Key Triggers of Osteoclast-Related Diseases and Available Strategies for Targeted Therapies: A Review
 Haidi Bi
Haidi Bi Xing Chen2†
Xing Chen2†
 Xiaolong Yu
Xiaolong Yu Jun Xiao
Jun Xiao Xuqiang Liu
Xuqiang Liu Min Dai
Min Dai- 1Department of Orthopaedics, The First Affiliated Hospital of Nanchang University, Artificial Joints Engineering and Technology Research Center of Jiangxi Province, Nanchang, China
- 2Department of Orthopaedics, The People’s Hospital of Changxing County, Huzhou, China
Osteoclasts, the only cells with bone resorption functions in vivo, maintain the balance of bone metabolism by cooperating with osteoblasts, which are responsible for bone formation. Excessive activity of osteoclasts causes many diseases such as osteoporosis, periprosthetic osteolysis, bone tumors, and Paget’s disease. In contrast, osteopetrosis results from osteoclast deficiency. Available strategies for combating over-activated osteoclasts and the subsequently induced diseases can be categorized into three approaches: facilitating osteoclast apoptosis, inhibiting osteoclastogenesis, and impairing bone resorption. Bisphosphonates are representative molecules that function by triggering osteoclast apoptosis. New drugs, such as tumor necrosis factor and receptor activator of nuclear factor kappa-B ligand (RANKL) inhibitors (e.g., denosumab) have been developed for targeting the receptor activator of nuclear factor kappa-B /RANKL/osteoprotegerin system or CSF-1/CSF-1R axis, which play critical roles in osteoclast formation. Furthermore, vacuolar (H+)-ATPase inhibitors, cathepsin K inhibitors, and glucagon-like peptide 2 impair different stages of the bone resorption process. Recently, significant achievements have been made in this field. The aim of this review is to provide an updated summary of the current progress in research involving osteoclast-related diseases and of the development of targeted inhibitors of osteoclast formation.
Introduction
The bone tissue in humans is renewed and reconstructed continuously with a dynamic balance between osteoblastic bone formation and osteoclastic bone resorption. Osteoclasts, the only cells with bone resorption function in vivo, maintain the balance of bone metabolism by cooperating with osteoblasts that are responsible for bone formation (1). During the process of osteoclast maturation, two hematopoietic factors, macrophage colony-stimulating factor (M-CSF, also called CSF-1) and receptor activator of nuclear factor kappa-B ligand (RANKL), are required (2, 3). Osteoclast differentiation and activation research have focused on tumor necrosis factor (TNF) receptor and TNF-like proteins, such as receptor activator of nuclear factor kappa-B (RANK), RANKL, and osteoprotegerin (OPG). The binding of RANKL to its receptor RANK activates signaling pathways that ultimately lead to osteoclastogenesis; however, this process can be suppressed by OPG, which is a soluble “decoy receptor” for RANKL (4, 5).
Functional disorders of osteoclasts and osteoblasts, particularly those related to the excessive activity of osteoclasts, cause many bone and joint diseases (Table 1) (6, 7). For example, osteoporosis, which occurs in people aged 40 and over and is more commonly found in postmenopausal women, presents as an imbalance in bone resorption and bone formation due to excessive osteoclast activation (8, 9). Excessive activation of osteoclasts induced by released wear particles also leads to periprosthetic osteolysis after artificial joint arthroplasty (10–12). In rheumatoid arthritis (RA), subchondral bone destruction is attributed to excessive bone absorption by osteoclasts after the differentiation and maturation induced by proinflammatory cytokines released by the autoimmune system (13, 14). The mechanism of bone metastases and bone destruction found in cancer is also related to the direct activation of osteoclasts by RANKL, which is secreted by cancer cells (15, 16). In addition, high expression of RANK (the RANKL receptor) on the osteoclast surface is an important factor in Paget’s disease (17). Considering the important roles osteoclasts play in the pathology of the above diseases, agents that modulate aberrant osteoclast differentiation and resorption would be useful in the development of bone-protective therapies (Table 2). Currently, approved and anti-resorptive agents include bisphosphonates (BPs), selective estrogen receptor modulators (SERMs), and monoclonal antibodies against RANKL (e.g., denosumab). Though neutralizing excessive osteoclasts have been partially or mostly mitigated with current therapies, they are far from ideal and still face enormous challenges because of their unexpected adverse effects. The long-term usage of BPs is limited due to the occurrence of severe gastrointestinal reactions, mandible necrosis, and atypical femur fractures (18, 19). In addition, treatment with SERMs is associated with increased risks of stroke and cardiovascular events (20). Finally, while mandible necrosis has been rarely observed in denosumab clinical trials (6 cases among 4,450 patients) (21), its safety and efficacy requires further evaluation. Thus, the identification and development of novel anti-resorptive agents are urgently needed. In-depth studies on new therapeutic targets that inhibit osteoclast formation and bone resorption have made important contributions to treatment and are of great socioeconomic value. The aim of this review is to provide an updated summary of the progress in research involving osteoclast-related diseases and targeted inhibitors.
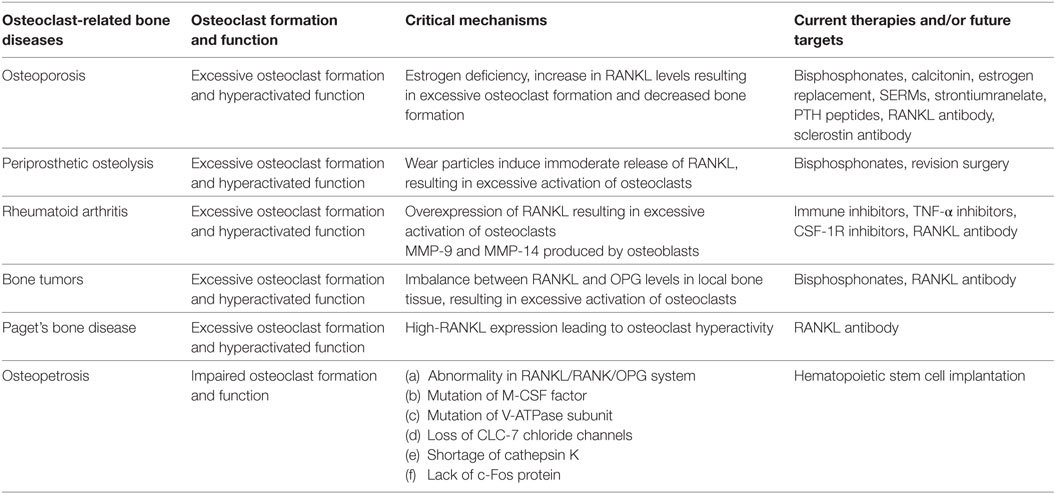
Table 1. Summary of osteoclast-related diseases and targeted inhibitors.
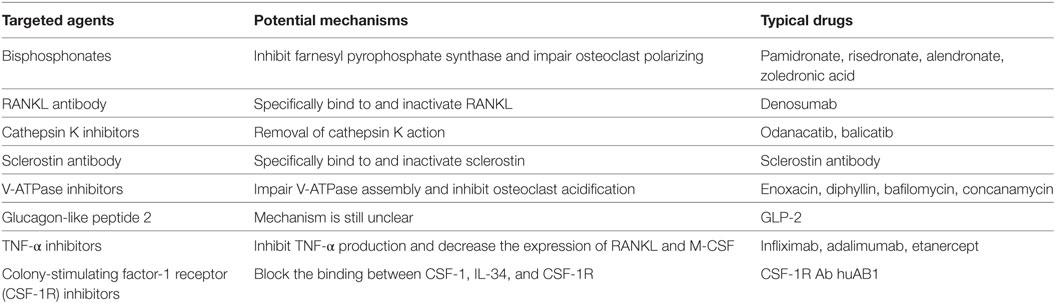
Table 2. Mechanisms of targeted inhibitors.
Ethics Statement
This review research was conducted according to the guiding principles of the Ethics Committee of Nanchang University.
