On the Importance of the Microbiome and Pathobiome in Coral Health and Disease
 Michael J. Sweet*
Michael J. Sweet*  Mark T. Bulling
Mark T. Bulling- Environmental Sustainability Research Centre, College of Life and Natural Sciences, University of Derby, Derby, UK
The term “microbiome” was first coined in 1988 and given the definition of a characteristic microbial community occupying a reasonably well defined habitat which has distinct physio-chemical properties. A more recent term has also emerged, taking this one step further and focusing on diseases in host organisms. The “pathobiome” breaks down the concept of “one pathogen = one disease” and highlights the role of the microbiome, more specifically certain members within the microbiome, in causing pathogenesis. The development of next generation sequencing has allowed large data sets to be amassed describing the microbial communities of many organisms and the field of coral biology is no exception. However, the choices made in the analytical process and the interpretation of these data can significantly affect the outcome and the overall conclusions drawn. In this review we explore the implications of these difficulties, as well as highlighting analytical tools developed in other research fields (such as network analysis) which hold substantial potential in helping to develop a deeper understanding of the role of the microbiome in disease in corals. We also make the case that standardization of methods will substantially improve the collective gain in knowledge across research groups.
Introduction
The term “microbiome” was first used in 1988 to describe microbial associates of plants (Whipps et al., 1988) and was defined as a characteristic microbial community occupying a well-defined habitat (i.e., a habitat with a distinct physio-chemical property). In this term the emphasis on the “biome” represents a community. In more recent years, a new term has evolved from that of the microbiome, one aimed at specifically describing the dynamics of the microbiome in response to stress and the onset of disease, and which has been coined the “pathobiome” (Ryan, 2013; Vayssier-Taussat et al., 2014). This new term has been used to describe the complex interactions of pathogenic microbes which may influence or drive disease processes and their relationship to the “normal” microbiome of the organism in question (Chow et al., 2011; Vayssier-Taussat et al., 2014). Both terms are now widely used throughout the literature, particularly in the medical domain, for example with respect to the human gut (Huttenhower and Human Microbiome Project Consortium, 2012; Krezalek et al., 2015; Lloyd-Price et al., 2016). However, these terms are less widely used in the environmental sciences, and studies focused on Scleractinian corals for example have more commonly utilized the terms “microbiota” or “microbial associates” to describe the microbial communities associated with these hosts. Only recently has the term “microbiome” been used by the coral research community (Bourne et al., 2013; Kimes et al., 2013; Meyer et al., 2014) and to date, as far as we can tell, the concept of the pathobiome has not been used at all. Scleractinian corals add an extra level of complexity to the microbiome concept due to their symbiosis with algal communities (Symbiodinium sp.). Combined with a diverse community of bacteria, archaea, viruses, protozoa, and fungi, which are all routinely found associated with these hosts, corals make ideal model host organisms for studying the specific relationships associated with the microbiome and the pathobiome, and how the host responds to changes and shifts in these communities.
In contrast to the corals' symbiotic algae, which have been well characterized with regard to their relationship with corals as well as their responses to variables of climate change, the other members of the coral microbiome remain less well characterized (Glasl et al., 2016). Recently it has been argued that the coral meta-organism or “holobiont” as it is often referred to, hosts three functionally distinct microbial sub-communities: a ubiquitous and stable core microbiome (consisting of very few symbiotic host-selected microbiota), a microbiome of spatially and/or regionally explicit core microbes each filling functional niches, and a highly variable microbial community that is responsive to biotic and abiotic processes across spatial and temporal scales (Hernandez-Agreda et al., 2016). Variations in the composition of the coral microbiome have been shown between large scale geographical regions, between different reef locations within the same geographical area, and between depths on the same reef (see Bourne and Webster, 2013 for a detailed review). The composition of the microbiome in corals has also been shown to respond to many biotic and abiotic factors. For example, shifts in the composition, richness, and abundance of the microbiome has been shown to vary due to biological events (e.g., algal competition, reproduction, and the onset of diseases or other health compromised states), coral characteristics (e.g., age of the colony), as well as changes in environmental variables (e.g., temperature, pH, nutrients, light, and dissolved organic carbon, Bourne and Webster, 2013; Sweet et al., 2013; Williams et al., 2015; Hernandez-Agreda et al., 2016). Furthermore, on a smaller scale, microbial communities have been shown to differ along a single host colony and between different compartments within a colony, such as the surface mucus layer (SML), the tissues, the skeleton and the endolithic algae (Sweet et al., 2011a; Bourne and Webster, 2013; Williams et al., 2015; Hernandez-Agreda et al., 2016). The microbes associated with the SML are a particularly interesting research topic as the SML is a highly dynamic environment and the microbes are thought to be representative of a transient community sourced mainly from the surrounding water column (Sweet et al., 2010, 2011b). Recently it has even been argued that corals could be “farming” their associated microbiota, maintaining a harvestable food source supplying nutrients to the host (Bourne and Webster, 2013). Regardless of the role of the SML in governing the coral microbiota, the above variation in the coral microbiome means great care is needed when designing microbiome studies in order to answer clear specific questions. For example, age of the coral colonies or colony size (where colony size is used as a proxy for age) should be taken into account when designing experiments to assess variation in the microbiomes of the same coral species with depth or location. The next step would be to explore which microbes cause such changes and variations. However, this is made difficult due to the extremely high microbial diversity usually found associated with these hosts. For example, studies have identified between 1000 and 6000 different bacterial operational taxonomic units (OTUs) or “phylotypes” associated with a wide range of coral species (Sunagawa et al., 2010; Ainsworth et al., 2015). In addition, numbers of these bacterial cells within coral mucus and tissue have been estimated to fall between the ranges of 1 × 106 ml−1 to 107 per cm2, respectively (Koren and Rosenberg, 2006; Garren and Azam, 2010; Bourne and Webster, 2013). Furthermore, the number of functional genes associated with these coral-associated bacteria may be in excess of 6700 (recorded in the coral Montastraea faveolata Kimes et al., 2013).
The development of high throughput sequencing methods has allowed researchers to explore the complexity of coral-microbial community interactions and dynamics. For example, a recent study by Neave et al. (2016) has explored a possible explanation for why species-species variations in the microbiome may occur over geographical spatial scales. In the study, the microbiomes of two closely related coral species, Stylophora pistillata and Pocillopora verrucosa, which can co-occur on the same reef habitats were assessed. Unsurprisingly, the study showed significant differences in their microbiomes. However, the study also highlighted further complexity in the spatial patterning, whereby the microbiome compositions associated with S. pistillata were much more strongly associated with geographical location than the microbiomes associated with P. verrucosa. Such variation was hypothesized to be driven by differences in the life histories of the respective corals. S. pistillata is a brooding coral, and corals exhibiting this life-history characteristic have been shown to pass microorganisms from parent to offspring (Sharp et al., 2012). Such a mechanism would increase spatial correlation in microbiome community composition. In contrast, P. verrucosa is a broadcast spawning coral, and such corals are known to gain their microorganisms horizontally from the water-column (Ceh et al., 2013). This would likely result in a decrease of the variability of the microbiome composition, particularly across smaller spatial scales.
