5. Superoxide formation followed HS− addition. The addition of HS− to 1.00 wt% suspensions of marsh sediments was followed by a rapid increase in superoxide at pH 7.00 and pH 8.25. The apparent stability of superoxide relative to its estimated half-life (max t1/2 ~ 100 s) indicated continuous generation during the experiment.
The stoichiometry of the conversion of reductive equivalents from HS− to hydrogen peroxide was investigated by comparing the outcome of integrated superoxide production vs. instantaneous hydrogen peroxide. The production of superoxide between any two time points (time a and b) was estimated by calculating the loss from time a to time b by dismutation and adding the difference between the new concentration and that observed (Equation 11):
where k = the conditional pH dependent dismutation value, t = the elapsed time between times a and b (0.5 s in this study) and assuming measurement times were close enough so not all superoxide was consumed between times a and b. The latter assumption was supported by the half-life calculations (Equation 10, vide supra) that demonstrated superoxide had a half life over an order of magnitude longer than the sampling interval for the superoxide technique. The approach allowed the overall estimation of the total moles of superoxide produced during the experiment. The accumulated superoxide was summed over the duration of the experiment (7200 s) and plotted against time to obtain a nearly linear estimated net increase in superoxide produced (r2 = 0.998 and 0.991 for pH 7.00 and 8.25 respectively, Figure 6). Given that superoxide production came at the cost of Fe(II) oxidation, the negatives of the slopes in Figure 6 were the rates of Fe(II) consumption at the two pHs and the reaction was zero order in Fe(II). This was consistent with the model of ferric and ferrous iron playing the role of a kinetically saturated catalyst for HS− oxidation. The net loss of Fe(II) in Figure 4 indicated that Fe(II)/Fe(III) cycling was not perfectly efficient and is speculatively a result of Fe(III) removal through precipitation. The stoichiometry of the process was investigated by assuming all superoxide produced was consumed by dismutation and comparing the measured hydrogen peroxide yield to that predicted from dismutation (Figures 7, 8 for pH 7.00 and 8.25, respectively). At pH 7.00 superoxide was consumed very rapidly with little hydrogen peroxide production and at pH 8.25 superoxide was converted nearly quantitatively to hydrogen peroxide. The insert plot of hydrogen peroxide predicted vs. measured in Figure 8 is notable for a slope of nearly 1. However, this model was inadequate for predicting hydrogen peroxide yield at pH 7 (Figure 7 and insert). The overall yields of hydrogen peroxide based on HS− consumed were <0.3% at pH 7.00 and ~1.6% at pH 8.25. Presumably this was a result of a pH dependent reaction (or reactions) that consumed either superoxide or hydrogen peroxide more effectively at the lower pH, such as the reaction of hydrogen peroxide with bisulfite or Fe(II), or more likely their reaction with an FeS species (Rush and Bielski, 1985; Warneck and Ziajka, 1995; Chirita and Schlegel, 2012; Giel et al., 2013; Theil et al., 2013; Duinea et al., 2016). It was also possible that at the lower pH some hydrogen peroxide or superoxide was consumed by reactions involving natural carbon in the sediments or through the Fenton reaction (Bielski, 1978; Bielski et al., 1985; Rush and Bielski, 1985; Deguillaume et al., 2005).
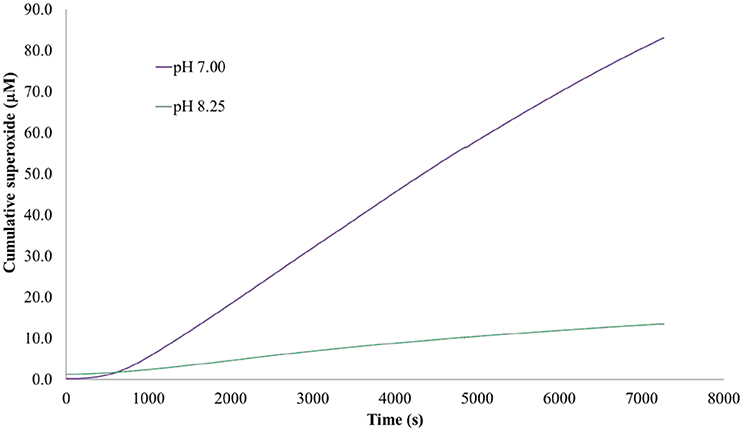
Figure 6. Estimated cumulative superoxide production. The measured, instantaneous concentration of superoxide was used as input with Equation (11) to solve for the total number of moles of superoxide generated over the course of the experiment at 7.00 and pH 8.25. The instantaneous concentration of superoxide was lower at pH 7.00 than 8.25 but the cumulative production was higher, correlating with an increased loss of superoxide from dismutation at the lower pH and consistent with Equation (8).
