Structural Characterization of Natural Nickel and Copper Binding Ligands along the US GEOTRACES Eastern Pacific Zonal Transect
 Rene M. Boiteau1,2
Rene M. Boiteau1,2  Claire P. Till3,4
Claire P. Till3,4  Angel Ruacho5
Angel Ruacho5  Randelle M. Bundy1
Randelle M. Bundy1  Nicholas J. Hawco1,2
Nicholas J. Hawco1,2  Amy M. McKenna6
Amy M. McKenna6  Katherine A. Barbeau5
Katherine A. Barbeau5  Kenneth W. Bruland3
Kenneth W. Bruland3  Mak A. Saito1
Mak A. Saito1  Daniel J. Repeta1*
Daniel J. Repeta1*- 1Department of Marine Chemistry and Geochemistry, Woods Hole Oceanographic Institution, Woods Hole, MA, USA
- 2Department of Earth, Atmospheric, and Planetary Sciences, Massachusetts Institute of Technology, Cambridge, MA, USA
- 3Ocean Sciences Department, University of California, Santa Cruz, CA, USA
- 4Chemistry Department, Humboldt State University, Arcata, CA, USA
- 5Scripps Institution of Oceanography, University of California, San Diego, La Jolla, CA, USA
- 6National High Magnetic Field Laboratory, Florida State University, Tallahassee, FL, USA
Organic ligands form strong complexes with many trace elements in seawater. Various metals can compete for the same ligand chelation sites, and the final speciation of bound metals is determined by relative binding affinities, concentrations of binding sites, uncomplexed metal concentrations, and association/dissociation kinetics. Different ligands have a wide range of metal affinities and specificities. However, the chemical composition of these ligands in the marine environment remains poorly constrained, which has hindered progress in modeling marine metal speciation. In this study, we detected and characterized natural ligands that bind copper (Cu) and nickel (Ni) in the eastern South Pacific Ocean with liquid chromatography tandem inductively coupled plasma mass spectrometry (LC-ICPMS), and high-resolution electrospray ionization mass spectrometry (ESIMS). Dissolved Cu, Ni, and ligand concentrations were highest near the coast. Chromatographically unresolved polar compounds dominated ligands isolated near the coast by solid phase extraction. Offshore, metal and ligand concentrations decreased, but several new ligands appeared. One major ligand was detected that bound both Cu2+ and Ni2+. Based on accurate mass and fragmentation measurements, this compound has a molecular formula of [C20H21N4O8S2+M]+ (M = metal isotope) and contains several azole-like metal binding groups. Additional lipophilic Ni complexes were also present only in oligotrophic waters, with masses of 649, 698, and 712 m/z (corresponding to the 58Ni metal complex). Molecular formulae of [C32H54N3O6S2Ni]+ and [C33H56N3O6S2Ni]+ were determined for two of these compounds. Addition of Cu and Ni to the samples also revealed the presence of additional compounds that can bind both Ni and Cu. Although these specific compounds represent a small fraction of the total dissolved Cu and Ni pool, they highlight the compositional diversity and spatial heterogeneity of marine Ni and Cu ligands, as well as variability in the extent to which different metals in the same environment compete for ligand binding.
Introduction
Strong organic ligands affect the solubility and reactivity of Cu and Ni in marine environments and impact the bioavailability of these essential micronutrient metals (Vraspir and Butler, 2009). Studies of Cu speciation using electrochemical methods show that more than 99% of dissolved Cu is complexed by strong organic ligands in the surface ocean (Coale and Bruland, 1988, 1990; Moffett et al., 1990; Donat and van den Berg, 1992; Campos and van den Berg, 1994; Moffett, 1995; Buck and Bruland, 2005; Jacquot et al., 2013; Thompson et al., 2014; Jacquot and Moffett, 2015). In comparison, a smaller fraction (30–50%) of dissolved Ni is complexed by strong organic ligands (van den Berg and Nimmo, 1987; Nimmo and van Den Berg, 1989; Zhang et al., 1990; Donat et al., 1994; Achterberg and Van Den Berg, 1997; Saito and Moffett, 2004; Saito et al., 2005). Although these electrochemical studies do not provide direct information on the chemical composition of ligands, they do suggest that structurally diverse ligands with distinct sources and binding strengths are present throughout the ocean (e.g., Buck and Bruland, 2005; Bundy et al., 2013).
Marine organisms are capable of producing a wide variety of Cu and Ni chelators that could potentially contribute to the natural ligand pool. Some Ni and Cu chelators such as glutathione, phytochelatins, and metallothioneins are produced to chelate metals as a detoxification mechanism (Ahner et al., 1994, 1997, 2002; Dupont et al., 2004; Freeman et al., 2004; Wei and Ahner, 2005; Devez et al., 2009). Other chelators such as methanobactins and staphylopines are produced to facilitate Cu or Ni acquisition (Kim et al., 2004; El Ghazouani et al., 2012; Ghssein et al., 2016). Ni is a constituent of some metabolite cofactors such as the tetrapyrrole F430, the prosthetic group of the enzyme involved in the final step of methanogenesis (Allen et al., 2014), while other pyrrole and azole-based modified peptides are known to have a strong affinity for Ni and Cu, although their physiological roles are unknown (Michael et al., 1993; Bertram and Pattenden, 2007; Comba et al., 2014). Although not directly produced by microbes, marine humic compounds, structurally heterogeneous refractory degradation products of organic matter decay, may also complex Cu and Ni (Baker and Khalili, 2003; Yang and Van den Berg, 2009; Whitby and Van den Berg, 2014; Abualhaija et al., 2015).
The chemical composition of Cu and Ni ligands determines their metal binding stability constants and turnover rates. In general, there is competition between the two metals for ligand binding sites, with Cu2+ usually forming more stable organic complexes compared to Ni2+, following the Irving Williams series (Irving and Williams, 1953). Since ligand concentrations are nearly always in excess of Cu concentrations, Cu tends to saturate stronger ligands, leaving weaker ligands free to coordinate Ni (and other cations). Thus, the speciation of metals depends on the relative binding strengths of co-occurring ligands for Cu and Ni. The rate at which these ligands approach equilibrium (i.e., kinetics of metal association and dissociation) also depends on ligand structure, as do the rates at which the ligands are produced and degraded in the environment.
Ligands also affect metal redox chemistry. The +II oxidation state of inorganic Cu and Ni are thermodynamically favored in oxic seawater (Turner et al., 1981). However, Cu can be reduced to the +I oxidation state by biological and photochemical processes (Moffett and Zika, 1983; Wuttig et al., 2013). Organic ligands possessing hard base electron donors such as nitrogen and oxygen can stabilize the +II oxidation state, while ligands with sulfur binding groups such as phytochelatin or methanobactin can stabilize the +I state, and can even catalyze Cu2+ reduction (Michael et al., 1993; Walsh and Ahner, 2013).
Structural characterization of organic ligands in seawater has remained challenging. Ligands occur at very low concentrations within a highly complex mixture of organic matter. Since high salinity matrices are unsuitable for most mass spectral techniques, solid phase extraction is typically used to remove salts and concentrate ligands. Solid phase extraction resins are available with a range of surface chemistries that can be selected to retain different fractions of the organic ligand pool (Waska et al., 2015). Liquid chromatography coupled to inductively coupled plasma mass spectrometry (LC-ICPMS) provides a means to detect and quantify metal ligand complexes directly, and to investigate compositional changes between samples (Mounicou et al., 2009; Boiteau et al., 2013). However, LC-ICPMS does not provide information on the chemical identity of ligands. Chromatographic separation combined with electrospray ionization mass spectral characterization (ESIMS) provides molecular weights for metal-containing compounds based on peak coherence between chromatographic features that match the isotopic fine structure of metals such as Fe, Cu and Ni (Mounicou et al., 2009; Baars et al., 2014, 2015; Boiteau and Repeta, 2015). Finally, high mass accuracy Fourier transform ion cyclotron resonance mass spectrometry (FT-ICRMS) can achieve resolving power and mass accuracy sufficient to assign elemental formulas to metal containing compounds (McKenna et al., 2014; Waska et al., 2015).
In this study, we combined these techniques to provide robust molecular formula assignments to several of the major nickel and copper binding compounds in the Eastern Pacific. This region exhibits strong surface gradients in metal concentrations and biological activity. The surface waters in the eastern upwelling region off Ecuador and Peru are some of the most productive in the world, fueled by the supply of nutrients and metals from deep waters (Pennington et al., 2006). In the western offshore region, metal concentrations are very low and can influence microbial community composition (Moore et al., 2013). We detected Ni and Cu ligands derived from seawater extracts using LC-ICPMS. We then used LC-ESIMS and metal isotope pattern detection algorithms to characterize metal binding compounds by molecular weight and mass fragmentation spectra, and determined molecular formulae through ESI FT-ICRMS. Finally, we investigated the effect of metal additions on the competition of metals for binding these compounds.
Methods
Materials and Reagents
Samples were collected and processed using protocols to minimize trace metal contamination (Boiteau et al., in press). All solutions were prepared using trace metal clean reagents and high purity water (18.2 MΩ cm, qH2O). LCMS grade methanol (MeOH; Optima, Fisher Scientific) used for chromatography was further purified by sub-boiling-point distillation in a Polytetrafluoroethylene (PTFE) still to reduce metal contamination. PTFE solid phase extraction (SPE) column hardware and vials were cleaned overnight with 0.1% detergent (Citranox), rinsed with qH2O, soaked in 1 N HCl (trace metal grade, Fisher Scientific) for 1–2 days before a final qH2O rinse. PTFE tubing and polyethersulfone capsule filters (0.2 μm Millipore) used to process seawater were flushed overnight with 1% HCl and rinsed with qH2O. Low-density polyethylene sample bottles for trace metal concentration measurements were cleaned in accordance with GEOTRACES sampling protocols (http://www.geotraces.org), stored in dilute (~0.1 N) HNO3, and rinsed three times with filtered sample before filling. Boric acid buffer (1.5 M) for electrochemical measurements of Cu binding ligands was prepared in 0.4 N aqueous NH4OH (Optima, Fisher Scientific). A 4 mM stock of salicylaldoxime (SA; > 98%) was prepared in methanol (Optima LCMS, Fisher Scientific) and was replaced every 6 months or as needed. Copper standards (100 nM to 10 μM) were diluted from an atomic adsorption standard (1000 ppm, Spex CertiPrep) into pH 2 qH2O (acidified with Optima grade HCl). For electrochemical Ni measurements, a 0.5 M EPPS buffer [N-(2-hydroxyethyl)piperazine-N-(3-propanesulfonic acid)] and a 1.5 M sodium nitrite solution were each prepared in qH2O and percolated through cleaned Chelex-100 resin (Biorad) before use. Dimethylglyoxime (DMG) was recrystallized as described by Saito and Moffett (2001) and added to Optima MeOH (Fisher Scientific) for a final concentration of 0.1 M. The Ni stock solution was diluted from an AA standard (CertiPrep; 1 mg L−1) to a final concentration of 1 μM. Ferrioxamine E and cyanocobalamin standards were purchased from Sigma Aldrich (St. Louis, Mo, USA).
