A Dormant Microbial Component in the Development of Preeclampsia
 Douglas B. Kell1,2,3*
Douglas B. Kell1,2,3*  Louise C. Kenny4,5
Louise C. Kenny4,5
- 1School of Chemistry, The University of Manchester, Manchester, UK
- 2The Manchester Institute of Biotechnology, The University of Manchester, Manchester, UK
- 3Centre for Synthetic Biology of Fine and Speciality Chemicals, The University of Manchester, Manchester, UK
- 4The Irish Centre for Fetal and Neonatal Translational Research (INFANT), University College Cork, Cork, Ireland
- 5Department of Obstetrics and Gynecology, University College Cork, Cork, Ireland
Preeclampsia (PE) is a complex, multisystem disorder that remains a leading cause of morbidity and mortality in pregnancy. Four main classes of dysregulation accompany PE and are widely considered to contribute to its severity. These are abnormal trophoblast invasion of the placenta, anti-angiogenic responses, oxidative stress, and inflammation. What is lacking, however, is an explanation of how these themselves are caused. We here develop the unifying idea, and the considerable evidence for it, that the originating cause of PE (and of the four classes of dysregulation) is, in fact, microbial infection, that most such microbes are dormant and hence resist detection by conventional (replication-dependent) microbiology, and that by occasional resuscitation and growth it is they that are responsible for all the observable sequelae, including the continuing, chronic inflammation. In particular, bacterial products such as lipopolysaccharide (LPS), also known as endotoxin, are well known as highly inflammagenic and stimulate an innate (and possibly trained) immune response that exacerbates the inflammation further. The known need of microbes for free iron can explain the iron dysregulation that accompanies PE. We describe the main routes of infection (gut, oral, and urinary tract infection) and the regularly observed presence of microbes in placental and other tissues in PE. Every known proteomic biomarker of “preeclampsia” that we assessed has, in fact, also been shown to be raised in response to infection. An infectious component to PE fulfills the Bradford Hill criteria for ascribing a disease to an environmental cause and suggests a number of treatments, some of which have, in fact, been shown to be successful. PE was classically referred to as endotoxemia or toxemia of pregnancy, and it is ironic that it seems that LPS and other microbial endotoxins really are involved. Overall, the recognition of an infectious component in the etiology of PE mirrors that for ulcers and other diseases that were previously considered to lack one.
Introduction
Preeclampsia
Preeclampsia is a multisystem disorder of pregnancy, characterized and indeed defined by the presence of hypertension after 20 weeks’ gestation and before the onset of labor, or postpartum, with either proteinuria or any multisystem complication (1–9). It is a common condition, affecting some 3–5% of nulliparous pregnant women (8, 10) and is characterized by high mortality levels (11–14). There is no known cure other than delivery, and consequently, preeclampsia (PE) also causes significant perinatal morbidity and mortality secondary to iatrogenic prematurity. There are a variety of known risk factors (Table 1) that may be of use in predicting a greater likelihood of developing PE, albeit there are so many, with only very modest correlations, that early-stage (especially the first-trimester) prediction of late-stage PE remains very difficult (8, 15–17).
It is striking that most of the “risk factors” of Table 1 are, in fact, risk factors for multiple vascular or metabolic diseases, i.e., they merely pre-dispose the individual to a greater likelihood of manifesting the disease or syndrome (in this case PE). Indeed, some of them are diseases. This would be consistent with the well-known comorbidities, e.g., between PE and later cardiovascular disease [e.g., Ref. (55–65)], between PE and intracerebral hemorrhage during pregnancy [OR 10.39 (66)], and between PE and stroke postpartum (67, 68). The penultimate row of Table 1 lists a series of diseases that amount to comorbidities, although our interest was piqued by the observation that one-third of patients with antiphospholipid syndrome have PE, and infectious agents with known cross-reacting antigens are certainly one original (external) source of the triggers that cause the antiphospholipid antibodies (43, 44, 47, 69) (and see below). Similarly, in the case of urinary tract infection (UTI), the “risk” factor is a genuine external trigger, a point [following the call by Mignini and colleagues (70) for systematic reviews] that we shall expand on considerably here. [A preprint has been lodged at bioRxiv (71).]
In recent decades, intense investigation has led to the development of a two-stage etiological model for PE, first proposed by Redman (72), in which inadequate remodeling of the spiral arteries in early gestation results in poor placental development (stage one) and the resultant ischemia/reperfusion injury and oxidative stress (73) eventually leads to maternal vascular endothelial cell dysfunction and the maternal manifestations of the disease (stage 2) (72, 74–77). However, many clinical inconsistencies challenge the simplicity of this model. For example, while the association between poor placentation and PE is well established, it is not specific. Poor placentation and fetal growth restriction (FGR) frequently present without maternal signs of PE. Moreover, FGR is not a consistent feature of PE. While it is commonly seen in PE presenting at earlier gestations, in PE presenting at term, neonates are not growth restricted and may even be large for dates (78).
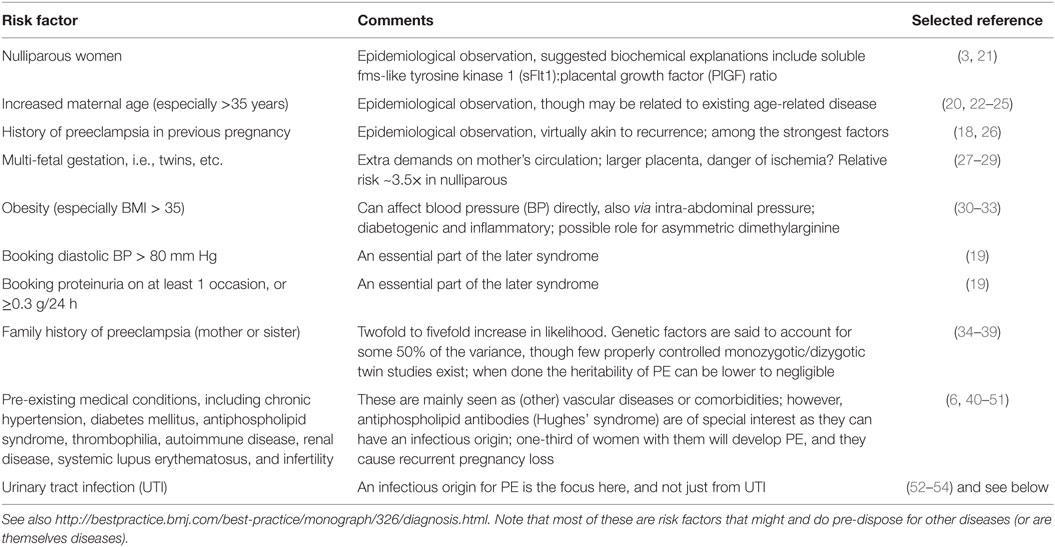
Thus, the two-stage model has been further refined by Roberts and others (77, 79, 80) to take into account the heterogeneous nature of PE and the varying contribution from mother and infant to the disorder. We now appreciate that normal pregnancy is characterized by a low-grade systemic inflammatory response and specific metabolic changes, and that virtually all of the features of normal pregnancy are simply exaggerated in PE (81–83). There is also widespread acceptance that maternal constitutional and environmental factors (such as obesity) can interact to modulate the risk of PE. Thus, with profoundly reduced placental perfusion (or significant “placental loading”), the generation of stage 2 may require very little contribution from the mother to provide sufficient stress to elicit the maternal syndrome. In this setting, almost any woman will develop PE. Conversely, the woman with extensive predisposing constitutional sensitivity could develop PE with very little reduced perfusion, or minimal “placental loading.” As with many complex disorders, multiple factors can affect disease development positively or negatively, with a convenient representation of the two main negative sources (fetal and maternal) being that of a see-saw (84), as in Figure 1.
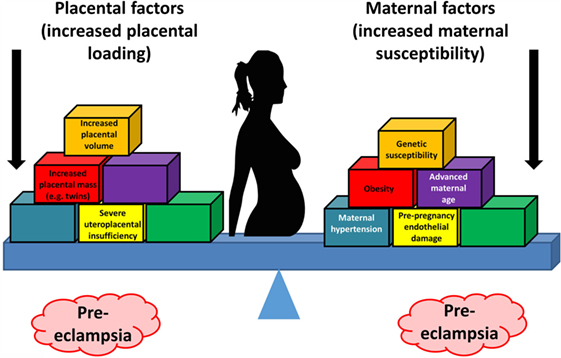
Figure 1. Two main sources (fetal and maternal) can drive a pregnancy toward preeclampsia.
While this explains the inconsistencies of the two-stage model, the precise mechanisms (1) underlying the initial poor placentation and (2) linking placental stress and the maternal syndrome have still not been fully elucidated.
Much recent research in PE has focused on various angiogenic factors, including the pro-angiogenic factors vascular endothelial growth factor (VEGF) and placental growth factor (PlGF) and the two anti-angiogenic proteins, such as soluble endoglin (sEng) and soluble fms-like tyrosine kinase 1 (sFlt-1). Recent data suggest that alterations in circulating angiogenic factors play a pathogenic role in PE. These angiogenic factors tightly regulate angiogenesis and are also essential for maintenance of normal vessel health. Consequently, the synthesis and action of these factors and their receptors in the uterine bed and placenta are essential for normal placental development and pregnancy (85, 86). In PE, increased levels of the anti-angiogenic sFlt-1 and sEng trap circulating VEGF, PlGF, and transforming growth factor-β (TGF-β), respectively. A myriad of data support the idea that circulating levels of these factors alone, or in combination, can be used to predict PE (87–89) (and see below under PE biomarkers), but in line with the heterogeneous nature of PE, these data are somewhat inconsistent and their performance as biomarkers seems limited to disease with significant placental loading (8). Therefore, angiogenic dysregulation would appear unlikely to be the sole link between the stressed placenta and endothelial dysfunction and the clinical manifestations of the disease.
Notwithstanding these many inconsistencies, the central role of the placenta as a source of “toxin,” in a condition regarded, and indeed often named, as “toxemia of pregnancy” (90–92) cannot be refuted. The uncertainty regarding the nature of the toxin continues, and other placental sources of endothelial dysfunction include syncytiotrophoblast basement membrane fragments (STBM) (93) and endothelial progenitor cells (EPC) (94); an increase of reactive oxygen species over scavenging by antioxidants (95, 96) has also been promoted.
The Bradford Hill criteria for causation of a disease Y by an environmental factor X (97) are as follows: (1) strength of association between X and Y, (2) consistency of association between X and Y, (3) specificity of association between X and Y, (4) experiments verify the relationship between X and Y, (5) modification of X alters the occurrence of Y, and (6) biologically plausible cause and effect relationship.
In general terms (98), if we see that two things (A and B) co-vary in different circumstances, we might infer that A causes B, that B causes A, or that something else (C) causes both B and A, whether in series or parallel. To disentangle temporal relations requires a longitudinal study. The job of the systems biologist doing systems medicine is to uncover the chief actors and the means by which they interact (99), in this way fulfilling the Bradford Hill postulates, a topic to which we shall return at the end.
In infection microbiology, and long predating the Bradford Hill criteria, the essentially equivalent metrics are known [widely, but somewhat inaccurately (100)] as the Koch or Henle–Koch postulates (i.e., criteria). They involve assessing the correlation of a culturable organism with the presence of a disease, the cure of the disease (and its symptoms) upon removal of the organism, and the development of the disease with (re)inoculation of the organism. They are of great historical importance but present us with three main difficulties here. The first is that we cannot apply the third of them to humans for obvious ethical reasons. The second (see also below) and related one is that we cannot usefully apply them in animal models because none of the existing models recapitulates human PE well. Finally, as widely recognized (100–107), they cannot be straightforwardly applied when dealing with dormant bacteria or bacteria that are otherwise refractory to culture.
Our solution to this is twofold: (i) we can assess the first two using molecular methods if culturing does not work and (ii) we exploit the philosophy of science principle known as “coherence” (108–112). This states that if a series of ostensibly unrelated findings are brought together into a self-consistent narrative and that narrative is thereby strengthened. Our systems approach purposely represents a “coherence” in the sense given.
Overall, known biochemical associations with PE come into four main categories, such as abnormal trophoblast invasion, oxidative stress, inflammation and altered immune response, and anti-angiogenic responses (Figure 2). Each of these can contribute directly to PE, and although they can interact with each other (black arrows), no external or causal source is apparent. Figure 2 has been redrawn from a very nice review by Pennington and colleagues (113), which indicates four main generally accepted “causes” (or at least accompaniments) of PE as the four outer colored circles. As illustrated with the black two-way arrows, many of these also interact with each other. What is missing, in a sense, is then what causes these causes, and that is the nub of our argument here. Since we now know (and describe below) that microbes can affect each of these four general mechanisms, we have added these routes to Figure 1 (using pink arrows) where dormant, resuscitating, or growing microbes are known to contribute.
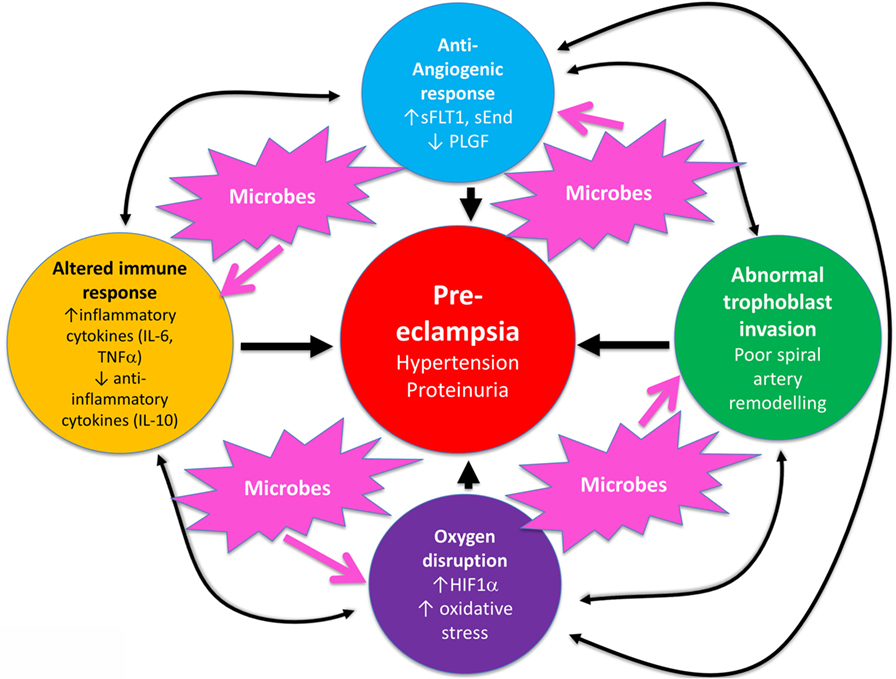
Figure 2. There are four main “causes” of preeclampsia, represented by the colored outer circles, and these can also interact with each other. That part of the figure is redrawn from Pennington et al. (113). In addition, we note here, as the theme of this review, that microbes can themselves cause each of the features in the outer colored circles to manifest.
In a similar vein, Magee and colleagues (114) have nicely set down their related analysis of the causes and consequences of PE, with a central focus (redrawn in Figure 3) on endothelial cell activation. While bearing much similarity in terms of overall content to the analysis of Pennington and colleagues (113), and ours above, it again lacks a microbial or infection component as a causative element, but importantly does note that infection and/or inflammation can serve to lower the threshold for PE in cases of inadequate placentation. In our view, microbes can also enter following normal placentation if their dormant microbiome begins to wake up and/or to shed inflammagens.
Figure 3. Another detailed representation of factors known to cause or accompany PE, redrawn from Magee et al. (114).
Heritability
The question of the extent of heritability of PE (susceptibility) is of interest. Although this seems to vary widely in different studies (Table 1), a number of candidate gene studies (37, 115–118) imply that a susceptibility to PE is at least partly heritable, consistent with the variance in all the other “risk factors” of Table 1 [and see Ref. (6)]. As with all the other gene association studies where phenotypic (“lifestyle”) information is absent (119–121), it is not possible to ascribe the heritability to genetics alone, as opposed to an interaction of a genetic susceptibility (e.g., in the HLA system) with environmental factors (117), such as cytomegalovirus infection (122).
Inflammation
Preeclampsia is accompanied by oxidative stress (123, 124) and inflammation and, thus, shares a set of observable properties with many other (and hence related) inflammatory diseases, be they vascular (e.g., atherosclerosis), neurodegenerative (e.g., Alzheimer’s, Parkinson’s), or “metabolic” (type 1 and 2 diabetes). It is thus at least plausible that they share some common etiologies, as we argue here, and that knowledge of the etiology of these diseases may give us useful clues for PE.
As well as raised levels of inflammatory cytokines that constitute virtually a circular definition of inflammation, we and others have noted that all of these diseases are accompanied by dysregulation of iron metabolism (84, 125, 126), hypercoagulability and hypofibrinolysis (127, 128), blood microparticles (126), and changes in the morphology of fibrin fibers [e.g., Ref. (129–134)] and of erythrocytes [e.g., Ref. (127, 132–137)].
In addition, we and others have recognized the extensive evidence for the role of a dormant blood and/or tissue microbiome in these (138–143) and related (144–147) diseases, coupled in part with the shedding of highly inflammagenic bacterial components such as Gram-negative lipopolysaccharide (LPS) and their Gram-positive cell wall equivalents such as lipoteichoic acids (148). (We shall often use the term “LPS” as a “shorthand,” to be illustrative of all of these kinds of highly inflammagenic molecules.)
The purpose of this review, outlined as a “mind map” in Figure 4, is thus to summarize the detailed and specific lines of evidence suggesting a very important role of a dormant microbial component in the etiology of PE [and see also Ref. (138)]. To do this, we must start by rehearsing what is meant by microbial dormancy.
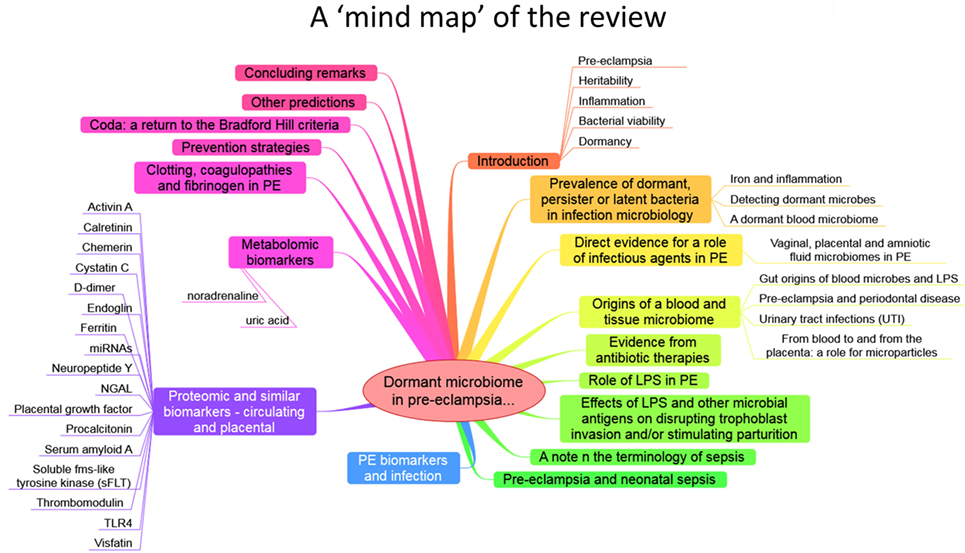
Figure 4. A mind map of the overall structure of the review.
Bacterial Viability
In microbiology, we usually consider microbes as being in one of the three “physiological macrostates” (Figure 5). The definition of a “viable” bacterium is normally based on its ability to replicate, i.e., “viability” = culturability (149–151). In this sense, classical microbiology has barely changed since the time of Robert Koch, with the presence of a “viable” microorganism in a sample being assessed via its ability to form a visible colony on an agar plate containing suitable nutrients. However, it is well known, especially in environmental microbiology [“the great plate count anomaly” (152)], that only a small percentage of cells observable microscopically is typically culturable on agar plates. In principle, this could be because they are/were “irreversibly” non-culturable (operationally “dead”), or because our culture media either kill them (153) or such media lack nutrients or signaling molecules necessary for their regrowth (154, 155) from an otherwise dormant state (156, 157). These statements are true even for microbes that appear in culture collections and (whose growth requirements) would be regarded as “known.”
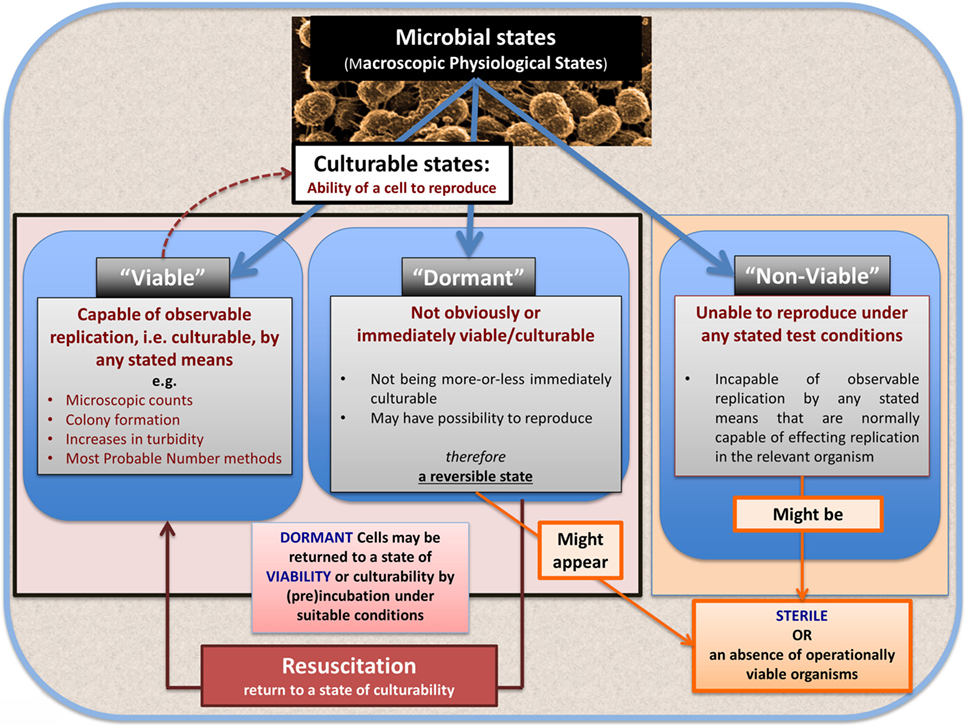
Figure 5. The chief physiological macrostates exhibited by microorganisms.
However, it is common enough in clinical microbiology that we detect the existence or presence of “novel” microbial pathogens with obscure growth requirements before we learn to culture them; this is precisely what happened in the case of Legionella pneumophila (158–161), Tropheryma whipplei [Whipple’s disease (162, 163)], and Coxiella burnetii [the causative agent of Q fever (164, 165)]. Even Helicobacter pylori was finally brought into culture on agar plates only because an unusually long Easter holiday break meant that the plates were incubated for an extended period of 5 days (rather than the normal 2) before being thrown out (166, 167)! Consequently, there is ample precedent for the presence of “invisible” microbes to go unremarked before they are discovered as the true cause of a supposedly non-infectious disease, even when they are perfectly viable (culturable) according to standard analyses.
Dormancy
Dormancy for a microbe is defined operationally as a state, commonly of low metabolic activity, in which the organism appears not to be viable in that it is unable to form a colony but where it is not dead in that it may revert to a state in which it can do so, via a process known as resuscitation (156, 157). However, an important issue (and see above) is that dormant bacteria do not typically fulfill the Koch–Henle postulates (100–103), and in order for them to do so it is necessary that they be grown or resuscitated. This is precisely what was famously done by Marshall and Warren when they showed that the supposedly non-infectious disease of gastric ulcers was, in fact, caused by a “novel” organism called H. pylori (168, 169). One of the present authors showed in laboratory cultures of actinobacteria that these too could enter a state of true dormancy (170, 171) [as is well known for Mycobacterium tuberculosis, e.g., Ref. (172–176)], and could be resuscitated by a secreted growth factor called Rpf (177–181). This RPF family has a very highly conserved motif that is extremely immunogenic (182, 183), and it is presently under trials as a vaccine against M. bovis.
Prevalence of Dormant, Persistent, or Latent Bacteria in Infection Microbiology
It is worth stressing here that the presence of dormant or latent bacteria in infection microbiology is well established; one-third of humans carry dormant M. tuberculosis [e.g., Ref. (175, 184–187)], most without reactivation, while probably 50–100% are infected with H. pylori, most without getting ulcers or worse (188, 189). As with the risk factors in Table 1, the organisms are merely or equivalently “risk factors” for these infectious diseases and are effectively seen as causative only when the disease is actually manifest.
In a similar vein, so-called persisters are phenotypic variants of infectious microbes that resist antibiotics and can effectively lie in hiding to resuscitate subsequently. This is also very well established [e.g., Ref. (139, 190–203)]. In many cases, they can hide intracellularly (204), where antibiotics often penetrate poorly (205) because the necessary transporters (206–209) are absent. This effectively provides for reservoirs of reinfection, e.g., for Staphylococcus aureus (210), Bartonella spp. (211), and – most pertinently here – for the Escherichia coli involved in urinary tract (re)infection (212–215). The same intracellular persistence is true for parasites such as Toxoplasma gondii (216).
Thus, the main point of the extensive prevalence of microbial dormancy and persistence is that microbes can appear to be absent when they are, in fact, present at high concentrations. This is true not only in cases where infection is recognized as the cause of disease but, as we here argue, such microbes may be an important part of diseases presently thought to lack an infectious component.
Iron and Inflammation
It is well known that [with the possible exception of Borrelia (217, 218)] a lack of free iron normally limits microbial growth in vivo [e.g., Ref. (219–243)], and we have reviewed previously (84, 125, 126) the very clear iron dysregulation accompanying PE [e.g., Ref. (90, 244–256)].
This has led to the recognition (128, 139, 141) that the source of the continuing inflammation might be iron-based resuscitation of dormant microbes that could release well known and highly potent inflammagens such as LPS. Indeed, we have shown that absolutely tiny (highly substoichiometric) amounts of LPS can have a massive effect on the blood clotting process (257), potentially inducing β-amyloid formation directly (258, 259) [something, interestingly, that can be mimicked in liquid crystals (260, 261)]. The overall series of interactions envisaged [see also Kell et al. (139)] is shown in Figure 6.
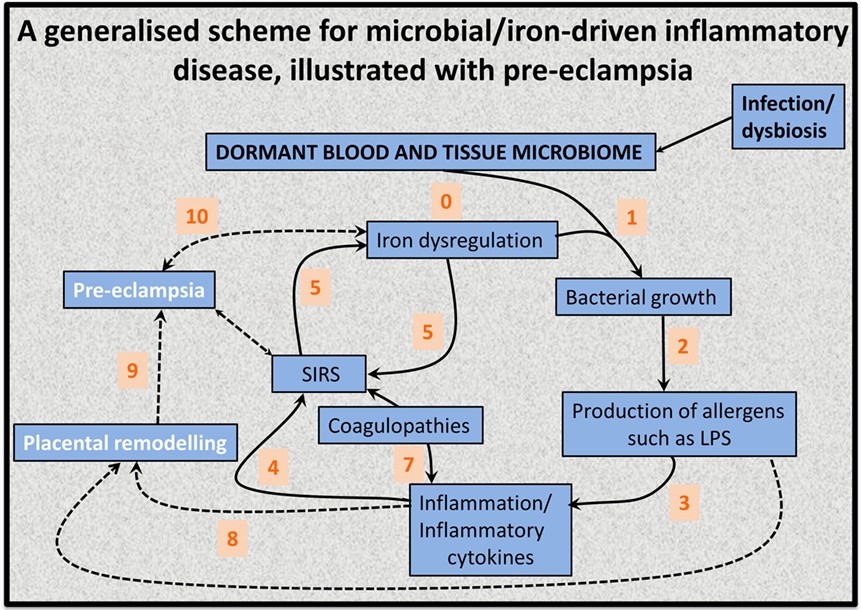
Figure 6. An 11-stage systems biology model of the factors that we consider cause initially formant microbes to manifest the symptoms (and disease) of preeclampsia.
As pointed out by a referee, worldwide, iron deficiency anemia is associated with increased perinatal morbidity and mortality, predominantly in low resource settings. This is most recently and comprehensively reviewed by Rahman and colleagues (262). This meta-analysis (262) found that in low- and middle-income countries, maternal hemoglobin concentrations <10 or <11 g dL−1 or hematocrit values <33 or <34% accounted for 12% of low birth weight, 19% of preterm births (PTBs), and 18% of perinatal mortality. However, the independent contribution of iron deficiency is difficult to define, especially since in low resource settings anemia is often not found in isolation but as part of a wider spectrum of nutritional insufficiencies (especially vitamins and folic acid) or as a result of parasitic infection. These can increase susceptibility to other insults such a hemorrhage or infection, also common in low resources settings. In addition, the exact speciation and hence availability of iron may be important (not just the total amount). Thus, the hypothesis presented here is that iron in excess or under certain physiological conditions can be, and indeed is, equally detrimental to maternal health. There is a likely a “Goldilocks” concentration of iron which is optimal for maternal and neonatal health, and the authors are proposing the selected use of treatments such as statins or iron chelators in the at-risk population, rather than as a broad panacea. Furthermore, the use of statins to prevent pregnancy complications is far from controversial (see also below). There are at least two registered current trials of statins to prevent PE (ISRCTN17787139 and ISRCTN23410175).
Detecting Dormant Microbes
By definition, dormant bacteria escape detection by classical methods of assessing viability that involve replication on agar plates. Other growth-associated methods include measurements involving changes in turbidity (263), including an important but now rather uncommon technique referred to as the “most probable number” (MPN). The MPN involves diluting samples serially and assessing by turbidity changes in the presence of growth/no growth. Look-up tables based on Poisson statistics enable estimation of the number of cells or propagules that were present. A particular virtue is that they allow dormant and “initially viable” cells to be discriminated via “dilution to extinction” (171), thereby avoiding many artifacts (157). As mentioned earlier, preincubation in a weak nutrient broth (171, 264) was instrumental in allowing the discovery (177) of an autocrine “wake-up” molecule necessary for the growth of many actinobacteria.
Other more classical means of detecting microbes, but not whether they were culturable, involved microscopy (190, 265–268) or flow cytometry (269) with or without various stains that reflected the presence or otherwise of an intact cell wall/membrane (170, 270–277). These stains are sometimes referred to as “viability” stains, but this is erroneous as they do not measure “culturability.” Readers may also come upon the term “viable-but-not-culturable”; however, since viable = culturable, this is an oxymoron that we suggest is best avoided (157). Other methods involved measurement of microbial products, e.g., CO2 (278, 279), or changes in the conductivity or impedance of the growth medium (263, 280–282).
Most importantly, however, dormant (as well as culturable) cells may be detected by molecular means, nowadays most commonly through PCR and/or sequencing of the DNA encoding their small subunit ribosomal RNA (colloquially “16S”) (283–297) or other suitable genes. It is clear that such methods will have a major role to play in detecting, identifying, and quantifying the kinds of microbes that we argue lie at the heart of PE etiology.
A Dormant Blood Microbiome
Of course, actual bacteremia, the presence of replicable bacteria in blood, is highly life-threatening (298), but – as emphasized – viability assays do not detect dormant bacteria. When molecular detection methods are applied to human blood, it turns out that blood does indeed harbor a great many dormant bacteria [e.g., Ref. (299–309)]; they may also be detected ultramicroscopically [e.g., Ref. (139–141, 190, 267, 300, 310)] or by flow cytometry (311), and dormant blood and tissue microbes probably underpin a great many chronic, inflammatory diseases normally considered to lack a microbial component (139–141, 144–147, 190, 267, 268, 302, 312–321). Multiple arguments serve to exclude “contaminants” as the source of the bacterial DNA (141): (1) there are significant differences between the blood microbiomes of individuals harboring disease states and nominally healthy controls, despite the fact that samples are treated identically; (2) the morphological type of organism (e.g., coccus vs. bacillus) seems to be characteristic of particular diseases; (3) in many cases, relevant organisms lurk intracellularly, which is hard to explain by contamination; (4) there are just too many diseases where bacteria have been found to play a role in the pathogenesis, that all of them may be caused by contamination; (5) the actual numbers of cells involved seem far too great to be explicable by contamination; given that blood contains ~5 × 109 erythrocytes mL−1, if there was just one bacterial cell per 50,000 erythrocytes this will equate to 105 bacteria mL−1. These are big numbers, and if the cells were culturable, that number of cells would be the same as that ordinarily defining bacteriuria.
A recent study by Damgaard and colleagues (306) is of particular interest here. Recognizing the strong mismatch between the likelihood of an infection post-transfusion [very high (306)] and the likelihood of detecting culturable microbes in blood bank units (negligible, ca. 0.1%) (306, 322), Damgaard et al. reasoned that our methods of detecting and culturing these microbes might be the problem. Certainly, taking cells from a cooled blood bag and placing them onto an agar plate at room temperature that is directly exposed to atmospheric levels of gaseous O2 is a huge stress leading to the production of “reactive oxygen species” (125, 323), that might plausibly kill any dormant, injured, or even viable microbes. Thus, they incubated samples from blood on a rich medium (trypticase soy agar) for a full week, both aerobically and anaerobically. Subsequent PCR and sequencing allowed them to identify specific microbes in some 35–53% of the samples. Thus, very careful methods need to be deployed to help resuscitate bacteria from physiological states that normally resist culture, even when these bacteria are well-established species. This is very much beginning to happen in environmental microbiology [e.g., Ref. (154, 324–326)], and such organisms are rightly seen as important sources of novel bioactives (327, 328).
As reviewed previously (139–143), the chief sources of these blood microbes are the gut microbiome, the oral microbiome [periodontitis (329)], and via UTIs. Consequently, if we are to argue that there is indeed a microbial component to PE, we should expect to see some literature evidence for it (53, 54, 138, 330–332). In what follows we shall rehearse the fact that it is voluminous.
Direct Evidence for a Role of Infectious Agents in PE
Although we recognize that many of the more molecular methods cannot distinguish culturable from dormant microbes, quite a number of studies have explicitly identified infection as a cause of PE (Table 2). The commonest microbe seems to be H. pylori; while it is most famously associated with gastric ulcers (168, 169, 333), there are many other extragastric manifestations [e.g., Ref. (334–342)]. The odds ratio of no less than 26 in PE vs. controls when the strains can produce CagA antigens is especially striking, not least because it provides a mechanistic link to poor trophoblast invasion via a mechanism involving host antibodies to CagA cross-reacting with trophoblasts (343, 344), and circulating (345) in microparticles (346) or endosomes (347, 348).
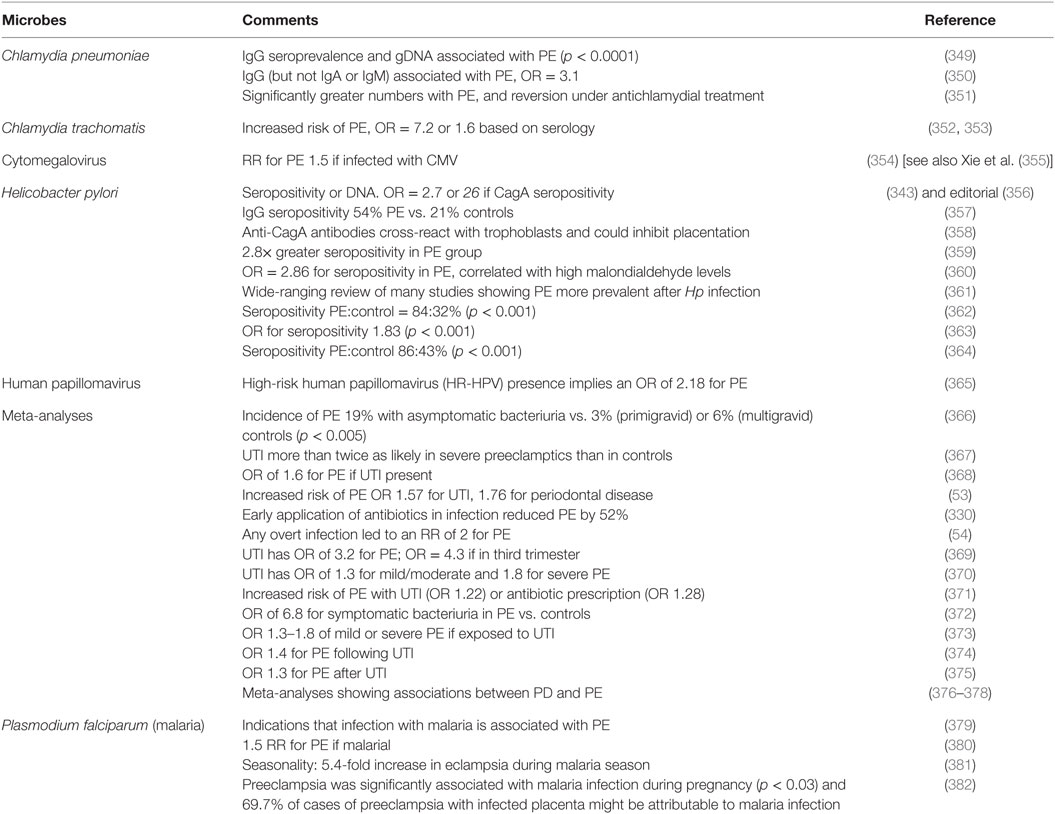
Table 2. Many studies have identified a much greater prevalence of infectious agents in the blood or urine of these exhibiting PE than in matched controls.
In contrast to the situation in PE, albeit severe PE is associated with iatrogenic PTBs, there is a widespread recognition [e.g., Ref. (383–410)] that infection is a common precursor to PTB in the absence of PE. The failure of antibiotics to help can be ascribed to their difficulty of penetrating to the trophoblasts and placental regions. Unfortunately, no proteomic biomarkers have yet been observed as predictive of PTB (411, 412). In a similar vein, and if we are talking about a time of parturition that is very much more “preterm,” we are in the realm of miscarriages and spontaneous abortions and stillbirths, where infection again remains a major cause (413–416). Here, we note that early or pre-emptive antibiotic therapy has also proved of considerable value in improving outcomes after multiple spontaneous abortions (417).
Vaginal, Placental, and Amniotic Fluid Microbiomes in PE
It might be natural to assume that the placenta is a sterile organ, like blood is supposed to be. However, various studies [modulo the usual issues of contamination (418)] have shown the presence of microbes in tissues including the placenta (400, 409, 419–432), vagina (393, 433–440), uterus (391, 441, 442), amniotic fluid (430, 443–448), and follicular fluid (449, 450), and how these may vary significantly in PE [we do not discuss other pregnancy disorders such as small for gestational age (SGA) and intrauterine growth restriction (IUGR)]. We list some of these in Table 3.

Table 3. Evidence for microbes in placental tissues, including those with PE.
Origins of a Blood and Tissue Microbiome
As assessed previously (139–141) over a large literature, the chief source of blood microbes is the gut (426), with another major entry point being via the oral microbiome (especially in periodontitis, see below). For rheumatoid arthritis (142, 458–460) and diseases of pregnancy, UTI (see below and Table TT) also provides a major source.
Gut Origins of Blood Microbes and LPS
We have recently rehearsed these issues elsewhere (139–141), so a brief summary will suffice. Clearly, the gut holds trillions of microbes, with many attendant varieties of LPS (461), so even low levels of translocation [e.g., Ref. (462–464)], typically via Peyer’s patches and M cells, provide a major source of the blood microbiome. This may be exacerbated by intra-abdominal hypertension and overeating (465–467) that can indeed stimulate the translocation of LPS (468). For reasons of space and scope, we do not discuss the origins and translocation of microbes in breast milk (469) nor the important question of the establishment of a well-functioning microbiome in the fetus and neonate (470), and the physiological role of the mother therein.
Preeclampsia and Periodontal Disease
One potential origin of microbes that might be involved in, or represent a major cause of, PE is the oral cavity, and in particular when there is oral disease (such as periodontitis and gum bleeding) that can allow microbes to enter the bloodstream. If this is a regular occurrence one would predict that PE would be much more prevalent in patients with pre-existing periodontitis [but cf. Ref. (471) for those in pregnancy] than in matched controls; this is indeed the case (Table 4). As with many of the tables herein, the odds ratios are far beyond anything that might weakly be referred to as an “association.”

Table 4. Periodontal disease (PD) and preeclampsia.
Urinary Tract Infections
A particular feature of UTIs is the frequency of reinfection (497–504). This is because the organisms can effectively “hide” in bladder epithelial cells as the so-called “quiescent intracellular reservoirs” (212, 501, 503, 505–509) of (presumably) dormant cells that can resuscitate. This is why reinfection is often from the same strains that caused the original infection (510–514). Other complications can include renal scarring (515). Bacteriuria (often asymptomatic) is a frequent occurrence in pregnancy [e.g., Ref. (373, 375, 473, 516–522)], and the frequency of UTI as a source of microbes causing PE is clear from Table 2.
From Blood to and from the Placenta: A Role for Microparticles
We and others have noted the fact that many chronic, inflammatory disease are accompanied by the shedding of various antigens and other factors; typically they pass through the bloodstream as microparticles (126, 140, 523–530), sometimes known as endosomes (345, 347, 348, 524, 531) [and see later under microRNAs (miRNAs)]. Similarly, LPS is normally bound to proteins such as the LPS-binding protein and apoE (140). Given their prevalence, their role in simply finding their way from maternal blood to placenta, and the fact that we discussed them extensively in two previous reviews (126, 140), we do not discuss them further here.
Evidence from Antibiotic Therapies
Antibiotic drug prescriptions (532–534) may be seen as a proxy for maternal infection, so if dormant (and resuscitating and growing) bacteria are a major part of PE etiology one might imagine an association between antibiotic prescriptions and PE. According to an opposite argument, antibiotics and antibiotic prescriptions given for nominally unrelated infections (UTI, chest, etc., and in particular diseases requiring long-term anti-infective medication that might even last throughout a pregnancy) might have the beneficial side-effect of controlling the proliferation of dormant cells as they seek to resuscitate. There is indeed some good evidence for both of these, implying that it is necessary to look quite closely at the nature, timing, and duration of the infections and of the anti-infective therapy relative to pregnancy. A summary is given in Table 5. A confounding factor can be that some (e.g., the antiretroviral) therapies are themselves quite toxic (535, 536); while the OR for avoiding PE was 15.3 in one study of untreated HIV-infected individuals vs. controls, implying (as is known) a strong involvement of the immune system in PE, the “advantage” virtually disappeared upon triple-antiretroviral therapy (537). Overall, it is hard to draw conclusions from antiretrovirals (538, 539). However, we have included one HIV study in the table. Despite a detailed survey, we found no reliable studies with diseases such as Lyme disease or tuberculosis, where treatment regimens are lengthy, that allowed a fair conclusion as to whether antibiotic treatment was protective against PE. However, we do highlight the absolutely stand-out study of Todros and colleagues (540), who noted that extended spiramycin treatment (of patients with T. gondii) gave a greater than 10-fold protection against PE, when the parasite alone had no effect (541). This makes such an endeavor (assessing the utility of early or pre-emptive antibiotics in PE) potentially highly worthwhile.

Table 5. Examples of decreased PE following antibiotic therapies given for various reasons.
Role of LPS in PE
It is exceptionally well known that LPS (sensu lato) is highly inflammagenic, and since one of us recently reviewed that literature in extenso (140) this is not directly rehearsed here. However, since we are arguing that it has a major role in PE naturally or in vivo, we do need to ask whether the literature is consistent with this more focused question. The answer is, of course, a resounding “yes.” Notwithstanding that only primates, and really only humans, are afflicted by “genuine” PE, so the genuine utility of rodent models is questionable (543), even if some can recapitulate elements of the disease (544, 545). Hence, it is somewhat ironic that there are a number of animal models in which LPS (also known as “endotoxin”) is used experimentally to induce a condition resembling PE [e.g., Ref. (546–551) and also see Ref. (552)]. We merely argue that it is not a coincidence that exogenous administration of LPS has these effects, because we consider that it is, in fact, normally one of the main mediators of PE. Also note, in the context of gestational diabetes, that serum levels of LPS are raised significantly in both type 1 (466, 553) and type 2 (554–556) diabetes.
The standard sequelae of LPS activation, e.g., TLR signaling and cytokine production, also occur in PE (557–559), bolstering the argument that this is precisely what is going on. In a similar vein, double-stranded RNA-mediated activation of TLR3 and TLR7/8 can play a key role in the development of PE (560–562). What is new here is our recognition that LPS and other inflammagens [e.g., Ref. (563–565)] may continue to be produced and shed by dormant and resuscitating bacteria that are generally invisible to classical microbiology.
Effects of LPS and Other Microbial Antigens on Disrupting Trophoblast Invasion and/or Stimulating Parturition
As with other cases of cross-reactivity such as that of various antigens in Proteus spp. that can cause disease in rheumatoid arthritis (458–460), the assumption is that various microbial antigens can lead to the production of (auto-)antibodies that attack the host, in the present case of interest by stopping the placentation by trophoblasts. This is commonly referred to as “molecular mimicry” [e.g., Ref. (566–569)] and may extend between molecular classes, e.g., peptide/carbohydrate (570, 571). Table 6 shows some molecular examples where this has been demonstrated.

Table 6. Molecular examples of bacterial antigens that can elicit antibodies that stop successful trophoblast implantation or stimulate parturition.
In many cases, the actual (and possibly microbial) antigens are unknown, and clearly the microbial elicitation of antibodies to anything that might contribute to PE points to multiple potential origins. To this end, we note that PE has also been associated with antibodies to angiotensin receptors (577–590), to smooth muscle (591, 592) [such blocking may be anti-inflammatory (593–595)], to adrenoceptors (596), to the M2 muscarinic receptor (597), and to Th17 (598) [and see Ref. (599)]. It is not unreasonable that epitope scanning of the antibody targets coupled with comparative sequence analysis of potential microbes might light up those responsible. In the case of angiotensin II type 1 receptor antibodies, the epitope is considered (600) to be AFHYESQ, an epitope that also appears on parvovirus B19 capsid proteins; in the event, parvoviruses seem not to be the culprits here (601). However, the role of these antibodies in activating the angiotensin receptor is also considered to underpin the lowering of the renin–angiotensin system that is commonly seen in PE (602–605), but which is typically raised during normal pregnancy.
Th-17 is of especial interest here, since these are the helper T (Th)-cell subset that produces IL-17. IL-17 is probably best known for its role in inflammation and autoimmunity (599, 606–610). However, it also has an important role in induction of the protective immune response against extracellular bacteria or fungal pathogens at mucosal surfaces (608, 611–623). Th17 cells seem to participate in successful pregnancy processes and can be lower in PE (624–626), although more studies show them as higher (599, 627–635) or unchanged (636, 637). One interpretation, consistent with the present thesis, is that the antimicrobial effects of placental IL-17 relative to Treg cells are compromised during PE (599, 633, 638).
A Note on the Terminology of Sepsis
As one may suppose from the name, sepsis (and the use of words like “antiseptic”) was originally taken to indicate the presence of culturable organisms in (or in a sample taken from) a host, e.g., as in bacteremia. Recognizing that it is the products of bacteria, especially cell wall components, that cause the cytokine storms that eventually lead to death from all kinds of infection (639–643), “sepsis” nowadays has more come to indicate the latter, as a stage (in the case of established infection) on a road that leads to septic shock and (eventually) to death [with a shockingly high mortality, and many failures of initially promising treatments, e.g., Ref. (644, 645), and despite the clear utility of iron chelation (84, 125, 646–648)]. In most cases, significant numbers of culturable microbes are either unmeasured or absent, and like most authors, we shall use “sepsis” to imply the results of an infection whether the organisms are detected or otherwise. Overall, it is possible to see the stages of PE as a milder form of the sepsis cascade on the left-hand side of Figure 7. Figure 7 compares the classical route of sepsis-induced death with the milder versions that we see in PE; they are at least consistent with the idea that PE is strongly related to the more classical sepsis in degree rather than in kind.
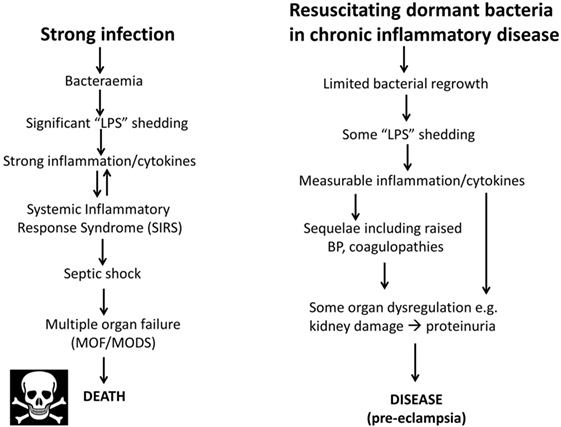
Figure 7. Preeclampsia bears some similarities to and may be considered as a milder form of, the changes that occur during genuine sepsis leading to a systematic inflammatory response syndrome, septic shock, and multiple organ dysfunction.
Preeclampsia and Neonatal Sepsis
If PE is really based on infectious agents, it is reasonable that one might expect to see a greater incidence of neonatal sepsis (i.e., infection) following PE. While there are clearly other possible explanations (e.g., simply a weakened immune system, sometimes expressed as neutropenia, after PE), there is certainly evidence that this is consistent with this suggestion (649–653).
