
- Center for Microbial Interface Biology, Division of Infectious Diseases, Department of Internal Medicine, The Ohio State University, Columbus, OH, USA
Tuberculosis is still a major health problem in the world. Initial interactions between Mycobacterium tuberculosis and the host mark the pathway of infection and the subsequent host inflammatory response. This inflammatory response is tightly regulated by both the host and the bacterium during different stages of infection. As infection progresses, the initial intense pro-inflammatory response observed is regulated by suppressive mediators balancing inflammation. In this environment, M. tuberculosis battles to survive interfering with the host inflammatory response. In this review we discuss the major effector molecules involved in inflammation in relation to the different stages of M. tuberculosis infection.
Introduction
Over the past 15 years as a result of a rigorous approach to treatment endorsed by the World Health Organization, close to 36 million people have been cured of tuberculosis (TB). However, although a great deal of effort has occurred for handling the TB pandemic around the world, there are still 1.8 million people dying from TB each year. Indeed, there are too many inherent factors involved in the surveillance of Mycobacterium tuberculosis (M.tb, the etiological agent of TB) including the nature of the spread of M.tb infection, the difficultly in obtaining the right drug treatment, and a complex therapeutic regime which still makes TB one of the major health challenges in the world. HIV and M.tb co-infection and the emergence of multi-drug and extensively drug resistant strains of M.tb are also adding to the burden of TB clinical cases in the world.
Tuberculosis pathogenesis is driven by a complex interplay between the host immune system and the survival strategies of the bacterium. The inflammatory response to M.tb infection is tightly regulated by both the host and the bacterium. The primary goal of this review is to discuss the major effector molecules involved in inflammation in relation to the different stages of M.tb infection.
Stages of Mycobacterium Tuberculosis Infection
Mycobacterium tuberculosis is mainly considered to be an airborne pathogen. The infection process of M.tb can be divided into three different but interrelated stages. The first stage is the aerosol transmission of droplets containing M.tb from an infected individual to a healthy individual. Once within the lungs, M.tb enters and resides within alveolar macrophages (AMs) and dendritic cells (DCs; Cooper, 2009). Though the AM ingests bacilli and often kills them, the bactericidal capacity of the AM is still not very well defined. In a given M.tb infection, the initial containment of the infection depends partially on the genetics of the human population (i.e., defined by the intrinsic microbicidal capacity of host phagocytes) and also on the inhaled M.tb strain (i.e., defined by innate virulent factors in each M.tb strain). In the primary infection M.tb multiplies in the lungs and causes mild inflammation. Although AMs are thought to be an effective barrier to contain pathogens, M.tb has evolved various mechanisms to evade the host immune response and survive in these cells. These survival mechanisms include triggering an anti-inflammatory response, blocking reactive oxygen and nitrogen intermediate (ROIs and RNIs, respectively) production, and reducing the acidification of the M.tb-containing phagosome (Flynn and Chan, 2001; Fenton et al., 2005; Cooper, 2009).
The next stage of infection is characterized by the emergence of cell-mediated immunity and the formation of granulomas (described below). M.tb bacilli that escape the bactericidal effects of the AM, will multiply and result in destruction of AMs. This will in turn attract blood monocytes and other inflammatory cells (i.e., neutrophils) to the site of infection. Monocytes mature to become antigen presenting AMs and DCs and ingest, but not effectively kill the bacteria. At this stage, M.tb grows under limited tissue damage. By 6–8 weeks post-infection, antigen presenting DCs have traveled to lymph nodes where T lymphocytes are activated and recruited. Activated T lymphocytes that migrate to the site of infection proliferate forming an early stage granuloma, where macrophages become activated to kill intracellular M.tb (Ulrichs and Kaufmann, 2006). However, continuing T cell activation leads to formation of granulomas that mark the persistence stage of the infection (latency), where the growth and spread of bacteria into additional tissue sites are limited. At this stage more than 90% of infected people remain asymptomatic, but M.tb may survive within AMs.
The third and final stage is when latent and controlled M.tb infection is reactivated. There are two main reasons described for a reactivation event to occur, a decline in the host’s immunity due to genetic or environmental cause; and a failure to develop and maintain immune signals. Under these circumstances, the granuloma structure disrupts and results in lung cavitation and pulmonary disease (Kaplan et al., 2003; Dheda et al., 2005; Ulrichs and Kaufmann, 2006; Russell, 2007). Among the genetic causes described that make a subject susceptible to TB are mutations in specific host C-type lectins, cytokines, chemokines, and their specific receptors disrupting critical signaling pathways involved in the immune response against M.tb. Compromised immune surveillance for reasons such as co-infection with HIV, where a host becomes immuno-compromised especially for CD4 T cells (the cell target for HIV), is the most important environmental or exogenous cause of susceptibility to TB (Geldmacher et al., 2010). The reactivation of M.tb infection can also be due to changes in host cytokine/chemokine networks, implicated in the inflammatory response against M.tb infection, that are a consequence of stress and/or old age (Turner, 2011). Earlier studies have also suggested that exogenous re-infection with another strain of M.tb (Sonnenberg et al., 2001; Behr, 2004) is an additional factor leading to active disease.
Development of the M.tb Granuloma
The hallmark in M.tb infection is the presence of granulomas within the lung. From the perspective of the bacterium, M.tb-induced granulomas are a collection of well organized immune cells that provide a safe microenvironment to establish latency. From the host perspective, granuloma formation restricts the spread of M.tb infection. The formation of a granuloma starts with a transient influx of neutrophils to the site of the infection, followed by activated macrophages and lymphocytes (reviewed in detail in Ulrichs and Kaufmann, 2006; Russell, 2007). An established granuloma is composed of infected AMs and epithelioid cells that form a necrotic central core which provides nutritional support to M.tb. The central necrotic core of the granuloma is composed by both host and M.tb factors. Surrounding this necrotic center there are activated macrophages and layers of CD4+ and CD8+ T cells defining a dense cellular wall that restricts the spread of the M.tb (Saunders and Cooper, 2002). In immunocompetent M.tb-infected people (Figure 1A), M.tb-containing granulomas are small, compact, and characterized by the presence of a large number of IFN-γ CD4-T cells; however, in immunodeficient M.tb-infected people (Figure 1B), granulomas are characterized by being large, rich in activated macrophages, and with few surrounding lymphocytes (Ulrichs et al., 2005). The main cause for tissue injury and clinical manifestation is the presence of large caseating granulomas and fibrotic scarring due to granulomatous inflammation (Daley, 2010); where the host Th1 response serves to try to contain the infection and prevent the development of active disease yet cannot eliminate M.tb (Saunders et al., 1999).
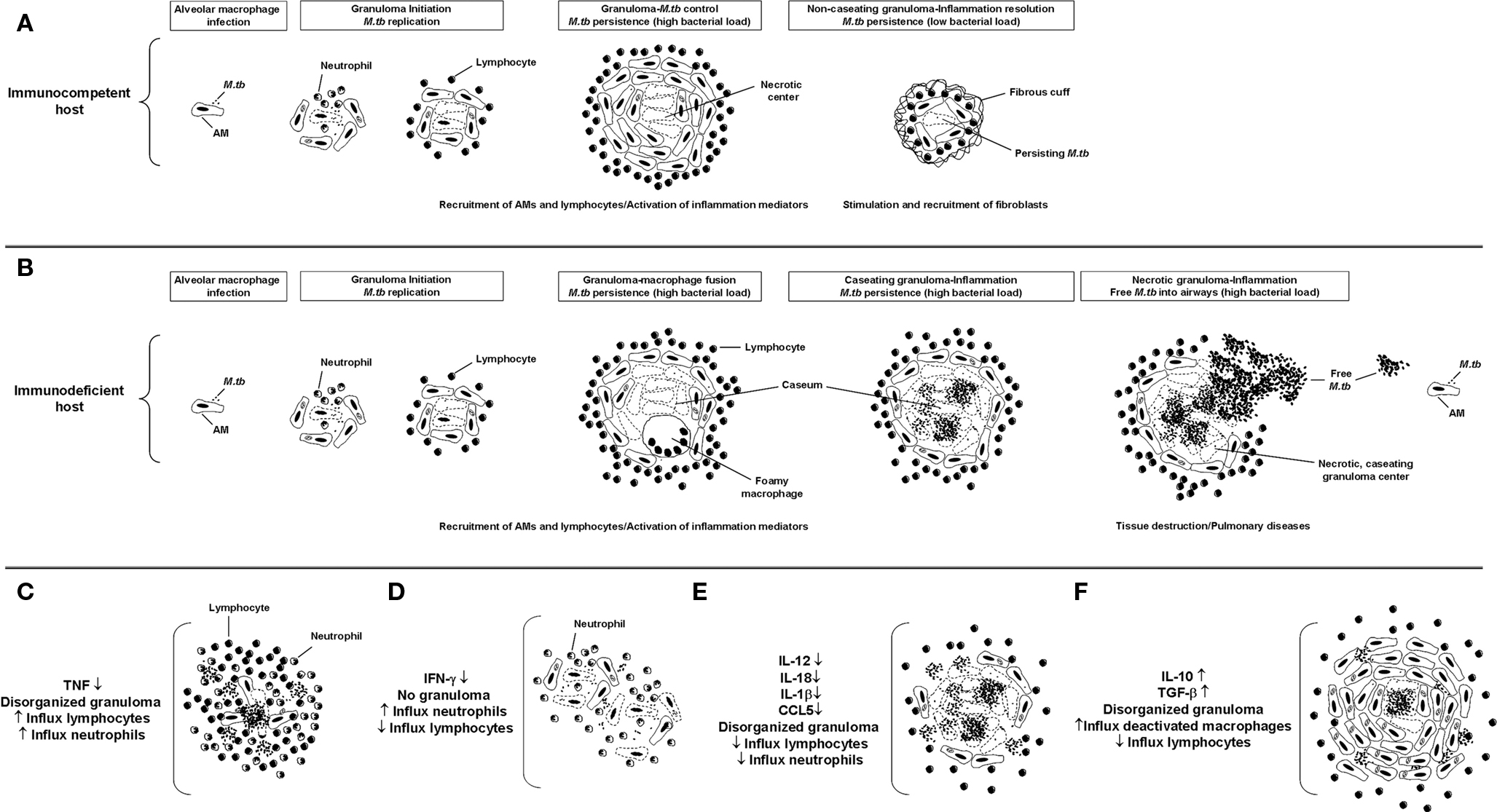
Figure 1. Granuloma formation. Schematics of lesion structure at each stage of granuloma formation in immunocompetent (A) or immunodeficient (B) M.tb-infected people. Once M.tb reaches the lungs, bacilli are internalized by AMs triggering the subsequent inflammatory response. In the early stage, the granuloma has a core of infected macrophages enclosed by mononuclear phagocytes, surrounded by lymphocytes. In the case of immunocompetent people, infection will be contained within small and compact granulomas characterized by the presence of a large number of IFN-γ CD4 T cells. For immunodeficient people, as the granuloma matures, it is characterized by being rich in activated macrophages and with few surrounding lymphocytes. Over time the granuloma caseous necrotic center liquefies and cavitates, spilling thousands of infectious M.tb bacilli into the airways. (C–F) Schematic of granulomas developed in the absence or presence of the main inflammatory mediators described in M.tb pathogenesis.
The mechanism and the products influencing granuloma formation are not very well established. In response to M.tb interaction with AMs and DCs, there is a release of inflammatory cytokines such as tumor necrosis factor-α(TNF), interleukin-(IL)-12, and IL-23 along with a variety of chemokines including (C-C) motif ligand 2 (CCL2), CCL5, and (C-X-C) motif ligand 8 (CXCL8). Production of IL-12 (Seder et al., 1993) and IL-23 (Oppmann et al., 2000) by DCs primes the Th1 T cell response, which is important for granuloma assembly. This flow of inflammatory events is regulated by the production of IFN-γ and IL-2 by activated T cells that reach the infection site (Cooper, 2009). Studies using CD4 deficient mice demonstrate that CD4 T cells are required for the recruitment of mononuclear cells to the infection site and the organization that is required for the long term survival of the host (Saunders and Cooper, 2002). Along with CD4 T cells, CD8 T cells (Saunders and Britton, 2007) and CD1-restricted NKT-cells (Chackerian et al., 2002; Co et al., 2004) are also key components of developing granuloma. A significant characteristic common between mouse and human pulmonary TB is also the presence of B-cell lymphocyte clusters (Turner et al., 2001; Ulrichs et al., 2004; Tsai et al., 2006). Later studies indicate that B-cell aggregates may serve different purposes in both species as in mice macrophages encircle B-cells; however, in humans B-cell aggregates recruit T cells that are evenly distributed in the clusters (Tsai et al., 2006). Interestingly, studies looking at the ratio between the necrotic core and the neighboring layers of dense leukocytes suggest that the necrotic core expands at the expense of the surrounding layers of cells and not because of leukocyte infiltration (Ulrichs and Kaufmann, 2006).
Although it seems contradictory, it is understood that the initial inflammatory response to M.tb is crucial for granuloma generation but also for M.tb long term survival within the host. In this context, pro-inflammatory cytokines reduce the bacterial burden, regulating the activity of other cytokines and chemokines, and generating and maintaining organized granulomas. Infection of macrophages by M.tb primarily induces the production of TNF, IFN-γ, IL-12, and RNIs and ROIs (Flynn and Chan, 2001; Cooper, 2009), which are considered to be key regulatory factors in the formation and/or maintenance of the granuloma structure. The importance of TNF in granuloma formation has been shown by using TNF neutralizing antibodies and TNF deficient mice in vivo. These studies showed that TNF and its receptor are critical in granuloma formation and subsequent protection against M.tb infection, as well as their involvement in RNI production by macrophages during early infection (Flynn et al., 1995a; Kaneko et al., 1999). TNF can also induce macrophages to release chemokines that guide cells to the site of infection and prevent their migration away from the site (Flynn and Chan, 2005). Recent studies performed in Cynomolgus macaques using TNF neutralizing drugs before M.tb infection demonstrated uncontrolled, disseminated infection by 8 weeks of infection. This lack of TNF also induced a high rate of reactivation TB among macaques with latent infection. Importantly, the granuloma architecture in non-human primates after treatment with neutralizing TNF agents was similar to that seen in active TB in humans (Capuano III et al., 2003; Lin et al., 2010). Histological studies performed on human lung biopsies from patients receiving TNF blockade reconfirm the role of TNF in granuloma formation by showing granuloma disorganization due to extensive lymphocytic infiltrations (Keane et al., 2001; Figure 1C). In addition to TNF, IFN-γ is also important in the development of granulomas (Flynn et al., 1993). IFN-γ deficient mice were found to be incapable of developing granulomas following aerosol infection and their lungs were found to be infiltrated with neutrophils resulting in cellular necrosis instead of granuloma formation (Cooper et al., 1993; Pearl et al., 2001; Figure 1D).
