
- Department of Microbiology, University of Virginia, Charlottesville, VA, USA
Infection with the human-specific bacterial pathogen Neisseria gonorrhoeae triggers a potent, local inflammatory response driven by polymorphonuclear leukocytes (neutrophils or PMNs). PMNs are terminally differentiated phagocytic cells that are a vital component of the host innate immune response and are the first responders to bacterial and fungal infections. PMNs possess a diverse arsenal of components to combat microorganisms, including the production of reactive oxygen species and release of degradative enzymes and antimicrobial peptides. Despite numerous PMNs at the site of gonococcal infection, N. gonorrhoeae can be cultured from the PMN-rich exudates of individuals with acute gonorrhea, indicating that some bacteria resist killing by neutrophils. The contribution of PMNs to gonorrheal pathogenesis has been modeled in vivo by human male urethral challenge and murine female genital inoculation and in vitro using isolated primary PMNs or PMN-derived cell lines. These systems reveal that some gonococci survive and replicate within PMNs and suggest that gonococci defend themselves against PMNs in two ways: they express virulence factors that defend against PMNs’ oxidative and non-oxidative antimicrobial components, and they modulate the ability of PMNs to phagocytose gonococci and to release antimicrobial components. In this review, we will highlight the varied and complementary approaches used by N. gonorrhoeae to resist clearance by human PMNs, with an emphasis on gonococcal gene products that modulate bacterial-PMN interactions. Understanding how some gonococci survive exposure to PMNs will help guide future initiatives for combating gonorrheal disease.
Gonorrheal Disease
Gonorrhea is a major global health problem, with greater than 62 million cases estimated to occur worldwide per year (Anonymous, 2001). Numbers of reported cases in the United States have remained at approximately 330,000 annually, but it is estimated that the actual number is at least twice as high, and rates of gonorrhea are rapidly increasing among men who have sex with men and young adults (Workowski and Berman, 2010). The cause of gonorrhea is the Gram-negative diplococcus Neisseria gonorrhoeae (the gonococcus or Gc). Gc is a human-specific pathogen that is transmitted via close sexual contact with an infected individual. Gonorrhea presents as an acute urethritis in men and cervicitis in women, but the pharynx and rectum can also be infected (Wiesner and Thompson, 1980). Because of the frequently asymptomatic nature of female infection, gonorrhea is a major cause of pelvic inflammatory disease, characterized by abdominal pain and tubal scarring that results in ectopic pregnancy and infertility; untreated infections in men also lead to sterility. Disseminated Gc infections can cause arthritis–dermatitis syndrome, endocarditis, and meningitis. Gc can also be vertically transmitted during childbirth and is still a leading cause of infectious neonatal blindness in the developing world (Wiesner and Thompson, 1980). Gc remains a major public health problem due to rapid acquisition of resistance to multiple antibiotics (Tapsall, 2009) and its ability to phase and antigenically vary its surface structures, preventing infected individuals from developing a protective immune response and hindering development of a protective vaccine (Virji, 2009).
Regardless of the anatomic site that is infected, Gc promotes an inflammatory response that is characterized by the recruitment of PMNs (Figure 1). In men, PMNs appear in urethral swabs and urine several days after infection and immediately prior to the onset of symptoms (Cohen and Cannon, 1999). The purulent exudate produced by infected men, described in the Bible and by Galen, is the best-known aspect of gonorrheal disease and is reflected in the translation of “gonorrhea” from Greek as “flow of seed” (Edwards and Apicella, 2004). The cervical secretions of women with gonorrhea also contain PMNs (Evans, 1977). Bacteria in gonorrheal secretions are attached to and within PMNs (Ovcinnikov and Delektorskij, 1971; Farzadegan and Roth, 1975; Evans, 1977; King et al., 1978; Apicella et al., 1996). PMNs are the primary innate immune responders to bacterial and fungal infection and are capable of phagocytosing and killing a variety of microorganisms (Borregaard, 2010). Yet in spite of the numerous PMNs at the site of gonorrheal infection, viable Gc can be cultured from the exudates of infected individuals (Wiesner and Thompson, 1980), and a subset of Gc remain viable when Gc are exposed to PMNs in vitro (see below). We interpret these results to show that the PMN-driven innate immune response to Gc is ineffective at clearing a gonorrheal infection. The persistence of Gc in the presence of PMNs facilitates Gc’s long-term colonization of its obligate human hosts, creating enhanced opportunity for dissemination and transmission of gonorrhea. In this review we will highlight our current knowledge about Gc resistance to PMN clearance, a critical aspect of the virulence of Gc.
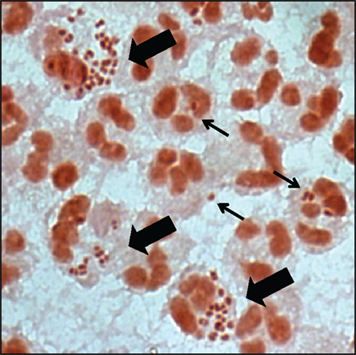
Figure 1. Gonorrhoeal exudates contain numerous PMNs with associated Gc. Gram stain of the urethral exudate from a male with uncomplicated gonorrhea. Some PMNs associate with single diplococci (thin arrow), while others have multiple adherent and internalized Gc (thick arrow). Note that the majority of PMNs in the exudate are uninfected.