Osteoporosis
Osteoporosis is a systemic skeletal disease characterized by a loss of bone mass and the destruction of bone microstructure, leading to fragility and fractures (22). It is considered to be a multifactorial disease potentially caused by genetic mutations, endocrine disorders, and nutritional deficiency. Hormones, such as estrogen, calcitonin, parathyroid hormone (PTH), and vitamin D, act to maintain the normal bone metabolism (23). PTH functions to improve the production of activated vitamin D and calcium absorption. In contrast, osteoclast activity can be accelerated through the stimulation of PTH, causing further bone resorption. Calcitonin exerts bone-protective effects by transferring calcium into bone tissues when binding to its receptor. In addition, estrogen deficiency reduces the rate of bone remodeling and increases osteoclast formation and resorption (24). Inhibition of the wingless-type and integrase 1 (Wnt) and bone morphogenetic protein signaling pathways, which play critical roles in regulating osteoblast formation, leads to decreased bone formation in postmenopausal women (22). In research on osteoclasts, the RANKL/RANK/OPG system represents an important discovery that has occurred in recent years. RANKL, produced by osteoblasts and bone matrix cells, is the key cytokine that stimulates osteoclast precursor cells to differentiate into mature osteoclasts (7, 25–27). It binds to RANK on the surfaces of osteoclast precursor cells and mature osteoclasts. Through this process, bone resorption is induced by the formation and differentiation of osteoclasts. As a decoy receptor that can block interactions between RANKL and RANK, OPG is also produced by osteoblasts and bone matrix cells. This RANKL/RANK/OPG system plays an important role in the occurrence of osteoporosis (28, 29). Estrogen deficiency is involved in the pathogenesis of osteoporosis in the elderly population, especially in postmenopausal women who are commonly found to have osteoporosis. In postmenopausal women, estrogen deficiency causes a decrease in OPG levels, which leads to an increase in RANKL levels; increased RANKL levels over-activate osteoclasts and result in loss of bone mass (22). These data indicate that the RANKL/RANK/OPG system represents a potential therapeutic target in the prevention and treatment of osteoporosis.
Periprosthetic Osteolysis
Arthroplasty is a reliable treatment used in cases of severe trauma, end-stage arthritis, and periarticular tumors. Knee and hip arthroplasties are being performed around the world at an increasing rate. However, the long-term use of artificial joints exhibits a major limitation in terms of periprosthetic osteolysis and the loosening induced by the wear particles released from the surface of the prosthesis (30, 31). Although the mechanism of wear particle-induced periprosthetic osteolysis is not clear, it is generally agreed that the excessive activation of osteoclasts caused by wear particles plays a critical role in this process (10–12). As foreign bodies, wear particles can stimulate monocytes/macrophages, fibroblasts, T lymphocytes, etc. to produce large amounts of inflammatory cytokines such as TNF-α, interleukin 1 (IL-1), IL-6, IL-11, IL-17, prostaglandin E2, and monocyte colony-stimulating factor (M-CSF). These inflammatory factors can induce local aseptic inflammation, but can also stimulate osteoblasts to express and release large amounts of RANKL, resulting in excessive activation of osteoclasts and periprosthetic osteolysis (32–34). This periprosthetic osteolysis induces loosening; in time, the instability caused by the loosening may further increase mechanical wear and produce more wear particles, resulting in more severe osteolysis. This creates a vicious cycle between periprosthetic osteolysis and loosening in this pathological process (35). Although the relative motions of the components of artificial joints and material corrosion and degradation in vivo during the use of prostheses will inevitably lead to the generation of wear particles, the effective inhibition of osteoclast formation, and bone resorption may be an effective way to prevent the loosening of prostheses and therefore extend their lives.
Rheumatoid Arthritis
Rheumatoid arthritis is a chronic systemic autoimmune disease characterized by progressive irreversible damage of bone and cartilage. Although the detailed mechanism of bone and cartilage destruction in RA has not yet been elucidated, the formation and increased activity of osteoclasts caused by an imbalance in the ratio of RANKL and OPG is considered to be the main factor responsible. Recent studies have revealed the presence of several mature osteoclasts and osteoclast precursor cells in localized lesions in RA. The overexpression of RANKL by active lymphocytes, macrophages, osteoblasts, etc. leads to excessive proliferation and abnormal activation of osteoclasts caused by the binding of RANKL to RANK on the surface of osteoclast precursor cells and mature osteoclasts. In addition to the overexpression of RANKL in damaged joint bone tissue, RANKL mRNA is also expressed by fibroblasts in the synovial tissue, which leads to the production of the RANKL protein (36). Kotake et al. isolated multinucleated cells from the synovial lesions of RA patients and showed that they could form bone absorption pits, thus confirming them to be osteoclasts (36). The formation of bone pits can be inhibited by OPG, and the number of pits formed is closely related to the ratio of RANKL and OPG at the mRNA level. Therefore, quantitative analysis of the RANKL/OPG levels in the synovial tissue and synovial fluid may contribute to the early diagnosis of RA. Moreover, MMP-9 and MMP-14 produced by osteoblasts are also important factors that lead to the degradation of the cartilage matrix, pannus formation, and migration of osteoclasts to the bone surface. All of these factors contribute to the erosion of the articular cartilage, subchondral bone, and synovial surface in RA, where osteoclasts play a key role.
Bone Tumors
Primary or secondary tumors are commonly found in orthopedics, but the success of clinical therapy for such tumors is limited due to the characteristics of invasion, metastasis, and recurrence. In-depth studies in recent years have shown that the RANKL/RANK/OPG system affects tumor biology by regulating osteoclast activity (37–39), imbalances in RANKL and OPG levels in local bone tissues are the main reason for increases in osteoclast bone resorption (40, 41). A previous study showed that the expression levels of OPG and RANKL mRNA in giant cell tumors of the bone are much higher than those in normal bone tissues (42, 43). Sezer et al. also studied the expression of RANKL and RANK in biopsy specimens of multiple myeloma (44). Data from the study by Sezer et al. also revealed lower serum OPG levels in multiple myeloma patients compared with those in healthy humans and similar patients without bone destruction (44). Although there is sufficient evidence indicating the effect of the RANKL/RANK/OPG system in bone metastases, the mechanism of metastasis is not entirely clear. However, abnormal osteoclast activation, which is caused by an imbalance in RANKL and OPG levels, is considered to be responsible for most tumors.
Paget’s Bone Disease
Paget’s disease of the bone is a metabolic bone disease accompanied by increased bone resorption and abnormal bone formation. This results in an increased risk of fracture caused by structural disorder, leading to a decrease in the mechanical properties of the bone (45, 46). Some studies have indicated that high-RANKL expression leading to osteoclast hyperactivity is an important factor in Paget’s disease (47, 48). Roodman (49) and Roodman and Windle (50) also showed that the number of osteoclasts in patients with Paget’s bone disease is increased, the osteoclasts are larger, and the number of nuclei is hundreds of times higher than that in normal cultures. In addition, whether the point of origin of the disease is the bone marrow or peripheral blood, mononuclear cells always exhibit a high degree of sensitivity to RANKL, and differentiation to mature osteoclasts seems to be increased (47).
Osteopetrosis
Osteopetrosis is a metabolic bone disease characterized by increased bone mass caused by polygenic disorders. Disorders in osteoclast formation and loss of osteoclast function are the main reasons for decreased bone resorption and increased bone mass. Recent studies have suggested that decreased bone resorption could be caused by abnormalities in the RANKL/RANK/OPG system, lack of c-Fos protein, and mutations in M-CSF, while mutations in the vacuolar (H+)-ATPase (V-ATPase) subunit, loss of CLC-7 chloride channels, and a shortage of cathepsin K are the most common reasons for osteopetrosis caused by bone resorption disorders. Bone marrow transplantation and the subsequent differentiation of hematopoietic stem cells from the implanted new bone marrow into mature and functioning osteoclasts is a treatment option for osteopetrosis.
