 Siobhán C. Cowley* Karen L. Elkins
Siobhán C. Cowley* Karen L. Elkins
- Center for Biologics Evaluation and Research, U.S. Food and Drug Administration, Bethesda, MD, USA
In recent years, studies on the intracellular pathogen Francisella tularensis have greatly intensified, generating a wealth of new information on the interaction of this organism with the immune system. Here we review the basic elements of the innate and adaptive immune responses that contribute to protective immunity against Francisella species, with special emphasis on new data that has emerged in the last 5 years. Most studies have utilized the mouse model of infection, although there has been an expansion of work on human cells and other new animal models. In mice, basic immune parameters that operate in defense against other intracellular pathogen infections, such as interferon gamma, TNF-α, and reactive nitrogen intermediates, are central for control of Francisella infection. However, new important immune mediators have been revealed, including IL-17A, Toll-like receptor 2, and the inflammasome. Further, a variety of cell types in addition to macrophages are now recognized to support Francisella growth, including epithelial cells and dendritic cells. CD4+ and CD8+ T cells are clearly important for control of primary infection and vaccine-induced protection, but new T cell subpopulations and the mechanisms employed by T cells are only beginning to be defined. A significant role for B cells and specific antibodies has been established, although their contribution varies greatly between bacterial strains of lower and higher virulence. Overall, recent data profile a pathogen that is adept at subverting host immune responses, but susceptible to many elements of the immune system’s antimicrobial arsenal.
Introduction and Overview
Although Francisella tularensis is highly infectious and readily establishes disease at low doses in both humans and animals, it has long been recognized that human tularemia victims rarely if ever suffer a second episode of disease. The collective human infection experience therefore strongly suggests that natural infection engenders strong immune responses that are usually protective. The older literature contains a wealth of information on the pathogenesis and host response to Francisella drawn from studies in both humans and animals, including vaccination and challenge studies in humans that would be difficult if not impossible to replicate today. These studies have been recently reviewed extensively elsewhere (Conlan and Oyston, 2007; Elkins et al., 2007). But the history of studies on Francisella includes its development as a biowarfare pathogen (Dennis et al., 2001); as a result, the recent heightened interest in biodefense has produced a flood of new and exciting data, particularly on respiratory infection and mucosal immune responses to this pathogen. The upsurge in studies is impressive: from 1900 to 2005, a search of PubMed for “Tularemia or Francisella” yielded a total of 2921 references, but 858 citations from 2006 to 2011. Thus this review will focus primarily on developments in about the last 5 years.
Human in vivo Immune Responses to Francisella Infection and Vaccination
Important Components of Human Immune Responses Revealed by Francisella Infection
The most recent studies of the epidemiology of infection with F. tularensis subsp. tularensis, as well as subsp. holarctica, clearly indicate differences in virulence among clades of Type A F. tularensis subsp. tularensis. These are now denoted A1a, A1b, and A2, in addition to Type B F. tularensis subsp. holarctica (Kugeler et al., 2009). Type A1 infections of humans tend to have a fulminant course with a high mortality rate, while Type A2 and Type B infections are rarely if ever lethal in humans (Staples et al., 2006; Kugeler et al., 2008). In studies in the U.S., differences in the attack rates of immunocompromised people for the various clades and subspecies have been described; 11 of 108 (10%) of Type B infections, 6 of 133 (6%) of A1 infections (both subtypes), and none of 68 A2 infections were diagnosed in people with an underlying immunocompromising condition, including medical conditions as diverse as end stage renal disease (Staples et al., 2006) and chronic granulomatous disease (CGD; Maranan et al., 1997). Nonetheless, the collective evidence suggests that the differences in virulence are largely due to intrinsic properties of the bacterial strains, and not directly related to host gender, susceptibility, genetics, or otherwise failed immune responses (Kugeler et al., 2009).
Because infection with Francisella is relatively infrequent in nature, informative examples of infection of people with primary or acquired immunodeficiencies subjects are rare. The handful of such cases prior to 2006, including infection of AIDS patients with reduced CD4+ T cell counts, has been reviewed elsewhere (Elkins et al., 2007), and thus will not be repeated here. More recently, an interesting case involving a 58-year-old man with refractory rheumatoid arthritis was described. The patient had been treated for about a year with methotrexate and an anti-TNF-α therapeutic (Humira®, adalimumab), when he presented with fever, a leg wound, enlarged lymph nodes, and eventually skin fistula. Tuberculosis was suspected initially; the lesion was surgically removed, and found to contain necrotic epithelioid granulomas. Surprisingly, both serology and PCR of a biopsied lymph node diagnosed F. tularensis infection (Konstantinou et al., 2009). It is tempting to speculate that TNF-α deficiency, provided in this case by drug treatment, increased susceptibility to Francisella infection, similar to observations in animal models (Cowley et al., 2008) and in mycobacteria infections of humans (Gardam et al., 2003).
Of note, although only about 20 cases of human infection with F. philomiragia have been reported in the literature, disease caused by this species is usually associated with immune defects. These include corticosteroid treatment, CGD, and near-drowning episodes (Hollis et al., 1989; Sicherer et al., 1997). The association between Francisella and CGD, in which neutrophils fail to produce fully functional NADPH oxidase and thus reactive oxygen radicals, obviously suggests a role for these mediators in human resistance to infection.
The experimental literature now frequently refers to the immunosuppressive nature of infection with virulent Francisella (the topic of another chapter in this issue), but naturally infected patients as well as vaccines eventually develop robust and readily measurable T and B cell responses to the bacterium. These responses have been studied for many years, particularly in regions such as Scandinavia which have appreciable amounts of disease. An outbreak of ulceroglandular tularemia in Sweden in 2003–2004 provided a unique opportunity to obtain peripheral blood cells from patients within days of infection, and perform large scale transcriptional profiling of human gene expression by microarray (Andersson et al., 2006a). Within 2–3 days, there was clear evidence for increased expression of genes regulated by interferon gamma (IFN-γ) and related to apoptosis, but also indications of down-regulation of many genes related to both innate and adaptive immune responses. Thus infection in humans may be characterized by both appropriate and subversive types of host reactions.
The major means to diagnose tularemia is still based on four-fold increase of serum antibodies to the bacterium between acute and convalescent sera. Robust specific IgM, IgG, and IgA serum antibodies, much of them directed against Francisella lipopolysaccharide (LPS), can be detected roughly simultaneously about 6–10 days after the onset of symptoms, or about 2 weeks after infection. Antibody responses peak between 1 and 2 months after infection, and persist for about a decade before diminishing (Koskela and Salminen, 1985). Large scale efforts using 2-D gel blotting (Janovska et al., 2007) or protein chips with a library of Francisella proteins (Sundaresh et al., 2007), reacted with patient sera, are beginning to identify and catalog the bacterial proteins recognized. No obvious immunodominant B cell epitopes have been revealed, however, and as a result panels of antigens have been proposed for diagnostic purposes (Sundaresh et al., 2007).
Within about 2 weeks after infection, ex vivo production of typical Th1-type cytokines such as IFN-γ, TNF-α, and IL-2 by CD4+ and CD8+ T cells is readily detectable in human peripheral blood lymphocytes (PBLs) obtained from tularemia patients (Koskela and Herva, 1980; Surcel et al., 1991). Unlike antibody responses, however, human CD4+ and CD8+ T cell PBL responses persist for as long as 30 years after documented infection (Ericsson et al., 1994). The development of more sensitive assays has facilitated even earlier detection of cytokines such as IFN-γ, which may be produced by both innate immune cells and nascent specific T cells. As a result, recent biological assays being developed for diagnosis have been based on ex vivo stimulation of whole peripheral blood leukocytes with bacteria, followed by assessment of IFN-γ secretion (Eliasson et al., 2008).
A curious, and still unexplained, feature of natural infection is the large expansion of phosphoantigen-responsive Vγ9/Vδ2 T cells in peripheral blood within the first 7 days after onset of symptoms in tularemia patients (Poquet et al., 1998). This intriguing phenomenon, which can result in as many as 30% of all CD3+ cells in human peripheral blood being Vγ9/Vδ2 T cells, is discussed in more detail below.
Human Immune Responses Revealed by Vaccination Against Francisella
As noted above, and discussed in detail in another chapter in this issue, there is a long history of using various live attenuated strains of Francisella as vaccines, particularly in the former Soviet Union (Sändstrom, 1994). As a reference point for further discussion of immune responses generally, here we include a brief summary of human immune responses to the LVS strain. Usually administered by scarification, LVS has been studied in western countries and particularly in the U.S. These studies have mostly used investigational lots of LVS produced in the 1970s by the Department of the Army; more recently, LVS was re-derived from the original lots, and newly manufactured under modern GMP conditions (El Sahly et al., 2009). Microarray studies of gene changes in PBLs from five volunteers obtained between 1 and 14 days after vaccination with LVS indicated a robust up-regulation of expression of pro-inflammatory mediators and genes involved in dendritic cell (DC) function, which peaked within 2 days (Fuller et al., 2006, 2007). Similar to natural infection, humans vaccinated with LVS develop specific IgM, IgA, and IgG antibodies in serum about 2 weeks after vaccination that persist for at least 1.5 years (Waag et al., 1995; El Sahly et al., 2009). However, as noted at the outset, anti-Francisella serum antibodies titers in vaccinated individuals have not been predictive of protection against virulent tularemia, and vaccination with killed bacteria (that elicit anti-Francisella antibodies but no detectable cell-mediated immune responses) has provided little or no benefits in human studies (Francis and Felton, 1942; Foshay, 1950; Overholt et al., 1961; Saslaw et al., 1961a,b; Hambleton et al., 1974; Burke, 1977; Tärnvik, 1989). In addition to serum antibodies, stimulated PBLs, or enriched CD4+ and CD8+ T cells, obtained from volunteers 2–4 weeks or more after LVS vaccination proliferated and produced typical Th1-type cytokines, especially IFN-γ, ex vivo (Waag et al., 1995; El Sahly et al., 2009). In one study, the responding cells from both LVS vaccines and patients after natural infection were characterized as traditional CD4 and CD8 memory T cells, mostly with an effector memory phenotype (CD45RA−/+, CD62−; Salerno-Goncalves et al., 2009).
Animal Models of Human Immunity to Francisella
Detailed consideration of animal models of Francisella infection is well beyond the scope of this article, and a comprehensive recent review is available elsewhere (Lyons and Wu, 2007). Nonetheless, because the bulk of data on immune responses is currently being generated using animal models of infection, here we include brief summaries of recent developments as they relate to modeling the immune responses of humans (for summary, see Table 1).
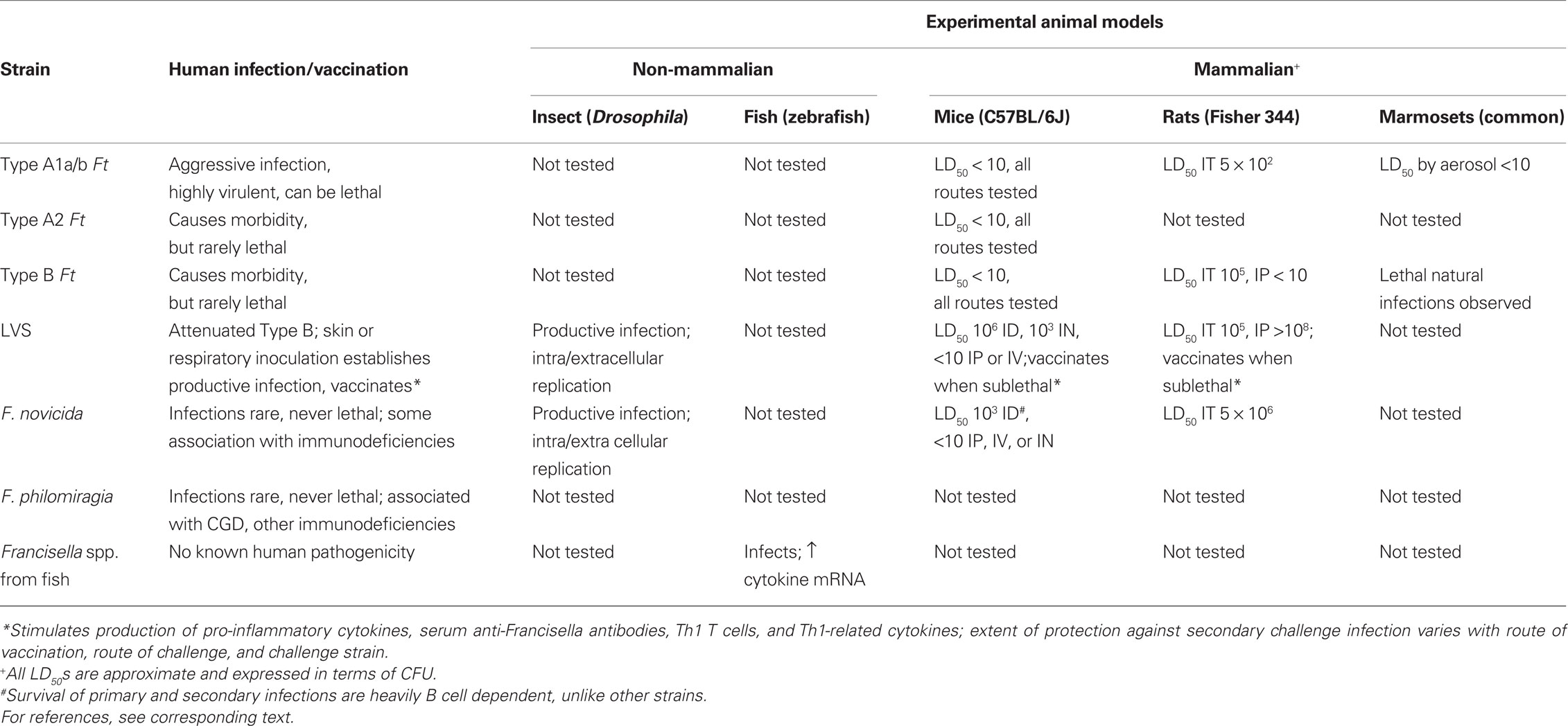
Table 1. Characteristics of human Francisella infections compared to current experimental animal models.
Non-Mammalian Infection Models
Two groups have explored insect models of Francisella infection. Both LVS (Vonkavaara et al., 2008) and F. novicida (Ahlund et al., 2010; Moule et al., 2010) productively infect and replicate in Drosophila melanogaster, both by infection of phagocytic fly hemocytes and by extracellular replication. Both efforts focused on the ability of the model to identify bacterial virulence factors; mutations in bacterial genes important to virulence in mouse models, notably pathogenicity island genes including mglA and others regulated by mglA, clearly contributed to virulence in flies. Given that deer flies are vectors of Francisella infection, however, it remains to be revealed whether the fly host biology discovered is applicable only to the vector, or also helpful in modeling human responses.
Initial efforts to establish Francisella infection of zebrafish, another genetically tractable system that has a more complex immune system than Drosophila, have recently been reported as well. In the last 3–4 years, new Francisella spp. have been isolated from diseased fish, both wild and cultivated. The zebrafish study used one of these strains, and demonstrated productive experimental infection followed by up-regulated expression of IL-1β, IFN-γ, and TNF-α, analogous to mammalian pro-inflammatory responses. There was no increase, and perhaps transient down-regulation, of iNOS, however (Vojtech et al., 2009).
Mammalian Infection Models
For all the obvious reasons, the majority of immunological studies of Francisella have used mouse models. The available data indicate that mice are a reasonable model of human immune responses, at least at a first approximation, but less satisfying as a model for pathogenicity. Inbred laboratory strains are readily susceptible to infection with all Francisella isolates tested to date; bacteria disseminate to the same target organs of the reticuloendothelial system, and infected tissues develop granulomatous pathologies that appear roughly comparable to lesions described in tissues of infected people. There is a major discrepancy between humans and mice in virulence and lethality, however. In mice, the LVS strain establishes a sublethal vaccinating infection when administered via skin inoculation, but kills mice at low doses when introduced by other routes, including intravenous (IV), intramuscular, or intraperitoneal (IP), and is intermediate for respiratory infections (Elkins et al., 2003). Importantly, infection with both Type A and Type B F. tularensis, as well as F. novicida, kills mice within a week with essentially any dose and when introduced by any route of infection. In contrast, as noted above, human Type B infections in particular are rarely lethal (Staples et al., 2006; Kugeler et al., 2008). Human F. novicida infections are quite rare, and when detected are sometimes associated with immunocompromised individuals (Hollis et al., 1989; Whipp et al., 2003; Leelaporn et al., 2008). To date, there is only one report of Francisella infection of HLA-DR4 transgenic mice, with the goal of uncovering antigens recognized by human T cells (Yu et al., 2010); but as expected, these mice were equally susceptible to intranasal LVS infection as wild type mice. It remains to be determined whether “humanized” mice created by engraftment of human stem cells exhibit susceptibilities that better approximate human infection profiles.
Over the years, other small animal models for Francisella infection have been developed using rats, guinea pigs, hamsters, voles, and rabbits. Several recent reports, coupled with older literature, indicate that infection of Fisher 344 rats with different Francisella strains may better approximate the phenotype of human infections that these other models. Both Fisher 344 and Lewis rats are much more resistant to F. novicida infection than mice, due at least in part to rapid production of nitric oxide from macrophages following recognition of the F. novicida LPS chemotype (Cowley et al., 1997; Ray et al., 2010). In a direct comparison, the intratracheal (IT) LD50 of Fisher 344 rats was recently reported to be 5 × 102 for Type A F. tularensis subsp. tularensis (SchuS4); 1 × 105 for Type B F. tularensis subsp. holarctica (OR960246); 5 × 106 for F. novicida; and greater than 107 for LVS (Ray et al., 2010). Of note, however, Fisher 344 rats were highly susceptible to IP infection with a different Type B F. tularensis subsp. holarctica strain (FSC108), although still quite resistant to IP LVS infection (Raymond and Conlan, 2009). Nonetheless, overall the hierarchy of susceptibility of rats appears to generally reflect human Francisella infections, and provide a more satisfying profile than that of mice. Equally important, Fisher 344 rats vaccinated IT, intradermally (ID), or subcutaneously (ID) with ∼107 LVS survived IT challenge with at least 100 LD50s of challenge with Type A F. tularensis subsp. tularensis (SchuS4; Wu et al., 2009; Ray et al., 2010). Thus this rat strain appears to provide both a useful model for both infection and immunological studies for further analyses.