Given the potential link between microbiome composition and such a key life-history component, it will be important for further studies to determine if such patterns hold more widely across species and geographical locations. Testing these potential links between the coral microbiome composition and life-history strategies could provide important insights into evolutionary drivers of coral-microbiome interactions. For example, corals in geographical locations where the planktonic-microbial variation is highly diverse would be exposed to a greater diversity of potential microbial symbionts to incorporate into its microbiome. This could result in increasing the plasticity of coral responses to climate change. However, the link between diversity and plasticity would also need to be tested in this regard. Importantly, if this did hold true, the above example suggests that such plasticity would be stronger in the broadcast spawning corals than in the brooding corals, potentially informing us of which coral species are more likely to be robust to the challenges of climate change.
A natural extension of the work examining patterns of the structures of the microbiome associated with corals has been the more recent focus on “core” microbiomes, i.e., sub-communities which remain common across space, time and/or individuals. However, definitions and methods of identifying such core microbiomes have varied considerably, potentially leading to inconsistencies when comparing findings across studies. That said, the search for a core microbiome represents the first step in trying to understand the dynamics of the interactions which may be occurring within the microbiome, as well as between the microbiome and the host corals. The hope is that this will lead to an understanding of the functional role of the microbiome, particularly the identification of key functional microbial species which play critical roles in a coral's resilience and response to climate change.
What is a Core Microbiome?
The core microbiome is broadly defined as the stable, consistent components across complex microbial assemblages from similar habitats (Table 1; Shade and Handelsman, 2012). However, such a definition is vague and this has resulted in a range of metrics used to determine which microbes belong to the core, with many studies further subdividing this “core” community. For example, some of the earlier studies discussing core microbiomes used sub-categories of the core microbiome under the justification that some “cores” may be shared only among subpopulations of hosts rather than all host individuals (Turnbaugh et al., 2009; Qin et al., 2010). In contrast, some authors believe that membership of the core microbiome should be reserved for those microbial species present throughout all samples of the same host species, regardless of specific habitat, geographical location and/or time period (Shade and Handelsman, 2012).

Table 1. Definitions of the various terms utilized in the current literature for microbiome and pathobiome studies.
A further complication results from the way sequence data is analyzed, and can substantially affect which microbes are identified as being core. For example, a typical approach currently utilized is to report either the relative abundance of microbial species found across localities from a similar habitat i.e., a coral reef in this instance and/or the presence or absence of the species. However, Shade and Handelsman (2012) illustrate that, depending on how one decides to utilize the sequence outputs, at least five different variations of a core microbiome could be determined from one set of data. We refer readers to Shade and Handelsman's (2012) review on this subject but, in brief, these variations can be summarized as:
(1) A core based on shared presence. Input operational taxonomic unit (OTU) table is presence/absence, and occurrences of shared presence are tallied across communities of interest.
(2) A core based on shared abundance. Input OTU table is relative abundance, and occurrences of dominance are highlighted as core members.
(3) A core based on shared composition. Only OTUs that are both shared and in similar proportions are counted toward a core (combination of 1 and 2 above).
(4) A core incorporating phylogenetic information. Related OTUs are counted as a single unit toward a core.
(5) A core based on interaction. Including only OTUs interacting (or presumed to be) with other members of its community (i.e., through the use of network analysis).
Options 1 and 2 are the more commonly utilized methods associated with the literature on this topic. However, option 2 runs the risk of ignoring the potential functional importance of rarer species in such communities. This could be particularly important in coral studies, where diversity of associated microbes is very high, and the majority of microbial species have relatively low abundance levels. Option 3 provides a more rigorous definition of the core microbiome, as it not only requires microbial species to be consistent in their presence, they must also be consistent in their levels of abundance. This option, therefore incorporates a requirement for consistency in community structure as part of identifying the core microbiome. Option 4 incorporates the idea of phylogenetic redundancy. Such redundancy occurs when multiple OTUs or phylotypes from the same lineage are present in a microbiome. This option should be specific to experiments which examine consistency across spatial and temporal scales as well as along environmental gradients. This is because it is dependent on having an appropriate experimental design and using profiling techniques which allow for large replication of samples from different areas, such as a number of geographical locations, or depths within the same location or locations with different levels of particular environmental stressors. It should be noted here that phylogenetic redundancy is not the same as “functional” redundancy, which occurs when multiple OTUs perform the same action within a microbiome (e.g., nitrogen fixation). Both types of redundancy are arguably important for defining and interpreting a core, as redundant microbes in a microbiome may be able to buffer against responses due to perturbations of the microbial community, i.e., a coral's ability to adapt and/or acclimatize to changes in environmental variables associated with climate change. In this respect, functional redundancy will offer greater utility, but it requires more involved work to characterize than phylogenetic redundancy, due to little being currently known about the true function of specific coral associated microbes. Once a greater understanding of the functional roles of the specific microbiota has been achieved, this could be incorporated into option 5, which explicitly tries to incorporate known (or presumed) microbial-microbial patterns of interactions as part of defining the core microbiome. That is, microbes must be interacting with other microbial associates if they are to be included in the core microbiome.
To assess which microbes are interacting with one another we can use inferential methods based on graph theory (e.g., Levy et al., 2015). Here, a network can be derived where OTUs are represented by vertices, and two vertices are connected by an edge if the OTUs which they represent are believed to interact with each other. These interactions can be either positive or negative in character. The inferential component in deriving these networks is based on examining similarity indices in the patterns of occurrence of pairs of OTUs across samples. Pearson and Spearman correlation coefficients are often used, with absolute values above a cut-off value indicating interaction between a pair of OTUs. Sparse multiple regression methods can also be used to detect more complex patterns of interaction involving more than two OTUs. Faust and Raes (2012) present a useful and informative overview of deriving microbial association networks and their potential uses in developing dynamic models. Such development of OTU interaction networks opens up the possibility of applying a vast array of network analysis tools which have been, and are continuing to be, developed (Newman, 2010). These metrics attempt to describe the overall structure of a network, and such metrics can help to establish the robustness of a network to perturbation as well as identifying likely key microbial species within the network structure. Such tools could be particularly important for identifying critical changes in the microbiome community structure associated with environmental changes or disturbance. In this review, we have used real data on coral microbiomes, which have been handled in the same way to illustrate some examples of visualizations of such derived networks and their use in coral biology (see Figure 1). Methods and sample descriptions have been described in brief within the legend. However, this is used as an example and is not intended to be a standalone piece of research.
