Genetic and Manual Survey Methods Yield Different and Complementary Views of an Ecosystem
 Ryan P. Kelly1*
Ryan P. Kelly1*  Collin J. Closek2
Collin J. Closek2  James L. O'Donnell1
James L. O'Donnell1  James E. Kralj1
James E. Kralj1  Andrew O. Shelton3
Andrew O. Shelton3  Jameal F. Samhouri3
Jameal F. Samhouri3- 1School of Marine and Environmental Affairs, University of Washington, Seattle, WA, USA
- 2Center for Ocean Solutions, Stanford University, Stanford, CA, USA
- 3Conservation Biology Division, Northwest Fisheries Science Center, National Marine Fisheries Service, National Oceanic and Atmospheric Administration, Seattle, WA, USA
Given the rapid rise of environmental DNA (eDNA) surveys in ecology and environmental science, it is important to be able to compare the results of these surveys to traditional methods of measuring biodiversity. Here we compare samples from a traditional method (a manual tow-net) to companion eDNA samples sequenced at three different genetic loci. We find only partial taxonomic overlap among the resulting datasets, with each reflecting a portion of the larger suite of taxa present in the sampled nearshore marine environment. In the larger context of eDNA sequencing surveys, our results suggest that primer amplification bias drives much of the taxonomic bias in eDNA detection, and that the baseline probability of detecting any given taxon with a broad-spectrum primer set is likely to be low. Whether catching fish with different nets or using different PCR primer sets, multiple data types can provide complementary views of a common ecosystem. However, it remains difficult to cross-validate eDNA sequencing techniques in the field, either for presence/absence or for abundance, particularly for primer sets that target very wide taxonomic ranges. Finally, our results highlight the breadth of diversity in a single habitat, and although eDNA does capture a richer sample of the community than traditional methods of sampling, a large number of eDNA primer sets focusing on different subsets of the biota would be necessary to survey any ecological community in a reasonably comprehensive way.
Introduction
Environmental DNA (eDNA)—genetic traces of organisms taken from a sample of water, soil, or other medium—is increasingly useful for ecological surveys, particularly where traditional sampling is difficult or expensive (Foote et al., 2012; Kelly et al., 2014b). While qPCR techniques have focused on single species detections to good effect (Mahon et al., 2013; Eichmiller et al., 2014), high-throughput sequencing using generalized PCR primers offers the promise of a more holistic view of an ecosystem, revealing hundreds or thousands of taxa present and potentially their abundance via cheap and relatively easy-to-collect environmental samples (e.g., Leray and Knowlton, 2015). Nevertheless, questions remain about sequence-based eDNA surveys, even as the technique becomes more common in the field. One key question is how to compare eDNA results to those of traditional (e.g., manually counted) ecological surveys, given that the two sets of methods can reveal quite different views of an ecological community (Baker et al., 2016).
A seemingly straightforward way of assessing the performance of the DNA surveys is to compare sequencing results to a known community (Kelly et al., 2014a; Evans et al., 2016) or to compare eDNA results to those from a traditional sampling technique (Lacoursière-Roussel et al., 2015; de Vargas et al., 2015; Port et al., 2016). These studies have tended to show a correlation between results obtained through traditional sampling methods and those using environmental DNA (but see Adams et al., 2013), although the relationship between traditionally collected results (e.g., manual counts or biomass) and relative eDNA sequence abundance clearly varies by taxon and with environmental context.
Results from eDNA need to be comparable to—and integrated with—information from other, more traditional sampling methods, and therefore we must be able to understand them in the context of results from methods that have been the basis of ecology and environmental science up to this point. However, developing such an intuitive and integrative understanding is challenging, especially because in many cases we should expect different methods of environmental sampling to yield different results. Like any survey method, eDNA detects only a portion of the biodiversity present in the sampled location. In the marine realm, sampling tools and techniques such as settlement plates, various nets, and aerial surveys reveal quite different sets of taxa, producing a variety of ecological patterns that are not easily compared. Similarly, eDNA surveys may detect distinct suites of species that are not easily comparable to results obtained through manual or visual surveys (Shelton et al., 2016).
As with traditional survey tools, eDNA studies should be explicit about the ways in which methodological choices determine the communities observed. One means of doing so is to survey multiple genetic markers (“loci”) simultaneously. Just as the results from any single genetic marker may differ from those of manual or visual sampling techniques, so too may the results differ among genetic markers (Cowart et al., 2015). Using multiple markers allows the researcher to assess the degree to which the detected ecological communities vary as a result of the primer set used, and where the taxa amplified by multiple markers do overlap, offers a chance to cross-validate detections of particular taxa of interest. To the extent that different survey techniques reveal non-overlapping portions of a fauna (and hence are not useful for cross-validation), they nevertheless may be used in combination to reveal a broader suite of taxa than would otherwise be possible.
Here, we present eDNA data from 3 genetic markers—16S, 18S, and COI—for water samples collected alongside a traditional tow-net survey of epifauna in nearshore eelgrass (Zostera marina) habitat in Puget Sound, Washington, USA. Taken together, our four data sources (three genes and one set of manually counted animal species) detected at least 366 taxonomic Families in 28 Phyla across 8 Kingdoms of Eukaryotes, including many conspicuous species commonly associated with this habitat. We compare the Families detected with different genetic loci to those collected in the manual tow-net. These comparisons then motivate a simulation of species detections with multiple eDNA loci as well as a discussion of the divergent and complementary views of an ecosystem that different collection methods afford.
Methods
Environmental Setting and Field Collections
We sampled 4 sites in nearshore eelgrass (Z. marina) habitats adjacent to watersheds along a gradient of urbanization in Puget Sound, Washington, USA, as part of a larger project focused on the effects of such urbanization (Kelly et al., 2016; Samhouri et al., in revision). Previous work identified imperviousness (i.e., surface areas that reduce infiltration and increase runoff; Schueler et al., 2009) as a variable that effectively reflected differences in upland watersheds associated with urbanization (Samhouri et al., in revision), and the urbanization gradient was also associated with changes in the eDNA community detected with 16S eDNA (Kelly et al., 2016). Here we focus on a subset of sites that reflect a range of impervious surface cover in upland watersheds (7–42%) to maximize the likely diversity of taxa captured, but our present purpose is to evaluate the results of different ecological sampling methods on a common pool of species rather than to reflect ecological trends along an urbanization gradient. Further site details and coordinates are given in Table S1.
As detailed in Samhouri et al. (in revision) and Kelly et al. (2016), we collected epibenthic macroinvertebrates along a 100 m length of coastline at each of the study sites (n = 3 transects per site, 4 sites). Collections occurred in July 2014 using a benthic sled (1 × 1 m opening, 1 mm mesh), towed over parallel, 10 m transects approximately 50 m apart (−0.5 m tidal elevation). Invertebrates were separated from the tow net in the field and preserved for quantification. Manual counts of invertebrates yielded 49 unique taxa—of which 36 were identified to Family level—including primarily malacostracan crustaceans, gastropods, and fish (Figure S2; Table S2).