Targeted Osteoclastic Inhibitors
There is a wide spectrum of diseases induced by osteoclast dysfunction, and excessive activation of osteoclasts plays a dominant role in most of these diseases. Therapies to inhibit osteoclast formation and bone resorption efficiently and safely are ideal approaches to combat such diseases. Frequent and long-term clinical use of BPs to reduce osteoclast formation is associated with serious complications including gastrointestinal reactions, mandible necrosis, and non-specific femur fractures (51–53). Monoclonal antibodies against RANKL, such as denosumab, are a new class of drugs used for the targeted inhibition of osteoclast formation. These act by blocking the RANK/RANKL/OPG regulatory system, and this has been a major discovery in the field of osteoblast research in recent years. Glucagon-like peptide 2 (GLP-2), cathepsin K, and V-ATPase inhibitors are also expected to be of use in inhibiting osteoclast formation, and other measures such as anti-TNF-α therapy can also be used.
Bisphosphonates
Bisphosphonates, such as alendronate and zoledronic acid, are anti-bone resorption drugs commonly used as a therapeutic choice for bone diseases including Paget’s disease of the bone and myeloma. Their ability to inhibit osteoclast resorption is the desired pharmacological effect. McClung (54) and Russell et al. (55) showed that BPs could effectively inhibit bone resorption by binding to hydroxyapatite (HAP) crystals, which results in blocking the prenylation process of proteins due to the inhibition of farnesyl pyrophosphate synthase. When prenylation is blocked, the osteoclast cytoskeleton cannot be rearranged and polarized as an enclosed area for adhering to the bone surface cannot be formed. Thus, although BPs have been confirmed to show an inhibitory effect on osteoclast resorption, this desired clinical effect is often limited by the above-mentioned complications. Due to the high affinity of BPs to HAP crystals in the bone matrix, novel bone-targeting agents have been synthesized based on the molecular structure of BPs. Toro et al. and Rivera et al. demonstrated that BP-enoxacin, a BP derivative, had an inhibitory effect on osteoclast formation and bone resorption and represented an ideal therapeutic agent for preventing orthodontic tooth movement (56, 57). Furthermore, our previous study showed a beneficial effect of BP-enoxacin on cortical bone mass and strength in ovariectomized rats (58). We speculate that it would be an exciting and insightful approach to explore bone-targeting agents that are BP “carriers.”
CSF-1/CSF-1R Axis Inhibitors
The binding of M-CSF (also called CSF-1) to its tyrosine kinase receptor (CSF-1R) promotes the differentiation of myeloid progenitors into monocytes, macrophages, dendritic cells, and osteoclasts. In vivo, circulating CSF-1 regulates the migration, proliferation, and survival of macrophages, which is beneficial to the innate and adaptive immune system, as well as osteoclastogenesis at multiple levels (59). Theoretically, targeting of the CSF-1/CSF-1R axis to modulate macrophage populations may result in potential therapeutic effects in four types of clinical diseases: inflammatory diseases, cancer, autoimmunity, and bone diseases (60). Antibodies against CSF-1 and its receptor, as well as specific inhibitors of CSF-1R kinase, have been evaluated either in animal models or in patients. As reported, the in vivo administration of CSF-1 exacerbated the inflammation and joint erosion in collagen-induced arthritis (61, 62) due to the role of CSF-1 in the pathology of osteoclastogenesis and subsequent osteolysis. Thus, anti-CSF-1 antibody or blockade of CSF-1R reduces inflammation in humans and in RA models (63, 64). According to the research of Cenci et al. (65), CSF-1 facilitates the process of bone loss in ovariectomized mice. In contrast, Gow et al. (66) observed an osteopetrosis phenotype in CSF-1-deficient animals due to the deficient production of bone-resorbing osteoclasts. Therefore, it is expected that anti-CSF-1 may be beneficial in treating human osteoporosis. Furthermore, it was demonstrated by Rietkotter et al. that anti-CSF-1 therapy was beneficial in preventing carcinoma invasion induced by monocyte-derived cells (67). Although an increasing number of studies have indicated the significant role of CSF-1 in osteoclastogenesis and the efficacy of anti-CSF-1/CSF-1R therapy in treating osteoclast-related diseases, further investigations to determine the safety and side effects of these methods still need to be conducted.
Interleukin-34 was first discovered to be a second ligand of CSF-1R in 2008 (68). It was reported that CSF-1 and IL-34 share structural homology and have largely overlapping effects in regulating monocyte survival and osteoclastogenesis (69, 70). Previous evidence had demonstrated that IL-34, both from giant cell tumors and gingival fibroblasts, plays a critical role in RANKL-induced osteoclast formation as a complete substitute for CSF-1 and that the systemic administration of IL-34 would result in a decrease in trabecular bone mass (71, 72). Cheng et al. (73) confirmed this opinion and demonstrated that IL-34 promotes the proliferation and differentiation of bone marrow macrophages by stimulating p-STAT3 expression, as well as inhibiting the expression of Smad7 in the absence of CSF-1. Regretfully, IL-34 has not been clinically tested for the potential existence of other receptors and adverse pathologies mediated by over-activated macrophages.
Anti-RANKL Monoclonal Antibody (e.g., Denosumab)
The RANKL/RANK/OPG axis is the key regulatory system that decides whether differentiation occurs. RANKL, a member of the TNF superfamily, is produced and secreted by osteoblasts, bone stromal cells, fibroblasts, and activated T cells. The interaction between RANKL and RANK (surface receptors on osteoclast precursor cells) promotes osteoclast differentiation and maturation and helps osteoclasts survive (74, 75). As a pseudo-receptor of RANKL, OPG can also inhibit osteoclast formation and accelerates apoptosis by binding to RANKL, which inhibits the interaction between RANKL and RANK. RANKL is therefore treated as an ideal target for inhibiting osteoclast formation based on the information obtained so far regarding the RANKL/RANK/OPG system. Denosumab, a synthetic IgG2 monoclonal antibody, can also specifically bind to and inactivate RANKL using the same action mechanism as OPG. In 2010, denosumab was approved for use in treating postmenopausal osteoporosis. A phase III trial, conducted over 3 years, has indicated that the incidence of hip and vertebral fracture decreased by 41 and 68% after administration of 60 mg of denosumab every 6 months (76). To evaluate the long-term efficacy and safety of denosumab use for up to 10 years, participants from the Fracture Reduction Evaluation of Denosumab in Osteoporosis every 6 Months (FREEDOM) trial were asked to join the 7-year FREEDOM Extension trial (clinicaltrials.gov: NCT00523341). This trial reported a sustained reduction in bone turnover markers and progressive increase in bone mineral density in the long-term denosumab treatment group, resulting in the maintenance of low-fracture rates (21, 77–79). However, several side effects of denosumab were also observed in this extension trial, including malignancy, eczema/dermatitis, pancreatitis, endocarditis, delayed fracture healing, and infections, especially the occasional occurrence of opportunistic infections, and these should be taken into serious consideration (79).
Anti-Sclerostin Monoclonal Antibody
Sclerostin is a small protein encoded by the SOST gene and produced in osteocytes. It responds to mechanical stress and targets the Wnt signaling cascade. When activated, sclerostin acts as a key negative regulator of bone anabolic metabolism and exhibits an inhibitory effect on osteoblast differentiation and bone formation (80, 81). Patients or gene mutation mice with consistently low levels of sclerostin due to rare skeletal disorders exhibit high-bone mineral density and low-fracture risk (81). Thus, anti-sclerostin therapy could potentially be used to treating bone metabolism diseases resulting in low-bone mass. Recently, humanized anti-sclerostin antibodies, such as romosozumab (AMG785), blosozumab (AMG167), and BSP804, have been synthesized and subjected to clinical trials. Increased bone mass at the spine and hip, along with modified bone turnover markers (increased bone formation markers and decreased resorption markers) have been observed in romosozumab and blosozumab clinical trials (82, 83). Following these trials, research has been conducted to clarify whether the increased bone mineral density resulted in an improvement in bone mechanical properties. Finite element analysis was employed to assess the strength of the spine (L1 vertebral body) and proximal femur under a simulated compression overload. Results showed that both spinal and femoral strength had increased from the baseline (27.3 versus −3.9%, P < 0.0001 and 3.6 versus −0.1%, P = 0.059, respectively) and that gains in bone mechanical properties corresponded to gains in bone mineral density (84). Additionally, research conducted on animal models or under other conditions indicated a potentially wide therapeutic application of anti-sclerostin antibodies in treating other bone and joint diseases, such as RA, osteoarthritis, and bone complications of type 2 diabetes mellitus (85). The most common side effects included elevated liver enzymes and injection site reactions. However, hypotheses regarding the relationship between sclerostin and cardiovascular events, intracranial pressure, and some tumors require further investigation (23).
