Protist Communities in Moored Long-Term Sediment Traps (Fram Strait, Arctic)–Preservation with Mercury Chloride Allows for PCR-Based Molecular Genetic Analyses
 Katja Metfies1,2,3*
Katja Metfies1,2,3*  Eduard Bauerfeind1
Eduard Bauerfeind1  Christian Wolf1
Christian Wolf1  Pim Sprong1
Pim Sprong1  Stephan Frickenhaus1 Lars Kaleschke4
Stephan Frickenhaus1 Lars Kaleschke4  Anja Nicolaus1
Anja Nicolaus1  Eva-Maria Nöthig1
Eva-Maria Nöthig1- 1Alfred -Wegener- Institut Helmholtz-Zentrum für Polar-und Meeresforschung, Bremerhaven, Germany
- 2Jacobs University Bremen, Bremen, Germany
- 3Helmholtz Institute for Functional Marine Biodiversity, Oldenburg, Germany
- 4Institute of Oceanography, University of Hamburg, Hamburg, Germany
Here we present a pilot study demonstrating, that preservation with mercury chloride allows the application of PCR-based molecular methods for the characterization of marine protist communities collected with moored long-term sediment traps. They can provide information on pelagic protist communities by collecting sinking plankton from the upper water column all year-round, even in remote polar oceans. Assessment of small protist species from the nano- and picoplankton fractions in sedimented material by microscopy is extremely challenging or almost impossible. Hence, comprehensive studies of variability in protist community composition in moored long-term sediment traps are scarce. Considering that marine nano- and picoeukaryotes are ecologically very important, new approaches are urgently needed to investigate protists in the smallest size-fractions of moored long-term sediment trap samples. We applied the quick and cost-effective Terminal Restriction Length Polymorphism (T-RFLP) on a set of selected samples that were collected between 2000 and 2010 in September at a depth of ~300 m in the area of the “LTER (Long-Term Ecological Research) site HAUSGARTEN” in the eastern Fram Strait (Arctic). The results of these analyses suggest a change in the trapped protist community after 2002 in this area. A comparison of 18S sequences obtained via 454-pyrosequencing from samples collected in the water column and mercury chloride preserved sediment traps in 2009 and 2010 suggests, that sediment traps might reflect the pelagic eukaryotic microbial biodiversity qualitatively. Furthermore, we have indication that preservation with mercury chloride does not severely change the nucleotide composition of 18S rRNA genes in long-term sediment traps. Overall, we suggest that preservation with mercury chloride is a key to open the door for molecular genetic analyses of long-term sediment trap samples, and that PCR-based molecular methods have a strong potential to become an important tool for comprehensive taxonomic analyses of protist- and bacterial communities in moored long-term sediment traps.
Introduction
The deployment of moored sediment traps provides valuable long-term information on particle export and composition of sedimenting particles. It facilitates an understanding of plankton dynamics in the upper water column all year-round. It has to be kept in mind, however, that there are uncertainties related to the use of these tools, such as trapping efficiency that might compromise the validity of the results (Butman, 1986; Gust et al., 1994; Buesseler et al., 2007). Nonetheless, they are an appropriate tool for gaining insights in vertical particle flux patterns. At present they are also the only tool that allows the continuous collection of sinking particles for further microscopic chemical and biochemical analyses over larger temporal scales. Time-series measurements represent an excellent approach to evaluate implications of environmental change on ecosystems, including the Arctic pelagic ecosystem (Glover et al., 2010; Wassmann et al., 2011). In 1999, the Alfred-Wegener-Institute Helmholtz Centre for Polar and Marine Research established the LTER (Long-Term Ecological Research) observatory HAUSGARTEN (Hausgarten) to carry out regular observations of the ecosystem in the eastern Fram Strait (Soltwedel et al., 2005, 2016). It is expected that global warming and the ensuing sea ice melt will strongly alter the Arctic pelagic environment. This could eventually result in modification of unicellular plankton species composition and biomass with changes in matter fluxes within the entire pelagic system. Thus, it is necessary to attain information about the temporal occurrences of phyto- and protozooplankton species and to understand their variability in relation to different environmental conditions. In the area of the “Hausgarten,” investigations involve year-round observation of vertical particle flux by using sediment traps. Measurements of bulk parameters like sedimenting matter and its components combined with light microscopy can provide an estimate of the pelagic protist community and its fate in the catchment area above the traps (Bauerfeind et al., 2009). However, assessment of small protist species from the nano- and picoplankton fraction in this way is almost impossible owing to their small size and simple morphology. As a general consequence, information on changes in protist communities collected with sediment traps is mainly limited to shell-forming taxa. Considering the ecological relevance of the nano- and picoplankton fractions, new approaches are needed that provide comprehensive information on changes in protist communities collected with long-term sediment traps.
Molecular methods have become an indispensable tool box in marine biology over the past three decades. These techniques allow comprehensive evaluation of marine protists, including the nano- and picoplankton fraction, because they are independent of organism size or morphology (Ebenezer et al., 2012). Among many others, these approaches include molecular fingerprints, such as terminal restriction lengths polymorphism (T-RFLP). Molecular fingerprinting approaches are well-established molecular genetic tools for quick comparative analyses targeting the rRNA-coding genes of microbial communities (Dunbar et al., 2000; Danovaro et al., 2006; Joo et al., 2010). Ribosomal genes, being universally present in all cellular organisms are well-suited for molecular surveillance of marine biodiversity (Woese, 1987). Over recent years, a considerable number of marine surveys have taken advantage of ribosomal sequence information to broaden our understanding of protist diversity and community structure (e.g., Medlin et al., 2006; Metfies et al., 2010). In this context, the assessment of the diversity of protist communities via T-RFLP fingerprints is based on sequence heterogeneity e.g., within the 18S rRNA gene of different taxa. The 18S rRNA gene is amplified with a fluorescently-labeled primer and digested with restriction enzymes (Marsh et al., 2000). The composition of differently sized restriction fragments in a sample acts as a characteristic fingerprint of a microbial community that allows qualitative comparisons of community composition. However, it does not provide information on species composition, abundance, or diversity in a sample (Bent et al., 2007). Nevertheless we have chosen this method as a representative PCR-based molecular method for our pilot study. It is quick and cost-efficient in comparison to other molecular methods, such as next generation sequencing of ribosomal genes. Thus, in case of a failure of our pilot study, financial loss would have been relatively small. In contrast, Next Generation Sequencing (NGS) of ribosomal genes, e.g., via 454-pyrosequencing or Illumina sequencing is more costly, but it allows high resolution, taxon-specific assessments of protist communities, including their smallest size fractions and the rare biosphere (Kilias et al., 2014; de Vargas et al., 2015).
Overall, molecular analyses of moored sediment trap samples are potentially promising tools for comprehensive long-term information on change in sinking protistan communities, even for the smallest protists. Molecular methods have been used to study microbial community composition in unpreserved samples collected with sediment traps (e.g., Amacher et al., 2009). However, time-series studies based on the deployment of long-term (annual) sediment traps require treatment of the collected particles with well-established and efficient fixatives like mercury chloride to prevent microbial degradation (Knauer et al., 1984). To our knowledge, to date there is no publication that describes the application of molecular methods to assess microbial community composition in preserved sample material collected with long-term sediment traps.
In this pilot study, we assessed the applicability of 18S T-RFLP fingerprinting and 454-pyrosequencing for analysis of mercury chloride-preserved protist communities collected with moored long-term sediment traps deployed in the eastern Fram Strait (Arctic). We used T-RFLP and 454-pyrosequencing as two methods among various other PCR-based molecular approaches, such as quantitative PCR or molecular sensors. The manuscript is based on the assumption that binding of mercury ions to nucleic acids is a reversible process that does not alter the sequence of the DNA (Yamane and Davidson, 1960). A further goal of this study was to address if differences in the observed fingerprinting patterns might reflect variability in the environmental conditions observed in the catchment area of the sediment traps.
