Experimental Evidence for Abiotic Sulfurization of Marine Dissolved Organic Matter
 Anika M. Pohlabeln
Anika M. Pohlabeln  Gonzalo V. Gomez-Saez
Gonzalo V. Gomez-Saez Beatriz E. Noriega-Ortega
Beatriz E. Noriega-Ortega Thorsten Dittmar
Thorsten Dittmar- Research Group for Marine Geochemistry (ICBM-MPI Bridging Group), Institute for Chemistry and Biology of the Marine Environment, University of Oldenburg, Oldenburg, Germany
Dissolved organic sulfur (DOS) is the largest pool of organic sulfur in the oceans, and as such it is an important component of the global sulfur cycle. DOS in the ocean is resistant against microbial degradation and turns over on a millennium time scale. However, sources and mechanisms behind its stability are largely unknown. Here, we hypothesize that in sulfate-reducing sediments sulfur is abiotically incorporated into dissolved organic matter (DOM) and released to the ocean. We exposed natural seawater and the filtrate of a plankton culture to sulfidic conditions. Already after 1-h at 20°C, DOS concentrations had increased 4-fold in these experiments, and 14-fold after 4 weeks at 50°C, indicating that organic matter does not need long residence times in natural sulfidic environments to be affected by sulfurization. Molecular analysis via ultrahigh-resolution mass spectrometry showed that sulfur was covalently and unselectively bound to DOM. Experimentally produced and natural DOS from sediments were highly similar on a molecular and structural level. By combining our data with published benthic DOC fluxes we estimate that 30–200 Tg DOS are annually transported from anaerobic and sulfate reducing sediments to the oceans. Uncertainties in this first speculative assessment are large. However, this first attempt illustrates that benthic DOS flux is potentially one order of magnitude larger than that via rivers indicating that this could balance the estimated global net removal of refractory DOS.
Introduction
Marine dissolved organic matter (DOM) is among the most complex molecular mixture known on Earth, consisting of at least hundreds of thousands of organic compounds (Zark et al., 2017). Many compounds within the DOM mixture contain sulfur (dissolved organic sulfur, DOS), and in sum they make up the largest reservoir of organic sulfur in the ocean (global inventory of >6.7 Pg S, Ksionzek et al., 2016). Despite the relevance of DOS in global biogeochemical cycles, knowledge on its sources and turnover, and its molecular composition is scarce (Lechtenfeld et al., 2011). Marine primary production is considered a major source of organic sulfur, explaining elevated DOS concentrations in surface waters (Ksionzek et al., 2016). A part of the marine organic sulfur is rapidly cycled and thus labile (Ksionzek et al., 2016), and may contribute little to refractory DOS in the oceans' interior (Dittmar et al., 2017). The source of the large refractory DOS pool which is evenly distributed in the water column is unknown (Levine, 2016).
In sulfidic sediments, organic matter abiotically reacts with inorganic reduced sulfur species during early diagenesis forming organic sulfur compounds (Sinninghe Damsté et al., 1989; Vairavamurthy et al., 1995; Schmidt et al., 2009). Sulfur can thereby be incorporated into different types of organic compounds, including alkenes, aldehydes (de Graaf et al., 1992; Krein and Aizenshtat, 1994; Schouten et al., 1994), carbohydrates and complex algal material (Kok et al., 2000; van Dongen et al., 2003). The incorporation of sulfur likely protects labile organic matter from microbial alterations (Anderson and Pratt, 1995). There are also indications that natural DOM may be sulfurized in sulfidic marine systems because DOS concentrations are distinctly higher in sulfidic environments than in oxic ones (Sleighter et al., 2014; Gomez-Saez et al., 2016). Benthic fluxes from anoxic marine sediments, where carbon oxidation rates are high, contribute major amounts of iron (Dale et al., 2015) and DOM (Burdige and Komada, 2014) to the ocean. If abiotic sulfurization of DOM takes place in these sediments, major benthic fluxes of DOS may also occur.
Here, we hypothesize that abiotic sulfurization of DOM occurs in marine sulfidic environments, making sediments with high carbon oxidation rates a potential formation site for DOS in the ocean. To test this hypothesis, we performed sulfurization experiments with natural DOM. DOS formation was quantitatively assessed, and sedimentary and experimental DOS were compared on a detailed molecular level. The molecular composition of the sulfurized samples was compared to DOM from sulfidic sediments in the German Wadden Sea (Janssand pore water) that is likely an area where natural sulfurization occurs (for details on the sample and sampling see Seidel et al., 2014). DOM comprises a largely inseparable mixture of compounds and is as such only partially accessible to conventional chromatographic analytical techniques. For the molecular analysis of DOM prior and after the sulfurization experiment we used ultrahigh-resolution Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS). FT-ICR-MS coupled to soft electrospray ionization enables the analysis of individual, intact molecules in the otherwise inseparable DOM mixture. The high mass accuracy of FT-ICR-MS allows the assignment of molecular formulae to the detected masses (Hertkorn et al., 2013). Additionally, we performed a series of analytical experiments prior to mass spectrometry analysis and within the FT-ICR-MS to determine the molecular structure of S-containing functional groups (Pohlabeln and Dittmar, 2015). Molecular analyses were accompanied by quantitative element analysis of dissolved organic carbon (DOC) and DOS. Because of the high concentrations of inorganic sulfur in all our samples, the samples were desalted via solid phase extraction (SPE) prior to molecular and elemental analysis. Thus, we refer here to the solid phase extractable fraction of DOM, i.e., SPE-DOC and SPE-DOS. For sample preparation and analysis, samples were exposed to atmospheric oxygen. Therefore, compounds that are easily oxidized and that would not survive for long in the oxic water column of the ocean were not targeted by our analysis.
Materials and Methods
Mesocosm Experiments and Samplings
For the sulfurization experiment, a freshly produced DOM sample from in-house mesocosms inoculated with a natural marine planktonic community was compared to a natural DOM sample from a coastal site in the North Sea (Germany). The planktonic mesocosm experiments were conducted as in Osterholz et al. (2015). The mesocosms were set up in triplicates (M1, M2, M3) each consisting of 4.95 L artificial nutrient-enriched seawater (DOC 18 μmol C L−1) (Osterholz et al., 2015) mixed with 0.05 L prefiltered (poresize: 100 μm) coastal North Sea water containing the natural communities of phyto- and bacterioplankton as inoculum (Spiekeroog, Germany, March 18th 2015, 53°01.30' N, 8°27.10'E, low tide, DOC 157 μmol C L−1) in acid-rinsed 5 L glass bottles (final DOC concentration of artificial seawater plus inoculum 19–20 μmol C L−1). The mesocosms were incubated at approximately 17°C and illuminated for 12-h per day (400–700 nm) while the water was constantly stirred using magnetic stirrers. After 18 days the algae that were dispersed in the water before had clustered together and the DOC concentration of the mesocosm water ranged between 100 and 150 μmol C L−1. At this time point the incubation was stopped and the mesocosm water was filtered sequentially through glass microfiber filters (2 μm, GMF, Whatman, USA) and glass fiber filters (0.7 μm, GFF, Whatman, USA) and acidified to pH 2 with hydrochloric acid (p.a., Merck, Germany) to stop any microbial activity.
The North Sea sample for the long-term sulfurization experiment was taken on March 3rd 2015 at low tide and the sample for the 1-h incubation on September 15th 2015 at high tide, both at the same location as the mesocosm inoculum near Spiekeroog Island, Germany. Samples were filtered and acidified as described above and stored at 4°C until the experiment.