Cathepsin K Inhibitors
Cathepsin K, which is specifically expressed and secreted by activated osteoclasts during bone resorption, is a key enzyme in the degradation of critical proteins in the bone matrix, including type I collagens (86). Bone resorption can be inhibited by the removal of cathepsin K from osteoclasts. Unlike other anti-resorptive drugs, cathepsin K inhibitors do not affect osteoclast activity, and osteogenic activity is maintained by the cross-coupling of osteoblasts to osteoclasts (87). Balicatib has been artificially synthesized as a specific cathepsin K inhibitor. However, phase II clinical trials for balicatib were discontinued because of morphea-like skin changes observed in the participants (88). Another new cathepsin K inhibitor, odanacatib, is orally selective (89). According to a 2-year randomized controlled study, 3.2 and 5.5% increases were observed in the BMD of the hip and spine, respectively, after odanacatib therapy (90). Recent research has indicated that the beneficial effect of odanacatib in improving bone mineral density is dose-dependent and persists for up to 5 years. Along with increased bone mineral density, the risk of fragility fracture was reduced, and the effect was similar to that of BPs and denosumab (91). Aside from its therapeutic efficacy, however, the safety of odanacatib should be studied and seriously evaluated. According to reported investigations, increased risks of stroke, arterial fibrillation, and atypical fractures were observed during the treatment procedures, although these types of agents are still under development (92).
V-ATPase Inhibitors
During the process of bone matrix degradation, an acidic microenvironment, which is created by V-ATPase that pump protons into the resorption lacuna, is necessary for osteoclast bone resorption. It has been shown that therapeutic interventions that involve modulating osteoclast V-ATPase activity would be reasonable for the treatment of osteoporosis and other osteolytic diseases (93). V-ATPases are protein complexes composed of at least 14 different protein subunits and are responsible for the active transmembrane transport of hydrogen ions in vivo. V-ATPases are organized into V1 and V0 domains, which have two different functions. The V1 domain is composed of eight different subunits (A–H), is located in the cytoplasm, and generates energy by ATP hydrolysis. The V0 domain contains six different subunits (a, c, c′, c′′, d, and e) and is involved in the active transmembrane transport of hydrogen ions (94, 95). In addition, the Ac45 and M8-9 auxiliary subunits found in mammal V-ATPases have a collaborative effect in facilitating hydrogen ion transport (96, 97). Some studies have shown that the dysfunction of V-ATPases may lead to the occurrence of many diseases such as osteopetrosis and tumor bone metastasis (98). V-ATPases are thus considered to be a potential target in the treatment of such osteoclast-hyperactive diseases such as osteoporosis and bone tumor metastasis. In 2002, two research groups reported that bafilomycin and concanamycin can inhibit the acidification of V-ATPases by affecting the c protein subunit in the V0 domain (99, 100), whereas diphyllin, as a new V-ATPase inhibitor, inhibits osteoclast resorption and apoptosis (101). In recent years, enoxacin has also been confirmed as a type of V-ATPase inhibitor that acts by blocking the binding of the B2 subunit to actin microfilaments, thus destroying suture zone formation in osteoclasts and inhibiting osteoclast acidification (102). Although there are many V-ATPase inhibitors, none has been subjected to a clinical trial. Thus, the mechanisms, targets, efficacy, and safety of these drugs in vivo remain to be studied in future research and feasibility studies.
Glucagon-Like Peptide 2
Glucagon-like peptide 2, whose secretion has a clear circadian rhythm and is regulated by diet, is a peptide hormone produced by intestinal mucosal cells (103). Henriksen et al. (104) found that bone remodeling also shows a circadian rhythm with a close relationship to food intake and eating time. Osteocalcin is a marker that is closely related to bone formation during the treatment of osteoporosis. Another study by Henriksen et al. (105) indicated that GLP-2 has no influence on bone formation due to the increase in bone resorption and the stable expression of osteocalcin, which occurs after GLP-2 treatment before sleeping. Although some studies have shown that GLP-2 can inhibit bone resorption and increase bone density, its mechanism is unclear, particularly with regard to its influence on the biological function of osteoclasts, which is still poorly understood.
TNF-α Inhibitors
The release of cytokines is closely related to RA and other bone destructive diseases. TNF-α is stimulated by activated T cells, macrophages, and synovial cells under inflammatory conditions and is the most critical inflammatory cytokine, causing excessive activation of osteoclasts (106, 107). The expression of TNF-α has several effects on osteoclastogenesis. RANKL is secreted is large quantities by osteoblasts and bone stromal cells (108), while the expression of RANK on the surface of osteoclast precursor cells increases and the sensitivity of RANK to RANKL is enhanced due to the recruitment of osteoclasts (109). Furthermore, TNF-α can stimulate the expression of another cytokine, M-CSF, which maintains the continuous formation and survival of osteoclasts (110). Based on the above-mentioned roles in bone destructive diseases, TNF-α therefore represents a candidate therapeutic target. Currently, biological targeted therapy using cytokines can be roughly divided into two categories: monoclonal antibodies and soluble receptors, each with different mechanisms. Monoclonal antibodies can be used as cytokines or cytokine receptors, while soluble receptors can pre-capture and inactivate cytokines before the connection between cytokines and cytokine receptors is established. Infliximab, adalimumab, and etanercept, representing TNF-α antagonists, have been investigated in clinical trials for use in the treatment of RA (111, 112).
Conclusion
Osteoclasts are responsible for the necessary function of bone resorption in vivo, but the clinical treatment of many diseases caused by osteoclast dysfunction, in particular by excessive activation of osteoclasts, faces enormous challenges. The development of new targeted drugs designed to inhibit osteoclast formation is urgently needed for clinical treatment. According to research on new drugs to inhibit osteoclast formation, the RANKL/RANK/OPG system, CSF-1/CSF-1R axis, cathepsin K, sclerostin, V-ATPases, and the cytokine TNF-α are currently considered as potential critical targets (Figure 1). The broad application prospects of denosumab, anti-sclerostin antibodies, cathepsin K inhibitors, and TNF-α inhibitors are currently being studied in phase II and phase III clinical trials. V-ATPases provide a theoretical advantage due to their important roles in osteoclast acidification and bone matrix degradation and the fact that they have been found to have no effect on osteogenic activity. However, although several V-ATPase inhibitors have been identified, no mature inhibitor has entered clinical trials because of limitations such as the associated toxicity, an unclear mechanism of action, and the lack of credible animal research models. GLP-2, a polypeptide produced by the body, has no physiological side effects and can regulate the circadian rhythm of bone remodeling and shorten bone resorption time, which indirectly prolongs osteogenic time. However, few studies have been conducted on the relationship between GLP-2 and bone metabolism, and the mechanism of action of GLP-2 remains unclear; therefore, its conversion and clinical application requires further research and feasibility studies.