We simultaneously collected 1-L water samples for eDNA analysis at each transect, immediately below the water surface using a ca. 3.3 m pole to hold the sampling bottle, with the goal of reducing human contamination. We kept these samples on ice until they could be processed in the lab (within hours of collection). We filtered the total volume of the samples (1 L) onto cellulose acetate filters (47 mm diameter; 0.45 um pore size) under vacuum pressure, and preserved the filter at room temperature in Longmire's buffer following (Renshaw et al., 2015). Deionized water (1-L) served as a negative control for filtering. We extracted total DNA from the filters using the phenol:chloroform:isoamyl alcohol protocol in Renshaw et al. (2015), resuspended the eluate in 200 uL water, and used 1 uL of diluted DNA extract (1:100) as template for PCR. See Supplemental Methods for additional sampling details.
eDNA Amplification and Sequencing
We used three primer sets for eDNA amplicon analysis, each targeting a different gene region, with the goal of detecting large numbers of taxa present in the sampled environment, which we could compare against the manual tow-net surveys of macroscopic animals. The primer sets amplified two mitochondrial loci [16S, ca. 116 bp, (Kelly et al., 2016; O'Donnell et al., 2016; Shelton et al., 2016), targeting metazoans exclusively; COI, ca. 315 bp, (Leray et al., 2013), targeting eukaryotes], and one nuclear ribosomal gene [18S, ca. 385 bp, (Stoeck et al., 2010), targeting eukaryotes]. We note that not all primers are expected to amplify all taxa, just as not all nets are expected to catch all fish; our purpose here is to compare the results of commonly used survey tools, rather than to ensure survey methods are likely to result in taxonomic overlap. We generated amplicons using a two-step PCR procedure, described in O'Donnell et al. (2016), to avoid the taxon-specific amplification bias that results from the use of differentially indexed PCR primers (commonly used to include multiple samples onto the same high-throughput sequencing run to minimize costs). The specific PCR protocol for each locus is included in Supplemental Methods.
We generated three (COI, 18S) or four (16S) PCR replicates for each of 18 water samples (3 samples per site, 6 sites) in order to assess variance due to amplification. We simultaneously sequenced four positive [Tilapia; Oreochromis niloticus, or Ostrich (Struthio camelus) tissue; selected because these taxa do not occur in the Puget Sound region and therefore could not be present in the field samples] and three negative controls (de-ionized water) with identical replication. Following library preparation according to manufacturers' protocols (KAPA Biosystems, Wilmington, MA, USA; NEXTflex DNA barcodes, BIOO Scientific, Austin, TX, USA), sequencing was carried out on an Illumina Nextseq (150 bp, paired-end; 16S) or MiSeq (250 bp, paired-end; 18S and COI) platform.
Sequence Processing, Bioinformatics, and Annotation
We processed the resulting sequence reads with a custom Unix-based script (O'Donnell, 2015), which calls third-party programs Swarm, Cutadapt, and PEAR (Martin, 2011; Zhang et al., 2014; Mahé et al., 2015) to move from raw sequence data to a quality-controlled dataset of counts of sequences from operational taxonomic units (OTUs). We controlled for contamination in three ways. First, we dropped samples that had highly dissimilar PCR replicates (Bray-Curtis dissimilarities > 0.8). To address putative contamination, we used a site-occupancy model to estimate the probability of OTU occurrence (Royle and Link, 2006), using multiple PCR replicates of each environmental sample as independent draws from a common binomial distribution. We eliminated from the dataset any OTU with <80% estimated probability of occurrence. Finally, we minimized the effect of potential cross-contamination among samples as follows: (1) we calculated the maximum proportional representation of each OTU across all control samples, considering these to be estimates of the proportional contribution of contamination to each OTU recovered from the field samples. (2) We then subtracted this proportion from the respective OTU in the field samples. Overall contamination was low; for example, 94–99.9% of positive control reads annotated to Oreochromis or Struthio. The result was a high-confidence dataset in which sample replicates and site replicates clustered together tightly in ordinal space following principal component analysis (Figure S1). Sequences are available as NCBI accession No. SAMN06173488.
We assigned a taxonomic name to each OTU sequence using blastn (Camacho et al., 2009) on a local version of the full NCBI nucleotide database (current as of July 2016), recovering up to 50 hits per query sequence and reconciling conflicts among equally good matches using the last common ancestor approach implemented in MEGAN (Huson et al., 2011).
For clarity, in the following we refer to the results for community composition from the three loci and the single manual counts as four “datasets.”
Community Composition and Diversity
We rarefied read counts of Family-level detections from each PCR replicate to allow for comparison across water samples and across datasets. For the three eDNA loci we generated 1000 rarefaction draws of each sample (ca. 3 × 104 reads each, reflecting the smallest sample size of the field samples) using the vegan package for R (Oksanen et al., 2015). We carried out subsequent analyses on a single, illustrative rarefaction draw; these did not vary substantially among the rarefaction replicates (the standard deviation of the number families among rarefaction replicates at each locus was 0.7, 1, and 2.5% of the mean number of families for 18s, COI, and 16s, respectively). To generate the Family-level accumulation curve, we sampled from a pool of all 1000 rarefaction replicates, to ensure adequate representation of the sampling error due to rarefaction. To assess variance among PCR replicates, among samples, and among sites with the eDNA data we used un-rarefied data.
We assessed the degree of taxonomic overlap among datasets by counting taxonomic Families occurring within and across datasets (Table S2). First, for each dataset, we asked whether a Family was detected at any of the sampled geographic sites in Puget Sound, and reported the number of Families detected in common across each of the 4 datasets. We then compared the value of adding geographic sites for each dataset individually (and for combinations of datasets) by constructing an accumulation curve of unique taxonomic Families. To do so, we took 1000 random ecological communities from the pool of 1000 rarefaction draws for each eDNA locus at each geographic site, and reported the accumulation of unique Families as the number of sites increased. These accumulation curves reflect the marginal benefit (in terms of additional Families detected) of sampling one additional geographic location with one or more collection methods or loci.
Different sampling methods capture different subsets of biodiversity, and each has taxonomic selectivity. To illustrate the biases of each of our methods, we mapped Family-level detections in each of our datasets onto a taxonomic tree. The taxonomic classification was determined using the NCBI Entrez Taxonomy Database (http://www.ncbi.nlm.nih.gov/taxonomy) and the phylogenetic tree was constructed using phyloT (phyloT: Phylogenetic Tree Generator, 2016). We then visualized the tree using the Interactive Tree of Life (Letunic and Bork, 2007, 2016) and overlaid data on locus-specific detections. The tree can be viewed at http://itol.embl.de/tree/1716783125293921474501988. We additionally plotted the proportion of annotated OTUs by Kingdom and by dataset (Figure S2).