PMN Antimicrobial Activities
PMNs are the most abundant white cells in the peripheral blood of humans. They are professional phagocytes and the first line of defense of the innate immune system (Borregaard, 2010). In response to peripheral infection or damage, PMNs follow chemotactic cues to extravasate from the bloodstream and migrate through tissues to reach the target site. Mucosal epithelial cells and resident immune release chemokines for PMNs, including interleukin-8, interleukin-6, tumor necrosis factor-α, and interleukin-1 (Borregaard, 2010). These chemokines are released during human Gc infection (Ramsey et al., 1995; Hedges et al., 1998).
PMNs possess receptors to bind and phagocytose complement- and antibody-opsonized particles [e.g., complement receptor 3 (CR3), FcRs]. They can also engulf unopsonized particles through lectin-like interactions or using receptors that are specific for ligands on the particle surface (Groves et al., 2008). Interaction between PMNs and a target particle results in the mobilization of different subsets of cytoplasmic granules to the plasma or phagosomal membrane (Figure 2). Granule fusion enables the degradation and killing of microorganisms both intracellularly and extracellularly (Borregaard et al., 2007). PMN mechanisms of microbial killing include production of reactive oxygen species (ROS) via the NADPH oxidase enzyme (the “oxidative burst”) as well as the oxygen-independent activities of degradative enzymes and antimicrobial peptides (Table 1). Human PMN granules are classified as azurophilic or primary granules, which contain myeloperoxidase, α-defensin peptides, and cathepsin G, among other antimicrobial components; specific or secondary granules containing the flavocytochrome b558 subunit of NADPH oxidase, LL-37 cathelicidin, lactoferrin, and CR3; and gelatinase or tertiary granules containing gelatinase (Borregaard et al., 2007). PMN granules release their contents in a set order. Initially, gelatinase granule contents degrade extracellular matrix, allowing PMNs to migrate across the tissues underlying the site of infection. Next, the release of specific granules at the target destination increases phagocytic potential due to presentation of CR3 on the PMN surface. Finally, the release of both specific and azurophilic granules creates an environment that is generally hostile to microbial survival (Lacy and Eitzen, 2008). PMNs also release neutrophil extracellular traps (NETs) composed of DNA, histones, and selected granule components, which trap and kill microbes without requiring phagocytosis (Papayannopoulos and Zychlinsky, 2009). Thus PMNs combine oxygen-dependent and -independent mechanisms to combat intracellular and extracellular microorganisms.
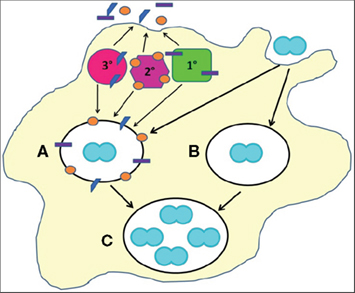
Figure 2. Cellular mechanisms of Gc survival after exposure to PMNs. Gc (blue diplococcus) attaches to the surface of PMNs and is engulfed into a phagosome (white oval). PMNs possess three classes of granules (1°, 2°, and 3°), each of which contains a unique subset of antimicrobial compounds. Granules fuse with the nascent phagosome or plasma membrane to deliver their contents to invading microorganisms. We propose two mechanisms that allow Gc to survive after exposure to PMNs. (For illustrative purposes, only intracellular Gc survival is depicted.) First, PMN granules release their contents at the plasma membrane or into phagosomes containing Gc (A). However, Gc virulence factors confer resistance to granules’ antimicrobial compounds. Second, Gc prevents PMN granules from releasing their contents at the plasma membrane or into phagosomes, allowing the bacteria to avoid encountering PMN antimicrobial compounds (B). Either mechanism would enable a fraction of Gc to survive and replicate in the presence of PMNs (C).

Table 1. Antimicrobial components housed in PMN granules.
The fact that gonorrheal exudates contain viable Gc indicates that PMNs are ineffective at completely clearing Gc infection. There are two mechanisms that could explain how Gc survives PMN challenge: Gc prevents PMNs from performing their normal antimicrobial functions (phagocytosis, granule content release), or Gc expresses defenses against oxidative and non-oxidative components produced by PMNs (Figure 2). As we will discuss, there is now substantial evidence for both mechanisms, which ultimately enable Gc to survive within a host and be transmitted to new individuals.
Model Systems for Examining PMNs During Gc Pathogenesis
Four experimental approaches have been used to investigate the involvement of PMNs in gonorrheal disease. Each has contributed to our understanding of how PMNs are recruited during acute gonorrhea and how Gc withstands this onslaught.