Sampling
Samples were collected during the GEOTRACES Eastern Pacific Zonal Transect cruise (October-December, 2013) from Manta, Ecuador to Papeete, Tahiti (GP16; Figure 1A). Seawater was collected from 3 m depth with a trace metal clean PTFE tow-fish pump system while steaming between stations (Bruland et al., 2005). Discrete samples for voltammetry and metal concentration measurements were filtered through a 0.2 μm capsule filter (Polyethersulfone, Pall acropak). Metal concentration samples were acidified at sea with the equivalent of 4 mL of 6 M quartz-distilled HCl per liter of seawater (resulting in a pH of 1.7–1.8). Samples for ligand titrations were stored at −20°C until analysis, in acid-clean fluorinated polyethylene bottles. To collect samples for trace metal ligand characterization by chromatography mass spectrometry, seawater was pumped from an under way tow-fish sampling system at a flow rate of 250 mL/min through a 0.2 μm polyethersulfone capsule filter followed by a custom PTFE extraction column (2 cm × 6.5 cm) packed with 20 g polystyrene divinylbenzene resin beads (ENV, Agilent Bondesil). After 600–800 L of water was passed through a column (representing a sampling interval of 1000–2000 km collected over 4–13 days), the column was frozen onboard and replaced with a fresh column for the next sample. Six samples were collected in total (Figure 1A). A sampling blank, collected by pumping only 200 mL of seawater through a column, was processed and analyzed along with the six samples. No Cu or Ni ligands were detected by LCMS in the sampling blank. Before sample collection, the extraction columns were activated by washing with three column volumes of MeOH followed by three column volumes of pH2 qH2O (acidified with HCl), before a final rinse with qH2O. Samples were stored at −20°C. Prior to analysis, columns were thawed, rinsed with 500 mL of qH2O to remove salts, and the organic compounds were eluted with 250 mL of MeOH. Methanol extracts were concentrated by rotary evaporation in an acid cleaned quartz flask and diluted to a final volume of 6 mL with qH2O. An aliquot of the sample was spiked with 1 μM cyanocobalamin (Sigma Aldrich) as an internal standard.
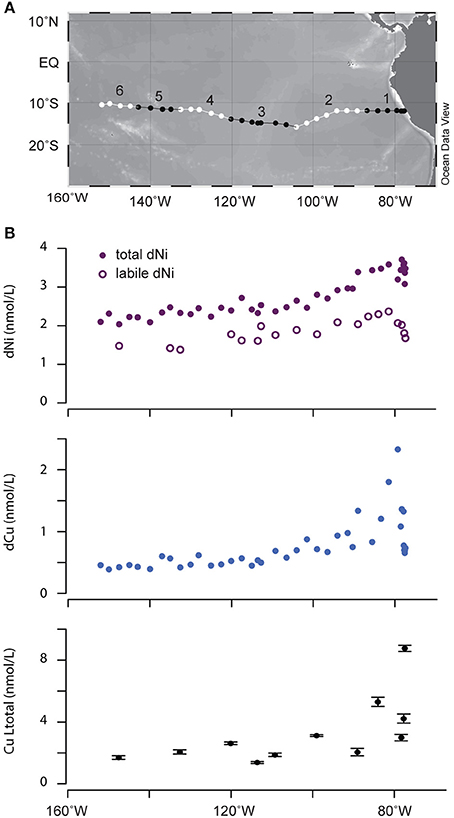
Figure 1. (A) Map of GEOTRACES EPZT section. Alternating black and white lines indicate the six legs over which integrated samples were collected. (B) Surface concentrations of dissolved Ni, Cu, and Cu ligands. Error bars represent ±1 standard deviation based on replicate measurements of Cu ligand concentrations.
Cu Ligand Electrochemical Titrations
Characterization of organic Cu binding ligands across the EPZT section was carried out via competitive ligand exchange adsorptive cathodic stripping voltammetry (CLE-ACSV). CLE-ACSV is an electrochemical method that employs the competition for Cu between a well-characterized ligand [here salicylaldoxime (SA)] and the natural seawater ligands to determine the thermodynamic stabilities of the natural ligands (Campos and van den Berg, 1994). Frozen samples were thawed in a refrigerator and vigorously shaken prior to analysis. The sample was then aliquoted (10 mL) into pre-conditioned PTFE vials and buffered with 1.5 M boric acid-ammonia to pH 8.2. The vials were spiked with increasing concentrations of Cu ranging from 0 to 25 nM, to saturate the natural ligands. The buffer and Cu were left to equilibrate with the natural ligands for 2 h after which the competing ligand, SA, was added at a concentration of 5 μM and left to equilibrate for 15 min. Eleven-point titrations were carried out in duplicate on a controlled growth mercury electrode (Bioanalytical Systems Incorporated) with electrochemical parameters as used previously (Buck and Bruland, 2005; Bundy et al., 2013), and the peaks detected represent SA-labile Cu complexes established during our equilibration period. Due to the short 15 min equilibration time, it is possible that the apparent natural ligand binding strength (logK) is overestimated compared to results from methods that use a longer equilibration time. Peaks generated from titrations were extracted using the ECDSoft software package and processed with ProMCC (Omanović et al., 2014) utilizing a single ligand model.
Electrochemical Determination of Labile Ni
Labile Ni was measured using cathodic stripping voltammetry with an Eco-Chemie μAutolab-III analyzer connected to a Metrohm 663 VA Stand with a hanging mercury drop electrode. Sample aliquots (8.5 mL) were placed in cleaned and conditioned PE vials, recrystallized DMG was added to a final concentration of 200 μM, and allowed to equilibrate overnight in the dark. Just prior to analysis, 100 μL EPPS buffer was added to each sample. Samples were then purged with ultra high purity N2 and conditioned at −0.6 V for 60 s while stirring. This was followed by a 10 s equilibration step and then a linear sweep from −0.6 V to −1.4 V with a 10 V s−1 scan rate. Labile Ni concentrations were determined using triplicate measurements of the initial concentrations followed by 4 standard additions of 1.0, 2.0, 3.0, and 4.0 nM Ni. Labile Ni concentrations were defined as the proportion of total dissolved Ni that was bound to DMG after the 12 h equilibration period, which likely represents weakly bound organic Ni compounds and free Ni2+. The remaining fraction of non-labile Ni represents strong organic Ni compounds with thermodynamic conditional stability constants (logK) ranging from 18.45 to 18.68 given the analytical window used in this study (van den Berg and Nimmo, 1987).
Trace Metal Concentration Measurements
Dissolved Cu and Ni were analyzed using previously described methods (Biller and Bruland, 2012; Parker et al., 2016). Briefly, metals of interest were preconcentrated by extraction from buffered seawater (pH 6.0) onto 2 cm columns of Nobias PA1 chelating resin, and eluted with 1N quartz-distilled HNO3. The eluent was analyzed on an Element XR Inductively Coupled Plasma Mass Spectrometer (Thermo Scientific) in counting mode. Cu and Ni were both analyzed in medium resolution and have recoveries >96%. Samples were UV irradiated prior to preconcentration.
LC-ICPMS-ESIMS Characterization
Organic ligand samples from solid phase extraction were separated by high pressure liquid chromatography (HPLC, Agilent bioinert 1260 series) using a C8 column (Hamilton, 2.1 × 100 mm, 3 μm particle size) and polyetheretherketone (PEEK) tubing and connectors. Compounds were separated over a gradient from 10% MeOH in H2O to 90% MeOH over 50 min followed by isocratic elution at 90% MeOH using 5 mM aqueous ammonium formate as a buffer, and a flow rate of 0.2 mL/min. The flow of the LC column was coupled directly to either ICPMS or ESIMS, using a PEEK post column splitter to reduce the flow to 50 μL/min.
ICPMS parameters were modified from Boiteau et al. (2013). The ICPMS was fitted with a perfluoroalkoxy micronebulizer (PFA-ST, Elemental Scientific), a cyclonic spray chamber cooled to 0°C, and platinum sample and skimmer cones. To prevent organic deposition onto the cones, oxygen gas was introduced at a rate of 25 mL/min. Measurements were made in kinetic energy discrimination mode using 4.2 mL/min He as a collision gas. 59Co, 58Ni, 60Ni, 63Cu, and 65Cu were monitored with an integration time of 0.05 s each. Concentrations of Ni and Cu ligands detected by LC-ICPMS were calculated by integrating peak areas for the 60Ni and 63Cu signals between 5 and 60 min, and converting this area to concentration based on a six point calibration curve of an Fe-ferrioxamine E standard solution (retention time = 19.8 min), with signal sensitivities intercalibrated between metals using ratios obtained by analyzing a 10 ppb Fe, Cu, and Ni solution in 5% nitric acid (Optima, Fisher Scientific).
To determine the mass of the Ni and Cu ligands, the flow from the LC was coupled to an Orbitrap Fusion mass spectrometer (Thermo Scientific) fitted with a heated electrospray ionization (ESI) source. ESI parameters were set to a capillary voltage of 3500 V, sheath, auxiliary and sweep gas flow rates of 12, 6, and 2 (arbitrary units), and ion transfer tube and vaporizer temperatures of 300 and 75°C. MS scans were collected in high resolution (450 K) positive ionization mode. Ions were trapped using a quadrupole isolation window of 1 m/z and were then fragmented using a high energy collision induced dissociation energy of 35%. LC-ESIMS data was converted to mzXML file format with the proteowizard msConvert toolkit (Chambers et al., 2012). Retention times between the LC-ICPMS and LC-ESIMS data were aligned by applying a constant time offset to match the retention time of the cyanocobalamin [M + H]2+ peak (m/z = 678) with the corresponding LC-ICPMS 59Co peak. An isotope pattern search algorithm was used to detect the masses of ions containing Cu and Ni by identifying m/z and intensity features from each scan that fit the mass differences and natural abundance ratios of 63Cu and 65Cu or 58Ni and 60Ni within a mass tolerance of 0.003 m/z and a ratio tolerance of 20%, and then align with the retention time of the LC-ICPMS peaks within 0.1 min. LC-ESIMS data is available as a MassIVE dataset (https://massive.ucsd.edu, accession MSV000080173).