Assessing Consistency among eDNA Markers
We assessed taxonomic consistency for each pair of genetic markers in two ways, both using nonparametric correlation. First, for Families detected in both of a pair of loci, we asked whether the number of OTUs in each Family (N = number of Families with detections in a pair of loci) was correlated across loci. Because the number of OTUs per taxonomic Family may vary between gene fragments as a result of locus-specific differences in lineage divergence rates (i.e., OTUs per taxonomic Family), read lengths, mutation rates, and differences in taxonomy within different organismal groups, we also assessed the number of sequence reads at each locus that were assigned to any given taxonomic rank. We then tested whether read-counts-per-taxon (e.g., Family, Order, Class, or Phylum) were correlated for each pair of genetic markers.
Partitioning Variance
We calculated the variance in OTU communities among PCR replicates (for the three eDNA loci), among samples within geographic sites, and among sites for each of our datasets with a PERMANOVA test on Jaccard distances among presence/absence versions of our non-normalized data. These variances are useful for parameterizing models that relate sequenced communities to biomass in the field (Shelton et al., 2016), as well as for developing eDNA sampling designs with adequate statistical power to test hypotheses of interest. To further examine the source of variance among technical (PCR) replicates, we also calculated Bray-Curtis dissimilarity among technical replicates for deciles of OTU abundance at each locus.
Occupancy Modeling
Species (or “site”) occupancy modeling provides an accessible framework for interpreting taxon detections in eDNA studies (Lahoz-Monfort et al., 2015; see also Ficetola et al., 2015; more broadly, see Miller et al., 2011; Royle and Link, 2006), and we used this method in a simple simulation to assess the value of using multiple eDNA loci for biodiversity surveys. Explicitly assessing the eDNA detection probabilities is especially important in contexts such as invasive (Jerde et al., 2011; Lodge et al., 2012) and endangered (Thomsen et al., 2012) species detection, where policy decisions might hinge on eDNA results.
Occupancy modeling is typically considered in a single taxon context. For example, in qPCR assays the probability of detection is strictly a function of the concentration of target DNA in a sample. By contrast, for sequencing studies occupancy models need to be extended to a multi-taxon community and therefore “detection” of a given taxon depends upon a series of analytical steps (from amplification to annotation) as well as on relative abundance of a sequence. Nevertheless, occupancy modeling can be applied to communities and is an appropriate framework for interpreting eDNA results from sequencing studies. An important component of interpreting the community observed from eDNA studies is then understanding how using multiple loci can affect the assessment of a given community. We performed simulations to compare the use of different loci singly or together in concert to characterize a community from eDNA. We note that this occupancy modeling is a special case of the more general framework elaborated in Shelton et al. (2016), which uses a Bayesian hierarchical model to estimate community structure from eDNA reads. However, for ease of computation our simulation below uses a version of binomial occupancy modeling.
Our simulation supposed 100 species were present in a sampled environment. We let the probability of detecting each of those 100 species vary among species and among each of 5 genetic loci (labeled loci A–E), drawing the values of true-positive detection rates (p11, i) for each locus i at random from five different beta distributions (p11 stands for the probability of a species being detected—1, as opposed to 0, which would signify non-detection or absence—given that it is present). These distributions broadly reflect the trends we observed in our empirical data, but do not precisely correspond to any real locus. Locus A amplified ~70% of the species present with a probability >0.01, but did so with a median p11_A = 0.1 (i.e., half of the species were detected <10% of the time, even when they are present). Locus B amplified a greater portion of the 100 species present (all have p11_B > 0.01), but with nearly the same median p11_B = 0.11. Locus C was similar to Locus B, but with a higher median p11_C = 0.19. Locus D amplified a few species very well, and most very poorly; Locus E amplified the species present much more poorly, with the median p11_E = 0.002 (distribution parameters given in Figure 4 caption). Thus, each simulated locus reflected characteristics of eDNA loci in practice ranging from very general but erratic loci in A and B to a highly specific and targeted locus in D. For each locus, we treated the frequency of false-positive detections (p10, following Royle and Link, 2006) as negligible given the quality-control of sequencing results that we and other researchers employ. Under such a scenario, any detection of a taxon at any locus is evidence of that taxon's presence in the field. Consequently, the number of species detected by any combination of loci was the sum of unique species detected by any locus.
We simulated a single biological replicate (for example, a single bottle of water to be analyzed for eDNA) as a single draw from a binomial distribution, with the probability of each species being detected by each locus given by the values of p11_A, p11_B, p11_C, p11_D, and p11_E, respectively. Multiple biological replicates represented additional independent binomial draws given those same detection probabilities. We then compared the number of taxa detected by increasing numbers of loci and numbers of replicates.
Results
Community Composition and Diversity
Our surveys revealed hundreds of taxa present across the sampled geographic sites in Puget Sound, representing many of the most common and characteristic animals from the nearshore habitats of the region. These included barnacles (Balanidae), mussels (Mytilidae), and snails (Littorinidae) of the upper intertidal zone, Cancer and kelp crabs (Cancridae, Epialtidae) of the lower intertidal zone, fishes (Embiotocidae, Cottidae, Syngnathidae, Salmonidae) and seals (Phocidae), subtidal clams (Hiatellidae, Veneridae), and many other iconic groups. In total, our samples featured 366 taxonomic Families, with each dataset providing a different subset of these taxa (Table S2).
Our tow-net surveys recovered 45 distinct animal taxa, of which 32 could be identified to Family level. eDNA surveys with PCR primers targeting three gene regions—16S, COI, and 18S—resulted in sequences detecting a total of 1.8 × 104 unique OTUs, of which a total of 1.3 × 104 could be classified by matching known sequences in GenBank (Table 1).
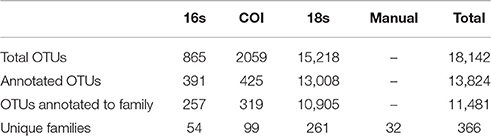
Table 1. OTUs and annotations by genetic locus, and manual annotations for comparison.
Of those that could be annotated, the eDNA samples represented a total 366 unique taxonomic Families across the Eukarya, including representatives from 33 Phyla from 9 Kingdoms (Table S2). The identities of taxa detected varied dramatically across genes, and between genetic vs. manual methods of sampling (Figure 1). No Family was detected in all four datasets, while four Families were detected by three data types, and 72 were detected by two data types. Hence, many of the Families detected were unique to a given data type (i.e., 290 Families detected only by a single data type), underscoring the differences in suites of taxa sampled by the different methods.
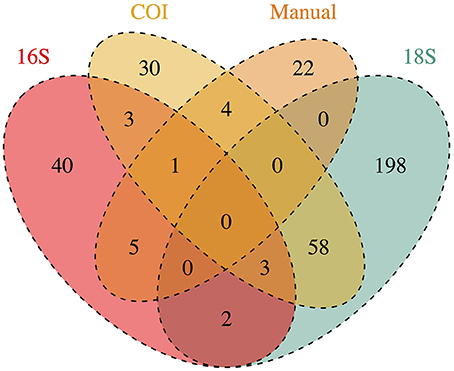
Figure 1. Taxonomic Families detected by each method of collecting ecological data (3 genetic markers and manual tow-net). Numbers at the intersections of each method reflect the number of Families detected in common. Numbers in the figure represent annotations from a single rarefaction draw; these numbers change only trivially across 1000 rarefaction draws from the raw datasets.