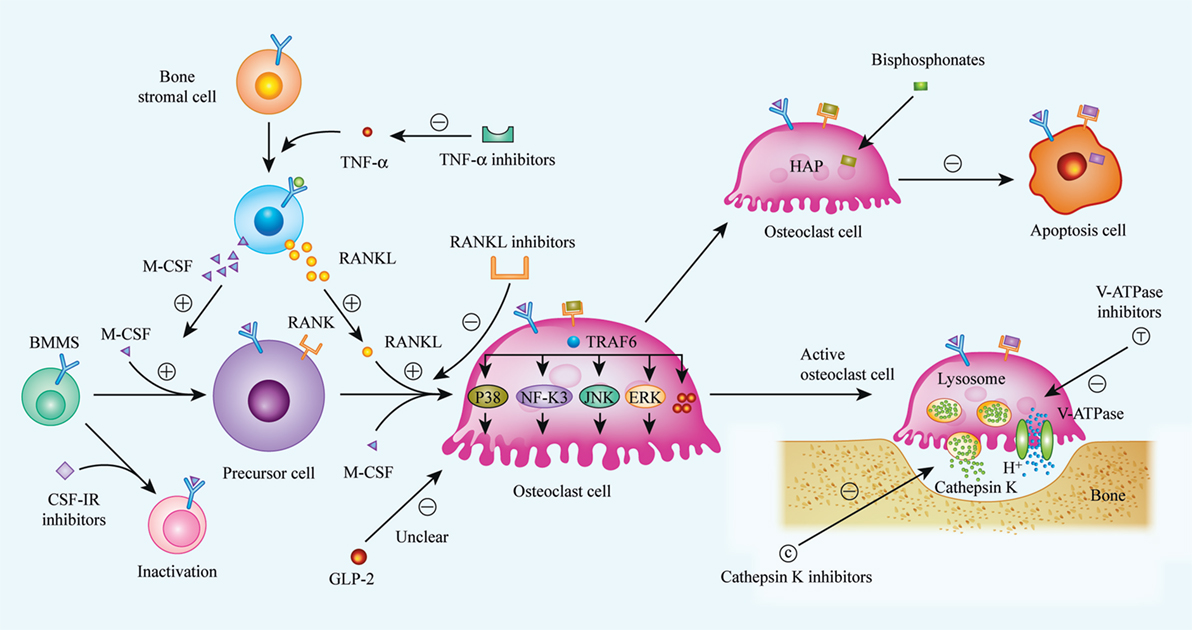
Figure 1. Biological procedures of osteoclast differentiation, bone resorption, and mechanisms of current or future therapeutic drugs. Osteoclasts matured from bone marrow hematopoietic stem cells (BMMs) with the stimulation of two critical factors, M-CSF (CSF-1) and receptor activator of nuclear factor kappa-B ligand (RANKL). When binding to its specific receptors [CSF-1R and receptor activator of nuclear factor kappa-B (RANK)] on BMMs membrane, a series of cascades are activated, and BMMs were then differentiated into matured osteoclast. Realizing the importance of M-CSF and RANKL in osteoclast differentiation, inhibitors to CSF-1R and RANKL were considered as available strategy to suppress over-activated osteoclasts. Bisphosphonates, a widely used anti-osteoporosis agent, can be absorbed by osteoclast and induce osteoclast apoptosis. Additionally, it has been indicated that GLP-2 is a negative regulator of osteoclast differentiation, thus, the exact mechanisms are still unclear. Bone resorption is demonstrated as specific function of osteoclast, and bone matrix degradation is induced by the release of cathepsin K, as well as H+, and the release of H+ is enabled by V-ATPase on the membrane of matured osteoclast. So that, cathepsin K and V-ATPase are considered as another two targets to impair osteoclast function, especially, inhibitors of cathepsin K, such as Odanacatib, Balicatib are undergoing clinical trials. (⊝ represents inhibitory or down-regulated effect, ⊕ represents facilitated or up-regulated effect).
Taken together, we propose that future research would be best served by focusing on two aspects. First, more studies should be conducted to explore and clarify the underlying mechanisms of each disease. Although, it has been reported that osteoclasts are the key factor triggering osteolytic diseases, the influence of osteoblast formation and activity should also be considered, as cross-talk between osteoclasts and osteoblasts exists in bone metabolic processes. Second, randomized, multicenter, controlled, and long-term studies are urgently needed to confirm the safety and efficacy of newly developed pharmacological agents. Anti-sclerostin antibody, which is associated with bone formation, increased bone mineral density, and suppressed bone resorption, is distinct from other anabolic agents and shows the potential for widespread application in treating diseases associated with aberrant bone metabolism. In addition, V-ATPases consist of many subunits, most of which are considered suitable targets for the development of novel agents, and the modulation of V-ATPases theoretically should not affect the formation of osteoclasts. Possibly, it would be the original source in drug discovery. Osteoporosis is the most prevalent bone metabolism disease, especially among postmenopausal women. Currently, developed new anti-osteoporosis drugs, such as denosumab, are clearly able to modify osteoporotic bone quality, enhance bone mechanical properties, and subsequently reduce the risk of fragility fractures due to accelerated bone turnover. There are high expectations for these newly discovered drugs, although most are still under development or awaiting approval.
Author Contributions
XL, MD, and BZ designed most of this review. HB, XC, SG, XL, and JX were primarily responsible for researching and the subsequent review of the research. XL and HB wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research was supported by grants from National Natural Science Foundation of China (no. 81660365) and National Natural Science Foundation for Youths (no. 81601912).
References
1. Pi C, Li YP, Zhou X, Gao B. The expression and function of microRNAs in bone homeostasis. Front Biosci (Landmark Ed) (2015) 20:119–38. doi:10.2741/4301
2. Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell (1998) 93(2):165–76. doi:10.1016/S0092-8674(00)81569-X
3. Yao GQ, Sun BH, Weir EC, Insogna KL. A role for cell-surface CSF-1 in osteoblast-mediated osteoclastogenesis. Calcif Tissue Int (2002) 70(4):339–46. doi:10.1007/s00223-001-1079-x
4. Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell (1997) 89(2):309–19. doi:10.1016/S0092-8674(00)80209-3
5. Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology (2001) 142(12):5050–5. doi:10.1210/endo.142.12.8536
6. Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science (2000) 289(5484):1508–14. doi:10.1126/science.289.5484.1508
7. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature (2003) 423:337–42. doi:10.1038/nature01658
8. Tang P, Xiong Q, Ge W, Zhang L. The role of microRNAs in osteoclasts and osteoporosis. RNA Biol (2014) 11(11):1355–63. doi:10.1080/15476286.2014.996462
9. Kwan P. Osteoporosis: from osteoscience to neuroscience and beyond. Mech Ageing Dev (2015) 145C:26–38. doi:10.1016/j.mad.2015.02.001
10. Ren W, Wu B, Peng X, Hua J, Hao HN, Wooley PH. Implant wear induces inflammation, but not osteoclastic bone resorption, in RANK(-/-) mice. J Orthop Res (2006) 24(8):1575–86. doi:10.1002/jor.20190
11. Purdue PE, Koulouvaris P, Potter HG, Nestor BJ, Sculco TP. The cellular and molecular biology of periprosthetic osteolysis. Clin Orthop Relat Res (2007) 454:251–61. doi:10.1097/01.blo.0000238813.95035.1b
12. Gallo J, Goodman SB, Konttinen YT, Wimmer MA, Holinka M. Osteolysis around total knee arthroplasty: a review of pathogenetic mechanisms. Acta Biomater (2013) 9(9):8046–58. doi:10.1016/j.actbio.2013.05.005
13. Romas E, Bakharevski O, Hards DK, Kartsogiannis V, Quinn JM, Ryan PF, et al. Expression of osteoclast differentiation factor at sites of bone erosion in collagen-induced arthritis. Arthritis Rheum (2000) 43(4):821–6. doi:10.1002/1529-0131(200004)43:4<821::AID-ANR12>3.0.CO;2-T
14. Jung SM, Kim KW, Yang CW, Park SH, Ju JH. Cytokine-mediated bone destruction in rheumatoid arthritis. J Immunol Res (2014) 2014:263625. doi:10.1155/2014/263625
15. Chu GC, Chung LW. RANK-mediated signaling network and cancer metastasis. Cancer Metastasis Rev (2014) 33(2–3):497–509. doi:10.1007/s10555-013-9488-7
16. Nagy V, Penninger JM. The RANKL-RANK story. Gerontology (2015). 16(6):534–42. doi:10.1159/000371845
17. Kurihara N, Reddy SV, Menaa C, Anderson D, Roodman GD. Osteoclasts expressing the measles virus nucleocapsid gene display a pagetic phenotype. J Clin Invest (2000) 105(5):607–14. doi:10.1172/JCI8489
18. Khosla S, Hofbauer LC. Osteoporosis treatment: recent developments and ongoing challenges. The lancet Diabetes & endocrinology (2017) 5(11):898–907. doi:10.1016/S2213-8587(17)30188-2