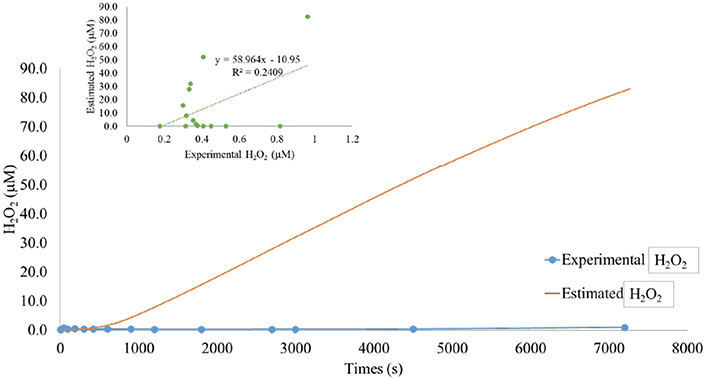
Figure 7. Modeled vs. predicted hydrogen peroxide, pH 7.00. Modeled superoxide was used to predict hydrogen peroxide observed in sediment suspensions. In marsh sediment suspensions at pH 7.00 the model over predicted hydrogen peroxide by approximately a factor of 100, indicating the presence of an unknown hydrogen peroxide sink. Correlation between model and data shown in the insert.
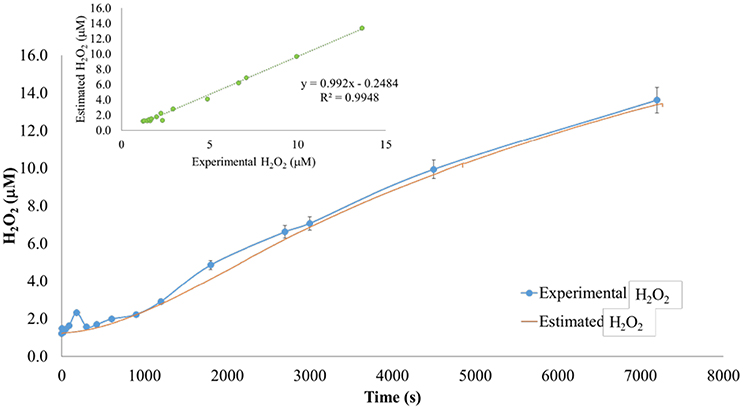
Figure 8. Modeled vs. predicted hydrogen peroxide, pH 8.25. Modeled superoxide was used to predict theoretical hydrogen peroxide observed in sediment suspensions. Theoretical concentrations were compared to measured concentrations to test the conservatism of hydrogen peroxide. In marsh sediment suspensions at pH 8.25 the model predicted measured hydrogen peroxide with an r2 = 0.992, indicating dismutation was the sink for superoxide. Better agreement between the modeled and observed data (insert) suggested the unknown peroxide sink exhibited a strong pH dependence.
The effects of sequential reductant and oxidant additions were measured in marsh sediments at pH 8.25. Additional hydrogen sulfide or hydrogen peroxide additions occurred at time = 1800, 3000, and 4500 s after the initiating hydrogen sulfide pulse, with each addition sufficient to bring the system to a nominal concentration of 300 × 10−6 M HS− or experience a net increase of 10 × 10−6 M hydrogen peroxide. Each additional HS− spike oxidized rapidly and the nominal concentrations were only directly observed in sediment-free controls. In sediment suspensions measured sulfide fell typically by 95% within the first 30 s after addition. The concentration of Fe(II) roughly followed the time profile of HS− over multiple additions, indicating the sediments sustained their ability to oxidize sulfide with very short reoxidation times (Figure 9). The ROS response of this system was also monitored. The sequential addition of HS− spikes to these samples resulted in an apparent decrease in superoxide immediately after each addition, however hydrogen peroxide tended to increase in concentration after the pulse while superoxide fell or plateaued (Figure 10). Hydrogen peroxide did not rise to higher levels than previously observed, indicating that consumption was occurring simultaneously with production. Regardless of brief changes in the relative slope of the time profile of superoxide or hydrogen peroxide, the introduction of multiple HS− pulses reduced the apparent plateau concentrations of superoxide by ~25% and hydrogen peroxide by ~50%. It is possible these reductions were an outcome of the accumulation of partially oxidized S species in the system such as S8, which coat sediment surfaces and inhibit their ability to act as catalysts or directly scavenge oxidants (Rickard et al., 1995; Rickard and Luther, 2007).
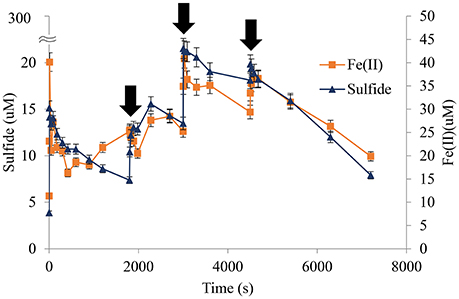
Figure 9. Sulfide spiked into sediments correlated with brief reappearances of Fe(II). Multiple aliquots of sulfide were added to sediments in the presence of oxygen. Sulfide was consumed rapidly in all cases with the nominal concentration of 300 × 10−6 M at each spike not detected. Dissolved Fe(II) increased slightly corresponding with each addition but was reoxidized on a similar timescale to that of the initiating pulse ( indicates time of sulfide addition, pH 8.25, 1.00 wt% marsh mud shown).
indicates time of sulfide addition, pH 8.25, 1.00 wt% marsh mud shown).
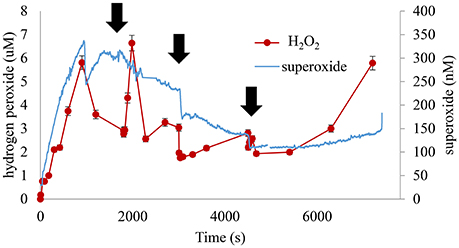
Figure 10. Hydrogen sulfide addition decreased instantaneous ROS concentrations. The repeated addition of separate aliquots of hydrogen sulfide resulted in initial declines in ROS followed by slow recovery to pre-spike level. However, the system was robustly catalytic for ROS production overall and the variance between the highest and lowest ROS concentrations was generally less than a factor of 2 ( indicates time of sulfide addition, pH 8.25, 1.00 wt% marsh mud shown, each addition sufficient for a net 300 × 10−6 M increase in sulfide).