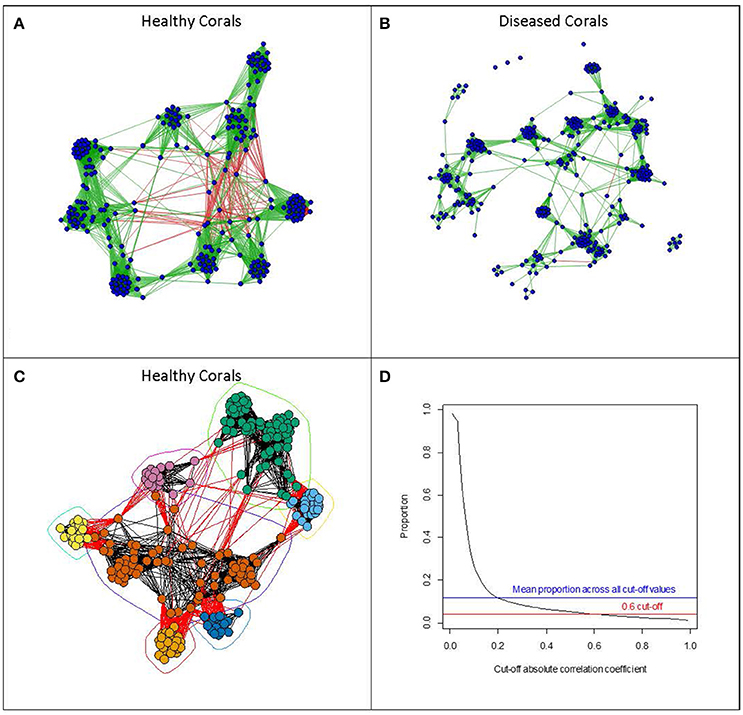
Figure 1. Examples of visualizations of microbial networks associated with Acorpora muricata collected at four different locations (Australia, Fiji, Maldives, and Solomon Islands). Networks with vertices representing individual OTUs, and edges representing absolute values of correlation coefficients of greater than 0.6, were derived from OTU abundances in (A) healthy corals and (B) diseased corals. Edges shown in green show positive correlations, whereas those shown in red represent negative correlations. Networks were derived using the qgraph function in the qgraph package (Epskamp et al., 2012). (C) Shows the network derived from the healthy coral arranged according to a community clustering algorithm (fastgreedy community from the igraph package; (Csardi and Nepusz, 2006)). The algorithm tries to identify clusters of OTUs which show similar patterns of abundance across samples. (D) Shows the relationship between the cut-off correlation value (x-axis) determining which of the correlations between OTU abundances should be included and the proportion of all the pair wise edges included in the network. All network analysis was done using the R statistical programming language (R Core Team, 2013). Methods: Acropora muricata samples were collected and preserved in 100% molecular grade ethanol (Sweet and Séré, 2016). All samples were extracted using the Qiagen DNeasy Extraction kit, amplified with primers 357 and 518 and sequenced on the 454 platform. All samples were pooled on the same run with barcodes for a complete list of methods including downstream processing of the sequencing run please refer to Sweet and Bythell (2015). For the full data set (OTU Table) used in this example please visit https://figshare.com/s/20918ac38006d83eac50
What is in a Coral Core Microbiome?
It should be noted that although corals have been shown to have various important relationships with a vast and variable microbiome (bacteria, viruses, fungi, etc.), the majority of the work examining core microbiome members has, to date, focussed on the bacterial cohort (Bayer et al., 2013; Ainsworth et al., 2015; Hernandez-Agreda et al., 2016). As a reflection of this, in this section the majority of research discussed will relate to results involving bacteria. However, future studies should start to examine patterns associated with all microbes (viruses, bacteria, fungi, archaea, and protozoa) when mapping changes in the microbiome of the coral holobiont.
Bayer et al. (2013) were amongst the first to attempt to map the core microbiome with the coral Eunicella cavolini. In their interpretation of a core, they required members to occur in all samples and found a total of only seven OTUs which were consistent across all samples assessed. These included; two Cyanobacteria, one Endozoicomonas, one Haliea, one Propioibacterium, one Arthrobacter, and one Elizabethkingia. Ainsworth et al. (2015) extended the resolution of the core microbiome concept by assessing if changes occurred in different compartments of the coral at three different depth ranges and five different sites. The authors assessed the microbiome in whole crushed coral samples, those only associated with the tissue, i.e., the endosymbionts and episymbionts (referred to as “core symbiotic” hereafter; Table 1), and those associated with the endosymbiont community, i.e., excluding the skeletal, mucus, surface, and loosely associated microbes (classed as the “core endosymbiont”; Table 1). Furthermore, the team also used a variation in cut off points to classify their core members, i.e., to be classed as a core member the bacterium must be present in 30, 50, or 75% of the samples. They justified the use of 30% as this was the minimum percentage of samples where a net change in core OTU abundance from the previous 2% was found to be zero (if a rate of change of zero, was found to occur at 30% representation). Using the 30% cut off, Ainsworth et al. (2015) reported that the core microbiome for Acropora granulosa consisted of 159 OTUs (out of 1508 sequenced) represented by 18 different families. Using this same 30% criterion, the team further split their definition of core members into those classed as “core endosymbiont” (which consisted of n = 41 OTUs) and “core symbiotic” (n = 39). Only 15 OTUs were shared between the three different types of tissue sampled (“core whole crushed,” “core symbiotic,” and “core endosymbiont”), highlighting the importance of carefully selecting appropriate tissue samples to address the hypothesis being tested. In the same study, two additional coral species were also studied, a Leptpseris sp. and Montipora capitata. These had larger core microbial communities containing 204 (out of 1242 sequenced) and 350 (out of 1433) OTUs, respectively. Importantly, the majority of the microbes which were described as core members for all three of these coral species were found in relatively low abundances, and significant differences in the microbiome as a whole were also observed, both between and within populations of the corals collected from the different sites. For example, in A. granulosa, 146 of the core microbiota had a relative abundance of less than 1%, and only two had a relative abundance greater than 5%. The authors did note the dependency of the numbers of OTUs described on the cut-off level utilized (i.e., 30%), and when the cut-off was increased to only include bacteria present in over 90% of the samples, only 0.09% of the total bacteria sequenced (i.e., only 1 OTU) was then able to be included in the core microbiome for A. granulosa.
The partitioning of sampling between the different coral tissue types by Ainsworth et al. (2015), provided valuable information about differences in the spatial distribution of the microbiome. For example, members of the microbiome including a Rhizobiales, a Caulobacterales, and a Burkholderiales were only detected within the “core symbiotic” microbiome, but not in the “core whole crushed” community. In contrast, a Rickettisales, a Rhodobacterales, and a Endozoicimonaceae were only amplified from the “core whole crushed” microbiome and not detected in the “core symbiotic” microbiome. This led Ainsworth and colleagues to conclude that, at least in the coral species studied, these microbes were likely restricted to the surface mucus layer and therefore likely not to be key players in the functioning of the coral's microbiome. The latter is particularly interesting as other studies have highlighted the importance of Endozoicimonaceae to coral health, and hypothesized that they may have been symbionts which have evolved with Scleractinian coral species (Bayer et al., 2013; Neave et al., 2016). Regarding Endozoicomonas and their relationships with corals, we are now starting to understand that different species of coral appear to show different relationships with these proposed endosymbionts. For example, Pocillopora verrucosa harbors the same Endozoicomonas over large spatial scales, whilst Stylophora pistillata appear to harbor geographically distinct genotypes of Endozoicomonas (Neave et al., 2016). In addition, a study by Meyer et al. (2014) highlighted that a reduction in abundance of these proposed endosymbionts correlated with an increase in abundances of opportunistic and potentially pathogenic bacteria associated with coral diseases. Endozoicomonas do indeed appear to be functionally important to corals, but it seems that the patterns are not universal (Ainsworth et al., 2015). Interestingly, Endozoicomonas sp. are known to have a transient part in their life cycle (Meyer et al., 2014) and the detection of this bacterium in the SML of the corals in Ainsworth et al.'s study is therefore not surprising. Furthermore, the SML has been shown to be a first line of defense for corals and therefore these microbes may still be playing a functional role of disease mitigation in the microbiome of this particular coral compartment.