Sulfurization Experiments
The sulfurization experiment was adapted from Kok et al. (2000). We chose this original setup to facilitate the comparison with published results for particulate organic matter. Sulfurization was tested by addition of NaSH and S to the samples as done previously for particulate organic matter (Kok et al., 2000). This experimental procedure was chosen because sulfide and S are present in sulfidic seawater and together form polysulfides (Adam et al., 1998). Polysulfides are the most nucleophilic species of reduced sulfur and likely the most important sulfur species for diagenetic sulfur incorporation into organic matter (Krein and Aizenshtat, 1994; Amrani et al., 2007). Long-term (4 weeks) sulfurization experiments were complemented by a very short (1-h) incubation. For the long-term incubation a temperature of 50°C was set in order to cope for the longer residence time that can be up to decades in coastal tidal flats (e.g., Janssand; Røy et al., 2008), in only 4 weeks of incubation. In addition, we conducted the short-term experiment at room temperature.
The filtered and acidified mesocosm and North Sea samples were transferred to 2.5 L amber glass bottles (Figure 1). The pH of all samples was adjusted to pH 8 with NaOH (p.a. Roth, Germany) to simulate the natural seawater pH and left overnight. The pH was checked at the next day and each sample was bubbled with argon for 20 min to expel all O2 from the bottles. No further treatments were conducted with the “control” samples. For the sulfurization approach, inorganic sulfur compounds (10 g NaSH and 0.29 g elemental sulfur) (Kok et al., 2000) were added to the samples that we refer to as “Sulf” samples in the following. Sulfur reagents were analytical grade (p.a. Sigma Aldrich). A reaction blank consisting of one liter of ultrapure water was bubbled with argon and 4 g NaSH and 0.12 g elemental sulfur were added. All bottles—controls, Sulfs and blank—were placed in ovens at 50°C for 4 weeks (Kok et al., 2000). The “Sulf” samples were shaken daily to disperse the inorganic sulfur compounds as the elemental sulfur does not dissolve in water but NaSH and elemental sulfur together form water soluble polysulfides over time. After 4 weeks at 50°C the samples were acidified and filtered as described above.
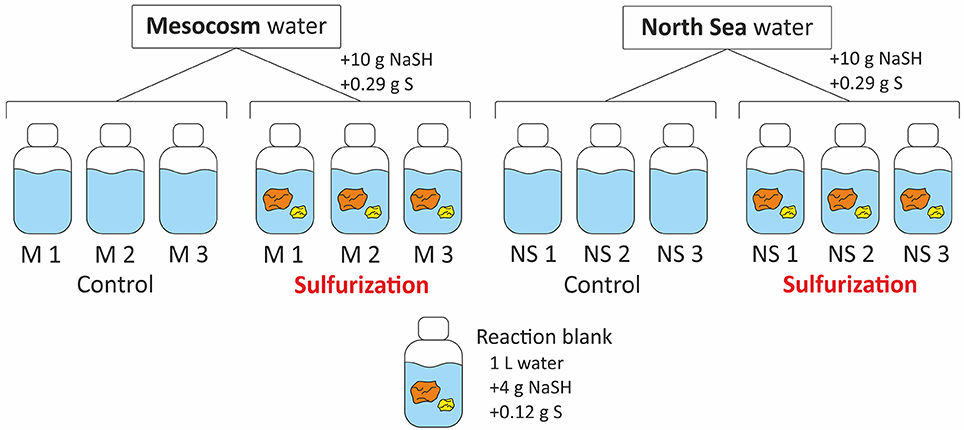
Figure 1. Scheme of the setup for the sulfurization experiment. Two types of DOM samples were used, each in triplicate, North Sea and freshly produced mesocosm DOM. Both were divided into controls and those that were mixed with NaSH and S. Additionally, a reaction blank was prepared that consisted of ultrapure water mixed with NaSH and S.
For the 1-h sulfurization experiment, the pH of the sample was also adjusted to pH 8 and the sample was split (540 ml each) in two controls and two “Sulf” samples. The samples were purged with Argon gas and 2 g NaSH and 60 mg sulfur were added to the “Sulf” samples. The solutions were kept in the dark at room temperature for 1-h with two intervals for shaking the samples, then they were filtered and acidified as described above.
An adduct test was conducted to ensure that the observed sulfurization products originate from covalent incorporation of inorganic sulfur into organic molecules and not only from hydrogen sulfide ion adducts. For this, the DOM methanol extract (see section Sample Preparation and Elemental and Molecular Formula Analyses) of sample “North Sea Control 1” was mixed with an aqueous solution of NaSH (DOC-to-sulfur ratio: 10:1, final DOC concentration: 1.25 mM) and immediately analyzed with the FT-ICR-MS. Potential sulfur-rich adducts of elemental sulfur or polysulfides could be ruled out in our sulfurization experiments because all detected DOS compounds contained not more than two sulfur atoms in their molecular formula (on the basis of our used molecular formulae assignment). A broad band mass spectrum was recorded after the fast addition of NaSH and additionally two nominal masses (m/z = 377 and 389) were fragmented. For comparison, the same FT-ICR-MS analysis was done with the “North Sea Control 1” extract without addition of inorganic sulfur.
Sample Preparation and Elemental and Molecular Formula Analyses
All samples including the blank were extracted using SPE on styrene divinyl benzene polymer filled cartridges (1 g, Agilent Bond Elut PPL, USA) (Dittmar et al., 2008). Bulk DOC concentrations of the samples prior to and after incubation were determined by high-temperature catalytic oxidation on a Shimadzu TOC-VCPH analyzer. Accuracy of the DOC determination was validated by analyzing the deep sea reference sample (Batch 10, Lot# 05–10), from the Consensus Reference Material (CRM) project, provided by D. Hansell and colleagues (University of Miami, USA). Accuracy was within 5%. DOC concentrations of all methanol extracts obtained by SPE were determined by taking an aliquot of the extract, removing the methanol by evaporation and dissolving the residue in 0.01 M HCl which was then analyzed on the same Shimadzu TOC-VCPH analyzer. From those values the extraction efficiencies were calculated. DOS concentrations of the SPE-extracts were measured on an Inductively Coupled Plasma Optical Emission Spectrometer (ICP-OES, iCAP 6000, Thermo Fisher Scientific GmbH, Bremen, Germany) and bulk DOS concentrations were estimated based on the extraction efficiency for DOC as done previously (Pohlabeln and Dittmar, 2015; Gomez-Saez et al., 2016; Ksionzek et al., 2016).
FT-ICR-MS measurements were performed with a solariX Fourier-transform ion cyclotron resonance mass spectrometer with a 15 T magnet system (Bruker Daltonik GmbH, Bremen, Germany). A Bruker Daltonik Apollo II atmospheric pressure electrospray ionization unit (ESI) was used as the external ion source in negative ionization mode. All samples were analyzed in a 1:1 volumetric mixture of methanol (or acetonitrile for derivatization experiment, see section Molecular Analysis of Sulfur Functional Groups) and ultrapure water (Pohlabeln and Dittmar, 2015). The DOC concentration was adjusted to 15 mg C L−1. The samples were directly infused into the ESI source at a flow rate of 120 μL h−1. For fragmentation experiments, the DOC concentration was adjusted to 100 mg C L−1, and a flow rate of 360 μL h−1 was used for ESI. Reproducibility was monitored by analyzing an in-house reference sample from North Equatorial Pacific Intermediate Water (Green et al., 2014) every morning and evening. Five hundred transient scans in broadband mode were accumulated for each run, covering the mass range of 150–2,000 Da. A method detection limit was applied to remove noise peaks from the data set (Riedel and Dittmar, 2014). All detected ions were singly charged. After internal calibration, the mass error was <100 ppb. At this high mass accuracy, molecular formulae were assigned with very high certainty to all compounds containing the elements C, H, O, S, N and P. Formula assignment for each detected mass was done following established procedures (Rossel et al., 2013) but the allowed number of nitrogen atoms was increased to four. Molecular formulae detected in the reaction blanks of the sulfurization experiments were disregarded from further consideration. The identified molecular formulae were tentatively assigned to compound groups based on their molar ratios, aromaticity index, and heteroatom content (Seidel et al., 2014; Stubbins et al., 2014). These compound groups were polycyclic aromatics, polyphenols, sugars, and peptides. Because of the multitude of possible isomers behind a given molecular formula, these assignments are not unambiguous but they provide a reasonable overview of possibly structures behind the cocktail of detected molecular formulae.