FT-ICR MS Analysis
Organic extract samples were diluted 1:1 in HPLC grade methanol (JT Baker Chemical Co., Centre Valley, PA) with 2% formic acid (by volume) for FT-ICRMS analysis. Sample solutions were analyzed with a custom-built FT-ICR mass spectrometer (Kaiser et al., 2011a) equipped with a 9.4 T horizontal 220 mm bore diameter superconducting solenoid magnet (Oxford Instruments, Abingdon, U.K.) operated at room temperature, and a modular ICR data station (Predator) (Blakney et al., 2011) facilitated instrument control, data acquisition, and data analysis. Solutions were infused via a microelectrospray source (Emmett et al., 1998) (50 μm i.d. fused silica emitter) at 500 nL/min by a syringe pump. Positive ions generated at atmospheric pressure enter the skimmer region (~2 Torr) through a heated metal capillary (7 A), pass through the first radio frequency (rf)-only quadrupole, and are accumulated (3–5 s) in an octopole equipped with tilted wire extraction electrodes for improved ion extraction and transmission (Wilcox et al., 2002). Helium gas introduced during accumulation collisionally cools ions prior to transfer through two rf-only quadrupoles (total length 127 cm) (2.0 MHz and 255 Vp−p amplitude) into a 7-segment open cylindrical ICR cell (Kaiser et al., 2011b) based on the Tolmachev configuration (Tolmachev et al., 2011). For each sample, quadrupole mass filtering within a defined m/z range (~5–30 Da) that corresponded to compounds A–D was applied prior to external ion accumulation in order to improve resolving power and ICR dynamic range (McKenna et al., 2013). Broadband frequency (chirp) excitation (700–70 kHz at a sweep rate of 50 Hz/μs and amplitude of 350 Vp−p) accelerated the ions to a cyclotron orbital radius detected by differential current induced between opposed 120° detection electrodes inside the ICR cell. Fifty individual transients of 6.8 s duration were averaged, apodized with a single-sided Hanning weight function, and zero-filled once prior to fast Fourier transformation. Due to increased complexity at higher m/z, broadband phase correction (Xian et al., 2010, 2012) was applied to each mass spectrum to increase resolution of isobaric species. Absorption-mode spectral resolving power is higher by a factor of up to 2 than magnitude-mode resolving power. The achieved mass spectral resolving power (m/Δm50%, in which Δm50% is the mass spectral peak width at half maximum peak height) was ~2,000,000–2,500,000 in mass isolated segments at m/z 500.
ICR frequencies were converted to ion masses based on the quadrupolar trapping potential approximation (Shi et al., 2000). Each m/z spectrum was internally calibrated with respect to an abundant homologous alkylation series whose members differ in mass by integer multiples of 14.01565 Da (mass of a CH2 unit) and further iterated based on the “walking” calibration equation (Savory et al., 2011). Calibration for quadrupole mass isolated segments typically relied on 30–60 calibration points with sub-ppm mass error (10–70 ppb) root-mean-square error achieved for all peaks with signal magnitude greater than six times the baseline noise.
LC-ICPMS Metal Exchange Experiment
To investigate metal binding competition among Ni and Cu ligands, splits of the sample extract were spiked with 1 mM citrate stock solutions containing (a) no metal, (b) 100 μM Cu, (c) 100 μM Ni, or (d) 100 μM Cu + 100 μM Ni. Citrate was used as a buffer (pH 6.4) and weak stabilizing ligand to keep the metals soluble. Metal stock solutions were prepared from 1000 ppm metal reference standard solutions in 2% nitric acid (Fisher scientific) diluted in qH2O containing 1 mM trisodium citrate dihydrate (Fisher bioreagents). Ninety microliters of sample extract was mixed with 10 μL of the metal stock solution and incubated at room temperature in the dark for 12 h prior to analysis. Samples were analyzed by LC-ICPMS as described above, but using a different chromatography system (UltiMate 3000, Dionex).
Results
Distributions of Cu, Ni, and Electrochemically Detected Cu and Ni Ligands in the Eastern Tropical South Pacific Ocean
Concentrations of dissolved Cu and Ni and metal binding ligands were measured across surface waters in the eastern tropical South Pacific Ocean (Table 1). Higher concentrations of both metals were observed near the coast, likely reflecting inputs from aerosols and from upwelling of subsurface waters that are rich in diagenetically remobilized metals released from coastal sediments (Jacquot et al., 2013; Baker et al., 2016). Moving offshore, Cu and Ni levels decreased until both metals reached relatively stable values west of 100°W (0.4 and 2 nM for Cu and Ni, respectively). For Cu, total ligand concentrations measured by CLE-ACSV consistently exceeded dissolved Cu concentrations across the section by a factor of 1.6–12 (Table 1). Cu ligand concentrations followed the same general trend as dissolved Cu, with higher concentrations near the coast. Ligand binding strength generally decreased moving offshore. These results suggest that the concentration of free (uncomplexed) dissolved Cu is <<1% throughout the surface of the eastern tropical South Pacific Ocean. In contrast, uncomplexed or weakly complexed dissolved Ni (measured as electrochemically labile dissolved Ni, Figure 1B) concentrations ranged between 48 and 78% of total dissolved Ni. The trend of decreasing concentration offshore was also observed for labile Ni, with a lower percentage of labile Ni in coastal stations (Stations 1–6; 57% average labile Ni) compared to the other stations in the transect (Stations 7–34; 68% average labile Ni). If the difference between total dissolved Ni and labile Ni represents the strong organic Ni-binding ligand concentrations, then Ni ligands ranged from 1 to 2 nM at Stations 1–6 and <1 nM in the remaining stations.
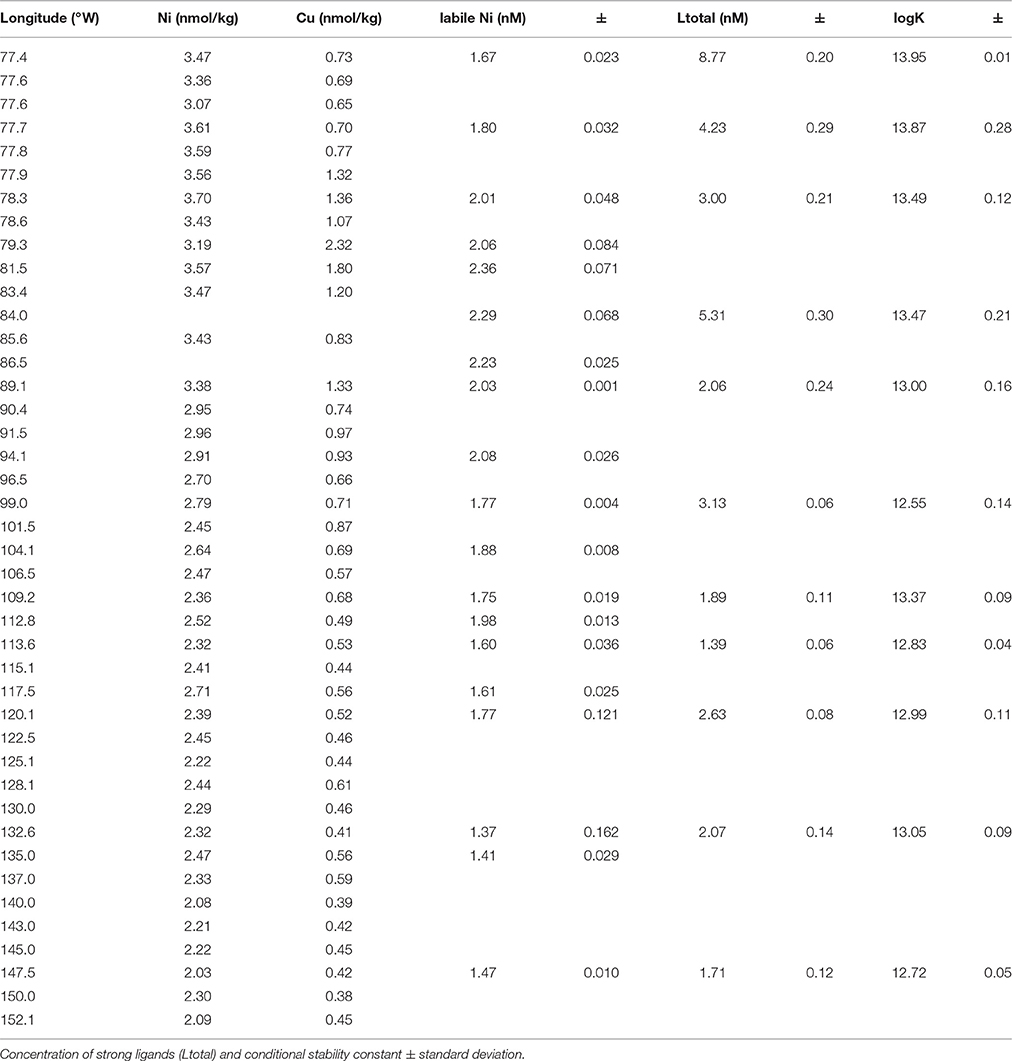
Table 1. Concentration of dissolved Ni, Cu, and Cu ligands in surface waters of the Eastern Pacific.
LCMS Ligand Characterization
Organic ligands complexed to Cu and Ni were isolated by solid phase extraction from surface seawater across six sections of the GEOTRACES cruise (Figure 1A). Ligands were separated by liquid chromatography, and the Ni and Cu associated with them was detected by ICPMS. Metal containing compounds appear in the resulting chromatograms as peaks in signal intensity (Figure 2). This method only detects the metal-complexed portion of the ligand pool, and peak intensity reflects the amount of metal associated with the complex. Retention time provides a measure of the polarity of a compound, with more polar compounds eluting earlier in the chromatogram.
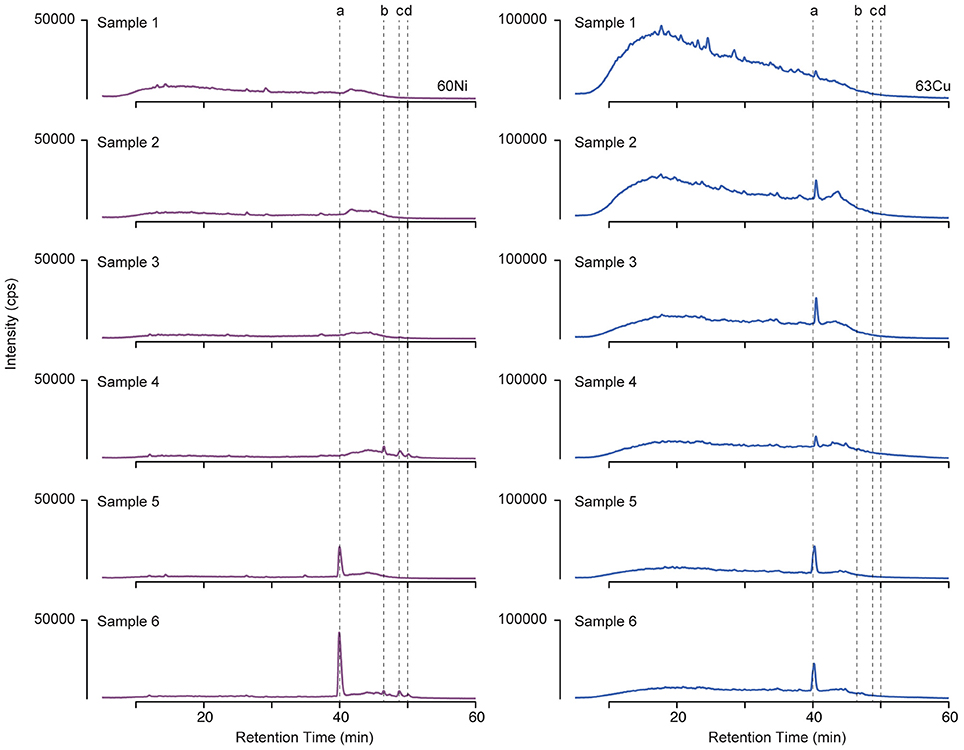
Figure 2. 60Ni (left) and 63Cu (right) LC-ICPMS chromatograms from the six samples collected along the transect from the coast of Peru (sample 1) to an oligotrophic station near Tahiti (sample 6). Peak intensities have been scaled to account for differences in preconcentration. Dashed lines indicate retention times of compounds (a–d) discussed in the text.