In contrast added hydrogen peroxide pulses did not have a statistically significant effect on measured HS− or Fe(II) under these conditions (Figure 11). However, superoxide appeared to experience a (50–100) × 10−9 M increase after each addition (Figure 12). The most interesting result from this experiment was the changing slope of hydrogen peroxide post-spike; it was evident that the initial spike was consumed rapidly while the latter two appeared to demonstrate more of a step function-like increase. Presumably this was due to consumption of oxidizable sulfur species, at least on the time scale of this study, so that later additions were more stable.
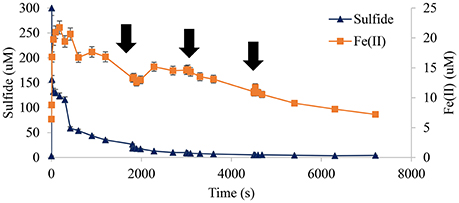
Figure 11. Contrasting sequential additions of hydrogen peroxide had no statistically significant effect on Fe(II) or HS−. Therefore, hydrogen peroxide was not a source of feedback or reductive equivalents that affected Fe(II) or the rate of HS− oxidation ( indicates time of peroxide addition pH, 8.25, 1.00 wt% marsh mud shown, each addition sufficient for a net 10 × 10−6 M increase in peroxide).
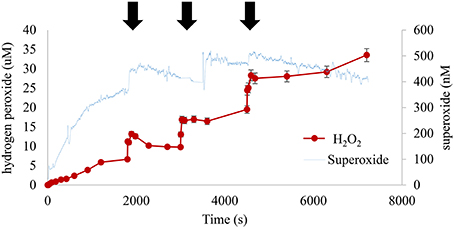
Figure 12. Sequential hydrogen peroxide spikes were increasingly stable. The initial hydrogen peroxide addition decayed rapidly while the latter two were essentially stable additions, indicating that peroxide-consuming reactions were still taking place at the time of first addition. ( indicates time of peroxide addition pH, 8.25, 1.00 wt% marsh mud shown, each addition sufficient for a net 10 × 10−6 M increase in peroxide).
Conclusions
This work demonstrated the potential for the global sulfide reoxidation flux to participate in ROS production in parallel to more recognized photoproduction of ROS. Specifically addition of hydrogen sulfide to oxic muds resulted in the rapid production of Fe(II) species, superoxide and hydrogen peroxide. The choice of hydrogen sulfide re-oxidation as an initial strategy to parameterize non-photochemical ROS production was justifiable because the speciation and redox characteristics of the sulfur cycle are well-established. It was particularly important that sulfate is very soluble in seawater and that there are no naturally occurring, known abiotic mechanisms for reducing sulfate to hydrogen sulfide in oxygenated solutions. In contrast, although the Fe cycle is well-studied, Fe(III) can be readily directly reduced by organic carbon or some ROS in the water column and its solubility is limited. These factors made it much more difficult to analyze reported Fe fluxes and extrapolate the number of reductive equivalents potentially transferred to O2.
The production of ROS was not quantitative and between 50 and 5% of the sulfide consumed appeared to contribute to ROS production in this system (based on estimated superoxide production and measured peroxide). These estimates are based on the assumption hydrogen peroxide was conservative in this system on the timescale of the experiment. Based on the sulfide re-oxidation budget posited in the introduction this suggests global HS− derived superoxide input on the order of 4.5–2.25 × 1012 moles/yr near the sediment/water column interface. In environments where the oxic/anoxic mixing zone is very near the sediment surface this implies an ROS production intensity comparable to photoderived ROS production in surface waters. Hydrous ferric oxides played the most significant role in promoting ROS formation over short time scales. The time scale of the experiments shown corresponds to previously measured efflux of the anoxic portion tidal prism through estuarine muds during the falling tide, suggesting that hydrous ferric oxides will be important sources of ROS in those ecosystems. They are also likely to be influential for ROS production in other episodic events, such as bioturbation, storm-driven agitation, dredging etc. It is notable that elevated levels of antioxidant enzymes are frequently observed in biota at environmental compartments that fall in this category, including hydrothermal vents (Bebianno et al., 2005; Geszvain et al., 2012; Genard et al., 2013), cold seeps (Bernhard and Bowser, 2008), and the surface sediments of many coastal salt marshes (Abele et al., 1998a,b; Company et al., 2006). These observations range from single-celled (planktonic) to complex multicellular organisms (limpets, worms) and suggest ROS may have an unexpected ecological importance even in niches that are reliably aphotic because of depth.
Author Contributions
SAM, SM, and JF conceived and designed the experiments; SAM, SM, DD, and BS performed the experiments; SAM, SM, and JF analyzed the data; SAM, TS, and JF co-wrote the paper. All authors reviewed the document prior to submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by the United States National Science Foundation, Grant CHE-1308801. The authors are grateful to Prof. Bill Miller for helpful comments during the preparation of the paper.