Materials and Methods
Sample Collection
Sediment Traps
Sedimenting particles including protist cells were sampled by modified automatic Kiel sediment traps, with a sampling area of ~0.5 m2, and 20 liquid-tight collector cups (Zeitzschel et al., 1978; Kremling et al., 1996). Here, we present results from the shallowest (~200–300 m below sea surface) sediment traps at the central station of the “Hausgarten” (79° N, 4° E; water depth 2,550 m) (Figure 1). The sampling time for the ten samples analyzed in this study varied between 8 and 20 days (one collector cup) during the first 2 weeks of September for ten years (Table 1). A gap of data in 2003 results from an electronic failure. The collector cups were filled with filtered sterile North Sea water. Salinity was adjusted with NaCl to 40 psu. The liquid in the collector cups (250 or 400 ml, depending on the sediment trap used) was spiked with mercury chloride (0.14% final concentration). After recovery of the moorings (~10 month after the collection period) the samples were stored refrigerated until further processing in the laboratory. A first batch of samples (2000–2008) was processed in 2010, while a second batch (2008–2010) was processed in 2013. The sample collected in 2008 was analyzed as a replicate in both batches. Samples were split by a wet splitting procedure after removal of zooplankton (swimmers) >0.5 mm, which were manually removed under a dissecting microscope at a magnification of 20 and 50. Subsequent molecular analyses are based on 1/64 splits of the original sediment trap sample. We collected cells for isolation of DNA by filtration of a fraction of the original sample onto a 0.2 μm Isopore GTTP membrane filter (Millipore, Schwalbach, Germany). Filters were washed with sterile North Sea water (~50 ml). The sterile sea water was applied and pumped over the filter while it was still kept in the filtration unit. This washing step was included in the protocol to remove residual mercury chloride from the samples. PCR-amplification from sediment trap material failed if this step was not included in the protocol (personal communication Stephan Thiele). Because of limited sample material we were not able to carry out a series of optimization experiments of this step. Nonetheless, the sediment trap sample collected in 2008 was analyzed twice (including the filtration step) to assess the reproducibility of the method. Furthermore, we aimed to elucidate if prolonged storage would affect the results of T-RFLP.
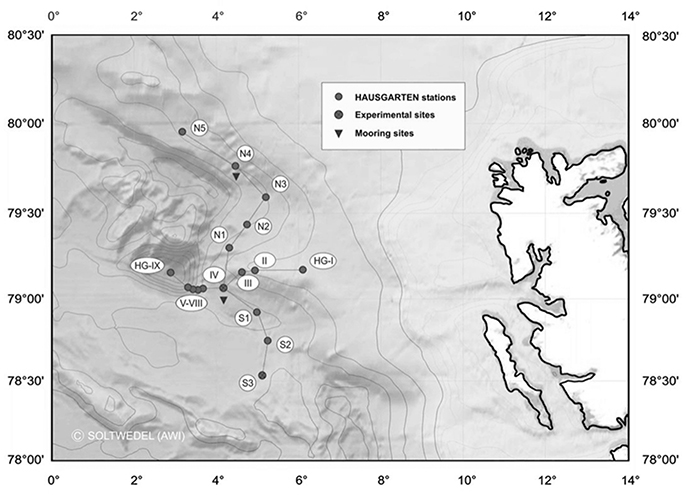
Figure 1. Map of the “Hausgarten” of the Alfred Wegener Institute for Polar and Marine Research in the Eastern Fram Strait. Black dots in the map mark the location of the stations. The central station (HG4) of the “LTER (Long-Term Ecological Research) observatory HAUSGARTEN” is labeled in the map as “IV” (copyright Soltwedel, AWI).
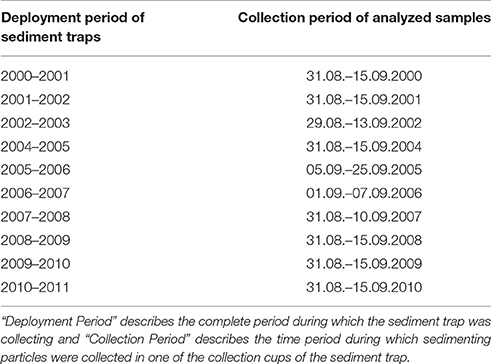
Table 1. Details of sediment trap moorings.
Water Column
Samples were taken in July during research cruises ARKXXIV (2009) and ARKXXV (2010) of RV Polarstern to Fram Strait. Water samples were collected during the upcasts at the vertical maximum of Chl a fluorescence determined during the downcasts. The sampling depths varied between 10 and 50 m. 2 liter subsamples were taken in PVC bottles from the Niskins. Particulate organic matter for molecular analyses was collected by sequential filtration of one water sample through three different mesh sizes (10 μm, 3 μm, 0.4 μm) on 45 mm diameter Isopore Membrane Filters at 200 mbar using a Millipore Sterifil filtration system (Millipore, USA).
Microscopic Counting of Phytoplankton
A split (1/8) of the original sediment trap sample was stored refrigerated in brown glass bottles in the fridge until microscopic counting. Aliquots of 3–10 ml were settled for 8–24 h, and a minimum of 50–100 cells of the dominant species or groups were counted with an inverted microscope at four different magnifications (100–400x) using phase contrast (Utermöhl, 1958).
DNA-Extraction
Genomic DNA was isolated from the samples with the E.Z.N.A Plant DNA Kit (Omega Bio-Tek Inc., Norcross, USA). We modified the original isolation protocol by insertion of an additional washing step using buffer “SPW wash buffer”. Following DNA extraction 2.4 μl of the genomic DNA [~15 ng/μL] isolated from the sediment traps was amplified with the REPLI-g Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol.
T-RFLP in silico Analyses
Protist communities collected from the depth of the chlorophyll a maximum (chl a max) during the expedition ARK XXVII-2 of RV Polarstern in 2010 to the Fram Strait were analyzed by 454-pyrosequencing the 18S V4-region (Kilias et al., 2013). Restriction sites of HaeIII (GGCC) were mapped in silico on the abundant 18S sequence reads (>1%) of all observed reads in the sample) observed in this data set. Representative sequences for these taxa were downloaded from Genbank. The mapping was carried out using MapDraw as implemented in the DNAstar software package (Lasergene, USA).
T-RFLP in vitro Analyses
PCR-Amplification
T-RFLP analysis is based on the amplification of ribosomal sequences via PCR (Treusch et al., 2009). A fragment of the small subunit ribosomal RNA gene was amplified with a universal primer set targeting marine protists. The amplification was based on the universal primer set 690F-FAM 5′-TCAGAGGTGAAATTCTTGGAT-3′ (Metfies and Medlin, 2008) and 1055R 5′-CGGCCATGCACCACCACCCAT-3′ (Metfies et al., 2007). We used this primer set because it amplifies parts of the 18S V4-region, which is the most variable region of this gene (Nickrent and Sargent, 1991). This primer set was chosen over other primer sets because other universal primers e.g., 528F 5′-GCGGTAATTCCAGCTCCAA-3′ (Medlin et al., 2006) that include the V4 region in the PCR-product have no mismatches against key metazoans that had to be excluded from the analyses. The forward primer (690F-FAM) has two mismatches to metazoan species like Calanus finmarchicus or Themisto libellula, which are frequently found in sediment traps deployed in Fram Strait. The 690F-FAM primer was labeled with phosphorarmidite fluorochrome 5-carboxy-fluorescein (FAM). The PCR cocktail contained 1 μL of a 1:20 dilution of the amplified genomic DNA as template, 1x amplification buffer (5 Prime, Hamburg, Germany), 0.2 μM dNTPs (5 Prime, Hamburg, Germany), 0.2 μM of each primer, and 1 U of Taq DNA polymerase (5 Prime, Hamburg, Germany). The amplification protocol was 5 min at 94°C, 1 min at 94°C, 2 min at 54°C, and 2 min at 72°C for 35 cycles and an extension for 10 min at 72°C. For some samples (2009 and 2010) the amplification efficiency was improved by addition of 0.016% BSA (Sigma, Hamburg, Germany) and 2.5% Polyethylenglycol (Sigma, Hamburg, Germany) to the PCR cocktail.