For both Cu and Ni, the highest ligand concentrations were detected by LC-ICPMS in the coastal region (Figure 3, sample 1), in agreement with electrochemical measurements of total Cu ligands. It should be noted that the LC-ICPMS and electrochemical methods employed here are not directly comparable in terms of which components of the ligand pool they target. Nonetheless, the agreement between the methods in terms of broad ligand distribution patterns is interesting. In the coastal sample, nearly all Cu and Ni eluted in a broad, unresolved feature appearing between 10 and 30 min. Small amounts of chromatographically resolved ligands appear as numerous sharp peaks atop the broad unresolved feature. These data suggests a very complex assemblage of structurally diverse ligands for both Cu and Ni. Moving offshore, ligands slowly decreased in concentration, but a larger fraction of the total ligand appeared as distinct compounds. Overall, the LCMS method used in this study captured 6–20% of the dissolved Cu and only 1–2% of total dissolved Ni across the transect, consistent with the findings of other studies that used similar solid phase extraction methods (Waska et al., 2015). The remaining metal is presumably either (1) unchelated or bound to very polar organic chelates that are not retained by SPE, (2) bound to nonpolar chelates that are strongly retained by SPE and do not elute with methanol, or (3) bound weakly such that they dissociate from the organic chelate during the extraction process.
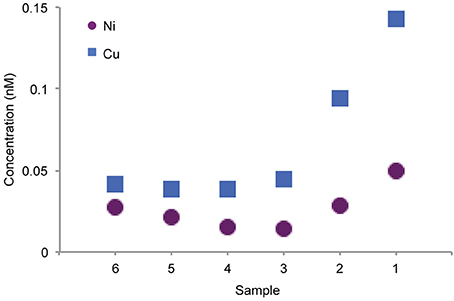
Figure 3. Concentration of Cu and Ni complexes detected by LC-ICPMS from the six integrated surface samples.
Several discrete Ni and Cu compounds were resolved and identified from the organic extracts (Figure 4). In samples 5 and 6, we characterized a compound eluting at 39.8 min that binds both Ni (m/z 567.015) and Cu (m/z 572.009; Table 2, compound a). A low intensity co-eluting peak with mass 511 m/z was also observed that matches the apo (metal free) form of the complex containing 2H+ in lieu of the metal ion, and suggests that Ni and Cu were in the 2+ oxidation state. MS2 fragmentation spectra of the Ni and Cu forms of compound a revealed similar fragments and neutral losses (Figure 5). Some additional major fragments were observed in the Cu complex fragmentation spectra (263.981, 393.997, and 527.440 m/z) but were absent from the Ni spectrum. These fragments could be derived from an isobaric interfering ion (571.410 m/z) that co-eluted with the 572.009 m/z ion and fell within the quadrupole mass isolation window (red values, Figure 5). The apo, Ni, and Cu forms of this compound were not detected in the ESIMS spectra of samples 1–4.
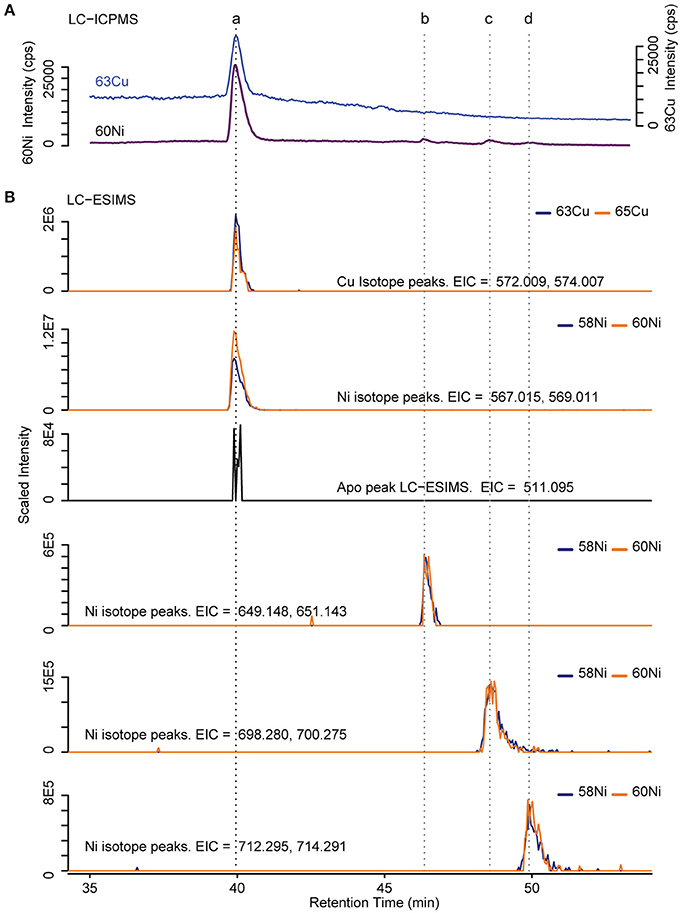
Figure 4. Determination of Cu and Ni compound mass. (A) ICPMS chromatogram of 63Cu and 60Ni. Dashed lines indicate retention times of identified compounds (B) Extracted ion chromatograms (EIC) from positive mode ESIMS. Blue and orange lines correspond to the light and heavy metal isotopologues respectively. The intensity of the heavy isotopologue has been scaled by the natural abundance ratio of 58Ni/60Ni or 63Cu/65Cu (multiplied by 2.6 or 2.2 respectively) so that the isotopologues overlap.
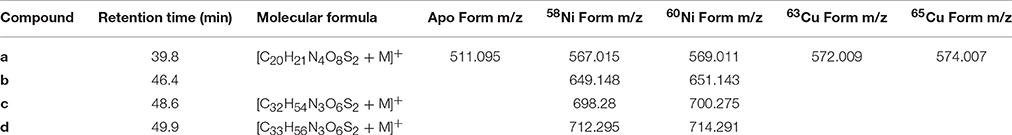
Table 2. Ni and Cu compounds detected across the GEOTRACES EPZT transect.
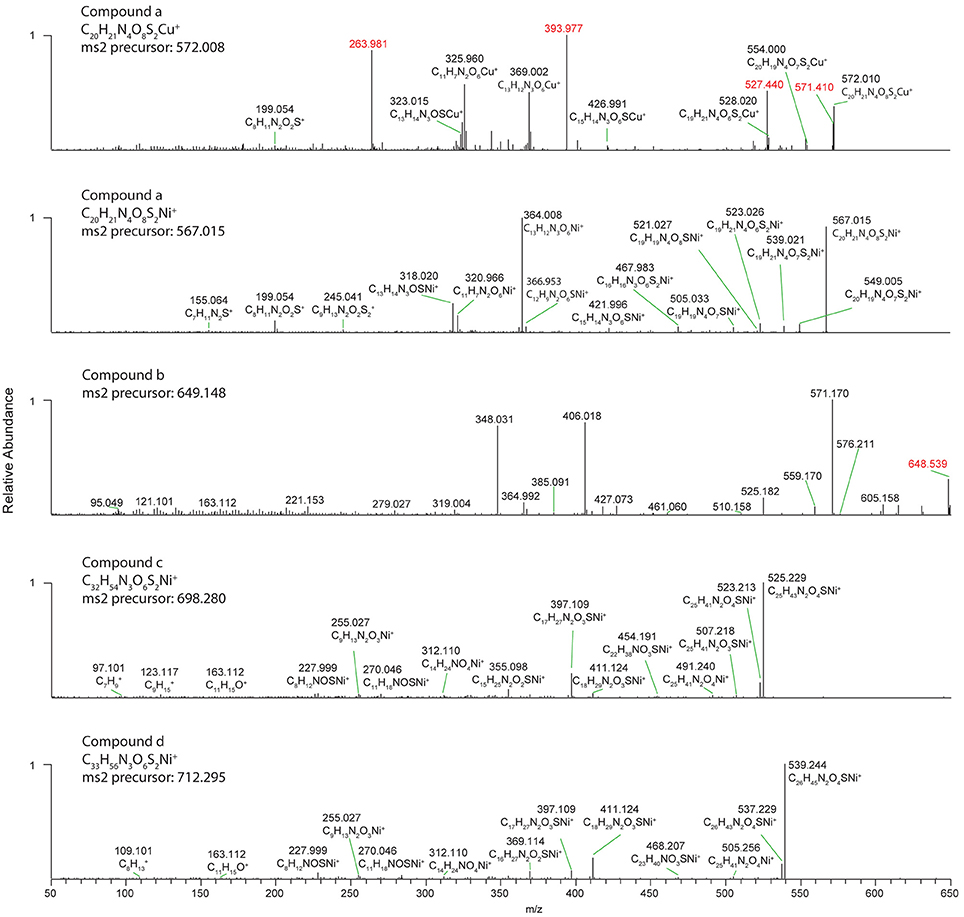
Figure 5. High resolution MS2 fragmentation spectra of Cu and Ni compounds detected in oligotrophic Eastern Tropical South Pacific. The first and second panels show spectra for the 63Cu and 58Ni isotopologues of the same ligand (compound a). The bottom three panels show spectra for the 58Ni isotopologues of compounds (b–d). Red masses are likely fragments from interfering isobaric ions.
Another major compound that bound only Cu eluted as a sharp peak at 40.2 min in the LC-ICPMS chromatograms of samples 1–4 (Figure 2). This peak increased in abundance in samples 2 and 3 and then decreased in samples collected farther offshore. Unfortunately, no ESIMS ion pair matching the Cu isotope pattern could be identified in these samples at this retention time, potentially due to matrix suppression effects, poor ionization of the compound, or a compound mass that was outside of the instrument's analytical range.