The Male Urethral Challenge Model
Experimental human infection is limited to male urethral inoculation, due to the potential for severe complications such as pelvic inflammatory disease in women with gonorrhea (Cohen and Cannon, 1999). Urethral infection of male volunteers results in the release of proinflammatory cytokines and appearance of PMNs in the urogenital tract 2–3 days after infection, similar to what is seen in natural cases of gonococcal urethritis (Cohen and Cannon, 1999). As in natural infections, exudates from males with experimental Gc infection contain PMNs with associated Gc and occasional exfoliated epithelial cells. Electron microscopic analysis of these exudates revealed that a subset of Gc inside PMNs appear intact, providing the initial evidence that Gc may survive within PMN phagosomes (Ovcinnikov and Delektorskij, 1971; Farzadegan and Roth, 1975; Apicella et al., 1996).
The Female Murine Genital Tract Model
Dr. Ann Jerse (Uniformed Services University of the Health Sciences) has developed a female mouse model of Gc genital tract infection, which allows gonorrheal infection to be examined in a genetically tractable host. In this model, estradiol-treated mice are inoculated vaginally with Gc, which allows over 80% of mice to be colonized with bacteria for over 1 week. Infected mice produce inflammatory cytokines, leading to rapid appearance of PMNs in the genital tract (Jerse, 1999). Experimental infection of female mice has provided insight into the selective advantage of opacity-associated (Opa) protein expression on Gc survival and the roles of Gc virulence factors conferring in vitro resistance to ROS and antimicrobial peptides in in vivo infection (Jerse, 1999; Jerse et al., 2003; Wu and Jerse, 2006; Wu et al., 2009; Cole et al., 2010). Because mice lack the human-specific receptors and other components that are likely to be important for gonorrheal disease, future studies could employ mice transgenic for human proteins of interest. Inbred mice that are transgenic for human carcinoembryonic antigen-related cellular adhesion molecules (CEACAMs) and CD46, receptors that are implicated in gonorrheal pathogenesis (Merz and So, 2000), have already been developed (Johansson et al., 2003; Gu et al., 2010), with additional mouse strains likely to be produced in the coming years.
Immortalized PMN-Like Cell Lines
The use of immortalized promyelocytic human cell lines to study the molecular mechanisms of Gc pathogenesis provides a system which is clonal, easy to maintain, and amenable to expression of transgenes. As one example, the leukemic HL-60 cell line can be differentiated into a PMN-like phenotype with retinoic acid (Collins et al., 1977; Newburger et al., 1979). Differentiated HL-60 cells can phagocytose and generate ROS in response to Opa-expressing Gc akin to primary human PMNs (Bauer et al., 1999; Pantelic et al., 2004). However, HL-60 cells do not possess the robust antimicrobial activity associated with primary cells, due in part to the absence of specific granules and other intracellular compartments (Le Cabec et al., 1997).
Primary PMNs
Research on the molecular mechanisms underlying Gc infection of PMNs has mostly relied upon primary human cells, purified from freshly isolated human blood. The abundance of PMNs in human blood and the ease of purification make PMNs amenable to infection with Gc in vitro. The limitations of working with primary PMNs include their short half-life, their limited capacity for genetic manipulation, and the person-to-person variability intrinsic to primary human cells. However, primary human PMNs have been used to measure binding and phagocytosis of Gc, quantify Gc survival after PMN exposure, and assess the roles of Gc virulence factors in bacterial defense against PMNs (see below). Gc infection of murine PMNs has also been conducted (Wu and Jerse, 2006; Soler-Garcia and Jerse, 2007). Future studies using primary PMNs along with cultured epithelial cells from relevant anatomic sites may provide a means to examine the complex interactions between host cells that occur during gonorrheal infection.
Gc interaction with PMNs is influenced by the physiological state of the PMNs being used. Initial experimentation with primary human PMNs utilized cells and Gc suspended in buffered saline solutions (Densen and Mandell, 1978; Rest et al., 1982), but this is unlikely to reflect the transmigrated, primed state of PMNs in the genitourinary tract during acute infection. Research from the laboratory of Dr. Richard Rest (Drexel University) demonstrated that when PMNs were allowed to adhere to tissue culture-treated dishes, they released granule components and bound significantly more Gc than PMNs in suspension (Farrell and Rest, 1990). Dr. Michael Apicella’s laboratory (University of Iowa) subsequently developed an assay using collagen-adherent PMNs, which generated a system for studying the role of selected Gc virulence factors in bacterial survival after PMN challenge (Seib et al., 2005; Simons et al., 2005). We adapted the Apicella protocol to include PMN treatment with the chemokine interleukin-8, which facilitates PMN activation (Borregaard, 2010). We used this system to demonstrate Gc survival inside PMNs and to identify Gc proteins that defend the bacteria from killing by PMNs (Stohl et al., 2005; Criss et al., 2009).