References
Abele, D., Burlando, B., Viarengo, A., and Portner, H. O. (1998a). Exposure to elevated temperatures and hydrogen peroxide elicits oxidative stress and antioxidant response in the Antarctic intertidal limpet Nacella concinna. Comp. Biochem. Phys. B 120, 425–435. doi: 10.1016/S0305-0491(98)10028-7
Abele, D., Grosspietsch, H., and Portner, H. O. (1998b). Temporal fluctuations and spatial gradients of environmental P-O2, temperature, H2O2 and H2S in its intertidal habitat trigger enzymatic antioxidant protection in the capitellid worm Heteromastus filiformis. Mar. Ecol. Prog. Ser. 163, 179–191. doi: 10.3354/meps163179
Afonso, M. D., and Stumm, W. (1992). Reductive dissolution of Iron(III) (Hydr)oxides by hydrogen-sulfide. Langmuir 8, 1671–1675. doi: 10.1021/la00042a030
Aller, J. Y., Aller, R. C., Kemp, P. F., Chistoserdov, A. Y., and Madrid, V. M. (2010). Fluidized muds: a novel setting for the generation of biosphere diversity through geologic time*. Geobiology 8, 169–178. doi: 10.1111/j.1472-4669.2010.00234.x
Aller, R. C., and Blair, N. E. (2006). Carbon remineralization in the Amazon-Guianas tropical mobile mudbelt: a sedimentary incinerator. Cont. Shelf Res. 26, 2241–2259. doi: 10.1016/j.csr.2006.07.016
Aller, R. C., Madrid, V., Chistoserdov, A., Aller, J. Y., and Heilbrun, C. (2010). Unsteady diagenetic processes and sulfur biogeochemistry in tropical deltaic muds: implications for oceanic isotope cycles and the sedimentary record. Geochim. Cosmochim. Acta 74, 4671–4692. doi: 10.1016/j.gca.2010.05.008
Anastacio, A. S., Harris, B., Yoo, H.-I., Fabris, J. D., and Stucki, J. W. (2008). Limitations of the ferrozine method for quantitative assay of mineral systems for ferrous and total iron. Geochim. Cosmochim. Acta 72, 5001–5008. doi: 10.1016/j.gca.2008.07.009
Bebianno, M. J., Company, R., Serafim, A., Camus, L., Cosson, R. P., and Fiala-Medoni, A. (2005). Antioxidant systems and lipid peroxidation in Bathymodiolus azoricus from Mid-Atlantic Ridge hydrothermal vent fields. Aquat. Toxicol. 75, 354–373. doi: 10.1016/j.aquatox.2005.08.013
Bernhard, J. M., and Bowser, S. S. (2008). Peroxisome proliferation in foraminifera inhabiting the chemocline: an adaptation to reactive oxygen species exposure? J. Eukaryot. Microbiol. 55, 135–144. doi: 10.1111/j.1550-7408.2008.00318.x
Bielski, B. H. J. (1978). Reevaluation of spectral and kinetic-properties of HO2 and free radicals. Photochem. Photobiol. 28, 645–649. doi: 10.1111/j.1751-1097.1978.tb06986.x
Bielski, B. H. J., Cabelli, D. E., Arudi, R. L., and Ross, A. B. (1985). Reactivity of HO2/ radicals in aqueous solution. J. Phys. Chem. Ref. Data 14, 1041–1100, doi: 10.1063/1.555739
Bottrell, S. H., and Newton, R. J. (2006). Reconstruction of changes in global sulfur cycling from marine sulfate isotopes. Earth-Sci. Rev. 75, 59–83. doi: 10.1016/j.earscirev.2005.10.004
Bowles, M. W., Mogollón, J. M., Kasten, S., Zabel, M., and Hinrichs, K. U. (2014). Global rates of marine sulfate reduction and implications for sub-sea-floor metabolic activities. Science 344, 889–891. doi: 10.1126/science.1249213
Burns, J. M., Craig, P. S., Shaw, T. J., and Ferry, J. L. (2010). Multivariate examination of Fe(II)/Fe(III) cycling and consequent hydroxyl radical generation. Environ. Sci. Technol. 44, 7226–7231. doi: 10.1021/es903519m
Burns, J. M., Craig, P. S., Shaw, T. J., and Ferry, J. L. (2011a). Short-term Fe cycling during Fe(II) oxidation: exploring joint oxidation and precipitation with a combinatorial system. Environ. Sci. Technol. 45, 2663–2669. doi: 10.1021/es102748p
Burns, J. M., Craig, P. S., Shaw, T. J., and Ferry, J. L. (2011b). Combinatorial parameter space as an empirical tool for predicting water chemistry: Fe(II) oxidation across a watershed. Environ. Sci. Technol. 45, 4023–4029. doi: 10.1021/es103631f
Cai, W. J., Luther, G. W., Cornwell, J. C., and Giblin, A. E. (2010). Carbon cycling and the coupling between proton and electron transfer reactions in aquatic sediments in lake champlain. Aquat. Geochem. 16, 421–446. doi: 10.1007/s10498-010-9097-9
Carey, E., and Taillefert, M. (2005). The role of soluble Fe(III) in the cycling of iron and sulfur in coastal marine sediments. Limnol. Oceanogr. 50, 1129–1141. doi: 10.4319/lo.2005.50.4.1129