Restriction Analyses
Subsequently ~500 ng of the resulting 18S fragment was subjected to a restriction analysis with the frequent cutter HaeIII (New England Biolabs, Ipswich, USA). We have chosen this restriction enzyme because it is a frequent cutter that cuts the 18S PCR-fragment in the motif GGCC downstream of a variable region that contains a number of insertion and deletions. The addition of another frequent cutter, such as Sau3aI (GATC) would have decreased the resolution of the analysis (Supplementary Figure 1). The restriction reaction was carried out in a reaction volume of 15 μL (500 ng DNA; 1.5 μg/μL BSA; 1x restriction buffer; 2 U HaeIII) for 1 h at 37°C. The restriction enzyme was inactivated by 20 min incubation of at 80°C. The sizes of the 18S terminal restriction fragments were determined by analysis with a capillary sequencer (ABI 3130XL, Applied Biosystems). The T-RFLP analysis was carried out in three technical replicates for each sample.
Statistical Analyses
In an 18S T-RFLP analysis the community is characterized by its composition (presence/absence) of differently sized DNA fragments, which are a result of a sequence specific restriction of the amplified 18S fragment with the restriction fragment HaeIII. Presence/absence matrices, reflecting the community profiles in the samples, were generated by binning the original data obtained after size separation with the capillary sequencer using the “Interactive Binner” (Ramette, 2009) (http://www.mpi-bremen.de/en/Software_2.html). Differences in the T-RFLP community profiles were estimated by calculating the Jaccard index. The Jaccard index is a statistical method used for comparing the similarity and diversity of sample sets. It determines the similarity between samples. The result of the analysis is a distance matrix of the samples in the data set. We visualized the resulting distances using multidimensional scaling (MDS) implemented in the vegan- software package (http://r-forge.r-project.org/projects/vegan/). Groups in the MDS plot were determined a priori. The significance of the grouping was tested by analyzing the similarity between groups with an ANOSIM analysis (Clarke, 1993). ANOSIM is a multivariate, non-parametric statistical method used for comparing community compositions among groups of samples. The correlation of environmental conditions and T-RFLP patterns was tested with a Mantel-test, comparing distance matrices of the environmental conditions and the T-RFLP profiles also using the vegan-software package. All statistical analyses were carried out within the R-package (R Development Core Team, 2011). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, URL http://www.R-project.org/).
454-Pyrosequencing and Sequence Analyses
For 454-Pyrosequencing, a ~670 bp fragment of the 18S rRNA gene containing the hypervariable V4 region was amplified separately from each filter fraction with the primer set 528F (GCG GTA ATT CCA GCT CCA A) and 1055R (ACG GCC ATG CAC CAC CAC CCA T). Details about the procedure, the PCR reaction mixture and the reaction conditions were described previously (Wolf et al., 2013). Pyrosequencing was performed on a Genome Sequencer FLX system (Roche, Germany) by GATC Biotech AG (Germany).
Data Analysis of Water Column
Raw sequence reads were processed using the analysis pipeline Quantitative insights into Microbial Ecology Version 1.8.0 (QIIME) (Caporaso et al., 2010). Reads with a length under 250 bp were excluded from further analysis to ensure including the complete V4 region in the analysis and to get rid of short reads. The quality score was set to 25 and eight homopolymeres and two primer mismatches were allowed. Chimeric sequences in the remaining data set were eliminated from further analyses based on an assessment using the software UCHIME (Edgar et al., 2011) within QIIME. The resulting high quality reads were grouped into operational taxonomic units (OTUs) at the 97% similarity level using Uclust (Edgar, 2010). OTUs comprised of less than 4 sequence reads were removed from the analysis. The remaining sequences were aligned using the SILVA reference database (SSU Ref 119). The raw sequences were deposited at the Sequence Read Archive of the European Nucleotide Archive (ENA) under Accession PRJEB21238.
Data Analysis of Sediment Traps
Raw sequence reads were processed as described previously (Wolf et al., 2014) to obtain high quality reads. Briefly, reads shorter than 300 bp and longer than 670 bp, reads with more than one uncertain base (N), chimeric reads, and reads belonging to metazoans were removed. The remaining high quality reads of all samples were clustered (furthest neighbor algorithm) into operational taxonomic units (OTUs) at the 97% similarity level using the software Lasergene 10 (DNASTAR, USA). OTUs comprised of only one sequence (singletons) were removed. Consensus sequences were generated for each OTU and their taxonomical affiliation was determined by placing them into a reference tree, containing about 1,250 high quality sequences of Eukarya from the SILVA reference database (SSU Ref 111), using the PhyloAssigner pipeline (Vergin et al., 2013). The compiled reference database is available on request in ARB-format. The raw sequences were deposited at the Sequence Read Archive of the European Nucleotide Archive (ENA) under Accession ERP024120.
Phylogenetic Sequence Comparison
Abundant OTUs obtained from water column samples were compared with OTUs from sediment traps via phylogenetic sequence comparison. Sequence alignment construction and phylogenetic tree calculation was done with the open software package MEGA (Kumar et al., 2016).
Environmental Parameters
Data on aerial sea ice extent were obtained from daily special sensor microwave imaging data. The number of days of ice coverage at the central station of the “Hausgarten” during the summer months (May–September) was calculated from these data (Bauerfeind et al., 2009). Average sea surface temperatures for the same period in the area of the “Hausgarten” were taken from the “Averaged Time Series, MODIS-Terra.R1.1 data set” (https://giovanni.gsfc.nasa.gov/giovanni/).
Results
T-RFLP Analyses
Prior to the in vitro analysis, we carried out an in silico restriction of ribosomal sequences originating from taxa that dominated 454-pyrosequencing libraries obtained from pelagic samples collected in the study area during a summer cruise (Kilias et al., 2013). The restriction fragments obtained in silico from the reference sequences were 383 bp for Gyrodinium sp. (JQ692033), 379 bp for Phaeocystis pouchetii (AJ278036), and 382 bp for the Arctic strain of Micromonas sp. (AY954999) and the 18S sequence of Bathycoccus sp. (JF794058).
The DNA-concentration obtained from the sediment trap samples was significantly correlated with concentrations of particulate organic carbon in the traps (R2 = 0.76). Subsequent to successful amplification of a ~400 bp PCR product (Figure 2), the T-RFLP analysis resulted in a total of 61 different 18S T-RFLP fragments in vitro. The average number of fragments per sample was rather low at 17 fragments. Eleven fragments were found mainly in the earlier phase of the observation period (2000–2002), while six fragments were mainly found after that period (Supplementary Table 1). All samples contained at least one T-RFLP fragment in the size range of 379–383 bp deduced in silico for the dominant pelagic taxa collected in Fram Strait in July 2010 (Kilias et al., 2013). Moreover, all samples contained a restriction fragment of 382 bp (Arctic Micromonas sp. CCMP 2099) and one of 383 bp (Gyrodinium sp.). The distances between the different T-RFLP profiles were visualized in a multidimensional scaling plot (metaMDS plot) (Figure 3). The plot suggests that the different community profiles segregate into four groups. The community profiles within a group are more similar to each other than to the community profiles of the other groups. An ANOSIM analysis to test the significance of the grouping resulted in an R-value of 0.9256 and p-value of 0.001, indicating highly significant differences between the groups. Group one is composed of samples collected in 2000–2002, group two of samples collected in 2004–2005, group three consists of samples collected in 2006–2007, and group four of samples collected in 2008 to 2010. The metaMDS-plot suggests that the protists communities in 2006–2010 are more similar to each other than to those of the years before. This observation might point toward a change in the protist community structure after 2002.
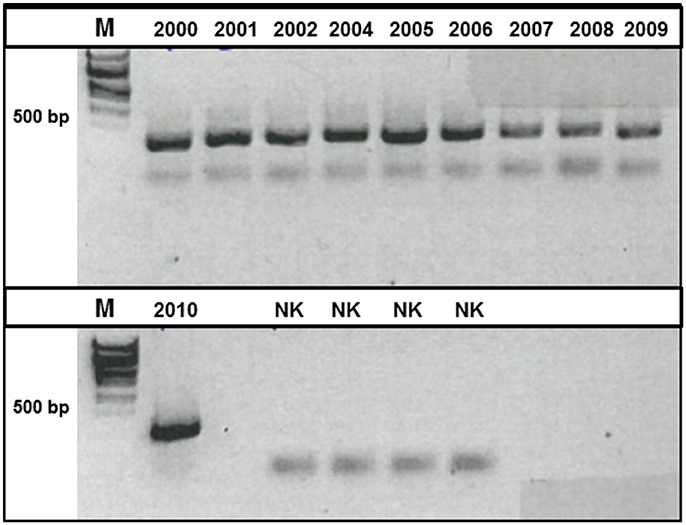
Figure 2. Picture of an agarose gel (1.5% [w/v]) to visualize the outcome of the amplification of a 18S DNA fragment via PCR. The size of the PCR-product was determined in comparison to a 1 kb size marker (M).