Gc Survival and Replication in the Presence of PMNs
Although the survival of Gc in association with PMNs was once hotly debated, there is now substantial evidence that gonococci survive and multiply within human phagocytes. Examination of urethral exudates by light and electron microscopy has repeatedly shown the presence of abundant PMNs with associated and internalized Gc (Ovcinnikov and Delektorskij, 1971; Farzadegan and Roth, 1975; King et al., 1978; Apicella et al., 1996). The fact that viable gonococci can be cultured from urethral exudates or cervical swabs is strongly suggestive of Gc survival in the presence of PMNs (Wiesner and Thompson, 1980). In vitro studies from the Apicella laboratory using adherent human PMNs demonstrated that over 50% of Gc internalized by PMNs remained viable for up to 6 h, as determined by viable bacterial counts and electron microscopy (Simons et al., 2005). Our group corroborated these findings and directly detected viable extracellular and intracellular Gc after PMN infection, using dyes that reveal the integrity of bacterial membranes (Criss et al., 2009). We conclude from these studies that a fraction of Gc can survive both extracellularly and intracellularly in the presence of PMNs.
There is evidence that Gc does not only persist within PMNs, but also uses the PMNs as a site for replication. Pioneering studies in the 1970s showed that Gc inside exudate-derived PMNs were sensitive to penicillin, which only kills replicating bacteria. In the presence of antimicrobial agents such as spectinomycin or pyocin that cannot permeate eukaryotic membranes, numbers of PMN-associated Gc increased over time, indicative of bacterial replication inside exudatous and in vitro-infected PMNs (Veale et al., 1976, 1979; Casey et al., 1979, 1980, 1986). Using electron microscopy and colony counts, the Apicella laboratory observed an increase in Gc within collagen-adherent human PMNs over a 6-h infection, results also suggestive of intracellular replication (Simons et al., 2005). Similarly, we used bacterial viability dyes to observe an increase in the number of viable Gc inside PMNs over time (Criss et al., 2009). While the advantage of Gc replicating inside terminally differentiated cells of a limited life span is questionable, the Apicella group showed that PMNs infected with Gc delay their spontaneous apoptosis (Simons et al., 2006). We anticipate that advances in cellular imaging will provide additional support for Gc replication inside PMNs and will give insight into the timing and extent of this event.
Binding and Phagocytosis of Gc by PMNs
Since gonorrheal secretions contain PMNs associated with viable intracellular and extracellular bacteria, Gc must possess factors that promote attachment and phagocytosis by PMNs. Opsonic and non-opsonic interactions are the two basic means of phagocytosis, both of which may be utilized by Gc (Groves et al., 2008; Figure 3).
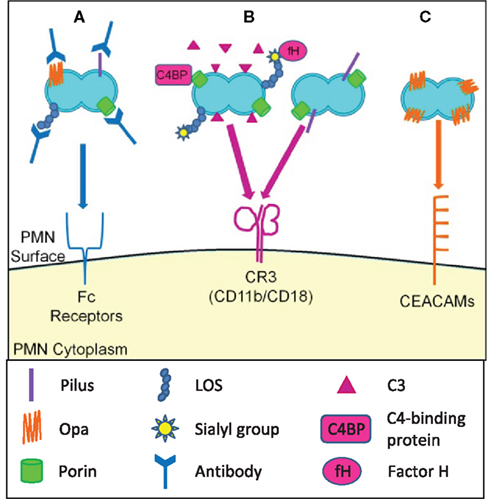
Figure 3. Opsonic and non-opsonic phagocytosis of Gc by PMNs. (A) Antibodies that recognize Gc surface structures opsonize the bacteria and allow for phagocytosis via Fc receptors. The efficacy of immunoglobulin-mediated phagocytosis is questionable given the extensive phase and antigenic variation of Gc surface structures. (B) Gc binds factor H and C4 binding protein, resulting in opsonization of Gc with C3 and other complement components. Gc is then phagocytosed via the CR3 receptor. Gc pili and porin can cooperatively interact with CR3, which may mediate the non-opsonic phagocytosis of Gc by PMNs. (C) Selected Opa proteins bind to CEACAM family receptors expressed on PMNs, leading to non-opsonic phagocytosis of Opa+ Gc.
Opsonic Uptake
The two major opsonins for PMN phagocytosis are immunoglobulins and complement, which bind to Fc receptors and complement receptors such as CR3, respectively (Groves et al., 2008). Patients with gonorrhea produce opsonic IgG and IgA directed against Gc surface-exposed components including porin, Opa proteins, pilin, iron-regulated outer membrane proteins, and lipooligosaccharide (LOS) (Brooks et al., 1976; McMillan et al., 1979; Tramont et al., 1980; Rice and Kasper, 1982; Siegel et al., 1982; Lammel et al., 1985; Schwalbe et al., 1985). Intriguingly, serum from individuals with no prior history of gonorrhea contains opsonic IgG against Gc porin and IgM against Gc LOS isotypes containing hexosamine; the non-Gc antigens recognized by these antibodies are not known (Sarafian et al., 1983; Griffiss et al., 1991). Many of the Gc surface structures that promote humoral immune responses are phase and antigenically variable and thus evade antibody-mediated immune surveillance (Virji, 2009). Also, Gc secretes an IgA protease that cleaves the polymeric IgA in mucosal secretions (Blake and Swanson, 1978). Thus complement rather than antibodies is likely to drive the opsonic phagocytosis of Gc by PMNs.