PE Biomarkers and Infection
Because of the lengthy development of PE during pregnancy, there has long been a search for biomarkers (somewhat equivalent to the “risk factors” discussed earlier) that might have predictive power, and some of these, at both metabolome (15, 654–661) and proteome (662–667) level, are starting to come forward. The typical experimental design is a case–control, in which markers that are raised or lowered significantly relative to the age-matched controls are considered to be candidate markers of PE. However, just as noted with leukocyte markers (81) and polycystic ovary syndrome (PCOS) (668) that does not mean that they might not also be markers for other things too, such as infection (669)!
Thus, one prediction is that if dormant and resuscitating bacteria are responsible for PE then at least some of these biomarkers should also be (known to be) associated with infection. However, one obvious point is that the markers may appear only after infection, and this may itself be after the first-trimester; clearly then these would not then be seen as “first-trimester” biomarkers! There are many well-known inflammatory biomarkers that are part of the innate [and possibly trained (670)] immune response, such as the inflammatory cytokines CRP [cf. Ref. (671, 672)], IL-6 (673), IL-1β (674), TNF-α (675), and macrophage migration inhibitory factor (MIF) (676), which are also all biomarkers of infection (677–681). Certainly, the fact that these increase in PE is consistent with a role for an infectious component. However, we shall mainly look at other biomarkers that are known to increase with PE, and see if they are also known to be biomarkers for (or at least changed in the presence of) infection (and see Th17/IL-17 above), and we next examine this. We shall see that pretty well every biomarker that is changed significantly in PE is also known to be changed following infection, a series of findings that we consider adds very strong weight to our arguments.
Proteomic and Similar Biomarkers – Circulating and Placental
What is really needed is a full systems biology strategy [see, e.g., Ref. (99, 682–684)] that brings together the actors that interact then parametrizes the nature of these interactions in a suitable encoding [e.g., SBML (685)] that permits their modeling, at least as an ODE model using software such as CellDesigner (686), COPASI (687), or Cytoscape (688). Thus, to take a small example, “agonistic autoantibodies against the angiotensin II type 1 receptor autoantibodies (AT1-AA) are described. They induce NADPH oxidase and the MAPK/ERK pathway leading to NF-κB and tissue factor activation. AT1-AA are detectable in animal models of PE and are responsible for elevation of soluble fms-related tyrosine kinase-1 (sFlt1) and soluble endoglin (sEng), oxidative stress, and endothelin-1, all of which are enhanced in pre-eclamptic women. AT1-AA can be detected in pregnancies with abnormal uterine perfusion” (589). Many such players have been invoked, and we next list some.
Activin A
Activin A is a member of the TGF-β superfamily. Its levels are raised significantly in PE (118, 689–691). However, activin A is also well established as a biomarker of infection (692–695).
Calretinin
In a proteomic study of preclamptic vs. normal placentas (696), calretinin was one of the most differentially upregulated proteins (p = 1.6 × 10−13 for preterm PE vs. controls, p = 8.9 × 10−7 for term PE vs. controls), and in a manner that correlated with the severity of disease. While calretinin [normally more expressed in neural tissue and mesotheliomas (697)] is not normally seen as a marker of infection, it is, in fact, raised significantly when Chlamydia pneumoniae infects human mesothelial cells (698).
Chemerin
Chemerin is a relatively recently discovered adipokine, whose level can increase dramatically in the first-trimester of preeclamptic pregnancies (699), and beyond (700). Its levels are related to the severity of the PE (701–703). Specifically, an ROC curve (704) analysis showed that a serum chemerin level >183.5 ng mL−1 predicted PE with 87.8% sensitivity and 75.7% specificity (AUC, 0.845; 95% CI, 0.811–0.875) (699). Papers showing that chemerin is also increased by infection (hence inflammation) include (705, 706); it even has antibacterial properties (707, 708) and was protective in a skin model of infection (709, 710). In a study of patients with sepsis (711), circulating chemerin was increased 1.69-fold compared with controls (p = 0.012) and was also protective as judged by survival. These seem like particularly potent argument for a role of chemerin as a marker of infection rather than of PE per se, and for the consequent fact that PE follows infection and not vice versa.
Copeptin
Copeptin, a glycosylated polypeptide consisting of the 39 C-terminal amino acids of arginine vasopressin, has been suggested as “a new biomarker that is specific for preeclampsia” (712), and certainly changes during its development (713). However, it turns out that it is also essentially a measure of all kinds of stresses and adverse events (714–719), including those caused by infection (720–729).
Cystatin C
Not least because kidney function is impaired in PE, low molecular weight (MW) proteins may serve as biomarkers for it. To this end, cystatin C (13 kDa) has been found to be raised significantly in PE (730–736); it also contributed to the marker set in the SCOPE study (8, 17). Notably, although it certainly can be raised during infection (737), it seems to be more of a marker of inflammation or kidney function (738, 739).
d-Dimer
“d-dimer” is a term used to describe quite varying forms of fibrin degradation products (740). Given that PE is accompanied by coagulopathies, it is probably not surprising that d-dimer levels are raised in PE (741–745), although this is true for many conditions (746), and some of the assays would bear improvement (747, 748). Needless to say, however, raised d-dimer levels are also a strong marker for infection (749, 750).
Endocan
Endothelial cell-specific molecule-1 (ESM-1), known as endocan, is a cysteine-rich dermatan sulfate proteoglycan expressed (and sometimes released) by endothelial cells. It has been suggested to be a new biomarker for endothelial dysfunction and PE (751, 752). It would appear, however, to be a rather less specific inflammatory biomarker (753, 754) and is associated with a variety of diseases, including chronic kidney disease (755, 756) and cardiovascular disease (755). Most pertinently from our perspective, it is also raised strongly during sepsis (757–760).
Endoglin
Endoglin is the product of a gene implicated (761, 762) in the rare disease Hereditary Hemorrhagic Telangiectasia. The role of endoglin remains somewhat enigmatic (763). However, endoglin levels were 2.5-fold higher in preeclamptic placentas compared to normal pregnancies (15.4 ± 2.6 vs. 5.7 ± 1.0, p < 0.01). After the onset of clinical disease, the mean serum level of sEng in women with preterm PE was 46.4 ng mL−1, as compared with 9.8 ng mL−1 in controls (p < 0.001) (88). Women with a particular endoglin polymorphism (AA) were 2.29 times more likely to develop PE than those with the GG genotype (p = 0.008) (764), and endoglin is seen as a reasonably good marker for PE (88, 691, 765–768) [cf. Ref. (769)]. Again, endoglin levels are raised following infection by a variety of organisms (770–773), with a particularly clear example that it is a marker of infection coming from the fact that there is raised endoglin only in infected vs. aseptic loosening in joints following arthroplasty (774). In general, it seems likely that these circulating (anti)angiogenic factors are more or less markers of endothelial cell damage, just as we have described for serum ferritin (126).
Ferritin
The natural iron transporter in blood is transferrin [e.g., Ref. (775–780)], present at ca. 1–2 g L−1, with ferritin being an intracellular iron storage molecule, so one is led to wonder why there is even any serum ferritin at all (126, 781). The answer is almost certainly that it is a leakage molecule from damaged cells (126), and when in serum it is found to have lost its iron content (782–785). Serum ferritin is, as expected, raised during PE (244, 246, 249, 253, 255, 786, 787) and in many other inflammatory diseases (126), including infection [e.g., Ref. (788, 789) and above].
microRNAs
microRNAs are a relatively novel and highly important class of ~22 nt non-coding, regulatory molecules (790–793). Some are placenta specific, and those in the circulation [often in endo/exosomes (794–796)] can be identified during pregnancy (797–800), potentially providing a minimally invasive readout of placental condition (801–803). There is aberrant expression of placenta-specific miRNAs in PE including miR-517a/b and miR-517c (804–810) and miR-1233 (811). C19MC is one of the largest miRNA gene clusters in humans, maps to chromosome 19q13.41, and spans a ~100 kb long region. C19MC miRNAs are processed from the cluster (812), are primate-specific, conserved in humans, and comprise 46 miRNA genes, including the miR-517 family (813). miR-517 is known to be antiviral (814, 815), while miR-517a overexpression is apoptotic (816) and can inhibit trophoblast invasion (817). Importantly for our argument, miR-517 molecules are overexpressed following infection (818, 819).
Neuropeptide Y
Although, as its name suggests, neuropeptide Y is a neurotransmitter, it is also correlated with stress. Certainly, it is related to noradrenaline (see below) that may itself be responsible for the raised blood pressure (BP) in PE (820). It is also raised in sepsis, where it is considered to counterbalance the vasodilation characteristic of septic shock [e.g., Ref. (821, 822)]. The apparent paradox of a raised BP in PE and a lowered one in septic shock is considered to be related to the very different concentrations of endotoxin involved (Figure 7).
NGAL (Lipocalin 2, Siderocalin)
Neutrophil gelatinase-associated lipocalin (NGAL) is a lipocalin that is capable of binding catecholate-based siderophores (125, 823, 824). As such it is antimicrobial and is also an inflammatory or sepsis biomarker (825, 826). Given our interest in iron, it is not surprising that it is changed during PE. While one study suggested it to be decreased in PE (827), a great many other studies showed it to be increased significantly in PE, and typically in a manner that correlated with PE severity (735, 828–837). Pertinently to PE, it is also well established as an early biomarker of acute kidney injury (AKI) (838–841). However, it is not a specific biomarker for AKI vs. sepsis (839, 842–850) and its origin in sepsis differs (851, 852). Of course, it can be the sepsis that leads to the AKI (853, 854). Fairly obviously, while it does tend to be increased during PE, we again see its direct role as an antimicrobial and marker of sepsis as highly supportive of our present thesis.
Placental Growth Factor
This is a member of the VEGF Family that despite its name has a great many activities (855). It is often considered in parallel with endoglin and sFlt, with a high sFlt:PlGF ratio being considered as especially discriminatory for PE (89, 856–869), i.e., a lower PlGF can be diagnostic of PE (769, 870–873). PlGF tends to be raised in sepsis unrelated to pregnancy (874, 875), while its lowering in PE may be due to the excess sFLT that decreases it (855, 876, 877). In one study of a patient with CMV infection and PE, it was, in fact, raised (878), while, in the case of IUGR, it was massively lowered (879). PlGF alone is thus probably not a useful general marker for either PE or sepsis if one is trying to disentangle them, although it has clear promise when PE is superimposed on CKD (872, 880).
Procalcitonin
Procalcitonin is the 116 amino acid polypeptide precursor of calcitonin, a calcium regulatory hormone. It is another marker that has been observed to be raised (according to severity) in preeclamptics (742, 881, 882) [but cf. Ref. (713)]. However, it is also a known marker of bacterial infections or sepsis (881, 883–891).
Serum Amyloid A
This is an inflammatory biomarker, which was shown to increase fourfold in PE in one study (892), was significantly raised in another (882), but not in a third (893). However, it is a well-established (and potent) biomarker for infection/sepsis [e.g., Ref. (894–907)]. Defective amyloid processing may be a hallmark of PE more generally (908), and of course amyloid can be induced by various microbes (317, 319, 909, 910) and their products (257).
Soluble fms-Like Tyrosine Kinase 1
The sFlt receptor is a splice variant of the VEGF receptor (766). It is raised considerably in PE (691, 767, 856, 862, 864, 911–914) and may be causal (545, 590, 915–918). Needless to say, by now, we can see that it is also a very clear marker of infection (767, 919, 920), whose levels even correlate with the severity of sepsis (921–923). Of particular note is the fact that sFLT is actually anti-inflammatory (922).
Thrombomodulin
Soluble thrombomodulin was recognized early as an endothelial damage biomarker and is raised in PE (924–934). Interestingly, it has been found to have significant efficacy in the treatment of sepsis [-based disseminated intravascular coagulation (DIC)] (935–943).
TLR4
TLR4 upregulation in preeclamptic placentas (944) is entirely consistent with infection and the “danger model” as applied to PE (945). As well as LPS activation [reviewed in Kell and Pretorius (140)], the heat shock protein 60 of Chlamydia also activates TLR4 (138).
Visfatin
Visfatin is another adipokine that is raised in PE, approximately 2-fold in the study of Fasshauer and colleagues (946), and 1.5-fold in that of Adali and colleagues (947). However, it was little different in a third study (948), while in a different study, it was rather lower in PE than in controls (949). This kind of phenomenon rather lights up the need for excellent quality studies, including ELISA reagents, when making assessments of this type.
Fairly obviously, the conclusion that this long list of biomarkers that are raised in PE might be specific “PE” biomarkers is challenged very strongly by the finding that they are, in fact, all known markers of infection, a finding that in our view strongly bolsters the case for an infectious component in PE.
In a similar vein, there are a number of other sepsis markers (where sepsis is varied via, or occurs as, an independent variable) that we would predict are likely to be visible as raised in PE patient. These might include (680, 950) PAI-1, sE-selectin (951), and sVCAM-1 (921). In particular, Presepsin looks like a potentially useful marker for sepsis (888, 889, 952–961), but we can find no literature on its use as a PE biomarker, where we predict that it may also be raised.
Metabolomic Biomarkers
For fundamental reasons connected with metabolic control and its formal, mathematical analysis (962–966), changes in the metabolome are both expected (967) and found (968–971) to be amplified relative to those in the transcriptome and proteome. For similar reasons, and coupled with evolution’s selection for robustness (972–978) (i.e., homeostasis) in metabolic networks, we do not normally expect to find single metabolic biomarkers for a complex disease or syndrome. Since our initial metabolomic analyzes (654), the technology has improved considerably (979–982), a full human metabolic network reconstruction has been published (978, 983–985) in the style of that done for yeast (986), and a number of candidate metabolomics biomarkers for PE have been identified reproducibly on an entirely separate validation set (15, 655).
This latter, LC-MS-based, study (15) found a cohort of 14 metabolites from the first-trimester that when combined gave an OR of 23 as being predictive of third-trimester PE. For convenience, we list them in Table 7. Note that because they were characterized solely via their mass, there are some uncertainties in the exact identification in some cases, and that untargeted metabolomics of this type has a moderately high limit of detection (maybe 10 μM) such that many potentially discriminatory metabolites are below the limit of detection.
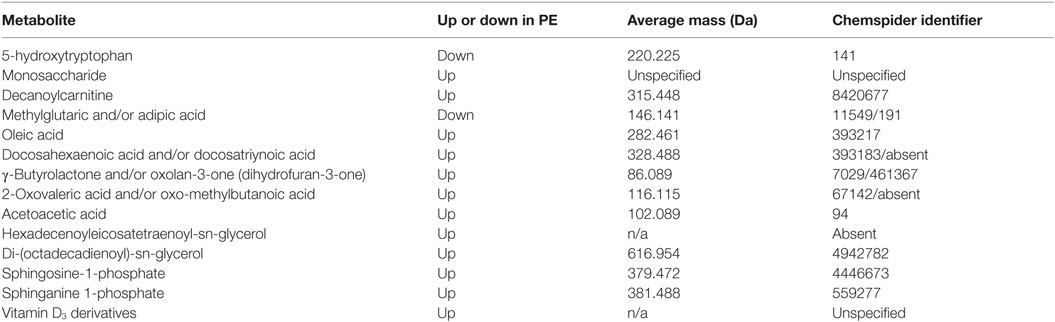
Table 7. Fourteen metabolites contributing to a preeclamptic “signature” (15).
A number of features of interest emerge from this:
1. All the markers save 5-hydroxytryptophan and adipic/methylglutaric acid that were raised in PE; 5-hydroxytryptophan is a precursor of serotonin [which in some studies (987) has been seen to be mildly elevated in PE].
2. Markers came from multiple classes of metabolite or areas of metabolism, including amino acids, carbohydrates, carnitines, dicarboxylic acids, fatty acids (especially), (phospho)lipids, and sterols.
3. γ-Butyrolactone derivatives can act as signaling molecules for a variety of bacteria (988, 989).
4. In common with many other inflammatory diseases (145), Vitamin D3 levels [usually measured as 25(OH)vitD or calcidiol] are often lower in PE (990–994) [cf. Ref. (995–997)], consistent with the levels of their derivatives being raised. However, the direction of causality inflammation ←→ vitamin D levels is not yet known (998) [see also Ref. (143, 145, 996)].
5. None of these metabolites was among four metabolites proposed as first-trimester biomarkers in two other (smaller) studies from different groups (656, 999).
6. Sphingolipid metabolism can be deranged in PE (1000) [also in Parkinson’s (1001)].
As well as the non-targeted metabolomics noted above, a number of other small molecule biomarkers have been turned up by more conventional measurements.
Noradrenaline (Norepinephrine)
An interesting early study (1002) found that venous plasma noradrenaline was raised by 67% in preeclamptics vs. controls. Similar data were found by others (1003). This is of particular interest in the present context since noradrenaline is well established as highly growth stimulatory to Gram-negative microorganisms [e.g., Ref. (1004–1008)], in part by acting as a siderophore (1009–1011). It also raises the levels of neuropeptide Y (820), and as a stress hormone (1012), is of course well known for its role in raising BP, a hallmark of PE.
There is relatively little metabolomics work in sepsis, but in one study, carnitine and sphingolipid metabolism were also modified during sepsis (1013), while in another (1014), a suite of molecules were decreased during acute sepsis. However, the patients involved here were quite close to death, so it is not clear that comparisons between the metabolome in PE and in dying patients are that worthwhile.
We also note a recent and rather interesting suggestion by Eggers (1015) that the maternal release of adrenaline (rather than noradrenaline) may have an important etiological role in PE, although as with the rest of our thesis here it is not there indicated as to what causes the adrenaline to rise (although infection and inflammation can of course do so).
Uric Acid
Hyperuricemia is a moderately common finding in preeclamptic pregnancies and may even be involved in its pathogenesis [see, e.g., Ref. (1016–1022)]. However, it does not seem to be very specific (1023–1027) and is seemingly not an early biomarker [and it did not appear in our own study (15)]. Its lack of specificity is illustrated by the fact that there is considerable evidence for the roles of purinergic signaling (1028), and especially the role of uric acid, in Alzheimer’s and Parkinson’s disease (1029–1031), as well as in a variety of other kinds of inflammatory processes, including pro-inflammatory cytokine production (1032, 1033), the Plasmodium falciparum-induced inflammatory response (1034), the mechanistic basis for the action of alum as an adjuvant (1035), and even peanut allergy (1036–1038). As is common in case–control studies when just one disease (e.g., PE) is studied, artificially high levels of sensitivity and (especially) specificity may appear when other patients with other diseases are not considered.
Clotting, Coagulopathies, and Fibrinogen in PE
In much of our previous work [e.g., Ref. (126–134)], we have noted that each of these chronic, inflammatory diseases is accompanied by changes in fibrin fiber morphologies, coagulopathies, and changes in erythrocytes that are both substantial and characteristic. They can variously be mimicked by adding unliganded iron or LPS. As is well known, LPS itself is a strong inducer of coagulation, whether via tissue factor or otherwise [e.g., Ref. (1039–1048)], and will bind to fibrin strongly (259, 1049). The morphological methods have not yet, to our knowledge, been performed on blood from preeclamptics, whether as a diagnostic or a prognostic, although we note that clotting factors came top in one GWAS looking for gene–PE associations (117). Fibrinogen itself is a TLR4 ligand (1050), is raised in PE (1051–1055), and we note the extensive evidence for coagulopathies during pregnancies with PE [e.g., Ref. (64, 128, 527, 741, 1056–1068)]. In the worst cases, these are the very frightening DIC (1047, 1069–1073) that can, of course, also emerge as a consequence of sepsis (1074–1080). Variations in the plasminogen activator inhibitor-1 may contribute to the hypofibrinolysis observed (1081–1083).
We recently showed that LPS can potently induce amyloid formation in fibrin (258, 259, 1084, 1085). Thus, in addition, we note the increasing recognition that amyloid proteins themselves, that may occur as a result of coagulopathies, are themselves both inflammatory [e.g., Ref. (565, 669, 1086–1091)] and cytotoxic [e.g., Ref. (257, 1092–1096)], and that this can of itself contribute strongly to the death of, e.g., trophoblasts.
Related to clotting parameters are three other “old” but easily measured variables that probably reflect inflammation (1097), that have been suggested to differ in PE from normotensives, and may have some predictive power. The first two are the erythrocyte sedimentation rate (ESR) (1098, 1099) and the red cell distribution width (RDW) (1100) [but cf. Ref. (1101)]. Interestingly, the former was the only variable that was predictive of a subsequent stroke following sub-arachnoid hemorrhage (1102). The third relates to the morphology of erythrocytes (that may in part underpin the other two). We and others have shown in a series of studies [e.g., Ref. (134–136, 1103–1106)] that erythrocyte morphology diverges very considerably from that “classical” discoid shape adopted by normal healthy cells, and that this can be a strong indicator of disease (137). In extreme cases [e.g., Ref. (133, 1107–1112)], including following infection (1113), this results in eryptosis, the suicidal death of erythrocytes. It is of interest that ceramide, a precursor of sphingosine-1-phosphate (S1P) (Table 7), is raised in various diseases such as Parkinson’s and may serve to stimulate eryptosis (1114). Although we know of no direct measurements to date, there is evidence that eryptosis may play a significant role in PE (1115).
Prevention Strategies
Apart from low-dose aspirin [that may have little effect (1116–1119) unless initiated relatively early in pregnancy (1120–1124)], and low-dose calcium (1125), there are relatively few treatment options in present use (1126–1129). [Magnesium sulfate (1130–1132) has been used as a treatment for eclampsia and, interestingly, prevents LPS-induced cell death in an in vitro model of the human placenta (1133).]
In the history of science or medicine, some treatments are empirical, while others are considered to have a mechanistic basis. The general assumption is that the more we know about the originating etiology of a disease or syndrome the more likely we are to be able to treat its causes effectively, and not just its symptoms. Clearly, also, clinicians are rightly loth to give complex and potentially teratogenic treatments to pregnant women when this can be avoided (1134–1137). However, the surprising lack of systematic data with antibiotics (1138), modulo one particularly spectacular success (540), suggests that we ought to be performing trials with safe antibiotics on women at special risk (1139). These must take care to avoid any Jarisch–Herxheimer reaction (1140–1143) due to the release from microbes induced by antibiotics of inflammagens such as LPS (1144–1147). A related strategy recognizes that some FDA-approved drugs can actually exert powerful antibiotic effects in vivo (but not on petri plates) by modifying the host (1148).
Because of the known oxidative stress accompanying PE, it had been assumed that antioxidants such as vitamin C (ascorbate) might be preventive; however, this turned out not to be the case (even the opposite) for ascorbate (1118, 1149). Probably, this is because in the presence of unliganded iron, ascorbate is, in fact, pro-oxidant (125). However, polyphenolic antioxidants that actually act by chelating iron (84, 125) seem to be more effective (1150).
Another area that we and others have previously highlighted recognizes the ability of non-siderophoric iron chelators to act as iron-withholding agents and thereby limit the growth of bacteria. Again, a prediction is that women with iron overload diseases should be more susceptible to PE, a prediction that is borne out for α-thalassemia (1151, 1152) though not apparently for hereditary hemochromatosis (1153). However, the extent of use of chelators and degree of control of free iron, thereby obtained is rarely recorded in any detail, so in truth it is difficult to draw conclusions.
The general benefits of nutritional iron chelators such as blueberries and other fruits and vegetables containing anthocyanins have been discussed elsewhere [e.g., Ref. (84, 125, 1154)].
How significant coagulopathies are to the etiology of PE development (as opposed to providing merely an accompaniment) is not entirely clear, but on the basis that they are then anticoagulants would potentially assist, just as thrombomodulin does in DIC accompanying sepsis (942, 943, 1079, 1080). Of course, one of many effects of low-dose aspirin is to act as an anticoagulant. There is also evidence for the efficacy of heparin (6, 1127, 1155–1160), which is especially interesting given our highlighting of the role of coagulopathies in PE. These anticoagulants that avoid bleeding (1161) are obviously of particular interest, while anything stopping the fibrin forming β-amyloid (258, 259) should serve as an especially useful anti-inflammatory anticoagulant.
With a change in focus from function-first to target-first-based drug discovery (976), there has been an assumption that because a drug is (i) found to bind potently to a molecular target and (ii) has efficacy at a physiological level in vivo, the first process is thus responsible for the second. This has precisely no basis in logic [it is a logical fault known variously as “affirming the consequent” or “post hoc ergo propter hoc” (1162)]. This is because the drug might be acting physiologically by any other means, since drug binding to proteins is typically quite promiscuous [e.g., Ref. (1163–1167)]. Indeed, the average known number of binding sites for marketed drugs is six (208, 1168). In particular, it is likely, from a network or systems pharmacology perspective [e.g., Ref. (978, 1169–1172)], that successful drugs (like aspirin) are successful precisely because they hit multiple targets. The so-called “statins” provide a particularly good case in point (125).
It had long been known that the enzyme HMGCoA reductase exerted strong control on the biosynthetic flux to cholesterol, and that inhibiting it might lower the flux and steady-state cholesterol levels (as indeed it does). Notwithstanding that cholesterol alone is a poor predictor of cardiovascular disease (1173–1175), especially in the normal range, HMGCoA reductase inhibitors have benefits in terms of decreasing the adverse events of various types of cardiovascular disease (1176). Following an original discovery of natural products such as compactin (mevastatin) and lovastatin containing a group related to hydroxymethylglutaric acid (rather than a CoA version) that inhibited the enzyme (1177), many variants with this (hydroxyl)methylglutaric substructure came to be produced, with the much larger “rest” of the molecule being considerably divergent [see Figure 8, where the MW values vary from 390.5 (mevastatin) to 558.6 (atorvastatin)]. Despite this wide structural diversity (Figure 8), they are still collectively known as “statins,” and despite the wildly illogical assumption that they might all work in the same way(s). The fact that different statins can cause a variety of distinct expression profiles (1178) is anyway utterly inconsistent with a unitary mode of action. In particular, in this latter study, statins clustered into whether they were (fluvastatin, lovastatin, and simvastatin) or were not (atorvastatin, pravastatin, and rosuvastatin) likely to induce the side-effect of rhabdomyolysis or any other myopathy. Clearly, any choice of “statin” should come from the latter group, with pravastatin and rosuvastatin being comparatively hydrophilic.
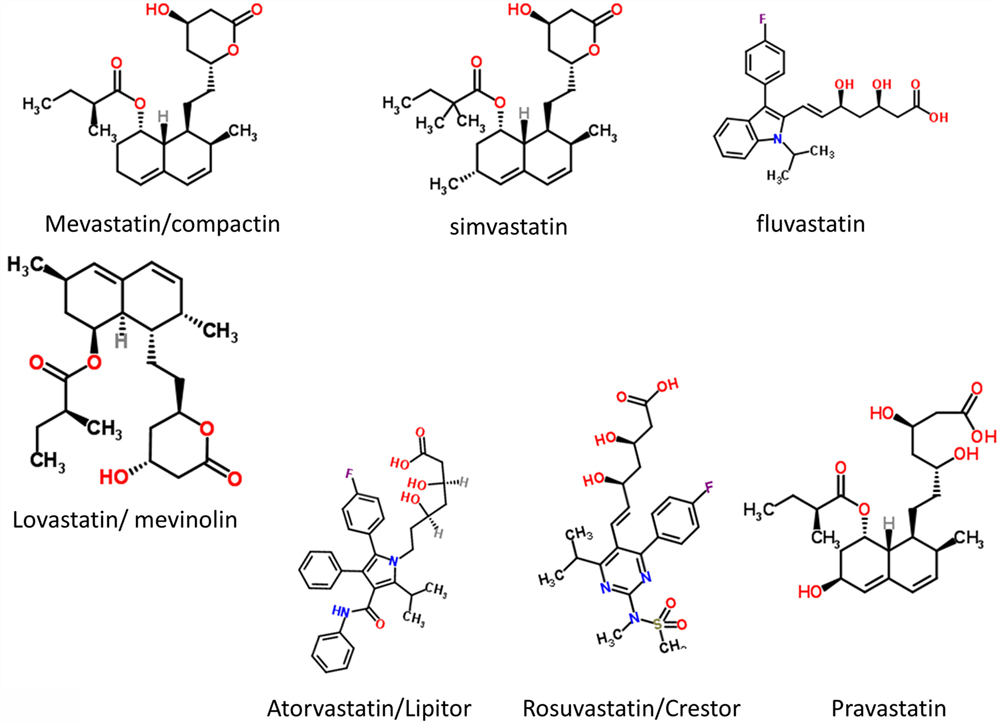
Figure 8. Some structures of various statins.
The epidemiological fact of improved survival despite the comparative irrelevance of cholesterol levels to atherosclerotic plaque formation and heart disease in the normal range provides an apparent paradox (1179). This is easily solved by the recognition [e.g., Ref. (1180–1193), and many other references and reviews] that “statins” are, in fact, anti-inflammatory. They may also be antimicrobial/antiseptic, whether directly or otherwise (1194–1198), and we also note the role of cholesterol in mopping up endotoxin (1199). Finally, here, it needs to be recognized that statins do themselves serve to lower iron levels (1200–1202), and (while oddly this seems not to have been tested directly) simple inspection of their structures (Figure 8) implies that the better ones (with their multiple OH groups) might, in fact, chelate iron directly.
In consequence, a number of authors have indicated the potential utility of statins in treating PE (113, 545, 1203–1213), and pravastatin has been the subject of a number of favorable studies (545, 1204, 1206, 1209, 1211, 1214, 1215), including in humans (1204, 1216–1218). Pravastatin seems more than ripe for a proper, randomized clinical trial (1203).
Another “vascular” class of drugs that has been proposed for treating PE is represented by those of the family of vasodilatory phosphodiesterase 5 inhibitors such as sildenafil (Viagra) and vardenafil (Levitra), as it is reasonable that they might improve endothelial function, especially if started early in pregnancy (1219). Thus, vardefanil restores endothelial function by increasing PlGF (1220), and sildenafil has shown promise in a number of animal studies (1221–1226) and in human tissues (1227, 1228), with a clinical trial ongoing (1229). In particular (1226), it was able to normalize the metabolomics changes observed in a mouse model (the COMT−/− model) of PE.
Anti-hypertensive therapy for PE has been reviewed by Abalos and colleagues (1230) and Magee and colleagues (114). Anti-hypertensives did halve the incidence of hypertension but had no effect on PE. Methyldopa is one of the most commonly used anti-hypertensives in pregnancy, but it may also stimulate eryptosis (1231); alternative drugs were considered to be better (1230) for hypertension. Nifedipine (1232) and labetalol (1233) are considered a reasonable choice. There was also a slight reduction in the overall risk of developing proteinuria/PE when beta blockers and calcium channel blockers considered together (but not alone) were compared with methyldopa (1230). In mice, olmesartan (together with captopril) proved usefully anti-hypertensive (1234); this is of interest because olmesartan is also an agonist of the vitamin D receptor (1235). However, it was not mentioned in either Ref. (1230) or Ref. (114).
Lipopolysaccharide itself has long been recognized as a target of inflammatory diseases. Unfortunately, despite initially promising trials of an anti-LPS antibody known as centoxin (1236), it was eventually withdrawn, apparently because of a combination of ineffectiveness (1237, 1238) and toxicity (1239, 1240). LPS is rather hydrophobic, and thus it is hard to make even monoclonal antibodies very selective for such targets, such that the toxicity was probably because of its lack of specificity between lipid A and other hydrophobic ligands (1241). Other possible treatments based on LPS, such as “sushi peptides” (1242–1249) [or variants (1250, 1251)], and LPS-binding protein were covered elsewhere (140).
If an aberrant or dysbiotic gut microbiome is the source of the microbes that underpin PE, it is at least plausible that the gut microbiome should be predictive of PE (378), but we know of no suitably powered study that has been done to assess this, and this would clearly be worthwhile. However, in a study of primiparous women, the OR for getting severe PE was only 0.6 if probiotic milk drinks containing lactobacilli were consumed daily (1252). This is a very significant effects, such that this too seems an area well worth following up.
From a metabolomics point of view, the molecules seen to be raised in PE may either be biomarkers of the disease etiology or of the body’s attempts to respond to the disease [and this is true generally (1253)]. Thus, it is of great interest that S1P was raised in PE [see Kenny (15) and Table 7]. S1P is mainly vasoconstrictive (1254, 1255), but agonists of the S1P-1 receptor (that is involved in endothelial cell function) seemed to have considerable value in combatting the cytokine storm that followed infection-driven sepsis (1256–1261). The detailed mechanism seems not to be known, but in the context of infection, a need for S1P and other sphingolipids for successful pregnancies (1262, 1263) [see also Parkinson’s (1001)], and the induction of PE by its disruption (1000, 1264–1268), some serious investigation of the potential protective effects of S1PR1 agonists seems highly warranted.
Among other small molecules, melatonin has shown some promise in the treatment of septic shock, by lowering inflammatory cytokine production (1269) [and see Gitto et al. (1270) for neonatal oxidative stress], and a trial is in prospect for PE (1271).
Lipoxin A4 (LXA4) is considered to be an endogenous stop signal in inflammation. While recognizing the difficulties with rodent PE models (above), we note that in one study, the effect of BML-111 (a synthetic analog of LXA4) was tested on experimental PE induced in rats by low-dose endotoxin (LPS), and showed highly beneficial effects (549).
Coda – A Return to the Bradford Hill Criteria
Returning to the Bradford Hill criteria for ascribing causation of a disease to an environmental factor (97), we can now ask whether a detectable (if largely dormant) microbiome X, that is more likely to replicate with free iron, and that can anyway secrete or shed a variety of inflammatory components such as LPS, represents a plausible and major etiological factor for PE (Y):
(1) what is the strength of association between X and Y? We found an overwhelming co-occurrence of microbes or their products and PE.
(2) what is the consistency of association between X and Y? Almost wherever we looked, whether via periodontal disease (PD), UTI, or other means of ingress, we could find a microbial component in PE.
(3) what is the specificity of association between X and Y? Insufficient data are available to ascribe PE solely to one type of organism; however, these data clearly indicate that a variety of microbes, each capable of shedding inflammatory molecules such as LPS, can serve to stimulate or exacerbate PE.
(4) experiments verify the relationship between X and Y. It is unethical to do these in humans in terms of purposely infecting pregnant women, but data from antibiotics show the expected improvements.
(5) modification of X alters the occurrence of Y; this is really as (4).
(6) biologically plausible cause and effect relationship. Yes, this is where we think the ideas set down here are entirely consistent with current thinking on the main causes of PE. What we add in particular is the recognition that bacteria (and other microbes) that may be invisible to culture are both present and responsible, by established means, for the inflammation and other sequelae (and especially the coagulopathies) seen as causative accompaniments to PE.
Other Predictions
Classical clinical microbiology, involving mainly replication-based methods, is evolving rapidly to assess the microbial content of samples on the basis of DNA sequences (296, 1272), including 16S rDNA (287, 288, 290, 292, 293, 295, 297, 1273), suitable protein-encoding housekeeping genes [e.g., Ref. (1274–1279)], and, increasingly, full genome sequences (1280). In the future, we can thus expect a considerable increase in molecular assessments of the microbiological content of blood, urine, and tissues, and this will obviously be a vital part of the experimental assessment and development of the ideas presented here. Molecular methods will also be used to assess maternal circulating DNA (1281–1283) and RNA (1284) in terms of both its presence and sequencing, as well as the use of digital PCR (1285).
Since PE has such a strong vascular component, we also predict that measurements designed to detect coagulopathies will increase in importance, for both diagnosis and prognosis, and for assessing treatments.
New drugs designed to kill non-growing bacteria (1286–1290) or to overcome amyloid coagulopathies (1291–1295) will be needed and will come to the fore.
Finally, we consider that real progress in understanding PE from a systems biology perspective means that it must be modeled accordingly, and this must be a major goal.
Concluding Remarks
We have brought together a large and widely dispersed literature to make the case that an important etiological role in PE is played by dormant microbes, or at least ones that are somewhat refractory to culture, and that these can awaken, shed inflammagens such as LPS, and thereby initiate inflammatory cascades. (The sequelae of these, involving cytokines, coagulopathies, and so on, are well enough accepted.) The case is founded on a large substructure of interlocking evidence, but readers might find the following elements as discussed above, especially persuasive and/or worthy of follow-up:
• the regular presence of detectable microbes in preeclamptic placentas [e.g., Ref. (400, 426, 427)].
• the fact that endotoxin (LPS) can act as such a mimic for invoking PE in experimental models.
• the fact that every known proteomic biomarker suggested for PE has also been shown to increase during infection.
• the significant number of papers reviewing a link between infection and PE [e.g., Ref. (53, 54, 138, 371)].
• the almost complete absence (one case) of PE in patients treated with spiramycin (540).
Any and all of these provide powerful strategies for testing whether PE is, in fact, such as gastric ulcers (166, 168, 169, 1296), essentially initiated as an infectious disease.
Author Notes
This is the paper number 8 of the series “The dormant blood microbiome in chronic, inflammatory diseases”. The other articles of the series can be found in the reference list with the numbers 141; 139; 140; 257; 147; 143; 1084; 258; 142.
Author Contributions
Both authors made substantial, direct, and intellectual contributions to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
DK thanks the Biotechnology and Biological Sciences Research Council (grant BB/L025752/1) for financial support. LK is a Science Foundation Ireland Principal Investigator (grant number 08/IN.1/B2083). LK is also The Director of the Science Foundation Ireland-funded INFANT Research Centre (grant no. 12/RC/2272).
References
1. Grill S, Rusterholz C, Zanetti-Dällenbach R, Tercanli S, Holzgreve W, Hahn S, et al. Potential markers of preeclampsia – a review. Reprod Biol Endocrinol (2009) 7:70. doi: 10.1186/1477-7827-7-70
2. Steegers EAP, Von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet (2010) 376:631–44. doi:10.1016/S0140-6736(10)60279-6
3. North RA, Mccowan LM, Dekker GA, Poston L, Chan EH, Stewart AW, et al. Clinical risk prediction for pre-eclampsia in nulliparous women: development of model in international prospective cohort. BMJ (2011) 342:d1875. doi:10.1136/bmj.d1875
4. Uzan J, Carbonnel M, Piconne O, Asmar R, Ayoubi JM. Pre-eclampsia: pathophysiology, diagnosis, and management. Vasc Health Risk Manag (2011) 7:467–74. doi:10.2147/VHRM.S20181
5. Kenny L. Improving diagnosis and clinical management of pre-eclampsia. MLO Med Lab Obs (2012) 44(12):14.
7. Chaiworapongsa T, Chaemsaithong P, Yeo L, Romero R. Pre-eclampsia part 1: current understanding of its pathophysiology. Nat Rev Nephrol (2014) 10:466–80. doi:10.1038/nrneph.2014.102
8. Kenny LC, Black MA, Poston L, Taylor R, Myers JE, Baker PN, et al. Early pregnancy prediction of preeclampsia in nulliparous women, combining clinical risk and biomarkers: the screening for pregnancy endpoints (SCOPE) international cohort study. Hypertension (2014) 64:644–52. doi:10.1161/HYPERTENSIONAHA.114.03578
9. Sircar M, Thadhani R, Karumanchi SA. Pathogenesis of preeclampsia. Curr Opin Nephrol Hypertens (2015) 24:131–8. doi:10.1097/MNH.0000000000000105
10. Abalos E, Cuesta C, Grosso AL, Chou D, Say L. Global and regional estimates of preeclampsia and eclampsia: a systematic review. Eur J Obstet Gynecol Reprod Biol (2013) 170:1–7. doi:10.1016/j.ejogrb.2013.05.005
11. Khan KS, Wojdyla D, Say L, Gulmezoglu AM, Van Look PF. WHO analysis of causes of maternal death: a systematic review. Lancet (2006) 367:1066–74. doi:10.1016/S0140-6736(06)68397-9
12. Duley L. The global impact of pre-eclampsia and eclampsia. Semin Perinatol (2009) 33:130–7. doi:10.1053/j.semperi.2009.02.010
13. Ghulmiyyah L, Sibai B. Maternal mortality from preeclampsia/eclampsia. Semin Perinatol (2012) 36:56–9. doi:10.1053/j.semperi.2011.09.011
14. Abalos E, Cuesta C, Carroli G, Qureshi Z, Widmer M, Vogel JP, et al. Pre-eclampsia, eclampsia and adverse maternal and perinatal outcomes: a secondary analysis of the World Health Organization Multicountry Survey on Maternal and Newborn Health. BJOG (2014) 121(Suppl 1):14–24. doi:10.1111/1471-0528.12629
15. Kenny LC, Broadhurst DI, Dunn W, Brown M, Francis-Mcintyre S, North RA, et al. Robust early pregnancy prediction of later preeclampsia using metabolomic biomarkers. Hypertension (2010) 56:741–9. doi:10.1161/HYPERTENSIONAHA.110.157297
16. Bartsch E, Medcalf KE, Park AL, Ray JG; High Risk of Pre-Eclampsia Identification Group. Clinical risk factors for pre-eclampsia determined in early pregnancy: systematic review and meta-analysis of large cohort studies. BMJ (2016) 353:i1753. doi:10.1136/bmj.i1753
17. Karumanchi SA, Granger JP. Preeclampsia and pregnancy-related hypertensive disorders. Hypertension (2016) 67:238–42. doi:10.1161/HYPERTENSIONAHA.115.05024
18. Duckitt K, Harrington D. Risk factors for pre-eclampsia at antenatal booking: systematic review of controlled studies. Br Med J (2005) 330:565–7. doi:10.1136/bmj.38380.674340.E0
20. Ananth CV, Keyes KM, Wapner RJ. Pre-eclampsia rates in the United States, 1980–2010: age-period-cohort analysis. BMJ (2013) 347:f6564. doi:10.1136/bmj.f6564
21. Bdolah Y, Elchalal U, Natanson-Yaron S, Yechiam H, Bdolah-Abram T, Greenfield C, et al. Relationship between nulliparity and preeclampsia may be explained by altered circulating soluble fms-like tyrosine kinase 1. Hypertens Pregnancy (2014) 33:250–9. doi:10.3109/10641955.2013.858745
22. Saftlas AF, Olson DR, Franks AL, Atrash HK, Pokras R. Epidemiology of preeclampsia and eclampsia in the United-States, 1979-1986. Am J Obstet Gynecol (1990) 163:460–5. doi:10.1016/0002-9378(90)91176-D
23. Zhang J, Zeisler J, Hatch MC, Berkowitz G. Epidemiology of pregnancy-induced hypertension. Epidemiol Rev (1997) 19:218–32. doi:10.1093/oxfordjournals.epirev.a017954
24. Lamminpää R, Vehvilainen-Julkunen K, Gissler M, Heinonen S. Preeclampsia complicated by advanced maternal age: a registry-based study on primiparous women in Finland 1997-2008. BMC Pregnancy Childbirth (2012) 12:47. doi:10.1186/1471-2393-12-47
25. Carolan M. Maternal age >= 45 years and maternal and perinatal outcomes: a review of the evidence. Midwifery (2013) 29:479–89. doi:10.1016/j.midw.2012.04.001
26. Trogstad L, Magnus P, Stoltenberg C. Pre-eclampsia: risk factors and causal models. Best Pract Res Clin Obstet Gynaecol (2011) 25:329–42. doi:10.1016/j.bpobgyn.2011.01.007
27. Coonrod DV, Hickok DE, Zhu KM, Easterling TR, Daling JR. Risk-factors for preeclampsia in twin pregnancies – a population-based cohort study. Obstet Gynecol (1995) 85:645–50. doi:10.1016/0029-7844(95)00049-W
28. Campbell DM, MacGillivray I. Preeclampsia in twin pregnancies: incidence and outcome. Hypertens Pregnancy (1999) 18:197–207. doi:10.3109/10641959909016193
29. Bdolah Y, Lam C, Rajakumar A, Shivalingappa V, Mutter W, Sachs BP, et al. Twin pregnancy and the risk of preeclampsia: bigger placenta or relative ischemia? Am J Obstet Gynecol (2008) 198:428.e1–6. doi:10.1016/j.ajog.2007.10.783
30. Bodnar LM, Ness RB, Harger GF, Roberts JM. Inflammation and triglycerides partially mediate the effect of prepregnancy body mass index on the risk of preeclampsia. Am J Epidemiol (2005) 162:1198–206. doi:10.1093/aje/kwi334
31. Roberts JM, Bodnar LM, Patrick TE, Powers RW. The role of obesity in preeclampsia. Pregnancy Hypertens (2011) 1:6–16. doi:10.1016/j.preghy.2010.10.013
32. Jeyabalan A. Epidemiology of preeclampsia: impact of obesity. Nutr Rev (2013) 71:S18–25. doi:10.1111/nure.12055
33. Sugerman HJ. Effect of obesity on incidence of preeclampsia. Am J Obstet Gynecol (2014) 210:375–375. doi:10.1016/j.ajog.2013.10.014
34. Thornton JG, MacDonald AM. Twin mothers, pregnancy hypertension and pre-eclampsia. Brit J Obstet Gynaecol (1999):570–5. doi:10.1111/j.1471-0528.1999.tb08326.x
35. Ros HS, Lichtenstein P, Lipworth L, Cnattingius S. Genetic effects on the liability of developing pre-eclampsia and gestational hypertension. Am J Med Genet (2000) 91:256–60. doi:10.1002/(SICI)1096-8628(20000410)91:4<256::AID-AJMG3>3.0.CO;2-T
36. Williams PJ, Broughton Pipkin F. The genetics of pre-eclampsia and other hypertensive disorders of pregnancy. Best Pract Res Clin Obstet Gynaecol (2011) 25:405–17. doi:10.1016/j.bpobgyn.2011.02.007
37. Valenzuela FJ, Perez-Sepulveda A, Torres MJ, Correa P, Repetto GM, Illanes SE. Pathogenesis of preeclampsia: the genetic component. J Pregnancy (2012) 2012:632732. doi:10.1155/2012/632732
38. Boyd HA, Tahir H, Wohlfahrt J, Melbye M. Associations of personal and family preeclampsia history with the risk of early-, intermediate- and late-onset preeclampsia. Am J Epidemiol (2013) 178:1611–9. doi:10.1093/aje/kwt189
39. Roten LT, Thomsen LCV, Gundersen AS, Fenstad MH, Odland ML, Strand KM, et al. The Norwegian preeclampsia family cohort study: a new resource for investigating genetic aspects and heritability of preeclampsia and related phenotypes. BMC Pregnancy Childbirth (2015) 15:319. doi:10.1186/s12884-015-0754-2
40. Asherson RA, Shoenfeld Y. The role of infection in the pathogenesis of catastrophic antiphospholipid syndrome – molecular mimicry? J Rheumatol (2000) 27:12–4.
41. Asherson RA, Cervera R. Antiphospholipid antibodies and infections. Ann Rheum Dis (2003) 62:388–93. doi:10.1136/ard.62.5.388
42. Blank M, Asherson RA, Cervera R, Shoenfeld Y. Antiphospholipid syndrome infectious origin. J Clin Immunol (2004) 24:12–23. doi:10.1023/B:JOCI.0000018058.28764.ce
43. Harel M, Aron-Maor A, Sherer Y, Blank M, Shoenfeld Y. The infectious etiology of the antiphospholipid syndrome: links between infection and autoimmunity. Immunobiology (2005) 210:743–7. doi:10.1016/j.imbio.2005.10.004
44. Shoenfeld Y, Blank M, Cervera R, Font J, Raschi E, Meroni PL. Infectious origin of the antiphospholipid syndrome. Ann Rheum Dis (2006) 65:2–6. doi:10.1136/ard.2005.045443
45. Clark EA, Silver RM, Branch DW. Do antiphospholipid antibodies cause preeclampsia and HELLP syndrome? Curr Rheumatol Rep (2007) 9:219–25. doi:10.1007/s11926-007-0035-9
46. Espinosa G, Cervera R, Asherson RA. Catastrophic antiphospholipid syndrome and sepsis. A common link? J Rheumatol (2007) 34:923–6.