In the core microbiome of the corals sampled in the study of Ainsworth et al. (2015), only two OTUs were found consistently in all three species of coral; an Actinomycelates (similar to a Propionibacterium sp.) and a Burkholderiales (similar to a Ralstonia sp.). The authors suggested that these OTUs, in particular, may be candidates for general coral symbionts, being present across many coral genera. Meta-analysis of a further 15 coral species over a wide geographical range lends further support to this theory as all coral species assessed were shown to harbor these two bacteria (Ainsworth et al., 2015). Finally, the same bacteria were localized within the tissues of the corals using taxa- and group-specific probes for fluorescent in situ hybridization, highlighting that these bacteria shared a close relationship with the coral's symbiotic algae.
In a similar study conducted by Hernandez-Agreda et al. (2016), the core microbiome of the coral Pachyseris speciose was assessed at two different geographical locations and at different depths. This study also recognized the potential sensitivity of the identification of the core microbiome to the cut-off value used and presented their findings based on OTUs found in 50, 80, and 90% of samples. At the 50% cut-off, 97 OTUs were included as core members (out of 173,690 sequenced), but this decreased to only 9 when the 90% cut-off was used. A search of other studies showed that out of the 97 OTUs included (with the 50% cut-off), 49 had been previously reported in other coral-microbe studies. However, for this review, we'll focus on the 9 OTUs described by Hernandez-Agreda et al. (2016) as core members found with the 90% cut-off as this represents the more stringent case. These OTUs included four members from three genera; Corynebacterium, Alteromonas, and Gluconacetobacter, one belonging to the phylum Bacteriodetes, one from the class Deltaproteobacteria, one from the class Alphaproteobacteria, and one each from the orders; Campylobacterales, and Rhodobacteraceae. This highlights another difficulty in identifying core microbiomes and making comparisons across different studies, i.e., the level of taxonomic classification reported or achieved due to the primer choice and/or platform utilized. For example, in the genus Corynebacterium there are currently 88 validly published species found across many different habitats including that of the human skin (Bernard, 2012). In contrast, there is only one species currently classified as belonging to the genus Alteromonas; that of A. macleodii. Although, sequences from this genera have been recorded in corals before (Ceh et al., 2013), A. macleodii appears to be extensive and is routinely detected in the water column across depths (López-Pérez et al., 2014). It should be noted that the repeated presence of A. macleodii in such studies could be simply a contaminant brought about during collection (in this instance) and could be associated more commonly with the surrounding water column rather than the coral itself. To remove such doubt, studies such as this, should routinely sample the water column and process the samples in the same way to account for any potential transient members from later downstream interpretation of the results. Regardless of potentially counting common microbial associates of the water column, the design of this study, taking into account different depths, did allow the authors to explore the potential of the presence of depth specific core microbiomes (Hernandez-Agreda et al., 2016). Indeed, the 9 core members identified (described above) were consistently found in 80% of the samples from the different depths studied. This was in addition to a further 14 OTUs which were found in 80% of the samples in the mesotrophic corals but not present in the shallower corals. The authors suggested that this could be an indication of a possible core community specific to deeper environments, and they referred to this as a “functional niche fraction” of core associated microbiota (Table 1).
In our own example, with the coral Acropora muricata as the host coral species (Figure 1), we identified a similar membership level composing the “core microbiome” as the three studies described above. Twelve OTUs were consistently detected in 90% of the samples across the four spatial locations (Australia, Fiji, Solomon Islands, and the Maldives) and included a Kocuria, a Propionibacterium, a Arcobacter, a Pseudomonas, three members of the Family Rhodobacteria (one of which was identified to the Genera Phaeobacter), one Flavobacteriaceae, two from the order Rickettsiales, and one from the order Stramenopiles.
As the focus on core microbes is a relatively new area for coral biology, we felt it worthwhile to highlight potential areas to which future work could be directed. To date, few studies have examined the potential effects of developmental stage or age of coral colonies on the composition of microbiomes as a whole, and none have attempted to assign core memberships to this important dimension of coral life history. In human biology, a further category of core microbiome has indeed been suggested, taking into account the age dimension (Saraswati and Sitaraman, 2015). The “temporal core” has been defined as those microbiota that are consistently found across developmental stages of the host (Table 1; Shade and Handelsman, 2012). Apprill et al. (2009) were amongst the first to show that there can be distinct microbial communities associated with the different life stages of corals by examining the microbiome of Pocillopora meandrina. Interestingly, they also highlighted that bacterial cells appeared to not be internally incorporated until the planulae were fully developed. Such evidence suggests that, unlike the Symbodinium which are vertically transmitted in the same coral species, bacteria are acquired via horizontal uptake. Furthermore, as these bacteria appear to be incorporated during late development of the planulae, it is possible that bacteria play a role in processes specific to this life stage such as benthic settlement (Mouchka et al., 2010). In a second example, the larvae of Porites astreoides appear to be dominated by two main phylotypes; Roseobacter, and Marinobacter, whilst Oceanospirillae dominate adults (Morrow et al., 2012; Sharp et al., 2012). Variation in the microbiome associated with age in settled colonies has also been assessed (Littman et al., 2009; Williams et al., 2015). In these latter two examples, corals which were younger (i.e., recruits) appeared to show the greatest variation in their microbiome at any given point in time and at any given location. Interestingly, although there were some bacterial OTUs which were shared between adults and juveniles, the vast majority of adult-associated bacteria were not found in juveniles in both studies (Littman et al., 2009; Williams et al., 2015). This suggests a succession process may well be occurring, whereby the microbiome of adult corals gradually replaces the diverse bacterial consortia of juveniles. Alternatively, the large variation in the juvenile microbiome may be present to enable corals greater plasticity in space and time. For example, a given microbial community may be beneficial under certain circumstances such as depth and/or in the face of climate change. The reduced microbial diversity in adult coral colonies could then be the result of survivorship of specific corals with a specific microbiome. Aging in corals has also been studied with regard to the separate compartments of corals, i.e., the SML. Glasl et al. (2016) suggested that one coral genera in particular (Porites) was an ideal model system to study such a process due to the well-defined aging process that proceeds periodic sloughing of the SML (Coffroth, 1991). In this study, Glasl and colleagues showed that the mucus-associated microbiome underwent significant changes throughout the mucus aging cycle. In brief, the SML was dominated by bacteria from Oxalobacteracea and Endozoicimonaceae, (in support of Ainsworth et al., 2015), whilst aged mucus had higher abundances of Verrucomicrobiaceae, Flammeovirgacea, Rhodobacteracea, and Vibrionaceae. After the detachment of the mucus sheet the bacterial community reverted back to being dominated by Oxalobacteracea and Endozoicimonaceae within 3–5 days. Such a natural fluctuation of mucus-associated bacteria taking place within a temporal scale of weeks has important implications for the design of studies assessing the community profiles of corals in both healthy and diseased states, as the microbiome may be dynamic over much shorter time scales than previously thought. Here the importance of the core microbiome may again shed further light on the role of certain members in aging and development of both the coral specifically, and certain compartments like the dynamic SML.