Each dataset showed a distinct phylogenetic selectivity, consistent with the idea that primer-site mismatches are likely to be driving the observed patterns (Figure 2). In particular, the manual tow-net surveys and our 16S primers detected metazoans exclusively (by design), while the 18S and COI primers also amplified dinoflagellates, diatoms, green plants, and a variety of other eukaryotic lineages.
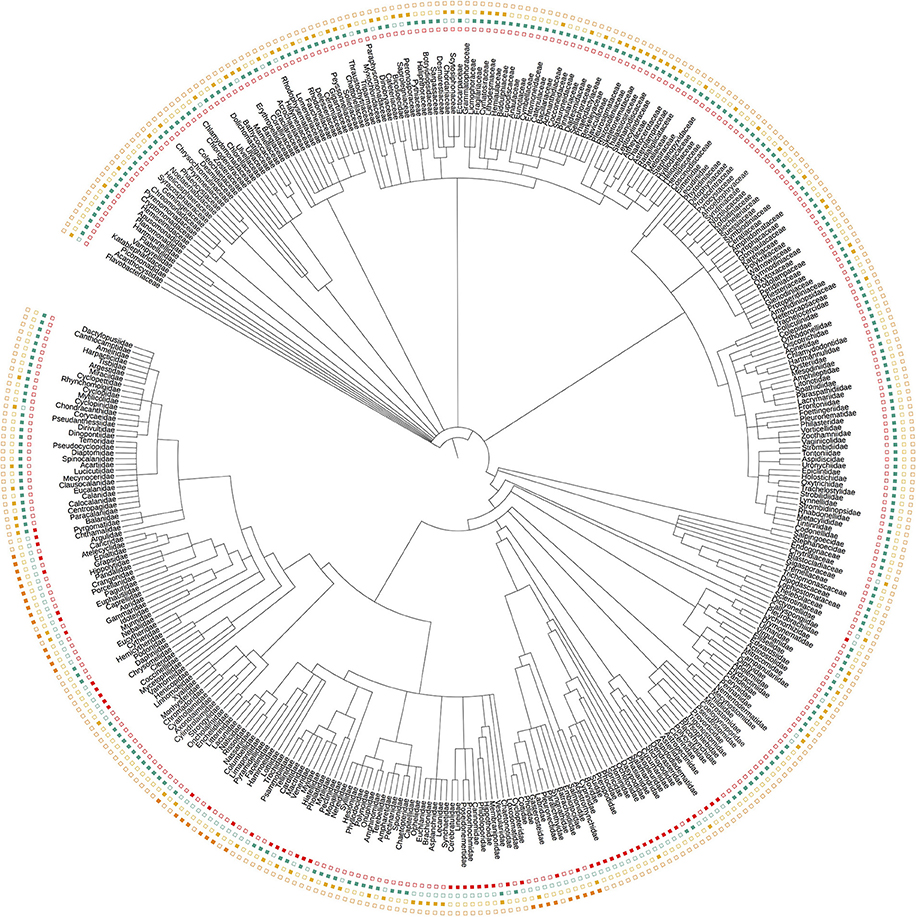
Figure 2. Taxonomic tree representing all Families detected across the three eDNA loci (16S, 18S, and COI) and by manual tow-net. Filled symbols indicate detection; empty symbols indicate non-detection. The tree illustrates the taxonomic selectivity of each sampling method, and does not contain all taxa (i.e., detected and undetected) from the relevant clades. Colors follow the convention in Figure 1.
Because each data type reveals a nearly non-overlapping set of taxa, the accumulation curves reflect the idea that adding data types dramatically increases taxonomic coverage (Figure 3). These curves demonstrate, in part, the taxonomic limitations of the data types. The manual tow-net survey focused exclusively on macroscopic epiphytic animals living on eelgrass. The 16S primer set targeted animal taxa exclusively, although it recovered animals with a broader set of ecologies—for example, benthic infauna and intertidal species—than did the manual counts, and its accumulation curve is consequently shifted upward. COI and 18S detected a wide range of animals, but also sampled from across the rest of the eukaryotic tree of life, and the accumulation curves for those two loci accordingly reflect this phylogenetic breadth. The rates of taxonomic accumulation accordingly vary significantly across datasets, but are strictly correlated with asymptotic values, suggesting that each might be useful for comparing biodiversity across systems or habitats.
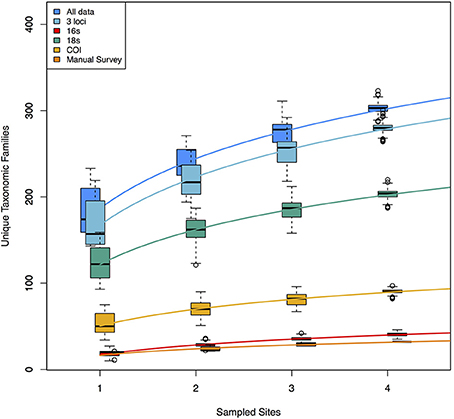
Figure 3. Accumulation curves for each of three molecular datasets, and for the manual dataset. Also shown are the curves for a combination of the three molecular datasets and from a combination of all data (molecular + manual). Molecular datasets are pools of 1000 rarefaction draws (of ca. 3 × 104 reads) from full datasets. The variance shown in each accumulation curve represents differences among 1000 random samples drawn from the available pool of data for each of 1–4 geographic sites. Rates of accumulation differ significantly for each of the six logarithmic trendlines shown.
Assessing Consistency among eDNA Markers
Where a taxon was detected by more than one eDNA marker, we asked whether the different eDNA markers yielded consistent estimates of diversity within the taxon. The only pair of markers that yielded consistent results among the taxa shared was COI-18S. Families detected by both 18S and COI had correlated numbers of OTUs (Spearman's rho = 0.37, p = 0.004; N = 61; data log-transformed), while 18S-16S (N = 5) and COI-16S (N = 7) co-detected Families had uncorrelated numbers of OTUs.
Between-marker correlations in read-counts-per-taxon mirrored the OTUs-per-Family results, with the 18S and COI primer sets reflecting correlated numbers of reads for the Phyla, Classes, and Families detected at both loci (rho = 0.74, 0.56, and 0.39, respectively; p < 0.01 for each comparison; log-transformed read-counts; OTUs-per-Order were nonsignificantly correlated). 16S read counts were uncorrelated with 18S or COI read counts for shared taxa at any rank.
Apportioning Variance in eDNA Sampling
Apportioning the variance of a given sampling method's results is critical to interpreting those results, to understanding the processes that might generate the observed data, and to comparing results across data types. Given the exponential nature of PCR reactions and the possibility of stochastic amplification of any given taxon with a given primer set, it is particularly important to assess the variability among PCR replicates in eDNA surveys.