Despite the practical appeal of small animal models, studies using non-human primates remain important not only for basic studies of host–pathogen interactions but for testing of drugs, vaccines, and therapeutics. Historically, monkeys have been considered to be even more susceptible than humans to Francisella infection. Outbreaks of tularemia in various species of monkeys in both zoo and experimental colonies have been reported repeatedly (Splettstoesser et al., 2007). There is an extensive older literature using monkeys, particularly Rhesus monkeys, for both natural history and vaccination studies (Lyons and Wu, 2007; Kugeler et al., 2008). Most recently, interest in product development has spurred renewed efforts to establish non-human primate models using species currently available for experimental studies. Marmosets (Callithrix jacchus) suffer lethal infection with as few as 10 CFU of Type A F. tularensis subsp. tularensis (SchuS4) administered by aerosol, with pathology that appears similar to that of humans. Infected marmosets further exhibited production of pro-inflammatory cytokines, as well as increased numbers of the major lymphoid and myeloid cell subpopulations in lungs and blood (Nelson et al., 2009, 2010). Similarly, the profile of aerosol infection of African green monkeys (Chlorocebus aethiops) given ∼700 CFU of SchuS4 was described recently (Twenhafel et al., 2009), and studies in cynomolgus monkeys as well as comparisons between species are underway (Wilder and Gelhaus, 2009). While it is premature to draw conclusions about the relative strengths and weaknesses of each of these approaches, further studies will no doubt provide data that informs the value of different non-human primate models for both pathogenesis and immunological studies.
In vivo Immune Responses to Francisella Novicida and Mutants Based on F. novicida
Although a comprehensive discussion of the relative virtues of conducting experimental studies using F. tularensis strains versus F. novicida strains is outside of the topic of this review, some general comments regarding immunological responses are pertinent here. For many years, only F. novicida was amenable to genetic manipulation; efforts to transform F. tularensis strains with transposons or develop mutants via allelic exchange using traditional techniques either failed, or were of such low efficiency as to be impractical. Following recent concerted efforts to develop better genetic tools for manipulation of F. tularensis, this situation has changed greatly. A number of transposon banks and deletion mutants of F. tularensis Type A, Type B, and LVS strains have been developed in the last ∼5 years. Nonetheless, manipulation of F. novicida remains considerably easier; further, this strain is exempt from Select Agent registration in the U.S., and generally used under BSL-2 laboratory conditions. Thus F. novicida remains popular for genetic studies, and mutants have frequently been used for in vivo infection studies, particularly those seeking virulence factors. However, when reading this literature, it is important to note that F. novicida’s name has been written as both F. novicida and later F. tularensis subsp. novicida, and the appropriate current designation remains controversial (Busse et al., 2010; Huber et al., 2010; Johansson et al., 2010).
Francisella tularensis subsp. tularensis and holarctica clearly have strong genetic homologies with F. novicida, and findings regarding pathogenesis and cell biology made using F. novicida and its mutants have often been applicable to the biology of F. tularensis. However, there are important biological differences between F. novicida and F. tularensis strains that appear particularly problematic for immunological studies. As discussed above, the virulence of F. novicida in both humans and various animal models is quite distinct from that of the F. tularensis subsp. In mice given F. novicida intranasally, the cell tropism in lungs is noticeably different from that for LVS and SchuS4. The latter two strains preferentially infect alveolar macrophages and later expand in macrophages, DCs, and neutrophils; in contrast, F. novicida starts in neutrophils and alveolar macrophages, and then expands in neutrophils while macrophages and DCs are lesser targets (Hall et al., 2008). Importantly, F. novicida expresses a structurally distinct chemotype of LPS that is more pro-inflammatory in mice than the dominant LPS chemotype expressed by F. tularensis strains (Cowley et al., 1996; Kieffer et al., 2003; Gunn and Ernst, 2007). Although not yet examined directly, the LPS bioactivity may contribute to the observed sepsis-like syndrome that follows intranasal infection of mice with F. novicida (Mares et al., 2008). Each chemotype of LPS also appears to play distinct roles in virulence of the respective bacteria and in contributing to protection in mouse models (Thomas et al., 2007).
Studies of immune responses to F. novicida in mice consistently reveal a prominent role for B cells and antibodies that is considerably more dramatic than LVS or fully virulent Francisella strains. The LD50 of F. novicida administered to inbred mice ID is about 103. The ID LD50 of F. novicida administered to B cell knockout mice is less than 5 × 101, and those that do survive vaccination are severely compromised compared to wild type mice for survival of secondary lethal IP challenge with LVS (Chou and Elkins, manuscript in preparation). Similarly, mice vaccinated IN with attenuated mutants in iglB or iglC of F. novicida have large amounts of both IgG1 and IgG2a serum anti-Francisella antibodies, which adoptively transfer protection against F. novicida challenge to recipients in the absence of primed T cells (Pammit et al., 2006; Powell et al., 2008). For the iglC mutant of F. novicida, vaccinated mice depleted of CD4+ T cells at the time of challenge with F. novicida survived, and thus protection depended on the presence of antibodies but not effector T cells (Powell et al., 2008). Collectively, the mechanistic data to date paints a picture that is quite distinct from studies using LVS as a vaccine or challenge with virulent Francisella.
Systemic Versus Mucosal Immunity to F. tularensis
Although Francisella infection can be initiated via multiple routes, historically the majority of studies in the Francisella murine model have focused on ID or SC exposure to the pathogen. In particular, ID infection with LVS has been utilized because this route approximates the most likely method of vaccination in humans (scarification or SC inoculation), and also conveniently allows for mechanistic studies of immunity after a sublethal infection in mice. More recent studies have focused on respiratory infections, given the interest in biodefense applications. Mice are much more susceptible to Francisella infections initiated via pulmonary routes as compared to the ID/SC route. For LVS, the IN LD50 is ∼102–104 bacteria, whereas infection via the ID/SC route exhibits an LD50 of ∼106–107 bacteria. For the more virulent Type A and Type B F. tularensis strains, both routes of primary infection are rapidly lethal, although protection against secondary respiratory challenge is much more difficult to achieve than protection against ID/SC challenge (Chen et al., 2003). Indeed, to date reasonable protection against virulent Type A respiratory challenge has only been achieved following mucosal (but not parenteral) LVS vaccination (Conlan et al., 2005; Wu et al., 2005; KuoLee et al., 2007). These observations recall theories of the “compartmentalization” of the mucosal immune system (Gill et al., 2010). In support of this concept, respiratory vaccination is not the only mucosal immunization route that is protective against respiratory challenge: LVS immunization via the oral route also results in survival of Type A pulmonary challenge, whereas ID/SC immunization does not (KuoLee et al., 2007). The immune mechanisms that are uniquely induced by mucosal – but not parenteral – vaccination remain to be identified. Recent data indicates that important cytokines such as IL-17A are preferentially produced in LVS-infected lungs following respiratory – but not parenteral – infection (Woolard et al., 2008; Cowley et al., 2010). Thus certain immune mediators may be of greater importance depending on the initial tissue encountered during vaccination and/or challenge; this possibility remains an interesting avenue of future investigation.
As noted above and in detail in another article in this issue, one theme that has emerged in recent years is the ability of Francisella to initially suppress or avoid induction of early immune responses following primary respiratory infection. Although multiple cell functions are clearly suppressed by both LVS and Type A F. tularensis, virulent Type A strains execute a broader range of immunosuppression that likely contributes to their increased virulence (Bosio et al., 2007). Further, some immunosuppressive functions – such as prostaglandin-E2 (PGE2) production or reduced levels of CD14 – may be operative primarily in certain tissues such as the lung (Woolard et al., 2008). Thus, both the route of inoculation and the strain of Francisella can have a widely different impact on the quantity and quality of immune responses measured. Differences in these factors can often make it difficult to draw comparisons between different studies.
Mediators of Innate Immune Responses
Complement
Recent studies documenting that F. tularensis and F. novicida can survive extracellularly in whole blood in vitro and in vivo during mouse infections (Forestal et al., 2007; Yu et al., 2008) have generated new perspectives on the role of extracellular mediators of immunity during infection. F. tularensis is clearly resistant to the bactericidal effects of sera from a variety of species, a feature that was initially associated with cell surface carbohydrate structures described as a capsule (Hood, 1977). More recently, however, studies using Francisella strains with targeted mutations in LPS biosynthesis genes have demonstrated that complement resistance is critically dependant upon LPS O antigen (Thomas et al., 2007; Clay et al., 2008). Although resistant to its bactericidal effects, Francisella clearly binds by complement components: complement-derived opsonins and complement receptors enhance phagocytic uptake of F. tularensis by a variety of cell types, including human and mouse monocytes and macrophages (Clemens et al., 2005; Pierini, 2006; Schulert and Allen, 2006), and human monocyte-derived DCs (Ben Nasr et al., 2006; Ben Nasr and Klimpel, 2008). Indeed, C3 complement components – but not the lethal C5b-C9 membrane attack complex – were shown to be deposited on the cell surface of F. tularensis after incubation in human sera. These C3-derived opsonins enhanced phagocytic uptake by human DCs via a process that promoted intracellular survival, bacterial growth, and DC death (Ben Nasr et al., 2006; Ben Nasr and Klimpel, 2008). This strategy of resistance to complement killing, and the use of complement opsonins to gain entry into an intracellular niche that supports bacterial growth, is likely an important virulence determinant of Francisella. In support of this hypothesis, Francisella O antigen mutants are attenuated in the mouse model of infection (Thomas et al., 2007).
Pattern-Recognition Receptors and Early Cytokine Production
Host cells express a variety of germline-encoded “pattern- recognition receptors” (PRRs), which recognize a number of evolutionarily conserved molecular patterns expressed only by pathogens. PRRs include the membrane-anchored Toll-like receptors (TLRs) and the cytosolic NOD-like receptors (NLRs). Because Francisella initially resides in a membrane-bound phagosome prior to escape into the cytosol, the bacterium has the potential to interact with both membrane and cytosolic PRRs. Indeed, recent evidence has shown that bacterial DNA engaged the cytoplasmic NLR sensor “absent in melanoma 2” (AIM2) within Francisella-infected macrophages, a process that was necessary to initiate inflammasome assembly, caspase-1 activation, and IL-1β release (Jones et al., 2010; Rathinam et al., 2010; Ulland et al., 2010). Since the role of the inflammasome in Francisella infection is reviewed in detail in another article in this issue, here we focus our discussion on the current knowledge of the contribution of TLRs to the initiation of in vivo host responses to Francisella.
Current in vitro and in vivo evidence indicates that TLR2 and MyD88 are critical mediators of inflammatory responses to Francisella. In vitro studies have demonstrated that TLR2 is required for murine DCs to activate NF-KB, produce TNF-α, and up-regulate maturation markers in response to LVS (Katz et al., 2006). Similarly, TLR2 was required for induction of a range of pro-inflammatory cytokine genes (e.g., TNF-α, IL-1β, KC, p40, RANTES, IFN-γ, IL-6, IFN-β, MCP-1, and iNOS) by peritoneal macrophages in response to LVS (Cole et al., 2007; Abplanalp et al., 2009). Full signaling via TLR2 in LVS-infected macrophages required the adaptor molecules MyD88 and TIRAP (Cole et al., 2010). Consistent with the membrane-bound nature of TLR2, LVS co-localized intracellularly within a TLR2/MyD88-containing phagosome (Cole et al., 2007). Interestingly, an LVS mutant that cannot escape the phagosome induced greatly increased expression of TLR2-dependent pro-inflammatory genes (such as TNF-α), and decreased expression of genes that rely on cytosolic recognition of bacteria (such as IFN-β; Cole et al., 2008). These data underscore the existence of the two separate arms of pathogen detection and early cytokine responses – those cytokines whose production is initiated solely via TLR signaling (TNF-α, IL-6), and those cytokines that require both TLR and NLR signaling (IFN-β, IL-1β).
In vitro data are supported by numerous in vivo studies that demonstrate increased susceptibility of TLR2 and MyD88 KO mice to LVS infection (Collazo et al., 2006; Malik et al., 2006; Abplanalp et al., 2009). Intranasal LVS infection of TLR2 KO mice resulted in increased mortality and decreased survival times as compared to their WT counterparts, accompanied by higher bacterial organ burdens and lower levels of TNF-α and IL-6 in the lungs (Malik et al., 2006; Abplanalp et al., 2009). Although TLR2 KO mice were significantly more susceptible to LVS infection than wild type mice via both the IN and ID routes, mice were consistently able to survive lower doses of LVS. This was in stark contrast to MyD88 KO mice, which were exquisitely susceptible to even the smallest doses of LVS delivered via both routes. This indicates a role for MyD88 in LVS infection that extends beyond its function as an adaptor for TLR2 signaling (Collazo et al., 2006; Abplanalp et al., 2009). Other MyD88-dependent molecules that have been tested in the LVS infection model include IL-18, IL-1R, TLR4, TLR1, TLR6, and TLR9 (for summary, see Table 2). Although most have not been exhaustively studied, mice singly deficient for these molecules did not exhibit notably increased susceptibility to LVS infection as compared to their WT counterparts (Collazo et al., 2006; Abplanalp et al., 2009). Similarly, TLR2/9 double KO mice readily survived doses of up to 105 LVS given ID (Chou and Elkins, manuscript in preparation). Further studies in mice deficient in multiple MyD88-dependent receptors would be needed to rule out the possibility of redundancy and/or compensatory effects.
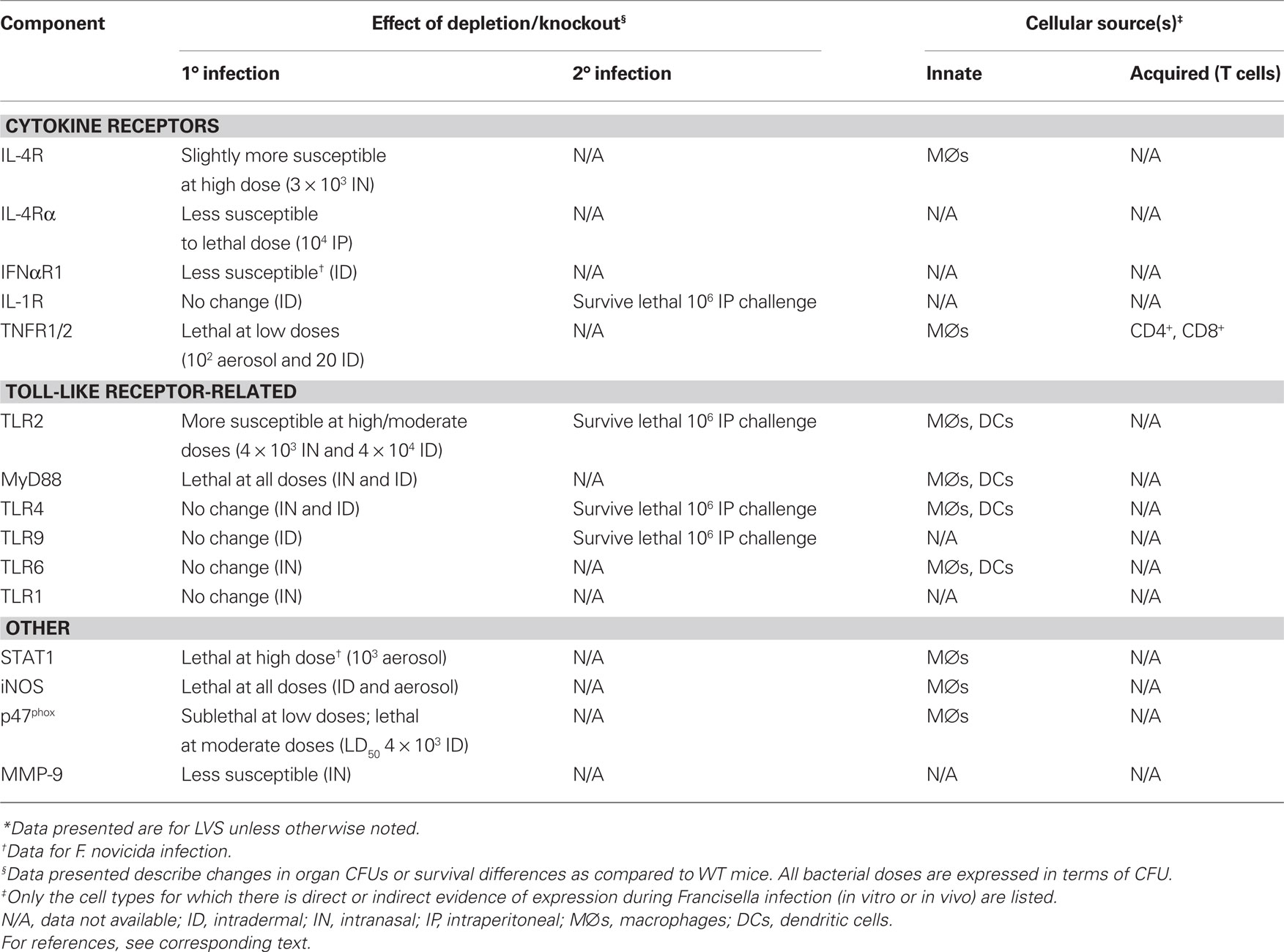
Table 2. The contribution of cytokine receptors, toll-like receptors, and other effector components of immune responses to in vivo murine infection with Francisella tularensis*.
Investigations of the bacterial ligands responsible for TLR recognition of Francisella indicate that LVS expresses lipoproteins that can activate HeLa cells transfected with either TLR2/TLR1 or TLR2/TLR6 heterodimers. Specifically, the lipoproteins Tul4 and FTT1103 stimulated activity via the TLR2/1 heterodimer, and induce expression of a panel of chemokines in both human peripheral blood mononuclear cell (PBMC) and mouse bone marrow-derived DCs (Thakran et al., 2008). A role for TLR6 in recognition of LVS was similarly demonstrated by the inability of bone marrow-derived DCs harvested from TLR6 KO mice to produce TNF-α in response to LVS (Katz et al., 2006). Conversely, in other studies, macrophages from TLR6 KO mice infected with LVS expressed higher levels of TNF-α, IL-6, and MCP-1 than their WT counterparts, and, as mentioned above, TLR6 KO mice exhibit no increased susceptibility to LVS infection (Abplanalp et al., 2009). Thus, it is possible that in some circumstances TLR6 and TLR1 are redundant in their abilities to recognize LVS ligands in concert with TLR2.