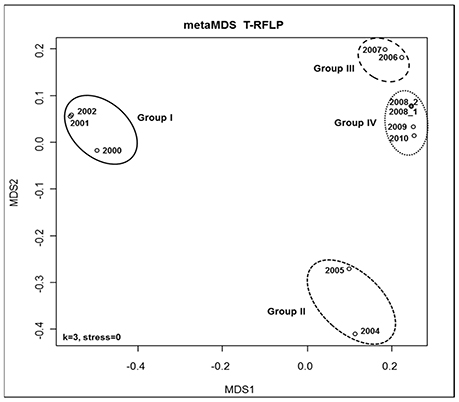
Figure 3. Multidimensional scaling plot (MDS-plot) to illustrate differences between the 18S T-RFLP patterns of the different protist communities in the sediment traps. The MDS-plot is based on the calculation of Jaccard indices for the data set derived from amplification of a 18S PCR fragment and subsequent digestion with the restriction enzyme HaeIII.
Correlation of Environmental Conditions with Molecular Fingerprints
Summer ice coverage at the sediment trap locations between 2000 and 2010 varied from a minimum total absence of ice (0 days) in 2006 to a maximum 113 days of ice coverage in 2008. Minimum and maximum average surface temperatures in the area of the central station “HGIV” (Figure 1) ranged from 1.33°C in 2008 to 3.37°C in 2006 (Table 2). Overall, we observed that the mean surface temperature during summer (May–September) was warmer during those years with less days of ice-coverage than in those years with longer periods of ice-coverage. We used the mean surface temperature and ice-coverage data from May to September for the statistical analyses, to ensure that the data cover the whole Arctic growth period. Based on a Mantel-test we did not discover a statistically significant correlation between the difference patterns of water temperature or ice-coverage and the difference patterns from the protist community fingerprinting profiles (data not shown).
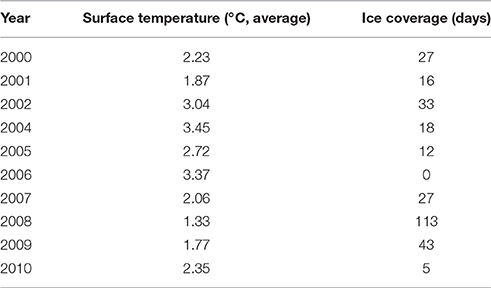
Table 2. Days of ice coverage over the sediment trap at the “Central Hausgarten Station” during the summer of the collection period and the average surface temperature in the “Hausgarten” during summer in the year of the collection period.
Phytoplankton Counts
Sedimentation patterns of phytoplankton cells encompassed with the light microscope reveal that during the first 3 years (2000–2002) high cell numbers of diatoms were found among the phytoplankton cells in the samples. In the following years the abundance of diatoms significantly decreased and they were present in only fairly low numbers in the sediment traps (Figure 4).
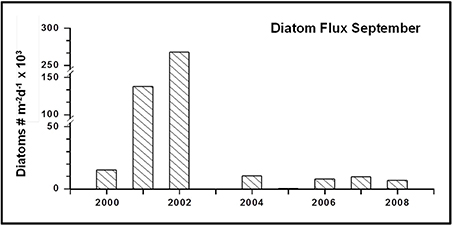
Figure 4. Microscopic counts of diatoms in the sediment trap samples.
Comparison of Sequence Assemblages Obtained from Unpreserved Pelagic Samples and Mercury Chloride Preserved Sediment Trap Samples
It would only make sense to use archived sediment trap samples to elucidate changes and variability in pelagic eukaryotic microbial biodiversity above the sediment traps if it is known to which degree the biodiversity found in the sediment traps is representative for pelagic biodiversity. We compared the abundant biosphere (>1% of total sequences) in 18S sequence libraries obtained from samples collected directly from the deep chlorophyll a maximum (DCM) with the abundant biosphere in 18S libraries generated from the mercury chloride preserved sediment trap samples. In this study the abundant biosphere represented for both sample sets 60–90% of all sequences identified in the individual samples. Sampling in the water column took place at four different stations within the observation area (HG4; S3; N4, and HG1) during summer of 2009 and 2010, and sediment traps were deployed at HG4 (Figure 1). For these years, the number of abundant operational taxonomic units (OTUs) in the two different sample types (water column, trap) was in a similar range. In the DCM samples we observed 45 abundant OTUs, while we detected 39 abundant OTUs in the sediment trap samples. At higher taxonomic level the OTUs of the DCM samples could be assigned to Mamiellophyceae, Phaeocystis sp., Arthropoda, other metazoan, Ciliophora, Dinophyceae, Syndiniales, MAST (Marine Stramenopiles), Bolidomonas sp. and diatoms (Table 3). The OTUs found in the sediment traps could be assigned to Mamiellophyceae, Chrysochromulina sp., Ichtyosporea, Metazoa, Fungi, Syndiniales and Rhizaria (Table 3). 18S sequences of Ciliophora were exclusively found in the water column sequence assemblages, while Ichtyosporea sequences were only found in the sediment trap sample of 2010. All other taxonomic groups were found in both, pelagic samples and sediment trap samples. Moreover, at the level of OTUs more than half of the abundant OTUs observed in the water column samples were also abundant in the sediment trap samples (24/39). This included sequences affiliating with Phaeocystis sp., Micromonas sp., diatoms, alveolates and copepods (Figure 5). Mamiellophyceae (mainly Micromonas sp.) and Syndiniales (alveolates) were significantly abundant in both sample sets. There was even agreement between the sample sets that the relative abundance of Micromonas sp. was higher in 2010 than in 2009. In contrast, OTUs assigned to Phaeocystis sp. were major contributors to the abundant biosphere of the DCM, while they were only found in the rare biosphere of the sediment trap samples. The data indicate the same trend for Dinophyceae that dominated the eukaryotic microbial sequence assemblage of the DCM, while they were found in the rare biosphere of the sediment traps. Only a third of the OTUs assigned to Dinophyceae observed in the abundant biosphere of the sediment traps were also detected in the abundant biosphere of the water column. Rhizaria and fungi dominated the eukaryotic microbial sequence assemblage of the sediment traps, while they were only found in the rare biosphere of the samples collected in the DCM above the sediment traps. Overall, most of the taxa found in our study were present in both kinds of samples, but their relative contribution to the eukaryotic microbial community differed between the DCM samples and the sediment trap samples.
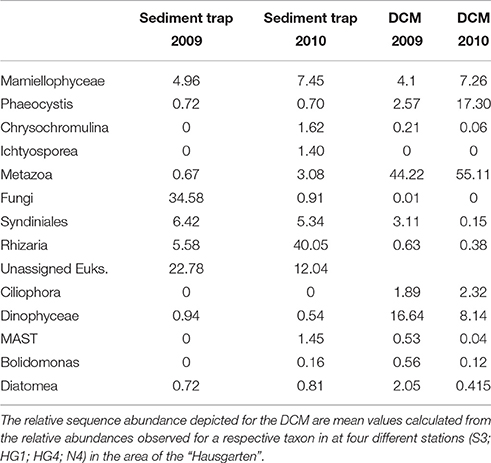
Table 3. Relative sequence abundance of taxonomic groups that contribute to the abundant biosphere in the sediment trap samples and the DCM samples analyzed in this study.
