Distinct Bacterial Microbiomes Associate with the Deep-Sea Coral Eguchipsammia fistula from the Red Sea and from Aquaria Settings
 Till Röthig
Till Röthig Anna Roik
Anna Roik Lauren K. Yum
Lauren K. Yum  Christian R. Voolstra
Christian R. Voolstra- Division of Biological and Environmental Science and Engineering, Red Sea Research Center, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
Microbial communities associated with deep-sea corals are beginning to be studied in earnest and the contribution of the microbiome to host organismal function remains to be investigated. In this regard, the ability of the microbiome to adjust to prevailing environmental conditions might provide clues to its functional importance. In this study, we characterized bacterial community composition associated with the deep-sea coral Eguchipsammia fistula under natural (in situ) and aquaria (ex situ) settings using 16S rRNA gene amplicon sequencing. We compared freshly collected Red Sea coral specimens with those reared for >1 year at conditions that partially differed from the natural environment, in particular regarding increased oxygen and food availability under ex situ conditions. We found substantial differences between the microbiomes associated with corals under both environmental settings. The core microbiome comprised only six bacterial taxa consistently present in all corals, whereas the majority of bacteria were exclusively associated either with freshly collected corals or corals under long-term reared aquaria settings. Putative functional profiling of microbial communities showed that corals in their natural habitat were enriched for processes indicative of a carbon- and nitrogen-limited environment, which might be reflective of differences in diet under in situ and ex situ conditions. The ability of E. fistula to harbor distinct microbiomes under different environmental settings might contribute to the flexibility and phenotypic plasticity of this cosmopolitan coral. Future efforts should further assess the role of these different bacteria in holobiont function, in particular since E. fistula is naturally present in markedly different environments.
Introduction
Corals are metaorganisms, so-called coral holobionts, consisting of the cnidarian animal host and a suite of microorganisms, most notably endosymbiotic algae of the genus Symbiodinium that provide nutrition via photosynthates (Muscatine and Porter, 1977) and a diverse array of bacteria (Rohwer et al., 2002). The acclimation capacity of a coral holobiont to environmental changes is determined by the coral animal (Todd, 2008), by Symbiodinium (in zooxanthellate corals; Brown, 1997), and likely by other associated microbes. In particular, adjustment of associated bacteria to prevailing environmental conditions has been suggested to support acclimatization of the coral holobiont (Reshef et al., 2006). This notion is supported by recent studies that show, at least in part, flexible bacterial microbiomes of the coral holobiont to environmental change (Jessen et al., 2013; Roder et al., 2015; Hernandez-Agreda et al., 2016; Röthig et al., 2016; Ziegler et al., 2016, 2017). However, our knowledge on the role of the bacterial microbiome to coral holobiont function is still limited (Bourne and Webster, 2013; Bourne et al., 2016), although bacteria are shown to contribute to coral health (Rosenberg et al., 2007; Krediet et al., 2013) and are involved in nutrient cycling (Rädecker et al., 2015; Bourne et al., 2016).
Deep-sea corals, similar to their shallow coral counterparts, provide structural habitats that are considered biodiversity hotspots (Roberts et al., 2006). However, they are generally restricted to light-limited and highly productive waters at low temperatures (<14°C) and are therefore commonly used synonymous with cold-water corals (Roberts et al., 2006; Naumann et al., 2014). The major differences between deep-sea corals and their shallow-water counterparts are consequently their temperature preference (shallow coral <30°C vs. deep coral <15°C in most places) and that deep-sea corals are azooxanthellate, which might be partially related to the temperature preference. Lacking Symbiodinium, the role of bacteria may be more substantial compared to shallow corals. For example, deep-sea coral associated bacteria are likely involved in fixing and recycling of nitrogen and carbon (Neulinger et al., 2008; Middelburg et al., 2015). However, mainly due to their limited accessibility, the knowledge on deep-sea corals is still scarce, especially in regard to their associated microbial communities. Few studies characterized deep-sea coral associated bacterial communities, focusing mainly on octocorals (Penn et al., 2006; Gray et al., 2011; Kellogg et al., 2016; Lawler et al., 2016) and two reef-building scleractinian corals, namely Lophelia pertusa and Madrepora oculata (Yakimov et al., 2006; Neulinger et al., 2008, 2009; Hansson et al., 2009; Kellogg et al., 2009, 2017; Schöttner et al., 2009, 2012; Galkiewicz et al., 2011; Emblem et al., 2012; van Bleijswijk et al., 2015; Meistertzheim et al., 2016). Findings include species-specific microbiomes, but also considerable spatial and temporal variations. Only very recently, the microbiomes of three further scleractinian corals (i.e., Eguchipsammia fistula, Dendrophyllia sp., and Rhizotrochus typus) from the Red Sea have been characterized (Röthig et al., 2017). The deep Red Sea with low oxygen (1–2 mg O2 L−1) and limited nutrient availability due to warm temperatures (>20°C) features markedly different conditions than otherwise common for deep-sea coral habitats (Roder et al., 2013). In line with other studies, Röthig et al. (2017) showed that these coral microbiomes were species-specific and suggested functional adaptation to their environment. These adaptations include in particular the presence of anaerobe bacterial taxa and potential hydrocarbon degraders. Interestingly, at least two of the three species investigated (i.e., E. fistula and R. typus) are not endemic to the Red Sea (van der Land, 2008). In line with its global presence (van der Land, 2008) and as a potential explanation to its wide distribution, E. fistula from the Red Sea shows a remarkable physiological plasticity displaying substantial tissue (re)growth and polyp budding during long-term rearing (>1 year) under conditions that only in part reflect its natural Red Sea habitat (Roik et al., 2015). More specifically, in comparison to their highly oligotrophic and low oxygen (i.e., 1–2 mg L−1) natural habitat in the Red Sea (Quadfasel, 2001; Roder et al., 2013; Qurban et al., 2014), aquaria-reared colonies of E. fistula were provided with a continuous but uniform diet under high oxygen conditions (i.e., >8 mg L−1). Besides assessment of phenotypic differenes, Roik et al. (2015), however, did not assess whether long-term aquaria rearing resulted in differences in bacteria associated with E. fistula that could either contribute to the acclimatization or be a result of it.
In this study, we set out to characterize bacterial community structure associated with the Red Sea deep-sea coral E. fistula after successful long-term (>1 year) rearing under aquaria settings and compared it to the bacteria associated with E. fistula in their native environment. This allowed us to assess coral microbiome differences under different environments and the putative capacity to adjust to markedly different environmental settings. In turn, this may provide insights to the functional importance of the bacterial microbiome to the coral host.
Materials and Methods
Coral Collection and Rearing
All E. fistula specimens used in this study were sampled by ROV from the central Red Sea in May 2013 on the R/V Aegaeo (KRSE2013L6). The Saudi Coastguard Authority under the auspices of KAUST issued sailing permits that include sample collection. In total, 6 coral samples from E. fistula were used for the bacterial microbiome analysis, three from their natural environment and three from long-term rearing in aquaria facilities. Each specimen consisted of one coral colony (~5–10 cm) of similar biomass, harboring several polyps and the coenosarc.
As described in Röthig et al. (2017), coral samples used to assess the native microbiome (in situ) were collected from between 314 and 320 m depth (N22°17.837, E38°53.811) with a custom-made scoop, transferred into a specifically designed two-compartment container, and preserved in RNAlater at depth. About 90 min later upon retrieval, samples were rinsed with sterile-filtered seawater, crushed on liquid nitrogen, and stored at −80°C. To assess the in situ microbiome 3 of the 4 E. fistula samples described in Röthig et al. (2017) were used in this study. To ensure an evenly distributed sample set and a high sequencing depth, one in situ sample from Röthig et al. (2017) presenting the lowest coverage was disregarded. Further, a water sample from the corals' direct vicinity was taken using Niskin bottles. One liter of the water sample was filtered over a 0.22 μm Durapore filter (Millipore, Billerica, USA), and filters were stored at −80°C until DNA extraction.