We used a permutation-based analysis of variance (PERMANOVA) to apportion the variance in Jaccard distances among our geographic sites (N = 4), among our biological samples within sites (3/geographic site) and among PCR replicates within each of those biological samples (3 or 4 PCRs/sample). Variance among sampling sites and among water samples (within sites) was significantly different for each of the eDNA loci (Table 2), although among-PCR-replicate variance was as high as among-site variance in the case of 18S.
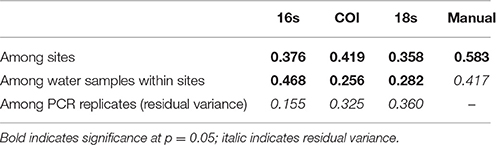
Table 2. Variance in Jaccard distances among PCR replicates (within samples), among samples (within sites), and among sites.
Variance among PCR replicates appears largely due to stochastic amplification and sequencing among rare OTUs. For example, the most common 10% of 16s OTUs had a mean among-replicate Bray-Curtis dissimilarity of 0.025 (SD = 0.022), while the least common 10% had a mean Bray-Curtis dissimilarity of 0.741 (SD = 0.026). We observed the same trend at each locus, with dissimilarity increasing in a saturating curve as OTUs became more rare (Figure S3).
Occupancy Modeling with Multiple Data Sources
Our empirical results provide reasonable starting values for a straightforward simulation of the number of taxa one might expect to recover with a given number of biological replicates and a given number of genetic loci. Two pertinent observations arise from our empirical data: first, the probability of detecting (p11) any given taxon with any given primer set is likely to be low for broad-spectrum primers used in eDNA/metabarcoding studies. Second, Figures 1, 2 show that these detection probabilities vary widely by primer set and by taxon, such that some primer-by-taxon combinations have high values of p11, while many other such combinations have p11 at or near zero (indicating that the primer set does not amplify the taxon of interest in the sampled environmental context at the read depth tested). We use these observations to parameterize our simulation, acknowledging that these are only starting values for exploration.
For a hypothetical community of 100 species, we simulated eDNA detections for each of five hypothetical genetic loci (see Section Methods). Each locus had a different probability of detecting each of the species present (i.e., p11 varied for each locus-by-species pairing, as in the real world; see Section Methods and Supplemental Methods for further model details). To assess the accumulation of species detections with multiple loci—and to examine the effect of replicate sampling on these species detections—we generated 1000 simulated eDNA communities for 1–5 loci carried out using 1–5 replicates (Figure 4).
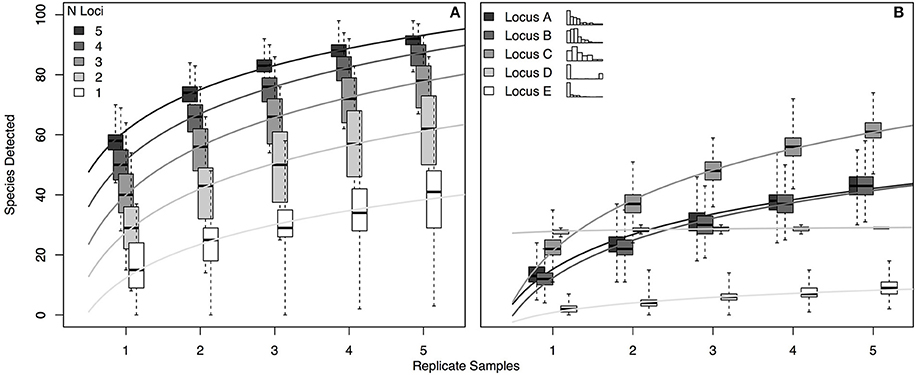
Figure 4. Occupancy detection simulations for each of 5 hypothetical eDNA loci. The probability of detection (p11) varied for each combination of 100 hypothetical species and the 5 loci, with values of p11 drawn from beta distributions as follows: p11_A~Beta (1, 5); p11_B~Beta (1.5, 10); p11_C~Beta (1.5, 5); p11_A~Beta (0.01, 0.04); p11_A~Beta (0.2, 10). (A) Accumulation of species detections by number of loci and by number of replicate samples. (B) Accumulation of species detections by locus and by number of replicate samples, where each replicate is an independent draw from the binomial distribution given probability of detection p11_i,j (where i is locus A–E and j is species 1–100). Inset histograms illustrate the distribution of p11 values for each locus across the 100 species, with frequency on the y-axis and the x-axis bounded by 0 and 1. For each panel, variance shown reflects the different numbers of species detected in each of 1000 simulations. R code for simulations is provided in Supplementary Material.
The simulation results suggest several practical conclusions for real-world eDNA surveys. First, increasing the number of loci used is likely to increase taxonomic coverage more effectively than increasing the number of biological replicates sequenced with a given locus. For example, using 2 loci rather than one approximately doubles the expected taxonomic coverage in a single replicate (median 29 species detected vs. 15; Figure 4A); the effect of using 2 replicates with a single locus depends strongly on the distribution of species detection rates for that locus, but generally results in a more modest increase in coverage (68% increase, Locus C; zero increase, Locus D. Figure 4B). Second, given idiosyncratic amplification of some species by some loci, using more replicates with a given locus does not guarantee more taxonomic coverage, as shown by the accumulation curve for locus D in the right-hand panel. This result is a function of the essentially binary distribution of p11 across species for locus D (see inset histogram of p11 values in Figure 4B): locus D detects a few species with nearly 100% efficiency, and otherwise very poorly, representing a locus with highly taxon-specific binding. Consequently, more replication does not result in more species detected. Finally, the simulation makes clear that even using 5 loci and 5 replicates—more than any current ecological eDNA study has done to our knowledge—leaves some number of species undetected. Even our most comprehensive simulated sampling detected only about 90% of species present.
Discussion
The attraction of eDNA sequencing for ecological surveys is the ability to detect hundreds of eukaryotic organisms from a water sample, but the interpretation of eDNA data relative to the results of established ecological sampling methods is a nascent endeavor (Shelton et al., 2016). Here, we have compared surveys of a nearshore marine environment using multiple eDNA markers and a traditional manual sampling technique. We find that each survey method reflects a distinct subset of organisms from the surrounding environment. As a result, sampling with multiple eDNA markers or survey methods is likely to yield a far richer and more holistic view of the ecosystem sampled, but the resulting datasets may have little taxonomic overlap, which can make it challenging to cross-validate datasets or combine information to make community-wide inferences. Below, we discuss the implications of multi-locus (or multi-method) sampling as it is relevant to core questions of ecological assessment, including community composition and diversity, likelihood of detection, and variance among samples.
Community Composition, Diversity, and Differences among eDNA Markers
Ecology and related disciplines depend upon techniques to sample and describe communities, ecosystems, and their properties. For example, larval settlement plates may capture barnacle larvae and bryozoans, but tell us nothing about the sea lions swimming nearby. Often this selectivity is quite intentional—perhaps we care about bryozoans, and not about sea lions—but where unintentional, such selectivity may provide a misleading picture of the community in ways that often remain unexplored.