For statistical analysis, the detected masses were normalized to the sum of all mass intensities of the corresponding sample. For presence-absence analysis only masses were considered when present in two out of three triplicates or in both of duplicates. Further multivariate statistical analysis was done on the normalized data (Bray-Curtis dissimilarity). For the analysis of the derivatization and hydrolysis alteration reactions a variance test was performed as described in the following. To evaluate whether sulfurization significantly increased the sulfur content of the samples, Student's t-tests were performed for the relevant parameters (Table 1, one-tailed t-test, assuming equal variance).
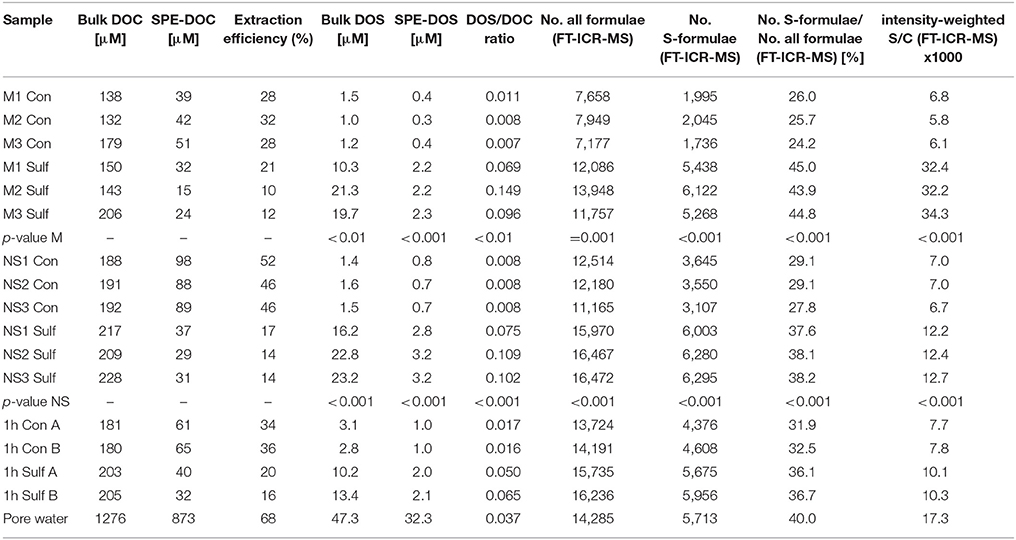
Table 1. Element concentrations and molar ratios for all samples obtained from elemental analysis and molecular formulae (from FT-ICR-MS): intensity weighted = number of carbon or sulfur atoms, respectively, for a detected molecular formula multiplied by its mass intensity divided by the sum of all mass intensities, the p-value represents the significance of Student's t-test of Sulf > Con (one-tailed t-test, assuming equal variance).
Molecular Analysis of Sulfur Functional Groups
Each sample from the sulfurization experiment, controls and “Sulfs,” were analyzed for their S-containing functional groups. For this purpose, functional group selective wet-chemical alteration reactions were conducted (Pohlabeln and Dittmar, 2015), and samples were analyzed in broadband mode on the FT-ICR-MS before and after the alteration reaction as described above. Each sample was analyzed at least in duplicate. In brief, to test for thiols SPE-DOM samples were dried and dissolved in an acetonitrile solution of the thiol-selective reagent 2-bromo-1,4-naphthoquinone for 30 min at 20°C (derivatization experiment). To test for thioesters, sulfonic acid esters and alkylsulfates, samples were hydrolyzed by dissolving the dried SPE-DOM samples in hydrochloric acid (25%, p.a., Merck, Germany) and heated to 110°C for 24-h. To test for non-aromatic thioethers and sulfoxides oxidation and deoxygenation experiments were done. For this purpuse, DOM samples were dissolved in acetonitrile and cyanuric chloride, and hydrogen peroxide or potassium iodide, respectively, were added. Then, the mixture was allowed to stand at 20°C for 2-h.
In addition, collision-induced fragmentation experiments were performed on selected nominal masses (Pohlabeln and Dittmar, 2015): 12 nominal masses (for M2 Sulf only 6 masses due to shortage of sample) consisting of three CH2-homologuous series were analyzed. For each of these 12 nominal masses, 4–7 sulfur-containing molecular formulae were fragmented.
For the statistical interpretation of the functional group selective alteration reactions a variance test was performed (Pohlabeln and Dittmar, 2015). With this approach, it was tested whether discrepancies between the mass spectra of the derivatization and hydrolysis experiments were actually based on the reaction processes or just due to measurement variations. Briefly, the spectra prior to and after reaction were screened for trends in the FT-ICR-MS signal intensities of the m/z-ratios (decreasing or increasing trend after reaction). The same analysis was done for the spectra of the reference sample that was measured every morning and evening, representing the instrument variability. The detected variance in the reference material was set as threshold. Only variance between treated and untreated DOM sample that were above the threshold (higher than instrument variability) were considered as reaction-induced. Compared to the derivatization and hydrolysis, the oxidation and deoxygenation experiments do not lead to a clear separation of reaction products. While there is a characteristic molecule addition in the thiol derivatization and molecule cleavage in hydrolysis, there is only a small (one or two oxygen atoms) increase or decrease in oxygen content for a S-containing molecule in the oxidation and deoxygenation experiment. To determine and visualize even slight differences between the spectra of the oxidation and deoxygenation experiments, the mass spectra were interpreted via multivariate statistical analysis (Bray Curtis dissimilarity; Figure S2).
Microbial Analysis
To verify the lack of microbial activity during sulfurization, samples for cell counts were taken prior to the experiments and afterwards. The samples were fixed with glutardialdehyde (1% final concentration, Carl Roth, Germany) and cells were counted with a BD accuri C6 Flow Cytometer (BD Biosciences, USA) using SYBRGreen (Invitrogen, United Kingdom) following Gasol and Del Giorgio (2000). Fluorescent microscopy was also used to verify the negative results from the flow cytometer. Samples were filtered (0.2 μm polycarbonate) and SYBRGreen was added. After a 30 min dark incubation, the filters were analyzed under the epifluorescence microscope AxioImager.Z2m (Carl Zeiss, Jena, Germany).
Potential Sulfurization Reactions Analysis
In this study, 20 potential sulfurization reactions were chosen (Gomez-Saez et al., 2016), following nine possibilities of S addition while adding/removing H and/or O: + S1; + S1/ − Hn; + HnS1On; + S1On; + S1On/ − Hn; + HnS1; +S1/ − HnOn; + S1/ − On; + HnS1/ − On. The corresponding potential reactions of S addition were proposed as the equivalent +H2S reaction. They were exchanging H2O, H2, and/or O2 by a H2S molecule and accordingly compounds with S1 were obtained (Table S1 and Figure S1). The effectiveness of the potential reactions was considered as a percentage of S1 formulae present in the sulfurized samples with one potential precursor following the corresponding reaction (Gomez-Saez et al., 2016). Additionally, two different groups of sulfur compounds were targeted: DOS formulae produced by sulfurization (Table S1A) and those DOS formulae already present in the samples before incubation (Table S1B). In this second case, two criteria were applied: 1) The intensity of the DOS compound's mass peaks increased after sulfurization and 2) the mass peaks of the corresponding CHO precursor of the DOS compounds decreased in intensity.