Finally, to end this section on the core microbiome, we felt it important to address an emerging topic area that we think will prove to be of great importance. That is, the role of bacterial predators within the microbiome. To date, few studies have focussed specifically on this aspect. Welsh and Vega Thurber (2016) is an important exception and is a study which also highlights a range of interesting directions for future research. Bacteria-bacteria predation is diverse in nature and can be epibiotic or periplasmic, and may be facultative or obligate in nature. Bacteria belonging to the genera Bdellovibrio and Halobacteriovorax are obligate periplasmic predators and importantly are thought to exert a top down control (or sideways) on microbial communities (Welsh and Vega Thurber, 2016). This would most likely play an influential role in the structuring of the microbiome as a whole. Controlled manipulation experiments with Halobacteriovorax for example, have shown this particular bacterial predator to be able to predate on a number of coral-microbial associates (Welsh and Vega Thurber, 2016). Importantly, when network analysis was applied to determine pairwise associations between the microbes, Halobacteriovorax were shown to co-occur with eight members of the coral microbiome, including Vibrionales, Cytophagales, and Alteromonadales, each of which have been shown to be opportunistic pathogens in previous studies (Sweet et al., 2011c). Thus, there is clearly substantial potential for bacterial predators to strongly influence the dynamics and structure of coral microbiomes, analogous to the roles played by top predators in terrestrial systems (Wallach et al., 2015). We therefore think that research in this area could offer a rich seam of findings for three main reasons. Firstly, there is already an established literature base in classical large scale ecology examining community structure and trophic levels, against which experimental work involving coral microbiomes could be compared. Secondly, theory suggests that predatory bacteria are likely to be highly influential in structuring the microbiome community, and therefore work in this area has the potential to give us a deep understanding of a key component of coral microbiome dynamics. And thirdly, very little work has been done in this area for coral microbiomes and therefore very little is known in this regard.
Introducing the Pathobiome
Recent studies of infectious agents have demonstrated that Koch's and Hill's fundamental postulates of “one microbe = one disease” has its limits (Vayssier-Taussat et al., 2014). Indeed, it is now established that many pathogens live and interact with other microorganisms in vast communities (Vayssier-Taussat et al., 2014), all generating and participating in complex interactions that may influence or drive the disease process (Chow et al., 2011; Vayssier-Taussat et al., 2014). Various different terms have been used to describe such processes, particularly in coral biology and these include; “opportunistic pathogens” associated with specific diseases, the definition of “polymicrobial diseases” and a microbial imbalance or maladaptation known as “dysbiosis.” However, the term “pathobiome” has to date not been utilized in coral studies, but has the potential to provide a powerful framework for developing an understanding of how the coral microbiome interacts with the disease process. However, as with the core microbiome, this concept is currently poorly defined in the literature. The pathobiome can be viewed as the collection or consortium of microbes which play a direct role in the causation of any given disease. However, the definition could be extended to include those microbes which have a direct role in mitigating disease in the coral (i.e., including both potential “opportunistic pathogens” and “health-associated” microbes which play some role in preventing disease). The definition could be extended even further to include microbes which indirectly impact on the abundance levels of pathogenic and disease mitigating microbes, that is microbes which affect the disease process but through indirect interactions with other microbial species. However, this latter definition, although recognizing the complex network of interactions between microbes, is so broad so as to be almost indistinguishable from that of the microbiome. Our recommendation would be to use the simplest definition of the pathobiome, i.e., a collection of microbial species which play a direct role in causing disease (Table 1). This is a parsimonious definition which allows for the recognition that microbes not included in the pathobiome may influence disease progression indirectly through microbe-microbe interactions, but avoids adding confusion by requiring a clear mechanistic link between a microbe and the causation of disease for the microbe to be included in the pathobiome.
Taking this definition of the pathobiome, understanding what the pathobiome is in any organism would therefore require four main steps: (1) the establishment of an accurate list of the microorganisms of which it is composed, (2) clear evidence of any effects this microbial community has on pathogenesis, (3) establishment of and/or an understanding of the impact of the microorganism community on persistence, transmission and evolution of pathogenic agents and (4) a gain of knowledge of the biotic and abiotic factors that may disrupt the microbiome (the healthy microbial community), allowing the actions of the pathobiome to lead to the onset of pathogensis (Vayssier-Taussat et al., 2014). This presents an initial problem. For such a concept to take a foothold a major shift, moving away from descriptive metagenomics and toward functional metagenomics, is required to establish the roles of specific microbes in coral health and disease, and to determine the extent to which variation in the microbiome and the pathobiome is driven by the environment. The transition from the view of one microbe = one disease to a broader pathobiome perspective involves revisiting the Koch's and Hill's postulates at the level of microbial communities and raises questions about the functioning and diversity of the microbiota present in a healthy coral individual, and of factors promoting the emergence of potentially disease-causing pathobiomes (Vayssier-Taussat et al., 2014).
To date there has been vigorous debate over the identity of pathogens in corals as well as the mechanisms by which diseases are caused. Some argue that there are well described pathogens such as Vibrio coralliilyticus, whilst others highlight the lack of evidence for a causal link with disease (reviewed in Sheridan et al., 2013). A particular concern of the latter group is related to the assessment of the histology of diseased corals with a lack of large numbers, if any, of the proposed pathogens found within the tissue of the diseased individuals (Work and Meteyer, 2014). Another controversial example involves a second member of the Vibrio genus, V. shiloi, which led Rosenberg et al. (2007) to propose the “hologenome theory of evolution.” This theory argues that corals can acquire beneficial (microbial) partners which can actively fight potential pathogens and/or that some pathogenic agents eventually lose their virulence. This theory was used to explain experimental results by Reshef et al. (2006) involving disease causing effects of V. shiloi. It is understandable why conclusions have been drawn about the importance of Vibrios in coral disease, as shifts in the relative dominance of Vibrios are often associated with diseased or thermally stressed corals (Vega Thurber et al., 2009). However, it has been argued that pathogenisis may be more related to the presence of virulence genes associated with certain Vibrios (and other potential pathogens) rather than the presence of specific individual Vibrio species themselves. That is, the causal agent of disease is at the gene level rather than at the microbial species level. If correct, the underlying mechanism leading to the selection of the virulence gene containing Vibrios (and other bacteria genera which contain the same or similar genes) remains unknown. However, it has been suggested that such a process could be related to virally mediated horizontal gene transfer of virulence factors observed in many different organisms (Vega Thurber et al., 2009). If true, this has important implications for the methods for establishing core members of the microbiome (described in the section “What is a core microbiome?” above, particularly with regard to option 4). Option 4 defines a core microbiome by utilizing phylogenetic information but, as phylogenetically related microbes can also contain diverse accessory genes critical for alternate functions such as niche expansion and opportunistic pathogenesis (Vega Thurber et al., 2009), this may not be a reliable method of identifying the core microbiome, from a functional point of view.