47. Sène D, Piette JC, Cacoub P. Antiphospholipid antibodies, antiphospholipid syndrome and infections. Autoimmun Rev (2008) 7:272–7. doi:10.1016/j.autrev.2007.10.001
48. Zinger H, Sherer Y, Goddard G, Berkun Y, Barzilai O, Agmon-Levin N, et al. Common infectious agents prevalence in antiphospholipid syndrome. Lupus (2009) 18:1149–53. doi:10.1177/0961203309345738
49. Kutteh WH. Antiphospholipid antibody syndrome and reproduction. Curr Opin Obstet Gynecol (2014) 26:260–5. doi:10.1097/Gco.0000000000000086
50. Kutteh WH, Hinote CD. Antiphospholipid antibody syndrome. Obstet Gyn Clin N Am (2014) 41:113–32. doi:10.1016/j.ogc.2013.10.004
51. O’Gorman N, Wright D, Syngelaki A, Akolekar R, Wright A, Poon LC, et al. Competing risks model in screening for preeclampsia by maternal factors and biomarkers at 11-13 weeks gestation. Am J Obstet Gynecol (2016) 214:103.e1–12. doi:10.1016/j.ajog.2015.08.034
52. Schieve LA, Handler A, Hershow R, Davis F. Urinary-tract infection during pregnancy – its association with maternal morbidity and perinatal outcome. Am J Public Health (1994) 84:405–10. doi:10.2105/Ajph.84.3.405
53. Conde-Agudelo A, Villar J, Lindheimer M. Maternal infection and risk of preeclampsia: systematic review and metaanalysis. Am J Obstet Gynecol (2008) 198:7–22. doi:10.1016/j.ajog.2007.07.040
54. Rustveld LO, Kelsey SF, Sharma R. Association between maternal infections and preeclampsia: a systematic review of epidemiologic studies. Matern Child Health J (2008) 12:223–42. doi:10.1007/s10995-007-0224-1
55. Irgens HU, Reisaeter L, Irgens LM, Lie RT. Long term mortality of mothers and fathers after pre-eclampsia: population based cohort study. BMJ (2001) 323:1213–7. doi:10.1136/bmj.323.7323.1213
56. Bellamy L, Casas JP, Hingorani AD, Williams DJ. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta-analysis. BMJ (2007) 335:974. doi:10.1136/bmj.39335.385301.BE
57. Craici IM, Wagner SJ, Hayman SR, Garovic VD. Pre-eclamptic pregnancies: an opportunity to identify women at risk for future cardiovascular disease. Womens Health (Lond Engl) (2008) 4:133–5. doi:10.2217/17455057.4.2.133
58. Romundstad PR, Magnussen EB, Smith GD, Vatten LJ. Hypertension in pregnancy and later cardiovascular risk: common antecedents? Circulation (2010) 122:579–84. doi:10.1161/CIRCULATIONAHA.110.943407
59. Powe CE, Levine RJ, Karumanchi SA. Preeclampsia, a disease of the maternal endothelium: the role of antiangiogenic factors and implications for later cardiovascular disease. Circulation (2011) 123:2856–69. doi:10.1161/CIRCULATIONAHA.109.853127
60. Skjaerven R, Wilcox AJ, Klungsøyr K, Irgens LM, Vikse BE, Vatten LJ, et al. Cardiovascular mortality after pre-eclampsia in one child mothers: prospective, population based cohort study. BMJ (2012) 345:e7677. doi:10.1136/bmj.e7677
61. Ananth CV, Lawrence Cleary K. Pre-eclampsia and cardiovascular disease: more questions than answers? BJOG (2013) 120:920–3. doi:10.1111/1471-0528.12215
62. Brennan LJ, Morton JS, Davidge ST. Vascular dysfunction in preeclampsia. Microcirculation (2014) 21:4–14. doi:10.1111/micc.12079
63. Chen CW, Jaffe IZ, Karumanchi SA. Pre-eclampsia and cardiovascular disease. Cardiovasc Res (2014) 101:579–86. doi:10.1093/cvr/cvu018
64. Tannetta DS, Hunt K, Jones CI, Davidson N, Coxon CH, Ferguson D, et al. Syncytiotrophoblast extracellular vesicles from pre-eclampsia placentas differentially affect platelet function. PLoS One (2015) 10:e0142538. doi:10.1371/journal.pone.0142538
65. Mol BWJ, Roberts CT, Thangaratinam S, Magee LA, De Groot CJM, Hofmeyr GJ. Pre-eclampsia. Lancet (2016) 387:999–1011. doi:10.1016/S0140-6736(15)00070-7
66. Bateman BT, Schumacher HC, Bushnell CD, Pile-Spellman J, Simpson LL, Sacco RL, et al. Intracerebral hemorrhage in pregnancy: frequency, risk factors, and outcome. Neurology (2006) 67:424–9. doi:10.1212/01.wnl.0000228277.84760.a2
67. Kajantie E, Eriksson JG, Osmond C, Thornburg K, Barker DJP. Pre-eclampsia is associated with increased risk of stroke in the adult offspring: the Helsinki birth cohort study. Stroke (2009) 40:1176–80. doi:10.1161/STROKEAHA.108.538025
68. Tang CH, Wu CS, Lee TH, Hung ST, Yang CY, Lee CH, et al. Preeclampsia-eclampsia and the risk of stroke among peripartum in Taiwan. Stroke (2009) 40:1162–8. doi:10.1161/STROKEAHA.108.540880
69. Sherer Y, Blank M, Shoenfeld Y. Antiphospholipid syndrome (APS): where does it come from? Best Pract Res Clin Rheumatol (2007) 21:1071–8. doi:10.1016/j.berh.2007.09.005
70. Mignini LE, Villar J, Khan KS. Mapping the theories of preeclampsia: the need for systematic reviews of mechanisms of the disease. Am J Obstet Gynecol (2006) 194:317–21. doi:10.1016/j.ajog.2005.08.065
71. Kell DB, Kenny LC. A dormant microbial component in the development of pre-eclampsia. BioRxiv preprint. bioRxiv (2016) 057356. doi:10.1101/057356
72. Redman CWG. Current topic: pre-eclampsia and the placenta. Placenta (1991) 12:301–8. doi:10.1016/0143-4004(91)90339-H
73. Genc H, Uzun H, Benian A, Simsek G, Gelisgen R, Madazli R, et al. Evaluation of oxidative stress markers in first trimester for assessment of preeclampsia risk. Arch Gynecol Obstet (2011) 284:1367–73. doi:10.1007/s00404-011-1865-2
74. Burton GJ, Jauniaux E. Placental oxidative stress: from miscarriage to preeclampsia. J Soc Gynecol Investig (2004) 11:342–52. doi:10.1016/j.jsgi.2004.03.003
75. Redman CWG, Sargent IL. Latest advances in understanding preeclampsia. Science (2005) 308:1592–4. doi:10.1126/science.1111726
76. Roberts JM, Bell MJ. If we know so much about preeclampsia, why haven’t we cured the disease? J Reprod Immunol (2013) 99:1–9. doi:10.1016/j.jri.2013.05.003
77. Redman CW, Sargent IL, Staff AC. IFPA Senior Award Lecture: making sense of pre-eclampsia – two placental causes of preeclampsia? Placenta (2014) 35(Suppl):S20–5. doi:10.1016/j.placenta.2013.12.008
78. Xiong X, Demianczuk NN, Saunders LD, Wang FL, Fraser WD. Impact of preeclampsia and gestational hypertension on birth weight by gestational age. Am J Epidemiol (2002) 155:203–9. doi:10.1093/aje/155.3.203
79. Roberts JM, Hubel CA. The two stage model of preeclampsia: variations on the theme. Placenta (2009) 30(Suppl A):S32–7. doi:10.1016/j.placenta.2008.11.009
80. Redman CWG. The six stages of pre-eclampsia. Pregnancy Hypertens (2014) 4:246. doi:10.1016/j.preghy.2014.04.020
81. Sacks GP, Studena K, Sargent K, Redman CWG. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol (1998) 179:80–6. doi:10.1016/S0002-9378(98)70254-6
82. Redman CWG, Sargent IL. Pre-eclampsia, the placenta and the maternal systemic inflammatory response – a review. Placenta (2003) 24(Suppl A):S21–7. doi:10.1053/plac.2002.0930
83. Hubel CA. Dyslipidemia and pre-eclampsia. In: Belfort MA, Lydall F, editors. Pre-Eclampsia-Aetiology and Clinical Practice. Cambridge: Cambridge University Press (2006). p. 164–82.
84. Kell DB. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch Toxicol (2010) 577:825–89. doi:10.1007/s00204-010-0577-x
85. Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature (2000) 407:242–8. doi:10.1038/35025215
86. Maynard S, Epstein FH, Karumanchi SA. Preeclampsia and angiogenic imbalance. Annu Rev Med (2008) 59:61–78. doi:10.1146/annurev.med.59.110106.214058
87. Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med (2004) 350:672–83. doi:10.1056/NEJMoa031884
88. Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med (2006) 355:992–1005. doi:10.1056/NEJMoa055352
89. Palomaki GE, Haddow JE, Haddow HR, Salahuddin S, Geahchan C, Cerdeira AS, et al. Modeling risk for severe adverse outcomes using angiogenic factor measurements in women with suspected preterm preeclampsia. Prenat Diagn (2015) 35:386–93. doi:10.1002/pd.4554
90. Entman SS, Richardson LD. Clinical applications of the altered iron kinetics of toxemia of pregnancy. Am J Obstet Gynecol (1983) 146:568–74. doi:10.1016/0002-9378(83)90804-9
91. MacDonald AB. Human fetal borreliosis, toxemia of pregnancy, and fetal death. Zentralbl Bakteriol Mikrobiol Hyg A (1986) 263:189–200.
92. Loudon I. Some historical aspects of toxaemia of pregnancy. A review. Br J Obstet Gynaecol (1991) 98:853–8. doi:10.1111/j.1471-0528.1991.tb13505.x
93. Meziani F, Tesse A, David E, Martinez MC, Wangesteen R, Schneider F, et al. Shed membrane particles from preeclamptic women generate vascular wall inflammation and blunt vascular contractility. Am J Pathol (2006) 169:1473–83. doi:10.2353/ajpath.2006.051304
94. Sugawara J, Mitsui-Saito M, Hayashi C, Hoshiai T, Senoo M, Chisaka H, et al. Decrease and senescence of endothelial progenitor cells in patients with preeclampsia. J Clin Endocrinol Metab (2005) 90:5329–32. doi:10.1210/jc.2005-0532
95. Wang Y, Walsh SW. Placental mitochondria as a source of oxidative stress in pre-eclampsia. Placenta (1998) 19:581–6. doi:10.1016/S0143-4004(98)90018-2
96. Schumacker PT. Current paradigms in cellular oxygen sensing. Adv Exp Med Biol (2003) 543:57–71. doi:10.1007/978-1-4419-8997-0_5
97. Bradford Hill A. Environment and disease: association or causation? Proc R Soc Med (1965) 58:295–300.
98. Karmon A, Pilpel Y. Biological causal links on physiological and evolutionary time scales. Elife (2016) 5:e14424. doi:10.7554/eLife.14424
99. Kell DB. Metabolomics, modelling and machine learning in systems biology: towards an understanding of the languages of cells. The 2005 Theodor Bücher lecture. FEBS J (2006) 273:873–94. doi:10.1111/j.1742-4658.2006.05136.x
100. Gradmann C. A spirit of scientific rigour: Koch’s postulates in twentieth-century medicine. Microbes Infect (2014) 16:885–92. doi:10.1016/j.micinf.2014.08.012
101. Falkow S. Molecular Koch’s postulates applied to microbial pathogenicity. Rev Infect Dis (1988) 10(Suppl 2):S274–6. doi:10.1093/cid/10.Supplement_2.S274
102. Fredricks DN, Relman DA. Sequence-based identification of microbial pathogens – a reconsideration of Koch’s postulates. Clin Microbiol Rev (1996) 9:18–33.
103. Falkow S. Molecular Koch’s postulates applied to bacterial pathogenicity – a personal recollection 15 years later. Nat Rev Microbiol (2004) 2:67–72. doi:10.1038/nrmicro799
104. Lowe AM, Yansouni CP, Behr MA. Causality and gastrointestinal infections: Koch, Hill, and Crohn’s. Lancet Infect Dis (2008) 8:720–6. doi:10.1016/S1473-3099(08)70257-3
105. Seal JB, Morowitz M, Zaborina O, An G, Alverdy JC. The molecular Koch’s postulates and surgical infection: a view forward. Surgery (2010) 147:757–65. doi:10.1016/j.surg.2010.03.008
106. Segre JA. What does it take to satisfy Koch’s postulates two centuries later? Microbial genomics and Propionibacteria acnes. J Invest Dermatol (2013) 133:2141–2. doi:10.1038/jid.2013.260
107. Byrd AL, Segre JA. Adapting Koch’s postulates. Science (2016) 351:224–6. doi:10.1126/science.aad6753
108. Thagard P. Explanatory coherence. Behav Brain Sci (1989) 12:435–502. doi:10.1017/S0140525X00057046
109. Thagard P, Verbeurgt K. Coherence as constraint satisfaction. Cogn Sci (1998) 22:1–24. doi:10.1207/s15516709cog2201_1
111. Thagard P. Coherence, truth, and the development of scientific knowledge. Philos Sci (2007) 74:28–47. doi:10.1086/520941
112. Thagard P. Explanatory coherence. Reasoning: Studies of Human Inference and Its Foundations. (2008). p. 471–513.
113. Pennington KA, Schlitt JM, Jackson DL, Schulz LC, Schust DJ. Preeclampsia: multiple approaches for a multifactorial disease. Dis Model Mech (2012) 5:9–18. doi:10.1242/dmm.008516
114. Magee LA, Pels A, Helewa M, Rey E, Von Dadelszen P; Canadian Hypertensive Disorders of Pregnancy Working Group. Diagnosis, evaluation, and management of the hypertensive disorders of pregnancy. Pregnancy Hypertens (2014) 4:105–45. doi:10.1016/j.preghy.2014.01.003
115. Goddard KAG, Tromp G, Romero R, Olson JM, Lu Q, Xu Z, et al. Candidate-gene association study of mothers with pre-eclampsia, and their infants, analyzing 775 SNPs in 190 genes. Hum Hered (2007) 63:1–16. doi:10.1159/000097926
116. Jebbink J, Wolters A, Fernando F, Afink G, van der Post J, Ris-Stalpers C. Molecular genetics of preeclampsia and HELLP syndrome – a review. Biochim Biophys Acta (2012) 1822:1960–9. doi:10.1016/j.bbadis.2012.08.004
117. Fong FM, Sahemey MK, Hamedi G, Eyitayo R, Yates D, Kuan V, et al. Maternal genotype and severe preeclampsia: a HuGE review. Am J Epidemiol (2014) 180:335–45. doi:10.1093/aje/kwu151
118. Williamson RD, O’Keeffe GW, Kenny LC. Activin signalling and pre-eclampsia: from genetic risk to pre-symptomatic biomarker. Cytokine (2015) 71:360–5. doi:10.1016/j.cyto.2014.11.017
120. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature (2009) 461:747–53. doi:10.1038/nature08494
121. Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci U S A (2012) 109:1193–8. doi:10.1073/pnas.1119675109
122. Carreiras M, Montagnani S, Layrisse Z. Preeclampsia: a multifactorial disease resulting from the interaction of the feto-maternal HLA genotype and HCMV infection. Am J Reprod Immunol (2002) 48:176–83. doi:10.1034/j.1600-0897.2002.01076.x
123. McCarthy CM, Kenny LC. Immunostimulatory role of mitochondrial DAMPs: alarming for pre-eclampsia? Am J Reprod Immunol (2016) 76:341–7. doi:10.1111/aji.12526
124. McCarthy CM, Kenny LC. Mitochondrial [dys]function; culprit in pre-eclampsia? Clin Sci (Lond) (2016) 130:1179–84. doi:10.1042/CS20160103
125. Kell DB. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med Genomics (2009) 2:2. doi:10.1186/1755-8794-2-2
126. Kell DB, Pretorius E. Serum ferritin is an important disease marker, and is mainly a leakage product from damaged cells. Metallomics (2014) 6:748–73. doi:10.1039/C3MT00347G
127. Pretorius E, Kell DB. Diagnostic morphology: biophysical indicators for iron-driven inflammatory diseases. Integr Biol (2014) 6:486–510. doi:10.1039/c4ib00025k
128. Kell DB, Pretorius E. The simultaneous occurrence of both hypercoagulability and hypofibrinolysis in blood and serum during systemic inflammation, and the roles of iron and fibrin(ogen). Integr Biol (2015) 7:24–52. doi:10.1039/c4ib00173g
129. Pretorius E, Oberholzer HM, van der Spuy WJ, Swanepoel AC, Soma P. Qualitative scanning electron microscopy analysis of fibrin networks and platelet abnormalities in diabetes. Blood Coagul Fibrinol (2011) 22:463–7. doi:10.1097/MBC.0b013e3283468a0d
130. Pretorius E, Steyn H, Engelbrecht M, Swanepoel AC, Oberholzer HM. Differences in fibrin fiber diameters in healthy individuals and thromboembolic ischemic stroke patients. Blood Coagul Fibrinolysis (2011) 22:696–700. doi:10.1097/MBC.0b013e32834bdb32
131. Pretorius E, Swanepoel AC, Oberholzer HM, van der Spuy WJ, Duim W, Wessels PF. A descriptive investigation of the ultrastructure of fibrin networks in thrombo-embolic ischemic stroke. J Thromb Thrombolysis (2011) 31:507–13. doi:10.1007/s11239-010-0538-5
132. Pretorius E, Du Plooy J, Soma P, Gasparyan AY. An ultrastructural analysis of platelets, erythrocytes, white blood cells, and fibrin network in systemic lupus erythematosus. Rheumatol Int (2014) 34:1005–9. doi:10.1007/s00296-013-2817-x
133. Pretorius E, Swanepoel AC, Buys AV, Vermeulen N, Duim W, Kell DB. Eryptosis as a marker of Parkinson’s disease. Aging (2014) 6:788–819. doi:10.18632/aging.100695
134. Pretorius E, Bester J, Vermeulen N, Alummoottil S, Soma P, Buys AV, et al. Poorly controlled type 2 diabetes is accompanied by significant morphological and ultrastructural changes in both erythrocytes and in thrombin-generated fibrin: implications for diagnostics. Cardiovasc Diabetol (2015) 13:30. doi:10.1186/s12933-015-0192-5
135. Bester J, Buys AV, Lipinski B, Kell DB, Pretorius E. High ferritin levels have major effects on the morphology of erythrocytes in Alzheimer’s disease. Front Aging Neurosci (2013) 5:00088. doi:10.3389/fnagi.2013.00088
136. Pretorius E, Bester J, Vermeulen N, Lipinski B, Gericke GS, Kell DB. Profound morphological changes in the erythrocytes and fibrin networks of patients with hemochromatosis or with hyperferritinemia, and their normalization by iron chelators and other agents. PLoS One (2014) 9:e85271. doi:10.1371/journal.pone.0085271
137. Pretorius E, Olumuyiwa-Akeredolu OO, Mbotwe S, Bester J. Erythrocytes and their role as health indicator: using structure in a patient-orientated precision medicine approach. Blood Rev (2016) 30:263–74. doi:10.1016/j.blre.2016.01.001
138. Todros T, Vasario E, Cardaropoli S. Preeclampsia as an infectious disease. Exp Rev Obstet Gynecol (2007) 2:735–41. doi:10.1586/17474108.2.6.735
139. Kell DB, Potgieter M, Pretorius E. Individuality, phenotypic differentiation, dormancy and ‘persistence’ in culturable bacterial systems: commonalities shared by environmental, laboratory, and clinical microbiology. F1000Research (2015) 4:179. doi:10.12688/f1000research.6709.1
140. Kell DB, Pretorius E. On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: the central roles of LPS and LPS-induced cell death. Integr Biol (2015) 7:1339–77. doi:10.1039/C5IB00158G
141. Potgieter M, Bester J, Kell DB, Pretorius E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol Rev (2015) 39:567–91. doi:10.1093/femsre/fuv013
142. Pretorius E, Akeredolu O-O, Soma P, Kell DB. Major involvement of bacterial components in rheumatoid arthritis and its accompanying oxidative stress, systemic inflammation and hypercoagulability. Exp Biol Med (Forthcoming 2016).
143. Pretorius E, Bester J, Kell DB. A bacterial component to Alzheimer-type dementia seen via a systems biology approach that links iron dysregulation and inflammagen shedding to disease. J Alzheimers Dis (2016) 53:1237–56. doi:10.3233/JAD-160318
144. Proal AD, Albert PJ, Marshall TG. The human microbiome and autoimmunity. Curr Opin Rheumatol (2013) 25:234–40. doi:10.1097/BOR.0b013e32835cedbf
145. Mangin M, Sinha R, Fincher K. Inflammation and vitamin D: the infection connection. Inflamm Res (2014) 63:803–19. doi:10.1007/s00011-014-0755-z
146. Proal AD, Albert PJ, Marshall TG. Inflammatory disease and the human microbiome. Discov Med (2014) 17:257–65.
147. Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Braak H, Bearer EL, et al. Microbes and Alzheimer’s disease. J Alzheimers Dis (2016) 51:979–84. doi:10.3233/JAD-160152
148. Morath S, Geyer A, Hartung T. Structure-function relationship of cytokine induction by lipoteichoic acid from Staphylococcus aureus. J Exp Med (2001) 193:393–7. doi:10.1084/jem.193.3.393
149. Postgate JR. Viability measurements and the survival of microbes under minimum stress. Adv Micro Physiol (1967) 1:1–23. doi:10.1016/S0065-2911(08)60248-9
150. Postgate JR. Viable counts and viability. Methods Microbiol (1969) 1:611–28. doi:10.1016/S0580-9517(08)70149-1
151. Postgate JR. Death in microbes and macrobes. In: Gray TRG, Postgate JR, editors. In the Survival of Vegetative Microbes. Cambridge: Cambridge University Press (1976). p. 1–19.
152. Staley JT, Konopka A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol (1985) 39:321–46. doi:10.1146/annurev.mi.39.100185.001541
153. Tanaka T, Kawasaki K, Daimon S, Kitagawa W, Yamamoto K, Tamaki H, et al. A hidden pitfall in the preparation of agar media undermines microorganism cultivability. Appl Environ Microbiol (2014) 80:7659–66. doi:10.1128/AEM.02741-14
154. Nichols D, Cahoon N, Trakhtenberg EM, Pham L, Mehta A, Belanger A, et al. Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl Environ Microbiol (2010) 76:2445–50. doi:10.1128/AEM.01754-09
155. Browne HP, Forster SC, Anonye BO, Kumar N, Neville BA, Stares MD, et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature (2016) 533:543–6. doi:10.1038/nature17645
156. Kaprelyants AS, Gottschal JC, Kell DB. Dormancy in non-sporulating bacteria. FEMS Microbiol Rev (1993) 10:271–86. doi:10.1111/j.1574-6968.1993.tb05871.x
157. Kell DB, Kaprelyants AS, Weichart DH, Harwood CL, Barer MR. Viability and activity in readily culturable bacteria: a review and discussion of the practical issues. Antonie Van Leeuwenhoek (1998) 73:169–87. doi:10.1023/A:1000664013047
158. Saito A, Rolfe RD, Edelstein PH, Finegold SM. Comparison of liquid growth media for Legionella pneumophila. J Clin Microbiol (1981) 14:623–7.
159. Meyer RD. Legionella infections – a review of 5 years of research. Rev Infect Dis (1983) 5:258–78. doi:10.1093/clinids/5.2.258
160. Barker J, Farrell ID, Hutchison JG. Factors affecting growth of Legionella pneumophila in liquid media. J Med Microbiol (1986) 22:97–100. doi:10.1099/00222615-22-2-97
161. Molinari J. Legionella and human disease: part 1: a path of scientific and community discovery. Compend Contin Educ Dent (1997) 18:556–9.
162. Maiwald M, Schuhmacher F, Ditton HJ, Vonherbay A. Environmental occurrence of the Whipple’s disease bacterium (Tropheryma whippelii). Appl Environ Microbiol (1998) 64:760–2.
163. Maiwald M, Relman DA. Whipple’s disease and Tropheryma whippelii: secrets slowly revealed. Clin Infect Dis (2001) 32:457–63. doi:10.1086/318512
164. Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, et al. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci U S A (2009) 106:4430–4. doi:10.1073/pnas.0812074106
165. Omsland A. Axenic growth of Coxiella burnetii. Adv Exp Med Biol (2012) 984:215–29. doi:10.1007/978-94-007-4315-1_11
166. Marshall BJ. One hundred years of discovery and rediscovery of Helicobacter pylori and its association with peptic ulcer disease. In: Mobley HLT, Mendz GL, Hazell SL, editors. Helicobacter pylori: Physiology and Genetics. Washington, DC: ASM Press (2001). p. 19–24.
168. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet (1984) 1:1311–5. doi:10.1016/S0140-6736(84)91816-6
169. Marshall BJ, Armstrong JA, Mcgechie DB, Glancy RJ. Attempt to fulfil Koch’s postulates for pyloric Campylobacter. Med J Aust (1985) 142:436–9.
170. Kaprelyants AS, Kell DB. Dormancy in stationary-phase cultures of Micrococcus luteus: flow cytometric analysis of starvation and resuscitation. Appl Environ Microbiol (1993) 59:3187–96.
171. Kaprelyants AS, Mukamolova GV, Kell DB. Estimation of dormant Micrococcus luteus cells by penicillin lysis and by resuscitation in cell-free spent medium at high dilution. FEMS Microbiol Lett (1994) 115:347–52. doi:10.1111/j.1574-6968.1994.tb06662.x
172. Wayne LG. Dormancy of Mycobacterium tuberculosis and latency of disease. Eur J Clin Microbiol Infect Dis (1994) 13:908–14. doi:10.1007/BF02111491
173. Kana BD, Gordhan BG, Downing KJ, Sung N, Vostroktunova G, Machowski EE, et al. The resuscitation-promoting factors of Mycobacterium tuberculosis are required for virulence and resuscitation from dormancy but are collectively dispensable for growth in vitro. Mol Microbiol (2008) 67:672–84. doi:10.1111/j.1365-2958.2007.06078.x
174. Gengenbacher M, Kaufmann SHE. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev (2012) 36:514–32. doi:10.1111/j.1574-6976.2012.00331.x
175. Alnimr AM. Dormancy models for Mycobacterium tuberculosis: a minireview. Braz J Microbiol (2015) 46:641–7. doi:10.1590/S1517-838246320140507
176. Shleeva M, Kondratieva T, Rubakova E, Vostroknutova G, Kaprelyants A, Apt A. Reactivation of dormant “non-culturable” Mycobacterium tuberculosis developed in vitro after injection in mice: both the dormancy depth and host genetics influence the outcome. Microb Pathog (2015) 78:63–6. doi:10.1016/j.micpath.2014.11.016
177. Mukamolova GV, Kaprelyants AS, Young DI, Young M, Kell DB. A bacterial cytokine. Proc Natl Acad Sci U S A (1998) 95:8916–21. doi:10.1073/pnas.95.15.8916
178. Mukamolova GV, Turapov OA, Kazarian K, Telkov M, Kaprelyants AS, Kell DB, et al. The rpf gene of Micrococcus luteus encodes an essential secreted growth factor. Mol Microbiol (2002) 46:611–21. doi:10.1046/j.1365-2958.2002.03183.x
179. Mukamolova GV, Turapov OA, Young DI, Kaprelyants AS, Kell DB, Young M. A family of autocrine growth factors in Mycobacterium tuberculosis. Mol Microbiol (2002) 46:623–35. doi:10.1046/j.1365-2958.2002.03184.x
180. Mukamolova GV, Murzin AG, Salina EG, Demina GR, Kell DB, Kaprelyants AS, et al. Muralytic activity of Micrococcus luteus Rpf and its relationship to physiological activity in promoting bacterial growth and resuscitation. Mol Microbiol (2006) 59:84–98. doi:10.1111/j.1365-2958.2005.04930.x
181. Mukamolova GV, Turapov O, Malkin J, Woltmann G, Barer MR. Resuscitation-promoting factors reveal an occult population of tubercle bacilli in sputum. Am J Respir Crit Care Med (2010) 181:174–80. doi:10.1164/rccm.200905-0661OC
182. Yeremeev VV, Kondratieva TK, Rubakova EI, Petrovskaya SN, Kazarian KA, Telkov MV, et al. Proteins of the Rpf family: immune cell reactivity and vaccination efficacy against tuberculosis in mice. Infect Immun (2003) 71:4789–94. doi:10.1128/IAI.71.8.4789-4794.2003
183. Zvi A, Ariel N, Fulkerson J, Sadoff JC, Shafferman A. Whole genome identification of Mycobacterium tuberculosis vaccine candidates by comprehensive data mining and bioinformatic analyses. BMC Med Genomics (2008) 1:18. doi:10.1186/1755-8794-1-18
184. Dye C, Scheele S, Dolin P, Pathania V, Raviglione RC. Global burden of tuberculosis – estimated incidence, prevalence, and mortality by country. JAMA (1999) 282:677–86. doi:10.1001/jama.282.7.677
185. Flynn JL, Chan J. Tuberculosis: latency and reactivation. Infect Immun (2001) 69:4195–201. doi:10.1128/IAI.69.7.4195-4201.2001
186. Wayne LG, Sohaskey CD. Nonreplicating persistence of Mycobacterium tuberculosis. Annu Rev Microbiol (2001) 55:139–63. doi:10.1146/annurev.micro.55.1.139
187. Gomez JE, McKinney JD. M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis (Edinb) (2004) 84:29–44. doi:10.1016/j.tube.2003.08.003
188. Amieva MR, El-Omar EM. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology (2008) 134:306–23. doi:10.1053/j.gastro.2007.11.009
189. Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology (2009) 136:1863–73. doi:10.1053/j.gastro.2009.01.073
190. Domingue GJ, Woody HB. Bacterial persistence and expression of disease. Clin Microbiol Rev (1997) 10:320–44.
191. Young D, Stark J, Kirschner D. Systems biology of persistent infection: tuberculosis as a case study. Nat Rev Microbiol (2008) 6:520–8. doi:10.1038/nrmicro1919
192. Lewis K. Persister cells. Annu Rev Microbiol (2010) 64:357–72. doi:10.1146/annurev.micro.112408.134306
193. Amato SM, Fazen CH, Henry TC, Mok WWK, Orman MA, Sandvik EL, et al. The role of metabolism in bacterial persistence. Front Microbiol (2014) 5:70. doi:10.3389/Fmicb.2014.00070
194. Conlon BP. Staphylococcus aureus chronic and relapsing infections: evidence of a role for persister cells: an investigation of persister cells, their formation and their role in S. aureus disease. Bioessays (2014) 36:991–6. doi:10.1002/bies.201400080
195. Kester JC, Fortune SM. Persisters and beyond: mechanisms of phenotypic drug resistance and drug tolerance in bacteria. Crit Rev Biochem Mol Biol (2014) 49:91–101. doi:10.3109/10409238.2013.869543
196. Levin BR, Concepción-Acevedo J, Udekwu KI. Persistence: a copacetic and parsimonious hypothesis for the existence of non-inherited resistance to antibiotics. Curr Opin Microbiol (2014) 21:18–21. doi:10.1016/j.mib.2014.06.016
197. Maisonneuve E, Gerdes K. Molecular mechanisms underlying bacterial persisters. Cell (2014) 157:539–48. doi:10.1016/j.cell.2014.02.050
198. Rank RG, Yeruva L. Hidden in plain sight: chlamydial gastrointestinal infection and its relevance to persistence in human genital infection. Infect Immun (2014) 82:1362–71. doi:10.1128/IAI.01244-13
200. Orman MA, Mok WWK, Brynildsen MP. Aminoglycoside-enabled elucidation of bacterial persister metabolism. Curr Protoc Microbiol (2015) 36:1–14. doi:10.1002/9780471729259.mc1709s36
201. Stepanyan K, Wenseleers T, Duéñez-Guzmán EA, Muratori F, Van Den Bergh B, Verstraeten N, et al. Fitness trade-offs explain low levels of persister cells in the opportunistic pathogen Pseudomonas aeruginosa. Mol Ecol (2015) 24:1572–83. doi:10.1111/mec.13127
203. Pu Y, Zhao Z, Li Y, Zou J, Ma Q, Zhao Y, et al. Enhanced efflux activity facilitates drug tolerance in dormant bacterial cells. Mol Cell (2016) 62:284–94. doi:10.1016/j.molcel.2016.03.035
204. Dehio C, Berry C, Bartenschlager R. Persistent intracellular pathogens. FEMS Microbiol Rev (2012) 36:513. doi:10.1111/j.1574-6976.2012.00336.x
205. Prideaux B, Via LE, Zimmerman MD, Eum S, Sarathy J, O’Brien P, et al. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med (2015) 21:1223–7. doi:10.1038/nm.3937
206. Dobson PD, Kell DB. Carrier-mediated cellular uptake of pharmaceutical drugs: an exception or the rule? Nat Rev Drug Discov (2008) 7:205–20. doi:10.1038/nrd2438
207. Kell DB, Dobson PD, Oliver SG. Pharmaceutical drug transport: the issues and the implications that it is essentially carrier-mediated only. Drug Discov Today (2011) 16:704–14. doi:10.1016/j.drudis.2011.05.010
208. Kell DB, Dobson PD, Bilsland E, Oliver SG. The promiscuous binding of pharmaceutical drugs and their transporter-mediated uptake into cells: what we (need to) know and how we can do so. Drug Discov Today (2013) 18:218–39. doi:10.1016/j.drudis.2012.11.008
209. Kell DB, Oliver SG. How drugs get into cells: tested and testable predictions to help discriminate between transporter-mediated uptake and lipoidal bilayer diffusion. Front Pharmacol (2014) 5:231. doi:10.3389/fphar.2014.00231
210. Thwaites GE, Gant V. Are bloodstream leukocytes Trojan horses for the metastasis of Staphylococcus aureus? Nat Rev Microbiol (2011) 9:215–22. doi:10.1038/nrmicro2508
211. Seubert A, Schulein R, Dehio C. Bacterial persistence within erythrocytes: a unique pathogenic strategy of Bartonella spp. Int J Med Microbiol (2002) 291:555–60. doi:10.1078/1438-4221-00167
212. Mysorekar IU, Hultgren SJ. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc Natl Acad Sci U S A (2006) 103:14170–5. doi:10.1073/pnas.0602136103
213. Blango MG, Mulvey MA. Persistence of uropathogenic Escherichia coli in the face of multiple antibiotics. Antimicrob Agents Chemother (2010) 54:1855–63. doi:10.1128/AAC.00014-10
214. Chen SL, Wu M, Henderson JP, Hooton TM, Hibbing ME, Hultgren SJ, et al. Genomic diversity and fitness of E. coli strains recovered from the intestinal and urinary tracts of women with recurrent urinary tract infection. Sci Transl Med (2013) 5:184ra160. doi:10.1126/scitranslmed.3005497
215. Goneau LW, Hannan TJ, Macphee RA, Schwartz DJ, Macklaim JM, Gloor GB, et al. Subinhibitory antibiotic therapy alters recurrent urinary tract infection pathogenesis through modulation of bacterial virulence and host immunity. MBio (2015) 6:e00356–15. doi:10.1128/mBio.00356-15
216. Sullivan WJ Jr, Jeffers V. Mechanisms of Toxoplasma gondii persistence and latency. FEMS Microbiol Rev (2012) 36:717–33. doi:10.1111/j.1574-6976.2011.00305.x
217. Posey JE, Gherardini FC. Lack of a role for iron in the Lyme disease pathogen. Science (2000) 288:1651–3. doi:10.1126/science.288.5471.1651
218. Aguirre JD, Clark HM, Mcilvin M, Vazquez C, Palmere SL, Grab DJ, et al. A manganese-rich environment supports superoxide dismutase activity in a Lyme disease pathogen, Borrelia burgdorferi. J Biol Chem (2013) 288:8468–78. doi:10.1074/jbc.M112.433540
219. Weinberg ED. Nutritional immunity. Host’s attempt to withold iron from microbial invaders. JAMA (1975) 231:39–41. doi:10.1001/jama.1975.03240130021018
220. Weinberg ED. Iron withholding: a defense against infection and neoplasia. Physiol Rev (1984) 64:65–102.
221. Marx JJM. Iron and infection: competition between host and microbes for a precious element. Best Pract Res Clin Haematol (2002) 15:411–26. doi:10.1053/beha.2002.0001
222. Fischbach MA, Lin HN, Liu DR, Walsh CT. How pathogenic bacteria evade mammalian sabotage in the battle for iron. Nat Chem Biol (2006) 2:132–8. doi:10.1038/nchembio771
223. Weinberg ED, Miklossy J. Iron withholding: a defense against disease. J Alzheimers Dis (2008) 13:451–63.
224. Reid DW, Anderson GJ, Lamont IL. Role of lung iron in determining the bacterial and host struggle in cystic fibrosis. Am J Physiol Lung Cell Mol Physiol (2009) 297:L795–802. doi:10.1152/ajplung.00132.2009
225. Weinberg ED. Iron availability and infection. Biochim Biophys Acta (2009) 1790:600–5. doi:10.1016/j.bbagen.2008.07.002
226. Chu BC, Garcia-Herrero A, Johanson TH, Krewulak KD, Lau CK, Peacock RS, et al. Siderophore uptake in bacteria and the battle for iron with the host; a bird’s eye view. Biometals (2010) 23:601–11. doi:10.1007/s10534-010-9361-x
227. Nairz M, Schroll A, Sonnweber T, Weiss G. The struggle for iron – a metal at the host-pathogen interface. Cell Microbiol (2010) 12:1691–702. doi:10.1111/j.1462-5822.2010.01529.x
228. Skaar EP. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog (2010) 6:e1000949. doi:10.1371/journal.ppat.1000949
229. Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol (2012) 10:525–37. doi:10.1038/nrmicro2836
230. Cassat JE, Skaar EP. Iron in infection and immunity. Cell Host Microbe (2013) 13:509–19. doi:10.1016/j.chom.2013.04.010
231. Deriu E, Liu JZ, Pezeshki M, Edwards RA, Ochoa RJ, Contreras H, et al. Probiotic bacteria reduce Salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe (2013) 14:26–37. doi:10.1016/j.chom.2013.06.007
232. Leal SM Jr, Roy S, Vareechon C, Carrion S, Clark H, Lopez-Berges MS, et al. Targeting iron acquisition blocks infection with the fungal pathogens Aspergillus fumigatus and Fusarium oxysporum. PLoS Pathog (2013) 9:e1003436. doi:10.1371/journal.ppat.1003436
233. Silva-Gomes S, Vale-Costa S, Appelberg R, Gomes MS. Iron in intracellular infection: to provide or to deprive? Front Cell Infect Microbiol (2013) 3:96. doi:10.3389/fcimb.2013.00096
234. Armitage AE, Drakesmith H. The battle for iron. Science (2014) 346:1299–300. doi:10.1126/science.aaa2468
235. Becker KW, Skaar EP. Metal limitation and toxicity at the interface between host and pathogen. FEMS Microbiol Rev (2014) 38:1235–49. doi:10.1111/1574-6976.12087
236. Diaz-Ochoa VE, Jellbauer S, Klaus S, Raffatellu M. Transition metal ions at the crossroads of mucosal immunity and microbial pathogenesis. Front Cell Infect Microbiol (2014) 4:2. doi:10.3389/fcimb.2014.00002
237. Potrykus J, Ballou ER, Childers DS, Brown AJP. Conflicting interests in the pathogen-host tug of war: fungal micronutrient scavenging versus mammalian nutritional immunity. PLoS Pathog (2014) 10:e1003910. doi:10.1371/journal.ppat.1003910
238. Barber MF, Elde NC. Buried treasure: evolutionary perspectives on microbial iron piracy. Trends Genet (2015) 31:627–36. doi:10.1016/j.tig.2015.09.001
239. Nairz M, Ferring-Appel D, Casarrubea D, Sonnweber T, Viatte L, Schroll A, et al. Iron regulatory proteins mediate host resistance to Salmonella infection. Cell Host Microbe (2015) 18:254–61. doi:10.1016/j.chom.2015.06.017
240. Raymond KN, Allred BE, Sia AK. Coordination chemistry of microbial iron transport. Acc Chem Res (2015) 48:2496–505. doi:10.1021/acs.accounts.5b00301
241. Subashchandrabose S, Mobley HLT. Back to the metal age: battle for metals at the host-pathogen interface during urinary tract infection. Metallomics (2015) 7:935–42. doi:10.1039/c4mt00329b
242. Wessling-Resnick M. Nramp1 and other transporters involved in metal withholding during infection. J Biol Chem (2015) 290:18984–90. doi:10.1074/jbc.R115.643973
243. Xia Y, Farah N, Maxan A, Zhou J, Lehmann C. Therapeutic iron restriction in sepsis. Med Hypotheses (2016) 89:37–9. doi:10.1016/j.mehy.2016.01.018
244. Entman SS, Richardson LD, Killam AP. Elevated serum ferritin in the altered ferrokinetics of toxemia of pregnancy. Am J Obstet Gynecol (1982) 144:418–22. doi:10.1016/0002-9378(82)90247-2
245. Samuels P, Main EK, Mennuti MT, Gabbe SG. The origin of increased serum iron in pregnancy-induced hypertension. Am J Obstet Gynecol (1987) 157:721–5. doi:10.1016/S0002-9378(87)80037-6
246. Raman L, Pawashe AB, Yasodhara P. Hyperferritinemia in pregnancy induced hypertension and eclampsia. J Postgrad Med (1992) 38:65–7.
247. Hubel CA, Kozlov AV, Kagan VE, Evans RW, Davidge ST, McLaughlin MK, et al. Decreased transferrin and increased transferrin saturation in sera of women with preeclampsia: implications for oxidative stress. Am J Obstet Gynecol (1996) 175:692–700. doi:10.1053/ob.1996.v175.a74252
248. Lao TT, Tam KF, Chan LY. Third trimester iron status and pregnancy outcome in non-anaemic women; pregnancy unfavourably affected by maternal iron excess. Hum Reprod (2000) 15:1843–8. doi:10.1093/humrep/15.8.1843
249. Rayman MP, Barlis J, Evans RW, Redman CW, King LJ. Abnormal iron parameters in the pregnancy syndrome preeclampsia. Am J Obstet Gynecol (2002) 187:412–8. doi:10.1067/mob.2002.123895
250. Serdar Z, Gür E, Develioğlu O. Serum iron and copper status and oxidative stress in severe and mild preeclampsia. Cell Biochem Funct (2006) 24:209–15. doi:10.1002/cbf.1235
251. Smith TG, Robbins PA. Iron, pre-eclampsia and hypoxia-inducible factor. BJOG (2007) 114:1581–2. doi:10.1111/j.1471-0528.2007.01490.x
252. Bhatla N, Kaul N, Lal N, Kriplani A, Agarwal N, Saxena R, et al. Comparison of effect of daily versus weekly iron supplementation during pregnancy on lipid peroxidation. J Obstet Gynaecol Res (2009) 35:438–45. doi:10.1111/j.1447-0756.2008.00972.x
253. Siddiqui IA, Jaleel A, Kadri HM, Saeed WA, Tamimi W. Iron status parameters in preeclamptic women. Arch Gynecol Obstet (2011) 284:587–91. doi:10.1007/s00404-010-1728-2
254. Fatima N, Islam F, Noor L, Das SR, Zeba D, Zesmin F. Serum ferritin in preeclampsia and eclampsia: a case control study. Faridpur Med Coll J (2013) 8:18–21. doi:10.3329/fmcj.v8i1.16892
255. Kandi S, Sudhakar T, Ramadevi C, Venugopal B, Rajkumar, Rafi M, et al. Pre eclampsia and iron status: a review. Am J Med Biol Res (2014) 2:121–3. doi:10.12691/ajmbr-2-6-1
256. Negi R, Pande D, Karki K, Kumar A, Khanna RS, Khanna HD. Association of oxidative DNA damage, protein oxidation and antioxidant function with oxidative stress induced cellular injury in pre-eclamptic/eclamptic mothers during fetal circulation. Chem Biol Interact (2014) 208:77–83. doi:10.1016/j.cbi.2013.11.010
257. Bester J, Soma P, Kell DB, Pretorius E. Viscoelastic and ultrastructural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget (2015) 6:35284–303. doi:10.18632/oncotarget.6074
258. Pretorius E, Mbotwe S, Bester J, Robinson C, Kell DB. Acute induction of anomalous blood clotting by highly substoichiometric levels of bacterial lipopolysaccharide (LPS). bioRxiv (2016) 053538. doi:10.1101/053538
259. Pretorius E, Mbotwe S, Bester J, Robinson CJ, Kell DB. Acute induction of anomalous and amyloidogenic blood clotting by molecular amplification of highly substoichiometric levels of bacterial lipopolysaccharide. J R Soc Interface (2016) 123:20160539. doi:10.1098/rsif.2016.0539
260. Lin IH, Miller DS, Bertics PJ, Murphy CJ, De Pablo JJ, Abbott NL. Endotoxin-induced structural transformations in liquid crystalline droplets. Science (2011) 332:1297–300. doi:10.1126/science.1195639
261. Miller DS, Abbott NL. Influence of droplet size, pH and ionic strength on endotoxin-triggered ordering transitions in liquid crystalline droplets. Soft Matter (2013) 9:374–82. doi:10.1039/C2SM26811F
262. Rahman MM, Abe SK, Rahman MS, Kanda M, Narita S, Bilano V, et al. Maternal anemia and risk of adverse birth and health outcomes in low- and middle-income countries: systematic review and meta-analysis. Am J Clin Nutr (2016) 103:495–504. doi:10.3945/ajcn.115.107896
263. Harris CM, Kell DB. The estimation of microbial biomass. Biosensors (1985) 1:17–84. doi:10.1016/0265-928X(85)85005-7
264. Kaprelyants AS, Kell DB. Do bacteria need to communicate with each other for growth? Trends Microbiol (1996) 4:237–42. doi:10.1016/0966-842X(96)10035-4
265. Domingue GJ. Electron dense cytoplasmic particles and chronic infection – a bacterial pleomorphy hypothesis. Endocytobiosis Cell Res (1995) 11:19–40.
266. Domingue GJ Sr. Cryptic bacterial infection in chronic prostatitis: diagnostic and therapeutic implications. Curr Opin Urol (1998) 8:45–9. doi:10.1097/00042307-199801000-00009
268. Domingue GJ. Demystifying pleomorphic forms in persistence and expression of disease: are they bacteria, and is peptidoglycan the solution? Discov Med (2010) 10:234–46.
269. Davey HM, Kell DB. Flow cytometry and cell sorting of heterogeneous microbial populations: the importance of single-cell analysis. Microbiol Rev (1996) 60:641–96.
270. Kaprelyants AS, Kell DB. Rapid assessment of bacterial viability and vitality using rhodamine 123 and flow cytometry. J Appl Bacteriol (1992) 72:410–22. doi:10.1111/j.1365-2672.1992.tb01854.x
271. Gant VA, Warnes G, Phillips I, Savidge GF. The application of flow cytometry to the study of bacterial responses to antibiotics. J Med Microbiol (1993) 39:147–54. doi:10.1099/00222615-39-2-147
272. Mason DJ, Shanmuganathan S, Mortimer FC, Gant VA. A fluorescent Gram stain for flow cytometry and epifluorescence microscopy. Appl Environ Microbiol (1998) 64:2681–5.
273. Nebe-Von-Caron G, Stephens PJ, Hewitt CJ, Powell JR, Badley RA. Analysis of bacterial function by multi-colour fluorescence flow cytometry and single cell sorting. J Microbiol Methods (2000) 42:97–114. doi:10.1016/S0167-7012(00)00181-0
274. Shapiro HM, Nebe-Von-Caron G. Multiparameter flow cytometry of bacteria. Methods Mol Biol (2004) 263:33–44. doi:10.1385/1-59259-773-4:033
275. Müller S, Nebe-Von-Caron G. Functional single-cell analyses: flow cytometry and cell sorting of microbial populations and communities. FEMS Microbiol Rev (2010) 34:554–87. doi:10.1111/j.1574-6976.2010.00214.x
276. Davey HM. Life, death, and in-between: meanings and methods in microbiology. Appl Environ Microbiol (2011) 77:5571–6. doi:10.1128/AEM.00744-11
277. Davey HM, Hexley P. Red but not dead? Membranes of stressed Saccharomyces cerevisiae are permeable to propidium iodide. Environ Microbiol (2011) 13:163–71. doi:10.1111/j.1462-2920.2010.02317.x
278. Kell DB, Markx GH, Davey CL, Todd RW. Real-time monitoring of cellular biomass: methods and applications. Trends Anal Chem (1990) 9:190–4. doi:10.1016/0165-9936(90)87042-K
279. Kell DB, Sonnleitner B. GMP – good modelling practice: an essential component of good manufacturing practice. Trends Biotechnol (1995) 13:481–92. doi:10.1016/S0167-7799(00)89006-X
281. Harris CM, Todd RW, Bungard SJ, Lovitt RW, Morris JG, Kell DB. The dielectric permittivity of microbial suspensions at radio frequencies: a novel method for the estimation of microbial biomass. Enzyme Microbiol Technol (1987) 9:181–6. doi:10.1016/0141-0229(87)90075-5
282. Kell DB, Davey CL. Conductimetric and impedimetric devices. In: Cass AEG, editor. Biosensors: A Practical Approach. Oxford: IRL Press (1990). p. 125–54.
283. Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A (1977) 74:5088–90. doi:10.1073/pnas.74.11.5088
284. Petti CA. Detection and identification of microorganisms by gene amplification and sequencing. Clin Infect Dis (2007) 44:1108–14. doi:10.1086/512818
285. Klouche M, Schröder U. Rapid methods for diagnosis of bloodstream infections. Clin Chem Lab Med (2008) 46:888–908. doi:10.1515/CCLM.2008.157
286. Woo PCY, Lau SKP, Teng JLL, Tse H, Yuen KY. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect (2008) 14:908–34. doi:10.1111/j.1469-0691.2008.02070.x
287. Cherkaoui A, Emonet S, Ceroni D, Candolfi B, Hibbs J, Francois P, et al. Development and validation of a modified broad-range 16S rDNA PCR for diagnostic purposes in clinical microbiology. J Microbiol Methods (2009) 79:227–31. doi:10.1016/j.mimet.2009.09.014
288. Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A (2011) 108(Suppl 1):4516–22. doi:10.1073/pnas.1000080107
289. Kramski M, Gaeguta AJ, Lichtfuss GF, Rajasuriar R, Crowe SM, French MA, et al. Novel sensitive real-time PCR for quantification of bacterial 16S rRNA genes in plasma of HIV-infected patients as a marker for microbial translocation. J Clin Microbiol (2011) 49:3691–3. doi:10.1128/JCM.01018-11
290. Kämpfer P. Systematics of prokaryotes: the state of the art. Antonie Van Leeuwenhoek (2012) 101:3–11. doi:10.1007/s10482-011-9660-4
291. Ohlin A, Bäckman A, Ewald U, Schollin J, Björkqvist M. Diagnosis of neonatal sepsis by broad-range 16S real-time polymerase chain reaction. Neonatology (2012) 101:241–6. doi:10.1159/000334655
292. Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol (2013) 31:814–21. doi:10.1038/nbt.2676
293. Mizrahi-Man O, Davenport ER, Gilad Y. Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: evaluation of effective study designs. PLoS One (2013) 8:e53608. doi:10.1371/journal.pone.0053608
294. Valencia-Shelton F, Loeffelholz M. Nonculture techniques for the detection of bacteremia and fungemia. Future Microbiol (2014) 9:543–59. doi:10.2217/fmb.14.8
295. Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer KH, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol (2014) 12:635–45. doi:10.1038/nrmicro3330
296. Zumla A, Al-Tawfiq JA, Enne VI, Kidd M, Drosten C, Breuer J, et al. Rapid point of care diagnostic tests for viral and bacterial respiratory tract infections – needs, advances, and future prospects. Lancet Infect Dis (2014) 14:1123–35. doi:10.1016/S1473-3099(14)70827-8
297. D’Amore R, Ijaz UZ, Schirmer M, Kenny JG, Gregory R, Darby AC, et al. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genomics (2016) 17:55. doi:10.1186/s12864-015-2194-9
298. Versalovic J, Carroll KC, Funke G, Jorgensen JH, Landry ML, Warnock DW. Manual of Clinical Microbiology. 10th ed. Washington: American Society of Microbiology (2011).