Corals for long-term rearing were collected in close proximity on a separate ROV dive during the same expedition (KRSE2013L6). Coral specimens were retrieved from the same habitat using the scoop and transferred into a plastic basket (Roik et al., 2015). The live E. fistula specimens were transferred into an aquaria system and transported to aquaria facilities at the King Abdullah University of Science and Technology (KAUST). Coral polyps were then attached onto reef cement sockets (Reef Construct, Aqua Medic) and transferred into an open flow batch system equipped with chiller, skimmer, trickling filter, and current pumps. Temperature was set to 21.3 ± 0.3°C and water was exchanged regularly. While water temperature resembled the in situ environment, oxygen levels were markedly higher (7.7–8.7 mg L−1 ex situ vs. 1–2 mg L−1 in situ) and corals were fed with Mysis and Artemia twice a week. The corals were reared for >1 year with prominent tissue and polyp growth (Roik et al., 2015). To assess bacterial communities associated with long-term reared (ex situ) E. fistula, three coral specimens were selected for DNA extraction. Feeding was ceased for 5 days, corals were rinsed with filtered seawater, wrapped in aluminum foil, and flash frozen in liquid nitrogen and transferred to −80°C. A one liter water sample was taken from the aquaria system and processed as described above.
DNA Isolation and Sequencing
DNA from flash frozen ex situ coral specimens was isolated using the AllPrep DNA/RNA Mini kit (Qiagen, Hilden, Germany). To do this, frozen specimens were unwrapped on ice, transferred to sterile zip lock bags, and dosed in 5 mL Qiagen RLT buffer. Buffer and coral tissue was carefully blasted off using tap air pressure and barrier pipette tips. The tissue-buffer mixture was then transferred into 15 mL Falcon tubes and vortexed. A 500 μL aliquot was used for DNA extraction following the manual (AllPrep Mini kit). DNA from crushed in situ corals was isolated according to the same manual. Water filters were cut in strips, Qiagen RLT buffer was added (1,200 μL to the deep-sea and 400 μL to the aquaria water samples), the samples were incubated for 20 min on a wheel, and further extraction followed the Qiagen AllPrep manual.
DNA libraries were generated according to the Illumina 16S metagenomics library preparation manual (Illumina, San Diego, CA, USA). The primers 784F and 1061R (Andersson et al., 2008) with Illumina adapter overhangs (underlined below) were used to amplify the variable regions 5 and 6 of the 16S rRNA gene: forward [5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGAGGATTAGATACCCTGGTA-3′] and reverse (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCRRCACGAGCTGACGAC-3′). These primers are shown to amplify well with coral DNA (Bayer et al., 2013b). All PCRs were performed in triplicates using ~1–5 ng DNA from water samples and ~50–100 ng DNA from coral samples, Qiagen Multiplex PCR master mix, and 0.25 μM of each primer. The total volume was adjusted to 20 μL with RNAse-free water. The amplification PCR was one cycle at 95°C for 15 min, 25 cycles each at 95°C for 30 s, 55°C for 90 s, and 72°C for 30 s; a final extension step at 72°C for 10 min. Successful amplification was visualized on the Bioanalyzer (Agilent Technologies, Santa Clara, USA) for the in situ samples, and 10 μL from each of the ex situ coral samples was used for a visual check via gel electrophoresis. The triplicate PCRs were pooled and cleaned with the Agencourt AMPure XP magnetic bead system (Beckman Coulter, Brea, CA, USA). Following the Illumina metagenomics sequencing library preparation protocol, PCR products underwent indexing PCR amplification. A 2% ultrapure agarose gel (Ultrapure Agarose, Life Technologies) was used for final size selection with the Zymoclean DNA large fragment recovery kit (Zymo Research, Irvine, USA) with two elutions at 10 μl to purify the PCR products from the gel. The libraries were sequenced with 25% phiX control on the Illumina MiSeq, 2 × 300 bp paired-end version 3 chemistry according to the manufacturer's specifications at the KAUST Bioscience Core Lab. Of note, PCR-based bacterial metabarcoding methods are amenable to potential bias by choice of primer selection, differences in 16S rRNA gene copy number across bacterial taxa, and length of the amplicon.
Bacterial Community Analysis
We used mothur (v.1.36.1; Schloss et al., 2009) for amplicon analysis. Sequence reads were split according to barcodes, assembled to contigs, and quality trimmed. Unique sequences were identified and counted, and the number of total sequences for each sample was determined using the “count.seqs” command. Singleton sequences (i.e., sequences that occurred only once over all samples) were removed. The remaining sequences were aligned to the SILVA reference set (release 119; Pruesse et al., 2007) and sequences that did not align were disregarded. A pre-cluster step with 3 bp difference was performed (Huse et al., 2010), and chimeras were detected using UCHIME as implemented in mothur (Edgar et al., 2011). Chimeras were removed and remaining sequences were classified with the Greengenes database (release 13_5) using a bootstrap of 60 (McDonald et al., 2012). Next, chloroplasts, mitochondria, archaea, eukaryotes, and unknown sequences were removed, and the bacterial composition of samples was compared on the taxonomic level of bacterial families using R (R Core Team, 2014). For further analyses, all samples were subsampled to 10,000 sequences and then clustered into OTUs with a cutoff ≤0.03. Alpha diversity indices [i.e., Chao1 (Chao, 1984), Simpson Evenness, and Inverse Simpson Index (Simpson, 1949)], beta diversity analyses [i.e., Principle Coordinate Analysis (PCoA) calculated with a Bray-Curtis dissimilarity matrix], and Analysis of MOlecular VAriance (AMOVA; Excoffier et al., 1992) were performed in mothur. PCoA results were plotted using SigmaPlot 11 (SYSTAT Software, Point Richmond, CA, USA).
To characterize the core microbiome of E. fistula, only OTUs present in all coral samples (including all in situ and all ex situ coral samples) were considered. For the in situ, microbiome, OTUs present in all coral samples collected from the natural environment were considered, and accordingly, for the ex situ microbiome, OTUs present in all corals from the long-term rearing were selected. Of note, members from the in situ and the ex situ microbiome are not exclusive and can be present in both the in situ and ex situ microbiome.
We used METAGENassist (Arndt et al., 2012) for automated taxonomic-to-phenotypic mapping in order to assess putative functional profiles based on the 16S community composition of the coral samples. Input files were created in mothur (“make.shared” and “classify.otu”). During data processing in METAGENassist, all OTUs in the six coral samples were assigned, mapped, and condensed into 372 functional taxa. After filtering based on interquartile range (Hackstadt and Hess, 2009), the remaining 335 functional taxa were normalized over sample by sum and over taxa by Pareto scaling. Subsequently, we analyzed the data for “metabolism by phenotype” using Euclidean distance measure and a complete clustering algorithm to visualize in a heatmap the top 15 features selected by random forest classification.
Please refer to Supplementary Data Sheet 1 for commented workflows for analyses with mothur, METAGENassist, and R.
Results
Microbiome Composition in Corals and Water
To assess differences of the coral microbiome after long-term rearing (>1 year) in comparison to the in situ microbiome of E. fistula, we analyzed 8 16S rRNA gene libraries from 3 freshly collected corals (in situ), 3 long-term reared (ex situ) corals, 1 water sample from the corals' natural habitat (in situ), and 1 water sample from the rearing water (ex situ). The libraries yielded 1,260,862 sequences, which were subsequently trimmed, filtered, and error-corrected (i.e., removal of chimeras, singletons, and chloroplasts, among others) resulting in 363,265 sequences with an average length of 293 bp.