Bacterial LPS is usually one of the first Gram negative pathogen-associated molecular patterns to be detected by the immune system, specifically targeted by the PRR TLR4. However, studies of Francisella LPS over the years have found it to be biologically inactive, unable to induce production of pro-inflammatory cytokines from all cell types tested. Subsequent studies have shown that Francisella LPS is not recognized by either human or murine TLR4 or TLR2, and further cannot act as an antagonist to block binding of Salmonella LPS to TLR4 (Duenas et al., 2006; Hajjar et al., 2006). Elucidation of the structure of Francisella LPS demonstrated that it is uniquely tetra-acylated and monophosphorylated, both structural properties that can reduce the endotoxicity of LPS (Gunn and Ernst, 2007). Indeed, the only biological activity ascribed to F. tularensis LPS (Ft-LPS) thus far is its ability to bind and stimulate rapid antibody production by the “innate” B lymphocyte subset B1a cells, and this function was clearly TLR4-independent (Cole et al., 2009); see below. Although Francisella LPS clearly has little if any TLR4-dependent stimulatory activities (Duenas et al., 2006), one recent study revealed that the F. tularensis heat shock protein DnaK can initiate cytokine production by DCs through TLR4 in vitro (Ashtekar et al., 2008). Despite the potential for Francisella to stimulate TLR4 through non-LPS ligands, TLR4-deficient mice do not exhibit increased susceptibility to Francisella infection, indicating that this TLR is not critical for in vivo defense (Chen et al., 2004, 2005b; Collazo et al., 2006).
More recently, the downstream effects of TLR2/MyD88 signaling have been studied in more detail. LVS infection of bone marrow-derived macrophages (BMDMs) induced TLR2-dependent splicing of mRNA for XBP-1, a transcription factor necessary for production of TNF-α and IL-6 mRNA. The importance of XBP-1 in resistance to LVS infection was confirmed by the presence of higher levels of LVS in the organs of XBP-1-deficient mice as compared to WT mice after aerosol infection (Martinon et al., 2010). In a different study, F. novicida infection of human PBMCs and murine macrophages resulted in up-regulation of the micro-RNA miR-155 in a manner that was dependent on TLR2 and MyD88. This micro-RNA blocks translation of SHIP, a negative regulator of the PI3K/Akt pathway (Cremer et al., 2009). The net effect of the release of Akt from inhibition was an increase in pro-inflammatory gene expression. Indeed, mice that express a constitutively active form of Akt survive an otherwise lethal F. novicida infection (Rajaram et al., 2009). Interestingly, experiments using the virulent Type A F. tularensis SchuS4 indicated that SchuS4 had the opposite effect on miR-155 induction, namely down-regulating the pro-inflammatory cytokine response. These results thus reveal one mechanism by which virulent Francisella may inhibit early immune responses (Cremer et al., 2009). These data are also in agreement with Affymetrix microarray and Western blot analyses of human PBMCs that indicated down-regulation of the PI3K/Akt pathway and TLR2 after SchuS4, but not F. novicida, infection (Butchar et al., 2008a; Melillo et al., 2010).
Cytokines and Chemokines in Primary Infection with Francisella
Cytokine production during Francisella infection is a very active area of research, and our understanding of the important cytokine and chemokine mediators continues to evolve (for an overview, refer to Figure 1). Similar to other intracellular pathogens, rapid production of pro-inflammatory and Th1-type cytokines is critical for initial control of Francisella infection in all settings examined to date. However, as noted earlier, there is a striking lack of crucial pro-inflammatory cytokines during the first 48 h of murine pulmonary Francisella infection. It is not until after the first 48–72 h of murine infection that key cytokines and chemokines become readily detectable. During virulent F. tularensis respiratory infection of mice, mRNA levels of essential antimicrobial cytokines such as IFN-γ and TNF-α rose in the lungs between days 2 and 4 (Andersson et al., 2006b), and serum/distal organ levels of pro-inflammatory mediators such as RANTES, IL-6, and IL-1β became detectable on days 3–4 (Conlan et al., 2008). However, after 2 days of unrestricted bacterial growth, key organs such as the lungs and liver harbor extremely high bacterial burdens. Thus the relatively late up-regulation of antimicrobial host immune mechanisms appears to be too late to prevent death. Indeed, a number of studies support the hypothesis that augmenting production of pro-inflammatory cytokines very early during infection can be beneficial. For example, mice administered either a synthetic TLR4 agonist, the TLR3 agonist poly I:C, or recombinant IL-12 shortly before inhalation of Francisella exhibited diminished organ bacterial burdens, and enhanced survival (Duckett et al., 2005; Lembo et al., 2008; Pyles et al., 2010).
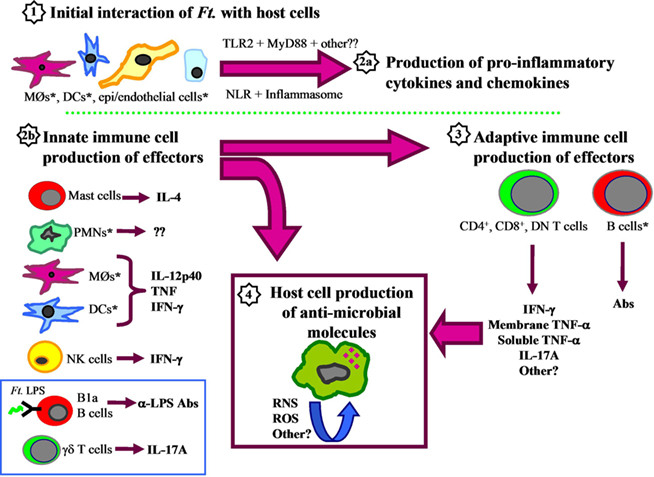
Figure 1. Components of murine innate and adaptive immune responses to Francisella. (1) The initial interaction of Francisella with host cells, such as macrophages, dendritic cells, epithelial cells, and endothelial cells, stimulates production of pro-inflammatory cytokines and chemokines (2a) in a manner that is dependent upon MyD88, TLR2, and other unidentified receptors that signal through MyD88. Bacterial DNA engagement of the NOD-like receptor (NLR) AIM2 may also be critical for inflammasome assembly and release of IL-1β. Simultaneously, important innate immune cells recruited to the area of infection produce effector cytokines such as IL-12p40, TNF-α, IFN-γ, and IL-17A (2b) that influence T cell development (3), and induce host cell production of antimicrobial molecules (4). In addition to the classic TH1-type cytokines, other mediators include mast cell production of IL-4, which can directly inhibit Francisella intramacrophage growth, and B-1a B cell production of anti-LPS antibodies that limit intraperitoneal infection. PMNs are essential for survival of Francisella infections initiated via some routes, but fail to eradicate intracellular organisms in vitro, so their contribution to infection remains unclear. After several days, activation and expansion of Francisella-specific T cells and B cells occurs (3). αβ T cells are essential for clearance of primary infection, and produce effector cytokines such as IL-17A and IFN-γ, as well as the membrane-bound and soluble forms of TNF-α. These factors presumably amplify and extend activation of infected host cells to limit Francisella intracellular growth through production of reactive oxygen and nitrogen intermediates, as well as other unidentified antimicrobials (4). Asterisks (*) indicate host cells that have been shown to harbor intracellular Francisella. The blue box indicates cell types that are neither fully innate nor adaptive, based on classical definitions.
The widespread up-regulation of multiple cytokines and chemokines by day 3 after murine F. novicida pulmonary infection resembles a “cytokine storm” that is associated with severe sepsis – a condition characterized by excessive production of pro-inflammatory cytokines that culminates in capillary leakage, tissue injury, and organ failure. One mediator of severe sepsis, the nuclear DNA-binding protein HMGB-1, was strongly up-regulated and localized extracellularly in mouse lungs by day 3 after F. novicida intranasal infection (Mares et al., 2008). Thus damage-associated molecular pattern molecules such as HMGB-1 are shed from dying host cells, and may be responsible for lethal severe sepsis. This striking outcome was age-dependent: older mice given a pulmonary F. novicida infection had significantly increased survival and an associated reduction in development of hypercytokinemia and cell death (Mares et al., 2008). Whether a similar intriguing phenomenon is responsible for mortality during LVS or virulent F. tularensis infection in vivo remains to be determined, but macrophages infected in vitro with SchuS4 also release HMGB-1 (Mares et al., 2008).
Consistent with the ability of virulent F. tularensis strains to initially suppress – and then ultimately overwhelm – the murine immune response, most studies examining the role of various cytokines and chemokines in this infection model have shown that mice deficient for such key cytokines as IFN-γ, TNF-α, lymphotoxin-α, and iNOS exhibit the same extreme susceptibility to infection via the pulmonary and parenteral routes as fully immunocompetent mice (Chen et al., 2004; Zhang et al., 2008). In contrast, the lower virulence of LVS allows for the determination of a spectrum of susceptibility for the different cytokines and chemokines that contribute to immunity. Thus far, the only cytokine-deficient mice that are exquisitely susceptible to all doses of LVS delivered via any route are those that lack either of the canonical Th1 cytokines, IFN-γ or TNF-α (for summary, see Table 3; Elkins et al., 2007). Mice deficient for either cytokine usually die within a week after inoculation, suggesting that early innate immune cell production of IFN-γ and TNF-α is critical for survival. A recent study of the cell types that produce IFN-γ after primary sublethal LVS ID infection revealed that a wide variety of liver and spleen innate immune cells produce IFN-γ during the first 7 days after infection, including NK cells, neutrophils, DCs, and cells that match the staining profile of “NK DCs” (De Pascalis et al., 2008). Given that both IFN-γ and TNF-α are important for macrophage production of RNI, and that RNI are effective mediators of inhibition of LVS intramacrophage growth in vitro, it is likely that induction of iNOS-derived products is one critical early role for these cytokines in vivo (Anthony et al., 1992; Lindgren et al., 2004, 2005). Indeed, iNOS-deficient mice die following sublethal LVS infections initiated via a variety of routes (Lindgren et al., 2004), although are not as dramatically impaired as IFN-γ or TNF-α knockout mice. Although the cells that produce TNF-α in response to ID LVS infection have not yet been systematically identified, the role of TNF-α during in vivo primary ID LVS infection clearly includes induction of reactive nitrogen species (RNS; Cowley et al., 2008). Further, membrane TNF-α is sufficient to partially mediate resistance to primary LVS ID infection, since mice that express only the membrane-bound form of TNF-α (and not the soluble form) exhibited an intermediate level of susceptibility to LVS infection, as well as intermediate levels of RNS (Cowley et al., 2008).

Table 3. The contribution of cytokines and chemokines to in vivo murine infection with Francisella tularensis*.
In contrast to the prototypic Th1 cytokines, Th2 mediators have not been examined in much detail during in vivo infection. Mice treated with anti-IL-4 antibodies and then infected with LVS ID exhibited an ID LD50 that was comparable to wild type mice, if not higher (Leiby et al., 1992), while IL-4 knockout mice were found to be only slightly more susceptible to IN LVS pulmonary infection (Ketavarapu et al., 2008), and IL-10 knockout mice were considerably more susceptible to IN LVS pulmonary infection (Metzger et al., 2007). Interestingly, as discussed in more detail below, IL-4 receptor α chain knockout mice were less susceptible to lethal IP LVS challenges (Shirey et al., 2008).
Unlike IFN-γ and TNF-α, the role for IL-12 in primary in vivo LVS infection is more nuanced. IL-12 is a heterodimer that consists of two distinct proteins, the p35 and p40 subunits. In addition to constituting one component of IL-12, the p40 subunit can also pair with another protein, denoted p19, to produce the IL-23 heterodimer. Thus, mice deficient for p35 lack IL-12, whereas mice deficient for p40 lack both IL-12 and IL-23. Although both p35- and p40-deficient mice can survive sublethal LVS ID infection, they are both clearly compromised. Whereas 35-deficient mice exhibit higher bacterial organ burdens and cleared the infection more slowly than WT mice, p40 KO mice were unable to clear ID LVS infection, exhibiting chronic high liver and spleen bacterial numbers (Elkins et al., 2002). Both types of LVS-infected knockout mice exhibit reduced levels of serum IFN-γ (Elkins et al., 2002), a finding that is consistent with the role of IL-12 in positive feedback regulation of IFN-γ production by T cells and NK cells. Interestingly, p40-deficient mice exhibited a greater defect in IFN-γ-production than their p35 counterparts, indicating that IL-23 may have an additional role in inducing IFN-γ production (Elkins et al., 2002). Indeed, recent studies found that IL-23 produced by Francisella-infected human monocytes induced NK cell production of IFN-γ, indicating that both IL-23 and IL-12 can positively regulate IFN-γ production (Butchar et al., 2007, 2008b). The unique phenotype of p40 knockout mice indicates that p40 – and by extension, IL-23 – has an as-yet unidentified role in the clearance of sublethal murine LVS ID infection. Whether a similar phenotype occurs in mice given LVS infection via other routes awaits comprehensive characterization: mice deficient for p35, p40, p19, and/or their associated receptors were more susceptible to pulmonary LVS infection, but the studies to date only used doses approaching the LD50 (Duckett et al., 2005; Lin et al., 2009).
IL-17A has an unexpected and critical role in primary LVS pulmonary murine infection. First detectable in mouse lungs by day 3 after infection, by days 6–7 the IL-17A-producing T cells identified in mouse lungs included CD4+ T cells, CD8+ T cells, double negative (DN) T cells, and γ/δ T cells (Lin et al., 2009; Cowley et al., 2010; Markel et al., 2010). The role of IL-17A in LVS pulmonary infection appears to be multi-fold; IL-17A stimulates LVS-infected DCs to up-regulate production of IL-12 and IFN-γ in vitro, and stimulates IFN-γ production by ovalbumin-specific transgenic T cells. Thus IL-17A appears poised to augment early IFN-γ production and aid in polarization of Th1 cells (Lin et al., 2009). However, IL-17A also has a role in the later stages of in vivo infection (days 10–21), when T cell-mediated immunity is critical for clearance of the infection: IL-17A knockout mice given a sublethal LVS pulmonary infection exhibit significantly increased bacterial organ burdens at these late time points (Cowley et al., 2010). Higher numbers of IFN-γ-producing CD4+ and DN T cells were present in the lungs of IL-17A KO mice than their WT counterparts at these later time points, indicating that although IL-17A has a role in inducing early Th1 immunity, IFN-γ-producing T cells are capable of responding to the infection at later time points. Importantly, the discovery that IL-17A can act in concert with IFN-γ to inhibit LVS intracellular growth in macrophages and alveolar type II epithelial cells (ATII) in vitro indicates that IL-17A can be a potent effector cytokine with more than just regulatory properties (Lin et al., 2009; Cowley et al., 2010).
IL-17A is perhaps best known for its ability to recruit neutrophils to the site of infection. In LVS pulmonary infection, IL-17A-deficient mice exhibited decreased levels of lung G-CSF that was accompanied by a reduction in the proportion of lung neutrophils at early time points (days 4 and 6) after infection (Lin et al., 2009; Cowley et al., 2010). Interestingly, type 1 Interferon receptor knockout mice exhibit increased resistance to ID F. novicida infection, a phenomenon that was associated with the ability of Type I IFNs to down-regulate the number of IL-17A+ γ/δ T cells and diminished recruitment of neutrophils to infected spleens (Henry et al., 2010). A similar Type I IFN-mediated negative regulation of γ/δ T cell IL-17A production was also observed in mice infected IN with SchuS4, as well as mice given IV L. monocytogenes infection. Thus, low levels of IL-17A production by γ/δ T cells during Francisella infection may be a consequence of negative regulation effected by Type I IFNs. This outcome was at least partially attributed to the ability of type I IFNs to induce IL-27 production by Francisella-infected macrophages (Henry et al., 2010).
The role of chemokines in host responses to Francisella infection is only beginning to be elucidated. Potent mediators of cell trafficking, chemokines are responsible for drawing critical cell types to the site of infection. Indeed, multiple cell types have been shown to produce chemokines in response to Francisella infection, including DCs, endothelial cells, alveolar type II cells, and macrophages. Despite the abundant production of many chemokines in response to Francisella infection, only a few have been directly examined to date. CCR2 knockout mice, which lack responses to MCP-1/3/5 group of macrophage chemotactic proteins, are quite susceptible to ID LVS infection compared to knockout mice, and fail to exhibit increases in numbers of responding lymphoid and myeloid cells typically found in the spleens of LVS-infected wild type mice (Meierovics and Elkins, manuscript in preparation). However, mice lacking Mig or its receptor CXCR3 did not exhibit increased susceptibility to primary sublethal ID or secondary lethal IP LVS challenges (Park et al., 2002). Similarly, mice deficient for the chemokine receptor CX3CR1 were not more susceptible to IN LVS infection, although they did exhibit modest but significant dysregulation in recruitment of monocytes, neutrophils, and DCs to the lungs (Hall et al., 2009). Chemokines and/or their receptors are functionally redundant, so future studies utilizing mice that are multiply deficient for these factors will no doubt be needed to better define the critical roles of chemokines during in vivo Francisella infection.
Innate Immune Responses Following Interactions of Francisella with Host Cells
Macrophages and Dendritic Cells
The virulence of Francisella has long been associated with its ability to exploit host phagocytic cells to support its own growth. In particular, the ability of macrophages and DCs from a variety of different host tissues to act as a replicative niche, as well as provide antimicrobial effector functions, has been intensively studied. Resident peritoneal macrophages from rats, mice, and guinea pigs, as well as human peripheral blood monocytes and monocyte-derived DCs, support Francisella growth when infected in vitro (Anthony et al., 1991; Ben Nasr et al., 2006; Katz et al., 2006; Chase et al., 2009; Chase and Bosio, 2010). Further, Francisella can grow in vitro in murine phagocytes harvested from a variety of different tissues, including elicited peritoneal macrophages, alveolar macrophages, BMDMs, bone marrow-derived DCs, and alveolar DCs (Anthony et al., 1991; Polsinelli et al., 1994; Bosio and Elkins, 2001; Bosio et al., 2007). In the absence of exogenous factors that induce activation, intracellular bacterial growth continues unrestricted until death of the host cell; at high multiplicities of LVS infection, this process results in caspase 3-dependent apoptosis of macrophages in vitro (Lai et al., 2001; Lai and Sjostedt, 2003). Although this phenomenon has not been demonstrated in vivo with LVS, virulent Type A Francisella induced widespread caspase 3-dependent apoptosis of macrophages in the organs of mice infected IN. Thus unrestricted growth of Francisella can induce apoptotic death of infected host cells in vivo as well as in vitro (Wickstrum et al., 2009).