299. Nikkari S, McLaughlin IJ, Bi W, Dodge DE, Relman DA. Does blood of healthy subjects contain bacterial ribosomal DNA? J Clin Microbiol (2001) 39:1956–9. doi:10.1128/JCM.39.5.1956-1959.2001
300. McLaughlin RW, Vali H, Lau PC, Palfree RGE, De Ciccio A, Sirois M, et al. Are there naturally occurring pleomorphic bacteria in the blood of healthy humans? J Clin Microbiol (2002) 40:4771–5. doi:10.1128/JCM.40.12.4771-4775.2002
301. Moriyama K, Ando C, Tashiro K, Kuhara S, Okamura S, Nakano S, et al. Polymerase chain reaction detection of bacterial 16S rRNA gene in human blood. Microbiol Immunol (2008) 52:375–82. doi:10.1111/j.1348-0421.2008.00048.x
302. Amar J, Serino M, Lange C, Chabo C, Iacovoni J, Mondot S, et al. Involvement of tissue bacteria in the onset of diabetes in humans: evidence for a concept. Diabetologia (2011) 54:3055–61. doi:10.1007/s00125-011-2329-8
303. Gaibani P, Mariconti M, Bua G, Bonora S, Sassera D, Landini MP, et al. Development of a broad-range 23S rDNA real-time PCR assay for the detection and quantification of pathogenic bacteria in human whole blood and plasma specimens. Biomed Res Int (2013) 2013:264651. doi:10.1155/2013/264651
304. Dinakaran V, Rathinavel A, Pushpanathan M, Sivakumar R, Gunasekaran P, Rajendhran J. Elevated levels of circulating DNA in cardiovascular disease patients: metagenomic profiling of microbiome in the circulation. PLoS One (2014) 9:e105221. doi:10.1371/journal.pone.0105221
305. Sato J, Kanazawa A, Ikeda F, Yoshihara T, Goto H, Abe H, et al. Gut dysbiosis and detection of “live gut bacteria” in blood of Japanese patients with type 2 diabetes. Diabetes Care (2014) 37:2343–50. doi:10.2337/dc13-2817
306. Damgaard C, Magnussen K, Enevold C, Nilsson M, Tolker-Nielsen T, Holmstrup P, et al. Viable bacteria associated with red blood cells and plasma in freshly drawn blood donations. PLoS One (2015) 10:e0120826. doi:10.1371/journal.pone.0120826
307. Gyarmati P, Kjellander C, Aust C, Kalin M, Öhrmalm L, Giske CG. Bacterial landscape of bloodstream infections in neutropenic patients via high throughput sequencing. PLoS One (2015) 10:e0135756. doi:10.1371/journal.pone.0135756
308. Gyarmati P, Kjellander C, Aust C, Song Y, Öhrmalm L, Giske CG. Metagenomic analysis of bloodstream infections in patients with acute leukemia and therapy-induced neutropenia. Sci Rep (2016) 6:23532. doi:10.1038/srep23532
309. Païssé S, Valle C, Servant F, Courtney M, Burcelin R, Amar J, et al. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion (2016) 56:1138–47. doi:10.1111/trf.13477
310. Domingue GJ, Schlegel JU. Novel bacterial structures in human blood: cultural isolation. Infect Immun (1977) 15:621–7.
311. Belstrøm D, Holmstrup P, Damgaard C, Borch TS, Skjødt MO, Bendtzen K, et al. The atherogenic bacterium Porphyromonas gingivalis evades circulating phagocytes by adhering to erythrocytes. Infect Immun (2011) 79:1559–65. doi:10.1128/IAI.01036-10
313. Price WA. Dental Infections Oral and Systemic, Being a Contribution to the Pathology of Dental Infections, Focal Infections and the Degenerative Diseases, Parts I and II. Cleveland: Penton Press (1923).
314. Miklossy J, Kis A, Radenovic A, Miller L, Forro L, Martins R, et al. Beta-amyloid deposition and Alzheimer’s type changes induced by Borrelia spirochetes. Neurobiol Aging (2006) 27:228–36. doi:10.1016/j.neurobiolaging.2005.01.018
315. Miklossy J. Chronic inflammation and amyloidogenesis in Alzheimer’s disease – role of spirochetes. J Alzheimers Dis (2008) 13:381–91.
316. Woolard MD, Frelinger JA. Outsmarting the host: bacteria modulating the immune response. Immunol Res (2008) 41:188–202. doi:10.1007/s12026-008-8021-5
317. Nicolson GL, Haier J. Role of chronic bacterial and viral infections in neurodegenerative, neurobehavioural, psychiatric, autoimmune and fatiguing illnesses: part 1. Br J Med Pract (2009) 2:20–8.
318. Proal AD, Albert PJ, Marshall T. Autoimmune disease in the era of the metagenome. Autoimmun Rev (2009) 8:677–81. doi:10.1016/j.autrev.2009.02.016
319. Nicolson GL, Haier J. Role of chronic bacterial and viral infections in neurodegenerative, neurobehavioural, psychiatric, autoimmune and fatiguing illnesses: part 2. Br J Med Pract (2010) 3:301–10.
320. Miklossy J. Alzheimer’s disease – a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation (2011) 8:90. doi:10.1186/1742-2094-8-90
321. Proal AD, Albert PJ, Blaney GP, Lindseth IA, Benediktsson C, Marshall TG. Immunostimulation in the era of the metagenome. Cell Mol Immunol (2011) 8:213–25. doi:10.1038/cmi.2010.77
322. Brecher ME, Hay SN. Bacterial contamination of blood components. Clin Microbiol Rev (2005) 18:195–204. doi:10.1128/CMR.18.1.195-204.2005
323. Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 4th ed. Oxford: Oxford University Press (2006).
324. Kaeberlein T, Lewis K, Epstein SS. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science (2002) 296:1127–9. doi:10.1126/science.1070633
325. Bollmann A, Lewis K, Epstein SS. Incubation of environmental samples in a diffusion chamber increases the diversity of recovered isolates. Appl Environ Microbiol (2007) 73:6386–90. doi:10.1128/AEM.01309-07
326. D’Onofrio A, Crawford JM, Stewart EJ, Witt K, Gavrish E, Epstein S, et al. Siderophores from neighboring organisms promote the growth of uncultured bacteria. Chem Biol (2010) 17:254–64. doi:10.1016/j.chembiol.2010.02.010
327. Lewis K, Epstein S, D’Onofrio A, Ling LL. Uncultured microorganisms as a source of secondary metabolites. J Antibiot (Tokyo) (2010) 63:468–76. doi:10.1038/ja.2010.87
328. Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, et al. A new antibiotic kills pathogens without detectable resistance. Nature (2015) 517:455–9. doi:10.1038/nature14098
329. Deschner J, Eick S, Damanaki A, Nokhbehsaim M. The role of adipokines in periodontal infection and healing. Mol Oral Microbiol (2014) 29:258–69. doi:10.1111/omi.12070
330. Herrera JA, Chaudhuri G, López-Jaramillo P. Is infection a major risk factor for preeclampsia? Med Hypotheses (2001) 57:393–7. doi:10.1054/mehy.2001.1378
331. López-Jaramillo P, Casas JP, Serrano N. Preeclampsia: from epidemiological observations to molecular mechanisms. Braz J Med Biol Res (2001) 34:1227–35. doi:10.1590/S0100-879X2001001000001
332. Von Dadelszen P, Magee LA. Could an infectious trigger explain the differential maternal response to the shared placental pathology of preeclampsia and normotensive intrauterine growth restriction? Acta Obstet Gynecol Scand (2002) 81:642–8. doi:10.1034/j.1600-0412.2002.810710.x
333. Marshall B. Helicobacter pylori: 20 years on. Clin Med (2002) 2:147–52. doi:10.1007/s102380200021
334. Suzuki H, Marshall BJ, Hibi T. Overview: Helicobacter pylori and extragastric disease. Int J Hematol (2006) 84:291–300. doi:10.1532/IJH97.06180
335. Prelipcean CC, Mihai C, Gogalniceanu P, Mitrica D, Drug VL, Stanciu C. Extragastric manifestations of Helicobacter pylori infection. Rev Med Chir Soc Med Nat Iasi (2007) 111:575–83.
336. Pellicano R, Franceschi F, Saracco G, Fagoonee S, Roccarina D, Gasbarrini A. Helicobacters and extragastric diseases. Helicobacter (2009) 14(Suppl 1):58–68. doi:10.1111/j.1523-5378.2009.00699.x
337. Figura N, Franceschi F, Santucci A, Bernardini G, Gasbarrini G, Gasbarrini A. Extragastric manifestations of Helicobacter pylori infection. Helicobacter (2010) 15(Suppl 1):60–8. doi:10.1111/j.1523-5378.2010.00778.x
338. Banić M, Franceschi F, Babić Z, Gasbarrini A. Extragastric manifestations of Helicobacter pylori infection. Helicobacter (2012) 17(Suppl 1):49–55. doi:10.1111/j.1523-5378.2012.00983.x
339. Roubaud Baudron C, Franceschi F, Salles N, Gasbarrini A. Extragastric diseases and Helicobacter pylori. Helicobacter (2013) 18(Suppl 1):44–51. doi:10.1111/hel.12077
340. Franceschi F, Tortora A, Gasbarrini G, Gasbarrini A. Helicobacter pylori and extragastric diseases. Helicobacter (2014) 19(Suppl 1):52–8. doi:10.1111/hel.12159
341. Franceschi F, Zuccala G, Roccarina D, Gasbarrini A. Clinical effects of Helicobacter pylori outside the stomach. Nat Rev Gastroenterol Hepatol (2014) 11:234–42. doi:10.1038/nrgastro.2013.243
342. Testerman TL, Morris J. Beyond the stomach: an updated view of Helicobacter pylori pathogenesis, diagnosis, and treatment. World J Gastroenterol (2014) 20:12781–808. doi:10.3748/wjg.v20.i36.12781
343. Ponzetto A, Cardaropoli S, Piccoli E, Rolfo A, Gennero L, Kanduc D, et al. Pre-eclampsia is associated with Helicobacter pylori seropositivity in Italy. J Hypertens (2006) 24:2445–9. doi:10.1097/HJH.0b013e3280109e8c
344. Franceschi F, Di Simone N, D’Ippolito S, Castellani R, Di Nicuolo F, Gasbarrini G, et al. Antibodies anti-CagA cross-react with trophoblast cells: a risk factor for pre-eclampsia? Helicobacter (2012) 17:426–34. doi:10.1111/j.1523-5378.2012.00966.x
345. Shimoda A, Ueda K, Nishiumi S, Murata-Kamiya N, Mukai SA, Sawada S, et al. Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci Rep (2016) 6:18346. doi:10.1038/srep18346
346. Raghupathy R. Cytokines as key players in the pathophysiology of preeclampsia. Med Princ Pract (2013) 22:8–19. doi:10.1159/000354200
347. Wurdinger T, Gatson NN, Balaj L, Kaur B, Breakefield XO, Pegtel DM. Extracellular vesicles and their convergence with viral pathways. Adv Virol (2012) 2012:767694. doi:10.1155/2012/767694
348. van Dongen HM, Masoumi N, Witwer KW, Pegtel DM. Extracellular vesicles exploit viral entry routes for cargo delivery. Microbiol Mol Biol Rev (2016) 80:369–86. doi:10.1128/MMBR.00063-15
349. Xie F, Hu Y, Magee LA, Money DM, Patrick DM, Brunham RM, et al. Chlamydia pneumoniae infection in preeclampsia. Hypertens Pregnancy (2010) 29:468–77. doi:10.3109/10641950903242642
350. Heine RP, Ness RB, Roberts JM. Seroprevalence of antibodies to Chlamydia pneumoniae in women with preeclampsia. Obstet Gynecol (2003) 101:221–6. doi:10.1016/S0029-7844(02)02591-7
351. El-Shourbagy MAA, El-Refaie TA, Sayed KKA, Wahba KAH, El-Din ASS, Fathy MM. Impact of seroconversion and antichlamydial treatment on the rate of pre-eclampsia among Egyptian primigravidae. Int J Gynaecol Obstet (2011) 113:137–40. doi:10.1016/j.ijgo.2010.11.014
352. Haggerty CL, Klebanoff MA, Panum I, Uldum SA, Bass DC, Olsen J, et al. Prenatal Chlamydia trachomatis infection increases the risk of preeclampsia. Pregnancy Hypertens (2013) 3:151–4. doi:10.1016/j.preghy.2013.03.002
353. Haggerty CL, Panum I, Uldum SA, Bass DC, Olsen J, Darville T, et al. Chlamydia trachomatis infection may increase the risk of preeclampsia. Pregnancy Hypertens (2013) 3:28–33. doi:10.1016/j.preghy.2012.09.002
354. Xie F, Hu Y, Magee LA, Money DM, Patrick DM, Krajden M, et al. An association between cytomegalovirus infection and pre-eclampsia: a case-control study and data synthesis. Acta Obstet Gynecol Scand (2010) 89:1162–7. doi:10.3109/00016349.2010.499449
355. Xie F, Von Dadelszen P, Nadeau J. CMV infection, TLR-2 and -4 expression, and cytokine profiles in early-onset preeclampsia with HELLP syndrome. Am J Reprod Immunol (2014) 71:379–86. doi:10.1111/aji.12199
356. Panarelli M, Sattar N. Pre-eclampsia associated with Helicobacter pylori seropositivity. J Hypertens (2006) 24:2353–4. doi:10.1097/HJH.0b013e3280113638
357. Mosbah A, Nabiel Y. Helicobacter pylori, Chlamydiae pneumoniae and trachomatis as probable etiological agents of preeclampsia. J Matern Fetal Neonatal Med (2016) 29:1607–12. doi:10.3109/14767058.2015.1056146
358. Tersigni C, Franceschi F, Todros T, Cardaropoli S, Scambia G, Di Simone N. Insights into the role of Helicobacter pylori infection in preeclampsia: from the bench to the bedside. Front Immunol (2014) 5:484. doi:10.3389/fimmu.2014.00484
359. Üstün Y, Engin-Üstün Y, Ozkaplan E, Otlu B, Sait Tekerekoğlu M. Association of Helicobacter pylori infection with systemic inflammation in preeclampsia. J Matern Fetal Neonatal Med (2010) 23:311–4. doi:10.3109/14767050903121456
360. Aksoy H, Ozkan A, Aktas F, Borekci B. Helicobacter pylori seropositivity and its relationship with serum malondialdehyde and lipid profile in preeclampsia. J Clin Lab Anal (2009) 23:219–22. doi:10.1002/jcla.20330
361. Cardaropoli S, Rolfo A, Todros T. Helicobacter pylori and pregnancy-related disorders. World J Gastroenterol (2014) 20:654–64. doi:10.3748/wjg.v20.i3.654
362. Pugliese A, Beltramo T, Todros T, Cardaropoli S, Ponzetto A. Interleukin-18 and gestosis: correlation with Helicobacter pylori seropositivity. Cell Biochem Funct (2008) 26:817–9. doi:10.1002/cbf.1503
363. Cardaropoli S, Giuffrida D, Piazzese A, Todros T. Helicobacter pylori seropositivity and pregnancy-related diseases: a prospective cohort study. J Reprod Immunol (2015) 109:41–7. doi:10.1016/j.jri.2015.02.004
364. Cardaropoli S, Rolfo A, Piazzese A, Ponzetto A, Todros T. Helicobacter pylori’s virulence and infection persistence define pre-eclampsia complicated by fetal growth retardation. World J Gastroenterol (2011) 17:5156–65. doi:10.3748/wjg.v17.i47.5156
365. McDonnold M, Dunn H, Hester A, Pacheco LD, Hankins GD, Saade GR, et al. High risk human papillomavirus at entry to prenatal care and risk of preeclampsia. Am J Obstet Gynecol (2014) 210(138):138.e1–5. doi:10.1016/j.ajog.2013.09.040
366. Hill JA, Devoe LD, Bryans CI Jr. Frequency of asymptomatic bacteriuria in preeclampsia. Obstet Gynecol (1986) 67:529–32.
367. Hsu CD, Witter FR. Urogenital infection in preeclampsia. Int J Gynaecol Obstet (1995) 49:271–5. doi:10.1016/0020-7292(95)02373-K
368. Mittendorf R, Lain KY, Williams MA, Walker CK. Preeclampsia. A nested, case-control study of risk factors and their interactions. J Reprod Med (1996) 41:491–6.
369. Easter SR, Cantonwine DE, Zera CA, Lim KH, Parry SI, Mcelrath TF. Urinary tract infection during pregnancy, angiogenic factor profiles, and risk of preeclampsia. Am J Obstet Gynecol (2016) 214:387.e1–7. doi:10.1016/j.ajog.2015.09.101
370. Mazor-Dray E, Levy A, Schlaeffer F, Sheiner E. Maternal urinary tract infection: is it independently associated with adverse pregnancy outcome? J Matern Fetal Neonatal Med (2009) 22:124–8. doi:10.1080/14767050802488246
371. Minassian C, Thomas SL, Williams DJ, Campbell O, Smeeth L. Acute maternal infection and risk of pre-eclampsia: a population-based case-control study. PLoS One (2013) 8:e73047. doi:10.1371/journal.pone.0073047
372. Rezavand N, Veisi F, Zangane M, Amini R, Almasi A. Association between asymptomatic bacteriuria and pre-eclampsia. Glob J Health Sci (2016) 8:235–9. doi:10.5539/gjhs.v8n7p235
373. Karmon A, Sheiner E. The relationship between urinary tract infection during pregnancy and preeclampsia: causal, confounded or spurious? Arch Gynecol Obstet (2008) 277:479–81. doi:10.1007/s00404-008-0643-2
374. Villar J, Carroli G, Wojdyla D, Abalos E, Giordano D, Ba’aqeel H, et al. Preeclampsia, gestational hypertension and intrauterine growth restriction, related or independent conditions? Am J Obstet Gynecol (2006) 194:921–31. doi:10.1016/j.ajog.2005.10.813
375. Bánhidy F, Ács N, Puhó EH, Czeizel AE. Pregnancy complications and birth outcomes of pregnant women with urinary tract infections and related drug treatments. Scand J Infect Dis (2007) 39:390–7. doi:10.1080/00365540601087566
376. López-Jaramillo P, Herrera JA, Arenas-Mantilla M, Jauregui IE, Mendoza MA. Subclinical infection as a cause of inflammation in preeclampsia. Am J Ther (2008) 15:373–6. doi:10.1097/MJT.0b013e318164c149
377. Ide M, Papapanou PN. Epidemiology of association between maternal periodontal disease and adverse pregnancy outcomes – systematic review. J Periodontol (2013) 84:S181–94. doi:10.1902/jop.2013.134009
378. Dunlop AL, Mulle JG, Ferranti EP, Edwards S, Dunn AB, Corwin EJ. Maternal microbiome and pregnancy outcomes that impact infant health: a review. Adv Neonatal Care (2015) 15:377–85. doi:10.1097/ANC.0000000000000218
379. Hlimi T. Association of anemia, pre-eclampsia and eclampsia with seasonality: a realist systematic review. Health Place (2015) 31:180–92. doi:10.1016/j.healthplace.2014.12.003
380. Brabin BJ, Johnson PM. Placental malaria and pre-eclampsia through the looking glass backwards? J Reprod Immunol (2005) 65:1–15. doi:10.1016/j.jri.2004.09.006
381. Anya SE. Seasonal variation in the risk and causes of maternal death in the Gambia: malaria appears to be an important factor. Am J Trop Med Hyg (2004) 70:510–3.
382. Sartelet H, Rogier C, Milko-Sartelet I, Angel G, Michel G. Malaria associated pre-eclampsia in Senegal. Lancet (1996) 347:1121. doi:10.1016/S0140-6736(96)90321-9
383. Romero R, Mazor M, Wu YK, Sirtori M, Oyarzun E, Mitchell MD, et al. Infection in the pathogenesis of preterm labor. Semin Perinatol (1988) 12:262–79.
384. Toth M, Witkin SS, Ledger W, Thaler H. The role of infection in the etiology of preterm birth. Obstet Gynecol (1988) 71:723–6.
385. Cassell GH, Waites KB, Watson HL, Crouse DT, Harasawa R. Ureaplasma urealyticum intrauterine infection: role in prematurity and disease in newborns. Clin Microbiol Rev (1993) 6:69–87. doi:10.1128/CMR.6.1.69
386. McGregor JA, French JI, Jones W, Milligan K, Mckinney PJ, Patterson E, et al. Bacterial vaginosis is associated with prematurity and vaginal fluid mucinase and sialidase: results of a controlled trial of topical clindamycin cream. Am J Obstet Gynecol (1994) 170:1048–59; discussion 1059–60. doi:10.1016/S0002-9378(94)70098-2
387. Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. N Engl J Med (2000) 342:1500–7. doi:10.1056/NEJM200005183422007
388. Gonçalves LF, Chaiworapongsa T, Romero R. Intrauterine infection and prematurity. Ment Retard Dev Disabil Res Rev (2002) 8:3–13. doi:10.1002/mrdd.10008
389. Gerber S, Vial Y, Hohlfeld P, Witkin SS. Detection of Ureaplasma urealyticum in second-trimester amniotic fluid by polymerase chain reaction correlates with subsequent preterm labor and delivery. J Infect Dis (2003) 187:518–21. doi:10.1086/368205
390. Gardella C, Riley DE, Hitti J, Agnew K, Krieger JN, Eschenbach D. Identification and sequencing of bacterial rDNAs in culture-negative amniotic fluid from women in premature labor. Am J Perinatol (2004) 21:319–23. doi:10.1055/s-2004-831884
391. Espinoza J, Erez O, Romero R. Preconceptional antibiotic treatment to prevent preterm birth in women with a previous preterm delivery. Am J Obstet Gynecol (2006) 194:630–7. doi:10.1016/j.ajog.2005.11.050
392. Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet (2008) 371:75–84. doi:10.1016/S0140-6736(08)60074-4
393. Check JH. A practical approach to the prevention of miscarriage. Part 4 – role of infection. Clin Exp Obstet Gynecol (2010) 37:252–5.
394. Bastek JA, Gómez LM, Elovitz MA. The role of inflammation and infection in preterm birth. Clin Perinatol (2011) 38:385–406. doi:10.1016/j.clp.2011.06.003
395. Johnson HL, Ghanem KG, Zenilman JM, Erbelding EJ. Sexually transmitted infections and adverse pregnancy outcomes among women attending inner city public sexually transmitted diseases clinics. Sex Transm Dis (2011) 38:167–71. doi:10.1097/OLQ.0b013e3181f2e85f
396. Rours GIJG, Duijts L, Moll HA, Arends LR, De Groot R, Jaddoe VW, et al. Chlamydia trachomatis infection during pregnancy associated with preterm delivery: a population-based prospective cohort study. Eur J Epidemiol (2011) 26:493–502. doi:10.1007/s10654-011-9586-1
397. Jefferson KK. The bacterial etiology of preterm birth. Adv Appl Microbiol (2012) 80:1–22. doi:10.1016/B978-0-12-394381-1.00001-5
398. Lee SYR, Leung CW. Histological chorioamnionitis – implication for bacterial colonization, laboratory markers of infection, and early onset sepsis in very-low-birth-weight neonates. J Matern Fetal Neonatal Med (2012) 25:364–8. doi:10.3109/14767058.2011.579208
399. Subramaniam A, Abramovici A, Andrews WW, Tita AT. Antimicrobials for preterm birth prevention: an overview. Infect Dis Obstet Gynecol (2012) 2012:157159. doi:10.1155/2012/157159
400. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med (2014) 6:237ra265. doi:10.1126/scitranslmed.3008599
401. Joergensen JS, Kjaer Weile LK, Lamont RF. The early use of appropriate prophylactic antibiotics in susceptible women for the prevention of preterm birth of infectious etiology. Expert Opin Pharmacother (2014) 15:2173–91. doi:10.1517/14656566.2014.950225
402. Allen-Daniels MJ, Serrano MG, Pflugner LP, Fettweis JM, Prestosa MA, Koparde VN, et al. Identification of a gene in Mycoplasma hominis associated with preterm birth and microbial burden in intraamniotic infection. Am J Obstet Gynecol (2015) 212:779.e1–13. doi:10.1016/j.ajog.2015.01.032
403. de Andrade Ramos B, Kanninen TT, Sisti G, Witkin SS. Microorganisms in the female genital tract during pregnancy: tolerance versus pathogenesis. Am J Reprod Immunol (2015) 73:383–9. doi:10.1111/aji.12326
404. Kacerovsky M, Vrbacky F, Kutova R, Pliskova L, Andrys C, Musilova I, et al. Cervical microbiota in women with preterm prelabor rupture of membranes. PLoS One (2015) 10:e0126884. doi:10.1371/journal.pone.0126884
405. Lamont RF. Advances in the prevention of infection-related preterm birth. Front Immunol (2015) 6:566. doi:10.3389/fimmu.2015.00566
406. Ueno T, Niimi H, Yoneda N, Yoneda S, Mori M, Tabata H, et al. Eukaryote-made thermostable DNA polymerase enables rapid PCR-based detection of mycoplasma, ureaplasma and other bacteria in the amniotic fluid of preterm labor cases. PLoS One (2015) 10:e0129032. doi:10.1371/journal.pone.0129032
407. Frey HA, Klebanoff MA. The epidemiology, etiology, and costs of preterm birth. Semin Fetal Neonatal Med (2016) 21:68–73. doi:10.1016/j.siny.2015.12.011
408. Nadeau HCG, Subramaniam A, Andrews WW. Infection and preterm birth. Semin Fetal Neonatal Med (2016) 21:100–5. doi:10.1016/j.siny.2015.12.008
409. Vinturache AE, Gyamfi-Bannerman C, Hwang J, Mysorekar IU, Jacobsson B; Preterm Birth International Collaborative (Prebic). Maternal microbiome – a pathway to preterm birth. Semin Fetal Neonatal Med (2016) 21:94–9. doi:10.1016/j.siny.2016.02.004
410. Yoneda S, Shiozaki A, Yoneda N, Ito M, Shima T, Fukuda K, et al. Antibiotic therapy increases the risk of preterm birth in preterm labor without intra-amniotic microbes, but may prolong the gestation period in preterm labor with microbes, evaluated by rapid and high-sensitive PCR system. Am J Reprod Immunol (2016) 75:440–50. doi:10.1111/aji.12484
411. Oliver RS, Lamont RF. Infection and antibiotics in the aetiology, prediction and prevention of preterm birth. J Obstet Gynaecol (2013) 33:768–75. doi:10.3109/01443615.2013.842963
412. Kacerovsky M, Lenco J, Musilova I, Tambor V, Lamont R, Torloni MR, et al. Proteomic biomarkers for spontaneous preterm birth: a systematic review of the literature. Reprod Sci (2014) 21:283–95. doi:10.1177/1933719113503415
413. McClure EM, Goldenberg RL. Infection and stillbirth. Semin Fetal Neonatal Med (2009) 14:182–9. doi:10.1016/j.siny.2009.02.003
414. Menezes EV, Yakoob MY, Soomro T, Haws RA, Darmstadt GL, Bhutta ZA. Reducing stillbirths: prevention and management of medical disorders and infections during pregnancy. BMC Pregnancy Childbirth (2009) 9(Suppl 1):S4. doi:10.1186/1471-2393-9-S1-S4
415. Nigro G, Mazzocco M, Mattia E, Di Renzo GC, Carta G, Anceschi MM. Role of the infections in recurrent spontaneous abortion. J Matern Fetal Neonatal Med (2011) 24:983–9. doi:10.3109/14767058.2010.547963
416. Giakoumelou S, Wheelhouse N, Cuschieri K, Entrican G, Howie SEM, Horne AW. The role of infection in miscarriage. Hum Reprod Update (2015) 22:116–33. doi:10.1093/humupd/dmv041
417. Toth A, Lesser ML, Brooks-Toth CW, Feiner C. Outcome of subsequent pregnancies following antibiotic therapy after primary or multiple spontaneous abortions. Surg Gynecol Obstet (1986) 163:243–50.
418. Lauder AP, Roche AM, Sherrill-Mix S, Bailey A, Laughlin AL, Bittinger K, et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome (2016) 4:29. doi:10.1186/s40168-016-0172-3
419. Onderdonk AB, Delaney ML, Dubois AM, Allred EN, Leviton A; Extremely Low Gestational Age Newborns Study Investigators. Detection of bacteria in placental tissues obtained from extremely low gestational age neonates. Am J Obstet Gynecol (2008) 198(110):e111–7. doi:10.1016/j.ajog.2007.05.044
420. Satokari R, Gronroos T, Laitinen K, Salminen S, Isolauri E. Bifidobacterium and Lactobacillus DNA in the human placenta. Lett Appl Microbiol (2009) 48:8–12. doi:10.1111/j.1472-765X.2008.02475.x
421. Stout MJ, Conlon B, Landeau M, Lee I, Bower C, Zhao Q, et al. Identification of intracellular bacteria in the basal plate of the human placenta in term and preterm gestations. Am J Obstet Gynecol (2013) 208(226):e221–7. doi:10.1016/j.ajog.2013.01.018
422. Cao B, Mysorekar IU. Intracellular bacteria in placental basal plate localize to extravillous trophoblasts. Placenta (2014) 35:139–42. doi:10.1016/j.placenta.2013.12.007
423. Cao B, Stout MJ, Lee I, Mysorekar IU. Placental microbiome and its role in preterm birth. Neoreviews (2014) 15:e537–45. doi:10.1542/neo.15-12-e537
424. Doyle RM, Alber DG, Jones HE, Harris K, Fitzgerald F, Peebles D, et al. Term and preterm labour are associated with distinct microbial community structures in placental membranes which are independent of mode of delivery. Placenta (2014) 35:1099–101. doi:10.1016/j.placenta.2014.10.007
425. Mysorekar IU, Cao B. Microbiome in parturition and preterm birth. Semin Reprod Med (2014) 32:50–5. doi:10.1055/s-0033-1361830
426. Abrahamsson TR, Wu RY, Jenmalm MC. Gut microbiota and allergy: the importance of the pregnancy period. Pediatr Res (2015) 77:214–9. doi:10.1038/pr.2014.165
427. Antony KM, Ma J, Mitchell KB, Racusin DA, Versalovic J, Aagaard K. The preterm placental microbiome varies in association with excess maternal gestational weight gain. Am J Obstet Gynecol (2015) 212: 653.e651–616. doi:10.1016/j.ajog.2014.12.041
428. Garmi G, Okopnik M, Keness Y, Zafran N, Berkowitz E, Salim R. Correlation between clinical, placental histology and microbiological findings in spontaneous preterm births. Fetal Diagn Ther (2015) 40:141–9. doi:10.1159/000441518
429. Zheng J, Xiao X, Zhang Q, Mao L, Yu M, Xu J. The placental microbiome varies in association with low birth weight in full-term neonates. Nutrients (2015) 7:6924–37. doi:10.3390/nu7085315
430. Collado MC, Rautava S, Aakko J, Isolauri E, Salminen S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep (2016) 6:23129. doi:10.1038/srep23129
431. Kuperman AA, Koren O. Antibiotic use during pregnancy: how bad is it? BMC Med (2016) 14:91. doi:10.1186/s12916-016-0636-0
432. Prince AL, Ma J, Kannan PS, Alvarez M, Gisslen T, Harris RA, et al. The placental microbiome is altered among subjects with spontaneous preterm birth with and without chorioamnionitis. Am J Obstet Gynecol (2016) 214:627.e1–16. doi:10.1016/j.ajog.2016.01.193
433. Viniker DA. Hypothesis on the role of sub-clinical bacteria of the endometrium (bacteria endometrialis) in gynaecological and obstetric enigmas. Hum Reprod Update (1999) 5:373–85. doi:10.1093/humupd/5.4.373
434. Svare JA, Schmidt H, Hansen BB, Lose G. Bacterial vaginosis in a cohort of Danish pregnant women: prevalence and relationship with preterm delivery, low birthweight and perinatal infections. BJOG (2006) 113:1419–25. doi:10.1111/j.1471-0528.2006.01087.x
435. Sheldon IM, Bromfield JJ. Innate immunity in the human endometrium and ovary. Am J Reprod Immunol (2011) 66(Suppl 1):63–71. doi:10.1111/j.1600-0897.2011.01034.x
436. Aagaard K, Riehle K, Ma J, Segata N, Mistretta TA, Coarfa C, et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS One (2012) 7:e36466. doi:10.1371/journal.pone.0036466
437. Walther-António MR, Jeraldo P, Berg Miller ME, Yeoman CJ, Nelson KE, Wilson BA, et al. Pregnancy’s stronghold on the vaginal microbiome. PLoS One (2014) 9:e98514. doi:10.1371/journal.pone.0098514
438. Huang YE, Wang Y, He Y, Ji Y, Wang LP, Sheng HF, et al. Homogeneity of the vaginal microbiome at the cervix, posterior fornix, and vaginal canal in pregnant Chinese women. Microb Ecol (2015) 69:407–14. doi:10.1007/s00248-014-0487-1
439. Witkin SS. The vaginal microbiome, vaginal anti-microbial defence mechanisms and the clinical challenge of reducing infection-related preterm birth. BJOG (2015):213–8. doi:10.1111/1471-0528.13115
440. Nelson DB, Rockwell LC, Prioleau MD, Goetzl L. The role of the bacterial microbiota on reproductive and pregnancy health. Anaerobe (2016) 42:67–73. doi:10.1016/j.anaerobe.2016.09.001
441. Payne MS, Bayatibojakhi S. Exploring preterm birth as a polymicrobial disease: an overview of the uterine microbiome. Front Immunol (2014) 5:595. doi:10.3389/fimmu.2014.00595
442. Verstraelen H, Vilchez-Vargas R, Desimpel F, Jauregui R, Vankeirsbilck N, Weyers S, et al. Characterisation of the human uterine microbiome in non-pregnant women through deep sequencing of the V1-2 region of the 16S rRNA gene. PeerJ (2016) 4:e1602. doi:10.7717/peerj.1602
443. Dong Y, St Clair PJ, Ramzy I, Kagan-Hallet KS, Gibbs RS. A microbiologic and clinical study of placental inflammation at term. Obstet Gynecol (1987) 70:175–82.
444. Bearfield C, Davenport ES, Sivapathasundaram V, Allaker RP. Possible association between amniotic fluid micro-organism infection and microflora in the mouth. BJOG (2002) 109:527–33. doi:10.1111/j.1471-0528.2002.01349.x
445. Combs CA, Gravett M, Garite TJ, Hickok DE, Lapidus J, Porreco R, et al. Amniotic fluid infection, inflammation, and colonization in preterm labor with intact membranes. Am J Obstet Gynecol (2014) 210:125.e1–15. doi:10.1016/j.ajog.2013.11.032
446. Wassenaar TM, Panigrahi P. Is a foetus developing in a sterile environment? Lett Appl Microbiol (2014) 59:572–9. doi:10.1111/lam.12334
447. Combs CA, Garite TJ, Lapidus JA, Lapointe JP, Gravett M, Rael J, et al. Detection of microbial invasion of the amniotic cavity by analysis of cervicovaginal proteins in women with preterm labor and intact membranes. Am J Obstet Gynecol (2015) 212:482.e481–e412. doi:10.1016/j.ajog.2015.02.007
448. Koleva PT, Kim JS, Scott JA, Kozyrskyj AL. Microbial programming of health and disease starts during fetal life. Birth Defects Res C Embryo Today (2015) 105:265–77. doi:10.1002/bdrc.21117
449. Pelzer ES, Allan JA, Cunningham K, Mengersen K, Allan JM, Launchbury T, et al. Microbial colonization of follicular fluid: alterations in cytokine expression and adverse assisted reproduction technology outcomes. Hum Reprod (2011) 26:1799–812. doi:10.1093/humrep/der108
450. Pelzer ES, Allan JA, Waterhouse MA, Ross T, Beagley KW, Knox CL. Microorganisms within human follicular fluid: effects on IVF. PLoS One (2013) 8:e59062. doi:10.1371/journal.pone.0059062
451. Barak S, Oettinger-Barak O, Machtei EE, Sprecher H, Ohel G. Evidence of periopathogenic microorganisms in placentas of women with preeclampsia. J Periodontol (2007) 78:670–6. doi:10.1902/jop.2007.060362
452. McDonagh S, Maidji E, Ma W, Chang HT, Fisher S, Pereira L. Viral and bacterial pathogens at the maternal-fetal interface. J Infect Dis (2004) 190:826–34. doi:10.1086/422330
453. Amarasekara R, Jayasekara RW, Senanayake H, Dissanayake VH. Microbiome of the placenta in pre-eclampsia supports the role of bacteria in the multifactorial cause of pre-eclampsia. J Obstet Gynaecol Res (2015) 41:662–9. doi:10.1111/jog.12619
454. Muehlenbachs A, Mutabingwa TK, Edmonds S, Fried M, Duffy PE. Hypertension and maternal-fetal conflict during placental malaria. PLoS Med (2006) 3:e446. doi:10.1371/journal.pmed.0030446
455. Duffy PE. Plasmodium in the placenta: parasites, parity, protection, prevention and possibly preeclampsia. Parasitology (2007) 134:1877–81. doi:10.1017/S0031182007000170
456. Vanterpool SF, Been JV, Houben ML, Nikkels PG, De Krijger RR, Zimmermann LJ, et al. Porphyromonas gingivalis within placental villous mesenchyme and umbilical cord stroma is associated with adverse pregnancy outcome. PLoS One (2016) 11:e0146157. doi:10.1371/journal.pone.0146157
457. Chaparro A, Blanlot C, Ramirez V, Sanz A, Quintero A, Inostroza C, et al. Porphyromonas gingivalis, Treponema denticola and toll-like receptor 2 are associated with hypertensive disorders in placental tissue: a case-control study. J Periodontal Res (2013) 48:802–9. doi:10.1111/jre.12074
458. Ebringer A, Rashid T, Wilson C. Rheumatoid arthritis, proteus, anti-CCP antibodies and Karl Popper. Autoimmun Rev (2010) 9:216–23. doi:10.1016/j.autrev.2009.10.006
460. Ebringer A, Rashid T. Rheumatoid arthritis is caused by a Proteus urinary tract infection. APMIS (2014) 122:363–8. doi:10.1111/apm.12154
461. Vatanen T, Kostic AD, D’Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell (2016) 165:842–53. doi:10.1016/j.cell.2016.04.007
462. Ferrier L, Mazelin L, Cenac N, Desreumaux P, Janin A, Emilie D, et al. Stress-induced disruption of colonic epithelial barrier: role of interferon-gamma and myosin light chain kinase in mice. Gastroenterology (2003) 125:795–804. doi:10.1016/S0016-5085(03)01057-6
463. Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol (2012) 30:759–95. doi:10.1146/annurev-immunol-020711-074937
464. Klatt NR, Funderburg NT, Brenchley JM. Microbial translocation, immune activation, and HIV disease. Trends Microbiol (2013) 21:6–13. doi:10.1016/j.tim.2012.09.001
465. Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr (2008) 87:1219–23.
466. Lassenius MI, Pietiläinen KH, Kaartinen K, Pussinen PJ, Syrjänen J, Forsblom C, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care (2011) 34:1809–15. doi:10.2337/dc10-2197
467. Vors C, Pineau G, Drai J, Meugnier E, Pesenti S, Laville M, et al. Postprandial endotoxemia linked with chylomicrons and lipopolysaccharides handling in obese versus lean men: a lipid dose-effect trial. J Clin Endocrinol Metab (2015) 100:3427–35. doi:10.1210/JC.2015-2518
468. Sawchuck DJ, Wittmann BK. Pre-eclampsia renamed and reframed: intra-abdominal hypertension in pregnancy. Med Hypotheses (2014) 83:619–32. doi:10.1016/j.mehy.2014.08.001
469. Perez PF, Dore J, Leclerc M, Levenez F, Benyacoub J, Serrant P, et al. Bacterial imprinting of the neonatal immune system: lessons from maternal cells? Pediatrics (2007) 119:e724–32. doi:10.1542/peds.2006-1649
470. Gensollen T, Iyer SS, Kasper DL, Blumberg RS. How colonization by microbiota in early life shapes the immune system. Science (2016) 352:539–44. doi:10.1126/science.aad9378
471. Newnham JP, Newnham IA, Ball CM, Wright M, Pennell CE, Swain J, et al. Treatment of periodontal disease during pregnancy: a randomized controlled trial. Obstet Gynecol (2009) 114:1239–48. doi:10.1097/AOG.0b013e3181c15b40
472. Huang X, Wang J, Liu J, Hua L, Zhang D, Hu T, et al. Maternal periodontal disease and risk of preeclampsia: a meta-analysis. J Huazhong Univ Sci Technolog Med Sci (2014) 34:729–35. doi:10.1007/s11596-014-1343-8
473. Gilbert GL, Garland SM, Fairley KF, Mcdowall DM. Bacteriuria due to ureaplasmas and other fastidious organisms during pregnancy: prevalence and significance. Pediatr Infect Dis (1986) 5:S239–43. doi:10.1097/00006454-198611010-00007
474. Ha JE, Jun JK, Ko HJ, Paik DI, Bae KH. Association between periodontitis and preeclampsia in never-smokers: a prospective study. J Clin Periodontol (2014) 41:869–74. doi:10.1111/jcpe.12281
475. Sgolastra F, Petrucci A, Severino M, Gatto R, Monaco A. Relationship between periodontitis and pre-eclampsia: a meta-analysis. PLoS One (2013) 8:e71387. doi:10.1371/journal.pone.0071387
476. Boggess KA, Edelstein BL. Oral health in women during preconception and pregnancy: implications for birth outcomes and infant oral health. Matern Child Health J (2006) 10:S169–74. doi:10.1007/s10995-006-0095-x
477. Boggess KA, Berggren EK, Koskenoja V, Urlaub D, Lorenz C. Severe preeclampsia and maternal self-report of oral health, hygiene, and dental care. J Periodontol (2013) 84:143–51. doi:10.1902/jop.2012.120079
478. Armitage GC. Bi-directional relationship between pregnancy and periodontal disease. Periodontol 2000 (2013) 61:160–76. doi:10.1111/j.1600-0757.2011.00396.x
479. Moura da Silva G, Coutinho SB, Piscoya MDBV, Ximenes RAA, Jamelli SR. Periodontitis as a risk factor for preeclampsia. J Periodontol (2012) 83:1388–96. doi:10.1902/jop.2012.110256
480. Jahromi BN, Adibi R, Adibi S, Salarian L. Periodontal disease as a risk factor for preeclampsia. Womens Health Bull (2014) 1:e18908. doi:10.17795/whb-18908
481. Lachat MF, Solnik AL, Nana AD, Citron TL. Periodontal disease in pregnancy: review of the evidence and prevention strategies. J Perinat Neonatal Nurs (2011) 25:312–9. doi:10.1097/JPN.0b013e31821072e4
482. Politano GT, Passini R, Nomura ML, Velloso L, Morari J, Couto E. Correlation between periodontal disease, inflammatory alterations and pre-eclampsia. J Periodontal Res (2011) 46:505–11. doi:10.1111/j.1600-0765.2011.01368.x
483. Nabet C, Lelong N, Colombier ML, Sixou M, Musset AM, Goffinet F, et al. Maternal periodontitis and the causes of preterm birth: the case-control Epipap study. J Clin Periodontol (2010) 37:37–45. doi:10.1111/j.1600-051X.2009.01503.x
484. Madianos PN, Bobetsis YA, Offenbacher S. Adverse pregnancy outcomes (APOs) and periodontal disease: pathogenic mechanisms. J Periodontol (2013) 84:S170–80. doi:10.1902/jop.2013.1340015
485. Bobetsis YA, Barros SP, Offenbacher S. Exploring the relationship between periodontal disease and pregnancy complications. J Am Dent Assoc (2006) 137(Suppl):7S–13S. doi:10.14219/jada.archive.2006.0403
486. Herrera JA, Parra B, Herrera E, Botero JE, Arce RM, Contreras A, et al. Periodontal disease severity is related to high levels of C-reactive protein in pre-eclampsia. J Hypertens (2007) 25:1459–64. doi:10.1097/HJH.0b013e3281139ea9
487. Ruma M, Boggess K, Moss K, Jared H, Murtha A, Beck J, et al. Maternal periodontal disease, systemic inflammation, and risk for preeclampsia. Am J Obstet Gynecol (2008) 198:389.e1–5. doi:10.1016/j.ajog.2007.12.002
488. Pralhad S, Thomas B, Kushtagi P. Periodontal disease and pregnancy hypertension: a clinical correlation. J Periodontol (2013) 84:1118–25. doi:10.1902/jop.2012.120264
489. Zi MY, Longo PL, Bueno-Silva B, Mayer MP. Mechanisms involved in the association between periodontitis and complications in pregnancy. Front Public Health (2014) 2:290. doi:10.3389/fpubh.2014.00290
490. Swati P, Thomas B, Vahab SA, Kapaettu S, Kushtagi P. Simultaneous detection of periodontal pathogens in subgingival plaque and placenta of women with hypertension in pregnancy. Arch Gynecol Obstet (2012) 285:613–9. doi:10.1007/s00404-011-2012-9
491. Shub A, Swain JR, Newnham JP. Periodontal disease and adverse pregnancy outcomes. J Matern Fetal Neonatal Med (2006) 19:521–8. doi:10.1080/14767050600797749
492. Xiong X, Buekens P, Fraser WD, Beck J, Offenbacher S. Periodontal disease and adverse pregnancy outcomes: a systematic review. BJOG (2006) 113:135–43. doi:10.1111/j.1471-0528.2005.00827.x
493. Ha JE, Oh KJ, Yang HJ, Jun JK, Jin BH, Paik DI, et al. Oral health behaviors, periodontal disease, and pathogens in preeclampsia: a case-control study in Korea. J Periodontol (2011) 82:1685–92. doi:10.1902/jop.2011.110035
494. Komine-Aizawa S, Hirohata N, Aizawa S, Abiko Y, Hayakawa S. Porphyromonas gingivalis lipopolysaccharide inhibits trophoblast invasion in the presence of nicotine. Placenta (2015) 36:27–33. doi:10.1016/j.placenta.2014.10.015
495. Contreras A, Herrera JA, Soto JE, Arce RM, Jaramillo A, Botero JE. Periodontitis is associated with preeclampsia in pregnant women. J Periodontol (2006) 77:182–8. doi:10.1902/jop.2006.050020
496. Oettinger-Barak O, Barak S, Ohel G, Oettinger M, Kreutzer H, Peled M, et al. Severe pregnancy complication (preeclampsia) is associated with greater periodontal destruction. J Periodontol (2005) 76:134–7. doi:10.1902/jop.2005.76.1.134
497. Foxman B. Recurring urinary tract infection: incidence and risk factors. Am J Public Health (1990) 80:331–3. doi:10.2105/AJPH.80.3.331
498. Foxman B. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Am J Med (2002) 113(Suppl 1A):5S–13S. doi:10.1016/S0002-9343(02)01054-9
499. Marrs CF, Zhang L, Foxman B. Escherichia coli mediated urinary tract infections: are there distinct uropathogenic E. coli (UPEC) pathotypes? FEMS Microbiol Lett (2005) 252:183–90. doi:10.1016/j.femsle.2005.08.028
500. Hannan TJ, Mysorekar IU, Hung CS, Isaacson-Schmid ML, Hultgren SJ. Early severe inflammatory responses to uropathogenic E. coli predispose to chronic and recurrent urinary tract infection. PLoS Pathog (2010) 6:e1001042. doi:10.1371/journal.ppat.1001042
501. Hunstad DA, Justice SS. Intracellular lifestyles and immune evasion strategies of uropathogenic Escherichia coli. Annu Rev Microbiol (2010) 64:203–21. doi:10.1146/annurev.micro.112408.134258
502. Ejrnaes K. Bacterial characteristics of importance for recurrent urinary tract infections caused by Escherichia coli. Dan Med Bull (2011) 58:B4187.