Finally, three compounds that only appeared in the Ni chromatogram were detected at 46.4, 48.6, and 49.9 min in samples 4 and 6 (Figure 4, compounds b–d). The 58Ni monoisotopic mass of these compounds was determined to be 649.148, 698.280, and 712.295 m/z. The MS2 spectra of compound b (m/z 649.148) exhibited three major neutral losses: 77.978 (likely CH2O2S), 243.130, and 301.117 m/z. However, a coeluting isobaric ion (m/z 648.539) fell within the quadrupole isolation window, and it is possible that some of the fragments are derived from this interfering ion. Compound d is structurally related to compound c based on their similar MS2 fragmentation patterns (Figure 5), differing only by the presence of an additional CH2. The masses of the apo forms of compounds b–d (calculated by substituting 2H+ for Ni2+) were not observed in the LC-ESIMS spectra.
To facilitate molecular formula assignments, we used FT-ICRMS to obtain sub-ppm mass measurement accuracy within the mass ranges of compounds a–d. Accurate masses were obtained for compounds a and c, while compounds b and d were below detection. The achieved ultrahigh resolving power of the FT-ICRMS shown in Figure 6 (m/Δm50% = 2,400,000 at m/z 569) was sufficient to resolve 34S isotopologues of compound a that remained unresolved at lower resolving power (LC-ESIMS). Molecular formula predictions for compounds a and c were first generated based on MS2 fragmentation data with the assistance of CSI-FingerID, which combines MS2 fragmentation tree computation and machine learning (Dührkop et al., 2015). To create a list of metal free fragment masses as an input to CSI-FingerID, we subtracted the mass of the metal from metal containing fragments that were identified based on their isotopic signature. We compared the top scoring molecular formula predictions to all molecular formulas containing C, H, N, O, P, and S within 2ppm of the exact mass determined by FT-ICRMS. [C20H21N4O8S2+M]+ (where M refers to an Ni or Cu isotopologue) and [C32H54N3O6S2Ni]+ were the only molecular formulae that were consistent with both the MS2 and accurate mass results for compounds a and c respectively. Compound d was then assigned a formula of [C33H56N3O6S2Ni]+ based on its structural similarity to compound c. Searches for the molecular formulae of the apo forms of compounds a, c, and d in the Pubchem database did not yield any matches that are consistent with the MS2 fragmentation patterns, suggesting that they are novel compounds. Obtaining full structures for these compounds requires additional characterization.
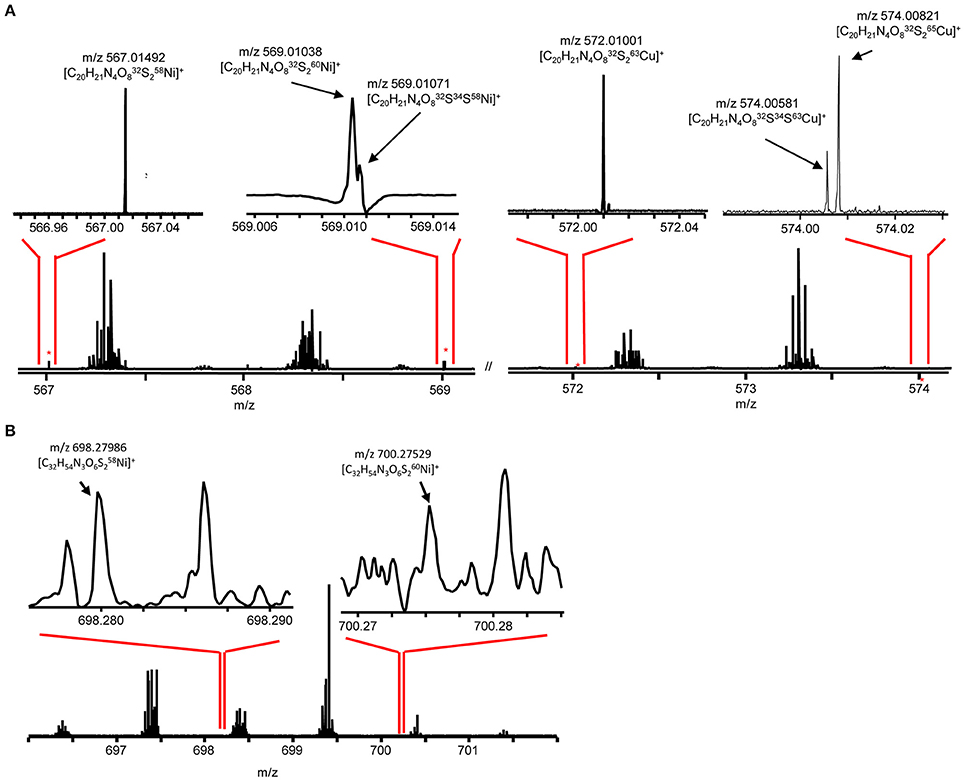
Figure 6. Direct infusion ESI FT-ICR MS at 9.4 Tesla of (A) compound a and (B) compound c. Major isotopologues are labeled.
Cu and Ni Addition Experiment
LC-ICPMS enables direct detection of the metals that are associated with each ligand, and could potentially be used to monitor the exchange of ligand bound metal. As a preliminary investigation of metal competition and exchange using LC-ICPMS, an aliquot of sample 6 was spiked with a large excess of Cu, Ni, and a combination of both metals, then allowed to equilibrate for 12–15 h prior to analysis by LC-ICPMS to monitor Cu and Ni. This treatment saturates uncomplexed ligands and drives metal exchange by changing the relative availability of Cu vs. Ni for ligand binding.
When Cu was added to the sample, there was a large increase in signal intensity in the LC-ICPMS chromatogram of Cu peaks relative to the unamended sample (Figure 7). Three additional chromatographic peaks with very high intensities appeared at 38.9, 39.7, and 43.0 min. This experiment demonstrates that there was a significant portion of uncomplexed ligand in the organic extract that became saturated with the addition of excess Cu. These results are consistent with electrochemical data that indicated free (uncomplexed) Cu ligands were at least 5x more abundant than Cu (Figure 1). The addition of Ni did not significantly affect the distribution of Cu containing compounds. The Cu baseline was slightly elevated in the Ni addition treatment, likely due to residual Cu carryover adsorbed to the walls of the injector flow-path or column from the preceding Cu addition analysis.

Figure 7. LC-ICPMS chromatograms of 58Ni and 63Cu after the addition of excess citrate bound Cu and Ni to sample 6 followed by incubation for 12 h. (A) Addition of Cu did not change the distribution of Ni ligands in this sample, while the addition of Ni resulted in the appearance of two Ni containing compounds. (B) Addition of Ni did not change the distribution of Cu compounds in the sample, while addition of Cu resulted in the appearance of a suite of Cu compounds.
Nickel addition did, however, cause new peaks to appear in the Ni ICPMS chromatogram at 37.1, and 38.5 min (Figure 7). Yet these peaks were absent when both Ni and Cu were added, indicating that Ni was outcompeted by Cu for binding to these ligands. The major uncomplexed Cu binding ligands at 38.9, 39.7, and 43.0 min did not appear in the Ni chromatogram after Ni addition. These three Cu binding ligands did not outcompete the added citrate for Ni binding, suggesting very weak Ni binding for these compounds and better selectivity for Cu. The metal addition treatments did not affect the intensity of the Ni or Cu peaks detected for compound a (39.8 min), showing that this compound was saturated with metal in the original sample and has slow metal exchange kinetics.
Discussion
These results provide a first glimpse of the distribution and chemical diversity of natural dissolved Cu and Ni complexes in the Eastern Pacific Ocean. The LC-ICPMS methods detected a fraction of the total dissolved metal (6–20% for Cu and 1–2% for Ni). LC-ICPMS only detects ligands if they are complexed to metals. It is important to note that despite efforts to minimize sample treatment, some changes in metal speciation are possible during extraction, since the final extract has a higher concentration of organic matter, a slightly lower pH (between 6 and 8), and much lower salt concentration than natural seawater. These differences can alter the kinetics of metal exchange compared to natural marine conditions but are unlikely to change the relative metal affinities of different ligands. Therefore, we believe the complexes observed in the LC-ICPMS chromatograms in Figure 2 probably represent strong ligands that bind these metals under native conditions.
Inshore Heterogeneous Ligands Detected by LC-ICPMS
The fraction of total dissolved Cu and Ni ligands in seawater detected by LC-ICPMS is particularly high inshore, between 80 and 90°W. In this region, ligands were predominantly polar, structurally heterogeneous compounds that eluted between 10 and 30 min (Figure 2). The LC-ICPMS chromatograms are qualitatively similar to chromatograms of coastal seawater organic extracts monitored by atomic fluorescence detection to investigate metal-organic speciation. These studies concluded that marine metal binding ligands are a complex mixture of many different compounds with a wide range of polarities (Mackey, 1983a; Mackey and O'sullivan, 1990). One potential source of these heterogeneous polar ligands is the organic matter upwelled from low oxygen subsurface water. Continental shelf sediments have been suggested as a major source of Cu ligands (Skrabal et al., 1997, 2000; Bundy et al., 2013) including humic materials that form as organic matter decays after burial (Whitby and Van den Berg, 2014; Abualhaija et al., 2015). Humic materials likely contribute to the broad, chromatographically unresolved baseline eluting between 10 and 30 min. In addition, there are small, discrete chromatographic peaks superimposed on the unresolved baseline suggesting that well defined compounds of biological origin are likely present as well. This is consistent with previous studies that observed strong Cu ligands in highly productive regions (Coale and Bruland, 1988, 1990; Moffett et al., 1990; Moffett, 1995). Unfortunately we were unable to detect Cu or Ni isotopologues at these retention times using LC-ESIMS due to interferences and ion suppression from the extremely complex matrix of coeluting compounds.
Structural Characterization of Defined Cu and Ni Ligands
In contrast to the broad suite of polar compounds found in the coastal upwelling sample, the well-defined Cu and Ni binding compounds a–d were only observed in the low-metal oligotrophic surface waters between 120 and 160°W. Based on the molecular formula assignments of the parent and fragment ions observed by ESIMS, these compounds are novel multi-dentate metal chelators. Compound a [C20H21N4O8S2+M]+ was particularly abundant, accounting for 4–5% of the Cu and 5–8% of Ni detected by LC-ICPMS in samples 5 and 6. The overall compound has 12 degrees of unsaturation, including rings plus double bonds. One domain of this molecule is represented by the ions [C11H7N2O6+M]+. The large number of heteroatoms and degrees of unsaturation in this fragment imply the presence of heavily conjugated cyclic azole-like functional groups. The nitrogen atoms of such groups serve as the electron donor of many strong Cu and Ni chelators including cyclic peptides and porphyrins. The remaining half of the molecule is represented by [C9H13N2O2S2]+. The sulfur atoms appear to be associated with thiols, as indicated by an observed neutral loss of CH2S and the fragment C6H10. Furthermore, the loss of CO2 between the fragments of C7H11N2S+ and C8H11N2O2S+ imply the presence of a labile carboxylic acid. This second half of the compound appears to have lower metal affinity, since the representative fragments were not observed in association with Ni or Cu. However, a fragment that retains one of the azole-like nitrogen groups [C13H14N3OS+M]+ does bind the metal, further implicating nitrogen as a key metal binding moiety. The closest structural analogs to compound a among well-studied biological Cu and Ni chelators may be other multidentate ligands that possess aromatic nitrogen based ligands, such as porphyrins or cyclic azole-based peptides.