Interleukin-12 is also related to granuloma formation by promoting the Th1 response and inducing IFN-γ positive CD4 T cells (Seder et al., 1993). Studies in mice established that neutralization of IL-12 by specific monoclonal antibodies results in a reduction in granuloma integrity and slowing of the capacity of the animal to control M.tb growth (Cooper et al., 1995; Figure 1E). Later human studies corroborated the importance of IL-12 in granuloma maintenance, where specific mutations in either the IL-12 p40 or IL-12Rβ1 (IL-12 receptor) gene showed reduced levels of IL-12 and IL-23, and subsequent low IFN-γ induced T cell responses in subjects susceptible to mycobacterial infections (Remus et al., 2001; Fieschi and Casanova, 2003). Other cytokines have also been implicated in the establishment of the granuloma; however, their specific role is still uncertain. This is the case for IL-10, where recent in vivo studies using mice over-expressing human IL-10 in their lungs found an increase in macrophage presence and bacterial burden in M. avium-containing granulomas (Feng et al., 2002; Figure 1F). This was related to a decrease in the levels of pro-inflammatory cytokines like TNF and IL-12 indicating that IL-10 may employ a suppressive effect on the control of mycobacterial infection by negatively controlling macrophage activation. Another regulatory molecule that has been indirectly implicated in granuloma formation is IL-27, which has been shown to activate naïve T cells after their encounter with macrophages and DCs (Owaki et al., 2005). In this context, IL-27 deficient mice have severe lung pathology due to the uncontrolled spread of M.tb infection (Holscher et al., 2005).
The exact role of chemokines in granuloma formation is still not very clear. Studies have shown some role for CC chemokine subfamily members particularly CCL5, CCL10, and CCL21 in granuloma formation (Mendez-Samperio, 2008). In vivo studies using mice have established that there is a certain level of redundancy in this model system. More recent studies showed the importance of CCL5 and CCL4 and their receptors in the establishment of early granulomas in mice by directly controlling the migration of IFN-γ positive CD4 T cells to the site of the infection (Vesosky et al., 2010). However in humans, chemokines like CCL2, CCL5, and CXCL10 may participate in the protection mechanism against M.tb infection (Algood et al., 2003).
Inflammasome-related cytokines like IL-1β (Juffermans et al., 2000; Sugawara et al., 2001) and IL-18 (Sugawara et al., 1999; Schneider et al., 2010) have also been shown to be involved in promoting granuloma formation and organization. In this context, the apoptotic speck-like protein containing a CARD domain (PYCARD), defined as an adaptor protein involved in IL-1β and IL-18 activation through caspase-1 processing, has been shown to play a major role in maintaining granuloma integrity during chronic M.tb infection (McElvania et al., 2010).
Host Receptors and Inflammation in M.tb Infection
The initial interaction between M.tb and the host dictates the pathway and outcome of infection (Figure 2). On the host side, there are specific receptors capable of recognizing M.tb. Normally this recognition is beneficial for the host by activating innate immunity; however, there are specific receptors that favor M.tb uptake, bypassing the pro-inflammatory response and leading to intracellular survival. In this section we will discuss host receptors involved in M.tb recognition and the subsequent inflammatory response.
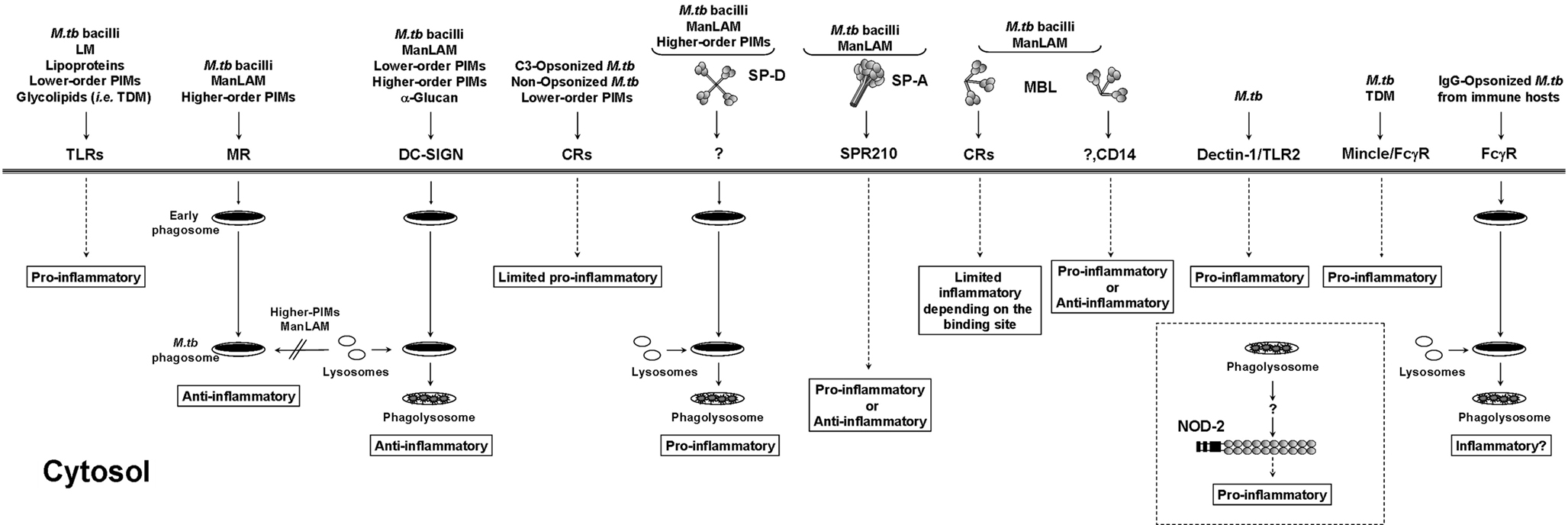
Figure 2. Antigen presenting cell receptors involved in M.tb infection. M.tb cell wall components associate with a subset of immune receptors initiating phagocytosis and mediating specific host cell responses. Interactions with certain receptors or soluble collectins lead to the activation of pro-/anti-inflammatory responses in the host that may influence the intracellular survival of M.tb. TLR, Toll-like receptor; MR, mannose receptor; DC-SIGN, dendritic cell-specific intercellular adhesion molecule-3 grabbing non-integrin; CR, complement receptor; SP-A and SP-D, surfactant protein-A and -D; MBL, mannose binding lectin; Dectin-1, dendritic cell-associated C-type lectin-1; Mincle, macrophage-inducible C-type lectin; NOD-2, nucleotide-binding oligomerization domain-like receptor 2.
Toll-Like Receptors
Toll-like receptor (TLR) signaling is essential for immunity to various intracellular pathogens. TLRs are a set of pattern recognition receptors (PRRs) that are expressed on many cell types but their function on antigen presenting cells (APCs) is particularly important (reviewed in Kawai and Akira, 2010). On macrophages TLRs are either expressed on the surface (like TLR2 and 4) or inside cell compartments (like TLR8 and 9; Kawai and Akira, 2010). TLRs detect a wide range of structures on M.tb (Figure 2), which aid in the activation of innate immunity and enhance adaptive immunity by mediating the secretion of various pro-inflammatory cytokines along with other anti-bacterial effector molecules. The relationship between TLRs and M.tb is a bit more complex than with other bacteria. Various studies performed indicate that different components of the bacteria interact with different TLRs. Among TLRs the key players in TB immunity are TLR2 (alone or as a heterodimer with TLR1 or TLR6), TLR9, and probably TLR4 (Harding and Boom, 2010). For TLR2 alone or in association with TLR1 or TLR6, several groups have reported a strong pro-inflammatory response induced by mycobacterial cell wall components. This is the case for the 19 KDa lipoprotein, phospho-myo-inositol-capped or non-capped lipoarabinomannan (PILAM and AraLAM respectively, both produced by less pathogenic mycobacteria), lower- and higher-order phosphatidyl-myo-inositol mannosides (PIM1, PIM2, and PIM6 families), lipomannan, and trehalose dimycolate (reviewed in Jo et al., 2007). A combination of studies using several M.tb ligands and mouse macrophages deficient in TLR2 and MyD88 (myeloid differentiation primary-response protein 88) determined conclusively that the strong pro-inflammatory response to M.tb infection observed via TLR2 signaling is mediated through its adaptor protein MyD88 (Quesniaux et al., 2004), where engagement of TLR2 triggers a nuclear factor kappa-light chain-enhancer of activated B-cell (NFκB) signaling cascade through the recruitment of MyD88 and TIRAP (toll-interleukin 1 receptor [TIR] domain containing adaptor protein; Kawai and Akira, 2010). Of no surprise is that the response observed depends on the mycobacterial ligand and the nature of the host cell type studied. As an example, uncapped AraLAM was shown to induce cell activation via TLR2 (Underhill et al., 1999) leading to M.tb killing in both murine and human macrophages but in a nitric oxide dependent and independent manner, respectively (Thoma-Uszynski et al., 2001).
Interestingly, an overwhelming stimulation of the TLR2 pathway may also be advantageous for M.tb. Recent studies have indicated that prolonged TLR2 signaling induced by M.tb lipoproteins LpqH, LpRG, and LpRA inhibit the expression of major histocompatibility complex (MHC) class II thereby decreasing antigen presentation in macrophages infected with M.tb (Harding and Boom, 2010). Related to this, more recent studies using mice also suggest that TLR2 may be one of many pathways exploited by M.tb to inhibit MHC-I antigen cross processing and presentation to CD8+ T cells (Harding and Boom, 2010). Studies performed on infected DCs also resulted in TLR2-dependent inhibition of TLR9-dependent IFN-α/β expression, leading to a decrease in induction of IFN-α/β-dependent MHC-I cross processing (Simmons et al., 2010). In this regard, during M.tb infection in vivo, it is still uncertain whether TLR2 dependent inhibition of the TLR9/IFN-α/β-dependent MHC-I processing pathway is of benefit to the host by limiting the harmful effects of excessive inflammation or to M.tb, as it may provide a path for M.tb to escape the host immune response. Overall, a negative regulatory feedback mechanism among TLR signaling networks during M.tb chronic infection may benefit the host by preventing detrimental effects of excessive inflammation (Simmons et al., 2010). In this context, studies performed to understand the role of TLR signaling in vivo using TLR deficient mice indicate that a balance in the TLR signaling network is necessary to control inflammation. This is indicated by studies where the exclusion of the TLR2 signaling pathway with a moderate dose of M.tb in vivo results in an exaggerated inflammatory response which may be detrimental for the host (Drennan et al., 2004).
TLR9 recognizes unmethylated CpG (cytosine phosphate guanosine motif) found in mycobacterial DNA (Kawai and Akira, 2010). As mentioned above, activation of TLR9 induces IFNα/β and MHC-I antigen cross processing (Simmons et al., 2010). Mycobacterial ligands for TLR4 are still undetermined; although recently, recombinant M.tb heat shock protein (hsp) 65 has been described as a ligand for TLR4 inducing NFκB via MyD88, TIRAP, TRIF (TIR-domain-containing adapter-inducing interferon-β), and TRAM (TRIF-related adaptor molecule)-dependent signaling pathways (Bulut et al., 2005). Studies using TLR4 deficient mice infected with M. bovis Bacillus Calmette–Guérin (BCG), showed that these mice are capable of controlling the infection; however, mice eventually showed body weight loss and increased local inflammation indicating that TLR4 plays a defined role in modulating inflammation in mycobacterial infections (Fremond et al., 2003).
The Mannose Receptor
The cell envelope of pathogenic mycobacteria like M.tb is particularly rich in mannose-containing biomolecules, including ManLAM, LM, higher-order PIMs, arabinomannan, mannan, and mannosylated glycoproteins. ManLAM, LM, and higher-order PIMs are incorporated into the M.tb plasma membrane and also exposed on its cell surface acting as ligands for host cell receptors contributing to the pathogenesis of M.tb (Torrelles and Schlesinger, 2010). ManLAM and higher-order PIMs contain terminal mannosyl units ideally located to interact with the MR on macrophages and DCs (Schlesinger et al., 1994; Torrelles and Schlesinger, 2010; Figure 2). The MR is defined as a homeostatic/clearance/immunomodulatory receptor for endogenous serum glycoproteins with N-linked high-mannose content normally elevated during inflammation (Martinez-Pomares et al., 2001). Evidence in the literature suggests that M.tb may exploit its mannosylated cell surface components to survive within the host by binding the MR. In this context, we recently demonstrated that ManLAM/MR and higher-order PIMs/MR phagocytic pathways lead to phagosome maturation arrest (Kang et al., 2005; Torrelles et al., 2006). Recognition of mannose residues by the MR has also been shown to reduce the microbicidal activities of the macrophage by inhibiting the production of nitric oxide, oxygen radicals, and pro-inflammatory cytokines and by blocking M.tb-induced apoptosis through modifying Ca2+-dependent signaling pathways (reviewed in Torrelles et al., 2008a). In addition, several studies have shown that ManLAM generates an anti-inflammatory response inhibiting the production of TNF and IL-12, and inducing the production of IL-10 and transforming growth factor β (TGF-β; Astarie-Dequeker et al., 1999; Nigou et al., 2001; Chieppa et al., 2003). Recent studies have also established an MR-specific signaling pathway in the pathogenesis of M.tb (Rajaram et al., 2010). Infection of human macrophages with virulent M.tb or the addition of ManLAM up-regulates a transcription factor named peroxisome proliferator-activated receptor-gamma (PPAR-γ), an important molecule that regulates the macrophage inflammatory response. PPAR-γ up-regulation via the MR leads to the simultaneous increase in the production of IL-8 (or CXCL-8), expression of cyclooxygenase 2 (COX2), and production of prostaglandin 2 (PGE2; Rajaram et al., 2010). This study also revealed that PPAR-γ deficiency in human macrophages leads to increased levels of TNF and decreased bacterial load, indicating that the MR negatively regulates protective macrophage inflammatory responses to M.tb infection. In murine macrophages, inhibition of PGE2 by M.tb infection prevents apoptosis and leads the infected cell to a necrotic pathway favoring the spread of the infection (Divangahi et al., 2010). In these type of studies we should carefully consider differences between mouse and human macrophages as well as the existence of receptor-dependent/specific regulatory mechanism(s) in these complex signaling networks. In this context, in addition to M.tb ManLAM blocking TNF production via the MR, unpublished data by Rajaram and Schlesinger identify a novel molecular and cellular mechanism underlying the ability of another major M.tb cell wall component, LM, to block TLR2 induced biosynthesis of TNF in macrophages, thereby allowing M.tb to subvert the host immune response and potentially increase its virulence (personal communication).
Dendritic Cell-Specific Icam-3-Grabbing Non-Integrin
Dendritic cell-specific ICAM-3-grabbing non-integrin (DC-SIGN) expressed on DCs (Geijtenbeek et al., 2000), strongly binds to M.tb through its surface-exposed mannose-containing molecules ManLAM, LM, and PIMs (reviewed in Ehlers, 2009). Recently, α-glucan found on the M.tb surface was also found to act as a ligand for DC-SIGN (Geurtsen et al., 2009). DC-SIGN actively participates in the phagocytosis of M.tb by DCs leading to bacterial killing by acidification of the M.tb-containing phagosome (Geijtenbeek et al., 2003) (Figure 2). However, the role of DC-SIGN in inflammation is controversial. Association of M.tb, α-glucan or its mannosylated cell wall components with DC-SIGN has been shown to induce production of anti-inflammatory mediators like IL-10 (Ehlers, 2009; Geurtsen et al., 2009). Recent in vivo studies using wild-type mice expressing human DC-SIGN homologs (Park et al., 2001; McGreal et al., 2005; Powlesland et al., 2006) or transgenic mice expressing human DC-SIGN, consider the possibility that DC-SIGN may act to dampen the immune response thereby promoting host protection by limiting tissue damage (Wieland et al., 2007; Schaefer et al., 2008; Tanne et al., 2009). Conversely, using genetically engineered M. marinum strains lacking essential mannosylated components on their cell surface revealed that the ManLAM–PIM/DC-SIGN pathway may not be significantly involved in the regulation of cytokine secretion (Appelmelk et al., 2008). However, as M. marinum does not reflect the overall mannosylation pattern observed in M.tb laboratory strains (where M.tb clinical isolates also present different degrees of mannosylation; Torrelles et al., 2008b), the direct functional consequences of DC-SIGN ligation in M.tb pathogenesis are still unclear.