The complement system is a key component of the innate immune system comprised of more than 30 proteins. The complement system can be activated by three routes: the classical, the alternative, and the lectin pathway, but all three routes normally proceed to proteolytic activation of the major complement protein C3 and assembly of the membrane attack complex (Ram et al., 2010). Gc has multiple ways of resisting the bactericidal activities of complement in normal human serum. Gc binds the complement regulatory proteins C4b-binding protein (C4BP) and factor H (fH) on its surface via porins and sialylated LOS (Ram et al., 1998a,b, 2001; Gulati et al., 2005). C4BP restricts the amount of C3 which can be deposited by the classical complement pathway. fH is a cofactor for factor I-mediated cleavage of C3b to the hemolytically inactive iC3b. In the alternative pathway fH irreversibly dissociates factor Bb to limit C3 deposition and subsequent C5 cleavage (Ram et al., 2010). C4BP and fH provide defense against direct complement-mediated killing but concomitantly increase iC3b deposition on the Gc surface. iC3b is a ligand for CR3 (CD11b/CD18), which in PMNs drives actin-dependent particle engulfment into degradative phagolysosomes and production of ROS (Groves et al., 2008). Although it is assumed that Gc is complement-opsonized at mucosal surfaces, how opsonization impacts Gc survival after PMN exposure remains to be explored.
Non-opsonic Uptake
In the absence of antibodies or complement, efficient binding and engulfment of Gc by PMNs is achieved via expression of colony Opa proteins (King and Swanson, 1978; Virji and Heckels, 1986; Fischer and Rest, 1988). Opa proteins, formerly known as “protein II,” are a family of closely related, 20–30 kD outer membrane proteins that facilitate Gc binding and internalization by human cells, including PMNs (Sadarangani et al., 2011). Gc strains possess approximately 11 opa genes encoding 7–8 antigenically distinct Opa proteins (Connell et al., 1990; Dempsey et al., 1991). Each opa gene is phase-variable due to slipped-strand mispairing in a pentameric nucleotide repeat that places the gene in our out of frame (Murphy et al., 1989), such that individual Gc can express zero, one, or any possible combination of Opa proteins. Differential expression of Opa proteins can influence bacterial tropism for host cell types and provides a mechanism of immune evasion (Sadarangani et al., 2011).
Opacity-associated proteins bind heparan sulfate proteoglycans (HSPGs) and/or CEACAMs. Only those Opa proteins that bind CEACAMs are reported to influence Gc interactions with PMNs (Sadarangani et al., 2011). The Opa-binding CEACAMs on PMNs are CEACAM1, CEACAM3, and CEACAM6, with CEACAM3 expression exclusively restricted to PMNs. CEACAM1 and CEACAM3 are transmembrane proteins, while CEACAM6 possesses a glycosylphosphatidylinositol anchor (Gray-Owen and Blumberg, 2006). Binding of Opa proteins to any of the three CEACAMs results in Gc internalization, but via different signaling events (McCaw et al., 2004).
Opacity-associated protein expression is selected for in the male urethra, the female cervix during the follicular phase of the menstrual cycle, and in the murine cervix (James and Swanson, 1978; Swanson et al., 1988; Jerse et al., 1994; Jerse, 1999). However, Opa− Gc survives better after exposure to PMNs in vitro than isogenic Opa+ Gc (Rest et al., 1982; Virji and Heckels, 1986; Criss et al., 2009). Opa protein expression increases Gc phagocytosis by PMNs and stimulates PMN ROS production, and both factors may influence bacterial survival after exposure to PMNs (Rest et al., 1982; Fischer and Rest, 1988).
Gc surface structures other than Opa proteins may contribute to adherence and phagocytosis by PMNs. Pili and porin cooperatively interact with CR3 on cervical epithelial cells (Edwards et al., 2002). It is not known if this interaction occurs on PMNs, but if it were to occur, it would drive non-opsonic uptake of Gc by PMNs. In vitro studies suggested that “type 1,” virulent, piliated Gc were resistant to phagocytosis and killing by PMNs compared to “type 4,” avirulent, non-piliated bacteria that arise after extensive laboratory passage (Ofek et al., 1974; Dilworth et al., 1975). We now know that type 1 and type 4 Gc vary in Opa expression as well as piliation, both of which could have contributed to these observations. Purified porins also decrease PMN actin polymerization, which may reduce the phagocytosis of Gc by PMNs (Bjerknes et al., 1995). Serogroup C strains of N. meningitidis with lacto-N-neotetraose (LNnT) on LOS are phagocytosed by neutrophils in an opsonin-independent manner (Estabrook et al., 1998); it has not been examined whether this LOS epitope on Gc affects phagocytosis by PMNs. Together, the combinatorial expression of Opa proteins, pili, porin, and LOS modulate Gc binding and internalization by PMNs.