For visualization of the bacterial composition in coral and water samples, we classified all sequences to the family level in a stacked column plot and summarized over replicates in pie charts (Figure 1). The bacterial communities of freshly collected (in situ) and reared (ex situ) corals were different from each other. In the bacterial communities of in situ corals, families of unclassified Gammaproteobacteria, Vibrionaceae, Rhodospirillaceae, and an unclassified family of Proteobacteria comprised on average about half (46%) of all sequences. Conversely, ex situ corals harbored different abundant families. Here, Brevibacteriaceae, Alteromonadaceae, Flavobacteriaceae, and Oceanospirillaceae constituted on average about half (48%) of all sequences. Both water samples were markedly different from the coral samples. The in situ water sample had >70% of the sequences belonging to only two bacterial families, namely Alteromonadaceae and Pseudoalteromonadaceae. The ex situ water sample was more diverse with bacteria from the Alteromonadaceae, Flavobacteriaceae, and Rhodobacteraceae making up >80% of the bacterial diversity.
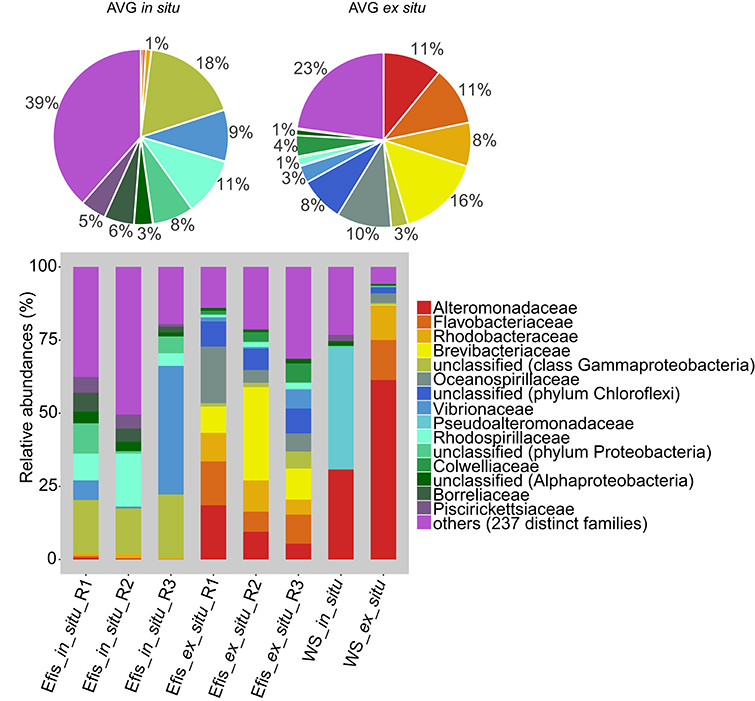
Figure 1. Bacterial diversity from freshly collected (in situ) and long-term reared (ex situ) Eguchipsammia fistula deep-sea corals from the Red Sea on the phylogenetic level of family (Greengenes database, bootstrap ≥60). Each color represents one of the 15 most abundant families across all samples. Less abundant taxa (comprising 237 distinct families) are grouped under the category “others.” Pie charts display average bacterial community composition of in situ (left) and ex situ (right) corals. Sequences that could not be classified on the family level are denoted at the next higher classified taxonomic level. AVG, average; Efis, E. fistula; WS, water sample; R, replicate.
To assess differences between bacterial communities of freshly collected (in situ) corals, reared (ex situ) corals, and water samples in more detail, we subsampled to 10,000 sequences and clustered into operational taxonomic units (OTUs). We identified 2,017 distinct OTUs across all samples, of which 1,750 OTUs were associated with corals and 458 with water samples (Supplementary Data Sheet 2). We next calculated alpha diversity indices. All coral samples contained more OTUs than the water samples, and alpha diversity indices on average were higher in in situ corals compared to their ex situ counterparts (Table 1). However, there were no significant differences for any alpha diversity measure between in situ and ex situ coral samples (all Pt-test > 0.05), which might be due to coral colonies displaying a pronounced inter-individual variability in alpha diversity indices. In particular, in situ reared specimens were highly variable, suggesting flexible microbial association (Table 1).
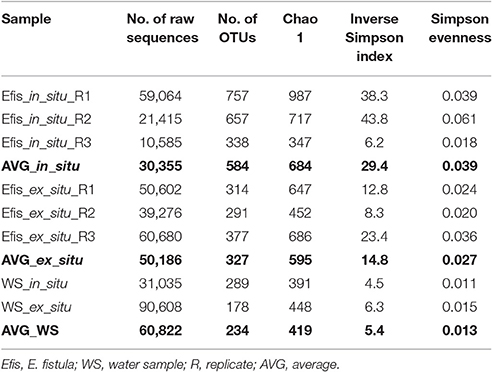
Table 1. Summary statistics of 16S rRNA gene amplicon sequencing of in situ and ex situ Eguchipsammia fistula and water samples.
To further assess differences in the bacterial communities of coral specimens and seawater samples, we plotted all samples in a principle coordinate analysis (PCoA) based on Bray-Curtis dissimilarity (Figure 2). We found the in situ and ex situ coral samples to cluster together. Further, they grouped away from each other and from the seawater samples. Both seawater samples were not clustering closely with any coral sample indicating distinct bacterial communities for corals and water. To assess differences between coral samples in more detail, we excluded both water samples from subsequent analyses.
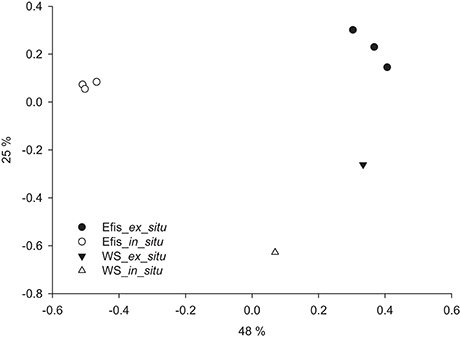
Figure 2. Differences in microbial communities of in situ and ex situ Eguchipsammia fistula and seawater based on Bray–Curtis dissimilarity of microbial taxon abundances in a principal coordinate analysis (PCoA) (R2 = 0.91). Efis, E. fistula; WS, water sample. Percentages on axes indicate variation explained by the two coordinates.
Distinct Bacterial Communities Associated with in situ and ex situ Corals
Based on OTU abundance, in situ corals and ex situ corals were significantly different from each other (PAMOVA < 0.05). Of the 1,750 coral associated OTUs, we found 70 to be present in samples from both groups. These shared OTUs generally displayed pronounced differences in their abundance between conditions, indicating strong dependence on environmental context (e.g., variability and composition of diet). Only 10 of these 70 OTUs were among the 50 most abundant OTUs. Further, the OTU abundances differed at least six-fold between both conditions (Supplementary Data Sheet 2). Hence, the majority of coral-associated bacterial taxa was present either in in situ or in ex situ samples, but not in both.
We identified 1,251 distinct OTUs associated with in situ corals, while reared specimens contained only 568 OTUs. Thus, bacterial richness was much higher in corals in their natural habitat, potentially reflecting the more stable environment and less diverse diet (with a consequently less diverse gut microbiome) under reared conditions (Supplementary Data Sheet 2). The 36 most abundant coral-associated OTUs (average abundance >50) included 26 that were exclusively associated with either the in situ or ex situ samples. The low number of shared taxa was even more pronounced among rare OTUs (<10 sequence counts across all coral samples). From 1,368 OTUs with an abundance <10 over all coral samples, 962 OTUs were only found associated with in situ corals, 388 exclusively in ex situ corals, and only 18 OTUs occurred in both groups (Supplementary Data Sheet 2).