Complement Receptors
Complement receptors (CRs) are expressed on all mononuclear phagocytes and mediate the phagocytosis of a diverse group of intracellular pathogens. Several studies have established the role of C3 opsonization and the contribution of CR1, CR3, and CR4 in the phagocytosis of M.tb (Fenton et al., 2005; Figure 2). C3 deposition on M.tb happens quickly and is initiated by activation of the classical (in low serum concentrations) and/or alternative pathways (in high serum concentrations) via covalent linkages to M.tb surface targets in the forms of C3b and C3bi (Ferguson et al., 2004). However, it is still unknown if C3 opsonization varies in form and amount among different stages of M.tb infection or tissue sites. M.tb surface-exposed polysaccharides and lower-order PIMs (i.e., PIM2) have also been shown to interact directly with the lectin domain of CR3 (Villeneuve et al., 2005) and thus, presumably mediate M.tb uptake by macrophages. Although CR3 mediates both opsonic and non-opsonic uptake of M.tb by macrophages, its role during human infection remains unknown. In vitro and in vivo studies did not show an altered phenotype between CR3 deficient and wild-type mice in bacterial burden and pathology (Hu et al., 2000). In this context, several studies have shown that CR4 is particularly abundant on cells that are involved in the uptake of M.tb leading to the conclusion that in the naïve host, CR4 may be the major player mediating M.tb uptake in the early stages of infection (Hirsch et al., 1994; Zaffran et al., 1998). This may correlate with the fact that CR4 and the MR are the most highly expressed receptors on AMs (Schlesinger et al., 2008).
Collectins and Their Specific Receptors
There are three major collectins within the C-type lectin family that recognize specific sugar moieties on the M.tb surface increasing the interaction between the host and the bacteria. They are surfactant proteins (SP)-A and SP-D, and mannose binding lectin (MBL). SP-A and SP-D are mainly located in the lung, being secreted by alveolar epithelial type II cells into the surfactant (Wright, 2005). Various studies have provided evidence suggesting a dual role model for SP-A and SP-D, as they help maintain a balance between pro- and anti-inflammatory responses to M.tb in the lung environment. In this context, SP-A may be considered to be the perfect liaison for M.tb. Apart from being characterized for its capacity to act as an opsonin, a regulator for cell receptor activity enhancing phagocytosis by macrophages, and a mediator of inflammation by regulating the synthesis of ROIs and RNIs and cytokine secretion (reviewed in Torrelles et al., 2008a; Figure 2); recent studies show that SP-A may be a contributor to the establishment of a successful M.tb infection. In this context, SP-A is capable of opsonizing M.tb and after being recognized by its specific receptor, SP-R210, leads to the secretion of anti-inflammatory cytokines like IL-10 and TGF-β mediating suppression of cell-mediated immunity against M.tb (Samten et al., 2008). Linked to this, is the establishment that SP-A also up-regulates macrophage expression of the MR (a receptor that leads to M.tb intracellular survival within the macrophage by limiting phagosome maturation; Beharka et al., 2002; Torrelles et al., 2008a). SP-A also regulates TLR surface expression and activity in human macrophages (Henning et al., 2008), where despite its ability to specifically up-regulate TLR2 expression, SP-A seems to dampen TLR2 and TLR4 signaling in these cells, and thus situates SP-A as a critical mediator in regulating lung inflammatory responses through TLRs (Henning et al., 2008). SP-D has been recently shown to bind in high avidity to M.tb ManLAM and PIMs (Ferguson et al., 1999; Carlson et al., 2009). SP-D opsonizes and subsequently aggregates M.tb thereby reducing its phagocytosis (Ferguson et al., 1999). Conversely, SP-D opsonized M.tb that is phagocytosed undergoes increased phagosome–lysosome fusion resulting in limited intracellular growth (Ferguson et al., 2006; Figure 2). Another soluble collectin considered in the establishment of M.tb infection is the MBL. MBL binds to M.tb promoting activation of the complement lectin pathway leading to C3bi deposition, as well as complement-independent phagocytosis which induces pro-inflammation with the release of TNF, IL-1β, and IL-6 (reviewed in Dommett et al., 2006; Figure 2). However, controversy regarding the model system (i.e., cell types and species specificity) studied still exists for determining if MBL is overall ultimately beneficial for M.tb or the host (Torrelles et al., 2008a).
Other Macrophage Surface Receptors
Other receptors (Figure 2) involved in M.tb recognition and inflammation are CD14 (Khanna et al., 1996) and the scavenger receptors SR-A (Zimmerli et al., 1996), which participate in the uptake of non-opsonized bacilli by tissue-specific macrophages. However, their role in inflammation varies depending on the species-specific cell type used. Dectin-1, a β-glucan receptor, in combination with TLR2 has also been shown to participate in the immune response against M.tb by inducing TNF production in macrophages only infected with attenuated M.tb strains (Yadav and Schorey, 2006). Recently, macrophage-inducible C-type lectin (Mincle; Yamasaki et al., 2008) on the macrophage surface, has been shown to recognize M.tb trehalose dimycolate, and working together with the Fcγ receptor transmembrane segment induces pro-inflammation (Ishikawa et al., 2009; Schoenen et al., 2010). Conversely, Fcγ receptors do not play a role in the phagocytosis of M.tb in the absence of specific antibody (Schlesinger et al., 1990).
Cytosolic Receptors: NOD2
Cytosolic regulators of the pro-inflammatory response known as nucleotide-binding oligomerization domain (NOD)-like receptors (Franchi et al., 2008) have been recently described to play a role during M.tb infection. NOD2, found in epithelial cells and APCs (Ogura et al., 2001; Gutierrez et al., 2002; Inohara and Nunez, 2003), regulates the production of inflammatory mediators in response to bacterial peptidoglycan components such as muramyl dipeptide (Inohara and Nunez, 2003). Human studies in patients infected with M.tb or M. leprae have linked a polymorphism in NOD2 to susceptibility to mycobacterial infections (Austin et al., 2008; Zhang et al., 2009). However, the role of NOD2 in the early stages of M.tb infection seems to be dependent on the model system studied. In this context, results obtained from in vitro and in vivo studies using the mouse model dispute the significance of NOD2 in controlling M.tb growth during the early stages of infection (Gandotra et al., 2007; Divangahi et al., 2008). Conversely, in vitro studies using human macrophages infected with M.tb have established that, in accordance with the human polymorphism studies, NOD2 plays a role in controlling pro-inflammation and M.tb intracellular growth (Brooks et al., 2010; Figure 2). How NOD2 intersects with signaling/trafficking networks is still unexplored. Although, NOD2 can synergize with other signaling pathways like TLRs, enhancing pro-inflammation (Ferwerda et al., 2005), its capability to interfere/associate with phagocytic receptor trafficking networks is not established. As cytosolic NOD2 appears to be associated with intracellular vesicles (Brooks et al., 2010), its role in triggering pro-inflammation may depend on vesicular fusion events controlled during M.tb phagocytosis.
The outcome of M.tb infection and the subsequent inflammatory response depends on the initial interaction(s) between M.tb and APCs. These interactions are mainly based on two factors: The nature and distribution of recognition receptors on the APC surface, and on the biochemical nature of the cell wall constituents of the M.tb bacilli recognized by APCs. As described above, published studies define the role of each individual receptor (or a combination of them) during infection in vitro or in vivo using different means. It still is unclear though, which receptors and how many of them are involved in the initial M.tb–host interaction in vivo. Our in vitro studies using single cell suspensions of M.tb and human macrophages show that M.tb has a predilection for infection of specific subpopulations of APCs. Do these APC subpopulations preferentially express a specific receptor(s) beneficial for M.tb recognition/infection? Does the genetic background of the host (man) predispose for surface expression of specific receptors on APCs that favor M.tb infection? If the M.tb cell wall determines the impact of the infection; why do different virulent strains of M.tb present different motifs on their surface? Are biochemical differences on the M.tb cell wall surface favoring M.tb long term persistence within the host? Conversely, are we accounting for how disruption of a specific receptor alters the complex inflammatory network in vivo? Human studies addressing receptor polymorphisms in TB patients may help address these still unanswered questions.
Chemokines in M.tb Infection
Due to their assistance in cell migration and subsequently granuloma formation, chemokines are essential but undefined players in the inflammatory response to M.tb infection. Their main function is to direct cell migration and immune homeostasis in the host. Current literature already has established a role for CC and CXC chemokines in the protective and immune host response to TB (reviewed in Mendez-Samperio, 2008). In vitro studies using M.tb-infected murine and human cells demonstrate that CC chemokines like CCL2, CCL3, CCL4, CCL5, CCL19, CCL20, CCL21, and CCL22 and their related chemokine receptors (i.e., CCR2 [for CCL2], CCR4 [for CCL2, CCL3, CCL5, and CCL22], CCR5 [for CCL2 to CCL5], CCR6 [for CCL20], and CCR7 [for CCL19, CCL21]) regulate migration and activation of various immune cells like monocytes, macrophages, DCs and T lymphocytes by chemotaxis (Mendez-Samperio, 2008). In particular, the use of CCL5 deficient mice confirmed the role of this chemokine in M.tb infection. CCL5 deficient mice infected with a low dose of aerosolized M.tb are described to present a decrease in recruitment of lymphocytes to TB granulomas site along with an impaired early acquired immunity (Vesosky et al., 2010). Human studies involving bronchoalveolar lavage fluid and tissue from TB patients also demonstrate the involvement of CXCL8 (or IL-8), CCL2, and CCL5 during the acute phase of this disease (Kurashima et al., 1997).
Three major cellular receptor-dependent intracellular signaling pathways regulating chemokine secretion in response to mycobacterial infections have been described (reviewed in detail in Mendez-Samperio, 2008). Mitogen-activated protein kinases (MAPKs) p38 and extracellular signal-regulated kinases (ERK)1/2 are shown to be phosphorylated by a variety of stimuli leading to a cell type and stimulus-specific dependent production of chemokines (Roach and Schorey, 2002). NFκB, a key molecule in inflammation regulation, is also involved in the regulation of chemokine gene transcription (Mendez-Samperio, 2008) and subsequent secretion. In this context, specific heat-stable cell wall components of M.tb are described to induce chemokine secretion in specific cell types differentially expressing defined receptors (Jones et al., 2001). This is the case for ManLAM, which induced chemokine secretion in monocytes, but not in AMs (Barnes et al., 1992). The fact that the MR, a major receptor for ManLAM, is present on AMs but not in monocytes, may favor ManLAM signaling through TLR2 and TLR4 in monocytes leading to chemokine secretion. Recent contradictory studies also established a role for TNF as a controller for chemokine secretion depending of the cell type and the mycobacterial strain (stimulus-specific) studied (Juffermans et al., 1999; Mendez-Samperio et al., 2003). For example, chemokine production induced by M. bovis BCG does not seem to be controlled by TNF (Mendez-Samperio et al., 2003), however, M.tb-infected macrophages reduce inflammatory chemokine secretion after TNF neutralization (Algood et al., 2004). The controversy also appears when the debate is who regulates who. Are cytokines regulating chemokines or contrary to the current view, are chemokines controlling the host response to infection? Kinetic studies closely evaluating levels of cytokines and chemokines in M.tb-infected host are necessary to address this issue. In this context, a recent study using M. bovis BCG showed that both cytokine and chemokine secretions seem to be tightly regulated, where a relationship between CCL2, CCL3, and TNF production exist in early stages of infection; and the same is described for CXCL10 and IL6 and IL10 in intermediate phases, and for CCL22 and IFN-γ in late stages of infection (Mendez-Samperio, 2008). As CCL2 has been recently shown to inhibit the pro-inflammatory cytokine IL-12p40 during M.tb infection (Flores-Villanueva et al., 2005), the regulatory role of chemokines controlling inflammation may dictate the establishment and outcome of M.tb infection. Current literature supporting this concept indicates that several chemokines trigger the activation of signaling cascades like MAPK/c-Jun N-terminal kinase (MAPK/JNK) and PI3K pathways associated with cell mitogenesis, chemotaxis, and cell activation (Mendez-Samperio, 2008). This is described for CCL2, which interacts with CCR2 activating different MAPK-ERK1/2 cascades involved in CCL2-associated cell immune responses against mycobacterial infections (Ashida et al., 2001). The importance of these regulatory pathways are emphasized in a recent study where MAPK-p38 and ERK1/2 activities can control intracellular mycobacterial replication within the macrophage independently of TNF and/or IL-10 mediated effects (Klug et al., 2010).
Other members of the CXC chemokine family are also implicated in controlling the inflammatory conditions following infection by M.tb. This is the case for CXCL1, CXCL7, CXCL8, CXCL9, and CXCL10 (reviewed in detail in Mendez-Samperio, 2008). In all cases, these CXC chemokines are, as in the case for CC chemokines, secreted depending on the stimulus and cell-type. Probably among the CXC chemokines CXCL8 (or IL-8) is the most studied in the regulation of M.tb infection. CXCL8 is secreted by different types of host cells in response to virulent M.tb infection recruiting inflammatory cells (i.e., neutrophils, lymphocytes, monocytes) to the infection site, and stimulating bactericidal non-oxidative mechanisms (Nibbering et al., 1993; Pace et al., 1999). Thus, chemokines triggered by M.tb infection regulate the innate immune events laying the foundation for the establishment of the subsequent host adaptive immune response.
Decoy Receptors
These rare receptors are structurally similar to conventional chemokine receptors but without the intrinsic capability to trigger signaling events. Although only three of these receptors have been identified in mammals, D6 has been intensely studied due to its capacity to be recognized by multiple chemokines during inflammation (reviewed in Di et al., 2009). In this context, studies challenging D6 deficient mice with complete Freund’s adjuvant described an extensive inflammation characterized by leukocyte infiltration with localized areas of severe necrosis (Martinez de la et al., 2005). M.tb-infected D6 deficient mice presented a similar phenotype, also characterized by a substantial leukocyte infiltration causing vast local and systemic production and accumulation of inflammatory chemokines and cytokines resulting in an overwhelming inflammation followed by severe tissue damage (Di et al., 2008). These findings support the initial concept that D6 is a receptor assisting with control of inflammation by scavenging circulating pro-inflammatory CC chemokines, but its action is dependent on the inflammatory conditions in the host environment. Another decoy receptor gaining interest in the field of TB research is Tir8. Tir8 is described to participate in the tight regulation of the immune response by inhibiting TLR/IL-1 NFκB activation (Polentarutti et al., 2003). In vivo studies using Tir8 deficient mice showed that, like D6 deficient mice, Tir8 play a role in damping the immure response against M.tb infection (Di et al., 2009).