Results and Discussion
Experimental Sulfurization of DOM
A distinct increase of bulk DOS concentrations determined by ICP-OES occurred in the 4 week sulfurization experiments with planktonic and North Sea DOM (increase by factor 14, on average, Table 1). The molar DOS/DOC ratio also increased strongly due to sulfurization. Consistently, the total number of non-S-containing compounds detected by FT-ICR-MS also increased (mesocosm: +16%; North Sea: +24%; Table 1). Up to 6000 sulfur-containing molecular formulae were identified in the sulfurized samples by FT-ICR-MS, about double than in the original and control samples (Figures 2, 3). A decrease of DOC extraction efficiency after sulfurization was observed (Table 1). As the bulk DOC concentrations did not decrease after sulfurization, this decrease in extraction efficiency cannot be due to coagulation with inorganic sulfur particles. It is possible though, that during sulfurization or due to second step rearrangement reactions like cyclisation reactions (Eglinton et al., 1994), small non-solid-phase-extractable organic compounds were formed. Nevertheless, the FT-ICR-MS signal intensity-weighted sulfur-to-carbon ratio (S/C) was higher after sulfurization (factor 5.3 for planktonic DOM, factor 1.8 for North Sea DOM). The sulfurization led to a higher number of shared DOS compounds between planktonic and North Sea DOM (Figures 4A,B), indicating a high similarity of DOS on a molecular level. These detected changes in DOS content must result from abiotic reactions as cell counts in the sulfurization experiment revealed no bacterial growth in any of the experiments (data not shown). Furthermore, we excluded the possibility of simple sulfur adducts formed in solution or during ionization by an adduct test where we added sulfide (NaSH) to the control samples immediately prior to FT-ICR-MS analysis. In this test no new DOS compounds were observed verifying the covalent incorporation of inorganic sulfur into organic matter in our sulfurization experiment as the presence of adducts could be ruled out.
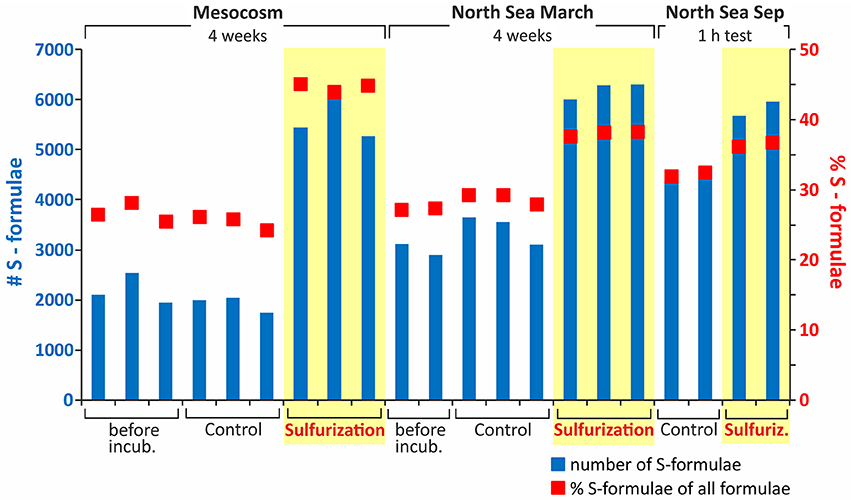
Figure 2. Number and proportion of DOS molecular formulae prior to and after sulfurization. The blue bars show the number of S-containing molecular formulae and the red dots the relative percentage of S-containing formulae of all assigned molecular formulae. Plotted are the short- and long-term sulfurization samples. A distinct increase in DOS concentration as well as number of DOS formulae is visible after sulfurization for mesocosm and North Sea DOM even after only 1-h of reaction time. In the original (untreated) sample of the 1-h experiment the total number of DOS compounds was higher compared to the 4-week approach, but this is only because the North Sea samples were taken at different seasons and tides. This difference reflects the highly dynamic nature of the coastal North Sea. For quantitative data from element analyses we refer to Table 1.
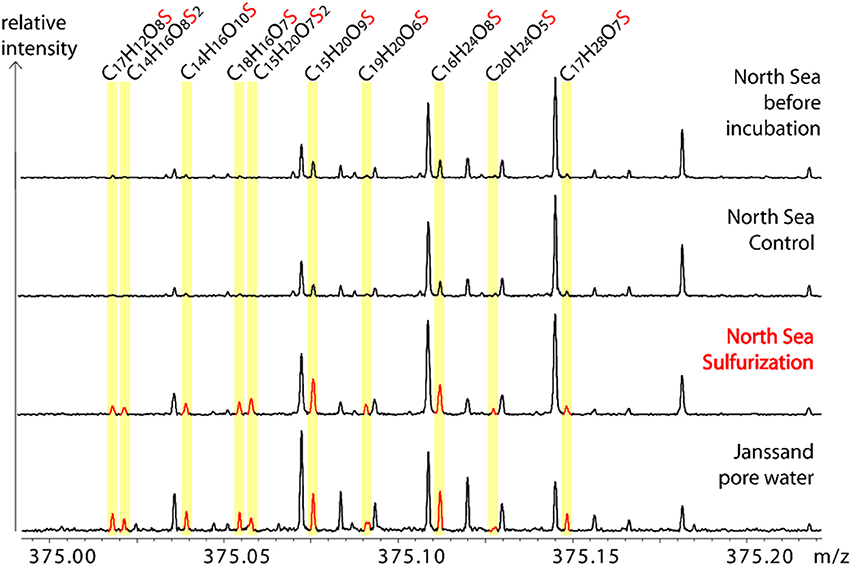
Figure 3. Molecular similarity of sulfurized and pore water DOM. Exemplary sections of the FT-ICR-MS spectra. Comparison of one nominal mass (m/z = 375) of the North Sea sample before incubation, control, and after sulfurization with the natural pore water sample (Janssand). The spectra are scaled to the same intensity level. Neutral S-containing molecular formulae of detected ions are assigned and the corresponding FT-ICR-MS peaks are highlighted.