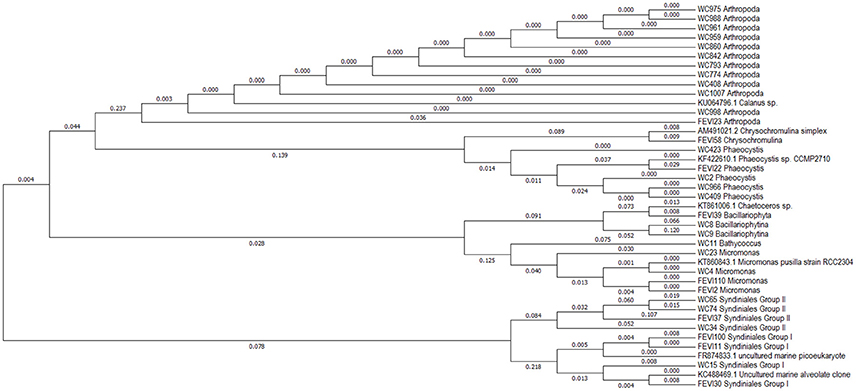
Figure 5. Phylogenetic tree (maximum likelihood) of 18S rRNA gene sequences shared by the abundant biospheres of the samples collected via long-term sediment traps and in the DCM (Deep Chlorophyll Maximum) above the traps. Names of sequences starting with WC (Water Column) represent sequences originating from the DCM, while names of sequences starting with FEVI (acronym for proper name of traps) represent sequences originating from sediment traps. The number adjacent to FEVI and WC is the identifier of the OTU in the respective dataset. The numbers attached to the branches indicate sequence dissimilarity.
The impact of preservation with mercury chloride on the nucleotide composition within the 18S rRNA gene of individual taxa was evaluated by comparing sequences of taxa that were found in the abundant biosphere of both, sediment trap and water column samples. In addition, one representative sequence for each taxon was downloaded from GenBank and included in the analyses. Overall, sequences retrieved from the sediment traps were highly similar to sequences obtained from the water column or GenBank. Any sequence that was observed in the abundant biosphere of the mercury chloride preserved sediment trap sample had max. 0.036% difference to either a sequence of this taxon observed in the water column or to a GenBank sequence (Figure 5). Most dissimilarity was in the range of 0.01%. Overall, the range of dissimilarity between the sequences of the abundant biospheres observed in unpreserved DCM samples and mercury chloride fixed sediment trap samples was in a range between <0.01 and 0.036%.
Discussion
Mercury Chloride Preservation
In this pilot study, we demonstrate for the first time the applicability of molecular methods on mercury chloride-preserved cells obtained by means of moored sediment traps. The use of long-term sediment traps means analyzing preserved organic material, which is often a challenging task. Prior to the molecular analyses the genomic DNA isolated from the preserved material was randomly amplified using a DNA-amplification kit in order to ensure that we would have sufficient amounts of genomic DNA for PCR-optimization and potential future molecular analyses. This method has proven to be applicable for reproducible random amplification of genomic DNA (Han et al., 2012), while highly GC-rich regions might be amplified with slightly less efficiency. The region amplified in this study is not particularly GC-rich. Therefore, the random amplification of this genomic region should not be affected by these problems. Other publications have noted difficulties with amplification of nucleic acids from tissues preserved with fixatives that contain mercury chloride stating that mercury chloride could be deposited and remain bound in tissues. This could inhibit Taq DNA polymerase activity during PCR (Oleary et al., 1994). Our protocols for the preparation of sample material for molecular analyses and nucleic acid isolation each involved an additional step of washing. For most of the samples this vigorous washing of the sample material and the isolated nucleic acids appeared to be sufficient to remove residual mercury chloride that could inhibit the PCR-amplification. However, for some samples (2009–2010) it was necessary to add the PCR enhancer bovine serum albumin (BSA) and polyethylenglycol (PEG) for successful amplification of an 18S PCR product. BSA is an agent used to increase DNA polymerase stability. BSA has also been shown to overcome the inhibitory effects on RT-PCR (Giambernardi et al., 1998). PEG has been shown to enhance PCR reactions in the presence of high concentrations of polysaccharides, which inhibit DNA polymerase efficiency (Pandey et al., 1996). These observations suggest that our DNA-extraction method does not always completely clean the DNA from potential PCR inhibitors present in the sediment trap material such as polysaccharides. However, it is not uncommon that PCR-amplification from sediment samples is hampered by inhibitors present in environmental samples e.g., from river sediments (Arbeli and Fuentes, 2007). Thus, individual optimizations of amplification protocols for single samples might be necessary for future PCR-based molecular analyses of long-term sediment trap samples.
PCR-Based Methods
As examples of PCR-based methods, T-RFLP analysis and meta-barcoding are prone to biases related to the PCR-amplification of ribosomal genes. Ideally, the relative contribution of taxa in a protist community should remain unchanged by PCR-amplification. However, there are indications that PCR in mixed communities leads to alterations in the original DNA-ratios and target gene compositions (Amacher et al., 2011) and the addition of the PCR-enhancer PEG might impact molecular fingerprinting patterns (Pandey et al., 1996). Amplification efficiency of the target gene in a sample is impacted by the fact that some taxa are amplified by a certain primer set with greater efficiency than others (Suzuki and Giovannoni, 1996; Polz and Cavanaugh, 1998). We used T-RFLP analysis and meta-barcoding to carry out qualitative comparisons of community compositions in sediment traps and the DCM. Thus, for this study it is probably of minor relevance that the 18S of some species is amplified with more efficiency than that from others. In any case, the PCR amplification for T-RFLP analysis in our study suggests reproducibility of the methodology. The eukaryotic microbial communities collected in 2008 were analyzed twice, including repetition of the filtration step and the amplification of genomic DNA. Amplification of the 18S from the samples of 2009 and 2010 was only possible in the presence of BSA and PEG. Therefore, the repetition of the analysis of the 2008 sample was also done in the presence of BSA and PEG. In the metaMDS-plot, the replicates are located in the same position. This result is of further interest, as the T-RFLP analyses were carried out in two batches. The first batch (2000–2008) was analyzed in 2010, while the second batch (2008–2010) was analyzed in 2013. This approach allowed us to elucidate if longer storage of the samples alters the T-RFLP results. The longest stored samples from 2000 to 2002 are most different from samples collected in later years. These data are supported by microscopic analyses that suggest a change in the community structure after 2002, reflected by a significant decline of diatoms (Figure 4). Therefore, it is likely that the differences in the T-RFLP profiles could be related to changes in the community structure, rather than to effects of loss of cells by filtration, washing steps, the addition of PCR-enhancers or long-term storage on the DNA composition. The superposition of the replicate samples in the metaMDS-plot suggests that neither the addition of PCR-enhancers, nor prolonged storage over three years alter the T-RFLP patterns obtained from the sediment trap samples. Certainly, based on the current data set and the limited amount of replicated samples, we cannot completely exclude the possibility that long-term storage impacts nucleic acid composition in the sediment trap samples. Therefore, we suggest repeating the analysis on a 5 years basis in order to monitor the impact of long-term storage on biodiversity patterns obtained from mercury chloride preserved protist communities collected in long-term sediment traps. Finally, the results also suggest that the amplification of genomic DNA prior to the analyses does not change the protist community structure in the sample.
T-RFLP Fragments and Species Compostion
Prior to T-RFLP analysis, expected fragment lengths were determined by in silico restriction of publically-available 18S sequences from taxa that dominated 454-pyrosequencing libraries obtained from pelagic samples collected in the eastern Fram Strait in summer 2010 (Kilias et al., 2013). These analyses were carried out to get a first impression of the T-RFLP fragment size spectrum that might be expected from in vitro analyses. We assumed that T-RFLP fragments in the size range determined in silico for abundant sequences (>1% of all sequence reads in a sequence library) observed in the water column should contribute to the protist communities in the sediment trap. The results of the in silico restriction were in agreement with the T-RFLP and meta-barcoding data. This could be an indication that the T-RFLP patterns obtained from the sediment traps might indeed reflect the protist composition in the traps. However, we cannot completely exclude the possibility that the presence of the restriction fragments could be based on the presence of a closely-related taxon with the same 18S restriction pattern. A T-RFLP fragment could represent the presence of a single species or of a variety of taxa (Supplementary Figure 1). Thus, T-RFLP is of very limited benefit for species identification and will probably be replaced by NG-sequencing in the future.