Protective Pro-Inflammatory Mediators
Inflammation and immune reaction in response to most pathogens is partly mediated by a group of secreted polypeptides known as cytokines. Not only is the inflammatory response generated by cytokines against M.tb helping in the control of the infection, but they also play an important role during the chronic infection stage dictating the pathogenesis of the disease (Flynn and Chan, 2001; Cooper, 2009). The fate of M.tb infection is determined by the interplay between various cytokines released at different time points of infection and the effect of these cytokines on the host cell and M.tb. In this section, we will present the main cytokines involved in chronic and cell-mediated inflammatory responses to M.tb infection.
Tumor Necrosis Factor
Tumor necrosis factor, an important component of innate immune mechanisms of the host against pathogens, is shown to be critical in the control of M.tb. TNF is an autocrine cytokine produced by macrophages, DCs and T cells, and performs functions like chemotaxis and granuloma formation. TNF, if not regulated can lead to tissue damage and cause immunopathology. The most widely studied models to understand the role of TNF in vivo are human TNF transgenic and TNF gene deficient mice. Studies using TNF and lymphotoxin-α (LT-α) deficient mice demonstrated that complete disruption of TNF and LT-α reduces host resistance to mycobacterial infection due to a delay and deficient granuloma formation leading to a widespread dissemination of M.tb (Bean et al., 1999; Jacobs et al., 2000). In this regard, TNF deficient mice are characterized for containing immature lymphocyte cells concentrated in the perivascular and peribronchial areas surrounding granulomas. Moreover, a deficiency in chemokine induction and cellular recruitment has also been established in this animal model system (Roach et al., 2002). Thus, disrupting the TNF balance leads to tissue damage and necrosis observed in many pathological features of disseminated TB. This TNF balance is critical to maintain as other studies showed that continued presence of TNF has also growth promoting effects on M.tb (Byrd, 1997).
Lymphotoxin
Studies done so far have been inconclusive about the unique role played by lymphotoxin in the host immune response to M.tb. However, a recent study done using both conventional and neo-free LT-α deficient mice indicates a non-essential role for LT-α in presence of unperturbed TNF expression in host defense to M.tb infection (Allie et al., 2010).
IL-12 Family
This family is composed of IL-12p40 (homodimer with p40 + p40 subunits), IL-12 or IL-12p70 (heterodimer with p40 + p35 subunits), IL-23 (heterodimer with p40 + p19 subunits), and IL-27 (heterodimer cytokine with Epstein–Barr virus-induced gene 3 [EBI3 or IL-27B] + p28 [known as IL-30]). IL-12Rβ1/IL-12Rβ2 complex is the receptor for IL-12, IL-12β1/IL-23R complex is the receptor for IL-23 while gp130/IL-27α complex is the receptor for IL-27 (Trinchieri et al., 2003; Hunter, 2005). Experimental models and human population studies have established that IL-12 plays an important role in both innate and adaptive responses against M.tb infection. IL-12 is shown to bind to its receptor IL-12R-β2 and activate the JAK-STAT pathway inducing IFN-γ to differentiate CD4+ T cells in Th1 effectors (Mendez-Samperio, 2010). Recent studies established a great level of complexity on the IL-12 family cytokines in their involvement in the immunological control of M.tb infection (reviewed in detail in Cooper et al., 2007). IL-12 is induced in macrophages and DCs after activation by microbial TLR ligands and other cytokines. Experiments using IL-12 deficient mice show an increase in bacterial burden and a drastic reduction in host survival (Flynn et al., 1995b; Cooper et al., 1997). These studies established IL-12 as a crucial cytokine in the development and maintenance of the type-1 cellular response to M.tb infection. IL-23 induces IL-17 production by memory T cells inducing the inflammatory response by Th17 cells (Khader and Cooper, 2008). IL-23 like IL-12 generates protective cellular responses but at lesser degree, however, IL-27 is shown to moderate inflammation by inducing the early Th1 differentiation and generation of IL-10-producing regulatory T cells, which results in limited tissue destruction but subsequently limited bacterial control (Khader and Cooper, 2008). Thus, the IL-12 family is implicated in maintaining the balance between inflammation and bacterial killing to minimize tissue damage. A new role for IL-12p35 has been recently described, where this cytokine induces inflammation by suppressing TGF-β and stimulating NKT cells (Park et al., 2010). Other recent studies also support this new role showing that IL-12p35 alone also drives neutrophil and CD4+ T cell infiltrations by increasing the levels of CCL2, CXCL2, CXCL3, and the angiogenic factor vascular endothelial growth factor (VEGF; Frank et al., 2010). Recently, it has been shown that the p35 subunit of IL-12 is forming a heterodimer with EBI3 resulting in IL-35. IL-35 is defined as an IL-12 family cytokine that is produced only by regulatory T cells contributing to immune suppression (Collison et al., 2007). The role of IL-35 in M.tb infection is still to be determined.
Interferon Family
The IFN are a heterogeneous family of cytokines divided in type I and type II on basis of their structure, function, and cell of origin. While type I IFN (IFN-α and IFN-β) is secreted by various cell types and through innate immune receptors, type II (IFN-γ) is mainly produced as a result of stimulation of T lymphocytes and NK cells.
Type I IFN regulates different aspects of the immune response, inducing cell-mediated immunity. Upon infection with M.tb, immune cells and receptors induce the production of IFN-α and INF-β promoting the priming of CD8+ and CD4+ T cells (Cho et al., 2002; Remoli et al., 2002). The effect of IFN-α/β may be both favorable and unfavorable to the infected host cell. Studies using exogenous administration of IFN-α/β have shown that increased production of IFN-α/β could contribute to the host susceptibility to M.tb infection due to an overwhelming inflammatory response (Manca et al., 2001; Bouchonnet et al., 2002). However, a recent study has demonstrated that M.tb inhibits the production of IFN-α/β in response to TLR9 signaling suggesting an evasion mechanism to control the immune response against M.tb that is beneficial to the host by controlling an excessive immune response (Simmons et al., 2010).
Type II IFN-γ released by AMs, CD4 T cells, CD8 T cells, NKT cells, γδ T cells, and NK cells (Boehm et al., 1997; Fenton et al., 1997; Wang et al., 1999; Vesosky et al., 2004) is considered as the key cytokine involved in the control of M.tb (Flynn and Chan, 2001; Cooper, 2009). IFN-γ activates macrophages enhancing their production of pro-inflammatory cytokines, and up-regulating their surface expression of cytokine/chemokine receptors, costimulatory and adhesion molecules, and MHC-I and II molecules enhancing macrophage antigen presentation to T cells (Boehm et al., 1997). IFN-γ also controls cellular immunity by regulating T cell-mediated apoptosis (Li et al., 2007) as well as accumulation of Th17 cells (Sher and Coffman, 1992), thus protecting the host from a massive T cell influx to the infection site. IFN-γ is defined as a correlate for host resistance to M.tb infection. In this scenario, antigen-specific IFN-γ is low in patients with clinical cases of active TB, when compared to latent TB (Zhang et al., 1995; Lin et al., 1996). It has also been reported that individuals defective for IFN-γ or IFN-γ receptors (IFNGR) present increased susceptibility to infection with severe disease manifestations (de Jong et al., 1998), suggesting that insufficient IFN-γ is associated with TB disease progression. Two independent studies using a sub-lethal infection with M.tb showed that IFN-γ deficient mice failed to produce nitric oxide due to inadequate iNOS expression, and exhibited unrestricted bacterial growth and tissue necrosis with mice succumbing to disease faster (Cooper et al., 1993; Flynn et al., 1993). These studies prove the essentiality of IFN-γ in protective cellular immunity to TB infection, where cessation of bacterial growth directly correlates with the release of IFN-γ (Cooper et al., 1993).
Other Pro-Inflammatory Cytokines
Other cytokines like IL-1, IL-6, IL-17, and IL-32 have been shown to be implicated to different degrees in M.tb infection control. IL-1β is a pro-inflammatory cytokine produced by monocytes, macrophages, DCs, B cells, and NK cells (Dinarello, 2009).
Interleukin-1β up-regulates essential mediators necessary for the control M.tb infection including iNOS and subsequent NO production (Chan et al., 2001), phagosomal acidification and maturation (Horsburgh Jr., 1991), adhesion molecules (Dinarello, 2009), and enzymatic activities like phospholipase A2 and cyclooxygenase (Hernandez-Pando et al., 2006; Yang et al., 2009). Human studies (Gomez et al., 2006; Hawn et al., 2006; Settas et al., 2007) and in vivo studies using IL-1Ra (receptor antagonist) and IL-1α/IL-1β deficient mice (Juffermans et al., 2000; Yamada et al., 2000; Fremond et al., 2007) established that the continuous effect of IL-1 is required for maintaining resistance to M.tb infection. IL-6 is mainly considered to be a pro-inflammatory cytokine involved in macrophage and cytotoxic T cell differentiation. Infection of IL-6 deficient mice with M.tb indicate that IL-6 is important for the control of M.tb infection, however, in humans it is considered a correlate for disease progression due to its role in inflammation and tissue damage (Appelberg, 1994; Ladel et al., 1997; Casarini et al., 1999; Tsao et al., 1999; Ilonidis et al., 2006).
Interleukin-17 is produced abundantly by Th17 CD4 T cells during early stages of M.tb infection (Lockhart et al., 2006). Studies using IL-17 deficient mice demonstrate that IL-17 is not essential to control M.tb infection, however, this cytokine plays a role in granuloma formation (Umemura et al., 2007). In this context, IL-17 has been implicated in the recruitment of Th1 cells (Happel et al., 2005; Khader et al., 2005; Wozniak et al., 2006), which produce antigen-specific IFN-γ, and inhibit M.tb growth (Khader et al., 2007). Human studies show that IL-17 is produced by TB patients and IL-17 is also induced by BCG vaccination, however, its protective role against M.tb infection in human remains to be elucidated.
Interleukin-32 is a cell-associated proinflammatory cytokine, which is specifically stimulated by mycobacteria through a caspase-1- and IL-18-dependent production of IFNγ (Netea et al., 2006).
Immunomodulatory Cytokines
Interleukin-10
Cell types producing IL-10 include monocytes, macrophages, DCs, regulatory CD4 T cells, and CD8 T cells (Sabat, 2010). The role of IL-10 is complicated to decipher as its regulatory properties depend on the cell type and infection model studied. For M.tb infection, IL-10 is thought to limit inflammation. In this scenario, IL-10 inhibits pro-inflammatory cytokines like IL-1, IL-6, IL-12, IL-18, and IFN-γ (Bogdan et al., 1991; de Waal Malefyt et al., 1991, 1993; Fiorentino et al., 1991; D’Andrea et al., 1993; Gruber et al., 1994; Aste-Amezaga et al., 1998); and chemokines like CCL3, CCL4, and CCL5 affecting cell recruitment, as well as blocking the generation of ROIs and NOIs (Bogdan et al., 1991; Cunha et al., 1992; Niiro et al., 1992; Cenci et al., 1993; Kuga et al., 1996; Roilides et al., 1997) that are important for M.tb control. In this process IL-10 is thought to work by interfering with intracellular signaling cascades such as suppressor of cytokine signaling-3 (SOCS3; Cassatella et al., 1999) and NFκB (Wang et al., 1995; Romano et al., 1996; Schottelius et al., 1999; Berrebi et al., 2003). IL-10 is also implicated in blocking antigen processing and presentation in different APCs, therefore diminishing T cell responses (Moore et al., 2001). Experimental data using the mouse model show that IL-10 alone plays a pivotal role during the chronic/latent stage of pulmonary TB potentially contributing to TB reactivation (Turner et al., 2002; Beamer et al., 2008). In this context, induction of IL-10 may be considered as preventing tissue damage during chronic M.tb infection. Studies using transgenic mice support this funding where induction of IL-10 is considered to prevent tissue damage but also contributes to M.tb growth (Murray et al., 1997). Studies using IL-10 deficient mice show no effect (North, 1998) or a protective effect when IL-10 is absent, which is also supported by studies blocking the action of IL-10 receptor (Beamer et al., 2008; Redford et al., 2010). These murine studies are supported by recent human population studies where presence of IL-10 correlates with susceptibility to mycobacterial infections (Boussiotis et al., 2000; de la Barrera et al., 2004). Recent in vitro reports suggest that IL-10 may interfere with M.tb persistence by stalling phagosome maturation in human macrophages (O’Leary et al., 2010). Thus, the benefits of IL-10 are controversial, as at the same time that IL-10 seems to limit tissue damage by suppressing inflammation, IL-10 also contributes to the host environment that allows M.tb to persist, and thus IL-10 directly contributes to reactivation of TB.
Transforming Growth Factor β
Transforming growth factor β is defined as a pluripotent cytokine that modulates the immune response by down-regulating acquired immunity and de-activating macrophages. TGF-β is secreted by a large number of cell types including monocytes, macrophages, DCs, and CD4+ regulatory T cells (Aung et al., 2005; Latchumanan et al., 2005). TGF-β is shown to synergize with IL-10 to promote immune tolerance and limit pathological inflammation (Oswald et al., 1992; Zeller et al., 1999; Clegg and Hughes, 2002; Chen et al., 2003). Similar to IL-10, TGF-β also suppresses APCs costimulatory molecules (Strobl and Knapp, 1999), NOS production, and indirectly down-regulates T cell function and proliferation by inhibiting cytokine production (Lee et al., 1997; Nakao et al., 1997; Bright and Sriram, 1998; Gorham et al., 1998). Studies have shown up-regulation of TGF-β in monocytes and macrophages in granuloma from patients with active TB (Toossi et al., 1995), and high levels of TGF-β is directly associated with severe stages of the disease. As TGF-β is considered an anti-inflammatory cytokine that helps to reduce the harmful inflammatory effects associated with T cell immunity to M.tb infection, regulating TGF-β effects may be critical to control M.tb infection. In this scenario, using TGF-β blockers like latency associated protein (Saharinen and Keski-Oja, 2000) and decorin (Hildebrand et al., 1994), which form inactive complexes with TGF-β should reduce TGF-β anti-inflammatory effects on T cells and macrophages, and may be a way to control inflammation and prevent M.tb infection spread.
Overall, the local response to M.tb infection is defined by an initial intense pro-inflammatory response followed by the production of anti-inflammatory mediators that serve to regulate tissue damage. Within this local environment M.tb battles to survive by interfering with the host inflammatory networks. It remains unclear whether the neutralization of anti-inflammatory mediators will benefit the host in contrast to the current goals of boosting Th1 mediated protective immunity.