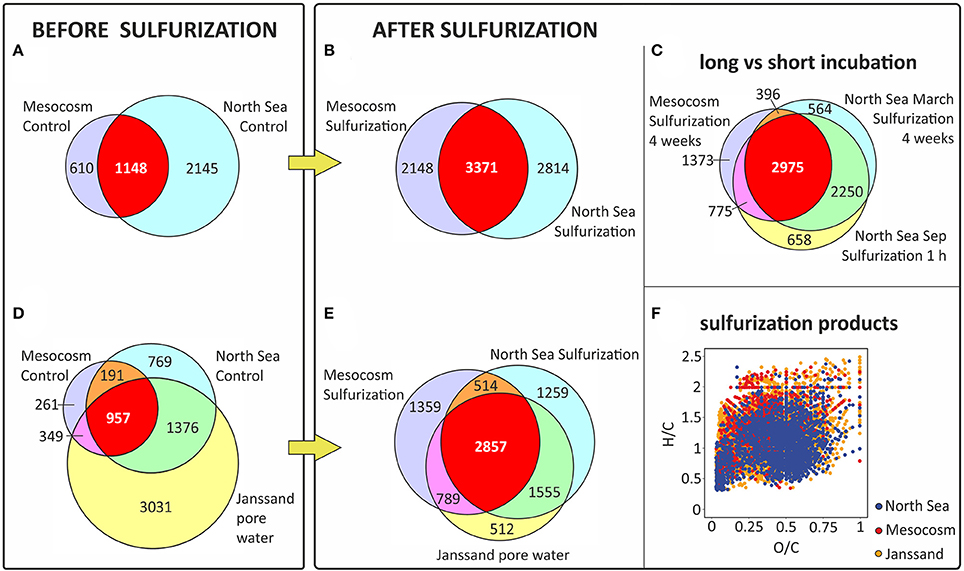
Figure 4. Molecular similarity of sulfurized and pore water DOM. Sulfurization increased the number of shared S-containing molecular formulae compared to DOS from sulfidic pore water (Janssand). Venn diagrams including only S-containing formulae of (A) mesocosms (planktonic DOM) and North Sea controls, (B) mesocosms sulfurized and North Sea sulfurized, (C) S-containing formulae that increased in relative intensity after sulfurization for long (4 weeks) and short (1-h) terms, (D) mesocosms controls, North Sea controls and pore water, and (E) mesocosms sulfurized, North Sea sulfurized and pore water. Only formulae were counted that appeared in at least two out of three replicates or in some cases for both duplicates. The number of shared DOS-formulae increases after sulfurization (red area) showing higher similarity among samples. (F) van Krevelen diagram (hydrogen-to-carbon (H/C) and oxygen-to-carbon (O/C) ratios) showing the S-containing formulae produced by sulfurization (present in the sulfurized but not in control samples) in the mesocosms and North Sea together with the S-containing formulae in the pore water.
In natural sulfidic environments as well as in our experiments, sulfide concentrations are in excess compared to the susceptible organic molecules and reach up to mM levels (e.g., Jansen et al., 2009; Seidel et al., 2014). In our experiments, we chose a higher reaction temperature than in nature to enhance the reaction rate. We did this to simulate the longer residence time of DOM in natural sulfidic environments, that can be up to decades in coastal tidal flats (e.g., Janssand; Røy et al., 2008), in only 4 weeks of incubation. To obtain general information on the speed of the sulfurization reaction of DOM, we incubated a North Sea water sample for only 1-h at 20°C in comparison to the 4 week incubation at 50°C. Even for the short-term sulfurization experiment an increase in bulk DOS concentration by factor 4 was detected (Table 1). Also the number of sulfur-containing molecular formulae increased by factor 1.3 (Figure 2), and the sulfurization products after 1-h of incubation were molecularly very similar to those after 4 weeks of incubation (Figure 4C). Thus, sulfur incorporation into DOM happens fast, indicating that organic matter does not need long residence times in natural sulfidic environments to be affected by sulfurization. A similar timescale was observed by Raven et al. (2016) for the sulfurization of sinking particles.
Molecular Similarity of Natural and Artificial DOS
Our experimental setup mimicked an aquatic, anoxic, and highly sulfidic environment. The molar DOS/DOC ratios of the sulfurized samples (mesocosms: 0.105, North Sea: 0.095) were higher than in the pore water (0.037) (Table 1). This is possibly due to the high sulfide concentration and the stable reaction conditions in the artificial sulfurization experiment. In nature, H2S concentrations in pore waters are dynamic (Jansen et al., 2009), and advective pore water transport across redox gradients or sediment reworking may temporarily interrupt sulfurization. Despite these higher reaction yields in our experiments, the artificial and natural DOS were very similar in their molecular composition. Sulfurization led to an almost undistinguishable molecular pattern compared to the pore water (Figures 3, 4F). Comprehensive, presence-absence analysis showed an increase in the number of shared DOS compounds of our samples and the pore water due to sulfurization (Figures 4D,E). In addition, a statistical Bray Curtis dissimilarity analysis was performed, taking into account also semi-quantitative FT-ICR-MS signal intensities. The sulfurized samples were more similar to the pore water than the controls (Figure S2A) (Bray Curtis dissimilarity indices: pore water vs. mesocosm control: 0.65; pore water vs. mesocosm sulfurized: 0.58; pore water vs. North Sea control: 0.52; pore water vs. North Sea sulfurized: 0.36).
For a comparison on a structural molecular level, we performed extensive analysis of the sulfur-containing functional groups (Pohlabeln and Dittmar, 2015). Even on a structural basis, the sulfurized samples and the pore water DOS showed a high level of similarity: sulfonic acids were the predominant functionality in all analyzed experimental and natural samples (Tables S2, S3), and none of the samples contained detectable or only minor amounts of thiols, thioesters, sulfonic acid esters, alkylsulfates, or sulfoxides. The samples differed in the fragmentation experiments by the occurrence of the neutral loss of H2S. These H2S fragments occurred in all sulfurized and the pore water samples, but not in any of the controls (Table S3). The loss of H2S is not indicative for an explicit functional group (Pretsch et al., 2009) but it is an indicator for reduced sulfur compounds (sulfur oxidation state ≤ 0). This trend is reasonable as reduced inorganic sulfur is incorporated into DOM during sulfurization.
Similar to the statistical comparison on a molecular formula level, we used the molecular fragmentation pattern as structural fingerprints for Bray Curtis dissimilarity analyses. The dissimilarity analysis was done with the FT-ICR-MS signal intensities of those fragments that lost SO3 and H2SO3 (from sulfonic acids) which were normalized to the intensity of the respective precursor-ion. Again, planktonic and North Sea DOS were more similar to the pore water after sulfurization than the controls (Figure S2B). This further confirms the structural similarity of DOS produced by artificial sulfurization compared to the DOS in natural pore water. The similarity of the artificially sulfurized DOM to the naturally sulfurized pore water supports the authenticity of our experimental setup.
Potential Reaction Pathways of Sulfur Incorporation
The detailed mechanism of sulfur incorporation in reduced sediments is unknown, but the most discussed possible mechanism for the sulfur incorporation is the Michael addition that would result in thiols (Krein and Aizenshtat, 1994; Movassagh and Shaygan, 2006; Amrani et al., 2007). As thiols were not detected here, it is likely that the thiols which are reactive nucleophiles themselves (Movassagh and Shaygan, 2006; Nair et al., 2014) incur a second Michael addition intra- or intermolecularly to form thioethers. However, thioethers were also not abundant in the sulfurization products which indicates additional reactions after the incorporation of sulfur. Possibly, sulfurized molecules react in an intramolecular fashion producing thiophenes which we are not able to unambiguously detect because thiophenes are too unreactive for selective alteration reactions and do not show characteristic neutral losses in fragmentation experiments. Potentially, this production of thiophenes resembles processes in later diagenesis or even catagenesis of organic material (Sinninghe Damsté and de Leeuw, 1990; Krein and Aizenshtat, 1994; Aizenshtat et al., 1995). Furthermore, the abundance of the sulfonic acid group and the essential lack of chemically oxidizable sulfur functionalities in all samples indicate almost complete secondary oxidation after sulfurization. Thiols and thioethers are in general unstable under the presence of oxygen (Dupont et al., 2006) and had likely been oxidized prior to analysis. In analogy, reduced sulfur-containing compounds are potentially quickly oxidized once they escape into the oxic open ocean (Gomez-Saez et al., 2016). The main oxidation products are apparently sulfonic acids which we found in all our so far analyzed environments even in the deep sea (Pohlabeln and Dittmar, 2015). Sulfonic acids are very stable compounds and it has been suggested that sulfur incorporation preserves organic matter from microbial degradation (Sinninghe Damsté and de Leeuw, 1990; Hansell, 2013).