To identify stable bacterial associates in E. fistula we determined the core microbiome, as defined by all OTUs that were present in all coral samples. The core microbiome comprised only 6 OTUs and was numerically dominated by Gammaproteobacteria (>73%), namely OTU0001 (Alteromonas sp.) and OTU0003 (Photobacterium angustum). The remaining 4 OTUs were less abundant and included Spirochaetes (OTU0013, unclassified Borreliaceae), Nitrospira (OTU0067, unclassified Nitrospiraceae), Betaproteobacteria (OTU0086, Pelomonas puraquae), and Cytophagia (OTU0111, Reichenbachiella sp.), which together comprised <27% of abundance counts of the core microbiome (Figure 3, Supplementary Data Sheet 2). Despite their consistent presence, core microbiome members were highly variable in abundance across coral samples (from 3 to 1,337 sequence counts, Supplementary Data Sheet 2).
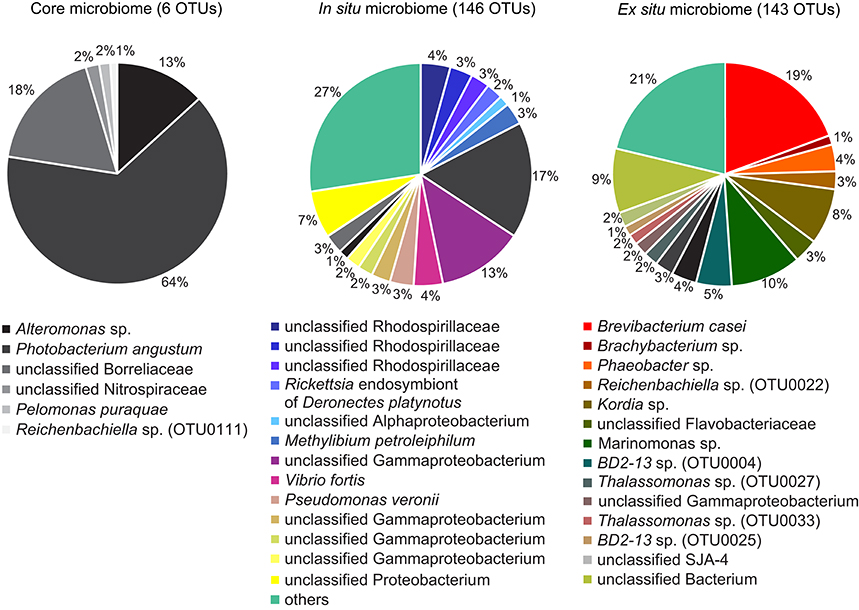
Figure 3. Eguchipsammia fistula core microbiome and microbiomes of freshly collected (in situ) and long-term reared (ex situ) coral specimens. Bacterial members were determined by assessing presence of OTUs over samples. OTUs present in all coral samples were considered to be members of the core microbiome, taxa represented in all in situ samples to be members of the in situ microbiome, and those occurring in all ex situ samples were considered taxa of the ex situ microbiome. Each color represents a distinct OTU, including all core microbiome members and the 16 most abundant taxa each of the in situ and ex situ microbiomes; 130 (in situ) and 127 (ex situ) rare OTUs have been summarized in the category “others,” respectively.
In the coral holobiont, distinct bacterial taxa may be functionally important under different environmental conditions (Roder et al., 2015; Röthig et al., 2016). This may impede the delineation of functionally relevant bacteria based on abundance, since rare bacteria under one environmental condition may become abundant under another. For this reason, we chose to look for bacterial taxa consistently present in all in situ or all ex situ samples (irrespective of relative abundance) to identify bacterial taxa that may provide functional insights. In all in situ corals, we identified 146 common OTUs, of which only 19 also occurred in reared samples. Similarly, in all ex situ corals, we found 143 common OTUs, of which only 27 occurred in freshly collected corals (Figure 3). OTUs exclusive to either condition comprised abundant and rare members. The pronounced differences in the microbiomes of in situ and ex situ corals highlight the flexibility of microbiome composition of E. fistula and suggest that abundant and rare bacterial members can change under prevailing environmental conditions, including taxa of putative functional importance.
Taxonomy-Based Functional Profiling of Bacterial Communities in E. fistula
To gain further insight into the functional importance of the pronounced microbiome differences, we assessed putative functional changes underlying the distinct bacterial communities in the freshly collected (in situ) and long-term reared (ex situ) corals using METAGENassist. As anticipated based on results from the OTU-based PCoA analysis (Figure 2), in situ coral samples clustered together and away from ex situ corals indicating functional differences between both groups as well as similarities within each group (Figure 4). Functional groups of in situ samples were represented by an enrichment of the processes “carbon fixation,” “chlorophenol degrading,” “napthalene degrading,” “sulfur metabolizing,” “dinitrogen-fixing,” and “chitin degradation.” In contrast, in ex situ corals we identified pronounced enrichment of the functions “sugars fermentor,” “lignin degrader,” “stores polyhydroxybutyrate,” “dehalogenation,” and “nitrite reducer.” Other processes like “xylan degrader,” “iron oxidizer,” and “sulfide oxidizer” were enriched for individual samples, but did not show overall trends related to the environment of origin.
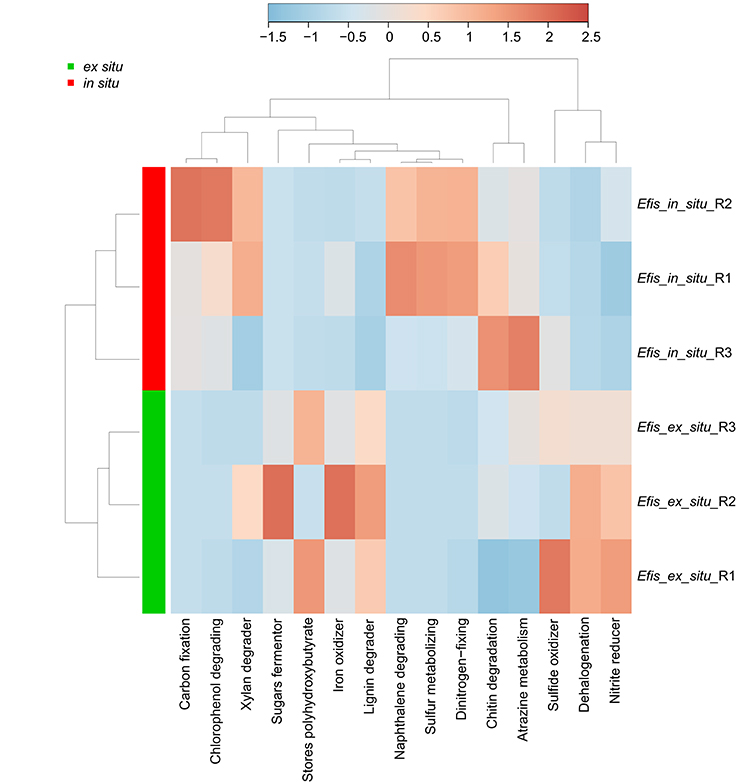
Figure 4. Taxonomy-based functional profiling of bacterial communities from freshly collected (in situ) and long-term reared (ex situ) Eguchipsammia fistula deep-sea coral specimens from the Red Sea. The heat map shows functional differences on a relative scale with enrichment in red and depletion in blue. Data were analyzed for “metabolism by phenotype” with Euclidean distance measure and a complete clustering algorithm. Efis, E. fistula; R, replicate.