Conclusion Remarks
The interplay between M.tb and the host inflammatory response depends on many factors. From the bacterial side, M.tb strains differ in the cell wall components exposed on their surface. Virulent laboratory strains H37Rv and Erdman are defined to be hypermannosylated on their surface; however, clinical isolates, some of them related to cases of hypervirulence and clinical outbreaks (i.e., HN878), present a more hydrophobic cell wall surface (Torrelles and Schlesinger, 2010). In this context, hypervirulent strains are characterized by the presence of other surface-exposed cell wall components (i.e., phenolic glycolipid, triacylglycerols), which regulate the cytokine response, and demonstrate rapid intracellular growth and marked tissue damage. These hypervirulent strains are associated with an unusual high proportion of active cases of disease and a high frequency of extrapulmonary disease. This has been directly attributed to host immune subversion. In contrast, M.tb strain CDC1551 is related to a low number of cases of active disease, followed by an unusually high rate of seroconversion, inducing a more rigorous immunologic response (reviewed in Torrelles and Schlesinger, 2010). Even within the same M.tb strain, it is likely not the case that all bacilli are identical replicas, and thus bacilli may differentially interact with the host. Thus, at an M.tb infection site we may find a mixture of events, where simplistically speaking, depending on its cell wall surface phenotype an M.tb bacillus may interact with a host cell receptor triggering pro-inflammation that promotes bacterial killing, or may interact with a host cell receptor triggering anti-inflammation and bacterial intracellular survival. From the host (man) perspective, genetic predisposition and environmental living conditions dictate the predilection for an M.tb infection. Even in the context of the same host (man) and within the same host cell population, differences between host cells in receptor expression, signaling and innate and adaptive immune functions exist (and we can extend these differences to the different model systems used to study M.tb infection in vitro and in vivo). Moreover, within the same host cell population (i.e., AMs), there are likely to be different host cell subpopulation(s) that differentially trigger the inflammatory response (i.e., AMs expressing more TLR2 vs. AMs expressing more MR). With these questions in mind, is the establishment of a successful M.tb infection just a question of chance or are we looking at the evolutionarily developed perfect storm of elements? In this review we have presented the major players involved in the tight regulation of the inflammatory response during M.tb infection. Some favor infection and others do not. However, upon the successful establishment of an M.tb infection, the ultimate goal of the host is to reduce inflammation and tissue destruction, and in this scenario, M.tb has learned to survive. Efforts to define the M.tb cell wall components produced in vivo as well as the properties of the M.tb host cell reservoir in vivo will establish the optimal M.tb and host cell phenotype/genotype combination that allows for the successful establishment of an M.tb infection in vivo. In this scenario, the understudied microenvironments that M.tb encounters during infection may be key elements to consider in future M.tb pathogenesis studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Joanne Turner and Larry S. Schlesinger for their careful reading of this review and helpful suggestions with the text. Work in the Torrelles lab is supported by grants to Jordi B. Torrelles from the NIH (AI-073856) and the Francis B. Parker Fellowship Program.
References
Algood, H. M., Chan, J., and Flynn, J. L. (2003). Chemokines and tuberculosis. Cytokine Growth Factor Rev. 14, 467–477.
Algood, H. M., Lin, P. L., Yankura, D., Jones, A., Chan, J., and Flynn, J. L. (2004). TNF influences chemokine expression of macrophages in vitro and that of CD11b+ cells in vivo during Mycobacterium tuberculosis infection. J. Immunol. 172, 6846–6857.
Allie, N., Keeton, R., Court, N., Abel, B., Fick, L., Vasseur, V., Vacher, R., Olleros, M. L., Drutskaya, M. S., Guler, R., Nedospasov, S. A., Garcia, I., Ryffel, B., Quesniaux, V. F., and Jacobs, M. (2010). Limited role for lymphotoxin alpha in the host immune response to Mycobacterium tuberculosis. J. Immunol. 185, 4292–4301.
Appelberg, R. (1994). Protective role of interferon gamma, tumor necrosis factor alpha and interleukin-6 in Mycobacterium tuberculosis and M. avium infections. Immunobiology 191, 520–525.
Appelmelk, B. J., den Dunnen, J., Driessen, N. N., Ummels, R., Pak, M., Nigou, J., Larrouy-Maumus, G., Gurcha, S. S., Movahedzadeh, F., Geurtsen, J., Brown, E. J., Eysink Smeets, M. M., Besra, G. S., Willemsen, P. T., Lowary, T. L., van, K. Y., Maaskant, J. J., Stoker, N. G., van der, L. P., Puzo, G., Vandenbroucke-Grauls, C. M., Wieland, C. W., Van Der, P. T., Geijtenbeek, T. B., van der Sar, A. M., and Bitter, W. (2008). The mannose cap of mycobacterial lipoarabinomannan does not dominate the Mycobacterium–host interaction. Cell. Microbiol. 10, 930–944.
Ashida, N., Arai, H., Yamasaki, M., and Kita, T. (2001). Distinct signaling pathways for MCP-1-dependent integrin activation and chemotaxis. J. Biol. Chem. 276, 16555–16560.
Astarie-Dequeker, C., N’Diaye, E. N., Le Cabec, V., Rittig, M. G., Prandi, J., and Maridonneau-Parini, I. (1999). The mannose receptor mediates uptake of pathogenic and nonpathogenic mycobacteria and bypasses bactericidal responses in human macrophages. Infect. Immun. 67, 469–477.
Aste-Amezaga, M., Ma, X., Sartori, A., and Trinchieri, G. (1998). Molecular mechanisms of the induction of IL-12 and its inhibition by IL-10. J. Immunol. 160, 5936–5944.
Aung, H., Wu, M., Johnson, J. L., Hirsch, C. S., and Toossi, Z. (2005). Bioactivation of latent transforming growth factor beta1 by Mycobacterium tuberculosis in human mononuclear phagocytes. Scand. J. Immunol. 61, 558–565.
Austin, C. M., Ma, X., and Graviss, E. A. (2008). Common nonsynonymous polymorphisms in the NOD2 gene are associated with resistance or susceptibility to tuberculosis disease in African Americans. J. Infect. Dis. 197, 1713–1716.
Barnes, P. F., Chatterjee, D., Abrams, J. S., Lu, S., Wang, E., Yamamura, M., Brennan, P. J., and Modlin, R. L. (1992). Cytokine production induced by Mycobacterium tuberculosis lipoarabinomannan. Relationship to chemical structure. J. Immunol. 149, 541–547.
Beamer, G. L., Flaherty, D. K., Assogba, B. D., Stromberg, P., Gonzalez-Juarrero, M., de Waal, M. R., Vesosky, B., and Turner, J. (2008). Interleukin-10 promotes Mycobacterium tuberculosis disease progression in CBA/J mice. J. Immunol. 181, 5545–5550.
Bean, A. G. D., Roach, D. R., Briscoe, H., France, M. P., Korner, H., Sedgwick, J. D., and Britton, W. J. (1999). Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J. Immunol. 162, 3504–3511.
Beharka, A. A., Gaynor, C. D., Kang, B. K., Voelker, D. R., McCormack, F. X., and Schlesinger, L. S. (2002). Pulmonary surfactant protein A up-regulates activity of the mannose receptor, a pattern recognition receptor expressed on human macrophages. J. Immunol. 169, 3565–3573.
Behr, M. A. (2004). Tuberculosis due to multiple strains: a concern for the patient? A concern for tuberculosis control? Am. J. Respir. Crit. Care Med. 169, 554–555.
Berrebi, D., Bruscoli, S., Cohen, N., Foussat, A., Migliorati, G., Bouchet-Delbos, L., Maillot, M. C., Portier, A., Couderc, J., Galanaud, P., Peuchmaur, M., Riccardi, C., and Emilie, D. (2003). Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood 101, 729–738.
Boehm, U., Klamp, T., Groot, M., and Howard, J. C. (1997). Cellular responses to interferon-gamma. Annu. Rev. Immunol. 15, 749–795.
Bogdan, C., Vodovotz, Y., and Nathan, C. (1991). Macrophage deactivation by interleukin 10. J. Exp. Med. 174, 1549–1555.
Bouchonnet, F., Boechat, N., Bonay, M., and Hance, A. J. (2002). Alpha/beta interferon impairs the ability of human macrophages to control growth of Mycobacterium bovis BCG. Infect. Immun. 70, 3020–3025.
Boussiotis, V. A., Tsai, E. Y., Yunis, E. J., Thim, S., Delgado, J. C., Dascher, C. C., Berezovskaya, A., Rousset, D., Reynes, J. M., and Goldfeld, A. E. (2000). IL-10-producing T cells suppress immune responses in anergic tuberculosis patients. J. Clin. Invest. 105, 1317–1325.
Bright, J. J., and Sriram, S. (1998). TGF-beta inhibits IL-12-induced activation of Jak-STAT pathway in T lymphocytes. J. Immunol. 161, 1772–1777.
Brooks, M. N., Rajaram, M. V., Azad, A. K., Amer, A. O., Valdivia-Arenas, M. A., Park, J. H., Nuñez, G., and Schlesinger, L. S. (2010). NOD2 controls the nature of the inflammatory response and subsequent fate of Mycobacterium tuberculosis and M. bovis BCG in human macrophages. Cell. Microbiol. doi: 10.1111/j.1462-5822.2010.01544.x. [Epub ahead of print].
Bulut, Y., Michelsen, K. S., Hayrapetian, L., Naiki, Y., Spallek, R., Singh, M., and Arditi, M. (2005). Mycobacterium tuberculosis heat shock proteins use diverse Toll-like receptor pathways to activate pro-inflammatory signals. J. Biol. Chem. 280, 20961–20967.
Byrd, T. F. (1997). Tumor necrosis factor alpha (TNFalpha) promotes growth of virulent Mycobacterium tuberculosis in human monocytes: iron-mediated growth suppression is correlated with decreased release of TNF-alpha from iron-treated infected monocytes. J. Clin. Invest. 99, 2518–2529.
Capuano, S. V. III, Croix, D. A., Pawar, S., Zinovik, A., Myers, A., Lin, P. L., Bissel, S., Fuhrman, C., Klein, E., and Flynn, J. L. (2003). Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect. Immun. 71, 5831–5844.
Carlson, T. K., Torrelles, J. B., Smith, K., Horlacher, T., Castelli, R., Seeberger, P. H., Crouch, E. C., and Schlesinger, L. S. (2009). Critical role of amino acid position 343 of surfactant protein-D in the selective binding of glycolipids from Mycobacterium tuberculosis. Glycobiology 19, 1473–1484.
Casarini, M., Ameglio, F., Alemanno, L., Zangrilli, P., Mattia, P., Paone, G., Bisetti, A., and Giosue, S. (1999). Cytokine levels correlate with a radiologic score in active pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 159, 143–148.
Cassatella, M. A., Gasperini, S., Bovolenta, C., Calzetti, F., Vollebregt, M., Scapini, P., Marchi, M., Suzuki, R., Suzuki, A., and Yoshimura, A. (1999). Interleukin-10 (IL-10) selectively enhances CIS3/SOCS3 mRNA expression in human neutrophils: evidence for an IL-10-induced pathway that is independent of STAT protein activation. Blood 94, 2880–2889.
Cenci, E., Romani, L., Mencacci, A., Spaccapelo, R., Schiaffella, E., Puccetti, P., and Bistoni, F. (1993). Interleukin-4 and interleukin-10 inhibit nitric oxide-dependent macrophage killing of Candida albicans. Eur. J. Immunol. 23, 1034–1038.
Chackerian, A., Alt, J., Perera, V., and Behar, S. M. (2002). Activation of NKT cells protects mice from tuberculosis. Infect. Immun. 70, 6302–6309.
Chan, E. D., Chan, J., and Schluger, N. W. (2001). What is the role of nitric oxide in murine and human host defense against tuberculosis? Current knowledge. Am. J. Respir. Cell Mol. Biol. 25, 606–612.
Chen, Z. M., O’Shaughnessy, M. J., Gramaglia, I., Panoskaltsis-Mortari, A., Murphy, W. J., Narula, S., Roncarolo, M. G., and Blazar, B. R. (2003). IL-10 and TGF-beta induce alloreactive CD4+ CD25− T cells to acquire regulatory cell function. Blood 101, 5076–5083.
Chieppa, M., Bianchi, G., Doni, A., Del Prete, A., Sironi, M., Laskarin, G., Monti, P., Piemonti, L., Biondi, A., Mantovani, A., Introna, M., and Allavena, P. (2003). Cross-linking of the mannose receptor on monocyte-derived dendritic cells activates an anti-inflammatory immunosuppressive program. J. Immunol. 171, 4552–4560.
Cho, H. J., Hayashi, T., Datta, S. K., Takabayashi, K., Van Uden, J. H., Horner, A., Corr, M., and Raz, E. (2002). IFN-alpha beta promote priming of antigen-specific CD8+ and CD4+ T lymphocytes by immunostimulatory DNA-based vaccines. J. Immunol. 168, 4907–4913.
Clegg, S., and Hughes, K. T. (2002). FimZ is a molecular link between sticking and swimming in Salmonella enterica serovar Typhimurium. J. Bacteriol. 184, 1209–1213.
Co, D. O., Hogan, L. H., Kim, S. I., and Sandor, M. (2004). Mycobacterial granulomas: keys to a long-lasting host–pathogen relationship. Clin. Immunol. 113, 130–136.
Collison, L. W., Workman, C. J., Kuo, T. T., Boyd, K., Wang, Y., Vignali, K. M., Cross, R., Sehy, D., Blumberg, R. S., and Vignali, D. A. (2007). The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450, 566–569.
Cooper, A. M. (2009). Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 27, 393–422.
Cooper, A. M., Dalton, D. K., Stewart, T. A., Griffin, J. P., Russell, D. G., and Orme, I. M. (1993). Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 178, 2243–2247.
Cooper, A. M., Magram, J., Ferrante, J., and Orme, I. M. (1997). Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with Mycobacterium tuberculosis. J. Exp. Med. 186, 39–45.
Cooper, A. M., Roberts, A. D., Rhoades, E. R., Callahan, J. E., Getzy, D. M., and Orme, I. M. (1995). The role of interleukin-12 in acquired immunity to Mycobacterium tuberculosis infection. Immunology 84, 423–432.
Cooper, A. M., Solache, A., and Khader, S. A. (2007). Interleukin-12 and tuberculosis: an old story revisited. Curr. Opin. Immunol. 19, 441–447.
Cunha, F. Q., Moncada, S., and Liew, F. Y. (1992). Interleukin-10 (IL-10) inhibits the induction of nitric oxide synthase by interferon-gamma in murine macrophages. Biochem. Biophys. Res. Commun. 182, 1155–1159.
D’Andrea, A., Aste-Amezaga, M., Valiante, N. M., Ma, X., Kubin, M., and Trinchieri, G. (1993). Interleukin 10 (IL-10) inhibits human lymphocyte interferon gamma-production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J. Exp. Med. 178, 1041–1048.
Daley, C. L. (2010). “Tuberculosis latency in humans,” in Tuberculosis, eds W. N. Rom and S. M. Garay (Philadelphia: Lippincott Williams & Wilkins), 85–99.
de Jong, R., Altare, F., Haagen, I. A., Elferink, D. G., Boer, T., van Breda, V., Kabel, P. J., Draaisma, J. M., Van Dissel, J. T., Kroon, F. P., Casanova, J. L., and Ottenhoff, T. H. (1998). Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science 280, 1435–1438.
de la Barrera, S., Aleman, M., Musella, R., Schierloh, P., Pasquinelli, V., Garcia, V., Abbate, E., and Sasiain, M. C. (2004). IL-10 down-regulates costimulatory molecules on Mycobacterium tuberculosis-pulsed macrophages and impairs the lytic activity of CD4 and CD8 CTL in tuberculosis patients. Clin. Exp. Immunol. 138, 128–138.
de Waal Malefyt, R., Abrams, J., Bennett, B., Figdor, C. G., and De Vries, J. E. (1991). Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 174, 1209–1220.
de Waal Malefyt, R., Figdor, C. G., Huijbens, R., Mohan-Peterson, S., Bennett, B., Culpepper, J., Dang, W., Zurawski, G., and De Vries, J. E. (1993). Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes: comparison with IL-4 and modulation by IFN-gamma or IL-10. J. Immunol. 151, 6370–6381.