Another interesting result of our sulfurization experiment was the non-selectivity of sulfur incorporation. Sulfurization occurred irrespective of saturation, aromaticity, degree of oxidation or heteroelement content (e.g., nitrogen) of the precursor compounds (e.g., Figure 4F). This makes sulfurization fundamentally different from other abiotic transformations of DOM like photodegradation (selective toward aromatic compounds, Kujawinski et al., 2004) or adsorption onto iron minerals (selective toward carboxylic-rich aromates, Riedel et al., 2013). The complexity of the sulfurization process became also apparent when analyzing potential precursor-product-relationships among the molecular formulae (Table S1). In general, the same potential reaction patterns were observed between the planktonic and North Sea samples. The most effective sulfurization reactions (precursors for over 80% of DOS formulae) were those exchanging one or two H2 by a H2S molecule (Table S1) which would not represent the Michael addition mechanism. However, we also found strong indication for the Michael pathway (+H2S, ~70%, Table S1) and potential reactions supporting the high abundance of the sulfonic acid groups (+O2 or +H2O reactions, ~50–80%, Table S1).
Global Relevance of Sedimentary DOS Flux
Our experiments showed that natural marine and planktonic DOM is efficiently sulfurized under sulfidic conditions (e.g., Kok et al., 2000). Sulfurized DOM and DOM from sedimentary pore water were very similar on a molecular formula and molecular structural level (Figures 3, 4). Motivated by these consistent results we attempt a speculative assessment of the potential global relevance of sulfurization in marine sediments. Benthic fluxes of trace elements and DOM from reduced marine sediments are globally significant, and are related to carbon oxidation rates in these sediments. As a result, anaerobic sediments release more DOC to the ocean than more oxidized ones (Burdige and Komada, 2014). Intertidal sediments that are largely covered by salt marshes, mangroves, seagrass and macroalgae deliver 106–416 Tg DOC a−1 to the global ocean (Maher and Eyre, 2010). This estimates includes large uncertainties and may be an underestimate because it does not include all forms of advective transport through permeable sediments (Santos et al., 2012). Coastal and continental margin sediments deliver an additional 121–233 Tg C a−1 (Burdige et al., 1999; Burdige and Komada, 2014), and sediments >2,000 m water depth deliver ~100 Tg DOC a−1 (Dunne et al., 2007; Burdige and Komada, 2014). The latter estimate hinged on the assumption of a constant DOC release of 36% of the total organic carbon reaching the sediments, but this percentage varies widely (from 6 to 32%; Otto and Balzer, 1998). Furthermore, all these estimates are based on a very limited number of studies. Therefore, the uncertainties are inherently large, but taken together, the integrated benthic flux from marine sediments (Burdige and Komada, 2014) may exceed the annual flux of DOC delivered by rivers to the ocean (Hedges et al., 1997). The actual impact these fluxes have on the oceanic carbon cycle ultimately depends on the extent to which sediment-derived DOM is reactive in the water.
To derive DOS fluxes from these estimates, we make two reasonable, yet untested, assumptions. First, we assume that the stoichiometry and efficiency of the sulfurization reaction is globally similar and that the sedimentary DOS compounds are stable over extended time periods. The consistency of our experimental results and field observations is supportive for these assumptions. Second, we assume that benthic fluxes largely originate from sulfidic pore waters, for most intertidal (Dittmar et al., 2006; Roy et al., 2012; Seidel et al., 2014) and marine sediments (Burdige and Komada, 2014).
Based on these two assumptions we estimate the global sedimentary DOS flux by multiplying benthic DOC fluxes with measured DOS/DOC concentration ratios. On the one hand, the minimum DOS flux results from the lower estimate of benthic fluxes (327 Tg C a−1, which is the sum of values cited above) and the DOS/DOC ratio in Janssand pore water (0.037; molar ratio, Table 1). To the best of our knowledge, this is the only quantitative number available for S/C ratios in sedimentary DOM and is comparable to the few reported S/C ratios in sedimentary solid organic matter, at the Peru margin (0.038 ± 0.026) (Mossmann et al., 1991), at the Cariaco Basin (0.048 ± 0.011) (Quijada et al., 2016), at the coast of British Columbia (0.041 ± 0.011) (Francois, 1987), or the Delaware salt marsh (0.038 ± 0.020) (Ferdelman et al., 1991), and the DOS/DOC ratio of sulfidic hydrothermal fluids in Milos Island (0.035 ± 0.029) (Gomez-Saez et al., 2016). On the other hand, our upper flux estimate results from the upper estimate of benthic fluxes (749 Tg C a−1, which is the sum of values cited above) and the average DOS/DOC ratio obtained in our sulfurization experiments (planktonic DOM: 0.105, North Sea DOM: 0.095; molar ratios, Table 1). This results in an approximate global benthic DOS flux from marine sediments of 30–200 Tg DOS a−1.
The uncertainties in this first speculative assessment are large, mainly due to the lack of global data on benthic DOC fluxes, the element stoichiometry of sedimentary DOM and direct measurements of benthic DOS fluxes. However, this first attempt illustrates that benthic DOS flux is potentially one order of magnitude larger than the riverine organic sulfur input to the ocean (8 Tg S a−1, Ksionzek et al., 2016) and may be able to balance the estimated global net removal of refractory DOS (1.1 Tg S a−1, Ksionzek et al., 2016). Based on our estimate, we suggest that sulfurization in sulfidic environments is possibly an important source mechanism of refractory DOS to the oceans. This pathway is not considered in current models of the global sulfur cycle (Ksionzek et al., 2016). A significant lack of knowledge still exists with respect to the reactivity and stability of the various DOS fractions in sulfidic and open ocean waters, which should be target of future studies.