503. Hannan TJ, Totsika M, Mansfield KJ, Moore KH, Schembri MA, Hultgren SJ. Host-pathogen checkpoints and population bottlenecks in persistent and intracellular uropathogenic Escherichia coli bladder infection. FEMS Microbiol Rev (2012) 36:616–48. doi:10.1111/j.1574-6976.2012.00339.x
504. Foxman B. Urinary tract infection syndromes: occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect Dis Clin North Am (2014) 28:1–13. doi:10.1016/j.idc.2013.09.003
505. Justice SS, Hung C, Theriot JA, Fletcher DA, Anderson GG, Footer MJ, et al. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc Natl Acad Sci U S A (2004) 101:1333–8. doi:10.1073/pnas.0308125100
506. Bower JM, Eto DS, Mulvey MA. Covert operations of uropathogenic Escherichia coli within the urinary tract. Traffic (2005) 6:18–31. doi:10.1111/j.1600-0854.2004.00251.x
507. Rosen DA, Hooton TM, Stamm WE, Humphrey PA, Hultgren SJ. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med (2007) 4:e329. doi:10.1371/journal.pmed.0040329
508. Dhakal BK, Kulesus RR, Mulvey MA. Mechanisms and consequences of bladder cell invasion by uropathogenic Escherichia coli. Eur J Clin Invest (2008) 38(Suppl 2):2–11. doi:10.1111/j.1365-2362.2008.01986.x
509. Schwartz DJ, Chen SL, Hultgren SJ, Seed PC. Population dynamics and niche distribution of uropathogenic Escherichia coli during acute and chronic urinary tract infection. Infect Immun (2011) 79:4250–9. doi:10.1128/IAI.05339-11
510. Brauner A, Jacobson SH, Kuhn I. Urinary Escherichia coli causing recurrent infections – a prospective follow-up of biochemical phenotypes. Clin Nephrol (1992) 38:318–23.
511. Russo TA, Stapleton A, Wenderoth S, Hooton TM, Stamm WE. Chromosomal restriction fragment length polymorphism analysis of Escherichia coli strains causing recurrent urinary tract infections in young women. J Infect Dis (1995) 172:440–5. doi:10.1093/infdis/172.2.440
512. Ikäheimo R, Siitonen A, Heiskanen T, Kärkkäinen U, Kuosmanen P, Lipponen P, et al. Recurrence of urinary tract infection in a primary care setting: analysis of a 1-year follow-up of 179 women. Clin Infect Dis (1996) 22:91–9. doi:10.1093/clinids/22.1.91
513. Rosen DA, Pinkner JS, Jones JM, Walker JN, Clegg S, Hultgren SJ. Utilization of an intracellular bacterial community pathway in Klebsiella pneumoniae urinary tract infection and the effects of FimK on type 1 pilus expression. Infect Immun (2008) 76:3337–45. doi:10.1128/IAI.00090-08
514. Luo Y, Ma Y, Zhao Q, Wang L, Guo L, Ye L, et al. Similarity and divergence of phylogenies, antimicrobial susceptibilities, and virulence factor profiles of Escherichia coli isolates causing recurrent urinary tract infections that persist or result from reinfection. J Clin Microbiol (2012) 50:4002–7. doi:10.1128/JCM.02086-12
515. Özlü T, Alçelik A, Çalışkan B, Dönmez ME. Preeclampsia: is it because of the asymptomatic, unrecognized renal scars caused by urinary tract infections in childhood that become symptomatic with pregnancy? Med Hypotheses (2012) 79:653–5. doi:10.1016/j.mehy.2012.08.002
516. Kincaid-Smith P, Bullen M. Bacteriuria in pregnancy. Lancet (1965) 1:395–9. doi:10.1016/S0140-6736(65)90001-2
517. Loh K, Sivalingam N. Urinary tract infections in pregnancy. Malays Fam Physician (2007) 2:54–7.
518. Macejko AM, Schaeffer AJ. Asymptomatic bacteriuria and symptomatic urinary tract infections during pregnancy. Urol Clin North Am (2007) 34:35–42. doi:10.1016/j.ucl.2006.10.010
519. Schnarr J, Smaill F. Asymptomatic bacteriuria and symptomatic urinary tract infections in pregnancy. Eur J Clin Invest (2008) 38(Suppl 2):50–7. doi:10.1111/j.1365-2362.2008.02009.x
520. Imade PE, Izekor PE, Eghafona NO, Enabulele OI, Ophori E. Asymptomatic bacteriuria among pregnant women. N Am J Med Sci (2010) 2:263–6. doi:10.4297/najms.2010.2263
521. Gilbert NM, O’Brien VP, Hultgren S, Macones G, Lewis WG, Lewis AL. Urinary tract infection as a preventable cause of pregnancy complications: opportunities, challenges, and a global call to action. Glob Adv Health Med (2013) 2:59–69. doi:10.7453/gahmj.2013.061
522. Glaser AP, Schaeffer AJ. Urinary tract infection and bacteriuria in pregnancy. Urol Clin North Am (2015) 42:547–60. doi:10.1016/j.ucl.2015.05.004
523. Germain SJ, Sacks GP, Sooranna SR, Sargent IL, Redman CWG. Systemic inflammatory priming in normal pregnancy and preeclampsia: the role of circulating syncytiotrophoblast microparticles. J Immunol (2007) 178:5949–56. doi:10.4049/jimmunol.178.9.5949
524. Redman CWG, Sargent IL. Circulating microparticles in normal pregnancy and pre-eclampsia. Placenta (2008) 29(Suppl A):S73–7. doi:10.1016/j.placenta.2007.11.016
525. Hahn S, Rusterholz C, Hösli I, Lapaire O. Cell-free nucleic acids as potential markers for preeclampsia. Placenta (2011) 32(Suppl):S17–20. doi:10.1016/j.placenta.2010.06.018
526. Rusterholz C, Messerli M, Hoesli I, Hahn S. Placental microparticles, DNA, and RNA in preeclampsia. Hypertens Pregnancy (2011) 30:364–75. doi:10.3109/10641951003599571
527. Redman CWG, Tannetta DS, Dragovic RA, Gardiner C, Southcombe JH, Collett GP, et al. Review: does size matter? Placental debris and the pathophysiology of pre-eclampsia. Placenta (2012) 33(Suppl):S48–54. doi:10.1016/j.placenta.2011.12.006
528. Goulopoulou S, Davidge ST. Molecular mechanisms of maternal vascular dysfunction in preeclampsia. Trends Mol Med (2015) 21:88–97. doi:10.1016/j.molmed.2014.11.009
529. Niccolai E, Emmi G, Squatrito D, Silvestri E, Emmi L, Amedei A, et al. Microparticles: bridging the gap between autoimmunity and thrombosis. Semin Thromb Hemost (2015) 41:413–22. doi:10.1055/s-0035-1549850
530. Souza ACP, Yuen PST, Star RA. Microparticles: markers and mediators of sepsis-induced microvascular dysfunction, immunosuppression, and AKI. Kidney Int (2015) 87:1100–8. doi:10.1038/ki.2015.26
531. Mitchell MD, Peiris HN, Kobayashi M, Koh YQ, Duncombe G, Illanes SE, et al. Placental exosomes in normal and complicated pregnancy. Am J Obstet Gynecol (2015) 213:S173–81. doi:10.1016/j.ajog.2015.07.001
532. de Jonge L, Bos HJ, Van Langen IM, De Jong-Van Den Berg LTW, Bakker MK. Antibiotics prescribed before, during and after pregnancy in the Netherlands: a drug utilization study. Pharmacoepidemiol Drug Saf (2014) 23:60–8. doi:10.1002/pds.3492
533. Bookstaver PB, Bland CM, Griffin B, Stover KR, Eiland LS, McLaughlin M. A review of antibiotic use in pregnancy. Pharmacotherapy (2015) 35:1052–62. doi:10.1002/phar.1649
534. Palmsten K, Hernández-Díaz S, Chambers CD, Mogun H, Lai S, Gilmer TP, et al. The most commonly dispensed prescription medications among pregnant women enrolled in the U.S. Medicaid program. Obstet Gynecol (2015) 126:465–73. doi:10.1097/AOG.0000000000000982
535. Mawson AR. Effects of antiretroviral therapy on occurrence of pre-eclampsia. Lancet (2003) 361:347–8. doi:10.1016/S0140-6736(03)12359-8
536. Suy A, Martínez E, Coll O, Lonca M, Palacio M, De Lazzari E, et al. Increased risk of pre-eclampsia and fetal death in HIV-infected pregnant women receiving highly active antiretroviral therapy. AIDS (2006) 20:59–66. doi:10.1097/01.aids.0000198090.70325.bd
537. Wimalasundera RC, Larbalestier N, Smith JH, De Ruiter A, Mc GTSA, Hughes AD, et al. Pre-eclampsia, antiretroviral therapy, and immune reconstitution. Lancet (2002) 360:1152–4. doi:10.1016/S0140-6736(02)11195-0
538. Hall DR. Is pre-eclampsia less common in patients with HIV/AIDS? J Reprod Immunol (2007) 76:75–7. doi:10.1016/j.jri.2007.04.005
539. Adams JW, Watts DH, Phelps BR. A systematic review of the effect of HIV infection and antiretroviral therapy on the risk of pre-eclampsia. Int J Gynaecol Obstet (2016) 133:17–21. doi:10.1016/j.ijgo.2015.08.007
540. Todros T, Verdiglione P, Oggè G, Paladini D, Vergani P, Cardaropoli S. Low incidence of hypertensive disorders of pregnancy in women treated with spiramycin for Toxoplasma infection. Br J Clin Pharmacol (2006) 61:336–40. doi:10.1111/j.1365-2125.2005.02572.x
541. Alvarado-Esquivel C, Vázquez-Alaníz F, Sandoval-Carrillo AA, Salas-Pacheco JM, Hernández-Tinoco J, Sanchez-Anguiano LF, et al. Lack of association between Toxoplasma gondii infection and hypertensive disorders in pregnancy: a case-control study in a Northern Mexican population. Parasit Vectors (2014) 7:167. doi:10.1186/1756-3305-7-167
542. Hall D, Gebhardt S, Theron G, Grové D. Pre-eclampsia and gestational hypertension are less common in HIV infected women. Pregnancy Hypertens (2014) 4:91–6. doi:10.1016/j.preghy.2013.11.008
543. McCarthy FP, Kingdom JC, Kenny LC, Walsh SK. Animal models of preeclampsia; uses and limitations. Placenta (2011) 32:413–9. doi:10.1016/j.placenta.2011.03.010
544. Aubuchon M, Schulz LC, Schust DJ. Preeclampsia: animal models for a human cure. Proc Natl Acad Sci U S A (2011) 108:1197–8. doi:10.1073/pnas.1018164108
545. Kumasawa K, Ikawa M, Kidoya H, Hasuwa H, Saito-Fujita T, Morioka Y, et al. Pravastatin induces placental growth factor (PGF) and ameliorates preeclampsia in a mouse model. Proc Natl Acad Sci U S A (2011) 108:1451–5. doi:10.1073/pnas.1011293108
546. Faas MM, Schuiling GA, Baller JF, Visscher CA, Bakker WW. A new animal model for human preeclampsia: ultra-low-dose endotoxin infusion in pregnant rats. Am J Obstet Gynecol (1994) 171:158–64. doi:10.1016/0002-9378(94)90463-4
547. Faas MM, Schuiling GA, Linton EA, Sargent IL, Redman CWG. Activation of peripheral leukocytes in rat pregnancy and experimental preeclampsia. Am J Obstet Gynecol (2000) 182:351–7. doi:10.1016/S0002-9378(00)70223-7
548. Sakawi Y, Tarpey M, Chen YF, Calhoun DA, Connor MG, Chestnut DH, et al. Evaluation of low-dose endotoxin administration during pregnancy as a model of preeclampsia. Anesthesiology (2000) 93:1446–55. doi:10.1097/00000542-200012000-00017
549. Lin F, Zeng P, Xu ZY, Ye DY, Yu XF, Wang N, et al. Treatment of lipoxin A4 and its analogue on low-dose endotoxin induced preeclampsia in rat and possible mechanisms. Reprod Toxicol (2012) 34:677–85. doi:10.1016/j.reprotox.2012.09.009
550. Cotechini T, Komisarenko M, Sperou A, MacDonald-Goodfellow S, Adams MA, Graham CH. Inflammation in rat pregnancy inhibits spiral artery remodeling leading to fetal growth restriction and features of preeclampsia. J Exp Med (2014) 211:165–79. doi:10.1084/jem.20130295
551. Xue PP, Zheng MM, Gong P, Lin CM, Zhou JJ, Li YJ, et al. Single administration of ultra-low-dose lipopolysaccharide in rat early pregnancy induces TLR4 activation in the placenta contributing to preeclampsia. PLoS One (2015) 10:e0124001. doi:10.1371/journal.pone.0124001
552. Kalkunte S, Boij R, Norris W, Friedman J, Lai Z, Kurtis J, et al. Sera from preeclampsia patients elicit symptoms of human disease in mice and provide a basis for an in vitro predictive assay. Am J Pathol (2010) 177:2387–98. doi:10.2353/ajpath.2010.100475
553. Nymark M, Pussinen PJ, Tuomainen AM, Forsblom C, Groop PH, Lehto M, et al. Serum lipopolysaccharide activity is associated with the progression of kidney disease in finnish patients with type 1 diabetes. Diabetes Care (2009) 32:1689–93. doi:10.2337/dc09-0467
554. Al-Attas OS, Al-Daghri NM, Al-Rubeaan K, Da Silva NF, Sabico SL, Kumar S, et al. Changes in endotoxin levels in T2DM subjects on anti-diabetic therapies. Cardiovasc Diabetol (2009) 8:20. doi:10.1186/1475-2840-8-20
555. Hawkesworth S, Moore SE, Fulford AJC, Barclay GR, Darboe AA, Mark H, et al. Evidence for metabolic endotoxemia in obese and diabetic Gambian women. Nutr Diabetes (2013) 3:e83. doi:10.1038/nutd.2013.24
556. Zaman GS, Zaman F. Relationship between postprandial endotoxemia in nonobese postmenopausal women and diabetic nonobese postmenopausal women. J Nat Sci Biol Med (2015) 6:89–93. doi:10.4103/0976-9668.149098
557. Rusterholz C, Hahn S, Holzgreve W. Role of placentally produced inflammatory and regulatory cytokines in pregnancy and the etiology of preeclampsia. Semin Immunopathol (2007) 29:151–62. doi:10.1007/s00281-007-0071-6
558. Xie F, Turvey SE, Williams MA, Mor G, Von Dadelszen P. Toll-like receptor signaling and pre-eclampsia. Am J Reprod Immunol (2010) 63:7–16. doi:10.1111/j.1600-0897.2009.00745.x
559. Anton L, Brown AG, Parry S, Elovitz MA. Lipopolysaccharide induces cytokine production and decreases extravillous trophoblast invasion through a mitogen-activated protein kinase-mediated pathway: possible mechanisms of first trimester placental dysfunction. Hum Reprod (2012) 27:61–72. doi:10.1093/humrep/der362
560. Ahn H, Park J, Gilman-Sachs A, Kwak-Kim J. Immunologic characteristics of preeclampsia, a comprehensive review. Am J Reprod Immunol (2011) 65:377–94. doi:10.1111/j.1600-0897.2010.00913.x
561. Pineda A, Verdin-Terán SL, Camacho A, Moreno-Fierros L. Expression of toll-like receptor TLR-2, TLR-3, TLR-4 and TLR-9 is increased in placentas from patients with preeclampsia. Arch Med Res (2011) 42:382–91. doi:10.1016/j.arcmed.2011.08.003
562. Chatterjee P, Weaver LE, Doersch KM, Kopriva SE, Chiasson VL, Allen SJ, et al. Placental toll-like receptor 3 and toll-like receptor 7/8 activation contributes to preeclampsia in humans and mice. PLoS One (2012) 7:e41884. doi:10.1371/journal.pone.0041884
563. Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, et al. Amyloid-DNA composites of bacterial biofilms stimulate autoimmunity. Immunity (2015) 42:1171–84. doi:10.1016/j.immuni.2015.06.002
564. Rapsinski GJ, Wynosky-Dolfi MA, Oppong GO, Tursi SA, Wilson RP, Brodsky IE, et al. Toll-like receptor 2 and NLRP3 cooperate to recognize a functional bacterial amyloid, curli. Infect Immun (2015) 83:693–701. doi:10.1128/IAI.02370-14
565. Spaulding CN, Dodson KW, Chapman MR, Hultgren SJ. Fueling the fire with fibers: bacterial amyloids promote inflammatory disorders. Cell Host Microbe (2015) 18:1–2. doi:10.1016/j.chom.2015.06.013
566. Wucherpfennig KW. Mechanisms for the induction of autoimmunity by infectious agents. J Clin Invest (2001) 108:1097–104. doi:10.1172/JCI14235
567. Wucherpfennig KW. Structural basis of molecular mimicry. J Autoimmun (2001) 16:293–302. doi:10.1006/jaut.2000.0499
568. Kohm AP, Fuller KG, Miller SD. Mimicking the way to autoimmunity: an evolving theory of sequence and structural homology. Trends Microbiol (2003) 11:101–5. doi:10.1016/S0966-842X(03)00006-4
569. Morris JA, Broughton SJ, Wessels Q. Microbes, molecular mimicry and molecules of mood and motivation. Med Hypotheses (2016) 87:40–3. doi:10.1016/j.mehy.2015.12.011
570. Jain D, Kaur KJ, Goel M, Salunke DM. Structural basis of functional mimicry between carbohydrate and peptide ligands of con A. Biochem Biophys Res Commun (2000) 272:843–9. doi:10.1006/bbrc.2000.2871
571. Goel M, Krishnan L, Kaur S, Kaur KJ, Salunke DM. Plasticity within the antigen-combining site may manifest as molecular mimicry in the humoral immune response. J Immunol (2004) 173:7358–67. doi:10.4049/jimmunol.173.12.7358
572. Uh A, Nicholson RC, Gonzalez GV, Simmons CF, Gombart A, Smith R, et al. Lipopolysaccharide stimulation of trophoblasts induces corticotropin-releasing hormone expression through MyD88. Am J Obstet Gynecol (2008) 199(317):e311–6. doi:10.1016/j.ajog.2008.06.091
573. Chen Q, Viall C, Kang Y, Liu B, Stone P, Chamley L. Anti-phospholipid antibodies increase non-apoptotic trophoblast shedding: a contribution to the pathogenesis of pre-eclampsia in affected women? Placenta (2009) 30:767–73. doi:10.1016/j.placenta.2009.06.008
574. Chen Q, Guo F, Hensby-Bennett S, Stone P, Chamley L. Antiphospholipid antibodies prolong the activation of endothelial cells induced by necrotic trophoblastic debris: implications for the pathogenesis of preeclampsia. Placenta (2012) 33:810–5. doi:10.1016/j.placenta.2012.07.019
575. Pantham P, Rosario R, Chen Q, Print CG, Chamley LW. Transcriptomic analysis of placenta affected by antiphospholipid antibodies: following the TRAIL of trophoblast death. J Reprod Immunol (2012) 94:151–4. doi:10.1016/j.jri.2012.03.487
576. Tong M, Viall CA, Chamley LW. Antiphospholipid antibodies and the placenta: a systematic review of their in vitro effects and modulation by treatment. Hum Reprod Update (2015) 21:97–118. doi:10.1093/humupd/dmu049
577. Dechend R, Homuth V, Wallukat G, Kreuzer J, Park JK, Theuer J, et al. AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation (2000) 101:2382–7. doi:10.1161/01.CIR.101.20.2382
578. Roberts JM. Angiotensin-1 receptor autoantibodies: a role in the pathogenesis of preeclampsia? Circulation (2000) 101:2335–7. doi:10.1161/01.CIR.101.20.2335
579. Wallukat G, Neichel D, Nissen E, Homuth V, Luft FC. Agonistic autoantibodies directed against the angiotensin II AT1 receptor in patients with preeclampsia. Can J Physiol Pharmacol (2003) 81:79–83. doi:10.1139/y02-160
580. Dechend R, Muller DN, Wallukat G, Homuth V, Krause M, Dudenhausen J, et al. AT1 receptor agonistic antibodies, hypertension, and preeclampsia. Semin Nephrol (2004) 24:571–9. doi:10.1016/j.semnephrol.2004.07.006
581. Dechend R, Homuth V, Wallukat G, Muller DN, Krause M, Dudenhausen J, et al. Agonistic antibodies directed at the angiotensin II, AT1 receptor in preeclampsia. J Soc Gynecol Investig (2006) 13:79–86. doi:10.1016/j.jsgi.2005.11.006
582. Hubel CA, Wallukat G, Wolf M, Herse F, Rajakumar A, Roberts JM, et al. Agonistic angiotensin II type 1 receptor autoantibodies in postpartum women with a history of preeclampsia. Hypertension (2007) 49:612–7. doi:10.1161/01.HYP.0000256565.20983.d4
583. Dechend R, Luft FC. Are we getting closer to a nobel prize for unraveling preeclampsia? Curr Cardiol Rep (2008) 10:440–7. doi:10.1007/s11886-008-0070-9
584. Herse F, Staff AC, Hering L, Müller DN, Luft FC, Dechend R. AT1-receptor autoantibodies and uteroplacental RAS in pregnancy and pre-eclampsia. J Mol Med (Berl) (2008) 86:697–703. doi:10.1007/s00109-008-0332-4
585. Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, et al. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med (2008) 14:855–62. doi:10.1038/nm.1856
586. Lorquet S, Pequeux C, Munaut C, Foidart JM. Aetiology and physiopathology of preeclampsia and related forms. Acta Clin Belg (2010) 65:237–41. doi:10.1179/acb.2010.051
587. Parrish MR, Murphy SR, Rutland S, Wallace K, Wenzel K, Wallukat G, et al. The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. Am J Hypertens (2010) 23:911–6. doi:10.1038/ajh.2010.70
588. Carbillon L. AT1-receptor autoantibody: a true causal factor of pre-eclampsia or only a marker of poor placentation? Am J Hypertens (2011) 24:375; author reply 376. doi:10.1038/ajh.2010.249
589. Herse F, LaMarca B. Angiotensin II type 1 receptor autoantibody (AT1-AA)-mediated pregnancy hypertension. Am J Reprod Immunol (2013) 69:413–8. doi:10.1111/aji.12072
590. Siddiqui AH, Irani RA, Zhang W, Wang W, Blackwell SC, Kellems RE, et al. Angiotensin receptor agonistic autoantibody-mediated soluble fms-like tyrosine kinase-1 induction contributes to impaired adrenal vasculature and decreased aldosterone production in preeclampsia. Hypertension (2013) 61:472–9. doi:10.1161/HYPERTENSIONAHA.111.00157
591. Pietarinen I, Kivinen S, Ylostalo P, Makitalo R, Laakso L. Smooth muscle antibodies in pre-eclampsia of pregnancy. Gynecol Obstet Invest (1982) 13:142–9. doi:10.1159/000299502
592. Alanen A. Serum IgE and smooth muscle antibodies in pre-eclampsia. Acta Obstet Gynecol Scand (1984) 63:581–2. doi:10.3109/00016348409155540
593. Fliser D, Buchholz K, Haller H, Olmesartan EUTO, Pravastatin In I, Atherosclerosis I. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation (2004) 110:1103–7. doi:10.1161/01.CIR.0000140265.21608.8E
594. Dagenais NJ, Jamali F. Protective effects of angiotensin II interruption: evidence for antiinflammatory actions. Pharmacotherapy (2005) 25:1213–29. doi:10.1592/phco.2005.25.9.1213
595. Platten M, Youssef S, Hur EM, Ho PP, Han MH, Lanz TV, et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc Natl Acad Sci U S A (2009) 106:14948–53. doi:10.1073/pnas.0903958106
596. Ma G, Li Y, Zhang J, Liu H, Hou D, Zhu L, et al. Association between the presence of autoantibodies against adrenoreceptors and severe pre-eclampsia: a pilot study. PLoS One (2013) 8:e57983. doi:10.1371/journal.pone.0057983
597. Li Y, Ma G, Zhang Z, Yue Y, Yuan Y, Wang Y, et al. Association of autoantibodies against the M2-muscarinic receptor with perinatal outcomes in women with severe preeclampsia. J Transl Med (2013) 11:285. doi:10.1186/1479-5876-11-285
598. Cornelius DC, Lamarca B. TH17- and IL-17-mediated autoantibodies and placental oxidative stress play a role in the pathophysiology of pre-eclampsia. Minerva Ginecol (2014) 66:243–9.
599. Darmochwał-Kolarz D, Kludka-Sternik M, Tabarkiewicz J, Kolarz B, Rolinski J, Leszczynska-Gorzelak B, et al. The predominance of Th17 lymphocytes and decreased number and function of Treg cells in preeclampsia. J Reprod Immunol (2012) 93:75–81. doi:10.1016/j.jri.2012.01.006
600. Wenzel K, Rajakumar A, Haase H, Geusens N, Hubner N, Schulz H, et al. Angiotensin II type 1 receptor antibodies and increased angiotensin II sensitivity in pregnant rats. Hypertension (2011) 58:77–84. doi:10.1161/HYPERTENSIONAHA.111.171348
601. Stepan H, Wallukat G, Schultheiss HP, Faber R, Walther T. Is parvovirus B19 the cause for autoimmunity against the angiotensin II type receptor? J Reprod Immunol (2007) 73:130–4. doi:10.1016/j.jri.2006.08.084
602. Irani RA, Zhang Y, Zhou CC, Blackwell SC, Hicks MJ, Ramin SM, et al. Autoantibody-mediated angiotensin receptor activation contributes to preeclampsia through tumor necrosis factor-alpha signaling. Hypertension (2010) 55:1246–53. doi:10.1161/HYPERTENSIONAHA.110.150540
603. Irani RA, Xia Y. Renin angiotensin signaling in normal pregnancy and preeclampsia. Semin Nephrol (2011) 31:47–58. doi:10.1016/j.semnephrol.2010.10.005
604. Mistry HD, Kurlak LO, Broughton Pipkin F. The placental renin-angiotensin system and oxidative stress in pre-eclampsia. Placenta (2013) 34:182–6. doi:10.1016/j.placenta.2012.11.027
605. Verdonk K, Visser W, Van Den Meiracker AH, Danser AH. The renin-angiotensin-aldosterone system in pre-eclampsia: the delicate balance between good and bad. Clin Sci (Lond) (2014) 126:537–44. doi:10.1042/CS20130455
606. Diveu C, Mcgeachy MJ, Cua DJ. Cytokines that regulate autoimmunity. Curr Opin Immunol (2008) 20:663–8. doi:10.1016/j.coi.2008.09.003
607. Dong C. Regulation and pro-inflammatory function of interleukin-17 family cytokines. Immunol Rev (2008) 226:80–6. doi:10.1111/j.1600-065X.2008.00709.x
608. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med (2009) 361:888–98. doi:10.1056/NEJMra0707449
609. Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov (2012) 11:763–76. doi:10.1038/nrd3794
610. Benedetti G, Miossec P. Interleukin 17 contributes to the chronicity of inflammatory diseases such as rheumatoid arthritis. Eur J Immunol (2014) 44:339–47. doi:10.1002/eji.201344184
611. Aujla SJ, Dubin PJ, Kolls JK. Th17 cells and mucosal host defense. Semin Immunol (2007) 19:377–82. doi:10.1016/j.smim.2007.10.009
612. Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol (2007) 19:409–17. doi:10.1016/j.smim.2007.10.011
613. Stockinger B, Veldhoen M, Martin B. Th17 T cells: linking innate and adaptive immunity. Semin Immunol (2007) 19:353–61. doi:10.1016/j.smim.2007.10.008
614. Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. The phenotype of human Th17 cells and their precursors, the cytokines that mediate their differentiation and the role of Th17 cells in inflammation. Int Immunol (2008) 20:1361–8. doi:10.1093/intimm/dxn106
615. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity (2008) 28:454–67. doi:10.1016/j.immuni.2008.03.004
616. Curtis MM, Way SS. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology (2009) 126:177–85. doi:10.1111/j.1365-2567.2008.03017.x
617. Eyerich K, Pennino D, Scarponi C, Foerster S, Nasorri F, Behrendt H, et al. IL-17 in atopic eczema: linking allergen-specific adaptive and microbial-triggered innate immune response. J Allergy Clin Immunol (2009) 123:59–66.e4. doi:10.1016/j.jaci.2008.10.031
618. Khader SA, Gaffen SL, Kolls JK. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol (2009) 2:403–11. doi:10.1038/mi.2009.100
619. Cypowyj S, Picard C, Marodi L, Casanova JL, Puel A. Immunity to infection in IL-17-deficient mice and humans. Eur J Immunol (2012) 42:2246–54. doi:10.1002/eji.201242605
620. Rubino SJ, Geddes K, Girardin SE. Innate IL-17 and IL-22 responses to enteric bacterial pathogens. Trends Immunol (2012) 33:112–8. doi:10.1016/j.it.2012.01.003
621. Weber A, Zimmermann C, Kieseier BC, Hartung HP, Hofstetter HH. Bacteria and their cell wall components uniformly co-activate interleukin-17-producing thymocytes. Clin Exp Immunol (2014) 178:504–15. doi:10.1111/cei.12414
622. Beringer A, Noack M, Miossec P. IL-17 in chronic inflammation: from discovery to targeting. Trends Mol Med (2016) 22:230–41. doi:10.1016/j.molmed.2016.01.001
623. Kumar P, Monin L, Castillo P, Elsegeiny W, Horne W, Eddens T, et al. Intestinal interleukin-17 receptor signaling mediates reciprocal control of the gut microbiota and autoimmune inflammation. Immunity (2016) 44:659–71. doi:10.1016/j.immuni.2016.02.007
624. Saito S, Nakashima A, Ito M, Shima T. Clinical implication of recent advances in our understanding of IL-17 and reproductive immunology. Expert Rev Clin Immunol (2011) 7:649–57. doi:10.1586/eci.11.49
625. Fu B, Tian Z, Wei H. TH17 cells in human recurrent pregnancy loss and pre-eclampsia. Cell Mol Immunol (2014) 11:564–70. doi:10.1038/cmi.2014.54
626. Ozkan ZS, Simsek M, Ilhan F, Deveci D, Godekmerdan A, Sapmaz E. Plasma IL-17, IL-35, interferon-gamma, SOCS3 and TGF-beta levels in pregnant women with preeclampsia, and their relation with severity of disease. J Matern Fetal Neonatal Med (2014) 27:1513–7. doi:10.3109/14767058.2013.861415
627. Santner-Nanan B, Peek MJ, Khanam R, Richarts L, Zhu E, Fazekas De St Groth B, et al. Systemic increase in the ratio between Foxp3+ and IL-17-producing CD4+ T cells in healthy pregnancy but not in preeclampsia. J Immunol (2009) 183:7023–30. doi:10.4049/jimmunol.0901154
628. Jianjun Z, Yali H, Zhiqun W, Mingming Z, Xia Z. Imbalance of T-cell transcription factors contributes to the Th1 type immunity predominant in pre-eclampsia. Am J Reprod Immunol (2010) 63:38–45. doi:10.1111/j.1600-0897.2009.00763.x
629. Saito S. Th17 cells and regulatory T cells: new light on pathophysiology of preeclampsia. Immunol Cell Biol (2010) 88:615–7. doi:10.1038/icb.2010.68
630. Toldi G, Rigó J, Stenczer B, Vásárhelyi B, Molvarec A. Increased prevalence of IL-17-producing peripheral blood lymphocytes in pre-eclampsia. Am J Reprod Immunol (2011) 66:223–9. doi:10.1111/j.1600-0897.2011.00987.x
631. Cornelius DC, Hogg JP, Scott J, Wallace K, Herse F, Moseley J, et al. Administration of interleukin-17 soluble receptor C suppresses TH17 cells, oxidative stress, and hypertension in response to placental ischemia during pregnancy. Hypertension (2013) 62:1068–73. doi:10.1161/HYPERTENSIONAHA.113.01514
632. Laresgoiti-Servitje E. A leading role for the immune system in the pathophysiology of preeclampsia. J Leukoc Biol (2013) 94:247–57. doi:10.1189/jlb.1112603
633. Perez-Sepulveda A, Torres MJ, Khoury M, Illanes SE. Innate immune system and preeclampsia. Front Immunol (2014) 5:244. doi:10.3389/fimmu.2014.00244
634. Cao W, Wang X, Chen T, Zhu H, Xu W, Zhao S, et al. The expression of notch/notch ligand, IL-35, IL-17, and Th17/Treg in preeclampsia. Dis Markers (2015) 2015:316182. doi:10.1155/2015/316182
635. Molvarec A, Czegle I, Szijártó J, Rigó J. Increased circulating interleukin-17 levels in preeclampsia. J Reprod Immunol (2015) 112:53–7. doi:10.1016/j.jri.2015.05.007
636. Wang H, Guo M, Liu F, Wang J, Zhou Z, Ji J, et al. Role of IL-17 variants in preeclampsia in Chinese Han women. PLoS One (2015) 10:e0140118. doi:10.1371/journal.pone.0140118
637. Vargas-Rojas MI, Solleiro-Villavicencio H, Soto-Vega E. Th1, Th2, Th17 and Treg levels in umbilical cord blood in preeclampsia. J Matern Fetal Neonatal Med (2016) 29:1642–5. doi:10.3109/14767058.2015.1057811
638. Darmochwał-Kolarz D, Oleszczuk J. The critical role of Th17 cells, Treg cells and co-stimulatory molecules in the development of pre-eclampsia. Dev Period Med (2014) 18:141–7.
639. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med (2006) 355:1018–28. doi:10.1056/NEJMoa063842
640. Wang H, Ma S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am J Emerg Med (2008) 26:711–5. doi:10.1016/j.ajem.2007.10.031
641. Tscherne DM, García-Sastre A. Virulence determinants of pandemic influenza viruses. J Clin Invest (2011) 121:6–13. doi:10.1172/JCI44947
642. D’Elia RV, Harrison K, Oyston PC, Lukaszewski RA, Clark GC. Targeting the “cytokine storm” for therapeutic benefit. Clin Vaccine Immunol (2013) 20:319–27. doi:10.1128/CVI.00636-12
643. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA (2016) 315:801–10. doi:10.1001/jama.2016.0287
644. Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med (2012) 366:2055–64. doi:10.1056/NEJMoa1202290
645. Lai PS, Matteau A, Iddriss A, Hawes JC, Ranieri V, Thompson BT. An updated meta-analysis to understand the variable efficacy of drotrecogin alfa (activated) in severe sepsis and septic shock. Minerva Anestesiol (2013) 79:33–43.
646. Vulcano M, Meiss RP, Isturiz MA. Deferoxamine reduces tissue injury and lethality in LPS-treated mice. Int J Immunopharmacol (2000) 22:635–44. doi:10.1016/S0192-0561(00)00026-6
647. Messaris E, Antonakis PT, Memos N, Chatzigianni E, Leandros E, Konstadoulakis MM. Deferoxamine administration in septic animals: improved survival and altered apoptotic gene expression. Int Immunopharmacol (2004) 4:455–9. doi:10.1016/j.intimp.2004.01.012
648. Islam S, Jarosch S, Zhou J, Del Carmen Parquet C, Toguri JT, Colp P, et al. Anti-inflammatory and anti-bacterial effects of iron chelation in experimental sepsis. J Surg Res (2016) 200:266–73. doi:10.1016/j.jss.2015.07.001
649. Doron MW, Makhlouf RA, Katz VL, Lawson EE, Stiles AD. Increased incidence of sepsis at birth in neutropenic infants of mothers with preeclampsia. J Pediatr (1994) 125:452–8. doi:10.1016/S0022-3476(05)83294-9
650. Kocherlakota P, La Gamma EF. Preliminary report: rhG-CSF may reduce the incidence of neonatal sepsis in prolonged preeclampsia-associated neutropenia. Pediatrics (1998) 102:1107–11. doi:10.1542/peds.102.5.1107
651. Bhaumik S, Ghosh S, Haldar KK, Mitra PK, Manna B. Risk of early onset neonatal septicemia in babies born to mothers with pre-eclampsia. Indian Pediatr (2000) 37:775–9.
652. Procianoy RS, Silveira RC, Mussi-Pinhata MM, Rugolo LMSS, Leone CR, Lopes JMD, et al. Sepsis and neutropenia in very low birth weight infants delivered of mothers with preeclampsia. J Pediatr (2010) 157:434–U118. doi:10.1016/j.jpeds.2010.02.066
653. Namdev S, Bhat V, Adhisivam B, Zachariah B. Oxidative stress and antioxidant status among neonates born to mothers with pre-eclampsia and their early outcome. J Matern Fetal Neonatal Med (2014) 27:1481–4. doi:10.3109/14767058.2013.860521
654. Kenny LC, Dunn WB, Ellis DI, Myers J, Baker PN; The Gopec Consortium, et al. Novel biomarkers for pre-eclampsia detected using metabolomics and machine learning. Metabolomics (2005) 1:227–34. doi:10.1007/s11306-005-0003-1
655. Kenny LC, Broadhurst D, Brown M, Dunn WB, Redman CWG, Kell DB, et al. Detection and identification of novel metabolomic biomarkers in preeclampsia. Reprod Sci (2008) 15:591–7. doi:10.1177/1933719108316908
656. Odibo AO, Goetzinger KR, Odibo L, Cahill AG, Macones GA, Nelson DM, et al. First-trimester prediction of preeclampsia using metabolomic biomarkers: a discovery phase study. Prenat Diagn (2011) 31:990–4. doi:10.1002/pd.2822
657. Bahado-Singh RO, Akolekar R, Mandal R, Dong E, Xia J, Kruger M, et al. Metabolomics and first-trimester prediction of early-onset preeclampsia. J Matern Fetal Neonatal Med (2012) 25:1840–7. doi:10.3109/14767058.2012.680254
658. Dunn WB, Brown M, Worton SA, Davies K, Jones RL, Kell DB, et al. The metabolome of human placental tissue: investigation of first trimester tissue and changes related to preeclampsia in late pregnancy. Metabolomics (2012) 8:579–97. doi:10.1007/s11306-011-0348-6
659. Woodham PC, O’Connell T, Grimes J, Haeri S, Eichelberger K, Baker A, et al. Metabolomics to predict severe preeclampsia in early pregnancy. Am J Obstet Gynecol (2012) 206:S348–348. doi:10.1016/j.ajog.2011.10.809
660. Kuc S, Koster MPH, Pennings JLA, Hankemeier T, Berger R, Harms AC, et al. Metabolomics profiling for identification of novel potential markers in early prediction of preeclampsia. PLoS One (2014) 9:e98540. doi:10.1371/journal.pone.0098540
661. Koster MPH, Vreeken RJ, Harms AC, Dane AD, Kuc S, Schielen PCJI, et al. First-trimester serum acylcarnitine levels to predict preeclampsia: a metabolomics approach. Dis Markers (2015) 2015:857108. doi:10.1155/2015/857108
662. Myers JE, Hart S, Armstrong S, Mires GJ, Beynon R, Gaskell SJ, et al. Evidence for multiple circulating factors in preeclampsia. Am J Obstet Gynecol (2007) 196(266):e261–6. doi:10.1016/j.ajog.2006.10.875
663. Kolla V, Jenö P, Moes S, Lapaire O, Hoesli I, Hahn S. Quantitative proteomic (iTRAQ) analysis of 1st trimester maternal plasma samples in pregnancies at risk for preeclampsia. J Biomed Biotechnol (2012) 2012:305964. doi:10.1155/2012/305964
664. Myers JE, Kenny LC, Mccowan LM, Chan EH, Dekker GA, Poston L, et al. Angiogenic factors combined with clinical risk factors to predict preterm pre-eclampsia in nulliparous women: a predictive test accuracy study. BJOG (2013) 120:1215–23. doi:10.1111/1471-0528.12195
665. Myers JE, Tuytten R, Thomas G, Laroy W, Kas K, Vanpoucke G, et al. Integrated proteomics pipeline yields novel biomarkers for predicting preeclampsia. Hypertension (2013) 61:1281–8. doi:10.1161/HYPERTENSIONAHA.113.01168
666. Polsani S, Phipps E, Jim B. Emerging new biomarkers of preeclampsia. Adv Chronic Kidney Dis (2013) 20:271–9. doi:10.1053/j.ackd.2013.01.001
667. Hahn S, Lapaire O, Than NG. Biomarker development for presymptomatic molecular diagnosis of preeclampsia: feasible, useful or even unnecessary? Expert Rev Mol Diagn (2015) 15:617–29. doi:10.1586/14737159.2015.1025757
668. Khan GH, Galazis N, Docheva N, Layfield R, Atiomo W. Overlap of proteomics biomarkers between women with pre-eclampsia and PCOS: a systematic review and biomarker database integration. Hum Reprod (2015) 30:133–48. doi:10.1093/humrep/deu268
669. Paulus P, Jennewein C, Zacharowski K. Biomarkers of endothelial dysfunction: can they help us deciphering systemic inflammation and sepsis? Biomarkers (2011) 16(Suppl 1):S11–21. doi:10.3109/1354750X.2011.587893
670. Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science (2016) 352:aaf1098. doi:10.1126/science.aaf1098
671. Savvidou MD, Lees CC, Parra M, Hingorani AD, Nicolaides KH. Levels of C-reactive protein in pregnant women who subsequently develop pre-eclampsia. BJOG (2002) 109:297–301. doi:10.1111/j.1471-0528.2002.01130.x
672. Kashanian M, Aghbali F, Mahali N. Evaluation of the diagnostic value of the first-trimester maternal serum high-sensitivity C-reactive protein level for prediction of pre-eclampsia. J Obstet Gynaecol Res (2013) 39:1549–54. doi:10.1111/jog.12105
673. Saito S, Shiozaki A, Nakashima A, Sakai M, Sasaki Y. The role of the immune system in preeclampsia. Mol Aspects Med (2007) 28:192–209. doi:10.1016/j.mam.2007.02.006
674. Rinehart BK, Terrone DA, Lagoo-Deenadayalan S, Barber WH, Hale EA, Martin JN Jr, et al. Expression of the placental cytokines tumor necrosis factor alpha, interleukin 1beta, and interleukin 10 is increased in preeclampsia. Am J Obstet Gynecol (1999) 181:915–20. doi:10.1016/S0002-9378(99)70325-X
675. Serin ÝS, Özçelik B, Bapbuð M, Kýlýç H, Okur D, Erez R. Predictive value of tumor necrosis factor alpha (TNF-alpha) in preeclampsia. Eur J Obstet Gynecol Reprod Biol (2002):143–5. doi:10.1016/S0301-2115(01)00484-5
676. Todros T, Bontempo S, Piccoli E, Ietta F, Romagnoli R, Biolcati M, et al. Increased levels of macrophage migration inhibitory factor (MIF) in preeclampsia. Eur J Obstet Gynecol Reprod Biol (2005) 123:162–6. doi:10.1016/j.ejogrb.2005.03.014
677. Mehr S, Doyle LW. Cytokines as markers of bacterial sepsis in newborn infants: a review. Pediatr Infect Dis J (2000) 19:879–87. doi:10.1097/00006454-200009000-00014
678. Roger T, Glauser MP, Calandra T. Macrophage migration inhibitory factor (MIF) modulates innate immune responses induced by endotoxin and Gram-negative bacteria. J Endotoxin Res (2001) 7:456–60. doi:10.1179/096805101101533089
679. Takala A, Nupponen I, Kylanpää-Bäck ML, Repo H. Markers of inflammation in sepsis. Ann Med (2002) 34:614–23. doi:10.1080/078538902321117841
680. Briassoulis G, Galani A. Prognostic markers of pediatric meningococcal sepsis. Expert Rev Anti Infect Ther (2014) 12:1017–20. doi:10.1586/14787210.2014.945431
681. Su H, Chang SS, Han CM, Wu KY, Li MC, Huang CY, et al. Inflammatory markers in cord blood or maternal serum for early detection of neonatal sepsis-a systemic review and meta-analysis. J Perinatol (2014) 34:268–74. doi:10.1038/jp.2013.186
682. Kell DB, Knowles JD. The role of modeling in systems biology. In: Szallasi Z, Stelling J, Periwal V, editors. System Modeling in Cellular Biology: From Concepts to Nuts and Bolts. Cambridge: MIT Press (2006). p. 3–18.
683. Palsson BØ. Systems Biology: Properties of Reconstructed Networks. Cambridge: Cambridge University Press (2006).
684. Palsson BØ. Systems Biology: Constraint-Based Reconstruction and Analysis. Cambridge: Cambridge University Press (2015).
685. Hucka M, Finney A, Sauro HM, Bolouri H, Doyle JC, Kitano H, et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics (2003) 19:524–31. doi:10.1093/bioinformatics/btg015
686. Funahashi A, Matsuoka Y, Jouraku A, Morohashi M, Kikuchi N, Kitano H. CellDesigner 3.5: a versatile modeling tool for biochemical networks. Proc IEEE (2008) 96:1254–65. doi:10.1109/JPROC.2008.925458
687. Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, et al. COPASI: a complex pathway simulator. Bioinformatics (2006) 22:3067–74. doi:10.1093/bioinformatics/btl485
688. Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics (2011) 27:431–2. doi:10.1093/bioinformatics/btq675
689. Madazli R, Kuseyrioglu B, Uzun H, Uludag S, Ocak V. Prediction of preeclampsia with maternal mid-trimester placental growth factor, activin A, fibronectin and uterine artery Doppler velocimetry. Int J Gynaecol Obstet (2005) 89:251–7. doi:10.1016/j.ijgo.2005.02.008
690. Diesch CH, Holzgreve W, Hahn S, Zhong XY. Comparison of activin A and cell-free fetal DNA levels in maternal plasma from patients at high risk for preeclampsia. Prenat Diagn (2006) 26:1267–70. doi:10.1002/pd.1606
691. Reddy A, Suri S, Sargent IL, Redman CW, Muttukrishna S. Maternal circulating levels of activin A, inhibin A, sFlt-1 and endoglin at parturition in normal pregnancy and pre-eclampsia. PLoS One (2009) 4:e4453. doi:10.1371/journal.pone.0004453
692. Phillips DJ, Jones KL, Scheerlinck JY, Hedger MP, De Kretser DM. Evidence for activin A and follistatin involvement in the systemic inflammatory response. Mol Cell Endocrinol (2001) 180:155–62. doi:10.1016/S0303-7207(01)00516-0
693. Hodges R, Salvador L, D’Antona D, Georgiou HM, Wallace EM. Activin A as a marker of intrauterine infection in women with preterm prelabour rupture of membranes. J Perinatol (2010) 30:22–6. doi:10.1038/jp.2009.109
694. Rosenberg VA, Buhimschi IA, Dulay AT, Abdel-Razeq SS, Oliver EA, Duzyj CM, et al. Modulation of amniotic fluid activin-A and inhibin-A in women with preterm premature rupture of the membranes and infection-induced preterm birth. Am J Reprod Immunol (2012) 67:122–31. doi:10.1111/j.1600-0897.2011.01074.x
695. Petrakou E, Fotopoulos S, Anagnostakou M, Anatolitou F, Samitas K, Semitekolou M, et al. Activin-A exerts a crucial anti-inflammatory role in neonatal infections. Pediatr Res (2013) 74:675–81. doi:10.1038/pr.2013.159
696. Shu C, Liu Z, Cui L, Wei C, Wang S, Tang JJ, et al. Protein profiling of preeclampsia placental tissues. PLoS One (2014) 9:e112890. doi:10.1371/journal.pone.0112890
697. Lugli A, Forster Y, Haas P, Nocito A, Bucher C, Bissig H, et al. Calretinin expression in human normal and neoplastic tissues: a tissue microarray analysis on 5233 tissue samples. Hum Pathol (2003) 34:994–1000. doi:10.1053/S0046-8177(03)00339-3
698. Rizzo A, Carratelli CR, De Filippis A, Bevilacqua N, Tufano MA, Buommino E. Transforming activities of Chlamydia pneumoniae in human mesothelial cells. Int Microbiol (2014) 17:185–93. doi:10.2436/20.1501.01.221
699. Xu QL, Zhu M, Jin Y, Wang N, Xu HX, Quan LM, et al. The predictive value of the first-trimester maternal serum chemerin level for pre-eclampsia. Peptides (2014) 62:150–4. doi:10.1016/j.peptides.2014.10.002
700. Stepan H, Philipp A, Roth I, Kralisch S, Jank A, Schaarschmidt W, et al. Serum levels of the adipokine chemerin are increased in preeclampsia during and 6 months after pregnancy. Regul Pept (2011) 168:69–72. doi:10.1016/j.regpep.2011.03.005
701. AL-Refai AA. Evaluation of serum levels of the adipokines chemerin and resistin in preeclampsia. Life Sci J (2012) 9:5143–51.
702. Duan DM, Niu JM, Lei Q, Lin XH, Chen X. Serum levels of the adipokine chemerin in preeclampsia. J Perinat Med (2012) 40:121–7. doi:10.1515/Jpm.2011.127
703. Wang LQ, Yang TL, Ding YL, Zhong Y, Yu L, Peng M. Chemerin plays a protective role by regulating human umbilical vein endothelial cell-induced nitric oxide signaling in preeclampsia. Endocrine (2015) 48:299–308. doi:10.1007/s12020-014-0286-y
704. Broadhurst D, Kell DB. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics (2006) 2:171–96. doi:10.1007/s11306-006-0037-z
705. Kukla M, Zwirska-Korczala K, Gabriel A, Waluga M, Warakomska I, Szczygiel B, et al. Chemerin, vaspin and insulin resistance in chronic hepatitis C. J Viral Hepat (2010) 17:661–7. doi:10.1111/j.1365-2893.2009.01224.x
706. Kukla M, Mazur W, Buldak RJ, Żwirska-Korczala K. Potential role of leptin, adiponectin and three novel adipokines-visfatin, chemerin and vaspin-in chronic hepatitis. Mol Med (2011) 17:1397–410. doi:10.2119/molmed.2010.00105
707. Kulig P, Kantyka T, Zabel BA, Banas M, Chyra A, Stefanska A, et al. Regulation of chemerin chemoattractant and antibacterial activity by human cysteine cathepsins. J Immunol (2011) 187:1403–10. doi:10.4049/jimmunol.1002352
708. Banas M, Zabieglo K, Kasetty G, Kapinska-Mrowiecka M, Borowczyk J, Drukala J, et al. Chemerin is an antimicrobial agent in human epidermis. PLoS One (2013) 8:e58709. doi:10.1371/journal.pone.0058709
709. Zabel BA, Kwitniewski M, Banas M, Zabieglo K, Murzyn K, Cichy J. Chemerin regulation and role in host defense. Am J Clin Exp Immunol (2014) 3:1–19.