The organisms we detected depended strongly upon the survey method (e.g., eDNA vs. manual counts) or the PCR primers used (e.g., 16S vs. 18S eDNA sampling), and each detection method had strong taxonomic selectivity. Most relevant to eDNA studies, our data suggest that different primer sets reveal different draws from a common pool of species represented in the sampled bottle of water. Given the taxonomic selectivity apparent among eDNA markers, our results indicate that differences in detections are most likely driven primarily by interactions between primer and template DNA rather than variation in environmental factors leading to abundance in eDNA, such as organismal DNA shedding rates. If differences in DNA shedding drive detection differences, we would expect the same taxa (those with high shedding rates) to be detected across different loci and to have correlated numbers of OTUs per locus; neither prediction is borne out in our data.
The different community-level views revealed by manual and eDNA sampling underscore the importance of complementary sampling methods for ecology, given that any one set of samples yields a necessarily selective view of the world; 10 different sampling methods can yield 10 different results even with small numbers of target taxa (Valentini et al., 2015). One advantage of using multiple data types is the ability to see far deeper into an ecosystem than would otherwise be possible, each providing a new window into a complex living world. Such breadth comes at a cost, however, because of the nearly non-overlapping suite of taxa these different datasets encompass, a small handful of datasets from the same ecosystem may not provide an opportunity for cross-validation. Neither do our present datasets give us mutually-informative estimates of abundance such as would be necessary for combining the different data types to yield a single view of the ecosystem (Shelton et al., 2016). A large number of independent surveys—perhaps using many PCR primers that differed only by one or a few base pairs—might serve both purposes, and in silico testing before field use might provide a starting point for estimating the extent of taxonomic overlap among such primer sets. Whatever the technique used, we view multi-method and multi-locus eDNA surveys as a significant advance over single-method ecological data, and perhaps as a first step toward being able to animate the responses of whole ecological communities to external phenomena.
Occupancy Modeling
We used the comparison between different survey methods to derive estimates of eDNA detection probabilities for animal taxa. Our results suggest that detection rates for particular taxa are likely to be low (p11 < 0.25) when using broad-spectrum (“universal”) PCR primers. For example, 6 of 32 (0.19) taxa caught in the net were also detected with 16S, and 5 of 32 (0.166) detected with COI. One implication is studies targeting a small number of known taxa—for example, invasive or endangered species—are more likely to have success with qPCR assays than with large-scale eDNA sequencing. Consistent with this idea, the most high-profile uses of eDNA to date have targeted particular taxa using qPCR (Thomsen et al., 2012; Mahon et al., 2013; Eichmiller et al., 2014; Laramie et al., 2015).
Our empirical datasets reflect the taxonomic selectivity of the different sampling methods and of the different eDNA primer sets. For example, the manual tow-net samples contained only macroscopic animal taxa, while animals are only one of at least eight Kingdoms detected by the 18S primer set. Hence, for a given sequencing depth, the 18S primer set is likely to detect only a small percentage of the traditionally sampled set of focal taxa (and hence p11_18S will necessarily be low for any given species). Because the 16S primer set is more narrowly targeted at metazoans, it is likely to reflect a higher proportion of any focal set of animals (i.e., p11_16S will be higher than p11_18S for any given animal species). We note that a previous eDNA sequencing study (Port et al., 2016) used a primer set targeting a much smaller suite of species (vertebrates alone), and consequently detected a high proportion of target taxa known to be present (0.917).
These detection estimates give us a starting point for developing an intuition for eDNA sequencing survey results. Namely, that primer-by-species p11 is likely to scale inversely as a function of the number of taxa amplified by the eDNA primer set, given a constant depth of sequencing (i.e., effort). More narrowly targeted primer sets—such as those targeting only a particular Family, Class, or Order, or in the extreme, qPCR primers targeting a single species—are likely to have much greater detection probabilities. It remains more difficult to estimate the probability of false-positive detections (p10), because nearly always there is no complete record of biological diversity in the sampled natural communities against which to compare eDNA results. Nevertheless, for well-characterized faunas, these rates seem likely to be much lower than p11 for any given group of interest, and moreover, common quality-control techniques (e.g., Ficetola et al., 2014) likely reduce p10 to near zero.
Multiple detections of the same taxon with different primer sets—or different sampling techniques—represent independent detections of that taxon. As a result, multiple data types are useful for evaluating the probability that a particular taxon is or is not present. One implication of this straightforward observation is that multiple eDNA markers can be useful for greatly improving estimates of the probability that a species is present, or for rapidly increasing taxonomic coverage. This is particularly useful when water samples are scarce or expensive; multiple genes provide an inferential benefit even when drawn from the same sample of water. Moreover, because our simulation assigned detection probabilities to each locus independently and with generally low probabilities, the result is a conservative estimate of the value of multi-locus data. The value of additional loci would be greater where loci are more taxon-specific.
We emphasize that the absolute values of detection probabilities are highly context-specific. For example, greater sequencing depth is likely to increase the probability of a true positive detection (p11) for any given species-by-primer set combination, and the calculation is highly sensitive to the completeness of the annotation database (e.g., GreenGenes, SILVA, or as here, the BLAST nucleotide database, which is not curated in the same way as are true annotation databases). Nevertheless, the values we estimate here provide a way of building intuition about reasonable expectations for eDNA sequencing surveys, and for making selectivity of traditional survey techniques explicit by putting their detection rates in context.
The occupancy simulations also underscore a final point about the use of multiple loci—or indeed, any set of multiple survey methods, molecular or not—to canvass biological diversity in an area. Our simulations suggest that some fraction of species will remain undetected even with intensive molecular surveys. This result of course depends upon the specified detection probabilities and species-by-primer interactions, but is a useful result to highlight the fact that no ecological sampling method is likely to reveal the whole of a community. We see this as a particularly relevant lesson as standardized techniques for biodiversity assessment (e.g., autonomous reef monitoring structures, ARMS) become more common around the world (Duffy et al., 2013).
Apportioning Variance in eDNA Sampling
Two salient observations arise from our assessment of variance in the eDNA datasets, particularly when combined with those reported for a fourth genetic marker in Port et al. (2016; 12S gene) in a different sampled environment. First, markers recovering a very broad suite of eukaryotic taxa (here, COI and 18S) can result in community-level samples that vary widely among PCR replicates. Somewhat more narrowly targeted markers (16S and 12S in Port et al., 2016) tend to be more consistent among technical replicates. This observation—if supported by data from a larger number of markers—suggests there is a tradeoff between consistency and taxonomic diversity recovered at any given sequencing depth.
Such a result is expected if the detectable diversity in a water sample is far greater than that which is amplified in any given PCR reaction, due to stochasticity in early PCR cycles. Primers only bind to a small fraction of the total number of potential taxonomic targets during the PCR reaction, and the result is a different sampling of taxa—even among technical replicates derived from the same field sample–upon sequencing. We would expect greater stochasticity among rare OTUs than among common ones as a result of this sampling effect, and our data and others' (Zhou et al., 2011) are consistent with this expectation. Taken together, these observations suggest that sequencing more PCR replicates is worthwhile for primer sets with greater taxonomic breadth.