Author Contributions
AP and TD conceived the study. AP performed laboratory work. AP, GG-S, BN-O, and TD analyzed data. AP, GG-S, and TD wrote the manuscript with input from BN-O.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to K. Klaproth for support in FT-ICR-MS analyses, B. Schnetger and E. Gründken for ICP-OES analyses, and to M. Friebe and I. Ulber for help with the experiments and DOC measurements. We thank M. Seidel for sharing the Janssand pore water sample, A. Braun for taking North Sea samples, A. Mentges for MATLAB help and M. Wolterink for support in cell counts. We also thank the editor and three reviewers whose comments helped to improve an earlier version of this manuscript. This study was financially supported by the Deutsche Forschungsgemeinschaft (DFG, DI 842/6-1).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2017.00364/full#supplementary-material
References
Adam, P., Phillippe, E., and Albrecht, P. (1998). Photochemical sulfurization of sedimentary organic matter: a widespread process occurring at early diagenesis in natural environments? Geochim. Cosmochim. Acta 62, 265–271. doi: 10.1016/S0016-7037(97)00332-3
Aizenshtat, Z., Krein, E. B., Vairavamurthy, M. A., and Goldstein, T. P. (1995). “Role of sulfur in the transformations of sedimentary organic matter: a mechanistic overview,” in Geochemical Transformations of Sedimentary Sulfur, eds M. A. Vairavamurthy, M. A. A. Schoonen, T. I. Eglinton, G. W. Luther, and B. Manowitz (Washington, DC: American Chemical Society), 16–37. doi: 10.1021/bk-1995-0612.ch002
Amrani, A., Turner, J. W., Ma, Q., Tang, Y., and Hatcher, P. G. (2007). Formation of sulfur and nitrogen cross-linked macromolecules under aqueous conditions. Geochim. Cosmochim. Acta 71, 4141–4160. doi: 10.1016/j.gca.2007.06.051
Anderson, T. F., and Pratt, L. M. (1995). “Isotopic evidence for the origin of organic sulfur and elemental sulfur in marine sediments,” in Geochemical Transformations of Sedimentary Sulfur, eds M. A. Vairavamurthy, M. A. A. Schoonen, T. I. Eglinton, G.W. Luther, and B. Manowitz (Washington, DC: American Chemical Society), 378–396. doi: 10.1021/bk-1995-0612.ch021
Burdige, D. J., and Komada, T. (2014). “Sediment pore waters,” in Biogeochemistry of Marine Dissolved Organic Matter, 2nd Edn, eds D. A. Hansell and C. A. Carlson (Elsevier Inc.), 535–577. Available online at: https://www.elsevier.com/books/biogeochemistry-of-marine-dissolved-organic-matter/hansell/978-0-12-405940-5
Burdige, D. J., Berelson, W. M., Coale, K. H., McManus, J., and Johnson, K. S. (1999). Fluxes of dissolved organic carbon from California continental margin sediments. Geochim. Cosmochim. Acta 63, 1507–1515. doi: 10.1016/S0016-7037(99)00066-6
Dale, A. W., Nickelsen, L., Scholz, F., Hensen, C., Oschlies, A., and Wallmann, K. (2015). A revised global estimate of dissolved iron fluxes from marine sediments. Global Biogeochem. Cycles 29, 691–707. doi: 10.1002/2014GB005017
de Graaf, W., Sinninghe Damsté, J. S., and de Leeuw, J. W. (1992). Laboratory simulation of natural sulphurization: I. formation of monomeric and oligomeric isoprenoid polysulphides by low-temperature reactions of inorganic polysulphides with phytol and phytadienes. Geochim. Cosmochim. Acta 56, 4321–4328. doi: 10.1016/0016-7037(92)90275-N
Dittmar, T., Hertkorn, N., Kattner, G., and Lara, R. J. (2006). Mangroves, a major source of dissolved organic carbon to the oceans. Global Biogeochem. Cycles 20, GB1012. doi: 10.1029/2005GB002570
Dittmar, T., Koch, B., Hertkorn, N., and Kattner, G. (2008). A simple and efficient method for the solid-phase extraction of dissolved organic matter (SPE-DOM) from seawater. Limnol. Ocean. Methods 6, 230–235. doi: 10.4319/lom.2008.6.230
Dittmar, T., Stubbins, A., Ito, T., and Jones, D. C. (2017). Comment on “Dissolved organic sulfur in the ocean: biogeochemistry of a petagram inventory.” Science 356, 813. doi: 10.1126/science.aam6039
Dunne, J. P., Sarmiento, J. L., and Gnanadesikan, A. (2007). A synthesis of global particle export from the surface ocean and cycling through the ocean interior and on the seafloor. Global Biogeochem. Cycles 21, GB4006. doi: 10.1029/2006GB002907
Dupont, C. L., Moffett, J. W., Bidigare, R. R., and Ahner, B. A. (2006). Distributions of dissolved and particulate biogenic thiols in the subarctic Pacific Ocean. Deep. Res. Part I 53, 1961–1974. doi: 10.1016/j.dsr.2006.09.003
Eglinton, T. I., Irvine, J. E., and Vairavamurthy, A. (1994). Formation and diagenesis of macromolecular organic sulfur in Peru margin sediments. Organic Geochem. 22, 781–799. doi: 10.1016/0146-6380(94)90139-2
Ferdelman, T. G., Church, T. M., and Luther, G. W. III (1991). Sulfur enrichment of humic substances in a Delaware salt marsh sediment core. Geochim. Cosmochim. Acta 55, 979–988. doi: 10.1016/0016-7037(91)90156-Y
Francois, R. (1987). A study of sulphur enrichment in the humic fraction of marine sediments during early diagenesis. Geochim. Cosmochim. Acta 51, 17–27. doi: 10.1016/0016-7037(87)90003-2
Gasol, J. M., and Del Giorgio, P. A. (2000). Using flow cytometry for counting natural planktonic bacteria and understanding the structure of planktonic bacterial communities. Sci. Mar. 64, 197–224. doi: 10.3989/scimar.2000.64n2197
Gomez-Saez, G. V., Niggemann, J., Dittmar, T., Pohlabeln, A. M., Lang, S. Q., Noowong, A., et al. (2016). Molecular evidence for abiotic sulfurization of dissolved organic matter in marine shallow hydrothermal systems. Geochim. Cosmochim. Acta 190, 35–52. doi: 10.1016/j.gca.2016.06.027
Green, N. W., Perdue, E. M., Aiken, G. R., Butler, K. D., Chen, H., Dittmar, T., et al. (2014). An intercomparison of three methods for the large-scale isolation of oceanic dissolved organic matter. Mar. Chem. 161, 14–19. doi: 10.1016/j.marchem.2014.01.012
Hansell, D. A. (2013). Recalcitrant dissolved organic carbon fractions. Ann. Rev. Mar. Sci. 5, 421–445. doi: 10.1146/annurev-marine-120710-100757
Hedges, J. I., Keil, R. G., and Benner, R. (1997). What happens to terrestrial organic matter in the ocean? Org. Geochem. 27, 195–212.
Hertkorn, N., Harir, M., Koch, B. P., Michalke, B., and Schmitt-Kopplin, P. (2013). High-field NMR spectroscopy and FTICR mass spectrometry: Powerful discovery tools for the molecular level characterization of marine dissolved organic matter. Biogeosciences 10, 1583–1624. doi: 10.5194/bg-10-1583-2013
Jansen, S., Walpersdorf, E., Werner, U., Billerbeck, M., Böttcher, M. E., and de Beer, D. (2009). Functioning of intertidal flats inferred from temporal and spatial dynamics of O2, H2S and pH in their surface sediment. Ocean Dyn. 59, 317–332. doi: 10.1007/s10236-009-0179-4
Kok, M. D., Schouten, S., and Sinninghe Damsté, J. S. (2000). Formation of insoluble, nonhydrolyzable, sulfur-rich macromolecules via incorporation of inorganic sulfur species into algal carbohydrates. Geochim. Cosmochim. Acta 64, 2689–2699. doi: 10.1016/S0016-7037(00)00382-3
Krein, E. B., and Aizenshtat, Z. (1994). The formation of isoprenoid sulfur compounds during diagenesis: simulated sulfur incorporation and thermal transformation. Org. Geochem. 21, 1015–1025. doi: 10.1016/0146-6380(94)90065-5
Ksionzek, K. B., Lechtenfeld, O. J., McCallister, S. L., Schmitt-Kopplin, P., Geuer, J. K., Geibert, W., et al. (2016). Dissolved organic sulfur in the ocean: biogeochemistry of a petagram inventory. Science 354, 456–459. doi: 10.1126/science.aaf7796
Kujawinski, E. B., Del Vecchio, R., Blough, N. V., Klein, G. C., and Marshall, A. G. (2004). Probing molecular-level transformations of dissolved organic matter: insights on photochemical degradation and protozoan modification of DOM from electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Mar. Chem. 92, 23–37. doi: 10.1016/j.marchem.2004.06.038
Lechtenfeld, O. J., Koch, B. P., Geibert, W., Ludwichowski, K.-U., and Kattner, G. (2011). Inorganics in organics: quantification of organic phosphorus and sulfur and trace element speciation in natural organic matter using HPLC-ICPMS. Anal. Chem. 83, 8968–8974. doi: 10.1021/ac201765a
Levine, N. M. (2016). Putting the spotlight on organic sulfur. Science 354, 418–419. doi: 10.1126/science.aai8650
Maher, D. T., and Eyre, B. D. (2010). Benthic fluxes of dissolved organic carbon in three temperate Australian estuaries: implications for global estimates of benthic DOC fluxes. J. Geophys. Res. 115, G04039. doi: 10.1029/2010JG001433
Mossmann, J.-R., Aplin, A. C., Curtis, C. D., and Coleman, M. L. (1991). Geochemistry of inorganic and organic sulphur in organic-rich sediments from the Peru Margin. Geochim. Cosmochim. Acta 55, 3581–3595. doi: 10.1016/0016-7037(91)90057-C
Movassagh, B., and Shaygan, P. (2006). Michael addition of thiols to α,β-unsaturated carbonyl compounds under solvent-free conditions. ARKIVOC 12, 130–137. doi: 10.3998/ark.5550190.0007.c15
Nair, D. P., Podgóroski, M., Chatani, S., Gong, T., Xi, W., Fenoli, C. R., et al. (2014). The thiol-Michael addition click reaction: a powerful and widely used tool in materials chemistry. Chem. Mater. 26, 724–744. doi: 10.1021/cm402180t
Osterholz, H., Niggemann, J., Giebel, H.-A., Simon, M., and Dittmar, T. (2015). Inefficient microbial production of refractory dissolved organic matter in the ocean. Nat. Commun. 6, 1–8. doi: 10.1038/ncomms8422
Otto, S., and Balzer, W. (1998). Release of dissolved organic carbon (DOC) from sediments of the NW European Continental Margin (Goban Spur) and its significance for benthic carbon cycling. Prog. Oceanogr. 42, 127–144.