710. Banas M, Zegar A, Kwitniewski M, Zabieglo K, Marczynska J, Kapinska-Mrowiecka M, et al. The expression and regulation of chemerin in the epidermis. PLoS One (2015) 10:e0117830. doi:10.1371/journal.pone.0117830
711. Horn P, Metzing UB, Steidl R, Romeike B, Rauchfuß F, Sponholz C, et al. Chemerin in peritoneal sepsis and its associations with glucose metabolism and prognosis: a translational cross-sectional study. Crit Care (2016) 20:39. doi:10.1186/s13054-016-12090-5
712. Cornelius DC. Copeptin: a new biomarker that is specific for preeclampsia? Hypertension (2014) 64:1189–91. doi:10.1161/HYPERTENSIONAHA.114.04255
713. Birdir C, Janssen K, Stanescu AD, Enekwe A, Kasimir-Bauer S, Gellhaus A, et al. Maternal serum copeptin, MR-proANP and procalcitonin levels at 11-13 weeks gestation in the prediction of preeclampsia. Arch Gynecol Obstet (2015) 292:1033–42. doi:10.1007/s00404-015-3745-7
714. Jochberger S, Morgenthaler NG, Mayr VD, Luckner G, Wenzel V, Ulmer H, et al. Copeptin and arginine vasopressin concentrations in critically ill patients. J Clin Endocrinol Metab (2006) 91:4381–6. doi:10.1210/jc.2005-2830
715. Morgenthaler NG, Struck J, Jochberger S, Dünser MW. Copeptin: clinical use of a new biomarker. Trends Endocrinol Metab (2008) 19:43–9. doi:10.1016/j.tem.2007.11.001
716. Katan M, Christ-Crain M. The stress hormone copeptin: a new prognostic biomarker in acute illness. Swiss Med Wkly (2010) 140:w13101. doi:10.4414/smw.2010.13101
717. Nickel CH, Bingisser R, Morgenthaler NG. The role of copeptin as a diagnostic and prognostic biomarker for risk stratification in the emergency department. BMC Med (2012) 10:7. doi:10.1186/1741-7015-10-7
718. Dobša L, Edozien KC. Copeptin and its potential role in diagnosis and prognosis of various diseases. Biochem Med (Zagreb) (2013) 23:172–90. doi:10.11613/BM.2013.021
719. Odermatt J, Bolliger R, Hersberger L, Ottiger M, Christ-Crain M, Briel M, et al. Copeptin predicts 10-year all-cause mortality in community patients: a 10-year prospective cohort study. Clin Chem Lab Med (2016) 54:1681–90. doi:10.1515/cclm-2016-0151
720. Jochberger S, Luckner G, Mayr VD, Wenzel V, Morgenthaler NG, Friesenecker BE, et al. Course of vasopressin and copeptin plasma concentrations in a patient with severe septic shock. Anaesth Intensive Care (2006) 34:498–500.
721. Müller B, Morgenthaler N, Stolz D, Schuetz P, Muller C, Bingisser R, et al. Circulating levels of copeptin, a novel biomarker, in lower respiratory tract infections. Eur J Clin Invest (2007) 37:145–52. doi:10.1111/j.1365-2362.2007.01762.x
722. Jochberger S, Dörler J, Luckner G, Mayr VD, Wenzel V, Ulmer H, et al. The vasopressin and copeptin response to infection, severe sepsis, and septic shock. Crit Care Med (2009) 37:476–82. doi:10.1097/CCM.0b013e3181957532
723. Krüger S, Ewig S, Kunde J, Hanschmann A, Marre R, Suttorp N, et al. C-terminal provasopressin (copeptin) in patients with community-acquired pneumonia – influence of antibiotic pre-treatment: results from the German competence network CAPNETZ. J Antimicrob Chemother (2009) 64:159–62. doi:10.1093/jac/dkp148
724. Limper M, Goeijenbier M, Wagenaar JF, Gasem MH, Isbandrio B, Kunde J, et al. Copeptin as a predictor of disease severity and survival in leptospirosis. J Infect (2010) 61:92–4. doi:10.1016/j.jinf.2010.03.029
725. Fluri F, Morgenthaler NG, Mueller B, Christ-Crain M, Katan M. Copeptin, procalcitonin and routine inflammatory markers-predictors of infection after stroke. PLoS One (2012) 7:e48309. doi:10.1371/journal.pone.0048309
726. Kolditz M, Halank M, Schulte-Hubbert B, Bergmann S, Albrecht S, Höffken G. Copeptin predicts clinical deterioration and persistent instability in community-acquired pneumonia. Respir Med (2012) 106:1320–8. doi:10.1016/j.rmed.2012.06.008
727. Du JM, Sang G, Jiang CM, He XJ, Han Y. Relationship between plasma copeptin levels and complications of community-acquired pneumonia in preschool children. Peptides (2013) 45:61–5. doi:10.1016/j.peptides.2013.04.015
728. Masajtis-Zagajewska A, Kurnatowska I, Wajdlich M, Nowicki M. Utility of copeptin and standard inflammatory markers in the diagnostics of upper and lower urinary tract infections. BMC Urol (2015) 15:67. doi:10.1186/s12894-015-0061-2
729. Wrotek A, Jackowska T, Pawlik K. Sodium and copeptin levels in children with community acquired pneumonia. Adv Exp Med Biol (2015) 835:31–6. doi:10.1007/5584_2014_41
730. Strevens H, Wide-Swensson D, Grubb A, Hansen A, Horn T, Ingemarsson I, et al. Serum cystatin C reflects glomerular endotheliosis in normal, hypertensive and pre-eclamptic pregnancies. BJOG (2003) 110:825–30. doi:10.1111/j.1471-0528.2003.02051.x
731. Yang X, Wang H, Wang Z, Dong M. Alteration and significance of serum cardiac troponin I and cystatin C in preeclampsia. Clin Chim Acta (2006) 374:168–9. doi:10.1016/j.cca.2006.07.006
732. Kristensen K, Wide-Swensson D, Schmidt C, Blirup-Jensen S, Lindström V, Strevens H, et al. Cystatin C, beta-2-microglobulin and beta-trace protein in pre-eclampsia. Acta Obstet Gynecol Scand (2007) 86:921–6. doi:10.1080/00016340701318133
733. Guo HX, Wang CH, Li ZQ, Gong SP, Zhou ZQ, Leng LZ, et al. The application of serum cystatin C in estimating the renal function in women with preeclampsia. Reprod Sci (2012) 19:712–7. doi:10.1177/1933719111431001
734. Novakov Mikic A, Cabarkapa V, Nikolic A, Maric D, Brkic S, Mitic G, et al. Cystatin C in pre-eclampsia. J Matern Fetal Neonatal Med (2012) 25:961–5. doi:10.3109/14767058.2011.601366
735. Xiao J, Niu J, Ye X, Yu Q, Gu Y. Combined biomarkers evaluation for diagnosing kidney injury in preeclampsia. Hypertens Pregnancy (2013) 32:439–49. doi:10.3109/10641955.2013.827203
736. Yalcin S, Ulas T, Eren MA, Aydogan H, Camuzcuoglu A, Kucuk A, et al. Relationship between oxidative stress parameters and cystatin C levels in patients with severe preeclampsia. Medicina (Kaunas) (2013) 49:118–23.
737. Odden MC, Scherzer R, Bacchetti P, Szczech LA, Sidney S, Grunfeld C, et al. Cystatin C level as a marker of kidney function in human immunodeficiency virus infection: the FRAM study. Arch Intern Med (2007) 167:2213–9. doi:10.1001/archinte.167.20.2213
738. Randers E, Kornerup K, Erlandsen EJ, Hasling C, Danielsen H. Cystatin C levels in sera of patients with acute infectious diseases with high C-reactive protein levels. Scand J Clin Lab Invest (2001) 61:333–5. doi:10.1080/00365510118007
739. Gupta SK, Kitch D, Tierney C, Melbourne K, Ha B, Mccomsey GA, et al. Markers of renal disease and function are associated with systemic inflammation in HIV infection. HIV Med (2015) 16:591–8. doi:10.1111/hiv.12268
740. Walker JB, Nesheim ME. The molecular weights, mass distribution, chain composition, and structure of soluble fibrin degradation products released from a fibrin clot perfused with plasmin. J Biol Chem (1999) 274:5201–12. doi:10.1074/jbc.274.8.5201
741. Bellart J, Gilabert R, Anglès A, Piera V, Miralles RM, Monasterio J, et al. Tissue factor levels and high ratio of fibrinopeptide A:d-dimer as a measure of endothelial procoagulant disorder in pre-eclampsia. Br J Obstet Gynaecol (1999) 106:594–7. doi:10.1111/j.1471-0528.1999.tb08330.x
742. Gulec UK, Ozgunen FT, Guzel AB, Buyukkurt S, Seydaoglu G, Urunsak IF, et al. An analysis of C-reactive protein, procalcitonin, and d-dimer in pre-eclamptic patients. Am J Reprod Immunol (2012) 68:331–7. doi:10.1111/j.1600-0897.2012.01171.x
743. Pinheiro MDB, Junqueira DRG, Coelho FF, Freitas LG, Carvalho MG, Gomes KB, et al. d-dimer in preeclampsia: systematic review and meta-analysis. Clin Chim Acta (2012) 414:166–70. doi:10.1016/j.cca.2012.08.003
744. Bozkurt M, Yumru AE, Sahin L, Salman S. Troponin I and d-dimer levels in preeclampsia and eclampsia: prospective study. Clin Exp Obstet Gynecol (2015) 42:26–31.
745. Rahman R, Begum K, Khondker L, Majumder NI, Nahar K, Sultana R, et al. Role of d-dimer in determining coagulability status in pre-eclamptic and normotensive pregnant women. Mymensingh Med J (2015) 24:115–20.
746. Di Castelnuovo A, De Curtis A, Costanzo S, Persichillo M, Olivieri M, Zito F, et al. Association of d-dimer levels with all-cause mortality in a healthy adult population: findings from the MOLI-SANI study. Haematologica (2013) 98:1476–80. doi:10.3324/haematol.2012.083410
747. Jennings I, Woods TAL, Kitchen DP, Kitchen S, Walker ID. Laboratory d-dimer measurement: improved agreement between methods through calibration. Thromb Haemost (2007) 98:1127–35. doi:10.1160/TH07-05-0377
748. Khalafallah AA, Morse M, Al-Barzan AM, Adams M, Dennis A, Bates G, et al. d-dimer levels at different stages of pregnancy in Australian women: a single centre study using two different immunoturbidimetric assays. Thromb Res (2012) 130:e171–7. doi:10.1016/j.thromres.2012.05.022
749. Rodelo JR, De La Rosa G, Valencia ML, Ospina S, Arango CM, Gómez CI, et al. d-dimer is a significant prognostic factor in patients with suspected infection and sepsis. Am J Emerg Med (2012) 30:1991–9. doi:10.1016/j.ajem.2012.04.033
750. Khalafallah A, Jarvis C, Morse M, Albarzan AM, Stewart P, Bates G, et al. Evaluation of the innovance d-dimer assay for the diagnosis of disseminated intravascular coagulopathy in different clinical settings. Clin Appl Thromb Hemost (2014) 20:91–7. doi:10.1177/1076029612454936
751. Hentschke MR, Lucas LS, Mistry HD, Pinheiro Da Costa BE, Poli-De-Figueiredo CE. Endocan-1 concentrations in maternal and fetal plasma and placentae in pre-eclampsia in the third trimester of pregnancy. Cytokine (2015) 74:152–6. doi:10.1016/j.cyto.2015.04.013
752. Cakmak M, Yilmaz H, Bağlar E, Darcin T, Inan O, Aktas A, et al. Serum levels of endocan correlate with the presence and severity of pre-eclampsia. Clin Exp Hypertens (2016) 38:137–42. doi:10.3109/10641963.2015.1060993
753. Mosevoll KA, Lindås R, Wendelbo Ø, Bruserud Ø, Reikvam H. Systemic levels of the endothelium-derived soluble adhesion molecules endocan and E-selectin in patients with suspected deep vein thrombosis. Springerplus (2014) 3:571. doi:10.1186/2193-1801-3-571
754. Yuksel MA, Tuten A, Oncul M, Acikgoz AS, Temel Yuksel I, Toprak MS, et al. Serum endocan concentration in women with pre-eclampsia. Arch Gynecol Obstet (2015) 292:69–73. doi:10.1007/s00404-014-3605-x
755. Balta S, Mikhailidis DP, Demirkol S, Ozturk C, Celik T, Iyisoy A. Endocan: a novel inflammatory indicator in cardiovascular disease? Atherosclerosis (2015) 243:339–43. doi:10.1016/j.atherosclerosis.2015.09.030
756. Pawlak K, Mysliwiec M, Pawlak D. Endocan – the new endothelial activation marker independently associated with soluble endothelial adhesion molecules in uraemic patients with cardiovascular disease. Clin Biochem (2015) 48:425–30. doi:10.1016/j.clinbiochem.2015.01.006
757. Filep JG. Endocan or endothelial cell-specific molecule-1: a novel prognostic marker of sepsis? Crit Care Med (2006) 34:574–5. doi:10.1097/01.CCM.0000199054.13823.A7
758. Scherpereel A, Depontieu F, Grigoriu B, Cavestri B, Tsicopoulos A, Gentina T, et al. Endocan, a new endothelial marker in human sepsis. Crit Care Med (2006) 34:532–7. doi:10.1097/01.CCM.0000198525.82124.74
759. Mihajlovic DM, Lendak DF, Brkic SV, Draskovic BG, Mitic GP, Novakov Mikic AS, et al. Endocan is useful biomarker of survival and severity in sepsis. Microvasc Res (2014) 93:92–7. doi:10.1016/j.mvr.2014.04.004
760. Pauly D, Hamed S, Behnes M, Lepiorz D, Lang S, Akin I, et al. Endothelial cell-specific molecule-1/endocan: diagnostic and prognostic value in patients suffering from severe sepsis and septic shock. J Crit Care (2016) 31:68–75. doi:10.1016/j.jcrc.2015.09.019
761. Shovlin CL, Hughes JMB, Scott J, Seidman CE, Seidman JG. Characterization of endoglin and identification of novel mutations in hereditary hemorrhagic telangiectasia. Am J Hum Genet (1997) 61:68–79. doi:10.1086/513906
762. Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev (2010) 24:203–19. doi:10.1016/j.blre.2010.07.001
763. Gregory AL, Xu G, Sotov V, Letarte M. Review: the enigmatic role of endoglin in the placenta. Placenta (2014) 35(Suppl):S93–9. doi:10.1016/j.placenta.2013.10.020
764. Bell MJ, Roberts JM, Founds SA, Jeyabalan A, Terhorst L, Conley YP. Variation in endoglin pathway genes is associated with preeclampsia: a case-control candidate gene association study. BMC Pregnancy Childbirth (2013) 13:82. doi:10.1186/1471-2393-13-82
765. Luft FC. Soluble endoglin (sEng) joins the soluble fms-like tyrosine kinase (sFlt) receptor as a pre-eclampsia molecule. Nephrol Dial Transplant (2006) 21:3052–4. doi:10.1093/ndt/gfl439
766. Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med (2006) 12:642–9. doi:10.1038/nm1429
767. Govender N, Moodley J, Gathiram P, Naicker T. Soluble fms-like tyrosine kinase-1 in HIV infected pre-eclamptic South African Black women. Placenta (2014) 35:618–24. doi:10.1016/j.placenta.2014.04.013
768. Pratt A, Da Silva Costa F, Borg AJ, Kalionis B, Keogh R, Murthi P. Placenta-derived angiogenic proteins and their contribution to the pathogenesis of preeclampsia. Angiogenesis (2015) 18:115–23. doi:10.1007/s10456-014-9452-3
769. Kleinrouweler CE, Wiegerinck MMJ, Ris-Stalpers C, Bossuyt PMM, van der Post JAM, Von Dadelszen P, et al. Accuracy of circulating placental growth factor, vascular endothelial growth factor, soluble fms-like tyrosine kinase 1 and soluble endoglin in the prediction of pre-eclampsia: a systematic review and meta-analysis. BJOG (2012) 119:778–87. doi:10.1111/j.1471-0528.2012.03311.x
770. Muenzner P, Rohde M, Kneitz S, Hauck CR. CEACAM engagement by human pathogens enhances cell adhesion and counteracts bacteria-induced detachment of epithelial cells. J Cell Biol (2005) 170:825–36. doi:10.1083/jcb.200412151
771. Clemente M, Núñez O, Lorente R, Rincón D, Matilla A, Salcedo M, et al. Increased intrahepatic and circulating levels of endoglin, a TGF-beta 1 co-receptor, in patients with chronic hepatitis C virus infection: relationship to histological and serum markers of hepatic fibrosis. J Viral Hepat (2006) 13:625–32. doi:10.1111/j.1365-2893.2006.00733.x
772. Silver KL, Conroy AL, Leke RG, Leke RJ, Gwanmesia P, Molyneux ME, et al. Circulating soluble endoglin levels in pregnant women in Cameroon and Malawi – associations with placental malaria and fetal growth restriction. PLoS One (2011) 6:e24985. doi:10.1371/journal.pone.0024985
773. Sasmito SD, Ulfiati A, Wardana A, Nugraheni F, Pradiptasari NF, Zulaifa Z, et al. Endoglin expression and the level of tgf-β are increased in the placental tissue and correlated with low fetal weight in malaria infected mice. J Trop Life Sci (2015) 5:1–7. doi:10.11594/jtls.05.01.01
774. Jansen P, Mumme T, Randau T, Gravius S, Hermanns-Sachweh B. Endoglin (CD105) expression differentiates between aseptic loosening and periprosthetic joint infection after total joint arthroplasty. Springerplus (2014) 3:561. doi:10.1186/2193-1801-3-561
775. Valerio LG. Mammalian iron metabolism. Toxicol Mech Methods (2007) 17:497–517. doi:10.1080/15376510701556690
776. Andrews NC. Forging a field: the golden age of iron biology. Blood (2008) 112:219–30. doi:10.1182/blood-2007-12-077388
777. Hower V, Mendes P, Torti FM, Laubenbacher R, Akman S, Shulaev V, et al. A general map of iron metabolism and tissue-specific subnetworks. Mol Biosyst (2009) 5:422–43. doi:10.1039/b816714c
778. Chifman J, Kniss A, Neupane P, Williams I, Leung B, Deng Z, et al. The core control system of intracellular iron homeostasis: a mathematical model. J Theor Biol (2012) 300:91–9. doi:10.1016/j.jtbi.2012.01.024
779. Mitchell S, Mendes P. A computational model of liver iron metabolism. PLoS Comput Biol (2013) 9:e1003299. doi:10.1371/journal.pcbi.1003299
780. Oliveira F, Rocha S, Fernandes R. Iron metabolism: from health to disease. J Clin Lab Anal (2014) 28:210–8. doi:10.1002/jcla.21668
781. Kell DB, Pretorius E. Interpreting raised serum ferritin levels. BMJ (2015) 351. doi:10.1136/bmj.h3692
782. Nielsen P, Günther U, Dürken M, Fischer R, Düllmann J. Serum ferritin iron in iron overload and liver damage: correlation to body iron stores and diagnostic relevance. J Lab Clin Med (2000) 135:413–8. doi:10.1067/mlc.2000.106456
783. Watanabe K, Yamashita Y, Ohgawara K, Sekiguchi M, Satake N, Orino K, et al. Iron content of rat serum ferritin. J Vet Med Sci (2001) 63:587–9. doi:10.1292/Jvms.63.587
784. Yamanishi H, Iyama S, Yamaguchi Y, Kanakura Y, Iwatani Y. Relation between iron content of serum ferritin and clinical status factors extracted by factor analysis in patients with hyperferritinemia. Clin Biochem (2002) 35:523–9. doi:10.1016/S0009-9120(02)00380-6
785. Konz T, Añôn Alvarez E, Montes-Bayon M, Sanz-Medel A. Antibody labeling and elemental mass spectrometry (inductively coupled plasma-mass spectrometry) using isotope dilution for highly sensitive ferritin determination and iron-ferritin ratio measurements. Anal Chem (2013) 85:8334–40. doi:10.1021/ac401692k
786. Entman SS, Richardson LD, Killam AP. Altered ferrokinetics in toxemia of pregnancy – a possible indicator of decreased red cell survival. Clin Exp Hypertens B (1983) 2:171–8. doi:10.3109/10641958309023469
787. Hubel CA, Bodnar LM, Many A, Harger G, Ness RB, Roberts JM. Nonglycosylated ferritin predominates in the circulation of women with preeclampsia but not intrauterine growth restriction. Clin Chem (2004) 50:948–51. doi:10.1373/clinchem.2003.030932
788. Visser A, van de Vyver A. Severe hyperferritinemia in Mycobacteria tuberculosis infection. Clin Infect Dis (2011) 52:273–4. doi:10.1093/cid/ciq126
789. Ishida JH, Johansen KL. Iron and infection in hemodialysis patients. Semin Dial (2014) 27:26–36. doi:10.1111/sdi.12168
790. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet (2011) 12:861–74. doi:10.1038/nrg3074
791. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res (2014) 42:D68–73. doi:10.1093/nar/gkt1181
792. Goulart LF, Bettella F, Sønderby IE, Schork AJ, Thompson WK, Mattingsdal M, et al. microRNAs enrichment in GWAS of complex human phenotypes. BMC Genomics (2015) 16:304. doi:10.1186/s12864-015-1513-5
793. Londin E, Loher P, Telonis AG, Quann K, Clark P, Jing Y, et al. Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc Natl Acad Sci U S A (2015) 112:E1106–15. doi:10.1073/pnas.1420955112
794. Luo SS, Ishibashi O, Ishikawa G, Ishikawa T, Katayama A, Mishima T, et al. Human villous trophoblasts express and secrete placenta-specific microRNAs into maternal circulation via exosomes. Biol Reprod (2009) 81:717–29. doi:10.1095/biolreprod.108.075481
795. Ouyang Y, Mouillet JF, Coyne CB, Sadovsky Y. Review: placenta-specific microRNAs in exosomes – good things come in nano-packages. Placenta (2014) 35(Suppl):S69–73. doi:10.1016/j.placenta.2013.11.002
796. Record M. Intercellular communication by exosomes in placenta: a possible role in cell fusion? Placenta (2014) 35:297–302. doi:10.1016/j.placenta.2014.02.009
797. Miura K, Miura S, Yamasaki K, Higashijima A, Kinoshita A, Yoshiura K, et al. Identification of pregnancy-associated microRNAs in maternal plasma. Clin Chem (2010) 56:1767–71. doi:10.1373/clinchem.2010.147660
798. Morales-Prieto DM, Markert UR. microRNAs in pregnancy. J Reprod Immunol (2011) 88:106–11. doi:10.1016/j.jri.2011.01.004
799. Mouillet JF, Ouyang Y, Coyne CB, Sadovsky Y. microRNAs in placental health and disease. Am J Obstet Gynecol (2015) 213:S163–72. doi:10.1016/j.ajog.2015.05.057
800. Jairajpuri DS, Almawi WY. microRNA expression pattern in pre-eclampsia (review). Mol Med Rep (2016) 13:2351–8. doi:10.3892/mmr.2016.4846
801. Kotlabova K, Doucha J, Hromadnikova I. Placental-specific microRNA in maternal circulation –identification of appropriate pregnancy-associated microRNAs with diagnostic potential. J Reprod Immunol (2011) 89:185–91. doi:10.1016/j.jri.2011.02.006
802. Zhao Z, Moley KH, Gronowski AM. Diagnostic potential for miRNAs as biomarkers for pregnancy-specific diseases. Clin Biochem (2013) 46:953–60. doi:10.1016/j.clinbiochem.2013.01.026
803. Tsochandaridis M, Nasca L, Toga C, Levy-Mozziconacci A. Circulating microRNAs as clinical biomarkers in the predictions of pregnancy complications. Biomed Res Int (2015) 2015:294954. doi:10.1155/2015/294954
804. Zhu XM, Han T, Sargent IL, Yin GW, Yao YQ. Differential expression profile of microRNAs in human placentas from preeclamptic pregnancies vs normal pregnancies. Am J Obstet Gynecol (2009) 200:661.e661–7. doi:10.1016/j.ajog.2008.12.045
805. Wang D, Song W, Na Q. The emerging roles of placenta-specific microRNAs in regulating trophoblast proliferation during the first trimester. Aust N Z J Obstet Gynaecol (2012) 52:565–70. doi:10.1111/j.1479-828X.2012.01481.x
806. Chen DB, Wang W. Human placental microRNAs and preeclampsia. Biol Reprod (2013) 88:130. doi:10.1095/biolreprod.113.107805
807. Dong F, Zhang Y, Xia F, Yang Y, Xiong S, Jin L, et al. Genome-wide miRNA profiling of villus and decidua of recurrent spontaneous abortion patients. Reproduction (2014) 148:33–41. doi:10.1530/REP-14-0095
808. Hromadnikova I, Kotlabova K, Hympanova L, Doucha J, Krofta L. First trimester screening of circulating C19MC microRNAs can predict subsequent onset of gestational hypertension. PLoS One (2014) 9:e113735. doi:10.1371/journal.pone.0113735
809. Anton L, Olarerin-George AO, Hogenesch JB, Elovitz MA. Placental expression of miR-517a/b and miR-517c contributes to trophoblast dysfunction and preeclampsia. PLoS One (2015) 10:e0122707. doi:10.1371/journal.pone.0122707
810. Yang S, Li H, Ge Q, Guo L, Chen F. Deregulated microRNA species in the plasma and placenta of patients with preeclampsia. Mol Med Rep (2015) 12:527–34. doi:10.3892/mmr.2015.3414
811. Ura B, Feriotto G, Monasta L, Bilel S, Zweyer M, Celeghini C. Potential role of circulating microRNAs as early markers of preeclampsia. Taiwan J Obstet Gynecol (2014) 53:232–4. doi:10.1016/j.tjog.2014.03.001
812. Bortolin-Cavaillé ML, Dance M, Weber M, Cavaillé J. C19MC microRNAs are processed from introns of large Pol-II, non-protein-coding transcripts. Nucleic Acids Res (2009) 37:3464–73. doi:10.1093/nar/gkp205
813. Morales-Prieto DM, Ospina-Prieto S, Chaiwangyen W, Schoenleben M, Markert UR. Pregnancy-associated miRNA-clusters. J Reprod Immunol (2013) 97:51–61. doi:10.1016/j.jri.2012.11.001
814. Delorme-Axford E, Donker RB, Mouillet JF, Chu T, Bayer A, Ouyang Y, et al. Human placental trophoblasts confer viral resistance to recipient cells. Proc Natl Acad Sci U S A (2013) 110:12048–53. doi:10.1073/pnas.1304718110
815. Bayer A, Delorme-Axford E, Sleigher C, Frey TK, Trobaugh DW, Klimstra WB, et al. Human trophoblasts confer resistance to viruses implicated in perinatal infection. Am J Obstet Gynecol (2015) 212(71):e71–8. doi:10.1016/j.ajog.2014.07.060
816. Olarerin-George AO, Anton L, Hwang YC, Elovitz MA, Hogenesch JB. A functional genomics screen for microRNA regulators of NF-kappaB signaling. BMC Biol (2013) 11:19. doi:10.1186/1741-7007-11-19
817. Song GY, Song WW, Han Y, Wang D, Na Q. Characterization of the role of microRNA-517a expression in low birth weight infants. J Dev Orig Health Dis (2013) 4:522–6. doi:10.1017/S204017441300024X
818. Mouillet JF, Ouyang Y, Bayer A, Coyne CB, Sadovsky Y. The role of trophoblastic microRNAs in placental viral infection. Int J Dev Biol (2014) 58:281–9. doi:10.1387/ijdb.130349ys
819. Moro L, Bardaji A, Macete E, Barrios D, Morales-Prieto DM, Espana C, et al. Placental microparticles and microRNAs in pregnant women with Plasmodium falciparum or HIV infection. PLoS One (2016) 11:e0146361. doi:10.1371/journal.pone.0146361
820. Khatun S, Kanayama N, Belayet HM, Bhuiyan AB, Jahan S, Begum A, et al. Increased concentrations of plasma neuropeptide Y in patients with eclampsia and preeclampsia. Am J Obstet Gynecol (2000) 182:896–900. doi:10.1016/S0002-9378(00)70342-5
821. Hauser GJ, Dayao EK, Zukowska-Grojec Z. Effect of neuropeptide Y on endotoxin-induced suppression of the response to various agonists in conscious rats. Life Sci (1995) 57:235–44. doi:10.1016/0024-3205(95)00266-9
822. Kuncová J, Sýkora R, Chvojka J, Švíglerová J, Štengl M, Kroužecký A, et al. Plasma and tissue levels of neuropeptide y in experimental septic shock: relation to hemodynamics, inflammation, oxidative stress, and hemofiltration. Artif Organs (2011) 35:625–33. doi:10.1111/j.1525-1594.2010.01154.x
823. Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell (2002) 10:1033–43. doi:10.1016/S1097-2765(02)00708-6
824. Bachman MA, Lenio S, Schmidt L, Oyler JE, Weiser JN. Interaction of lipocalin 2, transferrin, and siderophores determines the replicative niche of Klebsiella pneumoniae during pneumonia. mBio (2012) 3:e224–211. doi:10.1128/mBio.00224-11
825. Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature (2004) 432:917–21. doi:10.1038/nature03104
826. Chakraborty S, Kaur S, Guha S, Batra SK. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta (2012) 1826:129–69. doi:10.1016/j.bbcan.2012.03.008
827. Cemgil Arikan D, Ozkaya M, Adali E, Kilinc M, Coskun A, Ozer A, et al. Plasma lipocalin-2 levels in pregnant women with pre-eclampsia, and their relation with severity of disease. J Matern Fetal Neonatal Med (2010) 24:291–6. doi:10.3109/14767058.2010.487138
828. D’Anna R, Baviera G, Giordano D, Todarello G, Corrado F, Buemi M. Second trimester neutrophil gelatinase-associated lipocalin as a potential prediagnostic marker of preeclampsia. Acta Obstet Gynecol Scand (2008) 87:1370–3. doi:10.1080/00016340802464463
829. D’Anna R, Baviera G, Giordano D, Russo S, Dugo N, Santamaria A, et al. First trimester serum PAPP-A and NGAL in the prediction of late-onset pre-eclampsia. Prenat Diagn (2009) 29:1066–8. doi:10.1002/pd.2339
830. D’Anna R, Baviera G, Giordano D, Todarello G, Russo S, Recupero S, et al. Neutrophil gelatinase-associated lipocalin serum evaluation through normal pregnancy and in pregnancies complicated by preeclampsia. Acta Obstet Gynecol Scand (2010) 89:275–8. doi:10.3109/00016340903443676
831. Stepan H, Philipp A, Reiche M, Klostermann K, Schrey S, Reisenbuchler C, et al. Serum levels of the adipokine lipocalin-2 are increased in preeclampsia. J Endocrinol Invest (2010) 33:629–32. doi:10.3275/6884
832. Kim SM, Park JS, Norwitz ER, Jung HJ, Kim BJ, Park CW, et al. Circulating levels of neutrophil gelatinase-associated lipocalin (NGAL) correlate with the presence and severity of preeclampsia. Reprod Sci (2013) 20:1083–9. doi:10.1177/1933719113477480
833. Scazzochio E, Munmany M, Garcia L, Meler E, Crispi F, Gratacos E, et al. Prognostic role of maternal neutrophil gelatinase-associated lipocalin in women with severe early-onset preeclampsia. Fetal Diagn Ther (2013) 35:127–32. doi:10.1159/000356499
834. Karampas G, Eleftheriades M, Panoulis K, Rizou M, Haliassos A, Hassiakos D, et al. Maternal serum levels of neutrophil gelatinase-associated lipocalin (NGAL), matrix metalloproteinase-9 (MMP-9) and their complex MMP-9/NGAL in pregnancies with preeclampsia and those with a small for gestational age neonate. A longitudinal study. Prenat Diagn (2014) 34:726–33. doi:10.1002/pd.4337
835. Sachan R, Patel M, Gaurav A, Gangwar R, Sachan P. Correlation of serum neutrophil gelatinase associated lipocalin with disease severity in hypertensive disorders of pregnancy. Adv Biomed Res (2014) 3:223. doi:10.4103/2277-9175.145690
836. Simonazzi G, Capelli I, Curti A, Comai G, Rizzo N, La Manna G. Serum and urinary neutrophil gelatinase-associated lipocalin monitoring in normal pregnancy versus pregnancies complicated by pre-eclampsia. In Vivo (2015) 29:117–21.
837. Karampas GA, Eleftheriades MI, Panoulis KC, Rizou MD, Haliassos AD, Metallinou DK, et al. Prediction of pre-eclampsia combining NGAL and other biochemical markers with Doppler in the first and/or second trimester of pregnancy. A pilot study. Eur J Obstet Gynecol Reprod Biol (2016) 205:153–7. doi:10.1016/j.ejogrb.2016.08.034
838. Mishra J, Dent C, Tarabishi R, Mitsnefes MM, Ma Q, Kelly C, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet (2005) 365:1231–8. doi:10.1016/S0140-6736(05)74811-X
839. Soni SS, Cruz D, Bobek I, Chionh CY, Nalesso F, Lentini P, et al. NGAL: a biomarker of acute kidney injury and other systemic conditions. Int Urol Nephrol (2010) 42:141–50. doi:10.1007/s11255-009-9608-z
840. Clerico A, Galli C, Fortunato A, Ronco C. Neutrophil gelatinase-associated lipocalin (NGAL) as biomarker of acute kidney injury: a review of the laboratory characteristics and clinical evidences. Clin Chem Lab Med (2012) 50:1505–17. doi:10.1515/cclm-2011-0814
841. Patel M, Sachan R, Gangwar R, Sachan P, Natu S. Correlation of serum neutrophil gelatinase-associated lipocalin with acute kidney injury in hypertensive disorders of pregnancy. Int J Nephrol Renovasc Dis (2013) 6:181–6. doi:10.2147/IJNRD.S45523
842. Aydoğdu M, Gürsel G, Sancak B, Yeni S, Sari G, Taşyürek S, et al. The use of plasma and urine neutrophil gelatinase associated lipocalin (NGAL) and cystatin C in early diagnosis of septic acute kidney injury in critically ill patients. Dis Markers (2013) 34:237–46. doi:10.3233/DMA-130966
843. Mårtensson J, Bell M, Xu S, Bottai M, Ravn B, Venge P, et al. Association of plasma neutrophil gelatinase-associated lipocalin (NGAL) with sepsis and acute kidney dysfunction. Biomarkers (2013) 18:349–56. doi:10.3109/1354750X.2013.787460
844. Otto GP, Busch M, Sossdorf M, Claus RA. Impact of sepsis-associated cytokine storm on plasma NGAL during acute kidney injury in a model of polymicrobial sepsis. Crit Care (2013) 17:419. doi:10.1186/cc12540
845. Zughaier SM, Tangpricha V, Leong T, Stecenko AA, Mccarty NA. Peripheral monocytes derived from patients with cystic fibrosis and healthy donors secrete NGAL in response to Pseudomonas aeruginosa infection. J Investig Med (2013) 61:1018–25. doi:10.231/JIM.0b013e31829cbd14
846. MacDonald SPJ, Stone SF, Neil CL, Van Eeden PE, Fatovich DM, Arendts G, et al. Sustained elevation of resistin, NGAL and IL-8 are associated with severe sepsis/septic shock in the emergency department. PLoS One (2014) 9:e110678. doi:10.1371/journal.pone.0110678
847. Smertka M, Wroblewska J, Suchojad A, Majcherczyk M, Jadamus-Niebroj D, Owsianka-Podlesny T, et al. Serum and urinary NGAL in septic newborns. Biomed Res Int (2014) 2014:717318. doi:10.1155/2014/717318
848. Hjortrup PB, Haase N, Treschow F, Møller MH, Perner A. Predictive value of NGAL for use of renal replacement therapy in patients with severe sepsis. Acta Anaesthesiol Scand (2015) 59:25–34. doi:10.1111/aas.12427
849. Nga HS, Medeiros P, Menezes P, Bridi R, Balbi A, Ponce D. Sepsis and AKI in clinical emergency room patients: the role of urinary NGAL. Biomed Res Int (2015) 2015:413751. doi:10.1155/2015/413751
850. Suchojad A, Tarko A, Smertka M, Majcherczyk M, Brzozowska A, Wroblewska J, et al. Factors limiting usefulness of serum and urinary NGAL as a marker of acute kidney injury in preterm newborns. Ren Fail (2015) 37:439–45. doi:10.3109/0886022X.2014.996109
851. Kanda J, Mori K, Kawabata H, Kuwabara T, Mori KP, Imamaki H, et al. An AKI biomarker lipocalin 2 in the blood derives from the kidney in renal injury but from neutrophils in normal and infected conditions. Clin Exp Nephrol (2014) 19:99–106. doi:10.1007/s10157-014-0952-7
852. Mårtensson J, Bellomo R. The rise and fall of NGAL in acute kidney injury. Blood Purif (2014) 37:304–10. doi:10.1159/000364937
853. Srisawat N, Praditpornsilpa K, Patarakul K, Techapornrung M, Daraswang T, Sukmark T, et al. Neutrophil gelatinase associated lipocalin (NGAL) in leptospirosis acute kidney injury: a multicenter study in Thailand. PLoS One (2015) 10:e0143367. doi:10.1371/journal.pone.0143367
854. Leelahavanichkul A, Somparn P, Issara-Amphorn J, Eiam-Ong S, Avihingsanon Y, Hirankarn N, et al. Serum neutrophil gelatinase associated lipocalin (NGAL) outperforms serum creatinine in detecting sepsis-induced acute kidney injury, experiments on bilateral nephrectomy and bilateral ureter obstruction mouse models. Shock (2016) 45:570–6. doi:10.1097/SHK.0000000000000530
855. Dewerchin M, Carmeliet P. PlGF: a multitasking cytokine with disease-restricted activity. Cold Spring Harb Perspect Med (2012) 2:a011056. doi:10.1101/cshperspect.a011056
856. De Vivo A, Baviera G, Giordano D, Todarello G, Corrado F, D’Anna R. Endoglin, PlGF and sFlt-1 as markers for predicting pre-eclampsia. Acta Obstet Gynecol Scand (2008) 87:837–42. doi:10.1080/00016340802253759
857. Kaufmann I, Rusterholz C, Hosli I, Hahn S, Lapaire O. Can detection of late-onset PE at triage by sflt-1 or PlGF be improved by the use of additional biomarkers? Prenat Diagn (2012) 32:1288–94. doi:10.1002/pd.3995
858. Gómez-Arriaga PI, Herraiz I, López-Jiménez EA, Gómez-Montes E, Denk B, Galindo A. Uterine artery Doppler and sFlt-1/PlGF ratio: usefulness in diagnosis of pre-eclampsia. Ultrasound Obstet Gynecol (2013) 41:530–7. doi:10.1002/uog.12400
859. Ohkuchi A, Hirashima C, Takahashi K, Suzuki H, Matsubara S, Suzuki M. Onset threshold of the plasma levels of soluble fms-like tyrosine kinase 1/placental growth factor ratio for predicting the imminent onset of preeclampsia within 4 weeks after blood sampling at 19-31 weeks of gestation. Hypertens Res (2013) 36:1073–80. doi:10.1038/hr.2013.95
860. Hirashima C, Ohkuchi A, Takahashi K, Suzuki H, Matsubara S, Suzuki M. A novel three-step approach for predicting the imminent onset of preeclampsia within 4 weeks after blood sampling at 19-31 weeks of gestation. Hypertens Res (2014) 37:519–25. doi:10.1038/hr.2014.31
861. Lai J, Garcia-Tizon Larroca S, Peeva G, Poon LC, Wright D, Nicolaides KH. Competing risks model in screening for preeclampsia by serum placental growth factor and soluble fms-like tyrosine kinase-1 at 30-33 weeks’ gestation. Fetal Diagn Ther (2014) 35:240–8. doi:10.1159/000359968
862. Masoura S, Kalogiannidis I, Makedou K, Theodoridis T, Koiou K, Gerou S, et al. Biomarkers of endothelial dysfunction in preeclampsia and neonatal morbidity: a case-control study. Eur J Obstet Gynecol Reprod Biol (2014) 175:119–23. doi:10.1016/j.ejogrb.2014.01.012
863. Ohkuchi A, Hirashima C, Takahashi K, Shirasuna K, Suzuki H, Ariga H, et al. A trio of risk factors for the onset of preeclampsia in the second and early third trimesters. Pregnancy Hypertens (2014) 4:224–30. doi:10.1016/j.preghy.2014.04.024
864. Dröge L, Herraìz I, Zeisler H, Schlembach D, Stepan H, Küssel L, et al. Maternal serum sFlt-1/PlGF ratio in twin pregnancies with and without pre-eclampsia in comparison with singleton pregnancies. Ultrasound Obstet Gynecol (2015) 45:286–93. doi:10.1002/uog.14760
865. Rolfo A, Attini R, Tavassoli E, Neve FV, Nigra M, Cicilano M, et al. Is it possible to differentiate chronic kidney disease and preeclampsia by means of new and old biomarkers? A prospective study. Dis Markers (2015) 2015:127083. doi:10.1155/2015/127083
866. Stepan H, Herraiz I, Schlembach D, Verlohren S, Brennecke S, Chantraine F, et al. Implementation of the sFlt-1/PlGF ratio for prediction and diagnosis of pre-eclampsia in singleton pregnancy: implications for clinical practice. Ultrasound Obstet Gynecol (2015) 45:241–6. doi:10.1002/uog.14799
867. Khalil A, Maiz N, Garcia-Mandujano R, Penco JM, Nicolaides KH. Longitudinal changes in maternal serum placental growth factor and soluble fms-like tyrosine kinase-1 in women at increased risk of pre-eclampsia. Ultrasound Obstet Gynecol (2016) 47:324–31. doi:10.1002/uog.15750
868. Kim MY, Buyon JP, Guerra MM, Rana S, Zhang D, Laskin CA, et al. Angiogenic factor imbalance early in pregnancy predicts adverse outcomes in patients with lupus and antiphospholipid antibodies: results of the PROMISSE study. Am J Obstet Gynecol (2016) 214: 108.e101–e114. doi:10.1016/j.ajog.2015.09.066
869. Zeisler H, Llurba E, Chantraine F, Vatish M, Staff AC, Sennstrom M, et al. Predictive value of the sFlt-1:PlGF ratio in women with suspected preeclampsia. N Engl J Med (2016) 374:13–22. doi:10.1056/NEJMoa1414838
870. Levine RJ, Thadhani R, Qian C, Lam C, Lim KH, Yu KF, et al. Urinary placental growth factor and risk of preeclampsia. JAMA (2005) 293:77–85. doi:10.1001/jama.293.1.77
871. Odibo AO, Patel KR, Spitalnik A, Odibo L, Huettner P. Placental pathology, first-trimester biomarkers and adverse pregnancy outcomes. J Perinatol (2014) 34:186–91. doi:10.1038/Jp.2013.176
872. Bramham K, Seed PT, Lightstone L, Nelson-Piercy C, Gill C, Webster P, et al. Diagnostic and predictive biomarkers for pre-eclampsia in patients with established hypertension and chronic kidney disease. Kidney Int (2016) 89:874–85. doi:10.1016/j.kint.2015.10.012
873. Duckworth S, Griffin M, Seed PT, North R, Myers J, Mackillop L, et al. Diagnostic biomarkers in women with suspected preeclampsia in a prospective multicenter study. Obstet Gynecol (2016) 128:245–52. doi:10.1097/AOG.0000000000001508
874. Yano K, Liaw PC, Mullington JM, Shih SC, Okada H, Bodyak N, et al. Vascular endothelial growth factor is an important determinant of sepsis morbidity and mortality. J Exp Med (2006) 203:1447–58. doi:10.1084/jem.20060375
875. Yano K, Okada Y, Beldi G, Shih SC, Bodyak N, Okada H, et al. Elevated levels of placental growth factor represent an adaptive host response in sepsis. J Exp Med (2008) 205:2623–31. doi:10.1084/jem.20080398
876. Foidart JM, Schaaps JP, Chantraine F, Munaut C, Lorquet S. Dysregulation of anti-angiogenic agents (sFlt-1, PLGF, and sEndoglin) in preeclampsia – a step forward but not the definitive answer. J Reprod Immunol (2009) 82:106–11. doi:10.1016/j.jri.2009.09.001
877. Furuya M, Kurasawa K, Nagahama K, Kawachi K, Nozawa A, Takahashi T, et al. Disrupted balance of angiogenic and antiangiogenic signalings in preeclampsia. J Pregnancy (2011) 2011:123717. doi:10.1155/2011/123717
878. De Oliveira L, Sass N, Boute T, Moron AF. sFlt-1 and PlGF levels in a patient with mirror syndrome related to cytomegalovirus infection. Eur J Obstet Gynecol Reprod Biol (2011) 158:366–7. doi:10.1016/j.ejogrb.2011.04.049
879. Benton SJ, Hu Y, Xie F, Kupfer K, Lee SW, Magee LA, et al. Can placental growth factor in maternal circulation identify fetuses with placental intrauterine growth restriction? Am J Obstet Gynecol (2012) 206(163):e161–7. doi:10.1016/j.ajog.2011.09.019
880. Acharya A. Promising biomarkers for superimposed pre-eclampsia in pregnant women with established hypertension and chronic kidney disease. Kidney Int (2016) 89:743–6. doi:10.1016/j.kint.2016.01.008
881. Montagnana M, Lippi G, Albiero A, Scevarolli S, Salvagno GL, Franchi M, et al. Procalcitonin values in preeclamptic women are related to severity of disease. Clin Chem Lab Med (2008) 46:1050–1. doi:10.1515/CCLM.2008.199
882. Can M, Sancar E, Harma M, Guven B, Mungan G, Acikgoz S. Inflammatory markers in preeclamptic patients. Clin Chem Lab Med (2011) 49:1469–72. doi:10.1515/CCLM.2011.232
883. Simon L, Gauvin F, Amre DK, Saint-Louis P, Lacroix J. Serum procalcitonin and C-reactive protein levels as markers of bacterial infection: a systematic review and meta-analysis. Clin Infect Dis (2004) 39:206–17. doi:10.1086/421997
884. Ucar B, Yildiz B, Aksit MA, Yarar C, Colak O, Akbay Y, et al. Serum amyloid A, procalcitonin, tumor necrosis factor-alpha, and interleukin-1beta levels in neonatal late-onset sepsis. Mediators Inflamm (2008) 2008:737141. doi:10.1155/2008/737141
885. Riedel S, Melendez JH, An AT, Rosenbaum JE, Zenilman JM. Procalcitonin as a marker for the detection of bacteremia and sepsis in the emergency department. Am J Clin Pathol (2011) 135:182–9. doi:10.1309/AJCP1MFYINQLECV2
886. Riedel S. Procalcitonin and the role of biomarkers in the diagnosis and management of sepsis. Diagn Microbiol Infect Dis (2012) 73:221–7. doi:10.1016/j.diagmicrobio.2012.05.002
887. Schuetz P, Muller B, Christ-Crain M, Stolz D, Tamm M, Bouadma L, et al. Procalcitonin to initiate or discontinue antibiotics in acute respiratory tract infections. Cochrane Database Syst Rev (2012) 9:CD007498. doi:10.1002/14651858.CD007498.pub2
888. Ulla M, Pizzolato E, Lucchiari M, Loiacono M, Soardo F, Forno D, et al. Diagnostic and prognostic value of presepsin in the management of sepsis in the emergency department: a multicenter prospective study. Crit Care (2013) 17:R168. doi:10.1186/cc12847
889. Henriquez-Camacho C, Losa J. Biomarkers for sepsis. Biomed Res Int (2014) 2014:547818. doi:10.1155/2014/547818
890. Tian G, Pan SY, Ma G, Liao W, Su QG, Gu BC, et al. Serum levels of procalcitonin as a biomarker for differentiating between sepsis and systemic inflammatory response syndrome in the neurological intensive care unit. J Clin Neurosci (2014) 21:1153–8. doi:10.1016/j.jocn.2013.09.021
891. Westwood M, Ramaekers B, Whiting P, Tomini F, Joore M, Armstrong N, et al. Procalcitonin testing to guide antibiotic therapy for the treatment of sepsis in intensive care settings and for suspected bacterial infection in emergency department settings: a systematic review and cost-effectiveness analysis. Health Technol Assess (2015) 19:v–xxv, 1–236. doi:10.3310/hta19960
892. Engin-Üstün Y, Üstün Y, Karabulut AB, Özkaplan E, Meydanli MM, Kafkaslı A. Serum amyloid A levels are increased in pre-eclampsia. Gynecol Obstet Invest (2007) 64:117–20. doi:10.1159/000100329
893. Kristensen K, Wide-Swensson D, Lindström V, Schmidt C, Grubb A, Strevens H. Serum amyloid A protein and C-reactive protein in normal pregnancy and preeclampsia. Gynecol Obstet Invest (2009) 67:275–80. doi:10.1159/000214081
894. Malle E, De Beer FC. Human serum amyloid A (SAA) protein: a prominent acute-phase reactant for clinical practice. Eur J Clin Invest (1996) 26:427–35. doi:10.1046/j.1365-2362.1996.159291.x
895. Pizzini C, Mussap M, Plebani M, Fanos V. C-reactive protein and serum amyloid A protein in neonatal infections. Scand J Infect Dis (2000) 32:229–35. doi:10.1080/00365540050165848
896. Sipe JD. Serum amyloid A: from fibril to function. Current status. Amyloid (2000) 7:10–2. doi:10.3109/13506120009146815
897. Urieli-Shoval S, Linke RP, Matzner Y. Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states. Curr Opin Hematol (2000) 7:64–9. doi:10.1097/00062752-200001000-00012
898. Falsey AR, Walsh EE, Francis CW, Looney RJ, Kolassa JE, Hall WJ, et al. Response of C-reactive protein and serum amyloid A to influenza A infection in older adults. J Infect Dis (2001) 183:995–9. doi:10.1086/319275
899. Arnon S, Litmanovitz I, Regev RH, Bauer S, Shainkin-Kestenbaum R, Dolfin T. Serum amyloid A: an early and accurate marker of neonatal early-onset sepsis. J Perinatol (2007) 27:297–302. doi:10.1038/sj.jp.7211682
900. Bozinovski S, Hutchinson A, Thompson M, MacGregor L, Black J, Giannakis E, et al. Serum amyloid a is a biomarker of acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2008) 177:269–78. doi:10.1164/rccm.200705-678OC
901. Cicarelli DD, Vieira JE, Bensenor FE. Comparison of C-reactive protein and serum amyloid a protein in septic shock patients. Mediators Inflamm (2008) 2008:631414. doi:10.1155/2008/631414
902. Ebert EC, Nagar M. Gastrointestinal manifestations of amyloidosis. Am J Gastroenterol (2008) 103:776–87. doi:10.1111/j.1572-0241.2007.01669.x
903. Lannergård A, Larsson A, Friman G, Ewald U. Human serum amyloid A (SAA) and high sensitive C-reactive protein (hsCRP) in preterm newborn infants with nosocomial infections. Acta Paediatr (2008) 97:1061–5. doi:10.1111/j.1651-2227.2008.00814.x
904. Çetinkaya M, Özkan H, Köksal N, Çelebi S, Hacımustafaoğlu M. Comparison of serum amyloid A concentrations with those of C-reactive protein and procalcitonin in diagnosis and follow-up of neonatal sepsis in premature infants. J Perinatol (2009) 29:225–31. doi:10.1038/jp.2008.207
905. Lannergård A, Viberg A, Cars O, Karlsson MO, Sandström M, Larsson A. The time course of body temperature, serum amyloid A protein, C-reactive protein and interleukin-6 in patients with bacterial infection during the initial 3 days of antibiotic therapy. Scand J Infect Dis (2009) 41:663–71. doi:10.1080/00365540903127417
906. Yuan H, Huang J, Lv B, Yan W, Hu G, Wang J, et al. Diagnosis value of the serum amyloid A test in neonatal sepsis: a meta-analysis. Biomed Res Int (2013) 2013:520294. doi:10.1155/2013/520294
907. Derebe MG, Zlatkov CM, Gattu S, Ruhn KA, Vaishnava S, Diehl GE, et al. Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. Elife (2014) 3:e03206. doi:10.7554/eLife.03206
908. Buhimschi IA, Nayeri UA, Zhao G, Shook LL, Pensalfini A, Funai EF, et al. Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci Transl Med (2014) 6:245ra292. doi:10.1126/scitranslmed.3008808
909. Little CS, Hammond CJ, Macintyre A, Balin BJ, Appelt DM. Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol Aging (2004) 25:419–29. doi:10.1016/S0197-4580(03)00127-1
910. Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB. Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci Lett (2007) 429:95–100. doi:10.1016/j.neulet.2007.09.077
911. Veas CJ, Aguilera VC, Munoz IJ, Gallardo VI, Miguel PL, Gonzalez MA, et al. Fetal endothelium dysfunction is associated with circulating maternal levels of sE-selectin, sVCAM1, and sFlt-1 during pre-eclampsia. J Matern Fetal Neonatal Med (2011) 24:1371–7. doi:10.3109/14767058.2011.556204
912. Whitehead CL, Palmer KR, Nilsson U, Gao Y, Saglam B, Lappas M, et al. Placental expression of a novel primate-specific splice variant of sFlt-1 is upregulated in pregnancies complicated by severe early onset pre-eclampsia. BJOG (2011):1268–71. doi:10.1111/j.1471-0528.2011.02962.x
913. Higgins LE, Rey De Castro N, Addo N, Wareing M, Greenwood SL, Jones RL, et al. Placental features of late-onset adverse pregnancy outcome. PLoS One (2015) 10:e0129117. doi:10.1371/journal.pone.0129117
914. Hod T, Cerdeira AS, Karumanchi SA. Molecular mechanisms of preeclampsia. Cold Spring Harb Perspect Med (2015) 5:a023473. doi:10.1101/cshperspect.a023473
915. Thadhani R, Kisner T, Hagmann H, Bossung V, Noack S, Schaarschmidt W, et al. Pilot study of extracorporeal removal of soluble fms-like tyrosine kinase 1 in preeclampsia. Circulation (2011) 124:940–50. doi:10.1161/CIRCULATIONAHA.111.034793
916. Li F, Hagaman JR, Kim HS, Maeda N, Jennette JC, Faber JE, et al. eNOS deficiency acts through endothelin to aggravate sFlt-1-induced pre-eclampsia-like phenotype. J Am Soc Nephrol (2012) 23:652–60. doi:10.1681/ASN.2011040369
917. Carney EF. Hypertension: sFlt-1 removal seems to be beneficial in women with pre-eclampsia. Nat Rev Nephrol (2015) 11:690. doi:10.1038/nrneph.2015.168
918. Thadhani R, Hagmann H, Schaarschmidt W, Roth B, Cingoez T, Karumanchi SA, et al. Removal of soluble fms-like tyrosine kinase-1 by dextran sulfate apheresis in preeclampsia. J Am Soc Nephrol (2016) 27:903–13. doi:10.1681/ASN.2015020157
919. Shapiro NI, Yano K, Okada H, Fischer C, Howell M, Spokes KC, et al. A prospective, observational study of soluble FLT-1 and vascular endothelial growth factor in sepsis. Shock (2008) 29:452–7. doi:10.1097/SHK.0b013e31815072c1
920. Schuetz P, Jones AE, Aird WC, Shapiro NI. Endothelial cell activation in emergency department patients with sepsis-related and non-sepsis-related hypotension. Shock (2011) 36:104–8. doi:10.1097/SHK.0b013e31821e4e04
921. Shapiro NI, Schuetz P, Yano K, Sorasaki M, Parikh SM, Jones AE, et al. The association of endothelial cell signaling, severity of illness, and organ dysfunction in sepsis. Crit Care (2010) 14:R182. doi:10.1186/cc9290
922. Alves BE, Montalvao SA, Aranha FJ, Lorand-Metze I, De Souza CA, Annichino-Bizzacchi JM, et al. Time-course of sFlt-1 and VEGF-A release in neutropenic patients with sepsis and septic shock: a prospective study. J Transl Med (2011) 9:23. doi:10.1186/1479-5876-9-23
923. Xing K, Murthy S, Liles WC, Singh JM. Clinical utility of biomarkers of endothelial activation in sepsis – a systematic review. Crit Care (2012) 16:R7. doi:10.1186/cc11145
924. Hsu CD, Iriye B, Johnson TR, Witter FR, Hong SF, Chan DW. Elevated circulating thrombomodulin in severe preeclampsia. Am J Obstet Gynecol (1993) 169:148–9. doi:10.1016/0002-9378(93)90151-8
925. Minakami H, Takahashi T, Izumi A, Tamada T. Increased levels of plasma thrombomodulin in preeclampsia. Gynecol Obstet Invest (1993) 36:208–10. doi:10.1159/000292631
926. Bontis J, Vavilis D, Agorastos T, Zournatzi V, Konstantinidis T, Tagou K. Maternal plasma level of thrombomodulin is increased in mild preeclampsia. Eur J Obstet Gynecol Reprod Biol (1995) 60:139–41. doi:10.1016/0028-2243(95)02093-8
927. Hsu CD, Copel JA, Hong SF, Chan DW. Thrombomodulin levels in preeclampsia, gestational hypertension, and chronic hypertension. Obstet Gynecol (1995) 86:897–9. doi:10.1016/0029-7844(95)00326-M
928. Shaarawy M, Didy HE. Thrombomodulin, plasminogen activator inhibitor type 1 (PAI-1) and fibronectin as biomarkers of endothelial damage in preeclampsia and eclampsia. Int J Gynaecol Obstet (1996) 55:135–9. doi:10.1016/S0020-7292(96)02755-5
929. Boffa MC, Valsecchi L, Fausto A, Gozin D, Vigano’ D’Angelo S, Safa O, et al. Predictive value of plasma thrombomodulin in preeclampsia and gestational hypertension. Thromb Haemost (1998) 79:1092–5.