Discussion
In this study, we assessed differences in bacterial communities associated with the deep-sea coral E. fistula from the Red Sea from freshly collected (in situ) specimens and after >1 year rearing (ex situ) in aquaria facilities that only partly resemble natural conditions. We identified a major restructuring of the bacterial microbiome that aligns with distinct underlying putative functional profiles, suggesting that the bacterial compartment adjusts to prevailing environmental conditions. Whether this is a consequence of functional restructuring of the coral holobiont or a parallel response of the associated bacterial communities to differences in environmental conditions remains to be determined. Of note, in situ coral samples were preserved in RNAlater, whereas ex situ corals were flash frozen in liquid nitrogen, and it has been shown that RNAlater can bias samples regarding recovery of some bacterial taxa (Gray et al., 2013; Salter et al., 2014). Accordingly, some of the differences we found may be due to the different preservation methods used.
Microbiome Differences in E. fistula between Native Red Sea and Long-Term Rearing Environment
Studies on mucus microbiomes of shallow-water corals (Kooperman et al., 2007; Pratte et al., 2015) and on the microbiome of deep-sea corals (Schöttner et al., 2009) show that transfer and rearing of corals from their native environment to aquaria settings resulted in changes of the associated bacteria, but these were not accompanied by alterations in the appearance of the coral. In contrast, our previous study comparing in situ and ex situ long-term reared coral specimens of E. fistula (Roik et al., 2015) showed substantial morphological differences (i.e., enhanced tissue growth and polyp budding) of E. fistula under ex situ conditions that were characterized by higher oxygen and nutrition levels (among other parameters). Concomitant with the morphological changes, we hypothesized that long-term rearing may result in microbiome changes in E. fistula. Indeed, here we found pronounced differences in the coral bacterial microbiome of in situ and ex situ E. fistula. Changes in the microbial community based on environmental differences were shown before by Roder et al. (2015) where the microbiome of Ctenactis echinata was less structured and more diverse in lower populated coral habitats, indicating that preferred habitats align with more structured bacterial communities. A more extreme case of environmental adjustment was observed by Röthig et al. (2016) where associated bacteria of Fungia granulosa changed under high salinity exposure over 29 days and suggested a shift toward increased osmolyte production, sulfur oxidation, and nitrogen fixation. Similarly, Ziegler et al. (2017) reported on changes of the bacterial microbiome of Acropora hyacinthus in a highly variable and warm environment that align with increased heat tolerance of the coral host. In the current study, differences in the bacterial microbiome between in situ collected and ex situ reared corals were substantial and were representative of differences in prevailing environmental conditions.
In line with substantial differences between microbial community structure of in situ and ex situ coral specimens, we identified only 6 OTUs comprising the core microbiome (i.e., OTUs that were present in every coral sample). The core microbiome included abundant [OTU0001: average abundance (AVG) 161, OTU0003: AVG 786, and OTU0013: AVG 220] but also comparably rare members (OTU0067: AVG 24, OTU0086: AVG 20, and OTU0111: AVG 12; Supplementary Data Sheet 2). OTU0003 (P. angustum, previously Vibrio angustum/fischeri) was previously described as a squid endosymbiont and closely related taxa are shown to inhibit virulence gene expression (Thompson et al., 2009; Mansson et al., 2011), at least hypothetically indicating functional importance and a potential to form a symbiotic relationship with a metazoan host as suggested by Hernandez-Agreda et al. (2016). OTU0086 (P. puraquae) has been identified as a coral-derived endophytic bacterium (Deng et al., 2015). The previous occurrence of OTU0003 and OTU0086 in close relationship with marine metazoans provides a context of functional importance and/or a putatively symbiotic relationship.
All core microbiome members varied in their abundance between freshly collected and long-term reared corals. For example, OTU0001 displayed an average abundance of 320 in ex situ and only 3 in in situ corals, whereas the trend was reversed for OTU0003 with 236 reads in ex situ and 1337 in in situ coral samples (Supplementary Data Sheet 2). The small and highly variable core microbiome in E. fistula potentially indicates a limited functional importance of its members and highlights the microbiome flexibility of the E. fistula coral holobiont. Our analysis of in situ and ex situ microbiomes suggests that E. fistula is associated with largely different microbial communities, pending on the prevailing environmental conditions. More importantly, these microbiomes align functionally to the different environments (see below). As such, the notion of species-specific core microbiomes that are consistent over large geographical and environmental scales (Bayer et al., 2013a; Ainsworth et al., 2015; Roder et al., 2015; Hernandez-Agreda et al., 2016, 2017; Ziegler et al., 2016; Neave et al., 2017) is in contrast to the highly variable core microbiome in E. fistula. Rather, it may suggest a limited association with obligate bacterial symbionts and/or a high flexibility in regard to bacterial symbionts. At the same time, our ability to detect rare conserved OTUs is directly related to sequencing depth. Hence, we cannot positively exclude that we are missing conserved associations of OTUs that would become visible at a higher sequencing depth.
Putative Functional Differences between Native Red Sea and Long-Term Rearing Bacterial Microbiomes
To gain insight into putative functional differences associated with microbiome differences, we conducted taxonomy-based functional profiling. The identified differentially enriched processes revealed functional differences that might support the coral holobiont under two distinct environments. In particular changes in nutrition levels align with a decrease of “carbon fixation,” “dinitrogen-fixing,” and “chitin degradation,” but an increase in “nitrite reducer” in long-term reared ex situ corals. Deep-sea corals are dependent on heterotrophic feeding and therefore generally limited not only in nitrogen, but also in carbon supply (Kiriakoulakis et al., 2004; Roberts et al., 2006). To cover their demands, corals utilize a wide range of food sources including phytodetritus, phytoplankton, zooplankton, dissolved organic matter (Dodds et al., 2007; Gori et al., 2014), and even chemoautotrophic carbon-fixing has been suggested (Middelburg et al., 2015). In line with this, the bacterial microbiome of in situ E. fistula included bacterial taxa with the potential to fix nitrogen and carbon directly (i.e., “dinitrogen-fixing,” “carbon fixation”) or indirectly, e.g., from crustacean prey (i.e., “chitin degradation”). In captivity, an increased food supply may lead to increased growth as shown for M. oculata (Orejas et al., 2011) and E. fistula (Roik et al., 2015). The long-term reared E. fistula were fed regularly with crustaceans ad libitum (Roik et al., 2015). The stable supply of readily available nitrogen and carbon at least hypothetically reduced the need for bacterial fixed nitrogen and carbon from other sources, and enriched nitrite reducing bacterial taxa (i.e., “nitrite reducer”) in ex situ conditions might also be influenced by the provided nutrition. Expulsion of nitrogen (i.e., ammonium, nitrite, and nitrate) has been observed in L. pertusa (Maier et al., 2009, 2011), a process that may be increased by regular feeding in reared E. fistula. In this study, increased nitrite availability might enrich denitrifying bacteria in the microbiome. Taken together, microbiome differences correspond at least partly with the distinct environment and provide putative beneficial functions that might support the coral holobiont under the respective prevailing conditions. Future efforts should target analysis of the active fraction of bacteria, e.g., via RNA-based 16S gene profiling, and compare these data to DNA-based 16S gene profiling in order to disentangle active from total bacterial community composition.
Microbiome Flexibility Aligns with Phenotypic Plasticity
The microbiome is thought to adjust and support the coral holobiont under changing environmental conditions (Reshef et al., 2006). Coral microbiomes are shown to vary to a degree across spatial, depth, and environmental distributions (Pantos et al., 2015; Roder et al., 2015; Hernandez-Agreda et al., 2016; Glasl et al., 2017). Rapid adjustment of the microbiome has also recently been reported under stressful environmental conditions and suggested to support holobiont functioning (Röthig et al., 2016; Ziegler et al., 2017). Studies on deep-sea corals are scarcer, but findings are similar and indicate that environmental conditions influence coral-associated bacterial communities. For instance, diet of deep-sea corals is thought to influence the coral microbiome (Neulinger et al., 2008; Meistertzheim et al., 2016) and in turn the microbiome can unlock nutrient resources for the holobiont (Middelburg et al., 2015). In this context, L. pertusa has been suggested to be a more opportunistic feeder (based on fatty acid and δ15N analyses) than M. oculata (Meistertzheim et al., 2016). This may be based on a more variable bacterial microbiome compared to M. oculata, which also aligns with a higher flexibility and phenotypic plasticity reflected by higher thermal tolerance and a larger distribution range (Neulinger et al., 2008; Meistertzheim et al., 2016).