Dheda, K., Booth, H., Huggett, J. F., Johnson, M. A., Zumla, A., and Rook, G. A. (2005). Lung remodeling in pulmonary tuberculosis. J. Infect. Dis. 192, 1201–1209.
Di, L. D., Caccamo, N., Meraviglia, S., Guggino, G., La Manna, M. P., Sireci, G., Salerno, A., and Dieli, F. (2009). Tuning inflammation in tuberculosis: the role of decoy receptors. Microbes Infect. 11, 821–827.
Di, L. D., Locati, M., Caccamo, N., Vecchi, A., Meraviglia, S., Salerno, A., Sireci, G., Nebuloni, M., Caceres, N., Cardona, P. J., Dieli, F., and Mantovani, A. (2008). Role of the chemokine decoy receptor D6 in balancing inflammation, immune activation, and antimicrobial resistance in Mycobacterium tuberculosis infection. J. Exp. Med. 205, 2075–2084.
Dinarello, C. A. (2009). Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550.
Divangahi, M., Desjardins, D., Nunes-Alves, C., Remold, H. G., and Behar, S. M. (2010). Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat. Immunol. 11, 751–758.
Divangahi, M., Mostowy, S., Coulombe, F., Kozak, R., Guillot, L., Veyrier, F., Kobayashi, K. S., Flavell, R. A., Gros, P., and Behr, M. A. (2008). NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J. Immunol. 181, 7157–7165.
Dommett, R. M., Klein, N., and Turner, M. W. (2006). Mannose-binding lectin in innate immunity: past, present and future. Tissue Antigens 68, 193–209.
Drennan, M. B., Nicolle, D., Quesniaux, V. J., Jacobs, M., Allie, N., Mpagi, J., Fremond, C., Wagner, H., Kirschning, C., and Ryffel, B. (2004). Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am. J. Pathol. 164, 49–57.
Ehlers, S. (2009). DC-SIGN and mannosylated surface structures of Mycobacterium tuberculosis: a deceptive liaison. Eur. J. Cell Biol. 89, 95–101.
Feng, C. G., Kullberg, M. C., Jankovic, D., Cheever, A. W., Caspar, P., Coffman, R. L., and Sher, A. (2002). Transgenic mice expressing human interleukin-10 in the antigen-presenting cell compartment show increased susceptibility to infection with Mycobacterium avium associated with decreased macrophage effector function and apoptosis. Infect. Immun. 70, 6672–6679.
Fenton, M. J., Riley, L. W., and Schlesinger, L. S. (2005). “Receptor-mediated recognition of Mycobacterium tuberculosis by host cells,” in Tuberculosis and the Tubercle Bacillus, eds S. T. Cole, K. D. Eisenach, D. N. McMurray, and W. R. Jacobs Jr. (New York: ASM Press), 405–426.
Fenton, M. J., Vermeulen, M. W., Kim, S., Burdick, M., Strieter, R. M., and Kornfeld, H. (1997). Induction of gamma interferon production in human alveolar macrophages by Mycobacterium tuberculosis. Infect. Immun. 65, 5149–5156.
Ferguson, J. S., Martin, J. L., Azad, A. K., McCarthy, T. R., Kang, P. B., Voelker, D. R., Crouch, E. C., and Schlesinger, L. S. (2006). Surfactant protein D increases fusion of Mycobacterium tuberculosis-containing phagosomes with lysosomes in human macrophages. Infect. Immun. 74, 7005–7009.
Ferguson, J. S., Voelker, D. R., McCormack, F. X., and Schlesinger, L. S. (1999). Surfactant protein D binds to Mycobacterium tuberculosis bacilli and lipoarabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J. Immunol. 163, 312–321.
Ferguson, J. S., Weis, J. J., Martin, J. L., and Schlesinger, L. S. (2004). Complement protein C3 binding to Mycobacterium tuberculosis is initiated by the classical pathway in human bronchoalveolar lavage fluid. Infect. Immun. 72, 2564–2573.
Ferwerda, G., Girardin, S. E., Kullberg, B. J., Le Bourhis, L., de Jong, D. J., Langenberg, D. M., van Crevel, R., Adema, G. J., Ottenhoff, T. H., Van der Meer, J. W., and Netea, M. G. (2005). NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog. 1, 279–285. doi: 10.1371/journal.ppat.0010034
Fieschi, C., and Casanova, J. L. (2003). The role of interleukin-12 in human infectious diseases: only a faint signature. Eur. J. Immunol. 33, 1461–1464.
Fiorentino, D. F., Zlotnik, A., Mosmann, T. R., Howard, M., and O’Garra, A. (1991). IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 147, 3815–3822.
Flores-Villanueva, P. O., Ruiz-Morales, J. A., Song, C. H., Flores, L. M., Jo, E. K., Montano, M., Barnes, P. F., Selman, M., and Granados, J. (2005). A functional promoter polymorphism in monocyte chemoattractant protein-1 is associated with increased susceptibility to pulmonary tuberculosis. J. Exp. Med. 202, 1649–1658.
Flynn, J. L., and Chan, J. (2005). What’s good for the host is good for the bug. Trends Microbiol. 13, 98–102.
Flynn, J. L., Chan, J., Triebold, K. J., Dalton, D. K., Stewart, T. A., and Bloom, B. R. (1993). An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 178, 2249–2254.
Flynn, J. L., Goldstein, M. M., Chan, J., Triebold, K. J., Pfeffer, K., Lowenstein, C. J., Schreiber, R., Mak, T. W., and Bloom, B. R. (1995a). Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2, 561–572.
Flynn, J. L., Goldstein, M. M., Triebold, K. J., Sypek, J., Wolf, S., and Bloom, B. R. (1995b). IL-12 increases resistance of BALB/c mice to Mycobacterium tuberculosis infection. J. Immunol. 155, 2515–2524.
Franchi, L., Park, J. H., Shaw, M. H., Marina-Garcia, N., Chen, G., Kim, Y. G., and Nunez, G. (2008). Intracellular NOD-like receptors in innate immunity, infection and disease. Cell. Microbiol. 10, 1–8.
Frank, G. M., Divito, S. J., Maker, D. M., Xu, M., and Hendricks, R. L. (2010). A novel p40-independent function of IL-12p35 is required for progression and maintenance of herpes stromal keratitis. Invest. Ophthalmol. Vis. Sci. 51, 3591–3598.
Fremond, C. M., Nicolle, D. M., Torres, D. S., and Quesniaux, V. F. (2003). Control of Mycobacterium bovis BCG infection with increased inflammation in TLR4-deficient mice. Microbes Infect. 5, 1070–1081.
Fremond, C. M., Togbe, D., Doz, E., Rose, S., Vasseur, V., Maillet, I., Jacobs, M., Ryffel, B., and Quesniaux, V. F. (2007). IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J. Immunol. 179, 1178–1189.
Gandotra, S., Jang, S., Murray, P. J., Salgame, P., and Ehrt, S. (2007). Nucleotide-binding oligomerization domain protein 2-deficient mice control infection with Mycobacterium tuberculosis. Infect. Immun. 75, 5127–5134.
Geijtenbeek, T. B., Torensma, R., Van Vliet, S. J., van Duijnhoven, G. C., Adema, G. J., Van Kooyk, Y., and Figdor, C. G. (2000). Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell 100, 575–585.
Geijtenbeek, T. B., Van Vliet, S. J., Koppel, E. A., Sanchez-Hernandez, M., Vandenbroucke-Grauls, C. M., Appelmelk, B., and Van Kooyk, Y. (2003). Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 197, 7–17.
Geldmacher, C., Ngwenyama, N., Schuetz, A., Petrovas, C., Reither, K., Heeregrave, E. J., Casazza, J. P., Ambrozak, D. R., Louder, M., Ampofo, W., Pollakis, G., Hill, B., Sanga, E., Saathoff, E., Maboko, L., Roederer, M., Paxton, W. A., Hoelscher, M., and Koup, R. A. (2010). Preferential infection and depletion of Mycobacterium tuberculosis-specific CD4 T cells after HIV-1 infection. J. Exp. Med. 207, 2869–2881.
Geurtsen, J., Chedammi, S., Mesters, J., Cot, M., Driessen, N. N., Sambou, T., Kakutani, R., Ummels, R., Maaskant, J., Takata, H., Baba, O., Terashima, T., Bovin, N., Vandenbroucke-Grauls, C. M., Nigou, J., Puzo, G., Lemassu, A., Daffe, M., and Appelmelk, B. J. (2009). Identification of mycobacterial alpha-glucan as a novel ligand for DC-SIGN: involvement of mycobacterial capsular polysaccharides in host immune modulation. J. Immunol. 183, 5221–5231.
Gomez, L. M., Camargo, J. F., Castiblanco, J., Ruiz-Narvaez, E. A., Cadena, J., and Anaya, J. M. (2006). Analysis of IL1B, TAP1, TAP2 and IKBL polymorphisms on susceptibility to tuberculosis. Tissue Antigens 67, 290–296.
Gorham, J. D., Guler, M. L., Fenoglio, D., Gubler, U., and Murphy, K. M. (1998). Low dose TGF-beta attenuates IL-12 responsiveness in murine Th cells. J. Immunol. 161, 1664–1670.
Gruber, M. F., Williams, C. C., and Gerrard, T. L. (1994). Macrophage-colony-stimulating factor expression by anti-CD45 stimulated human monocytes is transcriptionally up-regulated by IL-1β and inhibited by IL-4 and IL-10. J. Immunol. 152, 1354–1361.
Gutierrez, O., Pipaon, C., Inohara, N., Fontalba, A., Ogura, Y., Prosper, F., Nunez, G., and Fernandez-Luna, J. L. (2002). Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J. Biol. Chem. 277, 41701–41705.
Happel, K. I., Lockhart, E. A., Mason, C. M., Porretta, E., Keoshkerian, E., Odden, A. R., Nelson, S., and Ramsay, A. J. (2005). Pulmonary interleukin-23 gene delivery increases local T-cell immunity and controls growth of Mycobacterium tuberculosis in the lungs. Infect. Immun. 73, 5782–5788.
Harding, C. V., and Boom, W. H. (2010). Regulation of antigen presentation by Mycobacterium tuberculosis: a role for Toll-like receptors. Nat. Rev. Microbiol. 8, 296–307.
Hawn, T. R., Dunstan, S. J., Thwaites, G. E., Simmons, C. P., Thuong, N. T., Lan, N. T., Quy, H. T., Chau, T. T., Hieu, N. T., Rodrigues, S., Janer, M., Zhao, L. P., Hien, T. T., Farrar, J. J., and Aderem, A. (2006). A polymorphism in Toll-interleukin 1 receptor domain containing adaptor protein is associated with susceptibility to meningeal tuberculosis. J. Infect. Dis. 194, 1127–1134.
Henning, L. N., Azad, A. K., Parsa, K. V., Crowther, J. E., Tridandapani, S., and Schlesinger, L. S. (2008). Pulmonary surfactant protein A regulates TLR expression and activity in human macrophages. J. Immunol. 180, 7847–7858.
Hernandez-Pando, R., Orozco-Esteves, H., Maldonado, H. A., Guilar-Leon, D., Vilchis-Landeros, M. M., Mata-Espinosa, D. A., Mendoza, V., and Lopez-Casillas, F. (2006). A combination of a transforming growth factor-beta antagonist and an inhibitor of cyclooxygenase is an effective treatment for murine pulmonary tuberculosis. Clin. Exp. Immunol. 144, 264–272.
Hildebrand, A., Romaris, M., Rasmussen, L. M., Heinegard, D., Twardzik, D. R., Border, W. A. and Ruoslahti, E. (1994). Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem. J. 302(Pt 2), 527–534.
Hirsch, C. S., Ellner, J. J., Russell, D. G., and Rich, E. A. (1994). Complement receptor-mediated uptake and tumor necrosis factor-alpha-mediated growth inhibition of Mycobacterium tuberculosis by human alveolar macrophages. J. Immunol. 152, 743–753.
Holscher, C., Holscher, A., Ruckerl, D., Yoshimoto, T., Yoshida, H., Mak, T., Saris, C., and Ehlers, S. (2005). The IL-27 receptor chain WSX-1 differentially regulates antibacterial immunity and survival during experimental tuberculosis. J. Immunol. 174, 3534–3544.
Horsburgh, C. R. Jr. (1991). Mycobacterium avium complex infection in the acquired immunodeficiency syndrome. N. Engl. J. Med. 324, 1332–1338.
Hu, C., Mayadas-Norton, T., Tanaka, K., Chan, J., and Salgame, P. (2000). Mycobacterium tuberculosis infection in complement receptor 3-deficient mice. J. Immunol. 165, 2596–2602.
Hunter, C. A. (2005). New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat. Rev. Immunol. 5, 521–531.
Ilonidis, G., Parapanisiou, E., Anogeianaki, A., Giavazis, I., Theofilogiannakos, E. K., Tsekoura, P., Kidonopoulou, K., Trakatelli, C., Polimenidis, Z., Conti, P., and Anogianakis, G. (2006). Interleukin-1beta (IL-1 beta), interleukin 6 (IL-6) and tumor necrosis factor (TNF) in plasma and pleural fluid of pneumonia, lung cancer and tuberculous pleuritis. J. Biol. Regul. Homeost. Agents 20, 41–46.
Inohara, N., and Nunez, G. (2003). NODs: intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 3, 371–382.
Ishikawa, E., Ishikawa, T., Morita, Y. S., Toyonaga, K., Yamada, H., Takeuchi, O., Kinoshita, T., Akira, S., Yoshikai, Y., and Yamasaki, S. (2009). Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J. Exp. Med. 206, 2879–2888.
Jacobs, M., Brown, N., Allie, N., and Ryffel, B. (2000). Fatal Mycobacterium bovis BCG infection in TNF-LT-alpha-deficient mice. Clin. Immunol. 94, 192–199.
Jo, E. K., Yang, C. S., Choi, C. H., and Harding, C. V. (2007). Intracellular signalling cascades regulating innate immune responses to mycobacteria: branching out from Toll-like receptors. Cell. Microbiol. 9, 1087–1098.
Jones, B. W., Heldwein, K. A., Means, T. K., Saukkonen, J. J., and Fenton, M. J. (2001). Differential roles of Toll-like receptors in the elicitation of proinflammatory responses by macrophages. Ann. Rheum. Dis. 60(Suppl. 3), iii6–iii12.
Juffermans, N. P., Florquin, S., Camoglio, L., Verbon, A., Kolk, A. H., Speelman, P., van Deventer, S. J., and Van Der, P. T. (2000). Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J. Infect. Dis. 182, 902–908.
Juffermans, N. P., Verbon, A., van Deventer, S. J., van, D. H., Belisle, J. T., Ellis, M. E., Speelman, P., and Van Der, P. T. (1999). Elevated chemokine concentrations in sera of human immunodeficiency virus (HIV)-seropositive and HIV-seronegative patients with tuberculosis: a possible role for mycobacterial lipoarabinomannan. Infect. Immun. 67, 4295–4297.
Kaneko, H., Yamada, H., Mizuno, S., Udagawa, T., Kazumi, Y., Sekikawa, K., and Sugawara, I. (1999). Role of tumor necrosis factor-alpha in Mycobacterium-induced granuloma formation in tumor necrosis factor-alpha-deficient mice. Lab. Invest. 79, 379–386.