19. Loyson T, Van Cann T, Schoffski P, Clement PM, Bechter O, Spriet I, et al. Incidence of osteonecrosis of the jaw in patients with bone metastases treated sequentially with bisphosphonates and denosumab. Acta Clin Belg (2017) 1–10. doi:10.1080/17843286.2017.1348001
20. Barrett-Connor E, Mosca L, Collins P, Geiger MJ, Grady D, Kornitzer M, et al. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med (2006) 355(2):125–37. doi:10.1056/NEJMoa062462
21. Bone HG, Chapurlat R, Brandi ML, Brown JP, Czerwinski E, Krieg MA, et al. The effect of three or six years of denosumab exposure in women with postmenopausal osteoporosis: results from the FREEDOM extension. J Clin Endocrinol Metab (2013) 98(11):4483–92. doi:10.1210/jc.2013-1597
22. Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest (2005) 115(12):3318–25. doi:10.1172/JCI27071
23. Tabatabaei-Malazy O, Salari P, Khashayar P, Larijani B. New horizons in treatment of osteoporosis. Daru (2017) 25(1):2. doi:10.1186/s40199-017-0167-z
24. Crockett JC, Rogers MJ, Coxon FP, Hocking LJ, Helfrich MH. Bone remodelling at a glance. J Cell Sci (2011) 124(Pt 7):991–8. doi:10.1242/jcs.063032
25. Long CL, Humphrey MB. Osteoimmunology: the expanding role of immunoreceptors in osteoclasts and bone remodeling. Bonekey Rep (2012) 1:59. doi:10.1038/bonekey.2012.59
26. Nakashima T, Hayashi M, Takayanagi H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol Metab (2012) 23(11):582–90. doi:10.1016/j.tem.2012.05.005
27. Xiong J, O’Brien CA. Osteocyte RANKL: new insights into the control of bone remodeling. J Bone Miner Res (2012) 27(3):499–505. doi:10.1002/jbmr.1547
28. Boyce BF, Xing L. The RANKL/RANK/OPG pathway. Curr Osteoporos Rep (2007) 5(3):98–104. doi:10.1007/s11914-007-0024-y
29. Liu W, Zhang X. Receptor activator of nuclear factor-kappaB ligand (RANKL)/RANK/osteoprotegerin system in bone and other tissues (Review). Mol Med Rep (2015) 11(5):3212–8. doi:10.3892/mmr.2015.3152
30. Harris WH. Wear and periprosthetic osteolysis: the problem. Clin Orthop Relat Res (2001) 393:66–70. doi:10.1097/00003086-200112000-00007
31. Teeny SM, York SC, Mesko JW, Rea RE. Long-term follow-up care recommendations after total hip and knee arthroplasty: results of the American Association of Hip and Knee Surgeons’ member survey. J Arthroplasty (2003) 18(8):954–62. doi:10.1016/j.arth.2003.09.001
32. Merkel KD, Erdmann JM, McHugh KP, Abu-Amer Y, Ross FP, Teitelbaum SL. Tumor necrosis factor-alpha mediates orthopedic implant osteolysis. Am J Pathol (1999) 154(1):203–10. doi:10.1016/S0002-9440(10)65266-2
33. Pioletti DP, Kottelat A. The influence of wear particles in the expression of osteoclastogenesis factors by osteoblasts. Biomaterials (2004) 25(27):5803–8. doi:10.1016/j.biomaterials.2004.01.053
34. Holding CA, Findlay DM, Stamenkov R, Neale SD, Lucas H, Dharmapatni AS, et al. The correlation of RANK, RANKL and TNFalpha expression with bone loss volume and polyethylene wear debris around hip implants. Biomaterials (2006) 27(30):5212–9. doi:10.1016/j.biomaterials.2006.05.054
35. Lin TH, Tamaki Y, Pajarinen J, Waters HA, Woo DK, Yao Z, et al. Chronic inflammation in biomaterial-induced periprosthetic osteolysis: NF-kappaB as a therapeutic target. Acta Biomater (2014) 10(1):1–10. doi:10.1016/j.actbio.2013.09.034
36. Kotake S, Udagawa N, Hakoda M, Mogi M, Yano K, Tsuda E, et al. Activated human T cells directly induce osteoclastogenesis from human monocytes: possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum (2001) 44(5):1003–12. doi:10.1002/1529-0131(200105)44:5<1003::AID-ANR179>3.0.CO;2-#
37. Chen G, Sircar K, Aprikian A, Potti A, Goltzman D, Rabbani SA. Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer (2006) 107(2):289–98. doi:10.1002/cncr.21978
38. Dougall WC. Molecular pathways: osteoclast-dependent and osteoclast-independent roles of the RANKL/RANK/OPG pathway in tumorigenesis and metastasis. Clin Cancer Res (2012) 18(2):326–35. doi:10.1158/1078-0432.CCR-10-2507
39. Peng X, Guo W, Ren T, Lou Z, Lu X, Zhang S, et al. Differential expression of the RANKL/RANK/OPG system is associated with bone metastasis in human non-small cell lung cancer. PLoS One (2013) 8(3):e58361. doi:10.1371/journal.pone.0058361
40. Kobayashi Y, Udagawa N, Takahashi N. Action of RANKL and OPG for osteoclastogenesis. Crit Rev Eukaryot Gene Expr (2009) 19(1):61–72. doi:10.1615/CritRevEukarGeneExpr.v19.i1.30
41. Jules J, Ashley JW, Feng X. Selective targeting of RANK signaling pathways as new therapeutic strategies for osteoporosis. Expert Opin Ther Targets (2010) 14(9):923–34. doi:10.1517/14728222.2010.511179
42. Atkins GJ, Kostakis P, Pan B, Farrugia A, Gronthos S, Evdokiou A, et al. RANKL expression is related to the differentiation state of human osteoblasts. J Bone Miner Res (2003) 18(6):1088–98. doi:10.1359/jbmr.2003.18.6.1088
43. Atkins GJ, Kostakis P, Vincent C, Farrugia AN, Houchins JP, Findlay DM, et al. RANK expression as a cell surface marker of human osteoclast precursors in peripheral blood, bone marrow, and giant cell tumors of bone. J Bone Miner Res (2006) 21(9):1339–49. doi:10.1359/jbmr.060604
44. Sezer O, Heider U, Jakob C, Zavrski I, Eucker J, Possinger K, et al. Immunocytochemistry reveals RANKL expression of myeloma cells. Blood (2002) 99(12):4646–7. doi:10.1182/blood-2002-01-0148
45. Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans (1877) 60(37–64):39.
46. Selby PL, Davie MW, Ralston SH, Stone MD, Bone and Tooth Society of Great Britain; National Association for the Relief of Paget’s Disease. Guidelines on the management of Paget’s disease of bone. Bone (2002) 31(3):366–73. doi:10.1016/S8756-3282(02)00817-7
47. Menaa C, Reddy SV, Kurihara N, Maeda H, Anderson D, Cundy T, et al. Enhanced RANK ligand expression and responsivity of bone marrow cells in Paget’s disease of bone. J Clin Invest (2000) 105(12):1833–8. doi:10.1172/JCI9133
48. Sun SG, Lau YS, Itonaga I, Sabokbar A, Athanasou NA. Bone stromal cells in pagetic bone and Paget’s sarcoma express RANKL and support human osteoclast formation. J Pathol (2006) 209(1):114–20. doi:10.1002/path.1953
49. Roodman GD. Mechanisms of abnormal bone turnover in Paget’s disease. Bone (1999) 24(5 Suppl):39S–40S. doi:10.1016/S8756-3282(99)00045-9
50. Roodman GD, Windle JJ. Paget disease of bone. J Clin Invest (2005) 115(2):200–8. doi:10.1172/JCI24281
51. Hagen JE, Miller AN, Ott SM, Gardner M, Morshed S, Jeray K, et al. Association of atypical femoral fractures with bisphosphonate use by patients with varus hip geometry. J Bone Joint Surg Am (2014) 96(22):1905–9. doi:10.2106/JBJS.N.00075
52. Kumar V, Shahi AK. Nitrogen containing bisphosphonates associated osteonecrosis of the jaws: a review for past 10 year literature. Dent Res J (2014) 11(2):147–53.