Similar to many other intracellular pathogens, treatment of infected macrophages ex vivo with IFN-γ inhibits Francisella growth. In quiescent murine and human macrophages, Francisella survives in these cells by rapidly escaping the phagosome to replicate in the cytosol, thereby circumventing phagosome–lysosome fusion and the associated bactericidal effects. LVS phagosomal escape was partially diminished in IFN-γ-treated murine peritoneal exudate cells (PECs; Lindgren et al., 2004). Similarly, control of F. novicida growth by IFN-γ-activated human monocyte-derived macrophages was attributed to its inability to disrupt phagosome–lysosome fusion in the activated cells (Santic et al., 2005). These data suggest that prevention of efficient escape from the phagosome is one mechanism by which IFN-γ inhibits Francisella intracellular growth. However, more recent studies examining LVS and SchuS4 growth in murine BMDMs and the murine macrophage-like cell line J774.1 demonstrated that IFN-γ-activation did not disrupt phagosomal escape (Bonquist et al., 2008; Edwards et al., 2010). These discrepancies are likely to be related to the different Francisella subsp. and host cells used for the different studies; regardless, although disruption of phagosomal escape is one potential mechanism by which IFN-γ-activation could diminish Francisella intracellular growth, it is certainly not the only mechanism at work.
The generation of reactive oxygen species (ROS) and RNS are two well-known mechanisms elicited by IFN-γ that can inhibit the growth of intracellular pathogens. Several studies have focused on defining the role of these two classic mechanisms in the control of Francisella growth both in vitro and in vivo. Optimal IFN-γ-induced inhibition of LVS growth in murine PECs required the combined action of Phox and iNOS, and was dependent upon the generation of peroxynitrite (Lindgren et al., 2005). These data are supported by the increased susceptibility of mice deficient for either iNOS or the phagocyte oxidase p47phox to ID LVS infection as compared to WT mice (Lindgren et al., 2004). In contrast, killing of virulent SchuS4 by IFN-γ-activated murine PECs was only partially dependent on iNOS, and correspondingly, SchuS4 was very resistant to killing by H2O2 and peroxynitrite compared to LVS (Lindgren et al., 2007). Further, a study using murine gp91phox/iNOS double-deficient BMDMs determined that control of SchuS4 growth by IFN-γ was independent of ROS and RNS, iron sequestration, and tryptophan depletion by indoleamine 2,3-dioxygenase (Edwards et al., 2010). Therefore, the IFN-γ-elicited bactericidal mechanism(s) involved in inhibition of SchuS4 growth in activated macrophages remain to be fully elucidated, while the less virulent LVS is more susceptible to the well-known bactericidal effects of ROS and RNS.
Interestingly, although LVS-infected murine macrophages initially produce pro-inflammatory cytokines in vitro, after several hours they begin to exhibit an “alternatively activated” phenotype – a condition characterized by mitigation of the pro-inflammatory response (including iNOS expression) and up-regulation of anti-inflammatory cytokines such as IL-4, IL-13, and TGF-β (Shirey et al., 2008). Differentiation of LVS-infected macrophages into the alternatively activated phenotype required TLR2, IL-4, and IL-13. These alternatively activated macrophages are evident in the peritoneum upon in vivo IP LVS infection, and mice deficient for the IL-4 receptor α chain (used by both IL-4 and IL-13 for signaling) exhibited increased survival following a normally lethal IP LVS infection. This increased resistance was associated with prolonged pro-inflammatory cytokine production and reduced expression of alternative activation markers (Shirey et al., 2008). Similarly, lethal pulmonary F. novicida infection resulted in the development of alternatively activated macrophages in mouse lungs (Mares et al., 2010). Alternative macrophage activation in the F. novicida model was exacerbated by the accumulation of dead cell debris in vitro and the poor ability of F. novicida-infected macrophages to perform efferocytosis (the uptake and clearing of dead cells; Mares et al., 2010). Thus, it appears that during lethal Francisella infections, alternative activation of macrophages diminishes pro-inflammatory cytokine production and the generation of macrophage effector mechanisms, and may contribute to the progression of lethal disease.
Using several different F. tularensis strains in the mouse model of pulmonary infection, studies have demonstrated that both DCs and macrophages in the lung were infected within 1 h after inoculation; these remained the predominant infected cell types by 24 h after infection (Bosio and Dow, 2005; Hall et al., 2008). Indeed, Francisella grows unrestricted within both human and mouse DCs in vitro until the host cells die (Ben Nasr et al., 2006; Bosio et al., 2007). In vitro and in vivo evidence indicates that Francisella-infected human and mouse DCs are actively immunosuppressed by the bacterium. Both are impaired in the ability to produce pro-inflammatory cytokines, and are refractory to stimulation with potent immunomodulators such as E. coli LPS (Bosio et al., 2007; Chase et al., 2009). This immunosuppression has been attributed to a variety of factors, including the ability of SchuS4 to increase production of the immunosuppressive cytokine TGF-β (an indicator of alternative macrophage activation, as described above), as well as the absence of CD14 expression by pulmonary DCs (Bosio et al., 2007; Chase and Bosio, 2010). IN administration of soluble CD14 to SchuS4-infected mice increased lung cytokine production, and reduced SchuS4 replication in the lungs and dissemination to the spleen (Chase and Bosio, 2010). Thus CD14 increased the capacity of DCs to “detect” SchuS4 infection, although paradoxically CD14 is not normally abundant in pulmonary tissues and thus its paucity may contribute to SchuS4 immune evasion. Interestingly, recent data has shown that DCs not only serve as a silent replicative niche for Francisella after respiratory infection, but they also transport the pathogen to the mediastinal lymph nodes, and therefore may play a prominent role in early pathogen dissemination (Bar-Haim et al., 2008).
Non-Phagocytic Cells
In recent years the spectrum of cell types infected by Francisella has broadened to include non-phagocytic cells, such as kidney epithelial cells, ATII, hepatocytes, and fibroblasts. The contribution of non-phagocytic cells to Francisella virulence was revealed by a recent study using a δpyrF SchuS4 mutant. Although this mutant grew less vigorously than WT bacteria in macrophages, it exhibited close to WT levels of growth in some non-macrophage cell types, and retained full virulence in the murine model of IT infection (Horzempa et al., 2010). Although not conclusive, this study suggests that Francisella growth in non-macrophage cell types contributes substantially to the virulence of the organism.
Alveolar epithelial cells, which form the interface between the outside environment and the host lung interior, are well positioned to interact with Francisella very early during respiratory infection. TEM micrographs showed LVS in contact with ATII cells in the airways of mice 2 h after an IN infection (Gentry et al., 2007). In vitro cultures of primary human ATII cells stimulated with SchuS4 and LVS produced high levels of IL-8, MCP-1, GRO-α, and GM-CSF (Gentry et al., 2007). Further, these conditioned culture supernatants induced transmigration of PMNs and DCs through cultured primary human pulmonary microvasculature endothelial cells (HVECs), a cell type that lines blood vessels and is found in close juxtaposition to ATII cells in vivo (Gentry et al., 2007). Studies of the direct interaction of Francisella with HVECs showed that LVS could be internalized – but did not replicate – in these cells, inducing a blunted pro-inflammatory response and transmigration of PMNs with a suppressed phenotype (Forestal et al., 2003; Moreland et al., 2009; Bublitz et al., 2010). It is not yet known whether PMNs that transmigrate across the endothelial layer in response to chemokines produced by Francisella-infected ATII cells are also functionally inhibited. Regardless, it is clear that, in addition to providing an early replicative niche for Francisella, ATII cells have the potential to play a vital role in initiation of inflammatory immune responses and recruitment of key immune cells.
The mechanisms exploited by Francisella for uptake and growth in these non-macrophage cell types – as well as the immune mechanisms that ultimately control this growth – are only beginning to be understood. Thus far it is clear that LVS uses host processes for invasion of murine ATII cells, and – similar to its growth in macrophages – escapes the initial phagosome to replicate in the cytoplasm (Craven et al., 2008). LVS infection of the human ATII cell line A549 resulted in up-regulation of the antimicrobial β-defensin molecule hBD-2, but not hBD-3, the β-defensin that had the most potent anti-Francisella activity in a cell-free system (Han et al., 2008). Therefore, Francisella avoids eliciting detrimental antimicrobial mechanisms in resting ATII cells. However, the combined action of recombinant IFN-γ and IL-17A limited LVS growth in a murine ATII cell line in vitro (Cowley et al., 2010), and thus ATII cells can be activated to produce antimicrobial activity against Francisella.
Neutrophils
Another important cell type that responds early to Francisella infection is the neutrophil. Neutrophils are key innate immune cells that use toxic ROS, cationic peptides, and degradative enzymes to kill ingested pathogens. In particular, the multicomponent enzyme NADPH oxidase, which catalyzes the conversion of molecular oxygen to superoxide anions, is a primary antimicrobial weapon in the neutrophil arsenal. Interestingly, although LVS and SchuS4 were readily phagocytosed by human neutrophils in vitro, they inhibited NADPH oxidase assembly and the associated production of ROS (McCaffrey and Allen, 2006; McCaffrey et al., 2010). Further, instead of being killed by neutrophils, Francisella escaped from the phagosome and persisted in the neutrophil cytosol (McCaffrey and Allen, 2006). Thus, instead of killing Francisella, neutrophils appear to provide a safe – if not replicative – niche for the organism. Nonetheless, in mice the in vivo importance of neutrophils in defense against systemic Francisella infection is clear: neutrophil-depleted mice are highly susceptible to otherwise sublethal parenteral LVS infections, succumbing quickly to an overwhelming disseminated infection (Sjostedt et al., 1994; Elkins et al., 1996; Conlan et al., 2002a). The inability of neutrophils to kill intracellular Francisella suggests that their critical contribution to early survival of systemic infection may lie in their ability to secrete cytokines and chemokines that recruit other important effector cells to the site of infection.
In contrast, the effect of neutrophil depletion on pulmonary LVS infection was much less striking than that noted for parenterally infected mice, with only a minor increase in bacterial burdens in the livers when using a dose just above the LD50 (Conlan et al., 2002a). Regardless, neutrophils are clearly actively recruited to Francisella-infected lungs: recent studies show that neutrophils constitute as much as 50% of the Francisella-infected cells in the lungs of mice 3 days after pulmonary LVS and SchuS4 infection (Hall et al., 2008), and depend at least partially on IL-17A for their early recruitment to the lungs (Lin et al., 2009; Cowley et al., 2010). However, excessive recruitment of neutrophils to the lung appears to contribute to Francisella pathogenesis: mice deficient for matrix metalloproteinase 9 – which generates neutrophil chemoattractants via cleavage of the extracellular matrix – exhibited reduced neutrophil numbers in the lungs after LVS pulmonary infection, and an associated reduction in bacterial burden that was accompanied by increased survival (Malik et al., 2007). Thus, the role of neutrophils in Francisella infection is likely to be a fine balance between aiding in control of infection and exacerbation of pathology.
Mast Cells
Mast cells are classically known for their involvement in allergic conditions associated with type I hypersensitivity reactions (Geha, 2003). However, recent data has revealed an unexpected and intriguing role for mast cells and IL-4 in control of respiratory Francisella infection. Mast cell-deficient mice were much more susceptible to pulmonary LVS infection than WT mice, exhibiting diminished production of IL-4 in the lungs (Ketavarapu et al., 2008). Further, IL-4-deficient mice were slightly more susceptible to LVS pulmonary infection. In vitro, mast cells inhibited LVS intramacrophage growth in a manner that was dependent on IL-4 (Ketavarapu et al., 2008). Overall, these data suggest that mast cells are capable of IL-4-dependent inhibition of LVS growth in macrophages, a phenomenon that was associated with increased macrophage cellular ATP levels and co-localization of LVS with acidified organelles (Rodriguez et al., 2010).
NK Cells
Numerous studies have shown that NK cell-deficient mice are not more susceptible to IN or ID LVS infection than fully immunocompetent mice (Lopez et al., 2004; Duckett et al., 2005; Bokhari et al., 2008), although clear conclusions from these studies are hampered by the inability to fully deplete mice of NK cells in vivo. Despite this limitation, it is clear that NK cells are important producers of IFN-γ during primary LVS infection initiated via both the ID and IN routes. During the first 96 h after an IN LVS infection, a subset of CD11b+DX5+NK1.1+ cells were primarily responsible for IFN-γ production in the lungs and livers (Lopez et al., 2004; Bokhari et al., 2008), while NK1.1+ cells were a large proportion of the IFN-γ producers in the spleens and livers of mice for the first 5–7 days after an ID LVS infection (De Pascalis et al., 2008). The NK1.1+ cell types that produce IFN-γ in response to LVS infection were quite heterogeneous, and could be sub-divided into populations phenotypically reminiscent of NK T cells and “NK DCs” (Bokhari et al., 2008; De Pascalis et al., 2008).
Further studies investigating the role of NK cell production of IFN-γ in Francisella infection have revealed some of its downstream impacts. For example, human monocyte production of IL-23 in responses to Francisella infection induced NK cells to produce IFN-γ; this IFN-γ subsequently up-regulated monocyte production of IL-23 and IL-12p70, establishing NK cells at the center of a positive feedback loop for IL-23 and IFN-γ production (Butchar et al., 2007, 2008b). In addition, NK cell production of IFN-γ was critical for hepatic granuloma formation during IN LVS infection; depletion of NK cells – although not lethal to the mice – resulted in “leaky” granulomas that poorly contained the infection (Bokhari et al., 2008).
Overall, although NK1.1+ cells clearly respond vigorously to early Francisella infection, much of their contribution identified thus far relates to IFN-γ-production; the role of the lytic activities of NK cells has not been studied directly. Since NK1.1+ cells are not the only innate immune cells capable of producing IFN-γ during these early critical time points (other cell types identified included DCs, PMNs, and macrophages; De Pascalis et al., 2008), it is likely that compensation by these other cells explains the dispensability of NK cells during survival of primary sublethal LVS infection. Indeed, mice depleted of NK cells during primary IN LVS infection exhibited only a 50% decrease in serum IFN-γ as compared to their WT counterparts (Bokhari et al., 2008).
Elements of the Adaptive Immune Response
Elucidation of the mechanisms involved in protection against intracellular pathogens is critical for successful vaccine development. Adaptive immune responses typically develop over a longer time frame than that of innate immunity, first requiring activation and clonal expansion of antigen-specific B and T cells. The resulting memory T and B cells respond rapidly to a second exposure to their cognate antigen, thus forming the basis of vaccine-induced immunity. For many intracellular pathogens, cellular immune responses have received the greatest attention; organisms are sequestered within cells and relatively inaccessible to antibodies, and as discussed above, serum antibody levels often do not correlate with protection. Thus T cell mechanisms including cytokine production and cytotoxicity are logical candidates for effecting pathogen eradication. However, recent data demonstrating significant extracellular phases for Francisella in vivo (Forestal et al., 2007; Yu et al., 2008), as well as a clear contribution of B cell-mediated functions to protection against secondary infections with Francisella strains of lower virulence, are leading to a new appreciation for how a combination of both B and T cell responses contribute to protective immunity against Francisella (for an overview, refer to Figure 1).
B Lymphocytes and Antibodies
As discussed above, while specific antibodies are clearly made during Francisella infection and following vaccination, their contribution to protection may be relatively limited, at least in isolation. Murine studies have used both passive transfer of immune sera and/or purified antibodies, as well as genetically deficient mice, to elucidate the role of B cells and antibodies in resistance to Francisella infection. Older studies demonstrated that immune serum transfer to naive animals does not confer protection against the highly virulent Type A Francisella strains (Thorpe and Marcus, 1967), although transfer of anti-LVS or anti-Francisella LPS antibodies conferred partial protection against LVS and the less virulent Type B Francisella challenges (Fortier et al., 1991; Fulop et al., 1995, 2001; Conlan et al., 2002b; Stenmark et al., 2003; Kirimanjeswara et al., 2007; Lavine et al., 2007). Similarly, efforts to stimulate protective immunity in mice through vaccination with Francisella LPS or its protein-conjugated derivatives have only met with success following challenge with LVS or the less virulent Type B Francisella (Fulop et al., 1995; Conlan et al., 2002b, 2003; Kieffer et al., 2003; Cole et al., 2009).
Although administration of inactivated preparations of Francisella or its components have historically been unable to confer protective immunity to virulent Francisella challenge, recent studies have challenged this dogma; further, some of these strategies are antibody-dependent. Vaccination with inactivated Francisella preparations augmented by co-administration with immunostimulatory compounds can induce antibody-dependent protection against challenge with the less virulent LVS. Mice given an IN vaccination of killed LVS and IL-12 survived a subsequent lethal IN LVS challenge in an IgA-dependent manner (Baron et al., 2007). Similarly, mice administered heat-killed LVS IP alongside an IL-12 expressing viral vector survived a high dose lethal IP LVS challenge in a manner that was mediated by antibodies (Lavine et al., 2007). The ability of IL-12 administration to augment humoral immunity and protect against virulent Francisella challenge remains to be explored. Further, mice vaccinated several times IP with Francisella outer membrane proteins or ethanol-inactivated LVS were significantly protected against subsequent IN Type A F. tularensis challenge (∼40–50% survival; Huntley et al., 2008). Intramuscular immunization of mice with killed LVS in conjunction with immunostimulatory complexes (ISCOMs) admixed with CpG protected 40% of mice against a low dose SC challenge with SchuS4 (Eyles et al., 2008). Further, intranasal immunization of mice with inactivated LVS and the mucosal adjuvant cholera toxin B subunit resulted in substantial protection against lethal IN LVS challenge, and partial protection against IN SchuS4 challenge (Bitsaktsis et al., 2009). However, in this case the presence of a mucosal adjuvant augmented Th1-type responses, such that the protection mediated against LVS challenge was fully operative in BKO mice; thus, the observed protection was not actually mediated by B cells or antibodies. Overall, it appears that antibody-mediated immune responses generated by immunization with live or killed LVS or antigenic Francisella preparations are, at best, partially protective against virulent strains of Francisella, and that T cell functions are necessary to achieve optimal protection. Interestingly, however, antisera from mice immunized IN with virulent SchuS4 and subsequently rescued by levofloxacin treatment was able to protect ∼90% of naive mice against a lethal SchuS4 IN challenge (Klimpel et al., 2008).