Kang, P. B., Azad, A. K., Torrelles, J. B., Kaufman, T. M., Beharka, A., Tibesar, E., Desjardin, L. E., and Schlesinger, L. S. (2005). The human macrophage mannose receptor directs Mycobacterium tuberculosis ipoarabinomannan-mediated phagosome biogenesis. J. Exp. Med. 202, 987–999.
Kaplan, G., Post, F. A., Moreira, A. L., Wainwright, H., Kreiswirth, B. N., Tanverdi, M., Mathema, B., Ramaswamy, S. V., Walther, G., Steyn, L. M., Barry, C. E., III, and Bekker, L. G. (2003). Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect. Immun. 71, 7099–7108.
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384.
Keane, J., Gershon, S., Wise, R. P., Mirabile-Levens, E., Kasznica, J., Schwieterman, W. D., Siegel, J. N., and Braun, M. M. (2001). Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N. Engl. J. Med. 345, 1098–1104.
Khader, S. A., Bell, G. K., Pearl, J. E., Fountain, J. J., Rangel-Moreno, J., Cilley, G. E., Shen, F., Eaton, S. M., Gaffen, S. L., Swain, S. L., Locksley, R. M., Haynes, L., Randall, T. D., and Cooper, A. M. (2007). IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 8, 369–377.
Khader, S. A., Pearl, J. E., Sakamoto, K., Gilmartin, L., Bell, G. K., Jelley-Gibbs, D. M., Ghilardi, N., deSauvage, F., and Cooper, A. M. (2005). IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 175, 788–795.
Khanna, K. V., Choi, C. S., Gekker, G., Peterson, P. K., and Molitor, T. W. (1996). Differential infection of porcine alveolar macrophage subpopulations by nonopsonized Mycobacterium bovis involves CD14 receptors. J. Leukoc. Biol. 60, 214–220.
Klug, K., Ehlers, S., Uhlig, S., and Reiling, N. (2010). Mitogen-activated protein kinases p38 and ERK1/2 regulated control of Mycobacterium avium replication in primary murine macrophages is independent of tumor necrosis factor-alpha and interleukin-10. Innate Immun. doi: 10.1177/1753425910377799 [Epub ahead of print].
Kuga, S., Otsuka, T., Niiro, H., Nunoi, H., Nemoto, Y., Nakano, T., Ogo, T., Umei, T., and Niho, Y (1996). Suppression of superoxide anion production by interleukin-10 is accompanied by a down-regulation of the genes for subunit proteins of NADPH oxidase. Exp. Hematol. 24, 151–157.
Kurashima, K., Mukaida, N., Fujimura, M., Yasui, M., Nakazumi, Y., Matsuda, T., and Matsushima, K. (1997). Elevated chemokine levels in bronchoalveolar lavage fluid of tuberculosis patients. Am. J. Respir. Crit. Care Med. 155, 1474–1477.
Ladel, C. H., Blum, C., Dreher, A., Reifenberg, K., Kopf, M., and Kaufmann, S. H. (1997). Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect. Immun. 65, 4843–4849.
Latchumanan, V. K., Balkhi, M. Y., Sinha, A., Singh, B., Sharma, P., and Natarajan, K. (2005). Regulation of immune responses to Mycobacterium tuberculosis secretory antigens by dendritic cells. Tuberculosis (Edinb.) 85, 377–383.
Lee, Y. J., Han, Y., Lu, H. T., Nguyen, V., Qin, H., Howe, P. H., Hocevar, B. A., Boss, J. M., Ransohoff, R. M., and Benveniste, E. N. (1997). TGF-beta suppresses IFN-gamma induction of class II MHC gene expression by inhibiting class II transactivator messenger RNA expression. J. Immunol. 158, 2065–2075.
Li, X., McKinstry, K. K., Swain, S. L., and Dalton, D. K. (2007). IFN-gamma acts directly on activated CD4+ T cells during mycobacterial infection to promote apoptosis by inducing components of the intracellular apoptosis machinery and by inducing extracellular proapoptotic signals. J. Immunol. 179, 939–949.
Lin, P. L., Myers, A., Smith, L., Bigbee, C., Bigbee, M., Fuhrman, C., Grieser, H., Chiosea, I., Voitenek, N. N., Capuano, S. V., Klein, E., and Flynn, J. L. (2010). Tumor necrosis factor neutralization results in disseminated disease in acute and latent Mycobacterium tuberculosis infection with normal granuloma structure in a cynomolgus macaque model. Arthritis Rheum. 62, 340–350.
Lin, Y. G., Zhang, M., Hofman, F. M., Gong, J. H., and Barnes, P. F. (1996). Absence of a prominent Th2 cytokine response in human tuberculosis. Infect. Immun. 64, 1351–1356.
Lockhart, E., Green, A. M., and Flynn, J. L. (2006). IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J. Immunol. 177, 4662–4669.
Manca, C., Tsenova, L., Bergtold, A., Freeman, S., Tovey, M., Musser, J. M., Barry, C. E., III, Freedman, V. H., and Kaplan, G. (2001). Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-α/B. Proc. Natl. Acad. Sci. U.S.A. 98, 5752–5757.
Martinez de la, T. Y., Locati, M., Buracchi, C., Dupor, J., Cook, D. N., Bonecchi, R., Nebuloni, M., Rukavina, D., Vago, L., Vecchi, A., Lira, S. A., and Mantovani, A. (2005). Increased inflammation in mice deficient for the chemokine decoy receptor D6. Eur. J. Immunol. 35, 1342–1346.
Martinez-Pomares, L., Linehan, S. A., Taylor, P. R., and Gordon, S. (2001). Binding properties of the mannose receptor. Immunobiology 204, 527–535.
McElvania, T. E., Allen, I. C., Hulseberg, P. D., Sullivan, J. T., McCann, J. R., Sandor, M., Braunstein, M., and Ting, J. P. (2010). Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS ONE 5, e12320. doi: 10.1371/journal.pone.0012320
McGreal, E. P., Miller, J. L., and Gordon, S. (2005). Ligand recognition by antigen-presenting cell C-type lectin receptors. Curr. Opin. Immunol. 17, 18–24.
Mendez-Samperio, P. (2008). Expression and regulation of chemokines in mycobacterial infection. J. Infect. 57, 374–384.
Mendez-Samperio, P. (2010). Role of interleukin-12 family cytokines in the cellular response to mycobacterial disease. Int. J. Infect. Dis. 14, e366–e371.
Mendez-Samperio, P., Vazquez, A., and Ayala, H. (2003). Infection of human monocytes with Mycobacterium bovis BCG induces production of CC-chemokines. J. Infect. 47, 139–147.
Moore, K. W., de Waal, M. R., Coffman, R. L., and O’Garra, A. (2001). Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19, 683–765.
Murray, P. J., Wang, L., Onufryk, C., Tepper, R. I., and Young, R. A. (1997). T cell-derived IL-10 antagonizes macrophage function in mycobacterial infection. J. Immunol. 158, 315–321.
Nakao, A., Afrakhte, M., Moren, A., Nakayama, T., Christian, J. L., Heuchel, R., Itoh, S., Kawabata, M., Heldin, N. E., Heldin, C. H., and ten, D. P. (1997). Identification of Smad7, a TGF-beta-inducible antagonist of TGF-beta signalling. Nature 389, 631–635.
Netea, M. G., Azam, T., Lewis, E. C., Joosten, L. A., Wang, M., Langenberg, D., Meng, X., Chan, E. D., Yoon, D. Y., Ottenhoff, T., Kim, S. H., and Dinarello, C. A. (2006). Mycobacterium tuberculosis induces interleukin-32 production through a caspase-1/IL-18/interferon-gamma-dependent mechanism. PLoS Med. 3, e277. doi: 10.1371/journal.pmed.0030277
Nibbering, P. H., Pos, O., Stevenhagen, A., and Van Furth, R. (1993). Interleukin-8 enhances nonoxidative intracellular killing of Mycobacterium fortuitum by human granulocytes. Infect. Immun. 61, 3111–3116.
Nigou, J., Zelle-Rieser, C., Gilleron, M., Thurnher, M., and Puzo, G. (2001). Mannosylated liparabinomannans inhibit IL-12 production b human dendritic cells: evidence for a negative signal delivered through the mannose receptor. J. Immunol. 166, 7477–7485.
Niiro, H., Otsuka, T., Abe, M., Satoh, H., Ogo, T., Nakano, T., Furukawa, Y., and Niho, Y. (1992). Epstein–Barr virus BCRF1 gene product (viral interleukin 10) inhibits superoxide anion production by human monocytes. Lymphokine Cytokine Res. 11, 209–214.
North, R. J. (1998). Mice incapable of making IL-4 or IL-10 display normal resistance to infection with Mycobacterium tuberculosis. Clin. Exp. Immunol. 113, 55–58.
Ogura, Y., Inohara, N., Benito, A., Chen, F. F., Yamaoka, S., and Nunez, G. (2001). Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J. Biol. Chem. 276, 4812–4818.
O’Leary, S., O’Sullivan, M. P., and Keane, J. (2010). IL-10 blocks phagosome maturation in Mycobacterium tuberculosis-infected human macrophages. Am. J. Respir. Cell Mol. Biol. doi: 10.1165/rcmb.2010-0319OC. [Epub ahead of print].
Oppmann, B., Lesley, R., Blom, B., Timans, J. C., Xu, Y., Hunte, B., Vega, F., Yu, N., Wang, J., Singh, K., Zonin, F., Vaisberg, E., Churakova, T., Liu, M., Gorman, D., Wagner, J., Zurawski, S., Liu, Y., Abrams, J. S., Moore, K. W., Rennick, D., Waal-Malefyt, R., Hannum, C., Bazan, J. F., and Kastelein, R. A. (2000). Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725.
Oswald, I. P., Gazzinelli, R. T., Sher, A., and James, S. L. (1992). IL-10 synergizes with IL-4 and transforming growth factor-beta to inhibit macrophage cytotoxic activity. J. Immunol. 148, 3578–3582.
Owaki, T., Asakawa, M., Morishima, N., Hata, K., Fukai, F., Matsui, M., Mizuguchi, J., and Yoshimoto, T. (2005). A role for IL-27 in early regulation of Th1 differentiation. J. Immunol. 175, 2191–2200.
Pace, E., Gjomarkaj, M., Melis, M., Profita, M., Spatafora, M., Vignola, A. M., Bonsignore, G., and Mody, C. H. (1999). Interleukin-8 induces lymphocyte chemotaxis into the pleural space. Role of pleural macrophages. Am. J. Respir. Crit. Care Med. 159, 1592–1599.
Park, C. G., Takahara, K., Umemoto, E., Yashima, Y., Matsubara, K., Matsuda, Y., Clausen, B. E., Inaba, K., and Steinman, R. M. (2001). Five mouse homologues of the human dendritic cell C-type lectin, DC-SIGN. Int. Immunol. 13, 1283–1290.
Park, Y., Kim, H. S., Ahn, J. Y., Yun, D., Cho, M. L., Hong, S., Kim, H. Y., and Chung, D. H. (2010). IL-12p35 promotes antibody-induced joint inflammation by activating NKT cells and suppressing TGF-beta. J. Immunol. 185, 1476–1484.
Pearl, J. E., Saunders, B., Ehlers, S., Orme, I. M., and Cooper, A. M. (2001). Inflammation and lymphocyte activation during mycobacterial infection in the interferon-gamma-deficient mouse. Cell. Immunol. 211, 43–50.
Polentarutti, N., Rol, G. P., Muzio, M., Bosisio, D., Camnasio, M., Riva, F., Zoja, C., Benigni, A., Tomasoni, S., Vecchi, A., Garlanda, C., and Mantovani, A. (2003). Unique pattern of expression and inhibition of IL-1 signaling by the IL-1 receptor family member TIR8/SIGIRR. Eur. Cytokine Netw. 14, 211–218.
Powlesland, A. S., Ward, E. M., Sadhu, S. K., Guo, Y., Taylor, M. E., and Drickamer, K. (2006). Widely divergent biochemical properties of the complete set of mouse DC-SIGN-related proteins. J. Biol. Chem. 281, 20440–20449.
Quesniaux, V. J., Nicolle, D. M., Torres, D., Kremer, L., Guerardel, Y., Nigou, J., Puzo, G., Erard, F., and Ryffel, B. (2004). Toll-like receptor 2 (TLR2)-dependent-positive and TLR2-independent-negative regulation of proinflammatory cytokines by mycobacterial lipomannans. J. Immunol. 172, 4425–4434.
Rajaram, M. V., Brooks, M. N., Morris, J. D., Torrelles, J. B., Azad, A. K., and Schlesinger, L. S. (2010). Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J. Immunol. 185, 929–942.
Redford, P. S., Boonstra, A., Read, S., Pitt, J., Graham, C., Stavropoulos, E., Bancroft, G. J., and O’Garra, A. (2010). Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur. J. Immunol. 40, 2200–2210.
Remoli, M. E., Giacomini, E., Lutfalla, G., Dondi, E., Orefici, G., Battistini, A., Uzé, G., Pellegrini, S., and Coccia, E. M. (2002). Selective expression of type I IFN genes in human dendritic cells infected with Mycobacterium tuberculosis. J. Immunol. 169, 366–374.
Remus, N., Reichenbach, J., Picard, C., Rietschel, C., Wood, P., Lammas, D., Kumararatne, D. S., and Casanova, J. L. (2001). Impaired interferon gamma-mediated immunity and susceptibility to mycobacterial infection in childhood. Pediatr. Res. 50, 8–13.
Roach, D. R., Bean, A. G., Demangel, C., France, M. P., Briscoe, H., and Britton, W. J. (2002). TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J. Immunol. 168, 4620–4627.
Roach, S. K., and Schorey, J. S. (2002). Differential regulation of the mitogen-activated protein kinases by pathogenic and nonpathogenic mycobacteria. Infect. Immun. 70, 3040–3052.
Roilides, E., Dimitriadou, A., Kadiltsoglou, I., Sein, T., Karpouzas, J., Pizzo, P. A., and Walsh, T. J. (1997). IL-10 exerts suppressive and enhancing effects on antifungal activity of mononuclear phagocytes against Aspergillus fumigatus. J. Immunol. 158, 322–329.
Romano, M. F., Lamberti, A., Petrella, A., Bisogni, R., Tassone, P. F., Formisano, S., Venuta, S., and Turco, M. C. (1996). IL-10 inhibits nuclear factor-kappa B/Rel nuclear activity in CD3-stimulated human peripheral T lymphocytes. J. Immunol. 156, 2119–2123.
Saharinen, J., and Keski-Oja, J. (2000). Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol. Biol. Cell 11, 2691–2704.
Samten, B., Townsend, J. C., Sever-Chroneos, Z., Pasquinelli, V., Barnes, P. F., and Chroneos, Z. C. (2008). An antibody against the surfactant protein A (SP-A)-binding domain of the SP-A receptor inhibits T cell-mediated immune responses to Mycobacterium tuberculosis. J. Leukoc. Biol. 84, 115–123.
Saunders, B. M., and Britton, W. J. (2007). Life and death in the granuloma: immunopathology of tuberculosis. Immunol. Cell Biol. 85, 103–111.