53. Parretta E, Sottosanti L, Sportiello L, Rafaniello C, Potenza S, D’Amato S, et al. Bisphosphonate-related osteonecrosis of the jaw: an Italian post-marketing surveillance analysis. Expert Opin Drug Saf (2014) 13(Suppl 1):S31–40. doi:10.1517/14740338.2014.951329
54. McClung MR. Bisphosphonates. Endocrinol Metab Clin North Am (2003) 32(1):253–71. doi:10.1016/S0889-8529(02)00079-8
55. Russell RG, Xia Z, Dunford JE, Oppermann U, Kwaasi A, Hulley PA, et al. Bisphosphonates: an update on mechanisms of action and how these relate to clinical efficacy. Ann N Y Acad Sci (2007) 1117:209–57. doi:10.1196/annals.1402.089
56. Toro EJ, Zuo J, Gutierrez A, La Rosa RL, Gawron AJ, Bradaschia-Correa V, et al. Bis-enoxacin inhibits bone resorption and orthodontic tooth movement. J Dent Res (2013) 92(10):925–31. doi:10.1177/0022034513501876
57. Rivera MF, Chukkapalli SS, Velsko IM, Lee JY, Bhattacharyya I, Dolce C, et al. Bis-enoxacin blocks rat alveolar bone resorption from experimental periodontitis. PLoS One (2014) 9(3):e92119. doi:10.1371/journal.pone.0092119
58. Liu X, Qu X, Nie T, Zhai Z, Li H, Ouyang Z, et al. The beneficial effects of bisphosphonate-enoxacin on cortical bone mass and strength in ovariectomized rats. Front Pharmacol (2017) 8:355. doi:10.3389/fphar.2017.00355
59. Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol (2013) 34(2):81–9. doi:10.1016/j.it.2012.08.006
60. Hume DA, MacDonald KP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood (2012) 119(8):1810–20. doi:10.1182/blood-2011-09-379214
61. Bischof RJ, Zafiropoulos D, Hamilton JA, Campbell IK. Exacerbation of acute inflammatory arthritis by the colony-stimulating factors CSF-1 and granulocyte macrophage (GM)-CSF: evidence of macrophage infiltration and local proliferation. Clin Exp Immunol (2000) 119(2):361–7. doi:10.1046/j.1365-2249.2000.01125.x
62. Campbell IK, Rich MJ, Bischof RJ, Hamilton JA. The colony-stimulating factors and collagen-induced arthritis: exacerbation of disease by M-CSF and G-CSF and requirement for endogenous M-CSF. J Leukoc Biol (2000) 68(1):144–50.
63. Paniagua RT, Chang A, Mariano MM, Stein EA, Wang Q, Lindstrom TM, et al. c-Fms-mediated differentiation and priming of monocyte lineage cells play a central role in autoimmune arthritis. Arthritis Res Ther (2010) 12(1):R32. doi:10.1186/ar2940
64. Garcia S, Hartkamp LM, Malvar-Fernandez B, van Es IE, Lin H, Wong J, et al. Colony-stimulating factor (CSF) 1 receptor blockade reduces inflammation in human and murine models of rheumatoid arthritis. Arthritis Res Ther (2016) 18:75. doi:10.1186/s13075-016-0973-6
65. Cenci S, Weitzmann MN, Gentile MA, Aisa MC, Pacifici R. M-CSF neutralization and egr-1 deficiency prevent ovariectomy-induced bone loss. J Clin Invest (2000) 105(9):1279–87. doi:10.1172/JCI8672
66. Gow DJ, Sester DP, Hume DA. CSF-1, IGF-1, and the control of postnatal growth and development. J Leukoc Biol (2010) 88(3):475–81. doi:10.1189/jlb.0310158
67. Rietkotter E, Bleckmann A, Bayerlova M, Menck K, Chuang HN, Wenske B, et al. Anti-CSF-1 treatment is effective to prevent carcinoma invasion induced by monocyte-derived cells but scarcely by microglia. Oncotarget (2015) 6(17):15482–93. doi:10.18632/oncotarget.3855
68. Lin H, Lee E, Hestir K, Leo C, Huang M, Bosch E, et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science (2008) 320(5877):807–11. doi:10.1126/science.1154370
69. Ma X, Lin WY, Chen Y, Stawicki S, Mukhyala K, Wu Y, et al. Structural basis for the dual recognition of helical cytokines IL-34 and CSF-1 by CSF-1R. Structure (2012) 20(4):676–87. doi:10.1016/j.str.2012.02.010
70. Felix J, Elegheert J, Gutsche I, Shkumatov AV, Wen Y, Bracke N, et al. Human IL-34 and CSF-1 establish structurally similar extracellular assemblies with their common hematopoietic receptor. Structure (2013) 21(4):528–39. doi:10.1016/j.str.2013.01.018
71. Baud’huin M, Renault R, Charrier C, Riet A, Moreau A, Brion R, et al. Interleukin-34 is expressed by giant cell tumours of bone and plays a key role in RANKL-induced osteoclastogenesis. J Pathol (2010) 221(1):77–86. doi:10.1002/path.2684
72. Chen Z, Buki K, Vaaraniemi J, Gu G, Vaananen HK. The critical role of IL-34 in osteoclastogenesis. PLoS One (2011) 6(4):e18689. doi:10.1371/journal.pone.0018689
73. Cheng X, Wan QL, Li ZB. AG490 suppresses interleukin-34-mediated osteoclastogenesis in mice bone marrow macrophages. Cell Biol Int (2017) 41(6):659–68. doi:10.1002/cbin.10771
74. Boyce BF, Xing L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther (2007) 9(Suppl 1):S1. doi:10.1186/ar2165
75. Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr Rev (2008) 29(2):155–92. doi:10.1210/er.2007-0014
76. Watts NB, Roux C, Modlin JF, Brown JP, Daniels A, Jackson S, et al. Infections in postmenopausal women with osteoporosis treated with denosumab or placebo: coincidence or causal association? Osteoporos Int (2012) 23(1):327–37. doi:10.1007/s00198-011-1755-2
77. Papapoulos S, Chapurlat R, Libanati C, Brandi ML, Brown JP, Czerwinski E, et al. Five years of denosumab exposure in women with postmenopausal osteoporosis: results from the first two years of the FREEDOM extension. J Bone Miner Res (2012) 27(3):694–701. doi:10.1002/jbmr.1479
78. Papapoulos S, Lippuner K, Roux C, Lin CJ, Kendler DL, Lewiecki EM, et al. The effect of 8 or 5 years of denosumab treatment in postmenopausal women with osteoporosis: results from the FREEDOM Extension study. Osteoporos Int (2015) 26(12):2773–83. doi:10.1007/s00198-015-3234-7
79. Bone HG, Wagman RB, Brandi ML, Brown JP, Chapurlat R, Cummings SR, et al. 10 Years of denosumab treatment in postmenopausal women with osteoporosis: results from the phase 3 randomised FREEDOM trial and open-label extension. Lancet Diabetes Endocrinol (2017) 5(7):513–23. doi:10.1016/S2213-8587(17)30138-9
80. Winkler DG, Sutherland MS, Ojala E, Turcott E, Geoghegan JC, Shpektor D, et al. Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J Biol Chem (2005) 280(4):2498–502. doi:10.1074/jbc.M400524200
81. Lewiecki EM. Role of sclerostin in bone and cartilage and its potential as a therapeutic target in bone diseases. Ther Adv Musculoskelet Dis (2014) 6(2):48–57. doi:10.1177/1759720X13510479
82. McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, Diez-Perez A, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med (2014) 370(5):412–20. doi:10.1056/NEJMoa1305224
83. Recknor CP, Recker RR, Benson CT, Robins DA, Chiang AY, Alam J, et al. The effect of discontinuing treatment with blosozumab: follow-up results of a phase 2 randomized clinical trial in postmenopausal women with low bone mineral density. J Bone Miner Res (2015) 30(9):1717–25. doi:10.1002/jbmr.2489