E. fistula possesses a wide distribution range including the Red Sea, Indo-Pacific, Australia, and New Zealand (van der Land, 2008). These deep-sea habitats differ substantially in their environmental conditions, specifically in temperature and oxygen level (Roder et al., 2013) suggesting a high flexibility of the coral E. fistula. The high phenotypic plasticity of E. fistula has recently been demonstrated by long-term rearing at conditions that only partly resemble the natural Red Sea habitat (Roik et al., 2015). After >1 year rearing under increased oxygen level and consistent food supply, E. fistula corals displayed enhanced skeletal and tissue growth. Our data shows that this phenotypic plasticity is accompanied by a remarkably flexible bacterial microbiome associated with E. fistula. The microbiome changes include the loss of and acquisition of hundreds of OTUs with only comparably few taxa occurring under both conditions (albeit at different abundances), to a point where the concept of a conserved core microbiome remains to be re-examined. As a notion of caution, at the same time, it is posited that rare taxa in particular respond to environmental changes (Jessen et al., 2013; Hernandez-Agreda et al., 2016), and our ability to detect those are directly related to sequencing depth. In their natural habitat, E. fistula may host a highly diverse microbiome to utilize a wide range of nutrients. Moreover, the more diverse diet under in situ conditions may be a source of microbial diversity itself (sensu Meistertzheim et al., 2016), and certain microbiome members may proliferate in response to carbon sources that become episodically available. By comparison, ex situ coral specimens were reared under highly stable environmental conditions and provided with a uniform diet, where the less diverse microbiome may reflect the uniform diet or may be a direct consequence of it. Taken together, phenotypic plasticity of E. fistula may be supported by its microbiome flexibility and more specifically by its ability to associate with distinct bacteria under different environmental conditions. However, specific implications of microbiome stability on coral holobiont functioning are still unknown at large for deep-sea and for shallow-water corals. Even though putative functional profiles indicate that microbiome changes support holobiont function in the respective environments, it remains to be determined if these changes are driven by the environment or a result of selection by the holobiont.
Conclusions
The bacterial microbiome of E. fistula is remarkably flexible and distinct between in situ and ex situ conditions. This is also reflected in the underlying putative functional profiles of the microbiomes that align at least in part with the environmental conditions. The associated bacterial communities may therefore be a result of or contribute to the high flexibility, phenotypical plasticity, and wide distribution of E. fistula. In this context it will be interesting to assess specific contributions of the different bacteria to holobiont functioning.
Data Accessibility
Sequence data determined in this study have been deposited on NCBI under BioProject accession no. PRJNA354830 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA354830) for in situ samples (Accession no.: Efis_in_situ_R1 = SRR5051585, Efis_in_situ_R2 = SRR5051579, Efis_in_situ_R3 = SRR5051575, WS_in_situ = SRR5051584) and under PRJNA383322 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA383322) for ex situ samples (Accession no.: Efis_ex_situ_R1 = SRR5458549, Efis_ex_situ_R2 = SRR5458548, Efis_ex_situ_R3 = SRR5458547, WS_ex_situ = SRR5458546).
Author Contributions
TR, AR, and CRV designed and conceived the experiments. TR, AR, and LY generated the data. TR and CRV analyzed and interpreted the data. CRV contributed reagents/materials/analysis tools. TR and CRV wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by baseline funds to CRV by King Abdullah University of Science and Technology (KAUST) and by the Center Competitive Funding (CCF) Program FCC/1/1973-18-01.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank CMOR and especially P. J. Müller for assistance and support in field operations and maintenance of the corals and C. Michell and KAUST Bioscience Core Lab (BCL) for MiSeq library generation and sequencing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fmars.2017.00259/full#supplementary-material
Supplementary Data Sheet 1. Commented workflow for the mothur and METAGENassist analysis; commented R script for stacked column charts plotting (Figure 1).
Supplementary Data Sheet 2. OTU abundance counts over samples with annotation, reference OTU sequence, and affiliation to core microbiome. Core microbiome members are displayed in bold. Efis, E. fistula; WS, water sample; R, replicate.
References
Ainsworth, T., Krause, L., Bridge, T., Torda, G., Raina, J. B., Zakrzewski, M., et al. (2015). The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9, 2261–2274. doi: 10.1038/ismej.2015.39
Andersson, A. F., Lindberg, M., Jakobsson, H., Bäckhed, F., Nyrén, P., and Engstrand, L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3:e2836. doi: 10.1371/journal.pone.0002836
Arndt, D., Xia, J., Liu, Y., Zhou, Y., Guo, A. C., Cruz, J. A., et al. (2012). METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res. 40, W88–W95. doi: 10.1093/nar/gks497
Bayer, T., Arif, C., Ferrier-Pagès, C., Zoccola, D., Aranda, M., and Voolstra, C. R. (2013a). Bacteria of the genus Endozoicomonas dominate the microbiome of the Mediterranean gorgonian coral Eunicella cavolini. Mar. Ecol. Prog. Ser. 479, 75–84. doi: 10.3354/meps10197
Bayer, T., Neave, M. J., Alsheikh-Hussain, A., Aranda, M., Yum, L. K., Mincer, T., et al. (2013b). The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated Endozoicomonas bacteria. Appl. Environ. Microb. 79, 4759–4762. doi: 10.1128/AEM.00695-13
Bourne, D. G., Morrow, K. M., and Webster, N. S. (2016). Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Ann. Rev. Microbiol. 70, 317–340. doi: 10.1146/annurev-micro-102215-095440
Bourne, D., and Webster, N. (2013). “Coral reef bacterial communities,” in The Prokaryotes, eds E. Rosenberg, E. Delong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin; Heidelberg: Springer), 163–187.
Brown, B. E. (1997). Adaptations of reef corals to physical environmental stress. Adv. Mar. Biol. 30, 221–299. doi: 10.1016/S0065-2881(08)60224-2
Chao, A. (1984). Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 11, 265–270.
Deng, Y., Hu, G., Chen, X., Liu, B., Dai, S., He, X., et al. (2015). Research on cyclo-dipeptides from the coral-derived endophytic bacteria pelomonas puraquae sp.nov of south China sea. Acta Sci. Nat. Univ. Sunyatseni 54, 80–84. doi: 10.13471/j.cnki.acta.snus.2015.03.014
Dodds, L. A., Roberts, J. M., Taylor, A. C., and Marubini, F. (2007). Metabolic tolerance of the cold-water coral Lophelia pertusa (Scleractinia) to temperature and dissolved oxygen change. J. Exp. Mar. Biol. Ecol. 349, 205–214. doi: 10.1016/j.jembe.2007.05.013
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Emblem, Å., Karlsen, B. O., Evertsen, J., Miller, D. J., Moum, T., and Johansen, S. D. (2012). Mitogenome polymorphism in a single branch sample revealed by SOLiD deep sequencing of the Lophelia pertusa coral genome. Gene 506, 344–349. doi: 10.1016/j.gene.2012.06.040
Excoffier, L., Smouse, P. E., and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491.