Gc Defenses Against PMN Antimicrobial Activities
Whether they remain extracellular or are phagocytosed by PMNs, Gc must contend with the variety of oxidative and non-oxidative antimicrobial components produced by PMNs (Figure 2). Gc isolated directly from human material or guinea pig subcutaneous chamber fluid display increased survival in the presence of phagocytes compared to Gc grown in vitro (Witt et al., 1976; Veale et al., 1977), suggesting that Gc possesses factors necessary for defending against phagocyte killing that are lost or altered with extended in vitro culture. These Gc factors aid Gc in resisting the toxic activities of PMNs in two ways. First, Gc prevents PMNs from producing or releasing antimicrobial components. Second, Gc expresses virulence factors that defend against these components. As we will describe, many Gc gene products have been identified that protect Gc from purified ROS, proteases, or antimicrobial peptides, but in most cases their roles in defense against PMNs have not yet been investigated.
Defenses Against Oxidative Damage
The major species of ROS include superoxide anion, hydrogen peroxide, and hydroxyl radical. These ROS have different reactivities and half-lives, but together they induce DNA, protein, and cell membrane damage that can lead to cell death (Fang, 2004). There are at least four potential sources of oxidative stress for Gc in vivo. (1) PMN NADPH oxidase transports electrons across the phagosomal or plasma membrane to generate superoxide, which spontaneously dismutates to hydrogen peroxide. In PMNs, the azurophilic enzyme myeloperoxidase uses hydrogen peroxide as a substrate to generate hypochlorous acid (bleach; Roos et al., 2003). Phagocytes can also produce reactive nitrogen species (RNS) such as nitric oxide and peroxynitrite, but RNS appear to be of limited importance in human PMN antimicrobial activity (Fang, 2004). (2) Enzymes related to phagocyte NADPH oxidase are expressed in epithelial cells, and the survival defect of Gc antioxidant mutants inside primary cervical cells implies that epithelial cells may also be an important source of oxidative stress for Gc (Wu et al., 2005, 2006; Achard et al., 2009; Potter et al., 2009). (3) Lactobacillus species that generate hydrogen peroxide are normally found in the vaginal flora of women (Eschenbach et al., 1989). Women with inhibitory lactobacilli are less likely to be infected with Gc (Saigh et al., 1978), and lactobacilli inhibit Gc growth in vitro (Saigh et al., 1978; Zheng et al., 1994; St Amant et al., 2002). However, it appears that effects of lactobacilli on Gc may be independent of hydrogen peroxide production, since mucosal secretions can effectively quench lactobacilli-derived ROS (O’Hanlon et al., 2010). (4) Gc generate ROS during aerobic respiration, although this may be less of an issue in vivo, where the oxygen tension in the genitourinary tract is low (Archibald and Duong, 1986). Gc defenses against oxidative stress involve manipulation of the PMN oxidative burst, detoxifying or repair of oxidative damage, and transcriptional upregulation of antioxidant gene products (Figure 4A).
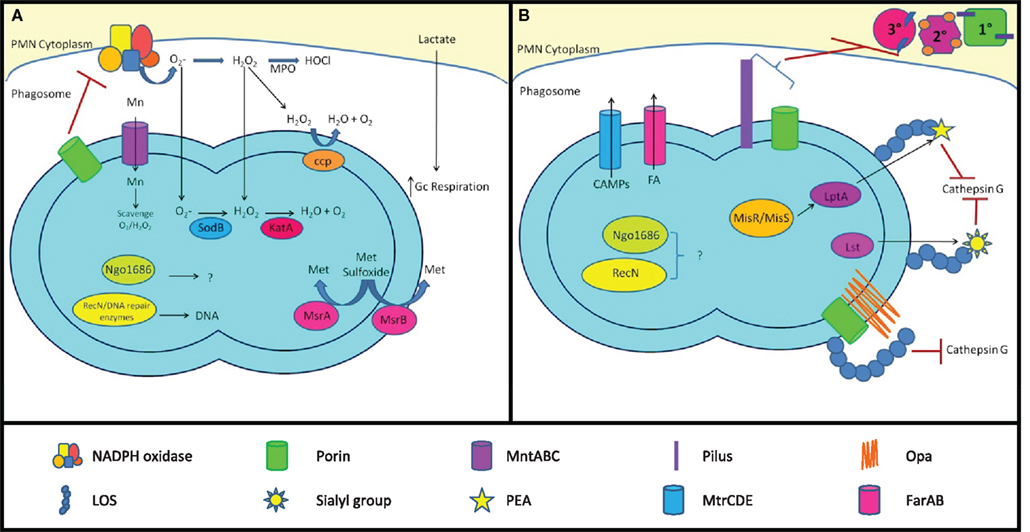
Figure 4. Mechanisms of Gc survival after exposure to antimicrobial compounds produced by PMNs. (A) Resistance to oxidative damage. PMN NADPH oxidase generates superoxide (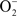 ) and hydrogen peroxide (H2O2) from O2, which are converted to hypochlorous acid (HOCl) by myeloperoxidase (MPO). Gc prevents PMNs from generating ROS by lactate-mediated increase in Gc O2 consumption and suppression of NADPH oxidase activity by porin or as-yet unidentified factors. Gc scavenges ROS through the activities of MntABC, superoxide dismutase (SodB), catalase (KatA), and cytochrome c peroxidase (Ccp). Gc can also repair damage due to ROS through DNA repair enzymes (RecN), protein reductases (MsrA/B), and other proteins (Ngo1686). (B) Resistance to PMN non-oxidative damage. Gc pili and/or porins prevent PMN granules from releasing non-oxidative antimicrobial components. LOS protects Gc outer membrane proteins such as Opa and porin from proteolysis by cathepsin G. Sialylation of LOS by Lst and PEA modification of LOS by LptA increases bacterial resistance to cathepsin G and other antimicrobials. The MisR/MisS two-component regulator increases expression of LptA and other gene products that confer resistance to PMN non-oxidative damage. Ngo1686 and RecN also protect Gc from PMN non-oxidative damage. The MtrCDE and FarAB efflux pumps export cationic antimicrobial peptides (CAMPs) and long-chain fatty acids (FA) from the Gc cytoplasm, respectively. In most cases, the contribution of these virulence factors to Gc survival after exposure to PMNs remains to be determined.