Compounds c and d are larger molecules with 7 degrees of unsaturation. As with compound a, multiple N and O electron donors appear to be more important for metal binding than S, as indicated by the fact that one or both sulfurs are missing from all of the fragments that retain Ni, such as C25H41N2O4Ni+ and C14H24NO4Ni+. In these molecules, sulfur is associated with domains represented by the major neutral loss of C7H11NOS and fragments C8H12NOSNi+ and C11H18NOSNi+. These compounds also possess a hydrocarbon domain represented by the fragment C11H15O+ that contribute to the hydrophobicity of these compounds and the late retention times.
These ligands provide insight into structural elements that are likely shared by other marine ligands with similar chemical properties (e.g., binding affinity, redox potential, bioavailability) and biological roles. However, fewer structures of marine Cu and Ni ligands are known compared to other ligand classes such as siderophores (Vraspir and Butler, 2009). While extracellular Cu binding ligands have been detected from a wide variety of ecologically abundant marine phytoplankton and bacterial taxa (Moffett and Brand, 1996; Croot et al., 1999, 2000; Gordon et al., 2000; Wiramanaden et al., 2008), few of these ligands have been structurally characterized. The combined LCMS approach we used here is well suited for rapid screening of laboratory pure cultures for Ni and Cu ligands, and offers the potential to expand the catalog of characterized ligands and their biological sources.
The appearance of compounds a–d in the western part of the transect suggest that they may be involved in metabolic processes that are unique to the oligotrophic ocean. One possibility is that these compounds are used to regulate intracellular free metal concentrations. As microbes concentrate iron, cobalt, and other scarce metals from the environment via transporters with imperfect specificity, they also concentrate more abundant metals including Cu and Ni (Waldron et al., 2007; Waldron and Robinson, 2009). Specialized chelators may be needed to prevent Cu or Ni from occupying binding sites in proteins that are meant for other metals. Alternatively, these Ni and Cu complexes may be cofactors used in metabolic processes that only occur in oligotrophic waters, perhaps as a substitution for scarce metals. Such cofactors are often tetrapyrroles such as the Ni cofactor F430 used by methane oxidizing archea (Allen et al., 2014). These intracellular metal compounds could be released into the dissolved ligand pool by cell lysis (Ahner et al., 1997; Dupont et al., 2004; Wei and Ahner, 2005). Another possibility is that these compounds are released by microbes in order to enhance metal uptake. Such strategies may provide an advantage to microbes that inhabit the oligotrophic Eastern tropical Pacific ocean where free Cu could potentially be in short supply (Jacquot et al., 2013).
Additional metal complexes were also observed for which masses could not detected by LC-ESIMS due to ion suppression from coeluting compounds. Improving the separation of these metal bound compounds from the organic background using multiple dimensions of chromatography may resolve this issue. Furthermore, the saturation of free ligands with added Ni and Cu gave rise to intense chromatographic peaks. Future work will focus on the LC-ESIMS characterization of these compounds, which likely correspond to weaker ligands in the samples. Some of these appear to have affinity for Cu but not Ni while others bind Ni as well, and these diverse metal binding behaviors may reflect different electron donating functional domains.
Other methods of extracting organic compounds from seawater may offer additional analytical windows for ligand characterization. The solid phase extraction method used in this study was selected due to the low organic background of the ENV resin and stability across a wide range of pH values. Other hydrophobic resins or other elution solvents may recover a different fraction of the total dissolved metal and ligands (Mills et al., 1982; Mackey, 1983b; Waska et al., 2015). In addition, other extraction approaches such as immobilized metal affinity chromatography have been used to extract weak or uncomplexed metal ligands from seawater (Ross et al., 2003). The composition of the ligands retained by these other extraction methods remains poorly constrained, but the LC-ICPMS-ESIMS approaches described here can be adapted to survey the composition of these extracts.
Slow Dissociation Kinetics of Compound a
The presence of both Ni and Cu complexes of compound a raises questions about the dynamics of Ni and Cu competition for metal binding sites. Two factors are important—the binding affinity of the chelator to different metals as the system approaches equilibrium, and the kinetics of metal exchange. Although Cu2+ ligand complexes are typically more stable than Ni2+ complexes in aqueous solutions (Irving and Williams, 1953), compound a did not release any bound Ni2+ after the addition of excess Cu2+ in our experiments. This suggests that the dissociation kinetics of compound a with Ni are much slower than the 12 h equilibration time used in this study. For such compounds, the metal that they bind in the ocean may reflect metal association rates rather than equilibrium binding constants. The kinetics of metal association depend on free metal ion concentrations and the loss rate of water from the inner coordination sphere of those metals (Hering and Morel, 1989, 1990). In this case of Ni and Cu competition, the significantly slower water loss rate of Ni compared to Cu may be offset by the higher free metal ion concentration of Ni (inferred from high labile Ni measurements), since over 99% of Cu is complexed by strong organic ligands throughout the transect, while <50% of dissolved Ni is strongly complexed. Alternatively, the metal speciation of these compounds may reflect the intracellular environment where metal-ligand complexes can form, likely in the presence of very low intracellular metal abundances (Rae et al., 1999), and subsequent slow dissociation kinetics as these metal-ligand complexes are released. Slow dissociation kinetics may be common for strong ligands of other metals as well. Nickel can impact the solubility and bioavailability of scarce micronutrient Co by competing for the same ligand sites, and the result of this competition may well be controlled by kinetics rather than thermodynamics (Saito and Moffett, 2001). Furthermore, strong iron binding ligands such as siderophores can have lifetimes of several months before dissociation (Witter et al., 2000), although the compounds themselves are likely taken up or degraded on shorter timescales similar to other peptidic compounds. These examples of slow dissociation kinetics are an important consideration for experiments and models of metal speciation, particularly for strong ligands that are capable of binding multiple metals.
Conclusions
In this study, we used a mass spectrometry based approach to detect and characterize Cu and Ni ligands in the US GEOTRACES section across the eastern tropical South Pacific Ocean. Using both mass spectrometric and electrochemical approaches, we found that the concentrations of Cu and Ni ligands are highest in the coastal upwelling region of the section, where ligands occur as a very complex mixture of chromatographically unresolved compounds. Ligand concentrations were lower in offshore oligotrophic surface waters, where structurally well-defined ligands become relatively more important. The partial structural characterization of these compounds reveals that these are multi-dentate ligands with hard base electron donors that bind metals in the 2+ oxidation state. Discovering the biological source of these ligands is the key to understanding their distinct distributions across the surface ocean as well as their role in biogeochemical metal cycles. The analyses presented here are well suited for the compound specific discovery and investigation of other metal chelators. As we learn more about the compounds that bind these metals in the ocean, structural similarities will continue to emerge and facilitate future identifications. This information will help generate specific molecular targets and model compounds that can be tracked through the environment and provide greater insight into mechanisms of metal cycling and connections with microbial ecology.
Author Contributions
RMBO and DR designed the study, NH collected the samples, RMBO, and AM determined ligand composition, CT measured metal concentrations. AR measured Cu ligand concentrations and binding strengths, RMBU measured labile nickel concentrations, AM performed FTICRMS analyses, RMBO, NH, RMBU, KAB, MS, and DR analyzed the field data, RMBO and DR wrote the manuscript with contributions from all authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank chief scientists James Moffett and Chris German, as well as Alan Shiller, Cheryl Zurbrick, and Geoffrey Smith for assistance with sample collection, and the scientists and crew of the R/V Thomas G. Thompson. Support was provided by the National Science Foundation (NSF) program in Chemical Oceanography (OCE-1356747, OCE-1233261, OCE-1233733, OCE-1233502, and OCE-1237034), the NSF Science and Technology Center for Microbial Oceanography Research and Education (C-MORE; DBI-0424599), the Gordon and Betty Moore Foundation (#3298 and 3934), and the Simons Foundation (#329108, DR). A portion of this work was performed at the National High Magnetic Field Laboratory, which is supported by NSF Division of Materials Research through DMR-11-57490, and the State of Florida.
References
Abualhaija, M. M., Whitby, H., and van den Berg, C. M. G. (2015). Competition between copper and iron for humic ligands in estuarine waters. Mar. Chem. 172, 46–56. doi: 10.1016/j.marchem.2015.03.010
Achterberg, E. P., and Van Den Berg, C. M. G. (1997). Chemical speciation of chromium and nickel in the western Mediterranean. Deep Sea Res. Part II Top. Stud. Oceanogr. 44, 693–720. doi: 10.1016/S0967-0645(96)00086-0
Ahner, B. A., Morel, F. M. M., and Moffett, J. W. (1997). Trace metal control of phytochelatin production in coastal waters. Limnol. Oceanogr. 42, 601–608. doi: 10.4319/lo.1997.42.3.0601
Ahner, B. A., Price, N. M., and Morel, F. M. (1994). Phytochelatin production by marine phytoplankton at low free metal ion concentrations: laboratory studies and field data from Massachusetts Bay. Proc. Natl. Acad. Sci. U.S.A. 91, 8433–8436. doi: 10.1073/pnas.91.18.8433
Ahner, B. A., Wei, L., Oleson, J. R., and Ogura, N. (2002). Glutathione and other low molecular weight thiols in marine phytoplankton under metal stress. Mar. Ecol. Prog. Ser. 232, 93–103. doi: 10.3354/meps232093
Allen, K. D., Wegener, G., and White, R. H. (2014). Discovery of multiple modified F430 coenzymes in methanogens and anaerobic methanotrophic archaea suggests possible new roles for F430 in nature. Appl. Environ. Microbiol. 80, 6403–6412. doi: 10.1128/AEM.02202-14
Baars, O., Morel, F. M. M., and Perlman, D. H. (2014). ChelomEx: isotope-assisted discovery of metal chelates in complex media using high-resolution LC-MS. Anal. Chem. 86, 11298–11305. doi: 10.1021/ac503000e
Baars, O., Zhang, X., Morel, F. M. M., and Seyedsayamdost, M. R. (2015). The siderophore metabolome of Azotobacter vinelandii. Appl. Environ. Microbiol. 82, 27–39. doi: 10.1128/AEM.03160-15
Baker, A. R., Thomas, M., Bange, H. W., and Plasencia Sanchez, E. (2016). Soluble trace metals in aerosols over the tropical south-east Pacific offshore of Peru. Biogeosciences 13, 817–825. doi: 10.5194/bg-13-817-2016
Baker, H., and Khalili, F. (2003). Comparative study of binding strengths and thermodynamic aspects of Cu(II) and Ni(II) with humic acid by Schubert's ion-exchange method. Anal. Chim. Acta 497, 235–248. doi: 10.1016/j.aca.2003.08.036
Bertram, A., and Pattenden, G. (2007). Marine metabolites: metal binding and metal complexes of azole-based cyclic peptides of marine origin. Nat. Prod. Rep. 24, 18–30. doi: 10.1039/b612600f
Biller, D. V., and Bruland, K. W. (2012). Analysis of Mn, Fe, Co, Ni, Cu, Zn, Cd, and Pb in seawater using the Nobias-chelate PA1 resin and magnetic sector inductively coupled plasma mass spectrometry (ICP-MS). Mar. Chem. 130–131, 12–20. doi: 10.1016/j.marchem.2011.12.001
Blakney, G. T., Hendrickson, C. L., and Marshall, A. G. (2011). Predator data station: a fast data acquisition system for advanced FT-ICR MS experiments. Int. J. Mass Spectrom. 306, 246–252. doi: 10.1016/j.ijms.2011.03.009
Boiteau, R. M., Fitzsimmons, J. N., Repeta, D. J., and Boyle, E. A. (2013). Detection of iron ligands in seawater and marine cyanobacteria cultures by high-performance liquid chromatography-inductively coupled plasma-mass spectrometry. Anal. Chem. 85, 4357–4362. doi: 10.1021/ac3034568
Boiteau, R. M., Hawco, N. J., Mende, D. R., McIlvin, M. R., Fitzsimmons, J. N., Sedwick, P. N., et al. (in press). Siderophore-based microbial adaptations to iron scarcity across the eastern Pacific Ocean. PNAS.