Catrouillet, C., Davranche, M., Dia, A., Bouhnik-Le Coz, M., Marsac, R., Pourret, O., et al. (2014). Geochemical modeling of Fe(II) binding to humic and fulvic acids. Chem. Geol. 372, 109–118. doi: 10.1016/j.chemgeo.2014.02.019
Chirita, P., and Schlegel, M. L. (2012). Reaction of FeS with Fe(III)-bearing acidic solutions. Chem. Geol. 334, 131–138. doi: 10.1016/j.chemgeo.2012.10.015
Chirita, P., and Schlegel, M. L. (2015). Oxidative dissolution of iron monosulfide (FeS) in acidic conditions: the effect of solid pretreatment. Int. J. Miner. Process. 135, 57–64. doi: 10.1016/j.minpro.2015.02.001
Cline, J. D. (1969). Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 14, 454–458. doi: 10.4319/lo.1969.14.3.0454
Coates, J. D., Councell, T., Ellis, D. J., and Lovley, D. R. (1998). Carbohydrate oxidation coupled to Fe(III) reduction, a novel form of anaerobic metabolism. Anaerobe 4, 277–282. doi: 10.1006/anae.1998.0172
Company, R., Serafim, A., Cosson, R., Fiala-Medioni, A., Dixon, D., and Bebianno, M. J. (2006). Temporal variation in the antioxidant defence system and lipid peroxidation in the gills and mantle of hydrothermal vent mussel Bathymodiolus azoricus. Deep-Sea Res. Part I-Oceanogr. Res. Pap. 53, 1101–1116. doi: 10.1016/j.dsr.2006.05.008
Cunha, I. T., Teixeira, I. F., Albuquerque, A. S., Ardisson, J. D., Macedo, W. a. A., Oliveira, H. S., et al. (2016). Catalytic oxidation of aqueous sulfide in the presence of ferrites (MFe2O4, M = Fe, Cu, Co). Catal. Today 259, 222–227. doi: 10.1016/j.cattod.2015.07.023
Deguillaume, L., Leriche, M., and Chaurnerliac, N. (2005). Impact of radical versus non-radical pathway in the Fenton chemistry on the iron redox cycle in clouds. Chemosphere 60, 718–724. doi: 10.1016/j.chemosphere.2005.03.052
Dollhopf, M. E., Nealson, K. H., Simon, D. M., and Luther, G. W. (2000). Kinetics of Fe(III) and Mn(IV) reduction by the Black Sea strain of Shewanella putrefaciens using in situ solid state voltammetric Au/Hg electrodes. Mar. Chem. 70, 171–180. doi: 10.1016/S0304-4203(00)00021-9
Duinea, M. I., Costas, A., Baibarac, M., and Chiriţǎ, P. (2016). Mechanism of the cathodic process coupled to the oxidation of iron monosulfide by dissolved oxygen. J. Colloid Interface Sci. 467, 51–59. doi: 10.1016/j.jcis.2016.01.010
Field, M. P., and Sherrell, R. M. (2000). Dissolved and particulate Fe in a hydrothermal plume at 9 degrees 45 ' N, East Pacific Rise: slow Fe (II) oxidation kinetics in Pacific plumes. Geochim. Cosmochim. Acta 64, 619–628. doi: 10.1016/S0016-7037(99)00333-6
Fukuto, J. M., Carrington, S. J., Tantillo, D. J., Harrison, J. G., Ignarro, L. J., Freeman, B. A., et al. (2012). Small molecule signaling agents: the integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem. Res. Toxicol. 25, 769–793. doi: 10.1021/tx2005234
Gartman, A., Yucel, M., Madison, A. S., Chu, D. W., Ma, S. F., Janzen, C. P., et al. (2011). Sulfide oxidation across diffuse flow zones of hydrothermal vents. Aquat. Geochem. 17, 583–601. doi: 10.1007/s10498-011-9136-1
Genard, B., Marie, B., Loumaye, E., Knoops, B., Legendre, P., Zal, F., et al. (2013). Living in a hot redox soup: antioxidant defences of the hydrothermal worm Alvinella pompejana. Aquat. Biol. 18, 217–228. doi: 10.3354/ab00498
Geszvain, K., Butterfield, C., Davis, R. E., Madison, A. S., Lee, S. W., Parker, D. L., et al. (2012). The molecular biogeochemistry of manganese(II) oxidation. Biochem. Soc. Trans. 40, 1244–1248. doi: 10.1042/BST20120229
Giel, J. L., Nesbit, A. D., Mettert, E. L., Fleischhacker, A. S., Wanta, B. T., and Kiley, P. J. (2013). Regulation of iron-sulphur cluster homeostasis through transcriptional control of the Isc pathway by 2Fe-2S -IscR in Escherichia coli. Mol. Microbiol. 87, 478–492. doi: 10.1111/mmi.12052
Giggenbach, W. (1972). Optical spectra and equilibrium distribution of polysulfide ions in aqueous solution at 20 degrees. Inorg. Chem. 11, 1201–1207. doi: 10.1021/ic50112a009
Godrant, A., Rose, A. L., Sarthou, G., and Waite, T. D. (2009). New method for the determination of extracellular production of superoxide by marine phytoplankton using the chemiluminescence probes MCLA and red-CLA. Limnol. Oceanogr. Methods 7, 682–692. doi: 10.4319/lom.2009.7.682