Although some diseases in some organisms are undoubtedly caused by a single pathogenic agent, this appears to not always be the case, and assessing larger data sets would allow researchers to map patterns and trends in the microbiome and/or pathobiome of healthy and diseased organisms. Indeed, one study attempted to do exactly this with regard to corals. Mouchka et al. (2010) highlighted three main bacterial groups which appeared to increase in abundance in the majority of diseased states which included Rhodobacter, Clostridia, and Cyanobacteria, whilst others such as Oceanospirillum were shown to decrease in abundance. Bleached corals in contrast had higher proportions of the r-selected opportunistic generas, Vibrio and Acidobacteria. However, such cross study comparisons are difficult for many reasons and it is this we address in our final section below.
Difficulties with Cross Study Comparisons of Microbiomes and Pathobiomes
In the sections above discussing microbiomes, we highlighted the difficulties in identifying the core microbiome and the sensitivity of the composition of the core microbiome to the particular selection criteria applied. These difficulties hold equally for the pathobiome. Thus, valid comparison of findings between studies is made difficult by variations in methodologies between these studies. However, it should be noted that complicating issues for such comparisons go back further downstream in the methodological processes. In fact, they go right back to the storage and extraction of the samples in the studies. The search for the most suitable combination of fixatives, extraction method, primer pairs, and sequencing platforms is ongoing. To make matters more difficult, those few studies which have attempted to assess the influence of differences in these methodological aspects have often come to contrasting conclusions (McOrist et al., 2002; Salonen et al., 2010; Fouhy et al., 2016). For example, a recent study used a mock community of bacterial DNA to assess commonly utilized extraction procedures, primer pairs, and platforms (Fouhy et al., 2016). The authors concluded that there was little variation in the outcome from the different extraction methods used. However, this similarity in results may be due to the restricted range of methods used, as other studies have found significant differences in the outcome when using a greater variety of extraction methods (McOrist et al., 2002; Salonen et al., 2010). Furthermore, primer choice can vary between studies and the best option again remains unknown. Fouhy et al. (2016) highlighted that the V4–V5 region run on the Ion PGM platform gave the least biased result, which supports a similar study by Tremblay et al. (2015). However, both studies went on to conclude that, although this is currently the most suitable region, it is not optimal and that the use of a shotgun metagenomics approach (not yet feasible for large scale projects due to cost) would result in a marked improvement. Interestingly, although the V4–V5 sequence region gave the most consistent results across platforms, the same primer set gave skewed abundances as well as detecting false hits (i.e., a certain percentage of reads achieved were not as expected based on the known composition of the mock DNA) (Fouhy et al., 2016). These false hits ranged from 6 to 80% of the total OTUs detected depending on the primers and the sequencing platforms used. Therefore, both these studies concluded that protocol consistency affects a given study outcome to a greater degree than either primer or sequencing platform choice. However, as mentioned above, although standardization of methodologies would enable reliable, within study comparisons, it is less likely to be achieved for comparisons between studies, as each group of researchers often have their own preferred combination of protocols. Indeed many argue the need for the selection and use of just one DNA extraction method, primer choice, and platform. That said, identifying the optimal combination of choices remains an open debate. Furthermore, in addition to the above considerations, sequencing depth has also been shown to be capable of changing the interpretation of the proportion of core OTUs. For example, Qin et al. (2010) showed that 3x sequencing depth can increase the core microbiome of a study by 25% compared to the more standard 1x sequencing depth.
This lack of consistency in methodologies applied in different studies is a significant issue as we cannot be sure that differences between studies in microbiome composition and dynamics are genuine, or due to methodological reasons. As outlined in previous sections in this article, even when a study has OTU abundance data there are choices and complexities involved in the analysis of these data which will potentially affect the legitimacy of making comparisons between studies. The coral holobiont is complex and dynamic and the microbial communities associated with this holobiont is a key component to be understood if we are going to move the field of coral disease forward.
If we refer back to our example utilized throughout this review, we can assess potential core members of the pathobiome in a similar way to that used to describe the microbiome in healthy corals. For example, we can identify 10 OTUs which were consistently detected in 90% of the samples across four different geographical locations. These included a Saprospira, an Arcobacter, an Oleibacter, and a Vibrio, a member of the Family Flavobacteriaceae, three Rhodobacteraceae (one of which was identified as from the genera Phaeobacter), a Stramenopile and one unidentified bacterium from the Class Gammaproteobacteria. The three Rhodobacteraceae were also detected in the microbiome of healthy corals and are therefore unlikely to be associated with the diseased state, with the same holding true for the Arcobacter, the Flavobacteriaceae, and the Stramenopile. This leaves potential opportunistic pathogens, such as in this instance the Saprospira, the Oleibacter, and the Vibrio, along with the unidentified Gammaproteobacteria. Also the loss of the Kocuria, the Propionibacterium, the Pseudomonas, and the two Rickettsiales (all consistently present in the microbiome of healthy corals) is also worthy of note and may be indicative of a reduction in these microbes which, together with the increase/presence of those associated with the disease state, could compose an interacting pathobiome community. However, it is not our intention in this review to make such conclusions, we use this only as an example to illustrate the complexity of the structure and dynamics of the microbiome and the pathobiome related to coral health and disease. Further work should be conducted in this instance to confirm or deny this working hypothesis. Interestingly, the presence of the Vibrio in this example lends some credibility to the role that this genus plays in this disease across geographical regions. That is alongside the findings from studies which have utilized antibiotics to treat these coral diseases which also point to a bacterial cause (Sweet et al., 2014; Sweet and Bythell, 2015).