Boiteau, R. M., and Repeta, D. J. (2015). An extended siderophore suite from Synechococcus sp. PCC 7002 revealed by LC-ICPMS-ESIMS. Metallomics 7, 877–884. doi: 10.1039/C5MT00005J
Bruland, K. W., Rue, E. L., Smith, G. J., and DiTullio, G. R. (2005). Iron, macronutrients and diatom blooms in the Peru upwelling regime: brown and blue waters of Peru. Mar. Chem. 93, 81–103. doi: 10.1016/j.marchem.2004.06.011
Buck, K. N., and Bruland, K. W. (2005). Copper speciation in San Francisco Bay: a novel approach using multiple analytical windows. Mar. Chem. 96, 185–198. doi: 10.1016/j.marchem.2005.01.001
Bundy, R. M., Barbeau, K. A., and Buck, K. N. (2013). Sources of strong copper-binding ligands in Antarctic Peninsula surface waters. Deep. Res. Part II Top. Stud. Oceanogr. 90, 134–146. doi: 10.1016/j.dsr2.2012.07.023
Campos, M. L. A. M., and van den Berg, C. M. G. (1994). Determination of copper complexation in sea water by cathodic stripping voltammetry and ligand competition with salicylaldoxime. Anal. Chim. Acta 284, 481–496. doi: 10.1016/0003-2670(94)85055-0
Chambers, M. C., Maclean, B., Burke, R., Amodei, D., Ruderman, D. L., Neumann, S., et al. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920. doi: 10.1038/nbt.2377
Coale, K. H., and Bruland, K. W. (1988). Copper complexation in the Northeast Pacific. Limnol. Oceanogr. 33, 1084–1101. doi: 10.4319/lo.1988.33.5.1084
Coale, K. H., and Bruland, K. W. (1990). Spatial and temporal variability in copper complexation in the North Pacific. Deep Sea Res. Part A Oceanogr. Res. Pap. 37, 317–336. doi: 10.1016/0198-0149(90)90130-N
Comba, P., Dovalil, N., Gahan, L. R., Hanson, G. R., and Westphal, M. (2014). Cyclic peptide marine metabolites and CuII. Dalt. Trans. 43, 1935–1956. doi: 10.1039/C3DT52664J
Croot, P. L., Moffett, J. W., and Brand, L. E. (2000). Production of extracellular Cu complexing ligands by eucaryotic phytoplankton in response to Cu stress. Limnol. Oceanogr. 45, 619–627. doi: 10.4319/lo.2000.45.3.0619
Croot, P. L., Moffett, J. W., and Luther, G. W. (1999). Polarographic determination of half-wave potentials for copper-organic complexes in seawater. Mar. Chem. 67, 219–232. doi: 10.1016/S0304-4203(99)00054-7
Devez, A., Achterberg, E., and Gledhill, M. (2009). Metal ion-binding properties of phytochelatins and related ligands. Met. Ions Life Sci. 5, 441–481. doi: 10.1039/9781847559531-00441
Donat, J. R., Lao, K. A., and Bruland, K. W. (1994). Speciation of dissolved copper and nickel in South San Francisco Bay: a multi-method approach. Anal. Chim. Acta 284, 547–571. doi: 10.1016/0003-2670(94)85061-5
Donat, J. R., and van den Berg, C. M. G. (1992). A new cathodic stripping voltammetric method for determining organic copper complexation in seawater. Mar. Chem. 38, 69–90. doi: 10.1016/0304-4203(92)90068-L
Dührkop, K., Shen, H., Meusel, M., Rousu, J., and Böcker, S. (2015). Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. U.S.A. 112, 12580–12585. doi: 10.1073/pnas.1509788112
Dupont, C. L., Nelson, R. K., Bashir, S., Moffett, J. W., and Ahner, B. A. (2004). Novel copper-binding and nitrogen-rich thiols produced and exuded by Emiliania huxleyi. Limnol. Oceanogr. 49, 1754–1762. doi: 10.4319/lo.2004.49.5.1754
El Ghazouani, A., Baslé, A., Gray, J., Graham, D. W., Firbank, S. J., and Dennison, C. (2012). Variations in methanobactin structure influences copper utilization by methane-oxidizing bacteria. Proc. Natl. Acad. Sci. U.S.A. 109, 8400–8404. doi: 10.1073/pnas.1112921109
Emmett, M. R., White, F. M., Hendrickson, C. L., Shi, S. D., and Marshall, A. G. (1998). Application of micro-electrospray liquid chromatography techniques to FT-ICR MS to enable high-sensitivity biological analysis. J. Am. Soc. Mass Spectrom. 9, 333–340. doi: 10.1016/S1044-0305(97)00287-0
Freeman, J. L., Persans, M. W., Nieman, K., Albrecht, C., Peer, W., Pickering, I. J., et al. (2004). Increased glutathione biosynthesis plays a role in nickel tolerance in thlaspi nickel hyperaccumulators. Plant Cell 16, 2176–2191. doi: 10.1105/tpc.104.023036
Ghssein, G., Brutesco, C., Ouerdane, L., Fojcik, C., Izaute, A., Wang, S., et al. (2016). Biosynthesis of a broad-spectrum nicotianamine-like metallophore in Staphylococcus aureus. Science 352, 1105–1109. doi: 10.1126/science.aaf1018
Gordon, A. S., Donat, J. R., Kango, R. A., Dyer, B. J., and Stuart, L. M. (2000). Dissolved copper-complexing ligands in cultures of marine bacteria and estuarine water. Mar. Chem. 70, 149–160. doi: 10.1016/S0304-4203(00)00019-0
Hering, J. G., and Morel, F. M. M. (1989). Slow coordination reactions in seawater. Geochim. Cosmochim. Acta 53, 611–618. doi: 10.1016/0016-7037(89)90004-5
Hering, J., and Morel, F. (1990). Kinetics of trace metal complexation: ligand-exchange reactions. Environ. Sci. Technol. 252, 242–252. doi: 10.1021/es00072a014
Irving, H., and Williams, R. (1953). The stability of transition-metal complexes. J. Chem. Soc. 3, 3192–3210. doi: 10.1039/jr9530003192
Jacquot, J. E., Kondo, Y., Knapp, A. N., and Moffett, J. W. (2013). The speciation of copper across active gradients in nitrogen-cycle processes in the eastern tropical South Pacific. Limnol. Oceanogr. 58, 1387–1394. doi: 10.4319/lo.2013.58.4.1387
Jacquot, J. E., and Moffett, J. W. (2015). Copper distribution and speciation across the International GEOTRACES Section GA03. Deep. Res. Part II Top. Stud. Oceanogr. 116, 187–207. doi: 10.1016/j.dsr2.2014.11.013
Kaiser, N. K., Quinn, J. P., Blakney, G. T., Hendrickson, C. L., and Marshall, A. G. (2011a). A novel 9.4 tesla FTICR mass spectrometer with improved sensitivity, mass resolution, and mass range. J. Am. Soc. Mass Spectrom. 22, 1343–1351. doi: 10.1007/s13361-011-0141-9
Kaiser, N. K., Savory, J. J., McKenna, A. M., Quinn, J. P., Hendrickson, C. L., and Marshall, A. G. (2011b). Electrically compensated fourier transform ion cyclotron resonance cell for complex mixture mass analysis. Anal. Chem. 83, 6907–6910. doi: 10.1021/ac201546d
Kim, H. J., Graham, D. W., DiSpirito, A. A., and Alterman, M. A. (2004). Methanobactin, a copper-acquisition compound from methane-oxidizing bacteria. Science 305, 16–19. doi: 10.1126/science.1098322
Mackey, D. J. (1983a). Metal-organic complexes in seawater - An investigation of naturally occurring complexes of Cu, Zn, Fe, Mg, Ni, Cr, Mn and Cd using high-performance liquid chromatography with atomic fluorescence detection. Mar. Chem. 13, 169–180. doi: 10.1016/0304-4203(83)90012-9
Mackey, D. J. (1983b). The strong complexing capacity of seawater - an investigation of south-eastern australian coastal waters. Mar. Chem. 14, 73–87. doi: 10.1016/0304-4203(83)90071-3
Mackey, D. J., and O'sullivan, J. E. (1990). Metal - organic interactions in sea water: an ecosystem experiment. Anal. Chim. Acta 232, 161–170.