Natural Pelagic Community and Mercury Chloride Preserved Trap Samples with Ecological Implications
In this study we used 454-pyrosequencing to evaluate to what extent the eukaryotic microbial biodiversity in the upper water column is reflected by the biodiversity of this group of organisms observed in mercury chloride preserved sediment trap samples. Furthermore, we addressed if preservation with mercury chloride severely impacts nucleotide sequence composition of the 18S rRNA gene of different taxa in the sediment traps. 454 pyrosequencing provides high resolution taxon-specific information on variability in sequence composition in the samples (Wolf et al., 2013). The untreated samples from the DCM were collected in 2009 and 2010 at four different stations of “Hausgarten” in order to acknowledge that the catchment area of the sediment-traps is large and very variable because of the highly dynamic oceanographic environment in Fram Strait. The integration of four different sampling sites was supposed to give a representative insight into the protist community composition in the DCM in the catchment area of the traps. The taxon composition of those sequences found in the abundant biosphere of the DCM reflects very well what was observed previously in large-scale biodiversity studies focusing on Arctic pelagic protists (Kilias et al., 2014; Metfies et al., 2016). In accordance with these studies, sequences affiliating with Micromonas sp., Dinophyceae and Phaeocystis sp. contributed significantly to the abundant biosphere of the 18S libraries generated from the DCM. Surprisingly the picoeukaryote Micromonas sp. was also significantly contributing to the abundant biosphere of the sediment trap samples, while Dinophyceae (Gymnodiniphycidae) and Phaeocystis sp. were only present in the rare biosphere of the sediment trap samples. Thus, the sequencing data are in accordance with the T-RFLP data of this study that suggest the presence of Micromonas sp. and Gyrodinium sp. (Gymnodiniphycidae) in the sediment trap samples. Overall, most taxa, and even picoeukaryote taxa were present in both sequence sample types. This finding suggests that most species of the photic zone might be exported to deeper water layers, while the mechanisms of transport remain to be elucidated. However, there are most probably differences in the export efficiency of different taxa due to a variety of factors and processes impacting the export efficiency of each taxon differently (Amacher et al., 2009). Thus, molecular based information on eukaryotic microbial biodiversity obtained from mercury chloride preserved sediment trap samples might provide valuable qualitative information on pelagic eukaryotic biodiversity in the catchment area, but it is most probably from limited value in respect to the relative contributions of taxa to the eukaryotic microbial communities above the traps. In this study we observed significant differences in the relative sequence contribution of some taxa. Our study suggests limited export of Phaeocystis sp., which is in agreement with a previous study that also reports limited sinking of Phaeocystis sp. in the Southern Ocean (Wolf et al., 2016). In contrast, sequences assigned to fungi were mainly found in the sediment traps. Marine fungi can dominate biomass on marine snow in the bathypelagic realm (Bochdansky et al., 2017). Thus, the marine fungi sequences observed in the sediment traps might originate from marine snow floating in the water column between the DCM and the sediment trap.
In this study the 18S rRNA gene sequences obtained from the mercury chloride preserved sediment trap samples were highly homologs to sequences obtained from the DCM or retrieved from a public database. The range of intra specific sequence variability observed in this study was in the range of 0.01–0.036 %, which is reasonable considering natural intra-specific variability and sequencing errors of NGS approaches (Behnke et al., 2011; Miranda et al., 2012). Overall, our data suggest that preservation with mercury chloride does not significantly impact the nucleotide composition of 18S sequences or qualitative information on community composition determined with molecular methods. Thus, molecular data obtained from mercury chloride preserved sediment trap material might be suited to provide meaningful information on quantitative changes and variability in pelagic and exported eukaryotic microbes, including organisms of the smallest size fraction such as Micromonas sp.
Time-Series Aspects
The value of the new approach was confirmed by testing the approach in the framework of LTER “Hausgarten.” In our data set the T-RFLP profiles point toward a shift or at least strong variability in protist community composition in the sediment trap samples during the observation period 2000–2010. The data suggest that in September the trapped protist communities in the latter part of the study are significantly different than those trapped at the beginning of the observation period in 2000–2002. Based on the choice of samples we cannot exclude that differences in bloom timing, plankton phenology or local patchiness might be the reason for the observed differences in the T-RFLP patterns in September during the observation period. A more comprehensive ecologically focused NGS-study that includes additional parameter, and all samples collected all year round at different locations and depths in the area of “Hausgarten” has to elucidate this uncertainty. Nonetheless, our findings are supported by microscopic analyses that suggest a change in the community structure after 2003, reflected by a significant decline of diatoms (Figure 4), and all year round studies of other parameters for example zooplankton composition and fecal pellets similarly imply a shift during the observation period (Kraft et al., 2013; Lalande et al., 2013). Considering these results, and assuming trophic interactions, it seems that T-RFLP analysis is well suited to identify long-term variability and shifts in protist communities collected with moored sediment traps. Admittedly, it does not provide information on the extent or nature of the variability, but this was not the scope of this study. Statistical analyses suggest that the observed variability cannot be significantly explained by differences in summer sea surface temperature and ice-coverage over the trap at the “LTER observatory HAUSGARTEN.” Long-term measurements across Fram Strait showed an anomalous incursion of warm Atlantic water into Fram Strait during 2005–2007 (Beszczynska-Möller et al., 2012). At the same time, summer sea ice-coverage in the area is determined by sea ice export from the central Arctic Ocean (Smedsrud et al., 2011). The failure to correlate the variability of T-RFLP patterns obtained from the sediment trap samples with differences in sea surface temperature and ice-coverage in eastern Fram Strait may not be too surprising given their complex causes, and given that they are independent processes.
Comments and Recommendations
The successful application of T-RFLP analysis and 454-pyrosequencing on our long-term sediment trap samples suggests that mercury chloride is appropriate to preserve long-term sediment trap samples for molecular genetic investigations that have the potential to provide meaningful information on pelagic eukaryotic biodiversity above the sediment traps. We also introduce T-RFLP as a cost-efficient, informative and reliable tool for quick qualitative assessments of protist communities. Finally, we suggest that other PCR-based methods, like quantitative PCR (Toebe et al., 2013) and molecular sensors (Metfies and Medlin, 2007; Diercks et al., 2008) could also be applied to analyze the composition of microbes (eukaryotes and prokaryotes) in moored long-term sediment traps. The application of next generation sequencing techniques, as demonstrated in this publication has the potential to provide qualitative taxon specific information on eukaryotic microbial biodiversity (Stoeck et al., 2010; Wolf et al., 2013). In contrast, quantitative PCR and molecular sensors could provide information on the presence and abundance of selected taxa in the sediment trap samples (Metfies et al., 2007). Currently, for various reasons these techniques are not applied routinely in marine ecology and long-term observation programs. But we expect, that technical progress make it feasible that some of these techniques might be applied on a routinely basis for the analysis of marine protist communities in the future. Overall, we expect that molecular methods become an important tool for comprehensive taxonomic analyses of protist- and bacterial communities in moored long-term sediment trap.
Author Contributions
KM and EN had the idea for the project and designed the experiments. AN carried out the practical work related to the analyses. KM analyzed the data and carried out statistical tests in collaboration with SF. EB was responsible for the sampling of protists via long-term moored sediment traps and contributed significantly to the evaluation of the data in the context of environmental conditions. LK contributed data on the ice-coverage for the observation periods. EN contributed microscopic counts of diatoms to the manuscript. CW contributed the 454-pyrosequencing data for the sediment traps. PS contributed the 454-pyrosequencing data for the water column. KM wrote the manuscript and all other authors contributed by critically reading and optimizing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by institutional funds of the Alfred -Wegener- Institut Helmholtz-Zentrum für Polar-und Meeresforschung, Germany and funds of the Helmholtz Association for financing the Helmholtz-University Young Investigators Group Planktosens (Grant VH-NG-500) and within the framework of project Frontiers in Arctic Marine Monitoring (FRAM). We greatly thank the crew of R.V. Polarstern for excellent support during the work at sea. Furthermore we thank Annika Schröer and Christiane Lorenzen for great technical assistance. Annegret Müller and Uwe John are acknowledged for excellent technical support of the fragment analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fmars.2017.00301/full#supplementary-material
Supplementary Table 1. Presence/Absence matrix of T-RFLP fragments. Fragments that appear mainly 2000–2002 are labeled in yellow, while fragments that appear mainly after 2006 are labeled in green.