A recent study reveals an interesting mechanism by which virulent Francisella may evade the protective effects of anti-Francisella antibodies. The highly virulent Type A strain SchuS4 – but not LVS – can directly bind plasmin, a host serine protease that degrades opsonizing antibodies, thus inhibiting antibody-mediated uptake of SchuS4 by macrophages (Crane et al., 2009). Importantly, antibody-opsonized SchuS4 elicited increased production of TNF-α and IL-6 from macrophages, an effect that was reduced by the addition of plasmin. Thus, the ability of SchuS4 to bind plasmin likely contributes to its capacity to evade the host antibodies response. Further, the in vivo protective effects provided by adoptively transferred immune serum against IN LVS challenge were dependent upon the Fcγ receptor and alveolar macrophages, indicating a role for opsonophagocytosis in antibody-mediated protection to LVS (Kirimanjeswara et al., 2007). Control of LVS growth by IFN-γ-treated alveolar macrophages was significantly greater when the bacteria had been serum opsonized prior to uptake (Kirimanjeswara et al., 2007). Since LVS does not bind plasmin, it is subject to opsonophagocytosis by macrophages that are stimulated to increase their production of pro-inflammatory mediators and control intracellular growth more potently. These phenomena may at least partially explain the aforementioned ability of adoptively transferred sera to protect against LVS, but not SchuS4, challenges.
A novel role for “innate immune B cells” in immunity to Francisella has emerged in the last several years. Called B-1 lymphocytes, these cells are primarily located in the pleural cavity, intestinal mucosa, and the spleen of mice, and rapidly produce antibodies against T-independent antigens. In the Francisella murine infection model, these cells mediate a very rapid antibody response within 2–3 days of LVS or Ft-LPS vaccination. The B-1a B cell response and resulting antibody secretion provides substantial protection against moderate lethal LVS challenges administered IP, and was not dependent on the presence of T cells (Dreisbach et al., 2000; Cole et al., 2009). In the Ft-LPS-vaccinated mice, B1a cells proliferated in the spleen, and differentiated into plasma cells that produced Ft-LPS-specific antibodies that were detectable in the sera by days 3–4 after immunization. This protection did not depend on TLR4 and was not elicited by LVS lipid A, indicating that the epitope recognized by B1a cells is LVS O antigen. Whether these rapid B1a protective responses provide immunity to Francisella challenge via other routes of infection remains to be determined. Nonetheless, the discovery that the resultant anti-Ft-LPS antibodies were detectable at low levels in the serum for months after immunization suggests that they have the capacity to contribute to longer-term immune responses, in addition to rapid immunity.
It is important to note that the contributions of B cells to primary and secondary Francisella infections may be quite different. B cell-deficient mice (BKO) given a primary ID LVS infection controlled bacterial growth with kinetics similar to WT mice with a minimal impact on overall susceptibility, and similarly, BKO mice administered a primary low dose aerosol LVS infection also readily survived (Elkins et al., 1999; Chen et al., 2004). In contrast, LVS-vaccinated BKO mice were significantly more susceptible than their WT counterparts to IP LVS secondary challenge, in a manner that could be readily rescued by transfer of immune splenic B cells but not immune sera (Elkins et al., 1999). Thus, in addition to revealing a more significant role for B cells in secondary as compared to primary infection, these results further suggest that part but not all of the contribution of B cells to protective immunity during secondary LVS challenge is antibody-mediated. The role of other functions of B cells, such as production of cytokines and chemokines and antigen presentation, remains to be fully explored. Interestingly, recent evidence indicates that Francisella can grow inside B cells, ultimately inducing apoptosis of the infected cells (Krocova et al., 2008). Thus the potential for Francisella to subvert B cell functions from within remains an interesting avenue for future research.
T Lymphocytes: T Cell Subpopulations
Although B cells and their products have significant roles in protective immunity to Francisella, optimal protection to Francisella infection clearly requires T cell-mediated immunity. Mice deficient in T cells (such as nu/nu or α/β TCR-deficient mice) initially control a primary sublethal parenteral LVS infection, but the mice maintain high bacterial organ burdens and ultimately succumb after approximately a month. Both CD4+ and CD8+ T cells are sufficient to resolve this infection, as mice depleted of either population individually clear the bacteria from the tissues. In contrast, mice simultaneously depleted of both CD4+ and CD8+ T cells survive – but do not clear – LVS infection, instead developing a long-term chronic infection for many months that is characterized by steady levels of bacteria in the organs of the reticuloendothelial system. Control of LVS infection in these depleted mice has been attributed to an unusual CD4−CD8−NK1.1−TCRαβ+Thy1.2+ “DN” T cell population. Enriched populations of these cells potently control LVS intramacrophage growth in vitro (Cowley et al., 2005). Thus, a variety of T cells contribute to control of primary parenteral LVS infection in mice, although only CD4+ and CD8+ αβ T cells have the ability to fully resolve the infection.
The role of T cells in the control and clearance of murine primary sublethal respiratory LVS infection is considerably less well studied than parenteral infection. Mice simultaneously depleted of CD4+ and CD8+ T cells and infected with LVS IN also develop a long-term chronic infection, indicating that one or both of these T cell subsets are necessary for clearance of pulmonary infections (Cowley et al., 2010). However, the requirement for the individual T cell subsets, or αβ TCR+ T cells as a group, has not been systematically studied in pulmonary LVS infection. Since fully immunocompetent mice die following both parenteral and respiratory primary infections with virulent Francisella, it is not surprising that infected mice deficient in T cells die at the same time (Chen et al., 2004; Wu et al., 2005). Interestingly, lethal murine pulmonary infection with virulent type A F. tularensis has been shown to result in thymic atrophy and loss of CD4+CD8+ thymocytes (Chen et al., 2005a), whereas mice given a lethal pulmonary F. novicida infection exhibited a profound depletion of αβ T cells in their lungs that was associated with apoptosis (Sharma et al., 2009). Overall, these data suggest that primary lethal pulmonary Francisella infections share the ability to undermine the development and/or function of T cell-mediated immune responses.
T Lymphocytes: Effector Mechanisms
Efforts to determine the T cell mechanisms that contribute to defense against Francisella infection have revealed a small number of key cytokines that are produced by responding T cells. CD4+, CD8+, and DN T cells in the lungs of mice given a primary sublethal LVS pulmonary infection produce IFN-γ and IL-17A (Woolard et al., 2008; Cowley et al., 2010). Correspondingly, mice deficient in either IFN-γ or IL-17A exhibit increased susceptibility to primary LVS pulmonary and ID infections (Lin et al., 2009; Cowley et al., 2010; Markel et al., 2010). However, since both cytokines are also produced by innate immune cells (see above), it is difficult to determine the relative importance of production of these factors by T cells versus that of other cell types during primary in vivo infection. Greater success on this front has been achieved through the use of an in vitro co-culture system, which directly measures the ability of LVS-immune T cells to control LVS intramacrophage growth. Using this system, it is apparent that IFN-γ and TNF-α contribute to the ability of all three T cell subsets to control LVS intramacrophage growth. However, the different T cell subsets utilize these two factors differentially: LVS-immune CD4+ T cells exhibited the greatest reliance on IFN-γ to control LVS growth, while LVS-immune CD8+ and DN T cells displayed a greater degree of IFN-γ-independence (Cowley and Elkins, 2003). In contrast, LVS-immune CD4+ T cells did not require TNF-α to control LVS intramacrophage growth, while CD8+ T cells were almost completely reliant on TNF-α production (Cowley et al., 2007). Further, LVS-immune CD8+ T cells were capable of utilizing the membrane-bound form of TNF-α, in the absence of soluble TNF-α, to execute full control of LVS growth in vitro (Cowley et al., 2007).
Secondary immune responses to LVS vaccination in mice have typically been studied by administering either IP, IV, or pulmonary Francisella challenges at doses that are lethal for a naive mouse. For secondary LVS IP challenges, all three αβ T cell subsets – CD4+, CD8+, and DN T cells – are sufficient individually for survival and clearance of an LVS infection. However, mice with only DN T cells (i.e., simultaneously depleted of both CD4+ and CD8+ T cells) eradicated the infection more slowly. In contrast, full resistance to virulent F. tularensis pulmonary secondary challenges of vaccinated mice requires both CD4+ and CD8+ T cells, as depletion of either T cell subset significantly reduces survival (Conlan et al., 2005; Wu et al., 2005; Bakshi et al., 2008).
Elucidation of immune T cell effector mechanisms that promote survival of secondary challenges remains an ongoing area of active investigation. Such information is likely to be critical for derivation of protective correlates of immunity. Mice administered neutralizing anti-TNF-α antibodies during lethal IV LVS secondary challenge were highly susceptible to the infection, exhibiting bacterial burdens that exceeded those of mice similarly treated with anti-IFN-γ antibodies (Sjostedt et al., 1996). However, mice genetically engineered to produce only the membrane-bound form of TNF-α (and not the soluble form), were resistant to a high lethal secondary IP LVS challenge and generated similar numbers of CD44hi-responding T cells in their spleens. These data suggest that membrane TNF-α is sufficient for the generation of activated responding memory T cells (Cowley et al., 2007). In contrast, mice treated with anti-IFN-γ Abs at the time of secondary IP LVS challenges were only able to survive the lowest challenge doses, and T cells harvested from vaccinated IFN-γ-deficient mice failed to control LVS intramacrophage growth in vitro, indicating that IFN-γ is required for successful priming of LVS-immune T cells (Elkins et al., 2010). In addition to clear roles for IFN-γ and TNF-α, one recent study found that vaccine efficacy against SchuS4 challenge best correlated with pulmonary levels of IL-17 (Shen et al., 2010). Further, antibody neutralization of IL-17A during in vivo secondary challenge slightly enhanced SchuS4 growth in mouse lungs, indicating that IL-17A may participate in vaccine-induced immunity to Francisella (Shen et al., 2010).
Classic cytotoxic T cell mechanisms do not appear to play a significant role in survival of primary or secondary LVS infections in mice, and similarly do not contribute to the ability of LVS-immune murine T cells to control LVS intramacrophage growth in vitro (Cowley, manuscript in preparation). However, the recent finding that Francisella is susceptible to killing by granulysin, a lytic antimicrobial peptide component of human – but not mouse – cytolytic granules, suggests that granule cytotoxicity may be an effective part of the human T and NK cell arsenal that cannot be assessed in the mouse model (Endsley et al., 2009).
The study of T cell responses during Francisella infection has been hampered by a paucity of information on the precise epitopes recognized by antigen-specific T cells, and a corresponding lack of tools such as tetramers to probe T cell biology. Efforts to identify protein epitopes recognized by T cells during Francisella infection have only recently begun to come to fruition. To date, T cell hybridomas, bioinformatics, proteomics, and “immunoinformatics” have been applied to identify several Francisella T cell epitopes in both mice and humans (McMurry et al., 2007; Valentino and Frelinger, 2009; Valentino et al., 2009; Yu et al., 2010). One murine CD4+ T cell epitope has been identified as amino acids 86–99 of the lipoprotein Tul4 (Valentino et al., 2009). Constituting as much as 20% of the responding splenic CD4+ T cells at the height of an ID LVS infection, this epitope appears to be immunodominant in C57Bl/6 mice (Valentino et al., 2009). Other murine studies have identified the LVS proteins GroEl, KatG, and bacterioferritin (Bfr) as immunostimulatory during in vitro recall assays with splenocytes harvested from LVS-vaccinated BALB/c mice, although the exact epitopes present in these proteins have yet to be identified (Lee et al., 2006). The identification of human epitopes has been approached largely through bioinformatics analyses to select candidate promiscuous or supertype CD4+ and CD8+ epitopes in the SchuS4 genome; this effort yielded a large number of candidate epitopes that induced low levels of IFN-γ production by PBMCs from former tularemia patients (McMurry et al., 2007). Immunization with a pool of 13 of these epitopes provided some protective immunity against a two or five LD50 IT LVS challenge in HLA class II (DRB1*0401) transgenic mice (Gregory et al., 2009), but this effort utilized a difficult vaccination regimen. Further studies to identify alternate vehicles or routes of vaccination will no doubt be necessary to develop a peptide-based vaccination approach that is feasible for humans.
Despite apparently productive interactions between T cells and Francisella-infected macrophages in vitro, infected macrophages also have the capacity to directly inhibit T cell responses in vitro. PGE2 production by BMDMs infected with a high multiplicity of infection of LVS inhibited proliferation of T cell hybridomas specific for SIINFEKL and hen egg lysozyme antigens, skewing the T cell responses from IL-2 to IL-5 production (Woolard et al., 2007). In addition, macrophage production of a PGE2-dependent factor modulated antigen presentation by LVS-infected macrophages via ubiquitinization and degradation of macrophage MHC class II molecules (Wilson et al., 2009). Finally, F. novicida-infected macrophages inhibited expression of the IFN-γ receptor and diminished the ability of ovalbumin-specific T cells to produce IL-2 in response to their cognate antigen (Roth et al., 2009). The in vivo consequences of these phenomena remain to be fully explored, although inhibition of the PGE-2-producing enzyme cyclooxygenase in mice during LVS pulmonary infection increased the number of IFN-γ-producing T cells and decreased the bacterial burden in the lungs. Thus lung PGE2 production may have a significant immunosuppressive impact in vivo.
Similar to murine Francisella infections and as discussed above, human CD4+ and CD8+ T cell responses have been demonstrated. PBMCs from LVS vaccines and naturally infected tularemia patients produce typical Th1-type cytokines such as IL-2, IFN-γ, and TNF-α upon ex vivo re-stimulation with LVS or its antigens (Karttunen et al., 1991; Surcel et al., 1991; Salerno-Goncalves et al., 2009). Further, memory CD4+ and CD8+ T cells in re-stimulated PBMCs from LVS-vaccinated volunteers also produce the Th17 cytokines IL-17 and IL-22 (Paranavitana et al., 2010). One recent study further investigated the phenotype of LVS-induced recall responses in the PBMCs of tularemia patients and LVS vaccines, revealing that CD4+ and CD8+ T cell responders (as defined by IFN-γ-production and/or proliferation) largely exhibited an effector memory phenotype, and expressed cell surface markers that were indicative of cytolytic potential and the ability to home to both mucosal and non-mucosal sites (Salerno-Goncalves et al., 2009). These effector memory cells were long-lived and in some cases detected greater than 25 years after primary pneumonic tularemia.
In addition to CD4+ and CD8+ T cell responses, it is interesting to note that γ/δ?T cells respond vigorously to Francisella infection in humans. The Vγ9/Vδ2 γδ T cell subset is known for its ability to specifically respond to pathogen-produced non-peptidic phosphoesters known collectively as “phosphoantigens.” In tularemia patients, the Vγ9/Vδ2 subset expanded to constitute as much as 30.5% of all CD3+ cells in human peripheral blood within the first 7 days after the onset of symptoms (Poquet et al., 1998). These cells remained elevated for at least a month, and persisted in some individuals for as long as a year after infection (Kroca et al., 2000). Acellular extracts from a variety of Francisella strains, including LVS, contained phosphoantigens that activated Vγ9/Vδ2 T cell expansion and effector functions in human PBMCs. Curiously, despite the existence of activating phosphoantigens in LVS extracts, LVS vaccines do not exhibit the same Vγ9/Vδ2 T cell subset expansion as tularemia patients. Since mice do not express a γδ T cell receptor that is homologous to Vγ9/Vδ2, it has not been possible to further explore this phenomenon in the murine model of infection. To date, only a small number of studies have addressed the role of γδ T cells in murine tularemia. Mice deficient for the γδ T cell receptor did not exhibit notable increased susceptibility to primary sublethal ID or IN LVS infection, and only a small but detectable increase in susceptibility was evident upon secondary IP challenge (Yee et al., 1996; Markel et al., 2010). However, studies using the primary sublethal pulmonary infection model revealed that these cells respond in the lungs of LVS-infected mice by producing IL-17A (Lin et al., 2009; Markel et al., 2010). Thus, overall it appears that any responses provided by γδ T cells in the murine model are not essential for survival of LVS infection.
Conclusions and Perspectives
In the last 5 years, abundant new findings have added to an already rich literature regarding Francisella pathogenesis and immunity. Of necessity, some of it has been more descriptive than hypothesis-driven, in reporting new genetic systems, results of genomic mining, advances in establishing and characterizing novel animal models, and in cataloging responding host cell types and mediators. But the tools developed are now poised to have payoffs not only for understanding of human infection with Francisella per se, but for insights into the biology of intracellular pathogens in general. As an experimental model, Francisella is unique in establishing productive infection in a huge variety of insects and mammals by many routes. Between this convenient biology and the plethora of experimental options now available, the field is well poised to perform detailed mechanistic studies. To do so, immunologists, long biased toward studies in mice, may need to make room in their thinking and their animal colonies for species with advantages in modeling human infection, such as rats. The Francisella models seem to be particularly attractive for tackling some of the current themes regarding immunity to pathogens: comparing similarities and differences in systemic or mucosal immunity; understanding the tension between pathogen-induced immunosuppression and host immunoresponsiveness; appreciating the ongoing interplay between rapid innate reactions and later specific immunity; and getting past a tendency to think of adaptive immune responses as either antibody-centric or T cell dominated, when instead both may complement, augment, and regulate each other. Collectively, we expect studies on immunity to Francisella to continue to be exciting, novel, and perhaps pivotal in achieving a critical public health goal: the search for predictive correlates of protective immunity to intracellular pathogens.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abplanalp, A. L., Morris, I. R., Parida, B. K., Teale, J. M., and Berton, M. T. (2009). TLR-dependent control of Francisella tularensis infection and host inflammatory responses. PLoS ONE 4, e7920. doi: 10.1371/journal.pone.0007920
Ahlund, M. K., Ryden, P., Sjöstedt, A., and Stöven, S. (2010). Directed screen of Francisella novicida virulence determinants using Drosophila melanogaster. Infect. Immun. 78, 3118–3128.