The corollary observation is that higher-variance markers are likely to have lower detection probabilities (p11) for any given taxonomic group (again, sequencing depth being held constant), because of the same stochasticity in the PCR process. Each amplification reaction is likely to contain the same high-abundance amplicons (whether from numerically abundant taxa or taxa with very high primer-template affinity), but to differ among low-abundance amplicons, given that each PCR reaction will reflect an arbitrary draw of rare templates from the large pool of diversity present. Taken together, the possible associations among variance, taxonomic breadth, and detection probability suggest that there is a tradeoff between the number of taxa one can detect with a single eDNA marker and the confidence with which one can detect those same taxa.
Conclusion
In order to confidently interpret eDNA results in the context of existing ecological study, it is necessary to compare the results of this emerging technique with those of more established methods of ecological sampling. We find that (1) consistent with previous results, eDNA captures a far broader selection of taxa than the accompanying manual survey, (2) the ecological communities detected vary dramatically among eDNA markers and between survey techniques, and (3) despite detecting a total of over 300 taxonomic Families across three eDNA markers, the genetic survey did not come close to detecting most of the eukaryotic diversity present. For example, only about one-third of manually-collected Families were present in the eDNA survey, and the Family-accumulation curve suggests that many more markers would be necessary to carry out a near-exhaustive sampling.
These results highlight the value of using multiple methods in ecological surveys, given that any one sampling method—even eDNA, which can reveal hundreds of taxa present at a location—unavoidably reflects only a small fraction of the true biological diversity present in the environment. Consequently, a single method or genetic marker may reveal ecological trends important in some (detected) taxa, but these trends may not necessarily be general across groups. Accordingly, many microbial and eukaryotic studies grounded in genetics-based observations of the environment may have reached questionable conclusions to the extent these conclusions represent a small and non-random portion of much larger ecological communities. Our results put eDNA in the company of other ecological survey techniques, in that these emerging methods reveal large (but incomplete) swaths of biodiversity for which traditional surveys provide valuable context.
Data Accessibility
The article's supporting data, metadata, and analytical code can be accessed as supplemental information at https://github.com/invertdna/Multilocus_eDNA_Analysis, and in Genbank (accession number: SAMN06173488).
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, and analysis and interpretation of data, and all authors approved of the final manuscript version submitted for publication.
Funding
This work was supported by grant 2014-39827 from the David and Lucile Packard Foundation to RK and by NASA grant NNX14AP62A “National Marine Sanctuaries as Sentinel Sites for a Demonstration Marine Biodiversity Observation Network (MBON)” funded under the National Ocean Partnership Program (NOPP RFP NOAA-NOS-IOOS-2014-2003803 in partnership between NOAA, BOEM, and NASA), and the U.S. Integrated Ocean Observing System (IOOS) Program Office. This paper is a contribution to the MBON program.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank N. Lowell, S. Hennessey, G. Williams, and B. Feist for all of their help throughout; J. Port, L. Sassoubre, and A. Boehm; A. Stier, and P. Levin; M. Dethier, E. Heery, J. Toft, and J. Cordell; R. Morris and V. Armbrust; J. Kralj; A. Wong, E. Garrison, J. Levy, M. Klein, and E. Buckner; coastal property owners for access to field sites; and the Helen R. Whiteley Center at Friday Harbor Laboratories, and two reviewers. We also thank the Marine Biodiversity Observation Network (MBON) eDNA team. JS thanks H. Bolt for inspiration. The views expressed herein are those of the authors and do not necessarily reflect the views of BOEM, NASA, NOAA or any of their sub-agencies. The US government is authorized to reproduce this paper for governmental purposes.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fmars.2016.00283/full#supplementary-material
References
Adams, R. I., Amend, A. S., Taylor, J. W., and Bruns, T. D. (2013). A unique signal distorts the perception of species richness and composition in high-throughput sequencing surveys of microbial communities: a case study of fungi in indoor dust. Microb. Ecol. 66, 735–741. doi: 10.1007/s00248-013-0266-4
Baker, D. G. L., Eddy, T. D., McIver, R., Schmidt, A. L., Thériault, M.-H., Boudreau, M., et al. (2016). Comparative analysis of different survey methods for monitoring fish assemblages in coastal habitats. PeerJ. 4:e1832. doi: 10.7717/peerj.1832
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Cowart, D. A., Pinheiro, M., Mouchel, O., Maguer, M., Grall, J., Miné, J., et al. (2015). Metabarcoding is powerful yet still blind: a comparative analysis of morphological and molecular surveys of seagrass communities. PLoS ONE 10:e0117562. doi: 10.1371/journal.pone.0117562
de Vargas, C., Audic, S., Henry, N., Decelle, J., Mahé, F., Logares, R., et al. (2015). Eukaryotic plankton diversity in the sunlit ocean. Science 348, 1–11. doi: 10.1126/science.1261605
Duffy, J. E., Amaral-zettler, L. A., Fautin, D. G., Paulay, G., Rynearson, T. A., Sosik, H. M., et al. (2013). Envisioning a marine biodiversity observation network. Bioscience 63, 350–361. doi: 10.1525/bio.2013.63.5.8
Eichmiller, J. J., Bajer, P. G., and Sorensen, P. W. (2014). The relationship between the distribution of common carp and their environmental DNA in a small lake. PLoS ONE 9:e112611. doi: 10.1371/journal.pone.0112611
Evans, N. T., Olds, B. P., Turner, C. R., Renshaw, M. A., Li, Y., Jerde, C. L., et al. (2016). Quantification of mesocosm fish and amphibian species diversity via eDNA metabarcoding. Mol. Ecol. Resour. 16, 25–41. doi: 10.1111/1755-0998.12433
Ficetola, G. F., Pansu, J., Bonin, A., Coissac, E., Giguet-Covex, C., De Barba, M., et al. (2014). Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Mol. Ecol. Resour. 15, 543–556. doi: 10.1111/1755-0998.12338
Ficetola, G. F., Pansu, J., Bonin, A., Coissac, E., Giguet-Covex, C., De Barba, M., et al. (2015). Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Mol. Ecol. Resour. 15, 543–556. doi: 10.1111/1755-0998.12338