Supplementary Figure 1. Alignment of a selected set of protist taxa. Restriction sites of HaeIII, Sau3aI, and the binding sites of the primer used in this study are indicated.
References
Amacher, J. S., Baysinger, C. W., and Neuer, S. (2011). Biases associated with DNA based molecular studies of marine protist diversity. J. Plankton Res. 33, 1762–1766. doi: 10.1093/plankt/fbr062
Amacher, J. S., Neuer, I., and Anderson Massana, R. (2009). Molecular approach to determine contributions of the protist community to particle flux. Deep Sea Res. I 56, 2206–2215. doi: 10.1016/j.dsr.2009.08.007
Arbeli, Z., and Fuentes, C. L. (2007). Improved purification and PCR amplification of DNA from environmental samples. FEMS Microbiol. Lett. 27, 269–275. doi: 10.1111/j.1574-6968.2007.00764.x
Bauerfeind, E., Nöthig, E. M., Beszczynska, A., Fahl, K., Kaleschke, L., Kreker, K., et al. (2009). Particle sedimentation patterns in the eastern Fram Strait during 2000-2005: results from the arctic long-term observatory HAUSGARTEN. Deep Sea Res. I 56, 1471–1487. doi: 10.1016/j.dsr.2009.04.011
Behnke, A., Engel, M., Christen, R., Nebel, M., Klein, R. R., and Stoeck, T. (2011). Depicting more accurate pictures of protistan community complexity using pyrosequencing of hypervariable SSU rRNA gene regions. Environ. Microbiol. 13, 340–349. doi: 10.1111/j.1462-2920.2010.02332.x
Bent, S. J., Pierson, J. D., and Forney, L. J. (2007). Measuring species richness based on microbial community fingerprints: the emperor has no clothes. Appl. Environ. Microbiol. 73, 2399–2401. doi: 10.1128/AEM.02383-06
Beszczynska-Möller, A., Fahrbach, E., Schauer, U., and Hansen, E. (2012). Variability in atlantic water temperature and transport at the entrance to the Arctic Ocean, 1997–2010. ICES J. Mar. Sci. 69, 852–863. doi: 10.1093/icesjms/fss056
Bochdansky, A. B., Clouse, M. A., and Herndl, G. J. (2017). Eukaryotic microbes, principally fungi and labyrinthulomycetes, dominate biomass on bathypelagic marine snow. ISME J. 11, 362–373. doi: 10.1038/ismej.2016.113
Buesseler, K. O., Antia, A. N., Chen, M., Fowler, S. W., Gardner, W. D., Gustafsson, O., et al. (2007). An assessment of the use of sediment traps for estimating upper ocean particle fluxes. J. Mar. Res. 65, 345–416. doi: 10.1357/002224007781567621
Butman, C. A. (1986). Sediment trap biases in turbulent flows – results from a laboraroty flume study. J. Mar. Res. 44, 645–693. doi: 10.1357/002224086788403051
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 335–336. doi: 10.1038/nmeth.f.303
Clarke, K. R. (1993). Non-parametric multivariate analysis of changes in community structure. Aust. J. Ecol. 18, 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
Danovaro, R., Luna, G. M., Dell'anno, A., and Pietrangeli, B. (2006). Comparison of two fingerprinting techniques, terminal restriction fragment length polymorphism and automated ribosomal intergenic spacer analysis, for determination of bacterial diversity in aquatic environments. Appl. Environ. Microbiol. 72, 5982–5989. doi: 10.1128/AEM.01361-06
de Vargas, C., Audic, S., Henry, N., Decelle, J., Mahe, F., Logares, R., et al. (2015). Eukaryotic plankton diversity in the sunlit ocean. Science 348:1261605. doi: 10.1126/science.1261605
Diercks, S., Medlin, L. K., and Metfies, K. (2008). Colorimetric detection of the toxic dinoflagellate Alexandrium minutum using sandwich hybridization in a microtiter plate assay. Harmful Algae 7, 137–145. doi: 10.1016/j.hal.2007.06.005
Dunbar, J., Ticknor, L. O., and Kuske, C. R. (2000). Assessment of microbial diversity in four southwestern United States soils by 16S rRNA gene terminal restriction fragment analysis. Appl. Environ. Microbiol. 66, 2943–2950. doi: 10.1128/AEM.66.7.2943-2950.2000
Ebenezer, V., Medlin, L. K., and Ki, J. S. (2012). Molecular detection, quantification, and diversity evaluation of microalgae. Mar. Biotechnol. 14, 129–142. doi: 10.1007/s10126-011-9427-y
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Giambernardi, T. A., Rodeck, U., and Klebe, R. J. (1998). Bovine serum albumin reverses inhibition of RT-PCR by melanin. Biotechniques 25, 564–566.
Glover, A. G., Gooday, A. J., Bailey, D. M., Billett, D. S. M., Chevaldonne, P., Colaco, A., et al. (2010). Temporal change in deep-sea benthic ecosystems: a review of the evidence from recent time-series studies. Adv. Mar. Biol. 58, 1–95. doi: 10.1016/B978-0-12-381015-1.00001-0
Gust, G., Michaelis, A. F., Johnson, R., Deuser, W. G., and Bowles, W. (1994). Mooring line motions and sediment trap hydromechanics: in situ intercomparison of three common deployment designs. Deep Sea Res. 41, 831–857. doi: 10.1016/0967-0637(94)90079-5
Han, T., Chang, C. W., Kwekel, J. C., Chen, Y., Ge, Y., Martinez-Murillo, F., et al. (2012). Characterization of Whole Genome Amplified (WGA) DNA for use in genotyping assay development. BMC Genomics 13:217. doi: 10.1186/1471-2164-13-217
Joo, S., Lee, S. R., and Park, S. (2010). Monitoring of phytoplankton community structure using terminal restriction fragment length polymorphism (T-RFLP). J. Microbiol. Methods 81, 61–68. doi: 10.1016/j.mimet.2010.01.025
Kilias, E., Kattner, G., Wolf, C., Frickenhaus, S., and Metfies, K. (2014). A molecular survey of protist diversity through the central Arctic Ocean. Polar Biol. 37, 1271–1287. doi: 10.1007/s00300-014-1519-5
Kilias, E., Wolf, C., Nöthig, E. M., Peeken, I., and Metfies, K. (2013). Protist Distribution in the western fram strait in summer 2010 based on 454-pyrosequencing of 18S rDNA. J. Phycol. 49, 996–1010. doi: 10.1111/jpy.12109
Knauer, G. A., Karl, D. M., Martin, J. H., and Hunter, C. N. (1984). In Situ effects of selected preservatives on total carbon, nitrogen and metals collected in sediment traps. J. Mar. Res. 42, 445–462. doi: 10.1357/002224084788502710
Kraft, A., Nöthig, E. M., Bauerfeind, E., Wildish, D. J., Pohle, G. W., Bathmann, U. V., et al. (2013). First evidence of reproductive success in a southern invader indicates possible community shifts among arctic zooplankton. Mar. Ecol. Prog. Ser. 493, 291–296. doi: 10.3354/meps10507
Kremling, K., Lentz, U., Zeitzschel, B., Schultz-Bull, D. E., and Duinker, J. C. (1996). New type of time-series sediment trap for the reliable collection of inorganic and organic trace chemical substances. Rev. Sci. Instrum. 67, 4360–4363. doi: 10.1063/1.1147582
Kumar, S., Stecher, G., and Tamura, K. (2016). Mega7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 doi: 10.1093/molbev/msw054
Lalande, C., Bauerfeind, E., Nöthig, E. M., and Beszczynska-Möller, A. (2013). Impact of a warm anomaly on export fluxes of biogenic matter in the eastern fram strait. Prog. Oceanogr. 109, 70–77. doi: 10.1016/j.pocean.2012.09.006
Marsh, T. L., Saxman, P., Cole, J., and Tiedje, J. (2000). Terminal restriction fragment length polymorphism analysis program, a web-based research tool for microbial community analysis. Appl. Environ. Microbiol. 66, 3616–3620. doi: 10.1128/AEM.66.8.3616-3620.2000