) and hydrogen peroxide (H2O2) from O2, which are converted to hypochlorous acid (HOCl) by myeloperoxidase (MPO). Gc prevents PMNs from generating ROS by lactate-mediated increase in Gc O2 consumption and suppression of NADPH oxidase activity by porin or as-yet unidentified factors. Gc scavenges ROS through the activities of MntABC, superoxide dismutase (SodB), catalase (KatA), and cytochrome c peroxidase (Ccp). Gc can also repair damage due to ROS through DNA repair enzymes (RecN), protein reductases (MsrA/B), and other proteins (Ngo1686). (B) Resistance to PMN non-oxidative damage. Gc pili and/or porins prevent PMN granules from releasing non-oxidative antimicrobial components. LOS protects Gc outer membrane proteins such as Opa and porin from proteolysis by cathepsin G. Sialylation of LOS by Lst and PEA modification of LOS by LptA increases bacterial resistance to cathepsin G and other antimicrobials. The MisR/MisS two-component regulator increases expression of LptA and other gene products that confer resistance to PMN non-oxidative damage. Ngo1686 and RecN also protect Gc from PMN non-oxidative damage. The MtrCDE and FarAB efflux pumps export cationic antimicrobial peptides (CAMPs) and long-chain fatty acids (FA) from the Gc cytoplasm, respectively. In most cases, the contribution of these virulence factors to Gc survival after exposure to PMNs remains to be determined.
Gc manipulation of the PMN oxidative burst
In the absence of Opa protein expression, Gc fails to induce the PMN oxidative burst (Rest et al., 1982; Virji and Heckels, 1986; Fischer and Rest, 1988; Criss and Seifert, 2008). Even in the presence of Opa+ Gc that induce ROS production in PMNs, the magnitude of ROS production is small relative to stimuli such as phorbol esters or other bacteria (Simons et al., 2005). Gc utilizes three mechanisms to reduce the amount of ROS produced by PMNs. First, exposure to lactate that is released from PMNs undergoing glycolysis stimulates the rate of Gc oxygen consumption, reducing the amount available to PMNs as a substrate for NADPH oxidase (Britigan et al., 1988). Second, purified Gc porin inhibits PMN ROS production in response to Gc, yeast particles, and latex beads (Lorenzen et al., 2000), but not formylated peptides (Haines et al., 1988; Bjerknes et al., 1995). Whether porin has this effect in the context of whole Gc bacteria remains to be examined. Third, we reported that Opa− Gc suppresses the PMN oxidative burst induced by serum opsonized staphylococci and formylated peptides by a process requiring bacterial protein synthesis and bacteria-PMN contact; the bacterial products mediating this effect are not known at this time (Criss and Seifert, 2008).
Detoxification and repair of oxidative damage
Bacteria respond to oxidative stress by catalysis of superoxide to hydrogen peroxide by superoxide dismutase (SOD), which is then converted to water and molecular oxygen by catalases and peroxidases (Seib et al., 2006). Gc possesses a single cytoplasmic superoxide dismutase (SodB), one cytoplasmic catalase (KatA), and several genes annotated as peroxidases. SodB activity is low in Gc and does not play a significant role in protection against oxidative stress (Tseng et al., 2001). In comparison, KatA is crucial to Gc defense against ROS. Gc has approximately 100-fold higher levels of catalase than E. coli (Hassett et al., 1990). Disruption of katA significantly reduces Gc survival to hydrogen peroxide and superoxide in vitro (Johnson et al., 1993; Soler-Garcia and Jerse, 2004; Stohl et al., 2005) and reduces the survival of some strains of Gc in the female murine genital tract (Wu et al., 2009). Gc also has high peroxidase Gc activity due to the periplasmic cytochrome c peroxidase encoded by ccp (Archibald and Duong, 1986). ccp mutant Gc show slight sensitivity to hydrogen peroxide, which is markedly enhanced when katA is also inactivated (Turner et al., 2003). Gc also imports Mn(II) into its cytoplasm via the MntABC transporter, where it scavenges superoxide and hydrogen peroxide by a mechanism independent of SodB and catalase (Tseng et al., 2001). This system is similar to the manganese transport system in Lactobacillus plantarum (Archibald and Duong, 1984).