Pohlabeln, A. M., and Dittmar, T. (2015). Novel insights into the molecular structure of non-volatile marine dissolved organic sulfur. Mar. Chem. 168, 86–94. doi: 10.1016/S0079-6611(98)00031-7
Pretsch, E., Bühlmann, P., and Badertscher, M. (2009). Structure Determination of Organic Compounds. Heidelberg, Springer.
Quijada, M., Riboulleau, A., Faure, P., Michels, R., and Tribovillard, N. (2016). Organic matter sulfurization on protracted diagenetic timescales: the possible role of anaerobic oxidation of methane. Mar. Geol. 381, 54–66. doi: 10.1016/j.margeo.2016.08.010
Raven, M. R., Sessions, A. L., Adkins, J. F., and Thunell, R. C. (2016). Rapid organic matter sulfurization in sinking particles from the Cariaco Basin water column. Geochim. Cosmochim. Acta 190, 175–190. doi: 10.1016/j.gca.2016.06.030
Riedel, T., and Dittmar, T. (2014). A method detection limit for the analysis of natural organic matter via Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 86, 8376–8382. doi: 10.1021/ac501946m
Riedel, T., Zak, D., Biester, H., and Dittmar, T. (2013). Iron traps terrestrially derived dissolved organic matter at redox interfaces. Proc. Natl. Acad. Sci. U.S.A. 110, 10101–10105. doi: 10.1073/pnas.1221487110
Rossel, P. E., Vähätalo, A. V., Witt, M., and Dittmar, T. (2013). Molecular composition of dissolved organic matter from a wetland plant (Juncus effusus) after photochemical and microbial decomposition (1.25 yr): common features with deep sea dissolved organic matter. Org. Geochem. 60, 62–71. doi: 10.1016/j.orggeochem.2013.04.013
Røy, H., Seong Lee, J., Jansen, S., and de Beer, D. (2008). Tide-driven deep pore-water flow in intertidal sand flats. Limnol. Ocean. 53, 1521–1530. doi: 10.4319/lo.2008.53.4.1521
Roy, M., Rouxel, O., Martin, J. B., and Cable, J. E. (2012). Iron isotope fractionation in a sulfide-bearing subterranean estuary and its potential influence on oceanic Fe isotope flux. Chem. Geol. 300–301, 133–142. doi: 10.1016/j.chemgeo.2012.01.022
Santos, I. R., Eyre, B. D., and Huettel, M. (2012). The driving forces of porewater and groundwater flow in permeable coastal sediments: a review. Estuar. Coast. Shelf Sci. 98, 1–15. doi: 10.1016/j.ecss.2011.10.024
Schmidt, F., Elvert, M., Koch, B. P., and Witt, M. (2009). Molecular characterization of dissolved organic matter in pore water of continental shelf sediments. Geochim. Cosmochim. Acta 73, 3337–3358. doi: 10.1016/j.gca.2009.03.008
Schouten, S., de Graaf, W., Sinninghe Damsté, J. S., van Driel, G. B., and de Leeuw, J. W. (1994). Laboratory simulation of natural sulphurization: II. reaction of multi-functionalized lipids with inorganic polysulphides at low temperatures. Org. Geochem. 22, 825–834. doi: 10.1016/0146-6380(94)90142-2
Seidel, M., Beck, M., Riedel, T., Waska, H., Suryaputra, I. G. N. A., Schnetger, B., et al. (2014). Biogeochemistry of dissolved organic matter in an anoxic intertidal creek bank. Geochim. Cosmochim. Acta 140, 418–434. doi: 10.1016/j.gca.2014.05.038
Sinninghe Damsté, J. S., and de Leeuw, J. W. (1990). Analysis, structure and geochemical significance of organically-bound sulfur in the geosphere: state of the art and future research. Org. Geochem. 16, 1077–1101. doi: 10.1016/0146-6380(90)90145-P
Sinninghe Damsté, J. S., Rijpstra, W. I. C., Kock-van Dalen, A. C., de Leeuw, J. W., and Schenck, P. A. (1989). Quenching of labile functionalized lipids by inorganic sulphur species: evidence for the formation of sedimentary organic sulphur compounds at the early stages of diagenesis. Geochim. Cosmochim. Acta 53, 1343–1355. doi: 10.1016/0016-7037(89)90067-7
Sleighter, R. L., Chin, Y. P., Arnold, W. A., Hatcher, P. G., McCabe, A. J., McAdams, B. C., et al. (2014). Evidence of incorporation of abiotic S and N into prairie wetland dissolved organic matter. Environ. Sci. Technol. Lett. 1, 345–350. doi: 10.1021/ez500229b
Stubbins, A., Lapierre, J. F., Berggren, M., Prairie, Y. T., Dittmar, T., and Del Giorgio, P. A. (2014). What's in an EEM? Molecular signatures associated with dissolved organic fluorescence in boreal Canada. Environ. Sci. Technol. 48, 10598–10606. doi: 10.1021/es502086e
Vairavamurthy, M. A., Schoonen, M. A. A., Eglinton, T. I., Luther, G. W. III., and Manowitz, M. (1995). Geochemical Transformations of Sedimentary Sulfur. Washington, DC: American Chemical Society.
van Dongen, B. E., Schouten, S., Baas, M., Geenevasen, J. A. J., and Sinninghe Damsté, J. S. (2003). An experimental study of the low-temperature sulfurization of carbohydrates. Org. Geochem. 34, 1129–1144. doi: 10.1016/S0146-6380(03)00060-3
Keywords: dissolved organic matter (DOM), dissolved organic sulfur (DOS), sulfurization, structural analysis, sulfidic sediments
Citation: Pohlabeln AM, Gomez-Saez GV, Noriega-Ortega BE and Dittmar T (2017) Experimental Evidence for Abiotic Sulfurization of Marine Dissolved Organic Matter. Front. Mar. Sci. 4:364. doi: 10.3389/fmars.2017.00364
Received: 15 June 2017; Accepted: 30 October 2017;
Published: 15 November 2017.
Edited by:
Christian Lønborg, Australian Institute of Marine Science, AustraliaReviewed by:
Krista Longnecker, Woods Hole Oceanographic Institution, United StatesMorgan Reed Raven, Washington University in St. Louis, United States
Joanna D. Kinsey, North Carolina State University, United States
Copyright © 2017 Pohlabeln, Gomez-Saez, Noriega-Ortega and Dittmar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gonzalo V. Gomez-Saez, gonzalo.gomez@uni-oldenburg.de
Thorsten Dittmar, thorsten.dittmar@uni-oldenburg.de