Medlin, L. K., Metfies, K., Mehl, H., Wiltshire, K., and Valentin, K. (2006). Picoeukaryotic plankton diversity at the Helgoland time series site as assessed by three molecular methods. Microb. Ecol. 52, 53–71. doi: 10.1007/s00248-005-0062-x
Metfies, K., Berzano, M., Mayer, C., Roosken, P., Gualerzi, C., Medlin, L., et al. (2007). An optimized protocol for the identification of diatoms, flagellated algae and pathogenic protozoa with phylochips. Mol. Ecol. Notes 7, 925–936. doi: 10.1111/j.1471-8286.2007.01799.x
Metfies, K., Gescher, C., Frickenhaus, S., Niestroy, R., Wichels, A., Gerdts, G., et al. (2010). Contribution of the class Cryptophyceae to phytoplankton structure in the German Bight. J. Phycol. 46, 1152–1160. doi: 10.1111/j.1529-8817.2010.00902.x
Metfies, K., and Medlin, L. K. (2007). Refining cryptophyte identification with DNA-microarrays. J. Plankton Res. 29, 1071–1075. doi: 10.1093/plankt/fbm080
Metfies, K., and Medlin, L. K. (2008). Feasibility of transferring fluorescent in situ hybridization probes to an 18S rRNA gene phylochip and mapping of signal intensities. Appl. Environ. Microbiol. 74, 2814–2821. doi: 10.1128/AEM.02122-07
Metfies, K., von Appen, W. J., Kilias, E., Nicolaus, A., and Nöthig, E. M. (2016). Biogeography and photosynthetic biomass of arctic marine pico-eukaryotes during summer of the record sea ice minimum 2012. PLoS ONE 11:E0148512. doi: 10.1371/journal.pone.0148512
Miranda, L. N., Zhuang, Y. Y., Zhang, H., and Lin, S. (2012). Phylogenetic analysis guided by intragenomic SSU rDNA polymorphism refines classification of “Alexandrium tamarense” species complex. Harmful Algae 16, 35–48. doi: 10.1016/j.hal.2012.01.002
Nickrent, D. L., and Sargent, M. L. (1991). An overview of the secondary structure of the V4-region of eukaryotic small-subunit ribosomal-RNA. Nucleic Acids Res. 19, 227–235. doi: 10.1093/nar/19.2.227
Oleary, J. J., Browne, G., Landers, R. J., Crowley, M., Healy, I. B., Street, J. T., et al. (1994). The importance of fixation procedures on DNA-template and its suitablility for solution-phase polymerase chain-reaction and PCR in situ hybridization. Histochem. J. 26, 337–346. doi: 10.1007/BF00157767
Pandey, R. N., Adams, R. P., and Flournoy, L. E. (1996). Inhibition of random amplified polymorphic DNAs (RAPDs) by plant polysaccharides. Plant Mol. Biol. Report. 14, 17–22. doi: 10.1007/BF02671898
Polz, M. F., and Cavanaugh, C. M. (1998). Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 64, 3724–3730.
Ramette, A. (2009). Quantitative community fingerprinting methods for estimating the abundance of operational taxonomic units in natural microbial communities. Appl. Environ. Microbiol. 75, 2495–2505. doi: 10.1128/AEM.02409-08
R Development Core Team (2011). R: A Language and Environment for Statistical Computing. Vienna: The R Foundation for Statistical Computing. Available online at: http://www.R-project.org/
Smedsrud, L. H., Sirevaag, A., Kloster, K., Sorteberg, A., and Sandven, S. (2011). Recent wind driven high sea ice area export in the Fram Strait contributes to Arctic sea ice decline. Cryosphere 5, 821–829. doi: 10.5194/tc-5-821-2011
Soltwedel, T., Bauerfeind, E., Bergmann, M., Bracher, A., Budaeva, N., Busch, K., et al. (2016). Natural variability or anthropogenically-induced variation? Insights from 15 years of multidisciplinary observations at the arctic open-ocean LTER site HAUSGARTEN. Ecol. Indic. 65, 89–102. doi: 10.1016/j.ecolind.2015.10.001
Soltwedel, T., Bauerfeind, E., Bergmann, M., Budaeva, N., Hoste, E., Jaeckisch, N., et al. (2005). HAUSGARTEN: multidisciplinary investigations at a deep-sea, long-term observatory in the Arctic Ocean. Oceanography 18, 46–63. doi: 10.5670/oceanog.2005.24
Stoeck, T., Bass, D., Nebel, M., Christen, R., Jones, M. D. M., Breiner, H. W., et al. (2010). Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19, 21–31. doi: 10.1111/j.1365-294X.2009.04480.x
Suzuki, M. T., and Giovannoni, S. J. (1996). Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 62, 625–630.
Toebe, K., Alpermann, T. J., Tillmann, U., Krock, B., Cembella, A., and John, U. (2013). Molecular discrimination of toxic and non-toxic Alexandrium species (Dinophyta) in natural phytoplankton assemblages from the Scottish coast of the North Sea. Eur. J. Phycol. 48, 12–26. doi: 10.1080/09670262.2012.752870
Treusch, A. H., Vergin, K. L., Finlay, L. A., Donatz, M. G., Burton, R. M., Carlson, C. A., et al. (2009). Seasonality and vertical structure of microbial communities in an ocean gyre. ISME J. 3, 1148–1163. doi: 10.1038/ismej.2009.60
Utermöhl, H. (1958). Zur Vervollkommnung der quantitativen Phytoplankton-Methodik. Int. Vereinigung Theor. Angwandte Limnol. 9, 1–38.
Vergin, K. L., Beszteri, B., Monier, A., Thrash, C., Temperton, B., Treusch, A. H., et al. (2013). High-resolution SAR11 ecotype dynamics at the Bermuda Atlantic Time-series Study site by phylogenetic placement of pyrosequences. ISME J. 7, 1322–1332. doi: 10.1038/ismej.2013.32
Wassmann, P., Duarte, C. M., Agusti, S., and Sejr, M. K. (2011). Footprints of climate change in the Arctic marine ecosystem. Glob. Chang. Biol. 17, 1235–1249. doi: 10.1111/j.1365-2486.2010.02311.x
Wolf, C., Frickenhaus, S., Kilias, E. S., Peeken, I., and Metfies, K. (2013). Regional variability in eukaryotic protist communities in the Amundsen Sea. Antarctic Sci. 25, 741–751. doi: 10.1017/S0954102013000229
Wolf, C., Frickenhaus, S., Kilias, E. S., Peeken, I., and Metfies, K. (2014). Protist community composition in the Pacific sector of the Southern Ocean during austral summer 2010. Polar Biol. 37, 375–389. doi: 10.1007/s00300-013-1438-x
Wolf, C., Iversen, M., Klaas, C., and Metfies, K. (2016). Limited sinking of Phaeocystis sp. during a 12 days sediment trap study. Mol. Ecol. 25, 3428–3435. doi: 10.1111/mec.13697
Yamane, T., and Davidson, N. (1960). On the Complexing of Desoxyribonucleic Acid (DNA) by Mercury Ion. J. Am. Chem. Soc. 83, 2599–2607. doi: 10.1021/ja01473a001
Keywords: mercury chloride, long-term sediment traps, 454-pyrosequencing, protist communities, terminal restriction length polymorphism, molecular analyses
Citation: Metfies K, Bauerfeind E, Wolf C, Sprong P, Frickenhaus S, Kaleschke L, Nicolaus A and Nöthig E-M (2017) Protist Communities in Moored Long-Term Sediment Traps (Fram Strait, Arctic)–Preservation with Mercury Chloride Allows for PCR-Based Molecular Genetic Analyses. Front. Mar. Sci. 4:301. doi: 10.3389/fmars.2017.00301
Received: 30 August 2016; Accepted: 04 September 2017;
Published: 21 September 2017.
Edited by:
Marcelino T. Suzuki, Sorbonne Universities (UPMC) and Centre National de la Recherche Scientifique, FranceReviewed by:
David Walsh, Concordia University, CanadaSusanne Neuer, Arizona State University, United States
Anna Vader, University Centre in Svalbard, Norway
Copyright © 2017 Metfies, Bauerfeind, Wolf, Sprong, Frickenhaus, Kaleschke, Nicolaus and Nöthig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katja Metfies, katja.metfies@awi.de