Saunders, B. M., and Cooper, A. M. (2002). Restraining mycobacteria: Role of granulomas in mycobacterial infections. Immunol. Cell Biol. 78, 334–341.
Saunders, B. M., Frank, A. A., and Orme, I. M. (1999). Granuloma formation is required to contain bacillus growth and delay mortality in mice chronically infected with Mycobacterium tuberculosis. Immunology 98, 324–328.
Schaefer, M., Reiling, N., Fessler, C., Stephani, J., Taniuchi, I., Hatam, F., Yildirim, A. O., Fehrenbach, H., Walter, K., Ruland, J., Wagner, H., Ehlers, S., and Sparwasser, T. (2008). Decreased pathology and prolonged survival of human DC-SIGN transgenic mice during mycobacterial infection. J. Immunol. 180, 6836–6845.
Schlesinger, L. S., Azad, A. K., Torrelles, J. B., Roberts, E., Vergne, I., and Deretic, V. (2008). “Determinants of phagocytosis, phagosome biogenesis and autophagy for Mycobacterium tuberculosis,” in Handbook of Tuberculosis. Immunology and Cell Biology, eds S. H. E. Kaufmann and W. J. Britton (Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA), 1–22.
Schlesinger, L. S., Bellinger-Kawahara, C. G., Payne, N. R., and Horwitz, M. A. (1990). Phagocytosis of Mycobacterium tuberculosis is mediated by human monocyte complement receptors and complement component C3. J. Immunol. 144, 2771–2780.
Schlesinger, L. S., Hull, S. R., and Kaufman, T. M. (1994). Binding of the terminal mannosyl units of lipoarabinomannan from a virulent strain of Mycobacterium tuberculosis to human macrophages. J. Immunol. 152, 4070–4079.
Schneider, B. E., Korbel, D., Hagens, K., Koch, M., Raupach, B., Enders, J., Kaufmann, S. H., Mittrucker, H. W., and Schaible, U. E. (2010). A role for IL-18 in protective immunity against Mycobacterium tuberculosis. Eur. J. Immunol. 40, 396–405.
Schoenen, H., Bodendorfer, B., Hitchens, K., Manzanero, S., Werninghaus, K., Nimmerjahn, F., Agger, E. M., Stenger, S., Andersen, P., Ruland, J., Brown, G. D., Wells, C., and Lang, R. (2010). Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J. Immunol. 184, 2756–2760.
Schottelius, A. J., Mayo, M. W., Sartor, R. B., and Baldwin, A. S. Jr. (1999). Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J. Biol. Chem. 274, 31868–31874.
Seder, R. A., Gazzinelli, R., Sher, A., and Paul, W. E. (1993). Interleukin 12 acts directly on CD4+ T cells to enhance priming for interferon gamma production and diminishes interleukin 4 inhibition of such priming. Proc. Natl. Acad. Sci. U.S.A. 90, 10188–10192.
Settas, L. D., Tsimirikas, G., Vosvotekas, G., Triantafyllidou, E., and Nicolaides, P. (2007). Reactivation of pulmonary tuberculosis in a patient with rheumatoid arthritis during treatment with IL-1 receptor antagonists (anakinra). J. Clin. Rheumatol. 13, 219–220.
Sher, A., and Coffman, R. L. (1992). Regulation of immunity to parasites by T cells and T cell-derived cytokines. Annu. Rev. Immunol. 10, 385–409.
Simmons, D. P., Canaday, D. H., Liu, Y., Li, Q., Huang, A., Boom, W. H., and Harding, C. V. (2010). Mycobacterium tuberculosis and TLR2 agonists inhibit induction of type I IFN and class I MHC antigen cross processing by TLR9. J. Immunol. 185, 2405–2415.
Sonnenberg, P., Murray, J., Glynn, J. R., Shearer, S., Kambashi, B., and Godfrey-Faussett, P. (2001). HIV-1 and recurrence, relapse, and reinfection of tuberculosis after cure: a cohort study in South African mineworkers. Lancet 358, 1687–1693.
Strobl, H., and Knapp, W. (1999). TGF-beta1 regulation of dendritic cells. Microbes Infect. 1, 1283–1290.
Sugawara, I., Yamada, H., Hua, S., and Mizuno, S. (2001). Role of interleukin (IL)-1 type 1 receptor in mycobacterial infection. Microbiol. Immunol. 45, 743–750.
Sugawara, I., Yamada, H., Kaneko, H., Mizuno, S., Takeda, K., and Akira, S. (1999). Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect. Immun. 67, 2585–2589.
Tanne, A., Ma, B., Boudou, F., Tailleux, L., Botella, H., Badell, E., Levillain, F., Taylor, M. E., Drickamer, K., Nigou, J., Dobos, K. M., Puzo, G., Vestweber, D., Wild, M. K., Marcinko, M., Sobieszczuk, P., Stewart, L., Lebus, D., Gicquel, B., and Neyrolles, O. (2009). A murine DC-SIGN homologue contributes to early host defense against Mycobacterium tuberculosis. J. Exp. Med. 206, 2205–2220.
Thoma-Uszynski, S., Stenger, S., Takeuchi, O., Ochoa, M., Engele, M., Sieling, P., Barnes, P., Rollinghoff, M., Bolcskei, P., Wagner, M., Akira, S., Norgard, M., Belisle, J., Godowski, P., Bloom, B., and Modlin, R. (2001). Induction of direct antimicrobial activity through mammalian Toll-like receptors. Science 291, 1544–1547.
Toossi, Z., Gogate, P., Shiratsuchi, H., Young, T., and Ellner, J. J. (1995). Enhanced production of TGF-β by blood monocytes from patients with active tuberculosis and presence of TGF-β in tuberculous granulomatous lung lesions. J. Immunol. 154, 465–473.
Torrelles, J. B., Azad, A. K., Henning, L. N., Carlson, T. K., and Schlesinger, L. S. (2008a). Role of C-type lectins in mycobacterial infections. Curr. Drug Targets 9, 102–112.
Torrelles, J. B., Azad, A. K., and Schlesinger, L. S. (2006). Fine discrimination in the recognition of individual species of phosphatidyl-myo-inositol mannosides from Mycobacterium tuberculosis by C-type lectin pattern recognition receptors. J. Immunol. 177, 1805–1816.
Torrelles, J. B., Knaup, R., Kolareth, A., Slepushkina, T., Kaufman, T. M., Kang, P. B., Hill, P., Brennan, P. J., Chatterjee, D., Belisle, J. T., Musser, J. M., and Schlesinger, L. S. (2008b). Identification of Mycobacterium tuberculosis clinical isolates with altered phagocytosis by human macrophages due to a truncated lipoarabinomannan. J. Biol. Chem. 283, 31417–31428.
Torrelles, J. B., and Schlesinger, L. S. (2010). Diversity in Mycobacterium tuberculosis mannosylated cell wall determinants impacts adaptation to the host. Tuberculosis 90, 84–93.
Trinchieri, G., Pflanz, S., and Kastelein, R. A. (2003). The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity 19, 641–644.
Tsai, M. C., Chakravarty, S., Zhu, G., Xu, J., Tanaka, K., Koch, C., Tufariello, J., Flynn, J., and Chan, J. (2006). Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell Microbiol. 8, 218–232.
Tsao, T. C., Hong, J., Huang, C., Yang, P., Liao, S. K., and Chang, K. S. (1999). Increased TNF-alpha, IL-1 beta and IL-6 levels in the bronchoalveolar lavage fluid with the upregulation of their mRNA in macrophages lavaged from patients with active pulmonary tuberculosis. Tuber. Lung Dis. 79, 279–285.
Turner, J. (2011). “Growing old and immunity to bacteria,” in The Immune Response to Infection, eds S. H. E. Kaufmann, B. T. Rouse, and D. L. Sacks (Washington, DC: ASM Press), 413–423.
Turner, J., Frank, A. A., Brooks, J. V., Gonzalez-Juarrero, M., and Orme, I. M. (2001). The progression of chronic tuberculosis in the mouse does not require the participation of B lymphocytes or interleukin-4. Exp. Gerontol. 36, 537–545.
Turner, J., Gonzalez-Juarrero, M., Ellis, D. L., Basaraba, R. J., Kipnis, A., Orme, I. M., and Cooper, A. M. (2002). In vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. J. Immunol. 169, 6343–6351.
Ulrichs, T., and Kaufmann, S. H. (2006). New insights into the function of granulomas in human tuberculosis. J. Pathol. 208, 261–269.
Ulrichs, T., Kosmiadi, G. A., Jorg, S., Pradl, L., Titukhina, M., Mishenko, V., Gushina, N., and Kaufmann, S. H. (2005). Differential organization of the local immune response in patients with active cavitary tuberculosis or with nonprogressive tuberculoma. J. Infect. Dis. 192, 89–97.
Ulrichs, T., Kosmiadi, G. A., Trusov, V., Jorg, S., Pradl, L., Titukhina, M., Mishenko, V., Gushina, N., and Kaufmann, S. H. (2004). Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J. Pathol. 204, 217–228.
Umemura, M., Yahagi, A., Hamada, S., Begum, M. D., Watanabe, H., Kawakami, K., Suda, T., Sudo, K., Nakae, S., Iwakura, Y., and Matsuzaki, G. (2007). IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J. Immunol. 178, 3786–3796.
Underhill, D. M., Ozinsky, A., Smith, K. D., and Aderem, A. (1999). Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. U.S.A. 96, 14459–14463.
Vesosky, B., Rottinghaus, E. K., Stromberg, P., Turner, J., and Beamer, G. (2010). CCL5 participates in early protection against Mycobacterium tuberculosis. J. Leukoc. Biol. 87, 1153–1165.
Vesosky, B., Turner, O. C., Turner, J., and Orme, I. M. (2004). Gamma interferon production by bovine gamma delta T cells following stimulation with mycobacterial mycolyl-arabinogalactan-peptidoglycan. Infect. Immun. 72, 4612–4618.
Villeneuve, C., Gilleron, M., Maridonneau-Parini, I., Daffe, M., Astarie-Dequeker, C., and Etienne, G. (2005). Mycobacteria use their surface-exposed glycolipids to infect human macrophages through a receptor-dependent process. J. Lipid Res. 46, 475–483.
Wang, J., Wakeham, J., Harkness, R., and Xing, Z. (1999). Macrophages are a significant source of type 1 cytokines during mycobacterial infection. J. Clin. Invest. 103, 1023–1029.
Wang, P., Wu, P., Siegel, M. I., Egan, R. W., and Billah, M. M. (1995). Interleukin (IL)-10 inhibits nuclear factor kappaB (NFkappaB) activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J. Biol. Chem. 270, 9558–9563.
Wieland, C. W., Koppel, E. A., den Dunnen, J., Florquin, S., McKenzie, A. N., van Kooyk, Y., Van Der, P. T., and Geijtenbeek, T. B. (2007). Mice lacking SIGNR1 have stronger T helper 1 responses to Mycobacterium tuberculosis. Microbes Infect. 9, 134–141.
Wozniak, T. M., Ryan, A. A., and Britton, W. J. (2006). Interleukin-23 restores immunity to Mycobacterium tuberculosis infection in IL-12p40-deficient mice and is not required for the development of IL-17-secreting T cell responses. J. Immunol. 177, 8684–8692.
Wright, J. R. (2005). Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 5, 58–68.
Yadav, M., and Schorey, J. S. (2006). The β-glucan receptor Dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood 108, 3168–3175.
Yamada, H., Mizumo, S., Horai, R., Iwakura, Y., and Sugawara, I. (2000). Protective role of interleukin-1 in mycobacterial infection in IL-1 alpha/beta double-knockout mice. Lab. Invest. 80, 759–767.
Yamasaki, S., Ishikawa, E., Sakuma, M., Hara, H., Ogata, K., and Saito, T. (2008). Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat. Immunol. 9, 1179–1188.
Yang, C. S., Yuk, J. M., Shin, D. M., Kang, J., Lee, S. J., and Jo, E. K. (2009). Secretory phospholipase A2 plays an essential role in microglial inflammatory responses to Mycobacterium tuberculosis. Glia 57, 1091–1103.
Zaffran, Y., Zhang, L., and Ellner, J. J. (1998). Role of CR4 in Mycobacterium tuberculosis-human macrophages binding and signal transduction in the absence of serum. Infect. Immun. 66, 4541–4544.
Zeller, J. C., Panoskaltsis-Mortari, A., Murphy, W. J., Ruscetti, F. W., Narula, S., Roncarolo, M. G., and Blazar, B. R. (1999). Induction of CD4+ T cell alloantigen-specific hyporesponsiveness by IL-10 and TGF-beta. J. Immunol. 163, 3684–3691.
Zhang, F. R., Huang, W., Chen, S. M., Sun, L. D., Liu, H., Li, Y., Cui, Y., Yan, X. X., Yang, H. T., Yang, R. D., Chu, T. S., Zhang, C., Zhang, L., Han, J. W., Yu, G. Q., Quan, C., Yu, Y. X., Zhang, Z., Shi, B. Q., Zhang, L. H., Cheng, H., Wang, C. Y., Lin, Y., Zheng, H. F., Fu, X. A., Zuo, X. B., Wang, Q., Long, H., Sun, Y. P., Cheng, Y. L., Tian, H. Q., Zhou, F. S., Liu, H. X., Lu, W. S., He, S. M., Du, W. L., Shen, M., Jin, Q. Y., Wang, Y., Low, H. Q., Erwin, T., Yang, N. H., Li, J. Y., Zhao, X., Jiao, Y. L., Mao, L. G., Yin, G., Jiang, Z. X., Wang, X. D., Yu, J. P., Hu, Z. H., Gong, C. H., Liu, Y. Q., Liu, R. Y., Wang, D. M., Wei, D., Liu, J. X., Cao, W. K., Cao, H. Z., Li, Y. P., Yan, W. G., Wei, S. Y., Wang, K. J., Hibberd, M. L., Yang, S., Zhang, X. J., and Liu, J. J. (2009). Genomewide association study of leprosy. N. Engl. J. Med. 361, 2609–2618.
Zhang, M., Lin, Y., Iyer, D. V., Gong, J., Abrams, J. S., and Barnes, P. F. (1995). T-cell cytokine responses in human infection with Mycobacterium tuberculosis. Infect. Immun. 63, 3231–3234.
Keywords: tuberculosis, inflammation, Mycobacterium tuberculosis, pattern recognition receptors
Citation: Sasindran SJ and Torrelles JB (2011) Mycobacterium tuberculosis infection and inflammation: what is beneficial for the host and for the bacterium? Front. Microbio. 2:2. doi: 10.3389/fmicb.2011.00002
Received: 13 December 2010;
Paper pending published: 28 December 2010;
Accepted: 05 January 2011;
Published online: 26 January 2011.
Edited by:
Amal Amer, The Ohio State University, USAReviewed by:
Jose A. Bengoechea, Fundacion Caubet-CIMERA Illes Balears, SpainAndrey P. Anisimov, State Research Center for Applied Microbiology and Biotechnology, Russia
Copyright: © 2011 Sasindran and Torrelles. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Jordi B. Torrelles, Center for Microbial Interface Biology, Division of Infectious Diseases, Department of Medicine, The Ohio State University, 460 West 12th Avenue, Biomedical Research Tower, Room 1016, Columbus, OH 43210, USA. e-mail: jordi.torrelles@osumc.edu