Conclusions
In this review, we explored the use of the terms “microbiome” and “pathobiome” in the field of coral biology. Although coral-microbial communities have been studied for decades with regard to health and disease, the onset of next generation sequencing saw a marked increase in the amount of publications associated with this field. With this advance in technology comes the reality that the microbiome of corals is very complex and we are now presented with new challenges of how to reliably analyse and interpret these data. Concentrating on the core microbiome has been suggested as a way of removing focus from more transient members of the coral holobiont and focussing on the key members. However, care needs to be taken over what the researcher defines as a core microbe (Table 1). The more recent concept of the pathobiome is, as far as we are aware, new to the field of coral biology and presents very interesting opportunities to increase our understanding of coral health and disease alongside the concept of the microbiome. However, this in turn requires a shift in thinking from the “one microbe = one disease” concept to the importance of a collection or consortium of microbiota which play a direct role in the causation of any given disease (Table 1). The use of macro-ecological framework tools, adapted to a micro scale, appears to be a useful way of presenting next generation data, and allowing us to explore how microbes interact with other members of the microbiome. Utilizing these frameworks with diseased corals can allow us to see if the changes in the core microbial community paves the way for the onset of disease and the increase in opportunistic pathogens, i.e., such analysis will allow us to explore the role of the pathobiome in coral diseases from now on. In this review we also touched on the importance of virulence genes in comparison to specific microbial phylotypes in coral diseases. As current metagenomics DNA-based analysis cannot differentiate between expressed and non-expressed genes, these approaches currently fail to reflect the actual activity or dynamics of microbial communities. The development of broader “metaomic” approaches such as in situ metatranscriptomics and metaproteomics will aid this field in the future.
Author Contributions
MS and MB wrote the paper and conducted the analysis associated with the examples used throughout.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ainsworth, T., Krause, L., Bridge, T., Torda, G., Raina, J.-B., Zakrzewski, M., et al. (2015). The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9, 2261–2274. doi: 10.1038/ismej.2015.39
Apprill, A., Marlow, H. Q., Martindale, M. Q., and Rappé, M. S. (2009). The onset of microbial associations in the coral Pocillopora meandrina. ISME J. 3, 685–699. doi: 10.1038/ismej.2009.3
Bayer, T., Arif, C., Ferrier-Pages, C., Zoccola, D., Aranda, M., and Voolstra, C. R. (2013). Bacteria of the genus Endozoicomonas dominate the microbiome of the Mediterranean gorgonian coral Eunicella cavolini. Mar. Ecol. Prog. Ser. 479, 75–84. doi: 10.3354/meps10197
Bernard, K. (2012). The genus Corynebacterium and other medically relevant coryneform-like bacteria. J. Clin. Microbiol. 50, 3152–3158. doi: 10.1128/JCM.00796-12
Bourne, D. G., Dennis, P. G., Uthicke, S., Soo, R. M., Tyson, G. W., and Webster, N. (2013). Coral reef invertebrate microbiomes correlate with the presence of photosymbionts. ISME J. 7, 1452–1458. doi: 10.1038/ismej.2012.172
Bourne, D., and Webster, N. S. (2013). “Coral Reef Bacterial Communities,” in Prokaryotes, Chapter 10, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin; Heidelberg: Springer Verlag), 163–187.
Ceh, J., Kilburn, M. R., Cliff, J. B., Raina, J. B., Van Keulen, M., and Bourne, D. G. (2013). Nutrient cycling in early coral life stages: Pocillopora damicornis larvae provide their algal symbiont (Symbiodinium) with nitrogen acquired from bacterial associates. Ecol. Evol. 3, 2393–2400. doi: 10.1002/ece3.642
Chow, J., Tang, H., and Mazmanian, S. K. (2011). Pathobionts of the gastrointestinal microbiota and inflammatory disease. Curr. Opin. Immunol. 23, 473–480. doi: 10.1016/j.coi.2011.07.010
Coffroth, M. A. (1991). Cyclical mucous sheet formation on poritid corals in the San Blas Islands, Panama. Mar. Biol. 109, 35–40. doi: 10.1007/BF01320229
Csardi, G., and Nepusz, T. (2006). The Igraph Software Package for Complex Network Research. InterJournal Complex Systems 1695. Available online at: http://igraph.org
Epskamp, S., Cramer, A. O. J., Waldorp, L. J., Schmittmann, V. D., and Borsboom, D. (2012). qgraph: network visualizations of relationships in psychometric data. J. Stat. Softw. 48, 1–18. doi: 10.18637/jss.v048.i04
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550. doi: 10.1038/nrmicro2832
Fouhy, F., Clooney, A. G., Stanton, C., Claesson, M. J., and Cotter, P. D. (2016). 16S rRNA gene sequencing of mock microbial populations- impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol. 16:123. doi: 10.1186/s12866-016-0738-z
Garren, M., and Azam, F. (2010). New method for counting bacteria associated with coral mucus. Appl. Environ. Microbiol. 76, 6128–6133. doi: 10.1128/AEM.01100-10
Glasl, B., Herndl, G. J., and Frade, P. R. (2016). The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 10, 2280–2292. doi: 10.1038/ismej.2016.9
Hernandez-Agreda, A., Leggat, W., Bongaerts, P., and Ainsworth, T. D. (2016). The microbial signature provides insight into the mechanistic basis of coral success across reef habitats. MBio 7, e00560–e00516. doi: 10.1128/mBio.00560-16
Huttenhower, C. and Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Kimes, N. E., Johnson, W. R., Torralba, M., Nelson, K. E., Weil, E., and Morris, P. J. (2013). The Montastraea faveolata microbiome: ecological and temporal influences on a Caribbean reef-building coral in decline. Environ. Microbiol. 15, 2082–2094. doi: 10.1111/1462-2920.12130
Koren, O., and Rosenberg, E. (2006). Bacteria associated with mucus and tissues of the coral Oculina patagonica in summer and winter. Appl. Environ. Microbiol. 72, 5254–5259. doi: 10.1128/AEM.00554-06
Krezalek, M. A., DeFazio, J., Zaborina, O., Zaborin, A., and Alverdy, J. C. (2015). The shift of an intestinal “Microbiome” to a “Pathobiome” governs the course and outcome of sepsis following surgical injury. Shock 45, 1. doi: 10.1097/SHK.0000000000000534
Levy, R., Carr, R., Kreimer, A., Freilich, S., and Borenstein, E. (2015). NetCooperate: a network-based tool for inferring host-microbe and microbe-microbe cooperation. BMC Bioinformatics 16:164. doi: 10.1186/s12859-015-0588-y
Littman, R. A., Willis, B. L., and Bourne, D. G. (2009). Bacterial communities of juvenile corals infected with different Symbiodinium (dinoflagellate) clades. Mar. Ecol. Prog. Ser. 389, 45–59. doi: 10.3354/meps08180