Andersson, H., Hartmanova, B., Back, E., Eliasson, H., Landfors, M., Naslund, L., Ryden, P., and Sjostedt, A. (2006a). Transcriptional profiling of the peripheral blood response during tularemia. Genes Immun. 7, 503–513.
Andersson, H., Hartmanova, B., Kuolee, R., Ryden, P., Conlan, W., Chen, W., and Sjostedt, A. (2006b). Transcriptional profiling of host responses in mouse lungs following aerosol infection with type A Francisella tularensis. J. Med. Microbiol. 55, 263–271.
Anthony, L. D., Burke, R. D., and Nano, F. E. (1991). Growth of Francisella spp. in rodent macrophages. Infect. Immun. 59, 3291–3296.
Anthony, L. S., Morrissey, P. J., and Nano, F. E. (1992). Growth inhibition of Francisella tularensis live vaccine strain by IFN-gamma-activated macrophages is mediated by reactive nitrogen intermediates derived from l-arginine metabolism. J. Immunol. 148, 1829–1834.
Ashtekar, A. R., Zhang, P., Katz, J., Deivanayagam, C. C., Rallabhandi, P., Vogel, S. N., and Michalek, S. M. (2008). TLR4-mediated activation of dendritic cells by the heat shock protein DnaK from Francisella tularensis. J. Leukoc. Biol. 84, 1434–1446.
Bakshi, C. S., Malik, M., Mahawar, M., Kirimanjeswara, G. S., Hazlett, K. R., Palmer, L. E., Furie, M. B., Singh, R., Melendez, J. A., Sellati, T. J., and Metzger, D. W. (2008). An improved vaccine for prevention of respiratory tularemia caused by Francisella tularensis SchuS4 strain. Vaccine 26, 5276–5288.
Bar-Haim, E., Gat, O., Markel, G., Cohen, H., Shafferman, A., and Velan, B. (2008). Interrelationship between dendritic cell trafficking and Francisella tularensis dissemination following airway infection. PLoS Pathog. 4, e1000211. doi: 10.1371/journal.ppat.1000211
Baron, S. D., Singh, R., and Metzger, D. W. (2007). Inactivated Francisella tularensis live vaccine strain protects against respiratory tularemia by intranasal vaccination in an immunoglobulin A-dependent fashion. Infect. Immun. 75, 2152–2162.
Ben Nasr, A., Haithcoat, J., Masterson, J. E., Gunn, J. S., Eaves-Pyles, T., and Klimpel, G. R. (2006). Critical role for serum opsonins and complement receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in phagocytosis of Francisella tularensis by human dendritic cells (DC): uptake of Francisella leads to activation of immature DC and intracellular survival of the bacteria. J. Leukoc. Biol. 80, 774–786.
Ben Nasr, A., and Klimpel, G. R. (2008). Subversion of complement activation at the bacterial surface promotes serum resistance and opsonophagocytosis of Francisella tularensis. J. Leukoc. Biol. 84, 77–85.
Bitsaktsis, C., Rawool, D. B., Li, Y., Kurkure, N. V., Iglesias, B., and Gosselin, E. J. (2009). Differential requirements for protection against mucosal challenge with Francisella tularensis in the presence versus absence of cholera toxin B and inactivated F. tularensis. J. Immunol. 182, 4899–4909.
Bokhari, S. M., Kim, K. J., Pinson, D. M., Slusser, J., Yeh, H. W., and Parmely, M. J. (2008). NK cells and gamma interferon coordinate the formation and function of hepatic granulomas in mice infected with the Francisella tularensis live vaccine strain. Infect. Immun. 76, 1379–1389.
Bonquist, L., Lindgren, H., Golovliov, I., Guina, T., and Sjostedt, A. (2008). MglA and Igl proteins contribute to the modulation of Francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect. Immun. 76, 3502–3510.
Bosio, C. M., Bielefeldt-Ohmann, H., and Belisle, J. T. (2007). Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J. Immunol. 178, 4538–4547.
Bosio, C. M., and Dow, S. W. (2005). Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J. Immunol. 175, 6792–6801.
Bosio, C. M., and Elkins, K. L. (2001). Susceptibility to secondary Francisella tularensis live vaccine strain infection in B-cell-deficient mice is associated with neutrophilia but not with defects in specific T-cell-mediated immunity. Infect. Immun. 69, 194–203.
Bublitz, D. C., Noah, C. E., Benach, J. L., and Furie, M. B. (2010). Francisella tularensis suppresses the proinflammatory response of endothelial cells via the endothelial protein C receptor. J. Immunol. 185, 1124–1131.
Burke, D. S. (1977). Immunization against tularemia: analysis of the effectiveness of live Francisella tularensis vaccine in prevention of laboratory-acquired tularemia. J. Infect. Dis. 135, 55–60.
Busse, H. J., Huber, B., Anda, P., Escudero, R., Scholz, H. C., Seibold, E., Splettstoesser, W. D., and Kampfer, P. (2010). Objections to the transfer of Francisella novicida to the subspecies rank of Francisella tularensis – response to Johansson et al. Int. J. Syst. Evol. Microbiol. 60, 1718–1720.
Butchar, J. P., Cremer, T. J., Clay, C. D., Gavrilin, M. A., Wewers, M. D., Marsh, C. B., Schlesinger, L. S., and Tridandapani, S. (2008a). Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PLoS ONE 3, e2924. doi: 10.1371/journal.pone.0002924
Butchar, J. P., Parsa, K. V., Marsh, C. B., and Tridandapani, S. (2008b). IFNgamma enhances IL-23 production during Francisella infection of human monocytes. FEBS Lett. 582, 1044–1048.
Butchar, J. P., Rajaram, M. V., Ganesan, L. P., Parsa, K. V., Clay, C. D., Schlesinger, L. S., and Tridandapani, S. (2007). Francisella tularensis induces IL-23 production in human monocytes. J. Immunol. 178, 4445–4454.
Chase, J. C., and Bosio, C. M. (2010). The presence of CD14 overcomes evasion of innate immune responses by virulent Francisella tularensis in human dendritic cells in vitro and pulmonary cells in vivo. Infect. Immun. 78, 154–167.
Chase, J. C., Celli, J., and Bosio, C. M. (2009). Direct and indirect impairment of human dendritic cell function by virulent Francisella tularensis Schu S4. Infect. Immun. 77, 180–195.
Chen, W., Kuolee, R., Austin, J. W., Shen, H., Che, Y., and Conlan, J. W. (2005a). Low dose aerosol infection of mice with virulent type A Francisella tularensis induces severe thymus atrophy and CD4+ CD8+ thymocyte depletion. Microb. Pathog. 39, 189–196.
Chen, W., Kuolee, R., Shen, H., Busa, M., and Conlan, J. W. (2005b). Toll-like receptor 4 (TLR4) plays a relatively minor role in murine defense against primary intradermal infection with Francisella tularensis LVS. Immunol. Lett. 97, 151–154.
Chen, W., KuoLee, R., Shen, H., and Conlan, J. W. (2004). Susceptibility of immunodeficient mice to aerosol and systemic infection with virulent strains of Francisella tularensis. Microb. Pathog. 36, 311–318.
Chen, W., Shen, H., Webb, A., KuoLee, R., and Conlan, J. W. (2003). Tularemia in BALB/c and C57BL/6 mice vaccinated with Francisella tularensis LVS and challenged intradermally, or by aerosol with virulent isolates of the pathogen: protection varies depending on pathogen virulence, route of exposure, and host genetic background. Vaccine 21, 3690–3700.
Clay, C. D., Soni, S., Gunn, J. S., and Schlesinger, L. S. (2008). Evasion of complement-mediated lysis and complement C3 deposition are regulated by Francisella tularensis lipopolysaccharide O antigen. J. Immunol. 181, 5568–5578.
Clemens, D. L., Lee, B. Y., and Horwitz, M. A. (2005). Francisella tularensis enters macrophages via a novel process involving pseudopod loops. Infect. Immun. 73, 5892–5902.
Cole, L. E., Laird, M. H., Seekatz, A., Santiago, A., Jiang, Z., Barry, E., Shirey, K. A., Fitzgerald, K. A., and Vogel, S. N. (2010). Phagosomal retention of Francisella tularensis results in TIRAP/Mal-independent TLR2 signaling. J. Leukoc. Biol. 87, 275–281.
Cole, L. E., Santiago, A., Barry, E., Kang, T. J., Shirey, K. A., Roberts, Z. J., Elkins, K. L., Cross, A. S., and Vogel, S. N. (2008). Macrophage proinflammatory response to Francisella tularensis live vaccine strain requires coordination of multiple signaling pathways. J. Immunol. 180, 6885–6891.
Cole, L. E., Shirey, K. A., Barry, E., Santiago, A., Rallabhandi, P., Elkins, K. L., Puche, A. C., Michalek, S. M., and Vogel, S. N. (2007). Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infect. Immun. 75, 4127–4137.
Cole, L. E., Yang, Y., Elkins, K. L., Fernandez, E. T., Qureshi, N., Shlomchik, M. J., Herzenberg, L. A., and Vogel, S. N. (2009). Antigen-specific B-1a antibodies induced by Francisella tularensis LPS provide long-term protection against F. tularensis LVS challenge. Proc. Natl. Acad. Sci. U.S.A. 106, 4343–4348.
Collazo, C. M., Sher, A., Meierovics, A. I., and Elkins, K. L. (2006). Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microbes Infect. 8, 779–790.
Conlan, J. W., KuoLee, R., Shen, H., and Webb, A. (2002a). Different host defences are required to protect mice from primary systemic vs pulmonary infection with the facultative intracellular bacterial pathogen, Francisella tularensis LVS. Microb. Pathog. 32, 127–134.
Conlan, J. W., Shen, H., Webb, A., and Perry, M. B. (2002b). Mice vaccinated with the O-antigen of Francisella tularensis LVS lipopolysaccharide conjugated to bovine serum albumin develop varying degrees of protective immunity against systemic or aerosol challenge with virulent type A and type B strains of the pathogen. Vaccine 20, 3465–3471.
Conlan, J. W., and Oyston, P. C. (2007). Vaccines against Francisella tularensis. Ann. N. Y. Acad. Sci. 1105, 325–350.
Conlan, J. W., Shen, H., Kuolee, R., Zhao, X., and Chen, W. (2005). Aerosol-, but not intradermal-immunization with the live vaccine strain of Francisella tularensis protects mice against subsequent aerosol challenge with a highly virulent type A strain of the pathogen by an alphabeta T cell- and interferon gamma-dependent mechanism. Vaccine 23, 2477–2485.
Conlan, J. W., Vinogradov, E., Monteiro, M. A., and Perry, M. B. (2003). Mice intradermally-inoculated with the intact lipopolysaccharide, but not the lipid A or O-chain, from Francisella tularensis LVS rapidly acquire varying degrees of enhanced resistance against systemic or aerogenic challenge with virulent strains of the pathogen. Microb. Pathog. 34, 39–45.
Conlan, J. W., Zhao, X., Harris, G., Shen, H., Bolanowski, M., Rietz, C., Sjostedt, A., and Chen, W. (2008). Molecular immunology of experimental primary tularemia in mice infected by respiratory or intradermal routes with type A Francisella tularensis. Mol. Immunol. 45, 2962–2969.
Cowley, S., Myltseva, S., and Nano, F. E. (1996). Phase variation in Francisella tularensis affecting intracellular growth, lipopolysaccharide antigenicity, and nitric oxide production. Mol. Microbiol. 20, 867–874.
Cowley, S. C., and Elkins, K. L. (2003). Multiple T cell subsets control Francisella tularensis LVS intracellular growth without stimulation through macrophage interferon gamma receptors. J. Exp. Med. 198, 379–389.
Cowley, S. C., Goldberg, M. F., Ho, J. A., and Elkins, K. L. (2008). The membrane form of tumor necrosis factor is sufficient to mediate partial innate immunity to Francisella tularensis live vaccine strain. J. Infect. Dis. 198, 284–292.
Cowley, S. C., Hamilton, E., Frelinger, J. A., Su, J., Forman, J., and Elkins, K. L. (2005). CD4-CD8-T cells control intracellular bacterial infections both in vitro and in vivo. J. Exp. Med. 202, 309–319.
Cowley, S. C., Meierovics, A. I., Frelinger, J. A., Iwakura, Y., and Elkins, K. L. (2010). Lung CD4-CD8-double-negative T cells are prominent producers of IL-17A and IFN-gamma during primary respiratory murine infection with Francisella tularensis live vaccine strain. J. Immunol. 184, 5791–5801.
Cowley, S. C., Myltseva, S. V., and Nano, F. E. (1997). Suppression of Francisella tularensis growth in the rat by co-infection with F. novicida. FEMS Microbiol. Lett. 153, 71–74.
Cowley, S. C., Sedgwick, J. D., and Elkins, K. L. (2007). Differential requirements by CD4+ and CD8+ T cells for soluble and membrane TNF in control of Francisella tularensis live vaccine strain intramacrophage growth. J. Immunol. 179, 7709–7719.
Crane, D. D., Warner, S. L., and Bosio, C. M. (2009). A novel role for plasmin-mediated degradation of opsonizing antibody in the evasion of host immunity by virulent, but not attenuated, Francisella tularensis. J. Immunol. 183, 4593–4600.
Craven, R. R., Hall, J. D., Fuller, J. R., Taft-Benz, S., and Kawula, T. H. (2008). Francisella tularensis invasion of lung epithelial cells. Infect. Immun. 76, 2833–2842.
Cremer, T. J., Ravneberg, D. H., Clay, C. D., Piper-Hunter, M. G., Marsh, C. B., Elton, T. S., Gunn, J. S., Amer, A., Kanneganti, T. D., Schlesinger, L. S., Butchar, J. P., and Tridandapani, S. (2009). MiR-155 induction by F. novicida but not the virulent F. tularensis results in SHIP down-regulation and enhanced pro-inflammatory cytokine response. PLoS ONE 4, e8508. doi: 10.1371/journal.pone.0008508
De Pascalis, R., Taylor, B. C., and Elkins, K. L. (2008). Diverse myeloid and lymphoid cell subpopulations produce gamma interferon during early innate immune responses to Francisella tularensis live vaccine strain. Infect. Immun. 76, 4311–4321.
Dennis, D. T., Inglesby, T. V., Henderson, D. A., Bartlett, J. G., Ascher, M. S., Eitzen, E., Fine, A. D., Friedlander, A. M., Hauer, J., Layton, M., Lillibridge, S. R., McDade, J. E., Osterholm, M. T., O’Toole, T., Parker, G., Perl, T. M., Russell, P. K., and Tonat, K. (2001). Tularemia as a biological weapon: medical and public health management. JAMA 285, 2763–2773.
Dreisbach, V. C., Cowley, S., and Elkins, K. L. (2000). Purified lipopolysaccharide from Francisella tularensis live vaccine strain (LVS) induces protective immunity against LVS infection that requires B cells and gamma interferon. Infect. Immun. 68, 1988–1996.
Duckett, N. S., Olmos, S., Durrant, D. M., and Metzger, D. W. (2005). Intranasal interleukin-12 treatment for protection against respiratory infection with the Francisella tularensis live vaccine strain. Infect. Immun. 73, 2306–2311.
Duenas, A. I., Aceves, M., Orduna, A., Diaz, R., Sanchez Crespo, M., and Garcia-Rodriguez, C. (2006). Francisella tularensis LPS induces the production of cytokines in human monocytes and signals via Toll-like receptor 4 with much lower potency than E. coli LPS. Int. Immunol. 18, 785–795.
Edwards, J. A., Rockx-Brouwer, D., Nair, V., and Celli, J. (2010). Restricted cytosolic growth of Francisella tularensis subsp. tularensis by IFN-gamma activation of macrophages. Microbiology 156, 327–339.
El Sahly, H. M., Atmar, R. L., Patel, S. M., Wells, J. M., Cate, T., Ho, M., Guo, K., Pasetti, M. F., Lewis, D. E., Sztein, M. B., and Keitel, W. A. (2009). Safety, reactogenicity and immunogenicity of Francisella tularensis live vaccine strain in humans. Vaccine 27, 4905–4911.
Eliasson, H., Olcen, P., Sjöstedt, A., Jurstrand, M., Bäck, E., and Andersson, S. (2008). Kinetics of the immune response associated with tularemia: comparison of an enzyme-linked immunosorbent assay, a tube agglutination test, and a novel whole-blood lymphocyte stimulation test. Clin. Vaccine Immunol. 15, 1238–1243.
Elkins, K. L., Bosio, C. M., and Rhinehart-Jones, T. R. (1999). Importance of B cells, but not specific antibodies, in primary and secondary protective immunity to the intracellular bacterium Francisella tularensis live vaccine strain. Infect. Immun. 67, 6002–6007.
Elkins, K. L., Colombini, S. M., Meierovics, A. I., Chu, M. C., Chou, A. Y., and Cowley, S. C. (2010). Survival of secondary lethal systemic Francisella LVS challenge depends largely on interferon gamma. Microbes Infect. 12, 28–36.
Elkins, K. L., Cooper, A., Colombini, S. M., Cowley, S. C., and Kieffer, T. L. (2002). In vivo clearance of an intracellular bacterium, Francisella tularensis LVS, is dependent on the p40 subunit of interleukin-12 (IL-12) but not on IL-12 p70. Infect. Immun. 70, 1936–1948.
Elkins, K. L., Cowley, S. C., and Bosio, C. M. (2003). Innate and adaptive immune responses to an intracellular bacterium, Francisella tularensis live vaccine strain. Microbes Infect. 5, 132–142.
Elkins, K. L., Cowley, S. C., and Bosio, C. M. (2007). Innate and adaptive immunity to Francisella. Ann. N. Y. Acad. Sci. 1105, 284–324.