Foote, A. D., Thomsen, P. F., Sveegaard, S., Wahlberg, M., Kielgast, J., Kyhn, L. A., et al. (2012). Investigating the potential use of environmental DNA (eDNA) for genetic monitoring of marine mammals. PLoS ONE 7:e41781. doi: 10.1371/journal.pone.0041781
Huson, D. H., Mitra, S., Ruscheweyh, H.-J., Weber, N., and Schuster, S. C. (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560. doi: 10.1101/gr.120618.111
Jerde, C. L., Mahon, A. R., Chadderton, W. L., and Lodge, D. M. (2011). “Sight-unseen” detection of rare aquatic species using environmental DNA. Conserv. Lett. 4, 150–157. doi: 10.1111/j.1755-263X.2010.00158.x
Kelly, R. P., O'Donnell, J. L., Lowell, N. C., Shelton, A. O., Samhouri, J. F., Hennessey, S. M., et al. (2016). Genetic signatures of ecological diversity along an urbanization gradient. PeerJ. 4:e2444. doi: 10.7717/peerj.2444
Kelly, R. P., Port, J. A., Yamahara, K. M., and Crowder, L. B. (2014a). Using environmental DNA to census marine fishes in a large mesocosm. PLoS ONE 9:e86175. doi: 10.1371/journal.pone.0086175
Kelly, R. P., Port, J. A., Yamahara, K. M., Martone, R. G., Lowell, N., Thomsen, P. F., et al. (2014b). Harnessing DNA to improve environmental management. Science 344, 1455–1456. doi: 10.1126/science.1251156
Lacoursière-Roussel, A., Côté, G., Leclerc, V., and Bernatchez, L. (2015). Quantifying relative fish abundance with eDNA: a promising tool for fisheries management. J. Appl. Ecol. 53, 1148–1157. doi: 10.1111/1365-2664.12598
Lahoz-Monfort, J. J., Guillera-Arroita, G., and Tingley, R. (2015). Statistical approaches to account for false positive errors in environmental DNA samples. Mol. Ecol. Resour. 16, 673–685. doi: 10.1111/1755-0998.12486
Laramie, M. B., Pilliod, D. S., and Goldberg, C. S. (2015). Characterizing the distribution of an endangered salmonid using environmental DNA analysis. Biol. Conserv. 183, 29–37. doi: 10.1016/j.biocon.2014.11.025
Leray, M., and Knowlton, N. (2015). DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proc. Natl. Acad. Sci. U.S.A. 112, 2076–2081. doi: 10.1073/pnas.1424997112
Leray, M., Yang, J. Y., Meyer, C. P., Mills, S. C., Agudelo, N., Ranwez, V., et al. (2013). A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents. Front. Zool. 10:34. doi: 10.1186/1742-9994-10-34
Letunic, I., and Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Lodge, D. M., Turner, C. R., Jerde, C. L., Barnes, M. A., Chadderton, L., Egan, S. P., et al. (2012). Conservation in a cup of water: estimating biodiversity and population abundance from environmental DNA. Mol. Ecol. 21, 2555–2558. doi: 10.1111/j.1365-294X.2012.05600.x
Mahé, F., Rognes, T., Quince, C., de Vargas, C., and Dunthorn, M. (2015). Swarm v2: highly-scalable and high-resolution amplicon clustering. PeerJ. 3:e1420. doi: 10.7717/peerj.1420
Mahon, A. R., Jerde, C. L., Galaska, M., Bergner, J. L., Chadderton, W. L., Lodge, D. M., et al. (2013). Validation of eDNA surveillance sensitivity for detection of Asian carps in controlled and field experiments. PLoS ONE 8:e58316. doi: 10.1371/journal.pone.0058316
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. doi: 10.14806/ej.17.1.200
Miller, D. A., Nichols, J. D., McClintock, B. T., Grant, E. H. C., Bailey, L. L., and Weir, L. A. (2011). Improving occupancy estimation when two types of observational error occur: non-detection and species misidentification. Ecology 92, 1422–1428. doi: 10.1890/10-1396.1
O'Donnell, J. L. (2015). Banzai: Scripts for Analyzing Illumina-Generated Environmental DNA Sequence Data. Available online at: https://github.com/jimmyodonnell/banzai
O'Donnell, J. L., Kelly, R. P., Lowell, N., and Port, J. A. (2016). Indexed PCR primers induce template-specific bias in large-scale DNA sequencing studies. PLoS ONE 11:e0148698. doi: 10.1371/journal.pone.0148698
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O'Hara, R. B., et al (2015). vegan: Community Ecology Package. Available online at: http://cran.r-project.org/package=vegan
Port, J. A., O'Donnell, J. L., Romero-Maraccini, O. C., Leary, P. R., Litvin, S. Y., Nickols, K. J., et al. (2016). Assessing vertebrate biodiversity in a kelp forest ecosystem using environmental DNA. Mol. Ecol. 25, 527–541. doi: 10.1111/mec.13481
phyloT: Phylogenetic Tree Generator (2016). Avaialble online at: http://phylot.biobyte.de/
Renshaw, M. A., Olds, B. P., Jerde, C. L., McVeigh, M. M., and Lodge, D. M. (2015). The room temperature preservation of filtered environmental DNA samples and assimilation into a phenol–chloroform–isoamyl alcohol DNA extraction. Mol. Ecol. Resour. 15, 168–176. doi: 10.1111/1755-0998.12281
Royle, J. A., and Link, W. A. (2006). Generalized site occupancy models allowing for false positive and false negative errors. Ecology 87, 835–841. doi: 10.1890/0012-9658(2006)87[835:GSOMAF]2.0.CO;2
Schueler, T. R., Fraley-McNeal, L., and Cappiella, K. (2009). Is impervious cover still important? Review of recent research. J. Hydrol. Eng. 14, 309–315. doi: 10.1061/(ASCE)1084-0699(2009)14:4(309)
Shelton, A. O., O'Donnell, J. L., Samhouri, J. F., Lowell, N., Williams, G. D., and Kelly, R. P. (2016). A framework for inferring biological communities from environmental DNA. Ecol. Appl. 26, 1645–1659. doi: 10.1890/15-1733.1
Stoeck, T., Bass, D., Nebel, M., Christen, R., Jones, M. D. M., Breiner, H.-W., et al. (2010). Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19, 21–31. doi: 10.1111/j.1365-294X.2009.04480.x
Thomsen, P. F., Kielgast, J., Iversen, L. L., Wiuf, C., Rasmussen, M., Gilbert, M. T. P., et al. (2012). Monitoring endangered freshwater biodiversity using environmental DNA. Mol. Ecol. 21, 2565–2573. doi: 10.1111/j.1365-294X.2011.05418.x
Valentini, A., Taberlet, P., Miaud, C., Civade, R., Herder, J., Thomsen, P. F., et al. (2015). Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol. Ecol. 25, 929–942. doi: 10.1111/mec.13428
Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A. (2014). PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. doi: 10.1093/bioinformatics/btt593
Keywords: metagenomics, metabarcoding, environmental monitoring, molecular ecology, marine, estuarine
Citation: Kelly RP, Closek CJ, O'Donnell JL, Kralj JE, Shelton AO and Samhouri JF (2017) Genetic and Manual Survey Methods Yield Different and Complementary Views of an Ecosystem. Front. Mar. Sci. 3:283. doi: 10.3389/fmars.2016.00283
Received: 29 September 2016; Accepted: 15 December 2016;
Published: 09 January 2017.
Edited by:
Sandie M. Degnan, University of Queensland, AustraliaCopyright © 2017 Kelly, Closek, O'Donnell, Kralj, Shelton and Samhouri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan P. Kelly, rpkelly@uw.edu