Lloyd-Price, J., Abu-Ali, G., and Huttenhower, C. (2016). The healthy human microbiome. Genome Med. 8, 51. doi: 10.1186/s13073-016-0307-y
López-Pérez, M., Gonzaga, A., Ivanova, E. P., and Rodriguez-Valera, F. (2014). Genomes of Alteromonas australica, a world apart. BMC Genomics 15:483. doi: 10.1186/1471-2164-15-483
McOrist, A. L., Jackson, M., and Bird, A. R. (2002). A comparison of five methods for extraction of bacterial DNA from human faecal samples. J. Microbiol. Methods 50, 131–139. doi: 10.1016/S0167-7012(02)00018-0
Meyer, J. L., Paul, V. J., and Teplitski, M. (2014). Community shifts in the surface microbiomes of the coral Porites astreoides with unusual lesions. PLoS ONE 9:e100316. doi: 10.1371/journal.pone.0100316
Morrow, K. M., Moss, A. G., Chadwick, N. E., and Liles, M. R. (2012). Bacterial associates of two caribbean coral species reveal species-specific distribution and geographic variability. Appl. Environ. Microbiol. 78, 6438–6449. doi: 10.1128/AEM.01162-12
Mouchka, M. E., Hewson, I., and Harvell, C. D. (2010). Coral-associated bacterial assemblages: current knowledge and the potential for climate-driven impacts. Integr. Comp. Biol. 50, 662–674. doi: 10.1093/icb/icq061
Neave, M. J., Rachmawati, R., Xun, L., Michell, C. T., Bourne, D. G., Apprill, A., et al. (2016). Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J. 11, 186–200. doi: 10.1038/ismej.2016.95
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
R Core Team (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://www.R-project.org/
Reshef, L., Koren, O., Loya, Y., Zilber-Rosenberg, I., and Rosenberg, E. (2006). The coral probiotic hypothesis. Environ. Microbiol. 8, 2068–2073. doi: 10.1111/j.1462-2920.2006.01148.x
Rosenberg, E., Koren, O., Reshef, L., Efrony, R., and Zilber-Rosenberg, I. (2007). The role of microorganisms in coral health, disease and evolution. Nat. Rev. Microbiol. 5, 355–362. doi: 10.1038/nrmicro1635
Ryan, E. T. (2013). The intestinal pathobiome: its reality and consequences among infants and young children in resource-limited settings. J. Infect. Dis. 208, 1732–1733. doi: 10.1093/infdis/jit509
Salonen, A., Nikkilä, J., Jalanka-Tuovinen, J., Immonen, O., Rajilić-Stojanović, M., Kekkonen, R. A., et al. (2010). Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 81, 127–134. doi: 10.1016/j.mimet.2010.02.007
Saraswati, S., and Sitaraman, R. (2015). Aging and the human gut microbiota-from correlation to causality. Front. Microbiol. 5:764. doi: 10.3389/fmicb.2014.00764
Shade, A., and Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12. doi: 10.1111/j.1462-2920.2011.02585.x
Sharp, K. H., Distel, D., and Paul, V. J. (2012). Diversity and dynamics of bacterial communities in early life stages of the Caribbean coral Porites astreoides. ISME J. 6, 790–801. doi: 10.1038/ismej.2011.144
Sheridan, C., Kramarsky-Winter, E., Sweet, M., Kushmaro, A., and Leal, M. C. (2013). Diseases in coral aquaculture: causes, implications and preventions. Aquaculture 396–399, 124–135. doi: 10.1016/j.aquaculture.2013.02.037
Sunagawa, S., Woodley, C. M., and Medina, M. (2010). Threatened corals provide underexplored microbial habitats. PLoS ONE 5:e9554. doi: 10.1371/journal.pone.0009554
Sweet, M. J., and Bythell, J. (2015). White syndrome in Acropora muricata: nonspecific bacterial infection and ciliate histophagy. Mol. Ecol. 24, 1150–1159. doi: 10.1111/mec.13097
Sweet, M. J., Croquer, A., and Bythell, J. C. (2010). Temporal and spatial patterns in waterborne bacterial communities of an island reef system. Aquat. Microb. Ecol. 61, 1–11. doi: 10.3354/ame01433
Sweet, M. J., Croquer, A., and Bythell, J. C. (2011a). Bacterial assemblages differ between compartments within the coral holobiont. Coral Reefs 30, 39–52. doi: 10.1007/s00338-010-0695-1
Sweet, M. J., Croquer, A., and Bythell, J. C. (2011b). Development of bacterial biofilms on artificial corals in comparison to surface-associated microbes of hard corals. PLoS ONE 6:e21195. doi: 10.1371/journal.pone.0021195
Sweet, M. J., Croquer, A., and Bythell, J. C. (2014). Experimental antibiotic treatment identifies potential pathogens of white band disease in the endangered Caribbean coral Acropora cervicornis. Proc. R. Soc. B. 281. doi: 10.1098/rspb.2014.0094
Sweet, M. J., Jones, R., and Bythell, J. C. (2011c). Coral diseases in aquaria and in nature. J. Mar. Biol. Assoc. U.K 92, 791–801.
Sweet, M. J., and Séré, M. G. (2016). Ciliate communities consistently associated with coral diseases. J. Sea Res. 113, 119–131. doi: 10.1016/j.seares.2015.06.008
Sweet, M. J., Smith, D., Bythell, J. C., and Craggs, J. (2013). Changes in microbial diversity associated with two coral species recovering from a stressed state in a public aquarium system. J. Zoo Aquarium Res. 1, 52–60. doi: 10.19227/JZAR.V1I2.32
Tremblay, J., Singh, K., Fern, A., Kirton, E. S., He, S., Woyke, T., et al. (2015). Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 6:771. doi: 10.3389/fmicb.2015.00771
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi: 10.1038/nature07540
Vayssier-Taussat, M., Albina, E., Citti, C., Cosson, J.-F., Jacques, M.-A., Lebrun, M.-H., et al. (2014). Shifting the paradigm from pathogens to pathobiome: new concepts in the light of meta-omics. Front. Cell. Infect. Microbiol. 4:29. doi: 10.3389/fcimb.2014.00029
Vega Thurber, R., Willner-Hall, D., Rodriguez-Mueller, B., Desnues, C., Edwards, R. A., Angly, F., et al. (2009). Metagenomic analysis of stressed coral holobionts. Environ. Microbiol. 11, 2148–2163. doi: 10.1111/j.1462-2920.2009.01935.x
Wallach, A. D., Ripple, W. J., and Carroll, S. P. (2015). Novel trophic cascades: apex predators enable coexistence. Trends Ecol. Evol. 30, 146–153. doi: 10.1016/j.tree.2015.01.003
Welsh, R. M., and Vega Thurber, R. L. (2016). Bacterial predators in host microbiomes. Microbe 11, 61–67. doi: 10.1128/microbe.11.61.1
Whipps, J. M., Lewis, K., and Cooke, R. C. (1988). “Mycoparasitism and plant disease control,” in Fungi in Biological Control Systems, ed N. M. Burge (Manchester University Press), 161–187.
Williams, A. D., Brown, B. E., Putchim, L., and Sweet, M. J. (2015). Age-related shifts in bacterial diversity in a reef coral. PLoS ONE 10:e0144902. doi: 10.1371/journal.pone.0144902
Keywords: gene, Vibrio, macro-ecology, network analysis, holobiont, microbiota, interaction, bacteria
Citation: Sweet MJ and Bulling MT (2017) On the Importance of the Microbiome and Pathobiome in Coral Health and Disease. Front. Mar. Sci. 4:9. doi: 10.3389/fmars.2017.00009
Received: 01 October 2016; Accepted: 09 January 2017;
Published: 20 January 2017.
Edited by:
Thomas K. Frazer, University of Florida, USAReviewed by:
Daniel Wangpraseurt, University of Cambridge, UKSuhelen Egan, University of New South Wales (UNSW), Australia
Copyright © 2017 Sweet and Bulling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael J. Sweet, m.sweet@derby.ac.uk