84. Keaveny TM, Crittenden DB, Bolognese MA, Genant HK, Engelke K, Oliveri B, et al. Greater gains in spine and hip strength for romosozumab compared with teriparatide in postmenopausal women with low bone mass. J Bone Miner Res (2017) 32(9):1956–62. doi:10.1002/jbmr.3176
85. MacNabb C, Patton D, Hayes JS. Sclerostin antibody therapy for the treatment of osteoporosis: clinical prospects and challenges. J Osteoporos (2016) 2016:6217286. doi:10.1155/2016/6217286
86. Vasiljeva O, Reinheckel T, Peters C, Turk D, Turk V, Turk B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr Pharm Des (2007) 13(4):387–403. doi:10.2174/138161207780162962
87. Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet (2011) 377(9773):1276–87. doi:10.1016/S0140-6736(10)62349-5
88. Runger TM, Adami S, Benhamou CL, Czerwinski E, Farrerons J, Kendler DL, et al. Morphea-like skin reactions in patients treated with the cathepsin K inhibitor balicatib. J Am Acad Dermatol (2012) 66(3):e89–96. doi:10.1016/j.jaad.2010.11.033
89. Gauthier JY, Chauret N, Cromlish W, Desmarais S, Duong le T, Falgueyret JP, et al. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg Med Chem Lett (2008) 18(3):923–8. doi:10.1016/j.bmcl.2007.12.047
90. Stoch SA, Zajic S, Stone J, Miller DL, Van Dyck K, Gutierrez MJ, et al. Effect of the cathepsin K inhibitor odanacatib on bone resorption biomarkers in healthy postmenopausal women: two double-blind, randomized, placebo-controlled phase I studies. Clin Pharmacol Ther (2009) 86(2):175–82. doi:10.1038/clpt.2009.60
91. Chapurlat RD. Odanacatib: a review of its potential in the management of osteoporosis in postmenopausal women. Ther Adv Musculoskelet Dis (2015) 7(3):103–9. doi:10.1177/1759720X15580903
92. Iniguez-Ariza NM, Clarke BL. Bone biology, signaling pathways, and therapeutic targets for osteoporosis. Maturitas (2015) 82(2):245–55. doi:10.1016/j.maturitas.2015.07.003
93. Yuan FL, Li X, Lu WG, Li CW, Li JP, Wang Y. The vacuolar ATPase in bone cells: a potential therapeutic target in osteoporosis. Mol Biol Rep (2010) 37(7):3561–6. doi:10.1007/s11033-010-0004-7
94. Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol (2007) 8(11):917–29. doi:10.1038/nrm2272
95. Toei M, Saum R, Forgac M. Regulation and isoform function of the V-ATPases. Biochemistry (2010) 49(23):4715–23. doi:10.1021/bi100397s
96. Supek F, Supekova L, Mandiyan S, Pan YC, Nelson H, Nelson N. A novel accessory subunit for vacuolar H(+)-ATPase from chromaffin granules. J Biol Chem (1994) 269(39):24102–6.
97. Ludwig J, Kerscher S, Brandt U, Pfeiffer K, Getlawi F, Apps DK, et al. Identification and characterization of a novel 9.2-kDa membrane sector-associated protein of vacuolar proton-ATPase from chromaffin granules. J Biol Chem (1998) 273(18):10939–47. doi:10.1074/jbc.273.18.10939
98. Hinton A, Bond S, Forgac M. V-ATPase functions in normal and disease processes. Pflugers Arch (2009) 457(3):589–98. doi:10.1007/s00424-007-0382-4
99. Bowman BJ, Bowman EJ. Mutations in subunit C of the vacuolar ATPase confer resistance to bafilomycin and identify a conserved antibiotic binding site. J Biol Chem (2002) 277(6):3965–72. doi:10.1074/jbc.M109756200
100. Huss M, Ingenhorst G, Konig S, Gassel M, Drose S, Zeeck A, et al. Concanamycin A, the specific inhibitor of V-ATPases, binds to the V(o) subunit c. J Biol Chem (2002) 277(43):40544–8. doi:10.1074/jbc.M207345200
101. Sorensen MG, Henriksen K, Neutzsky-Wulff AV, Dziegiel MH, Karsdal MA. Diphyllin, a novel and naturally potent V-ATPase inhibitor, abrogates acidification of the osteoclastic resorption lacunae and bone resorption. J Bone Miner Res (2007) 22(10):1640–8. doi:10.1359/jbmr.070613
102. Toro EJ, Zuo J, Ostrov DA, Catalfamo D, Bradaschia-Correa V, Arana-Chavez V, et al. Enoxacin directly inhibits osteoclastogenesis without inducing apoptosis. J Biol Chem (2012) 287(21):17894–904. doi:10.1074/jbc.M111.280511
103. Drucker DJ, Erlich P, Asa SL, Brubaker PL. Induction of intestinal epithelial proliferation by glucagon-like peptide 2. Proc Natl Acad Sci U S A (1996) 93(15):7911–6. doi:10.1073/pnas.93.15.7911
104. Henriksen DB, Alexandersen P, Hartmann B, Adrian CL, Byrjalsen I, Bone HG, et al. Disassociation of bone resorption and formation by GLP-2: a 14-day study in healthy postmenopausal women. Bone (2007) 40(3):723–9. doi:10.1016/j.bone.2006.09.025
105. Henriksen DB, Alexandersen P, Bjarnason NH, Vilsboll T, Hartmann B, Henriksen EE, et al. Role of gastrointestinal hormones in postprandial reduction of bone resorption. J Bone Miner Res (2003) 18(12):2180–9. doi:10.1359/jbmr.2003.18.12.2180
106. Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol (2001) 19:163–96. doi:10.1146/annurev.immunol.19.1.163
107. Radner H, Aletaha D. Anti-TNF in rheumatoid arthritis: an overview. Wien Med Wochenschr (2015) 165(1–2):3–9. doi:10.1007/s10354-015-0344-y
108. Quinn JM, Horwood NJ, Elliott J, Gillespie MT, Martin TJ. Fibroblastic stromal cells express receptor activator of NF-kappa B ligand and support osteoclast differentiation. J Bone Miner Res (2000) 15(8):1459–66. doi:10.1359/jbmr.2000.15.8.1459
109. Kitaura H, Sands MS, Aya K, Zhou P, Hirayama T, Uthgenannt B, et al. Marrow stromal cells and osteoclast precursors differentially contribute to TNF-alpha-induced osteoclastogenesis in vivo. J Immunol (2004) 173(8):4838–46. doi:10.4049/jimmunol.173.8.4838
110. Kitaura H, Zhou P, Kim HJ, Novack DV, Ross FP, Teitelbaum SL. M-CSF mediates TNF-induced inflammatory osteolysis. J Clin Invest (2005) 115(12):3418–27. doi:10.1172/JCI26132
111. Bruce ML, Peck B. New rheumatoid arthritis treatments. Holist Nurs Pract (2005) 19(5):197–204. doi:10.1097/00004650-200509000-00004
Keywords: osteoclast, osteoporosis, periprosthetic osteolysis, rheumatoid arthritis, Paget’s bonedisease, osteopetrosis
Citation: Bi H, Chen X, Gao S, Yu X, Xiao J, Zhang B, Liu X and Dai M (2017) Key Triggers of Osteoclast-Related Diseases and Available Strategies for Targeted Therapies: A Review. Front. Med. 4:234. doi: 10.3389/fmed.2017.00234
Received: 27 May 2017; Accepted: 04 December 2017;
Published: 20 December 2017
Edited by:
Peter N. Robinson, Jackson Laboratory for Genomic Medicine, United StatesReviewed by:
Carole Guillonneau, INSERM UMR1064 Centre de Recherche en Transplantation et Immunologie, FranceMelissa Anne Haendel, Oregon Health and Science University, United States
Copyright: © 2017 Bi, Chen, Gao, Yu, Xiao, Zhang, Liu and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuqiang Liu, shliuxuqiang@163.com;
Min Dai, daimin@medmail.com.cn
†These authors have contributed equally to this work.