Elkins, K. L., Rhinehart-Jones, T. R., Culkin, S. J., Yee, D., and Winegar, R. K. (1996). Minimal requirements for murine resistance to infection with Francisella tularensis LVS. Infect. Immun. 64, 3288–3293.
Endsley, J. J., Torres, A. G., Gonzales, C. M., Kosykh, V. G., Motin, V. L., Peterson, J. W., Estes, D. M., and Klimpel, G. R. (2009). Comparative antimicrobial activity of granulysin against bacterial biothreat agents. Open Microbiol. J. 3, 92–96.
Ericsson, M., Sandström, G., Sjöstedt, A., and Tärnvik, A. (1994). Persistence of cell-mediated immunity and decline of humoral immunity to the intracellular bacterium Francisella tularensis 25 years after natural infection. J. Infect. Dis. 170, 110–114.
Eyles, J. E., Hartley, M. G., Laws, T. R., Oyston, P. C., Griffin, K. F., and Titball, R. W. (2008). Protection afforded against aerosol challenge by systemic immunisation with inactivated Francisella tularensis live vaccine strain (LVS). Microb. Pathog. 44, 164–168.
Forestal, C. A., Benach, J. L., Carbonara, C., Italo, J. K., Lisinski, T. J., and Furie, M. B. (2003). Francisella tularensis selectively induces proinflammatory changes in endothelial cells. J. Immunol. 171, 2563–2570.
Forestal, C. A., Malik, M., Catlett, S. V., Savitt, A. G., Benach, J. L., Sellati, T. J., and Furie, M. B. (2007). Francisella tularensis has a significant extracellular phase in infected mice. J. Infect. Dis. 196, 134–137.
Fortier, A. H., Slayter, M. V., Ziemba, R., Meltzer, M. S., and Nacy, C. A. (1991). Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun. 59, 2922–2928.
Fuller, C. L., Brittingham, K. C., Hepburn, M. J., Martin, J. W., Petitt, P. L., Pittman, P. R., and Bavari, S. (2006). Dominance of human innate immune responses in primary Francisella tularensis live vaccine strain vaccination. J. Allergy Clin. Immunol. 117, 1186–1188.
Fuller, C. L., Brittingham, K. C., Porter, M. W., Hepburn, M. J., Petitt, P. L., Pittman, P. R., and Bavari, S. (2007). Transcriptome analysis of human immune responses following live vaccine strain (LVS) Francisella tularensis vaccination. Mol. Immunol. 44, 3173–3184.
Fulop, M., Manchee, R., and Titball, R. (1995). Role of lipopolysaccharide and a major outer membrane protein from Francisella tularensis in the induction of immunity against tularemia. Vaccine 13, 1220–1225.
Fulop, M., Mastroeni, P., Green, M., and Titball, R. W. (2001). Role of antibody to lipopolysaccharide in protection against low- and high-virulence strains of Francisella tularensis. Vaccine 19, 4465–4472.
Gardam, M. A., Keystone, E. C., Menzies, R., Manners, S., Skamene, E., Long, R., and Vinh, D. C. (2003). Anti-tumour necrosis factor agents and tuberculosis risk: mechanisms of action and clinical management. Lancet Infect. Dis. 3, 148–155.
Geha, R. S. (2003). Allergy and hypersensitivity. Nature versus nurture in allergy and hypersensitivity. Curr. Opin. Immunol. 15, 603–608.
Gentry, M., Taormina, J., Pyles, R. B., Yeager, L., Kirtley, M., Popov, V. L., Klimpel, G., and Eaves-Pyles, T. (2007). Role of primary human alveolar epithelial cells in host defense against Francisella tularensis infection. Infect. Immun. 75, 3969–3978.
Gill, N., Wlodarska, M., and Finlay, B. B. (2010). The future of mucosal immunology: studying an integrated system-wide organ. Nat. Immunol. 11, 558–560.
Gregory, S. H., Mott, S., Phung, J., Lee, J., Moise, L., McMurry, J. A., Martin, W., and DeGroot, A. S. (2009). Epitope-based vaccination against pneumonic tularemia. Vaccine 27, 5299–5306.
Gunn, J. S., and Ernst, R. K. (2007). The structure and function of Francisella lipopolysaccharide. Ann. N. Y. Acad. Sci. 1105, 202–218.
Hajjar, A. M., Harvey, M. D., Shaffer, S. A., Goodlett, D. R., Sjostedt, A., Edebro, H., Forsman, M., Bystrom, M., Pelletier, M., Wilson, C. B., Miller, S. I., Skerrett, S. J., and Ernst, R. K. (2006). Lack of in vitro and in vivo recognition of Francisella tularensis subspecies lipopolysaccharide by Toll-like receptors. Infect. Immun. 74, 6730–6738.
Hall, J. D., Kurtz, S. L., Rigel, N. W., Gunn, B. M., Taft-Benz, S., Morrison, J. P., Fong, A. M., Patel, D. D., Braunstein, M., and Kawula, T. H. (2009). The impact of chemokine receptor CX3CR1 deficiency during respiratory infections with Mycobacterium tuberculosis or Francisella tularensis. Clin. Exp. Immunol. 156, 278–284.
Hall, J. D., Woolard, M. D., Gunn, B. M., Craven, R. R., Taft-Benz, S., Frelinger, J. A., and Kawula, T. H. (2008). Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect. Immun. 76, 5843–5852.
Hambleton, P., Evans, C. G., Hood, A. M., and Strange, R. E. (1974). Vaccine potencies of the live vaccine strain of Francisella tularensis and isolated bacterial components. Br. J. Exp. Pathol. 55, 363–373.
Han, S., Bishop, B. M., and van Hoek, M. L. (2008). Antimicrobial activity of human beta-defensins and induction by Francisella. Biochem. Biophys. Res. Commun. 371, 670–674.
Henry, T., Kirimanjeswara, G. S., Ruby, T., Jones, J. W., Peng, K., Perret, M., Ho, L., Sauer, J. D., Iwakura, Y., Metzger, D. W., and Monack, D. M. (2010). Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J. Immunol. 184, 3755–3767.
Hollis, D. G., Weaver, R. E., Steigerwalt, A. G., Wenger, J. D., Moss, C. W., and Brenner, D. J. (1989). Francisella philomiragia comb. nov. (formerly Yersinia philomiragia) and Francisella tularensis biogroup novicida (formerly Francisella novicida) associated with human disease. J. Clin. Microbiol. 27, 1601–1608.
Horzempa, J., O’Dee, D. M., Shanks, R. M., and Nau, G. J. (2010). Francisella tularensis DeltapyrF mutants show that replication in nonmacrophages is sufficient for pathogenesis in vivo. Infect. Immun. 78, 2607–2619.
Huber, B., Escudero, R., Busse, H. J., Seibold, E., Scholz, H. C., Anda, P., Kampfer, P., and Splettstoesser, W. D. (2010). Description of Francisella hispaniensis sp. nov., isolated from human blood, reclassification of Francisella novicida (Larson et al. 1955) Olsufiev et al. 1959 as Francisella tularensis subsp. novicida comb. nov. and emended description of the genus Francisella. Int. J. Syst. Evol. Microbiol. 60, 1887–1896.
Huntley, J. F., Conley, P. G., Rasko, D. A., Hagman, K. E., Apicella, M. A., and Norgard, M. V. (2008). Native outer membrane proteins protect mice against pulmonary challenge with virulent type A Francisella tularensis. Infect. Immun. 76, 3664–3671.
Janovska, S., Pavkova, I., Reichelova, M., Hubaleka, M., Stulik, J., and Macela, A. (2007). Proteomic analysis of antibody response in a case of laboratory-acquired infection with Francisella tularensis subsp. tularensis. Folia Microbiol (Praha) 52, 194–198.
Johansson, A., Celli, J., Conlan, W., Elkins, K. L., Forsman, M., Keim, P. S., Larsson, P., Manoil, C., Nano, F. E., Petersen, J. M., and Sjöstedt, A. (2010). Objections to the transfer of Francisella novicida to the subspecies rank of Francisella tularensis. Int. J. Syst. Evol. Microbiol. 60, 1717–1718; author reply 1718–1720.
Jones, J. W., Kayagaki, N., Broz, P., Henry, T., Newton, K., O’Rourke, K., Chan, S., Dong, J., Qu, Y., Roose-Girma, M., Dixit, V. M., and Monack, D. M. (2010). Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U.S.A. 107, 9771–9776.
Karttunen, R., Surcel, H. M., Andersson, G., Ekre, H. P., and Herva, E. (1991). Francisella tularensis-induced in vitro gamma interferon, tumor necrosis factor alpha, and interleukin 2 responses appear within 2 weeks of tularemia vaccination in human beings. J. Clin. Microbiol. 29, 753–756.
Katz, J., Zhang, P., Martin, M., Vogel, S. N., and Michalek, S. M. (2006). Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infect. Immun. 74, 2809–2816.
Ketavarapu, J. M., Rodriguez, A. R., Yu, J. J., Cong, Y., Murthy, A. K., Forsthuber, T. G., Guentzel, M. N., Klose, K. E., Berton, M. T., and Arulanandam, B. P. (2008). Mast cells inhibit intramacrophage Francisella tularensis replication via contact and secreted products including IL-4. Proc. Natl. Acad. Sci. U.S.A. 105, 9313–9318.
Kieffer, T. L., Cowley, S., Nano, F. E., and Elkins, K. L. (2003). Francisella novicida LPS has greater immunobiological activity in mice than F. tularensis LPS, and contributes to F. novicida murine pathogenesis. Microbes Infect. 5, 397–403.
Kirimanjeswara, G. S., Golden, J. M., Bakshi, C. S., and Metzger, D. W. (2007). Prophylactic and therapeutic use of antibodies for protection against respiratory infection with Francisella tularensis. J. Immunol. 179, 532–539.
Klimpel, G. R., Eaves-Pyles, T., Moen, S. T., Taormina, J., Peterson, J. W., Chopra, A. K., Niesel, D. W., Carness, P., Haithcoat, J. L., Kirtley, M., and Nasr, A. B. (2008). Levofloxacin rescues mice from lethal intra-nasal infections with virulent Francisella tularensis and induces immunity and production of protective antibody. Vaccine 26, 6874–6882.
Konstantinou, M. P., Abecassis-Cotta, S., Valeyrie-Allanore, L., Ortonne, N., Maurin, M., Roujeau, J. C., Revuz, J., and Bagot, M. (2009). Severe tularaemia mimicking glandular tuberculosis during adalimumab therapy. Ann. Dermatol. Venereol. 136, 718–722.
Koskela, P., and Herva, E. (1980). Cell-mediated immunity against Francisella tularensis after natural infection. Scand. J. Infect. Dis. 12, 281–287.
Koskela, P., and Salminen, A. (1985). Humoral immunity against Francisella tularensis after natural infection. J. Clin. Microbiol. 22, 973–979.
Kroca, M., Tärnvik, A., and Sjöstedt, A. (2000). The proportion of circulating gamma/delta T cells increases after the first week of onset of tularaemia and remains elevated for more than a year. Clin. Exp. Immunol. 120, 280–284.
Krocova, Z., Hartlova, A., Souckova, D., Zivna, L., Kroca, M., Rudolf, E., Macela, A., and Stulik, J. (2008). Interaction of B cells with intracellular pathogen Francisella tularensis. Microb. Pathog. 45, 79–85.
Kugeler, K. J., Mead, P. S., Janusz, A. M., Staples, J. E., Kubota, K. A., Chalcraft, L. G., and Petersen, J. M. (2009). Molecular Epidemiology of Francisella tularensis in the United States. Clin. Infect. Dis. 48, 863–870.
Kugeler, K. J., Mead, P. S., McGowan, K. L., Burnham, J. M., Hogarty, M. D., Ruchelli, E., Pollard, K., Husband, B., Conley, C., Rivera, T., Kelesidis, T., Lee, W. M., Mabey, W., Winchell, J. M., Stang, H. L., Staples, J. E., Chalcraft, L. J., and Petersen, J. M. (2008). Isolation and characterization of a novel Francisella sp. from human cerebrospinal fluid and blood. J. Clin. Microbiol. 46, 2428–2431.
KuoLee, R., Harris, G., Conlan, J. W., and Chen, W. (2007). Oral immunization of mice with the live vaccine strain (LVS) of Francisella tularensis protects mice against respiratory challenge with virulent type A F. tularensis. Vaccine 25, 3781–3791.
Lai, X. H., Golovliov, I., and Sjostedt, A. (2001). Francisella tularensis induces cytopathogenicity and apoptosis in murine macrophages via a mechanism that requires intracellular bacterial multiplication. Infect. Immun. 69, 4691–4694.
Lai, X. H., and Sjostedt, A. (2003). Delineation of the molecular mechanisms of Francisella tularensis-induced apoptosis in murine macrophages. Infect. Immun. 71, 4642–4646.
Lavine, C. L., Clinton, S. R., Angelova-Fischer, I., Marion, T. N., Bina, X. R., Bina, J. E., Whitt, M. A., and Miller, M. A. (2007). Immunization with heat-killed Francisella tularensis LVS elicits protective antibody-mediated immunity. Eur. J. Immunol. 37, 3007–3020.
Lee, B. Y., Horwitz, M. A., and Clemens, D. L. (2006). Identification, recombinant expression, immunolocalization in macrophages, and T-cell responsiveness of the major extracellular proteins of Francisella tularensis. Infect. Immun. 74, 4002–4013.
Leelaporn, A., Yongyod, S., Limsrivanichakorn, S., Yungyuen, T., and Kiratisin, P. (2008). Francisella novicida bacteremia, Thailand. Emerging Infect. Dis. 14, 1935–1937.
Leiby, D. A., Fortier, A. H., Crawford, R. M., Schreiber, R. D., and Nacy, C. A. (1992). In vivo modulation of the murine immune response to Francisella tularensis LVS by administration of anticytokine antibodies. Infect. Immun. 60, 84–89.
Lembo, A., Pelletier, M., Iyer, R., Timko, M., Dudda, J. C., West, T. E., Wilson, C. B., Hajjar, A. M., and Skerrett, S. J. (2008). Administration of a synthetic TLR4 agonist protects mice from pneumonic tularemia. J. Immunol. 180, 7574–7581.
Lin, Y., Ritchea, S., Logar, A., Slight, S., Messmer, M., Rangel-Moreno, J., Guglani, L., Alcorn, J. F., Strawbridge, H., Park, S. M., Onishi, R., Nyugen, N., Walter, M. J., Pociask, D., Randall, T. D., Gaffen, S. L., Iwakura, Y., Kolls, J. K., and Khader, S. A. (2009). Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity 31, 799–810.
Lindgren, H., Shen, H., Zingmark, C., Golovliov, I., Conlan, W., and Sjostedt, A. (2007). Resistance of Francisella tularensis strains against reactive nitrogen and oxygen species with special reference to the role of KatG. Infect. Immun. 75, 1303–1309.
Lindgren, H., Stenman, L., Tarnvik, A., and Sjostedt, A. (2005). The contribution of reactive nitrogen and oxygen species to the killing of Francisella tularensis LVS by murine macrophages. Microbes Infect. 7, 467–475.
Lindgren, H., Stenmark, S., Chen, W., Tarnvik, A., and Sjostedt, A. (2004). Distinct roles of reactive nitrogen and oxygen species to control infection with the facultative intracellular bacterium Francisella tularensis. Infect. Immun. 72, 7172–7182.
Lopez, M. C., Duckett, N. S., Baron, S. D., and Metzger, D. W. (2004). Early activation of NK cells after lung infection with the intracellular bacterium, Francisella tularensis LVS. Cell. Immunol. 232, 75–85.
Lyons, R. C., and Wu, T. H. (2007). Animal models of Francisella tularensis infection. Ann. N. Y. Acad. Sci. 1105, 238–265.
Malik, M., Bakshi, C. S., McCabe, K., Catlett, S. V., Shah, A., Singh, R., Jackson, P. L., Gaggar, A., Metzger, D. W., Melendez, J. A., Blalock, J. E., and Sellati, T. J. (2007). Matrix metalloproteinase 9 activity enhances host susceptibility to pulmonary infection with type A and B strains of Francisella tularensis. J. Immunol. 178, 1013–1020.
Malik, M., Bakshi, C. S., Sahay, B., Shah, A., Lotz, S. A., and Sellati, T. J. (2006). Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect. Immun. 74, 3657–3662.
Maranan, M. C., Schiff, D., Johnson, D. C., Abrahams, C., Wylam, M., and Gerber, S. I. (1997). Pneumonic tularemia in a patient with chronic granulomatous disease. Clin. Infect. Dis. 25, 630–633.
Mares, C. A., Ojeda, S. S., Morris, E. G., Li, Q., and Teale, J. M. (2008). Initial delay in the immune response to Francisella tularensis is followed by hypercytokinemia characteristic of severe sepsis and correlating with upregulation and release of damage-associated molecular patterns. Infect. Immun. 76, 3001–3010.
Mares, C. A., Sharma, J., Li, Q., Rangel, E. L., Morris, E. G., Enriquez, M. I., and Teale, J. M. (2010). Defect in efferocytosis leads to alternative activation of macrophages in Francisella infections. Immunol. Cell Biol. doi: 10.1038/icb.2010.81. [Epub ahead of print].
Markel, G., Bar-Haim, E., Zahavy, E., Cohen, H., Cohen, O., Shafferman, A., and Velan, B. (2010). The involvement of IL-17A in the murine response to sub-lethal inhalational infection with Francisella tularensis. PLoS ONE 5, e11176. doi: 10.1371/journal.pone.0011176
Martinon, F., Chen, X., Lee, A. H., and Glimcher, L. H. (2010). TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11, 411–418.
McCaffrey, R. L., and Allen, L. A. (2006). Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J. Leukoc. Biol. 80, 1224–1230.
McCaffrey, R. L., Schwartz, J. T., Lindemann, S. R., Moreland, J. G., Buchan, B. W., Jones, B. D., and Allen, L. A. (2010). Multiple mechanisms of NADPH oxidase inhibition by type A and type B Francisella tularensis. J. Leukoc. Biol. 88, 791–805.
McMurry, J. A., Gregory, S. H., Moise, L., Rivera, D., Buus, S., and De Groot, A. S. (2007). Diversity of Francisella tularensis Schu4 antigens recognized by T lymphocytes after natural infections in humans: identification of candidate epitopes for inclusion in a rationally designed tularemia vaccine. Vaccine 25, 3179–3191.