McKenna, A. M., Marshall, A. G., and Rodgers, R. P. (2013). Heavy petroleum composition. 4. Asphaltene compositional space. Energy Fuels 27, 1257–1267. doi: 10.1021/ef301747d
McKenna, A. M., Williams, J. T., Putman, J. C., Aeppli, C., Reddy, C. M., Valentine, D. L., et al. (2014). Unprecedented ultrahigh resolution FT-ICR mass spectrometry and parts-per-billion mass accuracy enable direct characterization of nickel and vanadyl porphyrins in petroleum from natural seeps. Energy Fuels 28, 2454–2464. doi: 10.1021/ef5002452
Michael, J. P., Pattenden, G., and Michael, B. J. P. (1993). Marine metabolites and metal ion chelation: the facts and the fantasies. Angew. Chemie Int. Ed. 32, 1–130. doi: 10.1002/anie.199300013
Mills, G. L., Hanson, A. K., and Quinn, J. G. (1982). Chemical studies of copper-organic complexes isolated from estuarine waters using C18 Reverse-phase liquid chromatography. Mar. Chem. 11, 355–377. doi: 10.1016/0304-4203(82)90031-7
Moffett, J. W. (1995). Temporal and spatial variability of copper complexation by strong chelators in the Sargasso Sea. Deep. Res. Part I 42, 1273–1295. doi: 10.1016/0967-0637(95)00060-J
Moffett, J. W., and Brand, L. E. (1996). Production of strong, extracellular Cu chelators by marine cyanobacteria in response to Cu stress. Limnol. Oceanogr. 41, 388–395. doi: 10.4319/lo.1996.41.3.0388
Moffett, J. W., and Zika, R. G. (1983). Oxidation kinetics of Cu(I) in seawater: implications for its existence in the marine environment. Mar. Chem. 13, 239–251. doi: 10.1016/0304-4203(83)90017-8
Moffett, J. W., Zika, R. G., and Brand, L. E. (1990). Distribution and potential sources and sinks of copper chelators in the Sargasso Sea. Deep Sea Res. Part A, Oceanogr. Res. Pap. 37, 27–36. doi: 10.1016/0198-0149(90)90027-S
Moore, C. M., Mills, M. M., Arrigo, K. R., Berman-Frank, I., Bopp, L., Boyd, P. W., et al. (2013). Processes and patterns of oceanic nutrient limitation. Nat. Geosci. 6, 701–710. doi: 10.1038/ngeo1765
Mounicou, S., Szpunar, J., and Lobinski, R. (2009). Metallomics: the concept and methodology. Chem. Soc. Rev. 38, 1119–1138. doi: 10.1039/b713633c
Nimmo, M., and van Den Berg, C. M. G. (1989). The chemical speciation of dissolved nickel, copper, vanadium, and iron in liverpool bay, Irish Sea. Estuar. Coast. Shelf Sci. 29, 57–74. doi: 10.1016/0272-7714(89)90073-5
Omanović, D., Garnier, C., and Pižeta, I. (2014). ProMCC: an all-in-one tool for trace metal complexation studies. Mar. Chem. 173, 25–39. doi: 10.1016/j.marchem.2014.10.011
Parker, C. E., Brown, M. T., and Bruland, K. W. (2016). Scandium in the open ocean: a comparison with other group 3 trivalent metals. Geophys. Res. Lett. 43, 2758–2764. doi: 10.1002/2016gl067827
Pennington, J. T., Mahoney, K. L., Kuwahara, V. S., Kolber, D. D., Calienes, R., and Chavez, F. P. (2006). Primary production in the eastern tropical Pacific: a review. Prog. Oceanogr. 69, 285–317. doi: 10.1016/j.pocean.2006.03.012
Rae, T. D., Schmidt, P. J., Pufahl, R. A., Culotta, V. C., and O'halloran, T. V. (1999). Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284, 805–808. doi: 10.1126/science.284.5415.805
Ross, A. R., Ikonomou, M. G., and Orians, K. J. (2003). Characterization of copper-complexing ligands in seawater using immobilized copper(II)-ion affinity chromatography and electrospray ionization mass spectrometry. Mar. Chem. 83, 47–58. doi: 10.1016/S0304-4203(03)00095-1
Saito, M. A., and Moffett, J. W. (2001). Complexation of cobalt by natural organic ligands in the Sargasso Sea as determined by a new high-sensitivity electrochemical cobalt speciation method suitable for open ocean work. Mar. Chem. 75, 49–68. doi: 10.1016/S0304-4203(01)00025-1
Saito, M. A., and Moffett, J. W. (2004). Cobalt and nickel in the Peru upwelling region: a major flux of labile cobalt utilized as a micronutrient. Glob. Biogeochem. Cycles 18, 1–14. doi: 10.1029/2003GB002216
Saito, M. A., Rocap, G., and Moffett, J. W. (2005). Production of cobalt binding ligands in a Synechococcus feature at the Costa Rica upwelling dome. Limnol. Oceanogr. 50, 279–290. doi: 10.4319/lo.2005.50.1.0279
Savory, J. J., Kaiser, N. K., McKenna, A. M., Xian, F., Blakney, G. T., Rodgers, R. P., et al. (2011). Parts-per-billion fourier transform ion cyclotron resonance mass measurement accuracy with a “Walking” calibration equation. Anal. Chem. 83, 1732–1736. doi: 10.1021/ac102943z
Shi, S. D. H., Drader, J. J., Freitas, M. A., Hendrickson, C. L., and Marshall, A. G. (2000). Comparison and interconversion of the two most common frequency-to-mass calibration functions for Fourier transform ion cyclotron resonance mass spectrometry. Int. J. Mass Spectrom. 195–196, 591–598. doi: 10.1016/S1387-3806(99)00226-2
Skrabal, S. A., Donat, J. R., and Burdige, D. J. (1997). Fluxes of copper-complexing ligands from estuarine sediments. Limnol. Oceanogr. 42, 992–996. doi: 10.4319/lo.1997.42.5.0992
Skrabal, S. A., Donat, J. R., and Burdige, D. J. (2000). Pore water distributions of dissolved copper and copper-complexing ligands in estuarine and coastal marine sediments. Geochim. Cosmochim. Acta 64, 1843–1857. doi: 10.1016/S0016-7037(99)00387-7
Thompson, C. M., Ellwood, M. J., and Sander, S. G. (2014). Dissolved copper speciation in the Tasman Sea, SW Pacific Ocean. Mar. Chem. 164, 84–94. doi: 10.1016/j.marchem.2014.06.003
Tolmachev, A. V., Robinson, E. W., Wu, S., Smith, R. D., and Paša-Toli, L. (2011). Trapping radial electric field optimization in compensated FTICR cells. J. Am. Soc. Mass Spectrom. 22, 1334–1342. doi: 10.1007/s13361-011-0167-z
Turner, D., Whitfield, M., and Dickson, A. (1981). The equilibrium speciation of dissolved components in freshwater and sea water at 25°C and 1 atm pressure. Geochim. Cosmochim. Acta 45, 855–881. doi: 10.1016/0016-7037(81)90115-0
van den Berg, C. M. G., and Nimmo, M. (1987). Determination of interactions of nickel with dissolved organic material in seawater using cathodic stripping voltammetry. Sci. Total Environ. 60, 185–195. doi: 10.1016/0048-9697(87)90415-3
Vraspir, J. M., and Butler, A. (2009). Chemistry of marine ligands and siderophores. Ann. Rev. Mar. Sci. 1, 43–63. doi: 10.1146/annurev.marine.010908.163712
Waldron, K. J., and Robinson, N. J. (2009). How do bacterial cells ensure that metalloproteins get the correct metal? Nat. Rev. Microbiol. 7, 25–35. doi: 10.1038/nrmicro2057
Waldron, K. J., Tottey, S., Yanagisawa, S., Dennison, C., and Robinson, N. J. (2007). A periplasmic iron-binding protein contributes toward inward copper supply. J. Biol. Chem. 282, 3837–3846. doi: 10.1074/jbc.M609916200
Walsh, M. J., and Ahner, B. A. (2013). Determination of stability constants of Cu(I), Cd(II) and Zn(II) complexes with thiols using fluorescent probes. J. Inorg. Biochem. 128, 112–123. doi: 10.1016/j.jinorgbio.2013.07.012
Waska, H., Koschinsky, A., Ruiz Chancho, M. J., and Dittmar, T. (2015). Investigating the potential of solid-phase extraction and Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR-MS) for the isolation and identification of dissolved metal–organic complexes from natural waters. Mar. Chem. 173, 78–92. doi: 10.1016/j.marchem.2014.10.001
Wei, L., and Ahner, B. A. (2005). Sources and sinks of dissolved phytochelatin in natural seawater. Limnol. Oceanogr. 50, 13–22. doi: 10.4319/lo.2005.50.1.0013
Whitby, H., and Van den Berg, C. M. G. (2014). Evidence for copper-binding humic substances in seawater. Mar. Chem. 173, 282–290. doi: 10.1016/j.marchem.2014.09.011
Wilcox, B. E., Hendrickson, C. L., and Marshall, A. G. (2002). Improved ion extraction from a linear octopole ion trap: SIMION analysis and experimental demonstration. J. Am. Soc. Mass Spectrom. 13, 1304–1312. doi: 10.1016/S1044-0305(02)00622-0
Wiramanaden, C. I. E., Cullen, J. T., Ross, A. R. S., and Orians, K. J. (2008). Cyanobacterial copper-binding ligands isolated from artificial seawater cultures. Mar. Chem. 110, 28–41. doi: 10.1016/j.marchem.2008.02.003
Witter, A. E., Hutchins, D. A., Butler, A., and Luther, G. W. III. (2000). Determination of conditional stability constants and kinetic constants for strong model Fe-binding ligands in seawater. Mar. Chem. 69, 1–17. doi: 10.1016/S0304-4203(99)00087-0
Wuttig, K., Heller, M. I., and Croot, P. L. (2013). Pathways of superoxide () decay in the Eastern Tropical North Atlantic. Environ. Sci. Technol. 47, 10249–10256. doi: 10.1021/es401658t
Xian, F., Corilo, Y. E., Hendrickson, C. L., and Marshall, A. G. (2012). Baseline correction of absorption-mode Fourier transform ion cyclotron resonance mass spectra. Int. J. Mass Spectrom. 325–327, 67–72. doi: 10.1016/j.ijms.2012.06.007
Xian, F., Hendrickson, C. L., Blakney, G. T., Beu, S. C., and Marshall, A. G. (2010). Automated broadband phase correction of fourier transform ion cyclotron resonance mass spectra. Anal. Chem. 82, 8807–8812. doi: 10.1021/ac101091w
Yang, R., and Van den Berg, C. M. G. (2009). Metal complexation by humic substances in seawater. Environ. Sci. Technol. 43, 7192–7197. doi: 10.1021/es900173w
Keywords: copper, nickel, marine ligands, metal competition, GEOTRACES, Eastern Pacific
Citation: Boiteau RM, Till CP, Ruacho A, Bundy RM, Hawco NJ, McKenna AM, Barbeau KA, Bruland KW, Saito MA and Repeta DJ (2016) Structural Characterization of Natural Nickel and Copper Binding Ligands along the US GEOTRACES Eastern Pacific Zonal Transect. Front. Mar. Sci. 3:243. doi: 10.3389/fmars.2016.00243
Received: 01 August 2016; Accepted: 08 November 2016;
Published: 30 November 2016.
Edited by:
Christel Hassler, University of Geneva, SwitzerlandReviewed by:
Peter Croot, National University of Ireland, Galway, IrelandHannelore Waska, University of Oldenburg, Germany
Copyright © 2016 Boiteau, Till, Ruacho, Bundy, Hawco, McKenna, Barbeau, Bruland, Saito and Repeta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel J. Repeta, drepeta@whoi.edu