930. Kobayashi H, Sadakata H, Suzuki K, She MY, Shibata S, Terao T. Thrombomodulin release from umbilical endothelial cells initiated by preeclampsia plasma-induced neutrophil activation. Obstet Gynecol (1998) 92:425–30. doi:10.1016/S0029-7844(98)00217-8
931. Brenner B. Haemostatic changes in pregnancy. Thromb Res (2004) 114:409–14. doi:10.1016/j.thromres.2004.08.004
932. Bosco C, Parra M, Barja P, Rodrigo R, Fernández V, Suarez M, et al. Increased immunohistochemical expression of thrombomodulin at placental perivascular myofibroblast in severe preeclampsia (PE). Histol Histopathol (2005) 20:1045–55.
933. Wiwanitkit V. Correlation between thrombomodulin and severe preeclampsia: a summary. Clin Appl Thromb Hemost (2008) 14:99–101. doi:10.1177/1076029607303965
934. Turner RJ, Bloemenkamp KWM, Bruijn JA, Baelde HJ. Loss of thrombomodulin in placental dysfunction in preeclampsia. Arterioscler Thromb Vasc Biol (2016) 36:728–35. doi:10.1161/ATVBAHA.306780
935. Saito H, Maruyama I, Shimazaki S, Yamamoto Y, Aikawa N, Ohno R, et al. Efficacy and safety of recombinant human soluble thrombomodulin (ART-123) in disseminated intravascular coagulation: results of a phase III, randomized, double-blind clinical trial. J Thromb Haemost (2007) 5:31–41. doi:10.1111/j.1538-7836.2006.02267.x
937. Mimuro J, Takahashi H, Kitajima I, Tsuji H, Eguchi Y, Matsushita T, et al. Impact of recombinant soluble thrombomodulin (thrombomodulin alfa) on disseminated intravascular coagulation. Thromb Res (2013) 131:436–43. doi:10.1016/j.thromres.2013.03.008
938. Vincent JL, Ramesh MK, Ernest D, Larosa SP, Pachl J, Aikawa N, et al. A randomized, double-blind, placebo-controlled, phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, ART-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med (2013) 41:2069–79. doi:10.1097/CCM.0b013e31828e9b03
939. Shirahata A, Mimuro J, Takahashi H, Kitajima I, Tsuji H, Eguchi Y, et al. Recombinant soluble human thrombomodulin (thrombomodulin alfa) in the treatment of neonatal disseminated intravascular coagulation. Eur J Pediatr (2014) 173:303–11. doi:10.1007/s00431-013-2155-8
940. Levi M. Recombinant soluble thrombomodulin: coagulation takes another chance to reduce sepsis mortality. J Thromb Haemost (2015) 13:505–7. doi:10.1111/jth.12868
941. Yamakawa K, Aihara M, Ogura H, Yuhara H, Hamasaki T, Shimazu T. Recombinant human soluble thrombomodulin in severe sepsis: a systematic review and meta-analysis. J Thromb Haemost (2015) 13:508–19. doi:10.1111/jth.12841
942. Yoshimura J, Yamakawa K, Ogura H, Umemura Y, Takahashi H, Morikawa M, et al. Benefit profile of recombinant human soluble thrombomodulin in sepsis-induced disseminated intravascular coagulation: a multicenter propensity score analysis. Crit Care (2015) 19:78. doi:10.1186/s13054-015-0810-3
943. Hayakawa M, Yamakawa K, Saito S, Uchino S, Kudo D, Iizuka Y, et al. Recombinant human soluble thrombomodulin and mortality in sepsis-induced disseminated intravascular coagulation. A multicentre retrospective study. Thromb Haemost (2016) 115:1157–66. doi:10.1160/TH15-12-0987
944. Kim YM, Romero R, Oh SY, Kim CJ, Kilburn BA, Armant DR, et al. Toll-like receptor 4: a potential link between “danger signals,” the innate immune system and preeclampsia? Am J Obstet Gynecol (2005) 193:921–7. doi:10.1016/j.ajog.2005.06.053
945. Bonney EA. Preeclampsia: a view through the danger model. J Reprod Immunol (2007) 76:68–74. doi:10.1016/j.jri.2007.03.006
946. Fasshauer M, Waldeyer T, Seeger J, Schrey S, Ebert T, Kratzsch J, et al. Serum levels of the adipokine visfatin are increased in pre-eclampsia. Clin Endocrinol (Oxf) (2008) 69:69–73. doi:10.1111/j.1365-2265.2007.03147.x
947. Adali E, Yildizhan R, Kolusari A, Kurdoglu M, Bugdayci G, Sahin HG, et al. Increased visfatin and leptin in pregnancies complicated by pre-eclampsia. J Matern Fetal Neonatal Med (2009) 22:873–9. doi:10.1080/14767050902994622
948. Mazaki-Tovi S, Vaisbuch E, Romero R, Kusanovic JP, Chaiworapongsa T, Kim SK, et al. Maternal and neonatal circulating visfatin concentrations in patients with pre-eclampsia and a small-for-gestational age neonate. J Matern Fetal Neonatal Med (2010) 23:1119–28. doi:10.3109/14767050903572190
949. Hu W, Wang Z, Wang H, Huang H, Dong M. Serum visfatin levels in late pregnancy and pre-eclampsia. Acta Obstet Gynecol Scand (2008) 87:413–8. doi:10.1080/00016340801976012
950. Zonneveld R, Martinelli R, Shapiro NI, Kuijpers TW, Plotz FB, Carman CV. Soluble adhesion molecules as markers for sepsis and the potential pathophysiological discrepancy in neonates, children and adults. Crit Care (2014) 18:204. doi:10.1186/cc13733
951. de Pablo R, Monserrat J, Reyes E, Díaz D, Rodríguez-Zapata M, De La Hera A, et al. Circulating sICAM-1 and sE-selectin as biomarker of infection and prognosis in patients with systemic inflammatory response syndrome. Eur J Intern Med (2013) 24:132–8. doi:10.1016/j.ejim.2012.10.009
952. Mussap M, Noto A, Fravega M, Fanos V. Soluble CD14 subtype presepsin (sCD14-ST) and lipopolysaccharide binding protein (LBP) in neonatal sepsis: new clinical and analytical perspectives for two old biomarkers. J Matern Fetal Neonatal Med (2011) 24(Suppl 2):12–4. doi:10.3109/14767058.2011.601923
953. Okamura Y, Yokoi H. Development of a point-of-care assay system for measurement of presepsin (sCD14-ST). Clin Chim Acta (2011) 412:2157–61. doi:10.1016/j.cca.2011.07.024
954. Endo S, Suzuki Y, Takahashi G, Shozushima T, Ishikura H, Murai A, et al. Usefulness of presepsin in the diagnosis of sepsis in a multicenter prospective study. J Infect Chemother (2012) 18:891–7. doi:10.1007/s10156-012-0435-2
955. Endo S, Suzuki Y, Takahashi G, Shozushima T, Ishikura H, Murai A, et al. Presepsin as a powerful monitoring tool for the prognosis and treatment of sepsis: a multicenter prospective study. J Infect Chemother (2014) 20:30–4. doi:10.1016/j.jiac.2013.07.005
956. Nakamura Y, Ishikura H, Nishida T, Kawano Y, Yuge R, Ichiki R, et al. Usefulness of presepsin in the diagnosis of sepsis in patients with or without acute kidney injury. BMC Anesthesiol (2014) 14:88. doi:10.1186/1471-2253-14-88
957. Wu J, Hu L, Zhang G, Wu F, He T. Accuracy of presepsin in sepsis diagnosis: a systematic review and meta-analysis. PLoS One (2015) 10:e0133057. doi:10.1371/journal.pone.0133057
958. Ali FT, Ali MA, Elnakeeb MM, Bendary HN. Presepsin is an early monitoring biomarker for predicting clinical outcome in patients with sepsis. Clin Chim Acta (2016) 460:93–101. doi:10.1016/j.cca.2016.06.030
959. Klouche K, Cristol JP, Devin J, Gilles V, Kuster N, Larcher R, et al. Diagnostic and prognostic value of soluble CD14 subtype (presepsin) for sepsis and community-acquired pneumonia in ICU patients. Ann Intensive Care (2016) 6:59. doi:10.1186/s13613-016-0160-6
960. Song X, Song Y, Yuan Y, Zhang P, Zhang X. Prognostic value of presepsin for outcomes and complications in enterocutaneous fistula complicated by abdominal sepsis. Int J Surg (2016) 33(Pt A):96–101. doi:10.1016/j.ijsu.2016.07.070
961. Takahashi G, Shibata S, Fukui Y, Okamura Y, Inoue Y. Diagnostic accuracy of procalcitonin and presepsin for infectious disease in patients with acute kidney injury. Diagn Microbiol Infect Dis (2016) 86:205–10. doi:10.1016/j.diagmicrobio.2016.07.015
962. Kell DB, Westerhoff HV. Metabolic control theory: its role in microbiology and biotechnology. FEMS Microbiol Rev (1986) 39:305–20. doi:10.1111/j.1574-6968.1986.tb01863.x
963. Kell DB. Forces, fluxes and the control of microbial growth and metabolism. The twelfth Fleming lecture. J Gen Microbiol (1987) 133:1651–65.
964. Fell DA. Metabolic control analysis – a survey of its theoretical and experimental development. Biochem J (1992) 286:313–30. doi:10.1042/bj2860313
966. Kell DB, Oliver SG. The metabolome 18 years on: a concept comes of age. Metabolomics (2016) 12:148. doi:10.1007/s11306-016-1108-4
967. Oliver SG, Winson MK, Kell DB, Baganz F. Systematic functional analysis of the yeast genome. Trends Biotechnol (1998) 16:373–8. doi:10.1016/S0167-7799(98)01214-1
968. Cornish-Bowden A, Cárdenas ML. Silent genes given voice. Nature (2001) 409:571–2. doi:10.1038/35054646
969. Raamsdonk LM, Teusink B, Broadhurst D, Zhang N, Hayes A, Walsh M, et al. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat Biotechnol (2001) 19:45–50. doi:10.1038/83496
970. Goodacre R, Vaidyanathan S, Dunn WB, Harrigan GG, Kell DB. Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol (2004) 22:245–52. doi:10.1016/j.tibtech.2004.03.007
971. Kell DB. Metabolomics and systems biology: making sense of the soup. Curr Opin Microbiol (2004) 7:296–307. doi:10.1016/j.mib.2004.04.012
973. Kitano H, Oda K, Kimura T, Matsuoka Y, Csete M, Doyle J, et al. Metabolic syndrome and robustness tradeoffs. Diabetes (2004) 53(Suppl 3):S6–15. doi:10.2337/diabetes.53.suppl_3.S6
974. Wilhelm T, Behre J, Schuster S. Analysis of structural robustness of metabolic networks. Syst Biol (Stevenage) (2004) 1:114–20. doi:10.1049/sb:20045004
975. Grimbs S, Selbig J, Bulik S, Holzhutter HG, Steuer R. The stability and robustness of metabolic states: identifying stabilizing sites in metabolic networks. Mol Syst Biol (2007) 3:146. doi:10.1038/msb4100186
976. Kell DB. Finding novel pharmaceuticals in the systems biology era using multiple effective drug targets, phenotypic screening, and knowledge of transporters: where drug discovery went wrong and how to fix it. FEBS J (2013) 280:5957–80. doi:10.1111/febs.12268
977. Quinton-Tulloch MJ, Bruggeman FJ, Snoep JL, Westerhoff HV. Trade-off of dynamic fragility but not of robustness in metabolic pathways in silico. FEBS J (2013) 280:160–73. doi:10.1111/febs.12057
978. Kell DB, Goodacre R. Metabolomics and systems pharmacology: why and how to model the human metabolic network for drug discovery. Drug Discov Today (2014) 19:171–82. doi:10.1016/j.drudis.2013.07.014
979. Begley P, Francis-Mcintyre S, Dunn WB, Broadhurst DI, Halsall A, Tseng A, et al. Development and performance of a gas chromatography-time-of-flight mass spectrometry analysis for large-scale non-targeted metabolomic studies of human serum. Anal Chem (2009) 81:7038–46. doi:10.1021/ac9011599
980. Zelena E, Dunn WB, Broadhurst D, Francis-Mcintyre S, Carroll KM, Begley P, et al. Development of a robust and repeatable UPLC-MS method for the long-term metabolomic study of human serum. Anal Chem (2009) 81:1357–64. doi:10.1021/ac8019366
981. Dunn WB, Broadhurst D, Begley P, Zelena E, Francis-Mcintyre S, Anderson N, et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc (2011) 6:1060–83. doi:10.1038/nprot.2011.335
982. Dunn WB, Lin W, Broadhurst D, Begley P, Brown M, Zelena E, et al. Molecular phenotyping of a UK population: defining the human serum metabolome. Metabolomics (2015) 11:9–26. doi:10.1007/s11306-014-0707-1
983. Swainston N, Mendes P, Kell DB. An analysis of a ‘community-driven’ reconstruction of the human metabolic network. Metabolomics (2013) 9:757–64. doi:10.1007/s11306-013-0564-3
984. Thiele I, Swainston N, Fleming RMT, Hoppe A, Sahoo S, Aurich MK, et al. A community-driven global reconstruction of human metabolism. Nat Biotechnol (2013) 31:419–25. doi:10.1038/nbt.2488
985. Swainston N, Smallbone K, Hefzi H, Dobson PD, Brewer J, Hanscho M, et al. Recon 2.2: from reconstruction to model of human metabolism. Metabolomics (2016) 12:109. doi:10.1007/s11306-016-1051-4
986. Herrgård MJ, Swainston N, Dobson P, Dunn WB, Arga KY, Arvas M, et al. A consensus yeast metabolic network obtained from a community approach to systems biology. Nat Biotechnol (2008) 26:1155–60. doi:10.1038/nbt1492
987. Bolte AC, Van Geijn HP, Dekker GA. Pathophysiology of preeclampsia and the role of serotonin. Eur J Obstet Gynecol Reprod Biol (2001) 95:12–21. doi:10.1016/S0301-2115(00)00367-5
988. Kell DB, Kaprelyants AS, Grafen A. On pheromones, social behaviour and the functions of secondary metabolism in bacteria. Trends Ecol Evol (1995) 10:126–9. doi:10.1016/S0169-5347(00)89013-8
989. Takano E. Gamma-butyrolactones: Streptomyces signalling molecules regulating antibiotic production and differentiation. Curr Opin Microbiol (2006) 9:287–94. doi:10.1016/j.mib.2006.04.003
990. Hyppönen E, Cavadino A, Williams D, Fraser A, Vereczkey A, Fraser WD, et al. Vitamin D and pre-eclampsia: original data, systematic review and meta-analysis. Ann Nutr Metab (2013) 63:331–40. doi:10.1159/000358338
991. Tabesh M, Salehi-Abargouei A, Tabesh M, Esmaillzadeh A. Maternal vitamin D status and risk of pre-eclampsia: a systematic review and meta-analysis. J Clin Endocrinol Metab (2013) 98:3165–73. doi:10.1210/jc.2013-1257
992. Bakacak M, Serin S, Ercan O, Kostu B, Avci F, Kilinc M, et al. Comparison of vitamin D levels in cases with preeclampsia, eclampsia and healthy pregnant women. Int J Clin Exp Med (2015) 8:16280–6.
993. Barrera D, Diaz L, Noyola-Martinez N, Halhali A. Vitamin D and inflammatory cytokines in healthy and preeclamptic pregnancies. Nutrients (2015) 7:6465–90. doi:10.3390/nu7085293
994. Gargari BP, Tabrizi FPF, Sadien B, Jafarabadi MA, Farzadi L. Vitamin D status is related to oxidative stress but not high-sensitive C-reactive protein in women with pre-eclampsia. Gynecol Obstet Invest (2015) 81:308–14. doi:10.1159/000441781
995. Harvey NC, Holroyd C, Ntani G, Javaid K, Cooper P, Moon R, et al. Vitamin D supplementation in pregnancy: a systematic review. Health Technol Assess (2014) 18:1–190. doi:10.3310/hta18450
996. Kiely ME, Zhang JY, Kinsella M, Khashan AS, Kenny LC. Vitamin D status is associated with utero-placental dysfunction in a large prospective pregnancy cohort with low 25(OH)D3 and ubiquitous 3-epi-25(OH)D3 and 25(OH)D2. Am J Clin Nutr (2016) 102:354–61. doi:10.3945/ajcn.116.130419
997. van Weert B, Van Den Berg D, Hrudey EJ, Oostvogels AJJM, De Miranda E, Vrijkotte TGM. Is first trimester vitamin D status in nulliparous women associated with pregnancy related hypertensive disorders? Midwifery (2016) 34:–22. doi:10.1016/j.midw.2015.12.007
998. Palacios C, De-Regil LM, Lombardo LK, Peña-Rosas JP. Vitamin D supplementation during pregnancy: updated meta-analysis on maternal outcomes. J Steroid Biochem Mol Biol (2016) 164:148–55. doi:10.1016/j.jsbmb.2016.02.008
999. Bahado-Singh RO, Syngelaki A, Akolekar R, Mandal R, Bjondahl TC, Han B, et al. Validation of metabolomic models for prediction of early-onset preeclampsia. Am J Obstet Gynecol (2015) 213:530.e1–10. doi:10.1016/j.ajog.2015.06.044
1000. Melland-Smith M, Ermini L, Chauvin S, Craig-Barnes H, Tagliaferro A, Todros T, et al. Disruption of sphingolipid metabolism augments ceramide-induced autophagy in preeclampsia. Autophagy (2015) 11:653–69. doi:10.1080/15548627.2015.1034414
1001. Funke C, Schneider SA, Berg D, Kell DB. Genetics and iron in the systems biology of Parkinson’s disease and some related disorders. Neurochem Int (2013) 62:637–52. doi:10.1016/j.neuint.2012.11.015
1002. Manyonda IT, Slater DM, Fenske C, Hole D, Choy MY, Wilson C. A role for noradrenaline in pre-eclampsia: towards a unifying hypothesis for the pathophysiology. Br J Obstet Gynaecol (1998) 105:641–8. doi:10.1111/j.1471-0528.1998.tb10179.x
1003. Ferreira-de-Almeida JA, Amenta F, Cardoso F, Polonia JJ. Association of circulating endothelium and noradrenaline with increased calcium-channel binding sites in the placental bed in pre-eclampsia. Br J Obstet Gynaecol (1998) 105:1104–12. doi:10.1111/j.1471-0528.1998.tb09944.x
1004. Lyte M, Ernst S. Catecholamine-induced growth of Gram-negative bacteria. Life Sci (1992) 50:203–12. doi:10.1016/0024-3205(92)90273-R
1005. Freestone PP, Haigh RD, Williams PH, Lyte M. Stimulation of bacterial growth by heat-stable, norepinephrine-induced autoinducers. FEMS Microbiol Lett (1999) 172:53–60. doi:10.1111/j.1574-6968.1999.tb13449.x
1006. Reissbrodt R, Rienaecker I, Romanova JM, Freestone PPE, Haigh RD, Lyte M, et al. Resuscitation of Salmonella enterica serovar typhimurium and enterohemorrhagic Escherichia coli from the viable but nonculturable state by heat-stable enterobacterial autoinducer. Appl Environ Microbiol (2002) 68:4788–94. doi:10.1128/AEM.68.10.4788-4794.2002
1007. Lyte M. Microbial endocrinology and infectious disease in the 21st century. Trends Microbiol (2004) 12:14–20. doi:10.1016/j.tim.2003.11.004
1008. Lyte M. The effect of stress on microbial growth. Anim Health Res Rev (2014) 15:172–4. doi:10.1017/S146625231400019X
1009. Freestone PP, Lyte M, Neal CP, Maggs AF, Haigh RD, Williams PH. The mammalian neuroendocrine hormone norepinephrine supplies iron for bacterial growth in the presence of transferrin or lactoferrin. J Bacteriol (2000) 182:6091–8. doi:10.1128/JB.182.21.6091-6098.2000
1010. Freestone PPE, Haigh RD, Williams PH, Lyte M. Involvement of enterobactin in norepinephrine-mediated iron supply from transferrin to enterohaemorrhagic Escherichia coli. FEMS Microbiol Lett (2003) 222:39–43. doi:10.1016/S0378-1097(03)00243-X
1011. O’Donnell PM, Aviles H, Lyte M, Sonnenfeld G. Enhancement of in vitro growth of pathogenic bacteria by norepinephrine: importance of inoculum density and role of transferrin. Appl Environ Microbiol (2006) 72:5097–9. doi:10.1128/AEM.00075-06
1012. Kvetnansky R, Sabban EL, Palkovits M. Catecholaminergic systems in stress: structural and molecular genetic approaches. Physiol Rev (2009) 89:535–606. doi:10.1152/physrev.00042.2006
1013. Ribas Ripoll V, Romay E, Brunelli L, Pastorelli R, Goma G, Navas A, et al. Metabolite analysis in sepsis through conditional independence maps. Conf Proc IEEE Eng Med Biol Soc (2015) 2015:6477–80. doi:10.1109/EMBC.2015.7319876
1014. Su L, Huang Y, Zhu Y, Xia L, Wang R, Xiao K, et al. Discrimination of sepsis stage metabolic profiles with an LC/MS-MS-based metabolomics approach. BMJ Open Respir Res (2014) 1:e000056. doi:10.1136/bmjresp-2014-000056
1015. Eggers AE. A suggestion about the cause of inflammation in acute atherosis complicating poor placentation in preeclampsia. Med Hypotheses (2015) 85:718–9. doi:10.1016/j.mehy.2015.10.022
1016. D’Anna R, Baviera G, Scilipoti A, Leonardi I, Leo R. The clinical utility of serum uric acid measurements in pre-eclampsia and transient hypertension in pregnancy. Panminerva Med (2000) 42:101–3.
1017. Williams KP, Galerneau F. The role of serum uric acid as a prognostic indicator of the severity of maternal and fetal complications in hypertensive pregnancies. J Obstet Gynaecol Can (2002) 24:628–32. doi:10.1016/S1701-2163(16)30193-1
1018. Roberts JM, Bodnar LM, Lain KY, Hubel CA, Markovic N, Ness RB, et al. Uric acid is as important as proteinuria in identifying fetal risk in women with gestational hypertension. Hypertension (2005) 46:1263–9. doi:10.1161/01.HYP.0000188703.27002.14
1019. Bainbridge SA, Roberts JM. Uric acid as a pathogenic factor in preeclampsia. Placenta (2008) 29:S67–72. doi:10.1016/j.placenta.2007.11.001
1020. Koopmans CM, Van Pampus MG, Groen H, Aarnoudse JG, Van Den Berg PP, Mol BWJ. Accuracy of serum uric acid as a predictive test for maternal complications in pre-eclampsia: bivariate meta-analysis and decision analysis. Eur J Obstet Gynecol Reprod Biol (2009) 146:8–14. doi:10.1016/j.ejogrb.2009.05.014
1021. Agarwal V, Gupta BK, Vishnu A, Mamtatyagi, Shiprasolanki, Kiran J. Association of lipid profile and uric acid with pre-eclampsia of third trimester in nullipara women. J Clin Diagn Res (2014) 8:CC04–07. doi:10.7860/JCDR/2014/7901.4533
1022. Pereira KN, Knoppka CK, Da Silva JE. Association between uric acid and severity of pre-eclampsia. Clin Lab (2014) 60:309–14.
1023. Lim KH, Friedman SA, Ecker JL, Kao L, Kilpatrick SJ. The clinical utility of serum uric acid measurements in hypertensive diseases of pregnancy. Am J Obstet Gynecol (1998) 178:1067–71. doi:10.1016/S0002-9378(98)70549-6
1024. Cnossen JS, De Ruyter-Hanhijarvi H, van der Post JA, Mol BW, Khan KS, Ter Riet G. Accuracy of serum uric acid determination in predicting pre-eclampsia: a systematic review. Acta Obstet Gynecol Scand (2006) 85:519–25. doi:10.1080/00016340500342037
1025. Powers RW, Bodnar LM, Ness RB, Cooper KM, Gallaher MJ, Frank MP, et al. Uric acid concentrations in early pregnancy among preeclamptic women with gestational hyperuricemia at delivery. Am J Obstet Gynecol (2006) 194:160–6. doi:10.1016/j.ajog.2005.06.066
1026. Thangaratinam S, Ismail KM, Sharp S, Coomarasamy A, Khan KS; Tests in Prediction of Pre-Eclampsia Severity Review, Group. Accuracy of serum uric acid in predicting complications of pre-eclampsia: a systematic review. BJOG (2006) 113:369–78. doi:10.1111/j.1471-0528.2006.00908.x
1027. Chen Q, Lau S, Tong M, Wei J, Shen F, Zhao J, et al. Serum uric acid may not be involved in the development of preeclampsia. J Hum Hypertens (2016) 30:136–40. doi:10.1038/jhh.2015.47
1028. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med (2012) 367:2322–33. doi:10.1056/NEJMra1205750
1029. Kaddurah-Daouk R, Zhu H, Sharma S, Bogdanov M, Rozen SG, Matson W, et al. Alterations in metabolic pathways and networks in Alzheimer’s disease. Transl Psychiatry (2013) 3:e244. doi:10.1038/tp.2013.18
1030. McFarland NR, Burdett T, Desjardins CA, Frosch MP, Schwarzschild MA. Postmortem brain levels of urate and precursors in Parkinson’s disease and related disorders. Neurodegener Dis (2013) 12:189–98. doi:10.1159/000346370
1031. Esteve C, Jones EA, Kell DB, Boutin H, Mcdonnell LA. Mass spectral imaging shows major derangements in neurogranin and in purine metabolism in the triple-knockout 3xTg Alzheimer-like mouse model. Front Neurosci (Forthcoming 2016).
1032. Martinon F. Mechanisms of uric acid crystal-mediated autoinflammation. Immunol Rev (2010) 233:218–32. doi:10.1111/j.0105-2896.2009.00860.x
1033. Lyngdoh T, Vuistiner P, Marques-Vidal P, Rousson V, Waeber G, Vollenweider P, et al. Serum uric acid and adiposity: deciphering causality using a bidirectional Mendelian randomization approach. PLoS One (2012) 7:e39321. doi:10.1371/journal.pone.0039321
1034. Orengo JM, Leliwa-Sytek A, Evans JE, Evans B, van de Hoef D, Nyako M, et al. Uric acid is a mediator of the Plasmodium falciparum-induced inflammatory response. PLoS One (2009) 4:e5194. doi:10.1371/Journal.Pone.0005194
1035. Kool M, Soullie T, Van Nimwegen M, Willart MA, Muskens F, Jung S, et al. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med (2008) 205:869–82. doi:10.1084/jem.20071087
1036. Chalcraft KR. Comprehensive Metabolomics Analysis of Peanut Allergy and Peanut-Induced Anaphylaxis [Ph.D. Thesis] Open Access Dissertations and Theses. Paper 7733. Hamilton, ON: McMaster University (2013).
1037. Chalcraft KR, Kong J, Waserman S, Jordana M, Mccarry BE. Comprehensive metabolomic analysis of peanut-induced anaphylaxis in a murine model. Metabolomics (2014) 10:452–60. doi:10.1007/s11306-013-0589-7
1038. Kong J, Chalcraft K, Mandur TS, Jimenez-Saiz R, Walker TD, Goncharova S, et al. Comprehensive metabolomics identifies the alarmin uric acid as a critical signal for the induction of peanut allergy. Allergy (2015) 70:495–505. doi:10.1111/all.12579
1039. van Deventer SJH, Büller HR, Ten Cate JW, Aarden LA, Hack CE, Sturk A. Experimental endotoxemia in humans: analysis of cytokine release and coagulation, fibrinolytic, and complement pathways. Blood (1990) 76:2520–6.
1040. Johnson K, Aarden L, Choi Y, De Groot E, Creasey A. The proinflammatory cytokine response to coagulation and endotoxin in whole blood. Blood (1996) 87:5051–60.
1041. American College of Chest Physicians. From the bench to the bedside: the future of sepsis research. Executive summary of an American College of Chest Physicians, National Institute of Allergy and Infectious Disease, and National Heart, Lung, and Blood Institute Workshop. Chest (1997) 111:744–53.
1042. Pernerstorfer T, Stohlawetz P, Hollenstein U, Dzirlo L, Eichler HG, Kapiotis S, et al. Endotoxin-induced activation of the coagulation cascade in humans: effect of acetylsalicylic acid and acetaminophen. Arterioscler Thromb Vasc Biol (1999) 19:2517–23. doi:10.1161/01.ATV.19.10.2517
1043. Pernerstorfer T, Hollenstein U, Hansen JB, Stohlawetz P, Eichler HG, Handler S, et al. Lepirudin blunts endotoxin-induced coagulation activation. Blood (2000) 95:1729–34.
1044. Dellinger RP. Inflammation and coagulation: implications for the septic patient. Clin Infect Dis (2003) 36:1259–65. doi:10.1086/374835
1045. Schouten M, Wiersinga WJ, Levi M, van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol (2008) 83:536–45. doi:10.1189/jlb.0607373
1046. Anas AA, Wiersinga WJ, De Vos AF, van der Poll T. Recent insights into the pathogenesis of bacterial sepsis. Neth J Med (2010) 68:147–52.
1047. Saracco P, Vitale P, Scolfaro C, Pollio B, Pagliarino M, Timeus F. The coagulopathy in sepsis: significance and implications for treatment. Pediatr Rep (2011) 3:e30. doi:10.4081/pr.2011.e30
1048. Tsao CM, Ho ST, Wu CC. Coagulation abnormalities in sepsis. Acta Anaesthesiol Taiwan (2015) 53:16–22. doi:10.1016/j.aat.2014.11.002
1049. Armstrong MT, Rickles FR, Armstrong PB. Capture of lipopolysaccharide (endotoxin) by the blood clot: a comparative study. PLoS One (2013) 8:e80192. doi:10.1371/journal.pone.0080192
1050. Al-ofi E, Coffelt SB, Anumba DO. Fibrinogen, an endogenous ligand of toll-like receptor 4, activates monocytes in pre-eclamptic patients. J Reprod Immunol (2014) 103:23–8. doi:10.1016/j.jri.2014.02.004
1051. Chatterjee T, Maitra D, Chakravarty T, Datta AG. Studies on plasma fibrinogen level in pre-eclampsia and eclampsia. Experientia (1978) 34:562–3. doi:10.1007/BF01936958
1052. Manten GTR, Sikkema JM, Franx A, Hameeteman TM, Visser GHA, De Groot PG, et al. Increased high molecular weight fibrinogen in pre-eclampsia. Thromb Res (2003) 111:143–7. doi:10.1016/j.thromres.2003.08.025
1053. Üstün Y, Engin-Üstün Y, Kamacı M. Association of fibrinogen and C-reactive protein with severity of preeclampsia. Eur J Obstet Gynecol Reprod Biol (2005) 121:154–8. doi:10.1016/j.ejogrb.2004.12.009
1054. Sersam LW. Inflammatory markers in pre-eclampsia and related conditions. Int J Gynaecol Obstet (2009):69–70. doi:10.1016/j.ijgo.2008.11.016
1055. Sogani S, Sarakar PD. Evaluation of plasma fibrinogen and plasma fibrin degradation product (FDP) in preeclampsia. J Clin Biomed Sci (2013) 3:201–3.
1056. Perry KG, Martin JN. Abnormal hemostasis and coagulopathy in preeclampsia and eclampsia. Clin Obstet Gynecol (1992) 35:338–50. doi:10.1097/00003081-199235020-00015
1057. Metz J, Cincotta R, Francis M, Derosa L, Balloch A. Screening for consumptive coagulopathy in preeclampsia. Int J Gynecol Obstet (1994) 46:3–9. doi:10.1016/0020-7292(94)90301-8
1058. Gilabert J, Estellés A, Grancha S, España F, Aznar J. Fibrinolytic system and reproductive process with special reference to fibrinolytic failure in pre-eclampsia. Hum Reprod (1995) 10(Suppl 2):121–31. doi:10.1093/humrep/10.suppl_2.121
1059. He S, Bremme K, Blomback M. Acquired deficiency of antithrombin in association with a hypercoagulable state and impaired function of liver and/or kidney in preeclampsia. Blood Coagul Firbinolysis (1997) 8:232–8. doi:10.1097/00001721-199706000-00004
1060. Hladunewich M, Karumanchi SA, Lafayette R. Pathophysiology of the clinical manifestations of preeclampsia. Clin J Am Soc Nephrol (2007) 2:543–9. doi:10.2215/CJN.03761106
1061. Kher A, Bauersachs R, Nielsen JD. The management of thrombosis in pregnancy: role of low-molecular-weight heparin. Thromb Haemost (2007) 97:505–13. doi:10.1160/TH06-10-0606
1062. Jahromi BN, Rafiee SH. Coagulation factors in severe preeclampsia. Iran Red Crescent Med J (2009) 11:321–4.
1063. Ducloy-Bouthors AS. Clotting disorders and preeclampsia (in French). Ann Fr Anesth Reanim (2010) 29:e121–34. doi:10.1016/j.annfar.2010.03.012
1064. Martínez-Zamora MA, Tassies D, Carmona F, Espinosa G, Cervera R, Reverter JC, et al. Clot lysis time and thrombin activatable fibrinolysis inhibitor in severe preeclampsia with or without associated antiphospholipid antibodies. J Reprod Immunol (2010) 86:133–40. doi:10.1016/j.jri.2010.05.002
1065. Dusse LM, Rios DRA, Pinheiro MB, Cooper AJ, Lwaleed BA. Pre-eclampsia: relationship between coagulation, fibrinolysis and inflammation. Clin Chim Acta (2011) 412:17–21. doi:10.1016/j.cca.2010.09.030
1066. Godoi LC, Gomes KB, Alpoim PN, Carvalho MDG, Lwaleed BA, Sant’ana Dusse LM. Preeclampsia: the role of tissue factor and tissue factor pathway inhibitor. J Thromb Thrombolysis (2012) 34:1–6. doi:10.1007/s11239-012-0708-8
1067. Battinelli EM, Marshall A, Connors JM. The role of thrombophilia in pregnancy. Thrombosis (2013) 2013:516420. doi:10.1155/2013/516420
1068. Han L, Liu X, Li H, Zou J, Yang Z, Han J, et al. Blood coagulation parameters and platelet indices: changes in normal and preeclamptic pregnancies and predictive values for preeclampsia. PLoS One (2014) 9:e114488. doi:10.1371/journal.pone.0114488
1069. Koh SCL, Anandakumar C, Montan S, Ratnam SS. Plasminogen activators, plasminogen activator inhibitors and markers of intravascular coagulation in pre-eclampsia. Gynecol Obstet Invest (1993) 35:214–21. doi:10.1159/000292703
1070. Kaneko T, Wada H. Diagnostic criteria and laboratory tests for disseminated intravascular coagulation. J Clin Exp Hematop (2011) 51:67–76. doi:10.3960/jslrt.51.67
1071. Levi M, van der Poll T. Disseminated intravascular coagulation: a review for the internist. Int Emerg Med (2013) 8:23–32. doi:10.1007/s11739-012-0859-9
1072. Asakura H. Classifying types of disseminated intravascular coagulation: clinical and animal models. J Intensive Care (2014) 2:20. doi:10.1186/2052-0492-2-20
1073. Wada H, Matsumoto T, Yamashita Y, Hatada T. Disseminated intravascular coagulation: testing and diagnosis. Clin Chim Acta (2014) 436:130–4. doi:10.1016/j.cca.2014.04.020
1074. Zeerleder S, Hack CE, Wuillemin WA. Disseminated intravascular coagulation in sepsis. Chest (2005) 128:2864–75. doi:10.1378/chest.128.4.2864
1075. Xu M, Dong MQ, Cao FL, Liu ML, Wang YX, Dong HY, et al. Tanshinone IIA reduces lethality and acute lung injury in LPS-treated mice by inhibition of PLA2 activity. Eur J Pharmacol (2009) 607:194–200. doi:10.1016/j.ejphar.2009.02.003
1076. Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis-associated disseminated intravascular coagulation and thromboembolic disease. Mediterr J Hematol Infect Dis (2010) 2:e2010024. doi:10.4084/MJHID.2010.024
1077. Acikgoz S, Akduman D, Eskici ZM, Can M, Mungan G, Guven B, et al. Thrombocyte and erythrocyte indices in sepsis and disseminated intravascular coagulation. J Med Biochem (2012) 31:60–4. doi:10.2478/v10011-011-0043-1
1078. Wu LC, Lin X, Sun H. Tanshinone IIA protects rabbits against LPS-induced disseminated intravascular coagulation (DIC). Acta Pharmacol Sin (2012) 33:1254–9. doi:10.1038/aps.2012.84
1079. Iba T, Ito T, Maruyama I, Jilma B, Brenner T, Muller MC, et al. Potential diagnostic markers for disseminated intravascular coagulation of sepsis. Blood Rev (2016) 30:149–55. doi:10.1016/j.blre.2015.10.002
1080. Okamoto K, Tamura T, Sawatsubashi Y. Sepsis and disseminated intravascular coagulation. J Intensive Care (2016) 4:23. doi:10.1186/s40560-016-0149-0
1081. D’Elia AV, Fabbro D, Driul L, Barillari G, Marchesoni D, Damante G. Plasminogen activator inhibitor-1 gene polymorphisms in pre-eclampsia. Semin Thromb Hemost (2011) 37:97–105. doi:10.1055/s-0030-1270334
1082. Buurma AJ, Turner RJ, Driessen JH, Mooyaart AL, Schoones JW, Bruijn JA, et al. Genetic variants in pre-eclampsia: a meta-analysis. Hum Reprod Update (2013) 19:289–303. doi:10.1093/humupd/dms060
1083. Morgan JA, Bombell S, Mcguire W. Association of plasminogen activator inhibitor-type 1 (-675 4G/5G) polymorphism with pre-eclampsia: systematic review. PLoS One (2013) 8:e56907. doi:10.1371/journal.pone.0056907
1084. Kell DB, Pretorius E. Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting. Progr Biophys Mol Biol (2016). doi:10.1016/j.pbiomolbio.2016.08.006
1085. Kell DB, Pretorius E. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting. bioRxiv preprint. bioRxiv (2016) 054734. doi:10.1101/054734
1086. Akassoglou K, Adams RA, Bauer J, Mercado P, Tseveleki V, Lassmann H, et al. Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc Natl Acad Sci U S A (2004) 101:6698–703. doi:10.1073/pnas.0303859101
1087. Levi M, van der Poll T, Buller HR. Bidirectional relation between inflammation and coagulation. Circulation (2004) 109:2698–704. doi:10.1161/01.CIR.0000131660.51520.9A
1088. Flick MJ, Lajeunesse CM, Talmage KE, Witte DP, Palumbo JS, Pinkerton MD, et al. Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin alphaMbeta2 binding motif. J Clin Invest (2007) 117:3224–35. doi:10.1172/JCI30134
1089. Jennewein C, Paulus P, Zacharowski K. Linking inflammation and coagulation: novel drug targets to treat organ ischemia. Curr Opin Anaesthesiol (2011) 24:375–80. doi:10.1097/ACO.0b013e3283489ac0
1090. Jennewein C, Tran N, Paulus P, Ellinghaus P, Eble JA, Zacharowski K. Novel aspects of fibrin(ogen) fragments during inflammation. Mol Med (2011) 17:568–73. doi:10.2119/molmed.2010.00146
1091. Schuliga M. The inflammatory actions of coagulant and fibrinolytic proteases in disease. Mediators Inflamm (2015) 2015:437695. doi:10.1155/2015/437695
1092. Miranda S, Opazo C, Larrondo LF, Munoz FJ, Ruiz F, Leighton F, et al. The role of oxidative stress in the toxicity induced by amyloid beta-peptide in Alzheimer’s disease. Progr Neurobiol (2000) 62:633–48. doi:10.1016/S0301-0082(00)00015-0
1093. Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, De Calignon A, Rozkalne A, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature (2008) 451:720–4. doi:10.1038/nature06616
1094. Rival T, Page RM, Chandraratna DS, Sendall TJ, Ryder E, Liu B, et al. Fenton chemistry and oxidative stress mediate the toxicity of the beta-amyloid peptide in a Drosophila model of Alzheimer’s disease. Eur J Neurosci (2009) 29:1335–47. doi:10.1111/j.1460-9568.2009.06701.x
1095. Liu B, Moloney A, Meehan S, Morris K, Thomas SE, Serpell LC, et al. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J Biol Chem (2011) 286:4248–56. doi:10.1074/jbc.M110.158980
1096. Minter MR, Taylor JM, Crack PJ. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J Neurochem (2016) 136:457–74. doi:10.1111/jnc.13411
1097. Özcan F, Turak O, Durak A, Işleyen A, Uçar F, Giniş Z, et al. Red cell distribution width and inflammation in patients with non-dipper hypertension. Blood Press (2013) 22:80–5. doi:10.3109/08037051.2012.707336
1098. Heilmann L, Mattheck C, Kurz E. Changes in blood rheology and their influence on oxygen diffusion in normal and pathological pregnancies. Arch Gynäkol (1977) 223:283–98. doi:10.1007/Bf00667368
1099. Stoeff S, Dikov I, Vretenarska M, Jovtchev S, Trifonova N, Penev M. Quantitative utility assessment of some biophysical and rheological tests used in hypertension research. 1. Relevance analyses of the test parameters. C R Acad Bulg Sci (2003) 56:95–100.