Havig, J. R., McCormick, M. L., Hamilton, T. L., and Kump, L. R. (2015). The behavior of biologically important trace elements across the oxic/euxinic transition of meromictic Fayetteville Green Lake, New York, USA. Geochim. Cosmochim. Acta 165, 389–406. doi: 10.1016/j.gca.2015.06.024
Hoffmann, M. R., and Lim, B. C. (1979). Kinetics and mechanism of the oxidation of sulfide by oxygen - catalysis by homogeneous metal-phthalocyanine complexes. Environ. Sci. Technol. 13, 1406–1414. doi: 10.1021/es60159a014
Johnston, S. G., Keene, A. F., Bush, R. T., Burton, E. D., Sullivan, L. A., Isaacson, L., et al. (2011). Iron geochemical zonation in a tidally inundated acid sulfate soil wetland. Chem. Geol. 280, 257–270. doi: 10.1016/j.chemgeo.2010.11.014
King, D. W., Lounsbury, H. A., and Millero, F. J. (1995). Rates and mechanism of Fe(II) oxidation at nanomolar total iron concentrations. Environ. Sci. Technol. 29, 818–824. doi: 10.1021/es00003a033
Konovalov, S. K., Luther, G. W., and Yucel, M. (2007). Porewater redox species and processes in the Black Sea sediments. Chem. Geol. 245, 254–274. doi: 10.1016/j.chemgeo.2007.08.010
Kostka, J. E., and Luther, G. W. (1994). Partitioning and speciation of solid-phase iron in salt-marsh sediments. Geochim. Cosmochim. Acta 58, 1701–1710. doi: 10.1016/0016-7037(94)90531-2
Kostka, J. E., and Luther, G. W. (1995). Seasonal cycling of fe in salt-marsh sediments. Biogeochemistry 29, 159–181. doi: 10.1007/BF00000230
Kubo, K., Knittel, K., Amann, R., Fukui, M., and Matsuura, K. (2011). Sulfur-metabolizing bacterial populations in microbial mats of the Nakabusa hot spring, Japan. Syst. Appl. Microbiol. 34, 293–302. doi: 10.1016/j.syapm.2010.12.002
Larsen, O., Postma, D., and Jakobsen, R. (2006). The reactivity of iron oxides towards reductive dissolution with ascorbic acid in a shallow sandy aquifer - (Romo, Denmark). Geochim. Cosmochim. Acta 70, 4827–4835. doi: 10.1016/j.gca.2006.03.027
Lewis, B. L., Glazer, B. T., Montbriand, P. J., Luther, G. W., Nuzzio, D. B., Deering, T., et al. (2007). Short-term and interannual variability of redox-sensitive chemical parameters in hypoxic/anoxic bottom waters of the Chesapeake Bay. Mar. Chem. 105, 296–308. doi: 10.1016/j.marchem.2007.03.001
Lichtschlag, A., Kamyshny, A., Ferdelman, T. G., and deBeer, D. (2013). Intermediate sulfur oxidation state compounds in the euxinic surface sediments of the Dvurechenskii mud volcano (Black Sea). Geochim. Cosmochim. Acta 105, 130–145. doi: 10.1016/j.gca.2012.11.025
Lin, C., Larsen, E. I., Larsen, G. R., Cox, M. E., and Smith, J. J. (2012). Bacterially mediated iron cycling and associated biogeochemical processes in a subtropical shallow coastal aquifer: implications for groundwater quality. Hydrobiologia 696, 63–76. doi: 10.1007/s10750-012-1184-z
Lohmayer, R., Kappler, A., Lösekann-Behrens, T., and Planer-Friedrich, B. (2014). Sulfur species as redox partners and electron shuttles for ferrihydrite reduction by Sulfurospirillum deleyianum. Appl. Environ. Microb. 80, 3141–3149. doi: 10.1128/AEM.04220-13
Luther, G. W. (2010). The role of one- and two-electron transfer reactions in forming thermodynamically unstable intermediates as barriers in multi-electron redox reactions. Aquat. Geochem. 16, 395–420. doi: 10.1007/s10498-009-9082-3
Luther, G. W., Findlay, A. J., Macdonald, D. J., Owings, S. M., Hanson, T. E., Beinart, R. A., et al. (2011). Thermodynamics and kinetics of sulfide oxidation by oxygen: a look at inorganically controlled reactions and biologically mediated processes in the environment. Front. Microbiol. 2:62. doi: 10.3389/fmicb.2011.00062
Luther, G. W., Ferdelman, T. G., Kostka, J. E., Tsamakis, E. J., and Church, T. M. (1991). Temporal and spatial variability of reduced sulfur species (Fes2,S2o32-) and porewater parameters in salt-marsh sediments. Biogeochemistry 14, 57–88. doi: 10.1007/BF00000886
Luther, G. W., Rickard, D. T., Theberge, S., and Olroyd, A. (1996). Determination of metal (Bi)Sulfide stability constants of Mn2+, Fe2+, Co2+, Ni2+, Cu2+, and Zn2+ by voltammetric methods. Environ. Sci. Technol. 30, 671–679. doi: 10.1021/es950417i
Ma, S. F., Noble, A., Butcher, D., Trouwborst, R. E., and Luther, G. W. (2006). Removal of H2S via an iron catalytic cycle and iron sulfide precipitation in the water column of dead end tributaries. Estuar. Coast. Shelf Sci. 70, 461–472. doi: 10.1016/j.ecss.2006.06.033