Melillo, A. A., Bakshi, C. S., and Melendez, J. A. (2010). Francisella tularensis antioxidants harness reactive oxygen species to restrict macrophage signaling and cytokine production. J. Biol. Chem. 285, 27553–27560.
Metzger, D. W., Bakshi, C. S., and Kirimanjeswara, G. (2007). Mucosal immunopathogenesis of Francisella tularensis. Ann. N. Y. Acad. Sci. 1105, 266–283.
Moreland, J. G., Hook, J. S., Bailey, G., Ulland, T., and Nauseef, W. M. (2009). Francisella tularensis directly interacts with the endothelium and recruits neutrophils with a blunted inflammatory phenotype. Am. J. Physiol. Lung Cell Mol. Physiol. 296, L1076–L1084.
Moule, M. G., Monack, D. M., and Schneider, D. S. (2010). Reciprocal analysis of Francisella novicida infections of a Drosophila melanogaster model reveal host–pathogen conflicts mediated by reactive oxygen and imd-regulated innate immune response. PLoS Pathog. 6, e1001065. doi: 10.1371/journal.ppat.1001065
Nelson, M., Lever, M. S., Dean, R. E., Savage, V. L., Salguero, F. J., Pearce, P. C., Stevens, D. J., and Simpson, A. J. (2010). Characterization of lethal inhalational infection with Francisella tularensis in the common marmoset (Callithrix jacchus). J. Med. Microbiol. 59, 1107–1113.
Nelson, M., Lever, M. S., Savage, V. L., Salguero, F. J., Pearce, P. C., Stevens, D. J., and Simpson, A. J. (2009). Establishment of lethal inhalational infection with Francisella tularensis (tularaemia) in the common marmoset (Callithrix jacchus). Int. J. Exp. Pathol. 90, 109–118.
Overholt, E. L., Tigertt, W. D., Kadull, P. J., Ward, M. K., Charkes, N. D., Rene, R. M., Salzman, T. E., and Mallory, S. (1961). An analysis of forty-two cases of laboratory-acquired tularemia: treatment with broad-spectrum antibiotics. Am. J. Med. Sci. 30, 785–791.
Pammit, M. A., Raulie, E. K., Lauriano, C. M., Klose, K. E., and Arulanandam, B. P. (2006). Intranasal vaccination with a defined attenuated Francisella novicida strain induces gamma interferon- dependent antibody-mediated protection against tularemia. Infect. Immun. 74, 2063–2071.
Paranavitana, C., Zelazowska, E., DaSilva, L., Pittman, P. R., and Nikolich, M. (2010). Th17 cytokines in recall responses against Francisella tularensis in humans. J. Interferon Cytokine Res. 30, 471–476.
Park, M. K., Amichay, D., Love, P., Wick, E., Liao, F., Grinberg, A., Rabin, R. L., Zhang, H. H., Gebeyehu, S., Wright, T. M., Iwasaki, A., Weng, Y., DeMartino, J. A., Elkins, K. L., and Farber, J. M. (2002). The CXC chemokine murine monokine induced by IFN-gamma (CXC chemokine ligand 9) is made by APCs, targets lymphocytes including activated B cells, and supports antibody responses to a bacterial pathogen in vivo. J. Immunol. 169, 1433–1443.
Pierini, L. M. (2006). Uptake of serum-opsonized Francisella tularensis by macrophages can be mediated by class A scavenger receptors. Cell. Microbiol. 8, 1361–1370.
Polsinelli, T., Meltzer, M. S., and Fortier, A. H. (1994). Nitric oxide-independent killing of Francisella tularensis by IFN-gamma-stimulated murine alveolar macrophages. J. Immunol. 153, 1238–1245.
Poquet, Y., Kroca, M., Halary, F., Stenmark, S., Peyrat, M. A., Bonneville, M., Fournie, J. J., and Sjostedt, A. (1998). Expansion of Vgamma9 Vdelta2 T cells is triggered by Francisella tularensis-derived phosphoantigens in tularemia but not after tularemia vaccination. Infect. Immun. 66, 2107–2114.
Powell, H. J., Cong, Y., Yu, J. J., Guentzel, M. N., Berton, M. T., Klose, K. E., Murthy, A. K., and Arulanandam, B. P. (2008). CD4+ T cells are required during priming but not the effector phase of antibody-mediated IFN-gamma-dependent protective immunity against pulmonary Francisella novicida infection. Immunol. Cell Biol. 86, 515–522.
Pyles, R. B., Jezek, G. E., and Eaves-Pyles, T. D. (2010). Toll-like receptor 3 agonist protection against experimental Francisella tularensis respiratory tract infection. Infect. Immun. 78, 1700–1710.
Rajaram, M. V., Butchar, J. P., Parsa, K. V., Cremer, T. J., Amer, A., Schlesinger, L. S., and Tridandapani, S. (2009). Akt and SHIP modulate Francisella escape from the phagosome and induction of the Fas-mediated death pathway. PLoS ONE 4, e7919. doi: 10.1371/journal.pone.0007919
Rathinam, V. A., Jiang, Z., Waggoner, S. N., Sharma, S., Cole, L. E., Waggoner, L., Vanaja, S. K., Monks, B. G., Ganesan, S., Latz, E., Hornung, V., Vogel, S. N., Szomolanyi-Tsuda, E., and Fitzgerald, K. A. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402.
Ray, H. J., Chu, P., Wu, T. H., Lyons, C. R., Murthy, A. K., Guentzel, M. N., Klose, K. E., and Arulanandam, B. P. (2010). The Fischer 344 rat reflects human susceptibility to Francisella pulmonary challenge and provides a new platform for virulence and protection studies. PLoS ONE 5, e9952. doi: 10.1371/journal.pone.0009952
Raymond, C. R., and Conlan, J. W. (2009). Differential susceptibility of Sprague-Dawley and Fischer 344 rats to infection by Francisella tularensis. Microb. Pathog. 46, 231–234.
Rodriguez, A. R., Yu, J. J., Murthy, A. K., Guentzel, M. N., Klose, K. E., Forsthuber, T. G., Chambers, J. P., Berton, M. T., and Arulanandam, B. P. (2010). Mast cell/IL-4 control of Francisella tularensis replication and host cell death is associated with increased ATP production and phagosomal acidification. Mucosal Immunol. doi: 10.1038/mi.2010.59. [Epub ahead of print].
Roth, K. M., Gunn, J. S., Lafuse, W., and Satoskar, A. R. (2009). Francisella inhibits STAT1-mediated signaling in macrophages and prevents activation of antigen-specific T cells. Int. Immunol. 21, 19–28.
Salerno-Goncalves, R., Hepburn, M. J., Bavari, S., and Sztein, M. B. (2009). Generation of heterogeneous memory T cells by live attenuated tularemia vaccine in humans. Vaccine 28, 195–206.
Santic, M., Molmeret, M., and Abu Kwaik, Y. (2005). Modulation of biogenesis of the Francisella tularensis subsp. novicida-containing phagosome in quiescent human macrophages and its maturation into a phagolysosome upon activation by IFN-gamma. Cell. Microbiol. 7, 957–967.
Saslaw, S., Eigelsbach, H. T., Prior, J. A., Wilson, H. E., and Carhart, S. (1961a). Tularemia vaccine study. II. Respiratory challenge. Arch. Int. Med. 107, 134–146.
Saslaw, S., Eigelsbach, H. T., Wilson, H. E., Prior, J., and Carhart, S. (1961b). Tularemia vaccine study. I. Intracutaneous challenge. Arch. Int. Med. 107, 121–133.
Schulert, G. S., and Allen, L. A. (2006). Differential infection of mononuclear phagocytes by Francisella tularensis: role of the macrophage mannose receptor. J. Leukoc. Biol. 80, 563–571.
Sharma, J., Li, Q., Mishra, B. B., Pena, C., and Teale, J. M. (2009). Lethal pulmonary infection with Francisella novicida is associated with severe sepsis. J. Leukoc. Biol. 86, 491–504.
Shen, H., Harris, G., Chen, W., Sjostedt, A., Ryden, P., and Conlan, W. (2010). Molecular immune responses to aerosol challenge with Francisella tularensis in mice inoculated with live vaccine candidates of varying efficacy. PLoS ONE 5, e13349. doi: 10.1371/journal.pone.0013349
Shirey, K. A., Cole, L. E., Keegan, A. D., and Vogel, S. N. (2008). Francisella tularensis live vaccine strain induces macrophage alternative activation as a survival mechanism. J. Immunol. 181, 4159–4167.
Sicherer, S. H., Asturias, E. J., Winkelstein, J. A., Dick, J. D., and Willoughby, R. E. (1997). Francisella philomiragia sepsis in chronic granulomatous disease. Pediatr. Infect. Dis. J. 16, 420–422.
Sjostedt, A., Conlan, J. W., and North, R. J. (1994). Neutrophils are critical for host defense against primary infection with the facultative intracellular bacterium Francisella tularensis in mice and participate in defense against reinfection. Infect. Immun. 62, 2779–2783.
Sjostedt, A., North, R. J., and Conlan, J. W. (1996). The requirement of tumour necrosis factor-alpha and interferon-gamma for the expression of protective immunity to secondary murine tularaemia depends on the size of the challenge inoculum. Microbiology 142 (Pt 6), 1369–1374.
Splettstoesser, W. D., Matz-Rensing, K., Seibold, E., Tomaso, H., Al Dahouk, S., Grunow, R., Essbauer, S., Buckendahl, A., Finke, E. J., and Neubauer, H. (2007). Re-emergence of Francisella tularensis in Germany: fatal tularaemia in a colony of semi-free-living marmosets (Callithrix jacchus). Epidemiol. Infect. 135, 1256–1265.
Staples, J. E., Kubota, K. A., Chalcraft, L. G., Mead, P. S., and Petersen, J. M. (2006). Epidemiologic and molecular analysis of human tularemia, United States, 1964–2004. Emerging Infect. Dis. 12, 1113–1118.
Stenmark, S., Lindgren, H., Tarnvik, A., and Sjostedt, A. (2003). Specific antibodies contribute to the host protection against strains of Francisella tularensis subspecies holarctica. Microb. Pathog. 35, 73–80.
Sundaresh, S., Randall, A., Unal, B., Petersen, J. M., Belisle, J. T., Hartley, M. G., Duffield, M., Titball, R. W., Davies, D. H., Felgner, P. L., and Baldi, P. (2007). From protein microarrays to diagnostic antigen discovery: a study of the pathogen Francisella tularensis. Bioinformatics 23, i508–i518.
Surcel, H.-M., Syrjala, H., Karttunen, R., Tapaninaho, S., and Herva, E. (1991). Development of Francisella tularensis antigen responses measured as T-lymphocyte proliferation and cytokine production (tumor necrosis factor alpha, gamma interferon, and interleukin-2 and -4) during human tularemia. Infect. Immun. 59, 1948–1953.
Tärnvik, A. (1989). Nature of protective immunity to Francisella tularensis. Rev. Infect. Dis. 11, 440–451.
Thakran, S., Li, H., Lavine, C. L., Miller, M. A., Bina, J. E., Bina, X. R., and Re, F. (2008). Identification of Francisella tularensis lipoproteins that stimulate the toll-like receptor (TLR) 2/TLR1 heterodimer. J. Biol. Chem. 283, 3751–3760.
Thomas, R. M., Titball, R. W., Oyston, P. C., Griffin, K., Waters, E., Hitchen, P. G., Michell, S. L., Grice, I. D., Wilson, J. C., and Prior, J. L. (2007). The immunologically distinct O antigens from Francisella tularensis subspecies tularensis and Francisella novicida are both virulence determinants and protective antigens. Infect. Immun. 75, 371–378.
Thorpe, B. D., and Marcus, S. (1967). Phagocytosis and intracellular fate of Pasteurella tularensis: in vitro effects of exudate stimulants and streptomycin on phagocytic cells. J. Reticuloendothel. Soc. 4, 10–23.
Twenhafel, N. A., Alves, D. A., and Purcell, B. K. (2009). Pathology of inhalational Francisella tularensis spp. tularensis SCHU S4 infection in African green monkeys (Chlorocebus aethiops). Vet. Pathol. 46, 698–706.
Ulland, T. K., Buchan, B. W., Ketterer, M. R., Fernandes-Alnemri, T., Meyerholz, D. K., Apicella, M. A., Alnemri, E. S., Jones, B. D., Nauseef, W. M., and Sutterwala, F. S. (2010). Cutting edge: mutation of Francisella tularensis mviN leads to increased macrophage absent in melanoma 2 inflammasome activation and a loss of virulence. J. Immunol. 185, 2670–2674.
Valentino, M., and Frelinger, J. (2009). An approach to the identification of T cell epitopes in the genomic era: application to Francisella tularensis. Immunol. Res. 45, 218–228.
Valentino, M. D., Hensley, L. L., Skrombolas, D., McPherson, P. L., Woolard, M. D., Kawula, T. H., Frelinger, J. A., and Frelinger, J. G. (2009). Identification of a dominant CD4 T cell epitope in the membrane lipoprotein Tul4 from Francisella tularensis LVS. Mol. Immunol. 46, 1830–1838.
Vojtech, L. N., Sanders, G. E., Conway, C., Ostland, V., and Hansen, J. D. (2009). Host immune response and acute disease in a zebrafish model of Francisella pathogenesis. Infect. Immun. 77, 914–925.
Vonkavaara, M., Telepnev, M. V., Ryden, P., Sjöstedt, A., and Stöven, S. (2008). Drosophila melanogaster as a model for elucidating the pathogenicity of Francisella tularensis. Cell. Microbiol. 10, 1327–1338.
Waag, D. M., McKee, K. T. Jr., Sandstrom, G., Pratt, L. L., Bolt, C. R., England, M. J., Nelson, G. O., and Williams, J. C. (1995). Cell-mediated and humoral immune responses after vaccination of human volunteers with the live vaccine strain of Francisella tularensis. Clin. Diagn. Lab. Immunol. 2, 143–148.
Whipp, M. J., Davis, J. M., Lum, G., de Boer, J., Zhou, Y., Bearden, S. W., Petersen, J. M., Chu, M. C., and Hogg, G. (2003). Characterization of a novicida-like subspecies of Francisella tularensis isolated in Australia. J. Med. Microbiol. 52, 839–842.
Wickstrum, J. R., Bokhari, S. M., Fischer, J. L., Pinson, D. M., Yeh, H. W., Horvat, R. T., and Parmely, M. J. (2009). Francisella tularensis induces extensive caspase-3 activation and apoptotic cell death in the tissues of infected mice. Infect. Immun. 77, 4827–4836.
Wilder, J. A., and Gelhaus, H. C. (2009). “Characterization of aerosol infection with F. tularensis Schu S4 in cynomologus macaques and LD50 determination,” in Sixth International Tularemia Conference, Berlin, 127.
Wilson, J. E., Katkere, B., and Drake, J. R. (2009). Francisella tularensis induces ubiquitin-dependent major histocompatibility complex class II degradation in activated macrophages. Infect. Immun. 77, 4953–4965.
Woolard, M. D., Hensley, L. L., Kawula, T. H., and Frelinger, J. A. (2008). Respiratory Francisella tularensis live vaccine strain infection induces Th17 cells and prostaglandin E2, which inhibits generation of gamma interferon-positive T cells. Infect. Immun. 76, 2651–2659.
Woolard, M. D., Wilson, J. E., Hensley, L. L., Jania, L. A., Kawula, T. H., Drake, J. R., and Frelinger, J. A. (2007). Francisella tularensis-infected macrophages release prostaglandin E2 that blocks T cell proliferation and promotes a Th2-like response. J. Immunol. 178, 2065–2074.
Wu, T. H., Hutt, J. A., Garrison, K. A., Berliba, L. S., Zhou, Y., and Lyons, C. R. (2005). Intranasal vaccination induces protective immunity against intranasal infection with virulent Francisella tularensis biovar A. Infect. Immun. 73, 2644–2654.
Wu, T. H., Zsemlye, J. L., Statom, G. L., Hutt, J. A., Schrader, R. M., Scrymgeour, A. A., and Lyons, C. R. (2009). Vaccination of Fischer 344 rats against pulmonary infections by Francisella tularensis type A strains. Vaccine 27, 4684–4693.
Yee, D., Rhinehart-Jones, T. R., and Elkins, K. L. (1996). Loss of either CD4+ or CD8+ T cells does not affect the magnitude of protective immunity to an intracellular pathogen, Francisella tularensis strain LVS. J. Immunol. 157, 5042–5048.
Yu, J. J., Goluguri, T., Guentzel, M. N., Chambers, J. P., Murthy, A. K., Klose, K. E., Forsthuber, T. G., and Arulanandam, B. P. (2010). Francisella tularensis T-cell antigen identification using humanized HLA-DR4 transgenic mice. Clin. Vaccine Immunol. 17, 215–222.
Yu, J. J., Raulie, E. K., Murthy, A. K., Guentzel, M. N., Klose, K. E., and Arulanandam, B. P. (2008). The presence of infectious extracellular Francisella tularensis subsp. novicida in murine plasma after pulmonary challenge. Eur. J. Clin. Microbiol. Infect. Dis. 27, 323–325.
Keywords: Francisella, immunity, innate, adaptive, lymphocyte, cytokine
Citation: Cowley SC and Elkins KL (2011) Immunity to Francisella. Front. Microbio. 2:26. doi: 10.3389/fmicb.2011.00026
Received: 17 December 2010;
Accepted: 01 February 2011;
Published online: 16 February 2011.
Edited by:
Anders Sjostedt, Umeå University, SwedenReviewed by:
Dennis Metzger, Albany Medical College, USAShabaana Khader, University of Pittsburgh, USA
Copyright: © 2011 Cowley and Elkins. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Siobhán C. Cowley, Laboratory of Mycobacterial Diseases and Cellular Immunology, Division of Bacterial, Parasitic and Allergenic Products/ Office of Vaccines Research and Review/Center for Biologics Evaluation and Research, U.S. Food and Drug Administration, HFM-431, 29 Lincoln Drive, Room 516, Bethesda, MD 20892, USA. e-mail: siobhan.cowley@fda.hhs.gov