1100. Kurt RK, Aras Z, Silfeler DB, Kunt C, Islimye M, Kosar O. Relationship of red cell distribution width with the presence and severity of preeclampsia. Clin Appl Thromb Hemost (2015) 21:128–31. doi:10.1177/1076029613490827
1101. Abdullahi H, Osman A, Rayis DA, Gasim GI, Imam AM, Adam I. Red blood cell distribution width is not correlated with preeclampsia among pregnant Sudanese women. Diagn Pathol (2014) 9:29. doi:10.1186/1746-1596-9-29
1102. McMahon CJ, Hopkins S, Vail A, King AT, Smith D, Illingworth KJ, et al. Inflammation as a predictor for delayed cerebral ischemia after aneurysmal subarachnoid haemorrhage. J Neurointerv Surg (2013) 5:512–7. doi:10.1136/neurintsurg-2012-010386
1103. Swanepoel AC, Pretorius E. Scanning electron microscopy analysis of erythrocytes in thromboembolic ischemic stroke. Int J Lab Hematol (2012) 34:185–91. doi:10.1111/j.1751-553X.2011.01379.x
1104. Buys AV, Van Rooy MJ, Soma P, Van Papendorp D, Lipinski B, Pretorius E. Changes in red blood cell membrane structure in type 2 diabetes: a scanning electron and atomic force microscopy study. Cardiovasc Diabetol (2013) 12:25. doi:10.1186/1475-2840-12-25
1105. Pretorius E, Lipinski B. Iron alters red blood cell morphology. Blood (2013) 121:9. doi:10.1182/blood-2012-09-454793
1106. Pretorius E, Lipinski B. Thromboembolic ischemic stroke changes red blood cell morphology. Cardiovasc Pathol (2013) 22:241–2. doi:10.1016/j.carpath.2012.11.005
1107. Lang F, Lang KS, Lang PA, Huber SM, Wieder T. Mechanisms and significance of eryptosis. Antioxid Redox Signal (2006) 8:1183–92. doi:10.1089/ars.2006.8.1183
1108. Lang F, Gulbins E, Lerche H, Huber SM, Kempe DS, Föller M. Eryptosis, a window to systemic disease. Cell Physiol Biochem (2008) 22:373–80. doi:10.1159/000185448
1109. Lang F, Lang E, Foller M. Physiology and pathophysiology of eryptosis. Transfus Med Hemother (2012) 39:308–14. doi:10.1159/000342534
1110. Lang F, Qadri SM. Mechanisms and significance of eryptosis, the suicidal death of erythrocytes. Blood Purif (2012) 33:125–30. doi:10.1159/000334163
1111. Lang E, Lang F. Triggers, inhibitors, mechanisms, and significance of eryptosis: the suicidal erythrocyte death. Biomed Res Int (2015) 2015:513518. doi:10.1155/2015/513518
1112. Lang E, Zelenak C, Eberhard M, Bissinger R, Rotte A, Ghashghaeinia M, et al. Impact of cyclin-dependent kinase CDK4 inhibition on eryptosis. Cell Physiol Biochem (2015) 37:1178–86. doi:10.1159/000430241
1113. Qadri SM, Donkor DA, Bhakta V, Eltringham-Smith LJ, Dwivedi DJ, Moore JC, et al. Phosphatidylserine externalization and procoagulant activation of erythrocytes induced by Pseudomonas aeruginosa virulence factor pyocyanin. J Cell Mol Med (2016) 20:710–20. doi:10.1111/jcmm.12778
1114. Lang E, Bissinger R, Gulbins E, Lang F. Ceramide in the regulation of eryptosis, the suicidal erythrocyte death. Apoptosis (2015) 20:758–67. doi:10.1007/s10495-015-1094-4
1115. Hansson SR, Nääv Å, Erlandsson L. Oxidative stress in preeclampsia and the role of free fetal hemoglobin. Front Physiol (2014) 5:516. doi:10.3389/fphys.2014.00516
1116. Askie LM, Duley L, Henderson-Smart DJ, Stewart LA; Paris Collaborative Group. Antiplatelet agents for prevention of pre-eclampsia: a meta-analysis of individual patient data. Lancet (2007) 369:1791–8. doi:10.1016/S0140-6736(07)60712-0
1117. Roberts JM, Catov JM. Aspirin for pre-eclampsia: compelling data on benefit and risk. Lancet (2007) 369:1765–6. doi:10.1016/S0140-6736(07)60713-2
1118. Rossi AC, Mullin PM. Prevention of pre-eclampsia with low-dose aspirin or vitamins C and E in women at high or low risk: a systematic review with meta-analysis. Eur J Obstet Gynecol Reprod Biol (2011) 158:9–16. doi:10.1016/j.ejogrb.2011.04.010
1119. Villa PM, Kajantie E, Räikkönen K, Pesonen AK, Hämäläinen E, Vainio M, et al. Aspirin in the prevention of pre-eclampsia in high-risk women: a randomised placebo-controlled PREDO trial and a meta-analysis of randomised trials. BJOG (2013) 120:64–74. doi:10.1111/j.1471-0528.2012.03493.x
1120. Bujold E, Morency AM, Roberge S, Lacasse Y, Forest JC, Giguère Y. Acetylsalicylic acid for the prevention of preeclampsia and intra-uterine growth restriction in women with abnormal uterine artery Doppler: a systematic review and meta-analysis. J Obstet Gynaecol Can (2009) 31:818–26. doi:10.1016/S1701-2163(16)34300-6
1121. Roberge S, Giguere Y, Villa P, Nicolaides K, Vainio M, Forest JC, et al. Early administration of low-dose aspirin for the prevention of severe and mild preeclampsia: a systematic review and meta-analysis. Am J Perinatol (2012) 29:551–6. doi:10.1055/s-0032-1310527
1122. Roberge S, Demers S, Bujold E. Initiation of aspirin in early gestation for the prevention of pre-eclampsia. BJOG (2013) 120:773–4. doi:10.1111/1471-0528.12170
1123. Bergeron TS, Roberge S, Carpentier C, Sibai B, Mccaw-Binns A, Bujold E. Prevention of preeclampsia with aspirin in multiple gestations: a systematic review and meta-analysis. Am J Perinatol (2016) 33:605–10. doi:10.1055/s-0035-1570381
1124. Roberge S, Sibai B, McCaw-Binns A, Bujold E. Low-dose aspirin in early gestation for prevention of preeclampsia and small-for-gestational-age neonates: meta-analysis of large randomized trials. Am J Perinatol (2016) 33:781–5. doi:10.1055/s-0036-1572495
1125. Hofmeyr GJ, Belizán JM, Von Dadelszen P; Calcium Pre-Eclampsia Study Group. Low-dose calcium supplementation for preventing pre-eclampsia: a systematic review and commentary. BJOG (2014) 121:951–7. doi:10.1111/1471-0528.12613
1126. Leslie K, Thilaganathan B, Papageorghiou A. Early prediction and prevention of pre-eclampsia. Best Pract Res Clin Obstet Gynaecol (2011) 25:343–54. doi:10.1016/j.bpobgyn.2011.01.002
1127. Thangaratinam S, Langenveld J, Mol BW, Khan KS. Prediction and primary prevention of pre-eclampsia. Best Pract Res Clin Obstet Gynaecol (2011) 25:419–33. doi:10.1016/j.bpobgyn.2011.02.008
1128. Gillon TE, Pels A, Von Dadelszen P, Macdonell K, Magee LA. Hypertensive disorders of pregnancy: a systematic review of international clinical practice guidelines. PLoS One (2014) 9:e113715. doi:10.1371/journal.pone.0113715
1129. Cottrell EC, Sibley CP. From pre-clinical studies to clinical trials: generation of novel therapies for pregnancy complications. Int J Mol Sci (2015) 16:12907–24. doi:10.3390/ijms160612907
1130. Altman D, Carroli G, Duley L, Farrell B, Moodley J, Neilson J, et al. Do women with pre-eclampsia, and their babies, benefit from magnesium sulphate? The magpie trial: a randomised placebo-controlled trial. Lancet (2002) 359:1877–90. doi:10.1016/S0140-6736(02)08778-0
1131. Duley L. Evidence and practice: the magnesium sulphate story. Best Pract Res Clin Obstet Gynaecol (2005) 19:57–74. doi:10.1016/j.bpobgyn.2004.10.010
1132. Pratt JJ, Niedle PS, Vogel JP, Oladapo OT, Bohren M, Tunçalp Ö, et al. Alternative regimens of magnesium sulfate for treatment of preeclampsia and eclampsia: a systematic review of non-randomized studies. Acta Obstet Gynecol Scand (2016) 95:144–56. doi:10.1111/aogs.12807
1133. Williamson RD, McCarthy C, Kenny LC, O’Keeffe GW. Magnesium sulphate prevents lipopolysaccharide-induced cell death in an in vitro model of the human placenta. Pregnancy Hypertens (2016). doi:10.1016/j.preghy.2016.08.237
1134. Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med (1998) 338:1128–37. doi:10.1056/NEJM199804163381607
1135. Everett TR, Wilkinson IB, Lees CC. Drug development in preeclampsia: a ‘no go’ area? J Matern Fetal Neonatal Med (2012) 25:50–2. doi:10.3109/14767058.2011.557791
1136. Martinez de Tejada B. Antibiotic use and misuse during pregnancy and delivery: benefits and risks. Int J Environ Res Public Health (2014) 11:7993–8009. doi:10.3390/ijerph110807993
1137. Thinkhamrop J, Hofmeyr GJ, Adetoro O, Lumbiganon P, Ota E. Antibiotic prophylaxis during the second and third trimester to reduce adverse pregnancy outcomes and morbidity. Cochrane Database Syst Rev (2015) 1:CD002250. doi:10.1002/14651858.CD002250.pub2
1138. Vazquez JC, Abalos E. Treatments for symptomatic urinary tract infections during pregnancy. Cochrane Database Syst Rev (2011) CD002256. doi:10.1002/14651858.CD002256.pub2
1139. Mathew D, Khan K, Thornton JG, Todros T. Antibiotics for preventing hypertensive diseases in pregnancy (protocol). Cochrane Database Syst Rev (2007) CD006841. doi:10.1002/14651858.CD006841
1140. Pound MW, May DB. Proposed mechanisms and preventative options of Jarisch-Herxheimer reactions. J Clin Pharm Ther (2005) 30:291–5. doi:10.1111/j.1365-2710.2005.00631.x
1141. See S, Scott EK, Levin MW. Penicillin-induced Jarisch-Herxheimer reaction. Ann Pharmacother (2005) 39:2128–30. doi:10.1345/aph.1G308
1142. Belum GR, Belum VR, Chaitanya Arudra SK, Reddy BS. The Jarisch-Herxheimer reaction: revisited. Travel Med Infect Dis (2013) 11:231–7. doi:10.1016/j.tmaid.2013.04.001
1143. Guerrier G, D’Ortenzio E. The Jarisch-Herxheimer reaction in leptospirosis: a systematic review. PLoS One (2013) 8:e59266. doi:10.1371/journal.pone.0059266
1144. Prins JM, van Deventer SJH, Kuijper EJ, Speelman P. Clinical relevance of antibiotic-induced endotoxin release. Antimicrob Agents Chemother (1994) 38:1211–8. doi:10.1128/AAC.38.6.1211
1145. Kirikae T, Nakano M, Morrison DC. Antibiotic-induced endotoxin release from bacteria and its clinical significance. Microbiol Immunol (1997) 41:285–94. doi:10.1111/j.1348-0421.1997.tb01203.x
1146. Holzheimer RG. Antibiotic induced endotoxin release and clinical sepsis: a review. J Chemother (2001) 13(Spec No 1):159–72. doi:10.1179/joc.2001.13.Supplement-2.159
1147. Lepper PM, Held TK, Schneider EM, Bölke E, Gerlach H, Trautmann M. Clinical implications of antibiotic-induced endotoxin release in septic shock. Intensive Care Med (2002) 28:824–33. doi:10.1007/s00134-002-1330-6
1148. Andersson JA, Fitts EC, Kirtley ML, Ponnusamy D, Peniche AG, Dann SM, et al. New role for FDA-approved drugs in combating antibiotic-resistant bacteria. Antimicrob Agents Chemother (2016) 60:3717–29. doi:10.1128/AAC.00326-16
1149. Poston L, Briley AL, Seed PT, Kelly FJ, Shennan AH. Vitamin C and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): randomised placebo-controlled trial. Lancet (2006) 367:1145–54. doi:10.1016/S0140-6736(06)68433-X
1150. Ly C, Yockell-Lelievre J, Ferraro ZM, Arnason JT, Ferrier J, Gruslin A. The effects of dietary polyphenols on reproductive health and early development. Hum Reprod Update (2015) 21:228–48. doi:10.1093/humupd/dmu058
1151. Liang ST, Wong VC, So WW, Ma HK, Chan V, Todd D. Homozygous alpha-thalassaemia: clinical presentation, diagnosis and management. A review of 46 cases. Br J Obstet Gynaecol (1985) 92:680–4. doi:10.1111/j.1471-0528.1985.tb01447.x
1152. Tungwiwat W, Fucharoen S, Fucharoen G, Ratanasiri T, Sanchaisuriya K. Development and application of a real-time quantitative PCR for prenatal detection of fetal alpha(0)-thalassemia from maternal plasma. Ann N Y Acad Sci (2006) 1075:103–7. doi:10.1196/annals.1368.013
1153. Senden IP, De Groot CJ, Steegers EA, Bertina RM, Swinkels DW. Preeclampsia and the C282Y mutation in the hemochromatosis (HFE) gene. Clin Chem (2004) 50:973–4. doi:10.1373/clinchem.2004.031591
1154. Perron NR, Brumaghim JL. A review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem Biophys (2009) 53:75–100. doi:10.1007/s12013-009-9043-x
1155. Dodd JM, Mcleod A, Windrim RC, Kingdom J. Antithrombotic therapy for improving maternal or infant health outcomes in women considered at risk of placental dysfunction. Cochrane Database Syst Rev (2010) CD006780. doi:10.1002/14651858.CD006780.pub2
1156. Li Y, Wu Y, Gong X, Shi X, Qiao F, Liu H. Low molecular weight heparin decreases the permeability of glomerular endothelial cells when exposed to pre-eclampsia serum in vitro. Nephrology (Carlton) (2012) 17:754–9. doi:10.1111/j.1440-1797.2012.01657.x
1157. Dodd JM, Mcleod A, Windrim RC, Kingdom J. Antithrombotic therapy for improving maternal or infant health outcomes in women considered at risk of placental dysfunction. Cochrane Database Syst Rev (2013) 7:CD006780. doi:10.1002/14651858.CD006780.pub3
1158. Zhang Y, Liu F, Chen S, Zhong M. Low-molecular-weight heparin protects kidney through an anti-apoptotic mechanism in a rat pre-eclamptic model. Eur J Obstet Gynecol Reprod Biol (2015) 188:51–5. doi:10.1016/j.ejogrb.2015.02.001
1159. Darmochwał-Kolarz D, Kolarz B, Korzeniewski M, Kimber-Trojnar Z, Patro-Malysza J, Mierzynski R, et al. A prevention of pre-eclampsia with the use of acetylsalicylic acid and low-molecular weight heparin – molecular mechanisms. Curr Pharm Biotechnol (2016) 17:624–8. doi:10.2174/1389201017666160301103312
1160. Roberge S, Demers S, Nicolaides KH, Bureau M, Côté S, Bujold E. Prevention of pre-eclampsia by low-molecular-weight heparin in addition to aspirin: a meta-analysis. Ultrasound Obstet Gynecol (2016) 47:548–53. doi:10.1002/uog.15789
1161. Eggers AE. A hypothesis about how to achieve anticoagulation without bleeding. Med Hypotheses (2015) 85:720–2. doi:10.1016/j.mehy.2015.10.021
1162. Kell DB. How drugs pass through biological cell membranes – a paradigm shift in our understanding? Beilstein Mag (2016) 2. Available from: http://www.beilstein-institut.de/download/628/609_kell.pdf
1163. Park K, Lee S, Ahn HS, Kim D. Predicting the multi-modal binding propensity of small molecules: towards an understanding of drug promiscuity. Mol Biosyst (2009) 5:844–53. doi:10.1039/b901356c
1164. Pérez-Nueno VI, Venkatraman V, Mavridis L, Ritchie DW. Detecting drug promiscuity using Gaussian ensemble screening. J Chem Inf Model (2012) 52:1948–61. doi:10.1021/ci3000979
1165. Tarcsay A, Keserü GM. Contributions of molecular properties to drug promiscuity. J Med Chem (2013) 56:1789–95. doi:10.1021/jm301514n
1166. Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, et al. The promise and peril of chemical probes. Nat Chem Biol (2015) 11:536–41. doi:10.1038/nchembio.1867
1167. Niphakis MJ, Lum KM, Cognetta AB, Correia BE, Ichu TA, Olucha J, et al. A global map of lipid-binding proteins and their ligandability in cells. Cell (2015) 161:1668–80. doi:10.1016/j.cell.2015.05.045
1168. Mestres J, Gregori-Puigjané E, Valverde S, Solé RV. The topology of drug-target interaction networks: implicit dependence on drug properties and target families. Mol Biosyst (2009) 5:1051–7. doi:10.1039/b905821b
1169. Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol (2008) 4:682–90. doi:10.1038/nchembio.118
1170. Berger SI, Iyengar R. Network analyses in systems pharmacology. Bioinformatics (2009) 25:2466–72. doi:10.1093/bioinformatics/btp465
1171. van der Graaf PH, Benson N. Systems pharmacology: bridging systems biology and pharmacokinetics-pharmacodynamics (PKPD) in drug discovery and development. Pharm Res (2011) 28:1460–4. doi:10.1007/s11095-011-0467-9
1172. Cucurull-Sanchez L, Spink KG, Moschos SA. Relevance of systems pharmacology in drug discovery. Drug Discov Today (2012) 17:665–70. doi:10.1016/j.drudis.2012.01.015
1173. Prentice RL, Langer RD, Stefanick ML, Howard BV, Pettinger M, Anderson GL, et al. Combined analysis of women’s health initiative observational and clinical trial data on postmenopausal hormone treatment and cardiovascular disease. Am J Epidemiol (2006) 163:589–99. doi:10.1093/aje/kwj079
1174. Couzin J. Cholesterol veers off script. Science (2008) 322:220–3. doi:10.1126/science.322.5899.220
1175. Peterson RT. Chemical biology and the limits of reductionism. Nat Chem Biol (2008) 4:635–8. doi:10.1038/nchembio1108-635
1176. Robinson JG. Models for describing relations among the various statin drugs, low-density lipoprotein cholesterol lowering, pleiotropic effects, and cardiovascular risk. Am J Cardiol (2008) 101:1009–15. doi:10.1016/j.amjcard.2007.11.060
1177. Endo A. A historical perspective on the discovery of statins. Proc Jpn Acad Ser B Phys Biol Sci (2010) 86:484–93. doi:10.2183/pjab.86.484
1178. Wagner BK, Kitami T, Gilbert TJ, Peck D, Ramanathan A, Schreiber SL, et al. Large-scale chemical dissection of mitochondrial function. Nat Biotechnol (2008) 26:343–51. doi:10.1038/nbt1387
1179. Libby P, Aikawa M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat Med (2002) 8:1257–62. doi:10.1038/nm1102-1257
1180. Liao JK. Beyond lipid lowering: the role of statins in vascular protection. Int J Cardiol (2002) 86:5–18. doi:10.1016/S0167-5273(02)00195-X
1181. Undas A, Brozek J, Musial J. Anti-inflammatory and antithrombotic effects of statins in the management of coronary artery disease. Clin Lab (2002) 48:287–96.
1182. Weitz-Schmidt G. Statins as anti-inflammatory agents. Trends Pharmacol Sci (2002) 23:482–6. doi:10.1016/S0165-6147(02)02077-1
1183. Blanco-Colio LM, Tuñon J, Martin-Ventura JL, Egido J. Anti-inflammatory and immunomodulatory effects of statins. Kidney Int (2003) 63:12–23. doi:10.1046/j.1523-1755.2003.00744.x
1184. Kwak BR, Mulhaupt F, Mach F. Atherosclerosis: anti-inflammatory and immunomodulatory activities of statins. Autoimmun Rev (2003) 2:332–8. doi:10.1016/S1568-9972(03)00049-1
1185. Steffens S, Mach F. Anti-inflammatory properties of statins. Semin Vasc Med (2004) 4:417–22. doi:10.1055/s-2004-869599
1186. Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov (2005) 4:977–87. doi:10.1038/nrd1901
1187. Abeles AM, Pillinger MH. Statins as antiinflammatory and immunomodulatory agents: a future in rheumatologic therapy? Arthritis Rheum (2006) 54:393–407. doi:10.1002/art.21521
1188. Endres M. Statins: potential new indications in inflammatory conditions. Atheroscler Suppl (2006) 7:31–5. doi:10.1016/j.atherosclerosissup.2006.01.005
1189. Li JJ, Zheng X, Li J. Statins may be beneficial for patients with slow coronary flow syndrome due to its anti-inflammatory property. Med Hypotheses (2007) 69:333–7. doi:10.1016/j.mehy.2006.09.070
1190. Mira E, Manes S. Immunomodulatory and anti-inflammatory activities of statins. Endocr Metab Immune Disord Drug Targets (2009) 9:237–47. doi:10.2174/187153009789044383
1191. Dinarello CA. Anti-inflammatory agents: present and future. Cell (2010) 140:935–50. doi:10.1016/j.cell.2010.02.043
1192. Bu DX, Griffin G, Lichtman AH. Mechanisms for the anti-inflammatory effects of statins. Curr Opin Lipidol (2011) 22:165–70. doi:10.1097/MOL.0b013e3283453e41
1193. Antonopoulos AS, Margaritis M, Lee R, Channon K, Antoniades C. Statins as anti-inflammatory agents in atherogenesis: molecular mechanisms and lessons from the recent clinical trials. Curr Pharm Des (2012) 18:1519–30. doi:10.2174/138161212799504803
1194. Liappis AP, Kan VL, Rochester CG, Simon GL. The effect of statins on mortality in patients with bacteremia. Clin Infect Dis (2001) 33:1352–7. doi:10.1086/323334
1195. Terblanche M, Almog Y, Rosenson RS, Smith TS, Hackam DG. Statins: panacea for sepsis? Lancet Infect Dis (2006) 6:242–8. doi:10.1016/S1473-3099(06)70439-X
1196. Falagas ME, Makris GC, Matthaiou DK, Rafailidis PI. Statins for infection and sepsis: a systematic review of the clinical evidence. J Antimicrob Chemother (2008) 61:774–85. doi:10.1093/jac/dkn019
1197. Sun HY, Singh N. Antimicrobial and immunomodulatory attributes of statins: relevance in solid-organ transplant recipients. Clin Infect Dis (2009) 48:745–55. doi:10.1086/597039
1198. Kozarov E, Padro T, Badimon L. View of statins as antimicrobials in cardiovascular risk modification. Cardiovasc Res (2014) 102:362–74. doi:10.1093/cvr/cvu058
1199. Sandek A, Utchill S, Rauchhaus M. The endotoxin-lipoprotein hypothesis – an update. Arch Med Sci (2007) 3:S81–90.
1200. Ukinc K, Ersoz HO, Erem C, Hacihasanoglu AB, Karti SS. Effects of one year simvastatin and atorvastatin treatments on acute phase reactants in uncontrolled type 2 diabetic patients. Endocrine (2009) 35:380–8. doi:10.1007/s12020-009-9157-3
1201. Mascitelli L, Goldstein MR. Might the beneficial effects of statin drugs be related to their action on iron metabolism? QJM (2012) 105:1225–9. doi:10.1093/qjmed/hcs204
1202. Kanugula AK, Gollavilli PN, Vasamsetti SB, Karnewar S, Gopoju R, Ummanni R, et al. Statin-induced inhibition of breast cancer proliferation and invasion involves attenuation of iron transport: intermediacy of nitric oxide and antioxidant defence mechanisms. FEBS J (2014) 281:3719–38. doi:10.1111/febs.12893
1203. Lecarpentier E, Morel O, Fournier T, Elefant E, Chavatte-Palmer P, Tsatsaris V. Statins and pregnancy: between supposed risks and theoretical benefits. Drugs (2012) 72:773–88. doi:10.2165/11632010-000000000-00000
1204. Costantine MM, Cleary K; Eunice Kennedy Shriver National Institute of Child, Health, Human Development Obstetric – Fetal Pharmacology Research Units Network. Pravastatin for the prevention of preeclampsia in high-risk pregnant women. Obstet Gynecol (2013) 121:349–53. doi:10.1097/AOG.0b013e31827d8ad5
1205. Downing JW, Baysinger CL, Johnson RF, Paschall RL. Review: potential druggable targets for the treatment of early onset preeclampsia. Pregnancy Hypertens (2013) 3:203–10. doi:10.1016/j.preghy.2013.04.120
1206. Carver AR, Tamayo E, Perez-Polo JR, Saade GR, Hankins GDV, Costantine MM. The effect of maternal pravastatin therapy on adverse sensorimotor outcomes of the offspring in a murine model of preeclampsia. Int J Dev Neurosci (2014) 33:33–40. doi:10.1016/j.ijdevneu.2013.11.004
1207. Cindrova-Davies T. The therapeutic potential of antioxidants, ER chaperones, NO and H2S donors, and statins for treatment of preeclampsia. Front Pharmacol (2014) 5:119. doi:10.3389/fphar.2014.00119
1208. Girardi G. Can statins prevent pregnancy complications? J Reprod Immunol (2014) 10(1–102):161–7. doi:10.1016/j.jri.2013.07.005
1209. McDonnold M, Tamayo E, Kechichian T, Gamble P, Longo M, Hankins GDV, et al. The effect of prenatal pravastatin treatment on altered fetal programming of postnatal growth and metabolic function in a preeclampsia-like murine model. Am J Obstet Gynecol (2014) 210(542):e541–7. doi:10.1016/j.ajog.2014.01.010
1210. Ramma W, Ahmed A. Therapeutic potential of statins and the induction of heme oxygenase-1 in preeclampsia. J Reprod Immunol (2014) 10(1–102):153–60. doi:10.1016/j.jri.2013.12.120
1211. Saad AF, Kechichian T, Yin H, Sbrana E, Longo M, Wen M, et al. Effects of pravastatin on angiogenic and placental hypoxic imbalance in a mouse model of preeclampsia. Reprod Sci (2014) 21:138–45. doi:10.1177/1933719113492207
1212. Staff AC, Johnsen GM, Dechend R, Redman CWG. Preeclampsia and uteroplacental acute atherosis: immune and inflammatory factors. J Reprod Immunol (2014) 10(1–102):120–6. doi:10.1016/j.jri.2013.09.001
1213. Ahmed A, Ramma W. Unravelling the theories of pre-eclampsia: are the protective pathways the new paradigm? Br J Pharmacol (2015) 172:1574–86. doi:10.1111/bph.12977
1214. Bauer AJ, Banek CT, Needham K, Gillham H, Capoccia S, Regal JF, et al. Pravastatin attenuates hypertension, oxidative stress, and angiogenic imbalance in rat model of placental ischemia-induced hypertension. Hypertension (2013) 61:1103–10. doi:10.1161/HYPERTENSIONAHA.111.00226
1215. Carver AR, Andrikopoulou M, Lei J, Tamayo E, Gamble P, Hou Z, et al. Maternal pravastatin prevents altered fetal brain development in a preeclamptic CD-1 mouse model. PLoS One (2014) 9:e100873. doi:10.1371/journal.pone.0100873
1216. Lefkou E, Mamopoulos A, Fragakis N, Dagklis T, Vosnakis C, Nounopoulos E, et al. Clinical improvement and successful pregnancy in a preeclamptic patient with antiphospholipid syndrome treated with pravastatin. Hypertension (2014) 63:e118–9. doi:10.1161/HYPERTENSIONAHA.114.03115
1217. Brownfoot FC, Tong S, Hannan NJ, Binder NK, Walker SP, Cannon P, et al. Effects of pravastatin on human placenta, endothelium, and women with severe preeclampsia. Hypertension (2015) 66:687–97; discussion 445. doi:10.1161/HYPERTENSIONAHA.115.05445
1218. Chaiworapongsa T, Romero R, Korzeniewski SJ, Chaemsaithong P, Hernandez-Andrade E, Segars JH, et al. Pravastatin to prevent recurrent fetal death in massive perivillous fibrin deposition of the placenta (MPFD). J Matern Fetal Neonatal Med (2016) 29:855–62. doi:10.3109/14767058.2015.1022864
1219. Downing J. Sildenafil for the treatment of preeclampsia. Hypertens Pregnancy (2010) 29:248–50; author reply 251–2. doi:10.3109/10641951003634766
1220. Kakigano A, Tomimatsu T, Mimura K, Kanayama T, Fujita S, Minato K, et al. Drug repositioning for preeclampsia therapeutics by in vitro screening: phosphodiesterase-5 inhibitor vardenafil restores endothelial dysfunction via induction of placental growth factor. Reprod Sci (2015) 22:1272–80. doi:10.1177/1933719115574340
1221. Ramesar SV, Mackraj I, Gathiram P, Moodley J. Sildenafil citrate decreases sFlt-1 and sEng in pregnant l-NAME treated Sprague-Dawley rats. Eur J Obstet Gynecol Reprod Biol (2011) 157:136–40. doi:10.1016/j.ejogrb.2011.03.005
1222. Herraiz S, Pellicer B, Serra V, Cauli O, Cortijo J, Felipo V, et al. Sildenafil citrate improves perinatal outcome in fetuses from pre-eclamptic rats. BJOG (2012) 119:1394–402. doi:10.1111/j.1471-0528.2012.03430.x
1223. Nassar AH, Masrouha KZ, Itani H, Nader KA, Usta IM. Effects of sildenafil in Nomega-nitro-L-arginine methyl ester-induced intrauterine growth restriction in a rat model. Am J Perinatol (2012) 29:429–34. doi:10.1055/s-0032-1304823
1224. Stanley JL, Andersson IJ, Poudel R, Rueda-Clausen CF, Sibley CP, Davidge ST, et al. Sildenafil citrate rescues fetal growth in the catechol-O-methyl transferase knockout mouse model. Hypertension (2012) 59:1021–8. doi:10.1161/HYPERTENSIONAHA.111.186270
1225. George EM, Palei AC, Dent EA, Granger JP. Sildenafil attenuates placental ischemia-induced hypertension. Am J Physiol Regul Integr Comp Physiol (2013) 305:R397–403. doi:10.1152/ajpregu.00216.2013
1226. Stanley JL, Sulek K, Andersson IJ, Davidge ST, Kenny LC, Sibley CP, et al. Sildenafil therapy normalizes the aberrant metabolomic profile in the comt(-/-) mouse model of preeclampsia/fetal growth restriction. Sci Rep (2015) 5:18241. doi:10.1038/srep18241
1227. Karasu E, Kayacan N, Sadan G, Dinc B. Different effects of different phosphodiesterase type-5 inhibitors in pre-eclampsia. Pregnancy Hypertens (2011) 1:231–7. doi:10.1016/j.preghy.2011.04.002
1228. Karasu E, Kayacan N, Sadan G, Dinc B. Endothelial dysfunction in the human umbilical artery due to preeclampsia can be prevented by sildenafil. Clin Exp Hypertens (2012) 34:79–85. doi:10.3109/10641963.2011.628730
1229. Ganzevoort W, Alfirevic Z, Von Dadelszen P, Kenny L, Papageorghiou A, Van Wassenaer-Leemhuis A, et al. STRIDER: sildenafil therapy in dismal prognosis early-onset intrauterine growth restriction – a protocol for a systematic review with individual participant data and aggregate data meta-analysis and trial sequential analysis. Syst Rev (2014) 3:23. doi:10.1186/2046-4053-3-23
1230. Abalos E, Duley L, Steyn DW. Antihypertensive drug therapy for mild to moderate hypertension during pregnancy. Cochrane Database Syst Rev (2014) 2:CD002252. doi:10.1002/14651858.CD002252.pub3
1231. Mahmud H, Föller M, Lang F. Stimulation of erythrocyte cell membrane scrambling by methyldopa. Kidney Blood Press Res (2008) 31:299–306. doi:10.1159/000153250
1232. Firoz T, Magee LA, Macdonell K, Payne BA, Gordon R, Vidler M, et al. Oral antihypertensive therapy for severe hypertension in pregnancy and postpartum: a systematic review. BJOG (2014) 121:1210–8; discussion 1220. doi:10.1111/1471-0528.12737
1233. Magee LA, Namouz-Haddad S, Cao V, Koren G, Von Dadelszen P. Labetalol for hypertension in pregnancy. Expert Opin Drug Saf (2015) 14:453–61. doi:10.1517/14740338.2015.998197
1234. Ishimaru T, Ishida J, Nakamura S, Hashimoto M, Matsukura T, Nakamura A, et al. Short-term suppression of the renin-angiotensin system in mice associated with hypertension during pregnancy. Mol Med Rep (2012) 6:28–32. doi:10.3892/mmr.2012.886
1235. Marshall TG, Lee RE, Marshall FE. Common angiotensin receptor blockers may directly modulate the immune system via VDR, PPAR and CCR2b. Theor Biol Med Model (2006) 3:1. doi:10.1186/1742-4682-3-1
1236. Ziegler EJ, Fisher CJ Jr, Sprung CL, Straube RC, Sadoff JC, Foulke GE, et al. Treatment of Gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. The HA-1A Sepsis Study Group. N Engl J Med (1991) 324:429–36. doi:10.1056/NEJM199102143240701
1237. Baumgartner JD, Glauser MP. Immunotherapy of endotoxemia and septicemia. Immunobiology (1993) 187:464–77. doi:10.1016/S0171-2985(11)80357-8
1238. Derkx B, Wittes J, Mccloskey R. Randomized, placebo-controlled trial of HA-1A, a human monoclonal antibody to endotoxin, in children with meningococcal septic shock. European Pediatric Meningococcal Septic Shock Trial Study Group. Clin Infect Dis (1999) 28:770–7. doi:10.1086/515184
1239. Yentis SM, Soni N, Riches PG. In vitro effects of HA-1A (centoxin) on cytokine production in whole blood from intensive care unit patients. Br J Anaesth (1994) 73:805–11. doi:10.1093/bja/73.6.805
1240. Marks L. The birth pangs of monoclonal antibody therapeutics: the failure and legacy of centoxin. MAbs (2012) 4:403–12. doi:10.4161/mabs.19909
1241. Helmerhorst EJ, Maaskant JJ, Appelmelk BJ. Anti-lipid A monoclonal antibody centoxin (HA-1A) binds to a wide variety of hydrophobic ligands. Infect Immun (1998) 66:870–3.
1242. Tan NS, Ho B, Ding JL. High-affinity LPS binding domain(s) in recombinant factor C of a horseshoe crab neutralizes LPS-induced lethality. FASEB J (2000) 14:859–70. doi:10.1096/fj.1530-6860
1243. Tan NS, Ng ML, Yau YH, Chong PKW, Ho B, Ding JL. Definition of endotoxin binding sites in horseshoe crab factor C recombinant sushi proteins and neutralization of endotoxin by sushi peptides. FASEB J (2000) 14:1801–13. doi:10.1096/fj.99-0866com
1244. Yau YH, Ho B, Tan NS, Ng ML, Ding JL. High therapeutic index of factor C sushi peptides: potent antimicrobials against Pseudomonas aeruginosa. Antimicrob Agents Chemother (2001) 45:2820–5. doi:10.1128/AAC.45.10.2820-2825.2001
1245. Li P, Sun M, Wohland T, Ho B, Ding JL. The molecular mechanism of interaction between sushi peptide and Pseudomonas endotoxin. Cell Mol Immunol (2006) 3:21–8.
1246. Li P, Sun M, Wohland T, Yang D, Ho B, Ding JL. Molecular mechanisms that govern the specificity of sushi peptides for Gram-negative bacterial membrane lipids. Biochemistry (2006) 45:10554–62. doi:10.1021/bi0602765
1247. Ding JL, Li P, Ho B. The sushi peptides: structural characterization and mode of action against Gram-negative bacteria. Cell Mol Life Sci (2008) 65:1202–19. doi:10.1007/s00018-008-7456-0
1248. Leptihn S, Har JY, Chen J, Ho B, Wohland T, Ding JL. Single molecule resolution of the antimicrobial action of quantum dot-labeled sushi peptide on live bacteria. BMC Biol (2009) 7:22. doi:10.1186/1741-7007-7-22
1249. Leptihn S, Guo L, Frecer V, Ho B, Ding JL, Wohland T. One step at a time: action mechanism of sushi 1 antimicrobial peptide and derived molecules. Virulence (2010) 1:42–4. doi:10.4161/viru.1.1.10229
1250. Frecer V, Ho B, Ding JL. De novo design of potent antimicrobial peptides. Antimicrob Agents Chemother (2004) 48:3349–57. doi:10.1128/AAC.48.9.3349-3357.2004
1251. Bhattacharjya S. De novo designed lipopolysaccharide binding peptides: structure based development of antiendotoxic and antimicrobial drugs. Curr Med Chem (2010) 17:3080–93. doi:10.2174/092986710791959756
1252. Brantsaeter AL, Myhre R, Haugen M, Myking S, Sengpiel V, Magnus P, et al. Intake of probiotic food and risk of preeclampsia in primiparous women: the Norwegian Mother and Child Cohort Study. Am J Epidemiol (2011) 174:807–15. doi:10.1093/aje/kwr168
1253. Gluckman P, Beedle A, Hanson M. Principles of Evolutionary Medicine. Oxford: Oxford University Press (2009).
1254. Hemmings DG. Signal transduction underlying the vascular effects of sphingosine 1-phosphate and sphingosylphosphorylcholine. Naunyn Schmiedebergs Arch Pharmacol (2006) 373:18–29. doi:10.1007/s00210-006-0046-5
1255. Hemmings DG, Hudson NK, Halliday D, O’Hara M, Baker PN, Davidge ST, et al. Sphingosine-1-phosphate acts via rho-associated kinase and nitric oxide to regulate human placental vascular tone. Biol Reprod (2006) 74:88–94. doi:10.1095/biolreprod.105.043034
1256. Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell (2011) 146:980–91. doi:10.1016/j.cell.2011.08.015
1257. Oldstone MBA, Teijaro JR, Walsh KB, Rosen H. Dissecting influenza virus pathogenesis uncovers a novel chemical approach to combat the infection. Virology (2013) 435:92–101. doi:10.1016/j.virol.2012.09.039
1258. Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature (2014) 510:58–67. doi:10.1038/nature13475
1259. Oldstone MBA, Rosen H. Cytokine storm plays a direct role in the morbidity and mortality from influenza virus infection and is chemically treatable with a single sphingosine-1-phosphate agonist molecule. Curr Top Microbiol Immunol (2014) 378:129–47. doi:10.1007/978-3-319-05879-5_6
1260. Walsh KB, Teijaro JR, Brock LG, Fremgen DM, Collins PL, Rosen H, et al. Animal model of respiratory syncytial virus: CD8+ T cells cause a cytokine storm that is chemically tractable by sphingosine-1-phosphate 1 receptor agonist therapy. J Virol (2014) 88:6281–93. doi:10.1128/JVI.00464-14
1261. Arish M, Husein A, Kashif M, Saleem M, Akhter Y, Rub A. Sphingosine-1-phosphate signaling: unraveling its role as a drug target against infectious diseases. Drug Discov Today (2016) 21:133–42. doi:10.1016/j.drudis.2015.09.013
1262. Zhang J, Dunk CE, Lye SJ. Sphingosine signalling regulates decidual NK cell angiogenic phenotype and trophoblast migration. Hum Reprod (2013) 28:3026–37. doi:10.1093/humrep/det339
1263. Nagamatsu T, Iwasawa-Kawai Y, Ichikawa M, Kawana K, Yamashita T, Osuga Y, et al. Emerging roles for lysophospholipid mediators in pregnancy. Am J Reprod Immunol (2014) 72:182–91. doi:10.1111/aji.12239
1264. Romanowicz L, Bańkowski E. Altered sphingolipid composition in Wharton’s jelly of pre-eclamptic newborns. Pathobiology (2010) 77:78–87. doi:10.1159/000278289
1265. Romanowicz L, Bańkowski E. Lipid compounds of human Wharton’s jelly and their alterations in preeclampsia. Int J Exp Pathol (2010) 91:1–9. doi:10.1111/j.1365-2613.2009.00675.x
1266. Romanowicz L, Bańkowski E. Sphingolipids of human umbilical cord vein and their alteration in preeclampsia. Mol Cell Biochem (2010) 340:81–9. doi:10.1007/s11010-010-0403-z
1267. Baig S, Lim JY, Fernandis AZ, Wenk MR, Kale A, Su LL, et al. Lipidomic analysis of human placental syncytiotrophoblast microvesicles in adverse pregnancy outcomes. Placenta (2013) 34:436–42. doi:10.1016/j.placenta.2013.02.004
1268. Li Q, Pan Z, Wang X, Gao Z, Ren C, Yang W. miR-125b-1-3p inhibits trophoblast cell invasion by targeting sphingosine-1-phosphate receptor 1 in preeclampsia. Biochem Biophys Res Commun (2014) 453:57–63. doi:10.1016/j.bbrc.2014.09.059
1269. Carrillo-Vico A, Lardone PJ, Naji L, Fernández-Santos JM, Martín-Lacave I, Guerrero JM, et al. Beneficial pleiotropic actions of melatonin in an experimental model of septic shock in mice: regulation of pro-/anti-inflammatory cytokine network, protection against oxidative damage and anti-apoptotic effects. J Pineal Res (2005) 39:400–8. doi:10.1111/j.1600-079X.2005.00265.x
1270. Gitto E, Pellegrino S, Gitto P, Barberi I, Reiter RJ. Oxidative stress of the newborn in the pre- and postnatal period and the clinical utility of melatonin. J Pineal Res (2009) 46:128–39. doi:10.1111/j.1600-079X.2008.00649.x
1271. Hobson SR, Lim R, Gardiner EE, Alers NO, Wallace EM. Phase I pilot clinical trial of antenatal maternally administered melatonin to decrease the level of oxidative stress in human pregnancies affected by pre-eclampsia (PAMPR): study protocol. BMJ Open (2013) 3:e003788. doi:10.1136/bmjopen-2013-003788
1272. Westh H, Lisby G, Breysse F, Boddinghaus B, Chomarat M, Gant V, et al. Multiplex real-time PCR and blood culture for identification of bloodstream pathogens in patients with suspected sepsis. Clin Microbiol Infect (2009) 15:544–51. doi:10.1111/j.1469-0691.2009.02736.x
1273. Yang B, Wang Y, Qian PY. Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinformatics (2016) 17:135. doi:10.1186/s12859-016-0992-y
1274. Khamis A, Raoult D, La Scola B. Comparison between rpoB and 16S rRNA gene sequencing for molecular identification of 168 clinical isolates of Corynebacterium. J Clin Microbiol (2005) 43:1934–6. doi:10.1128/JCM.43.4.1934-1936.2005
1275. Adékambi T, Drancourt M, Raoult D. The rpoB gene as a tool for clinical microbiologists. Trends Microbiol (2009) 17:37–45. doi:10.1016/j.tim.2008.09.008
1276. Roux S, Enault F, Bronner G, Debroas D. Comparison of 16S rRNA and protein-coding genes as molecular markers for assessing microbial diversity (bacteria and archaea) in ecosystems. FEMS Microbiol Ecol (2011) 78:617–28. doi:10.1111/j.1574-6941.2011.01190.x
1277. Liu W, Li L, Khan MA, Zhu F. Popular molecular markers in bacteria. Mol Gen Microbiol Virol (2012) 27(3):103–7. doi:10.3103/S0891416812030056
1278. Větrovský T, Baldrian P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One (2013) 8:e57923. doi:10.1371/journal.pone.0057923
1279. Das S, Dash HR, Mangwani N, Chakraborty J, Kumari S. Understanding molecular identification and polyphasic taxonomic approaches for genetic relatedness and phylogenetic relationships of microorganisms. J Microbiol Methods (2014) 103:80–100. doi:10.1016/j.mimet.2014.05.013
1280. Cody AJ, McCarthy ND, Jansen Van Rensburg M, Isinkaye T, Bentley SD, Parkhill J, et al. Real-time genomic epidemiological evaluation of human Campylobacter isolates by use of whole-genome multilocus sequence typing. J Clin Microbiol (2013) 51:2526–34. doi:10.1128/JCM.00066-13
1281. Lazar L, Rigó J Jr, Nagy B, Balogh K, Makó V, Cervenak L, et al. Relationship of circulating cell-free DNA levels to cell-free fetal DNA levels, clinical characteristics and laboratory parameters in preeclampsia. BMC Med Genet (2009) 10:120. doi:10.1186/1471-2350-10-120
1282. Galbiati S, Causarano V, Pinzani P, Francesca S, Orlando C, Smid M, et al. Evaluation of a panel of circulating DNA, RNA and protein potential markers for pathologies of pregnancy. Clin Chem Lab Med (2010) 48:791–4. doi:10.1515/CCLM.2010.160
1283. Miranda ML, Macher HC, Munoz-Hernandez R, Vallejo-Vaz A, Moreno-Luna R, Villar J, et al. Role of circulating cell-free DNA levels in patients with severe preeclampsia and HELLP syndrome. Am J Hypertens (2013) 26:1377–80. doi:10.1093/ajh/hpt187
1284. Oudejans CBM. Maternal plasma RNA sequencing. Clin Biochem (2015) 48:942–7. doi:10.1016/j.clinbiochem.2015.03.004
1285. Hudecova I. Digital PCR analysis of circulating nucleic acids. Clin Biochem (2015) 48:948–56. doi:10.1016/j.clinbiochem.2015.03.015
1286. Coates ARM, Hu Y. New strategies for antibacterial drug design: targeting non-multiplying latent bacteria. Drugs R D (2006) 7:133–51. doi:10.2165/00126839-200607030-00001
1287. Hu Y, Shamaei-Tousi A, Liu Y, Coates A. A new approach for the discovery of antibiotics by targeting non-multiplying bacteria: a novel topical antibiotic for staphylococcal infections. PLoS One (2010) 5:e11818. doi:10.1371/journal.pone.0011818
1288. Coates AR, Halls G, Hu Y. Novel classes of antibiotics or more of the same? Br J Pharmacol (2011) 163:184–94. doi:10.1111/j.1476-5381.2011.01250.x
1289. Laxminarayan R, Duse A, Wattal C, Zaidi AK, Wertheim HF, Sumpradit N, et al. Antibiotic resistance – the need for global solutions. Lancet Infect Dis (2013) 13:1057–98. doi:10.1016/S1473-3099(13)70318-9
1290. Hu Y, Liu A, Ortega-Muro F, Alameda-Martin L, Mitchison D, Coates A. High-dose rifampicin kills persisters, shortens treatment duration, and reduces relapse rate in vitro and in vivo. Front Microbiol (2015) 6:641. doi:10.3389/fmicb.2015.00641
1291. Barber M, Tait RC, Scott J, Rumley A, Lowe GD, Stott DJ. Dementia in subjects with atrial fibrillation: hemostatic function and the role of anticoagulation. J Thromb Haemost (2004) 2:1873–8. doi:10.1111/j.1538-7836.2004.00993.x
1292. Murthy SB, Jawaid A, Qureshi SU, Schulz PE, Schulz PE. The apolipoprotein 2 allele in Alzheimer’s disease: suggestions for a judicious use of antiplatelet and anticoagulant medications. J Am Geriatr Soc (2009) 57:1124–5. doi:10.1111/j.1532-5415.2009.02284.x
1293. Ahn HJ, Glickman JF, Poon KL, Zamolodchikov D, Jno-Charles OC, Norris EH, et al. A novel abeta-fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer’s disease mice. J Exp Med (2014) 211:1049–62. doi:10.1084/jem.20131751
1294. Krishnan R, Tsubery H, Proschitsky MY, Asp E, Lulu M, Gilead S, et al. A bacteriophage capsid protein provides a general amyloid interaction motif (GAIM) that binds and remodels misfolded protein assemblies. J Mol Biol (2014) 426:2500–19. doi:10.1016/j.jmb.2014.04.015
1295. Ankarcrona M, Winblad B, Monteiro C, Fearns C, Powers ET, Johansson J, et al. Current and future treatment of amyloid diseases. J Intern Med (2016) 280:177–202. doi:10.1111/joim.12506
Keywords: preeclampsia, dormancy, infection, biomarkers, sepsis, coagulopathies, amyloidoses
Citation: Kell DB and Kenny LC (2016) A Dormant Microbial Component in the Development of Preeclampsia. Front. Med. 3:60. doi: 10.3389/fmed.2016.00060
Received: 27 June 2016; Accepted: 04 November 2016;
Published: 29 November 2016
Edited by:
Belinda Jim, Jacobi Medical Center, USAReviewed by:
Ana Claudia Zenclussen, Otto-von-Guericke University Magdeburg, GermanyZaleha Abdullah Mahdy, National University of Malaysia, Malaysia
Copyright: © 2016 Kell and Kenny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Douglas B. Kell, dbk@manchester.ac.uk