Michaud, E., Aller, R. C., and Stora, G. (2010). Sedimentary organic matter distributions, burrowing activity, and biogeochemical cycling: natural patterns and experimental artifacts. Estuar. Coast. Shelf Sci. 90, 21–34. doi: 10.1016/j.ecss.2010.08.005
Moore, T. S., Nuzzio, D. B., Di Toro, D. M., and Luther, G. W. (2009b). Oxygen dynamics in a well mixed estuary, the lower Delaware Bay, USA. Mar. Chem. 117, 11–20. doi: 10.1016/j.marchem.2009.08.003
Moore, T. S., Shank, T. M., Nuzzio, D. B., and Luther, G. W. (2009a). Time-series chemical and temperature habitat characterization of diffuse flow hydrothermal sites at 9 degrees 50 ′ N East Pacific Rise. Deep-Sea Res. Part II 56, 1616–1621. doi: 10.1016/j.dsr2.2009.05.008
Murphy, S. A., Solomon, B. M., Meng, S. N., Copeland, J. M., Shaw, T. J., and Ferry, J. L. (2014). Geochemical production of reactive oxygen species from biogeochemically reduced Fe. Environ. Sci. Technol. 48, 3815–3821. doi: 10.1021/es4051764
Peiffer, S., Behrends, T., Hellige, K., Larese-Casanova, P., Wan, M., and Pollok, K. (2015). Pyrite formation and mineral transformation pathways upon sulfidation of ferric hydroxides depend on mineral type and sulfide concentration. Chem. Geol. 400, 44–55 doi: 10.1016/j.chemgeo.2015.01.023
Perry, K. A., Kostka, J. E., Luther, G. W. III, and Nealson, K. H. (1993). Mediation of sulfur speciation by a black-sea facultative anaerobe. Science 259, 801–803. doi: 10.1126/science.259.5096.801
Poulton, S. W., Krom, M. D., and Raiswell, R. (2004). A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide. Geochim. Cosmochim. Acta 68, 3703–3715. doi: 10.1016/j.gca.2004.03.012
Powers, L. C., and Miller, W. L. (2014). Blending remote sensing data products to estimate photochemical production of hydrogen peroxide and superoxide in the surface ocean. Environ. Sci. Process Impacts 16, 792–806. doi: 10.1039/C3EM00617D
Precht, E., Franke, U., Polerecky, L., and Huettel, M. (2004). Oxygen dynamics in permeable sediments with wave-driven pore water exchange. Limnol. Oceanogr. 49, 693–705. doi: 10.4319/lo.2004.49.3.0693
Rickard, D. (2012). In Sulfidic Sediments and Sedimentary Rocks Vol. 65. Amsterdam: Developments in Sedimentology. Elsevier Science.
Rickard, D., and Luther, G. W. (1997). Kinetics of pyrite formation by the H2S oxidation of iron(II) monosulfide in aqueous solutions between 25 and 125 degrees C: the mechanism. Geochim. Cosmochim. Acta 61, 135–147. doi: 10.1016/S0016-7037(96)00322-5
Rickard, D., and Luther, G. W. (2007). Chemistry of iron sulfides. Chem. Rev. 107, 514–562. doi: 10.1021/cr0503658
Rickard, D., Schoonen, M. A. A., and Luther, G. W. (1995). Chemistry of iron sulfides in sedimentary environments. Geochem. Transform. Sediment. Sulfur 612, 168–193. doi: 10.1021/bk-1995-0612.ch009
Rickard, D. T. (1975). Kinetics and mechanism of pyrite formation at low-temperatures. Am. J. Sci. 275, 636–652. doi: 10.2475/ajs.275.6.636
Roden, E. E., Sobolev, D., Glazer, B., and Luther, G. W. (2004). Potential for microscale bacterial Fe redox cycling at the aerobic-anaerobic interface. Geomicrobiol. J. 21, 379–391. doi: 10.1080/01490450490485872
Rose, A. L., Moffett, J. W., and Waite, T. D. (2008). Determination of superoxide in seawater using 2-methyl-6-(4-methoxyphenyl)-3,7-dihydroimidazo[1,2-a]pyrazin-3(7H)-one chemiluminescence. Anal. Chem. 80, 1215–1227. doi: 10.1021/ac7018975
Rozan, T. F., Taillefert, M., Trouwborst, R. E., Glazer, B. T., Ma, S. F., Herszage, J., et al. (2002). Iron-sulfur-phosphorus cycling in the sediments of a shallow coastal bay: implications for sediment nutrient release and benthic macroalgal blooms. Limnol. Oceanogr. 47, 1346–1354. doi: 10.4319/lo.2002.47.5.1346
Rush, J. D., and Bielski, B. H. J. (1985). Pulse radiolytic studies of the reactions of HO2/ with Fe(II)/Fe(III) ions - the reactivity of HO2/ with ferric ions and its implication on the occurrence of the Haber-Weiss reaction. J. Phys. Chem. 89, 5062–5066. doi: 10.1021/j100269a035
Santana-Casiano, J. M., González-Dávila, M., and Millero, F. J. (2005). Oxidation of nanomolar levels of Fe(II) with oxygen in natural waters. Environ. Sci. Technol. 39, 2073–2079. doi: 10.1021/es049748y
Santos, I. R., Eyre, B. D., and Huettel, M. (2012). The driving forces of porewater and groundwater flow in permeable coastal sediments: a review. Estuar. Coast. Shelf Sci. 98, 1–15. doi: 10.1016/j.ecss.2011.10.024