Galkiewicz, J. P., Pratte, Z. A., Gray, M. A., and Kellogg, C. A. (2011). Characterization of culturable bacteria isolated from the cold-water coral Lophelia pertusa. FEMS Microbiol. Ecol. 77, 333–346. doi: 10.1111/j.1574-6941.2011.01115.x
Glasl, B., Bongaerts, P., Elisabeth, N. H., Hoegh-Guldberg, O., Herndl, G. J., and Frade, P. R. (2017). Microbiome variation in corals with distinct depth distribution ranges across a shallow–mesophotic gradient (15–85 m). Coral Reefs 36, 447–452. doi: 10.1007/s00338-016-1517-x
Gori, A., Grover, R., Orejas, C., Sikorski, S., and Ferrier-Pagès, C. (2014). Uptake of dissolved free amino acids by four cold-water coral species from the Mediterranean Sea. Deep Sea Res. II Top. Stud. Oceanogr. 99, 42–50. doi: 10.1016/j.dsr2.2013.06.007
Gray, M. A., Pratte, Z. A., and Kellogg, C. A. (2013). Comparison of DNA preservation methods for environmental bacterial community samples. FEMS Microbiol. Ecol. 83, 468–477. doi: 10.1111/1574-6941.12008
Gray, M. A., Stone, R. P., McLaughlin, M. R., and Kellogg, C. A. (2011). Microbial consortia of gorgonian corals from the Aleutian islands. FEMS Microbiol. Ecol. 76, 109–120. doi: 10.1111/j.1574-6941.2010.01033.x
Hackstadt, A. J., and Hess, A. M. (2009). Filtering for increased power for microarray data analysis. BMC Bioinform. 10:11. doi: 10.1186/1471-2105-10-11
Hansson, L., Agis, M., Maier, C., and Weinbauer, M. G. (2009). Community composition of bacteria associated with cold-water coral Madrepora oculata: within and between colony variability. Mar. Ecol. Prog. Ser. 397, 89–102. doi: 10.3354/meps08429
Hernandez-Agreda, A., Gates, R. D., and Ainsworth, T. D. (2017). Defining the core microbiome in corals' microbial soup. Trends Microbiol. 25, 125–140. doi: 10.1016/j.tim.2016.11.003
Hernandez-Agreda, A., Leggat, W., Bongaerts, P., and Ainsworth, T. D. (2016). The microbial signature provides insight into the mechanistic basis of coral success across reef habitats. mBio 7:e00560–16. doi: 10.1128/mBio.00560-16
Huse, S. M., Welch, D. M., Morrison, H. G., and Sogin, M. L. (2010). Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12, 1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x
Jessen, C., Villa Lizcano, J. F., Bayer, T., Roder, C., Aranda, M., Wild, C., et al. (2013). In-situ effects of eutrophication and overfishing on physiology and bacterial diversity of the red sea coral Acropora hemprichii. PLoS ONE 8:e62091. doi: 10.1371/annotation/be4a3168-5284-4083-b5ed-5cd0f4630823
Kellogg, C. A., Goldsmith, D. B., and Gray, M. A. (2017). Biogeographic comparison of Lophelia-associated bacterial communities in the Western Atlantic reveals conserved core microbiome. Front. Microbiol. 8:796. doi: 10.3389/fmicb.2017.00796
Kellogg, C. A., Lisle, J. T., and Galkiewicz, J. P. (2009). Culture-independent characterization of bacterial communities associated with the cold-water coral Lophelia pertusa in the Northeastern gulf of Mexico. Appl. Environ. Microbiol. 75, 2294–2303. doi: 10.1128/AEM.02357-08
Kellogg, C. A., Ross, S. W., and Brooke, S. D. (2016). Bacterial community diversity of the deep-sea octocoral Paramuricea placomus. PeerJ 4:e2529. doi: 10.7717/peerj.2529
Kiriakoulakis, K., Bett, B. J., White, M., and Wolff, G. A. (2004). Organic biogeochemistry of the Darwin Mounds, a deep-water coral ecosystem, of the NE Atlantic. Deep Sea Res. I Oceanogr. Res. Pap. 51, 1937–1954. doi: 10.1016/j.dsr.2004.07.010
Kooperman, N., Ben-Dov, E., Kramarsky-Winter, E., Barak, Z., and Kushmaro, A. (2007). Coral mucus-associated bacterial communities from natural and aquarium environments. FEMS Microbiol. Lett. 276, 106–113. doi: 10.1111/j.1574-6968.2007.00921.x
Krediet, C. J., Ritchie, K. B., Paul, V. J., and Teplitski, M. (2013). Coral-associated micro-organisms and their roles in promoting coral health and thwarting diseases. Proc. R. Soc. B 280:20122328. doi: 10.1098/rspb.2012.2328
Lawler, S. N., Kellogg, C. A., France, S. C., Clostio, R. W., Brooke, S. D., and Ross, S. W. (2016). Coral-associated bacterial diversity is conserved across two deep-sea anthothela species. Front. Microbiol. 7:458. doi: 10.3389/fmicb.2016.00458
Maier, C., De Kluijver, A., Agis, M., Brussaard, C. P. D., Van Duyl, F. C., and Weinbauer, M. G. (2011). Dynamics of nutrients, total organic carbon, prokaryotes and viruses in onboard incubations of cold-water corals. Biogeosciences 8, 2609–2620. doi: 10.5194/bg-8-2609-2011
Maier, C., Hegeman, J., Weinbauer, M. G., and Gattuso, J. P. (2009). Calcification of the cold-water coral Lophelia pertusa under ambient and reduced pH. Biogeosciences 6, 1671–1680. doi: 10.5194/bg-6-1671-2009
Mansson, M., Nielsen, A., Kjærulff, L., Gotfredsen, C. H., Wietz, M., Ingmer, H., et al. (2011). Inhibition of virulence gene expression in Staphylococcus aureus by novel Depsipeptides from a marine Photobacterium. Mar. Drugs 9:2537. doi: 10.3390/md9122537
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., Desantis, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
Meistertzheim, A. L., Lartaud, F., Arnaud-Haond, S., Kalenitchenko, D., Bessalam, M., Le Bris, N., et al. (2016). Patterns of bacteria-host associations suggest different ecological strategies between two reef building cold-water coral species. Deep Sea Res. I Oceanogr. Res. Pap. 114, 12–22. doi: 10.1016/j.dsr.2016.04.013
Middelburg, J. J., Mueller, C. E., Veuger, B., Larsson, A. I., Form, A., and Oevelen, D. (2015). Discovery of symbiotic nitrogen fixation and chemoautotrophy in cold-water corals. Sci. Rep. 5:17962. doi: 10.1038/srep17962
Muscatine, L., and Porter, J. W. (1977). Reef Corals: mutualistic symbioses adapted to nutrient-poor environments. BioScience 27, 454–460. doi: 10.2307/1297526
Naumann, M. S., Orejas, C., and Ferrier-Pagès, C. (2014). Species-specific physiological response by the cold-water corals Lophelia pertusa and Madrepora oculata to variations within their natural temperature range. Deep Sea Res. II Top. Stud. Oceanogr. 99, 36–41. doi: 10.1016/j.dsr2.2013.05.025
Neave, M. J., Rachmawati, R., Xun, L., Michell, C. T., Bourne, D. G., Apprill, A., et al. (2017). Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J. 11, 186–200. doi: 10.1038/ismej.2016.95
Neulinger, S. C., Gärtner, A., Järnegren, J., Ludvigsen, M., Lochte, K., and Dullo, W.-C. (2009). Tissue-associated “Candidatus Mycoplasma corallicola” and Filamentous bacteria on the cold-water coral Lophelia pertusa (Scleractinia). Appl. Environ. Microbiol. 75, 1437–1444. doi: 10.1128/AEM.01781-08
Neulinger, S. C., Järnegren, J., Ludvigsen, M., Lochte, K., and Dullo, W.-C. (2008). Phenotype-specific bacterial communities in the cold-water coral Lophelia pertusa (scleractinia) and their implications for the coral's nutrition, health, and distribution. Appl. Environ. Microbiol. 74, 7272–7285. doi: 10.1128/AEM.01777-08
Orejas, C., Ferrier-Pagès, C., Reynaud, S., Tsounis, G., Allemand, D., and Gili, J. M. (2011). Experimental comparison of skeletal growth rates in the cold-water coral Madrepora oculata Linnaeus, 1758 and three tropical scleractinian corals. J. Exp. Mar. Biol. Ecol. 405, 1–5. doi: 10.1016/j.jembe.2011.05.008
Pantos, O., Bongaerts, P., Dennis, P. G., Tyson, G. W., and Hoegh-Guldberg, O. (2015). Habitat-specific environmental conditions primarily control the microbiomes of the coral Seriatopora hystrix. ISME J. 9, 1916–1927. doi: 10.1038/ismej.2015.3
Penn, K., Wu, D., Eisen, J. A., and Ward, N. (2006). Characterization of bacterial communities associated with deep-sea corals on gulf of Alaska seamounts. Appl. Environ. Microbiol. 72, 1680–1683. doi: 10.1128/AEM.72.2.1680-1683.2006
Pratte, Z. A., Richardson, L. L., and Mills, D. K. (2015). Microbiota shifts in the surface mucopolysaccharide layer of corals transferred from natural to aquaria settings. J. Invertebr. Pathol. 125, 42–44. doi: 10.1016/j.jip.2014.12.009
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Quadfasel, D. R. (2001). “Red sea circulation,” in Encyclopedia of Ocean Sciences, eds J. H. Steele, S. A. Thorpe, and K. K. Turekian (Cambridge, MA: Academic Press), 2366–2376.