Sekar, R., and DiChristina, T. J. (2014). Microbially driven fenton reaction for degradation of the widespread environmental contaminant 1,4-dioxane. Environ. Sci. Technol. 48, 12858–12867. doi: 10.1021/es503454a
Sima, J., and Makanova, J. (1997). Photochemistry of iron(III) complexes. Coord. Chem. Rev. 160, 161–189. doi: 10.1016/S0010-8545(96)01321-5
Simpson, S. L. (2001). A rapid screening method for acid-volatile sulfide in sediments. Environ. Toxicol. Chem. 20, 2657–2661. doi: 10.1002/etc.5620201201
Snyder, M., Taillefert, M., and Ruppel, C. (2004). Redox zonation at the saline-influenced boundaries of a permeable surficial aquifer: effects of physical forcing on the biogeochemical cycling of iron and manganese. J. Hydrol. 296, 164–178. doi: 10.1016/j.jhydrol.2004.03.019
Stookey, L. L. (1970). Ferrozine - a new spectrophotometric reagent for iron. Anal. Chem. 42, 779–781. doi: 10.1021/ac60289a016
Sulzberger, B., and Laubscher, H. (1995). Reactivity of various types of iron(III) (hydr)oxides towards light-induced dissolution. Mar. Chem. 50, 103–115. doi: 10.1016/0304-4203(95)00030-U
Taillefert, M., Hover, V. C., Rozan, T. F., Theberge, S. M., and Luther, G. W. (2002). The influence of sulfides on soluble organic-Fe(III) in anoxic sediment porewaters. Estuaries 25, 1088–1096. doi: 10.1007/BF02692206
Theil, E. C., Behera, R. K., and Tosha, T. (2013). Ferritins for chemistry and for life. Coord. Chem. Rev. 257, 579–586. doi: 10.1016/j.ccr.2012.05.013
Trapp, J. M., and Millero, F. J. (2007). The oxidation of iron(II) with oxygen in NaCl brines. J. Solut. Chem. 36, 1479–1493. doi: 10.1007/s10953-007-9192-8
Vazquez, F., Zhang, J. Z., and Millero, F. J. (1989). Effect of metals on the rate of the oxidation of H2S in seawater. Geophys. Res. Lett. 16, 1363–1366. doi: 10.1029/GL016i012p01363
Veverica, T. J., Kane, E. S., Marcarelli, A. M., and Green, S. A. (2016). Ionic liquid extraction unveils previously occluded humic-bound iron in peat soil pore water. Soil Sci. Soc. Am. J. 80, 771–782. doi: 10.2136/sssaj2015.10.0377
Voelker, B. M., Morel, F. M. M., and Sulzberger, B. (1997). Iron redox cycling in surface waters: effects of humic substances and light. Environ. Sci. Technol. 31, 1004–1011. doi: 10.1021/es9604018
Wan, M. L., Shchukarev, A., Lohmayer, R., Planer-Friedrich, B., and Peiffer, S. (2014). Occurrence of surface polysulfides during the interaction between ferric (Hydr)oxides and aqueous sulfide. Environ. Sci. Technol. 48, 5076–5084. doi: 10.1021/es405612f
Warneck, P., and Ziajka, J. (1995). Reaction mechnanism of the iron(III) catalyzed autoxidation of bisulfite in aqueous solution - steady state description for benzene as radicl scavenger. Ber. Bunsen-Ges. Phys. Chem. Chem. Phys. 99, 59–65. doi: 10.1002/bbpc.19950990109
Watson, A., Lovelock, J. E., and Margulis, L. (1978). Methanogenesis, fires and the regulation of atmospheric oxygen. Biosystems 10, 293–298. doi: 10.1016/0303-2647(78)90012-6
Yucel, M., Mullaugh, K. M., and Luther, G. W. (2009). Post-eruption sulfide and iron content of hydrothermal vent fluids from East Pacific Rise, 9 degrees 50 ′ N. Geochim. Cosmochim. Acta 73, A1492–A1492.
Zepp, R. G., Callaghan, T. V., and Erickson, D. J. (1998). Effects of enhanced solar ultraviolet radiation on biogeochemical cycles. J. Photochem. Photobiol. B Biol. 46, 69–82. doi: 10.1016/S1011-1344(98)00186-9
Zhou, M. J., Diwu, Z. J., PanchukVoloshina, N., and Haugland, R. P. (1997). A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal. Biochem. 253, 162–168. doi: 10.1006/abio.1997.2391
Keywords: nanomaterial, ISCO, hydroxyl radical, mineralization, catalytic
Citation: Murphy SA, Meng S, Solomon BM, Dias DMC, Shaw TJ and Ferry JL (2016) Hydrous Ferric Oxides in Sediment Catalyze Formation of Reactive Oxygen Species during Sulfide Oxidation. Front. Mar. Sci. 3:227. doi: 10.3389/fmars.2016.00227
Received: 16 June 2016; Accepted: 31 October 2016;
Published: 17 November 2016.
Edited by:
William L. Miller, University of Georgia, USAReviewed by:
Elizabeth Swanner, Iowa State University, USAMartial Taillefert, Georgia Institute of Technology, USA
Copyright © 2016 Murphy, Meng, Solomon, Dias, Shaw and Ferry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John L. Ferry, ferry@sc.edu