Qurban, M. A., Krishnakumar, P. K., Joydas, T. V., Manikandan, K. P., Ashraf, T. T. M., Quadri, S. I., et al. (2014). In-situ observation of deep water corals in the northern Red Sea waters of Saudi Arabia. Deep Sea Res. I Oceanogr. Res. Pap. 89, 35–43. doi: 10.1016/j.dsr.2014.04.002
Rädecker, N., Pogoreutz, C., Voolstra, C. R., Wiedenmann, J., and Wild, C. (2015). Nitrogen cycling in corals: the key to understanding holobiont functioning? Trends Microbiol. 23, 490–497. doi: 10.1016/j.tim.2015.03.008
R Core Team (2014). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Reshef, L., Koren, O., Loya, Y., Zilber-Rosenberg, I., and Rosenberg, E. (2006). The Coral probiotic hypothesis. Environ. Microbiol. 8, 2068–2073. doi: 10.1111/j.1462-2920.2006.01148.x
Roberts, J. M., Wheeler, A. J., and Freiwald, A. (2006). Reefs of the deep: the biology and geology of cold-water coral ecosystems. Science 312, 543–547. doi: 10.1126/science.1119861
Roder, C., Bayer, T., Aranda, M., Kruse, M., and Voolstra, C. R. (2015). Microbiome structure of the fungid coral Ctenactis echinata aligns with environmental differences. Mol. Ecol. 24, 3501–3511. doi: 10.1111/mec.13251
Roder, C., Berumen, M. L., Bouwmeester, J., Papathanassiou, E., Al-Suwailem, A., and Voolstra, C. R. (2013). First biological measurements of deep-sea corals from the red sea. Sci. Rep. 3:2802. doi: 10.1038/srep02802
Rohwer, F., Seguritan, V., Azam, F., and Knowlton, N. (2002). Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 243, 1–10. doi: 10.3354/meps243001
Roik, A., Röthig, T., Roder, C., Müller, P. J., and Voolstra, C. R. (2015). Captive rearing of the deep-sea coral Eguchipsammia fistula from the red sea demonstrates remarkable physiological plasticity. PeerJ 3:e734. doi: 10.7717/peerj.734
Rosenberg, E., Koren, O., Reshef, L., Efrony, R., and Zilber-Rosenberg, I. (2007). The role of microorganisms in coral health, disease and evolution. Nat. Rev. Micro. 5, 355–362. doi: 10.1038/nrmicro1635
Röthig, T., Ochsenkühn, M. A., Roik, A., Van Der Merwe, R., and Voolstra, C. R. (2016). Long-term salinity tolerance is accompanied by major restructuring of the coral bacterial microbiome. Mol. Ecol. 25, 1308–1323. doi: 10.1111/mec.13567
Röthig, T., Yum, L. K., Kremb, S. G., Roik, A., and Voolstra, C. R. (2017). Microbial community composition of deep-sea corals from the red sea provides insight into functional adaption to a unique environment. Sci. Rep. 7:44714. doi: 10.1038/srep44714
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microb. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schöttner, S., Hoffmann, F., Wild, C., Rapp, H. T., Boetius, A., and Ramette, A. (2009). Inter- and intra-habitat bacterial diversity associated with cold-water corals. ISME J. 3, 756–759. doi: 10.1038/ismej.2009.15
Schöttner, S., Wild, C., Hoffmann, F., Boetius, A., and Ramette, A. (2012). Spatial scales of bacterial diversity in cold-water coral reef ecosystems. PLoS ONE 7:e32093. doi: 10.1371/journal.pone.0032093
Thompson, C. C., Vicente, A. C. P., Souza, R. C., Vasconcelos, A. T. R., Vesth, T., Alves, N., et al. (2009). Genomic taxonomy of vibrios. BMC Evol. Biol. 9:258. doi: 10.1186/1471-2148-9-258
Todd, P. A. (2008). Morphological plasticity in scleractinian corals. Biol. Rev. 83, 315–337. doi: 10.1111/j.1469-185X.2008.00045.x
van Bleijswijk, J. D. L., Whalen, C., Duineveld, G. C. A., Lavaleye, M. S. S., Witte, H. J., and Mienis, F. (2015). Microbial assemblages on a cold-water coral mound at the SE Rockall Bank (NE Atlantic): interactions with hydrography and topography. Biogeosciences 12, 4483–4496. doi: 10.5194/bg-12-4483-2015
van der Land, J. (2008). UNESCO-IOC Register of Marine Organisms. National Museum of Natural History.
Yakimov, M. M., Cappello, S., Crisafi, E., Tursi, A., Savini, A., Corselli, C., et al. (2006). Phylogenetic survey of metabolically active microbial communities associated with the deep-sea coral Lophelia pertusa from the Apulian plateau, Central Mediterranean Sea. Deep Sea Res. I Oceanogr. Res. Pap. 53, 62–75. doi: 10.1016/j.dsr.2005.07.005
Ziegler, M., Roik, A., Porter, A., Zubier, K., Mudarris, M. S., Ormond, R., et al. (2016). Coral microbial community dynamics in response to anthropogenic impacts near a major city in the central red sea. Mar. Pollut. Bull. 105, 629–640. doi: 10.1016/j.marpolbul.2015.12.045
Keywords: phenotypic plasticity, microbial community profiling, 16S rRNA gene, deep-sea ecosystems, climate change, acclimation
Citation: Röthig T, Roik A, Yum LK and Voolstra CR (2017) Distinct Bacterial Microbiomes Associate with the Deep-Sea Coral Eguchipsammia fistula from the Red Sea and from Aquaria Settings. Front. Mar. Sci. 4:259. doi: 10.3389/fmars.2017.00259
Received: 25 April 2017; Accepted: 28 July 2017;
Published: 10 August 2017.
Edited by:
Verena Schoepf, University of Western Australia, AustraliaReviewed by:
Christina A. Kellogg, United States Geological Survey, United StatesIsabelle Domart-Coulon, National Museum of Natural History, France
Copyright © 2017 Röthig, Roik, Yum and Voolstra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian R. Voolstra, christian.voolstra@kaust.edu.sa