Gc can also repair oxidative damage to proteins and DNA. Gc expresses two forms of methionine sulfoxide reductase, which reverses the oxidation of methionine residues in proteins. The MsrA protein is localized to the cytoplasm, while MsrB is secreted to the outer membrane. A msrAB mutant is more sensitive to hydrogen peroxide and superoxide in vitro than its wild-type parent (Skaar et al., 2002). Many Gc gene products involved in recombinational DNA repair, base excision repair, and nucleotide excision repair participate in Gc defense against ROS, such as the recombinase RecA and the DNA-binding protein RecN (Davidsen et al., 2005; Stohl et al., 2005; Stohl and Seifert, 2006; LeCuyer et al., 2010). The putative metalloprotease Ngo1686 helps protect Gc from hydrogen peroxide and the lipid oxidant cumene hydroperoxide, but the cellular targets with which Ngo1686 interacts are currently unknown (Stohl et al., 2005). Both ngo1686 and recN mutants have significant survival defects after exposure to primary human PMNs, but a recA mutant does not (Stohl et al., 2005; Criss et al., 2009).
Transcriptional induction of antioxidant gene products
Gc pre-exposed to hydrogen peroxide survives PMN challenge significantly better than unexposed Gc (Criss et al., 2009). This finding implies that Gc possesses complex transcriptional circuitry that is important for defenses against ROS and/or PMNs. The transcriptome of Gc exposed to sublethal concentrations of hydrogen peroxide was defined and revealed the upregulation of transcripts encoding RecN, Ngo1686, and other antioxidants after oxidative challenge (Stohl et al., 2005). Antioxidant gene expression is regulated by selected transcriptional repressors. The OxyR protein represses KatA expression, which is relieved following oxidative stress in order to increase catalase production (Tseng et al., 2003). PerR is responsive to Mn(II) levels and represses expression of MntC, part of the Mn(II) transporter (Wu et al., 2006). Finally, Ngo1427, a LexA homolog, represses expression of RecN, which is relieved when a cysteine residue is oxidized (Schook et al., 2011).
PMNs Primarily Direct Non-oxidative Antimicrobial Components against Gc
Although Gc has complex mechanisms for detecting oxidative damage and responding to it, the importance of these processes in Gc survival to PMNs appears to be limited. Mutants in katA, sodB, ccp, or mntABC, alone or in combination, do not affect the percentage of Gc that can survive PMN challenge (Seib et al., 2005; Criss et al., 2009). Moreover, Gc survival is similar between normal PMNs and ROS-deficient PMNs obtained from patients with chronic granulomatous disease (CGD; Rest et al., 1982; Criss and Seifert, 2008), and PMNs maintained in anoxic conditions, as are likely to be found in the upper reproductive tract of females, are not impaired for antigonococcal activity (Casey et al., 1986; Frangipane and Rest, 1992). Our group showed that Gc survival was unaffected after exposure to PMNs treated with diphenyleneiodonium (DPI), an inhibitor of NADPH oxidase. DPI treatment or CGD PMNs did not increase the percent survival of ngo1686 or recN Gc, nor did it enhance survival of Opa+ Gc that induce ROS from PMNs (Criss et al., 2009). From these results, we conclude that PMNs primarily direct non-oxidative antimicrobial activities against Gc. The functional redundancy in Gc antioxidant defenses may be sufficient to counter PMN-derived ROS; alternatively, PMNs may not generate enough ROS during infection to affect Gc survival.
Defenses Against Non-Oxidative Damage
Seminal research from the Rest and Shafer laboratories indicated that components found inside PMN granules display oxygen-independent antigonococcal activity (Rest, 1979; Casey et al., 1985; Rock and Rest, 1988). These components include the bactericidal/permeability-increasing protein (“hCAP57”), cathepsin G protease, and LL-37 antimicrobial peptide (Casey et al., 1985; Shafer et al., 1986, 1998). Unlike many Gram-negative bacteria, Gc are highly resistant (>0.2 mg/ml) to another class of antimicrobial peptides, the defensins (Qu et al., 1996), although the observed resistance varies depending on experimental conditions used (Porter et al., 2005). Of PMN non-oxidative granule components, cathepsin G and LL-37 have been the most actively studied for their effects on Gc.
Cathepsin G is a highly cationic serine protease which resides in PMN azurophilic granules. It enzymatically cleaves Gc outer membrane proteins including porin and Opa proteins (Rest and Pretzer, 1981; Shafer and Morse, 1987). However, heat and protease inhibitors do not impede cathepsin G’s ability to kill Gc in vitro, indicating its antigonococcal activity is independent of its proteolytic activity (Shafer et al., 1986). Cathepsin G can insert into Gc membranes, but killing does not appear to be due to changes in membrane permeability; instead, cathepsin G may impede peptidoglycan biosynthesis (Shafer et al., 1990).
LL-37 is the active form of an 18 kD protein precursor (“hCAP18”) which resides in specific granules. hCAP-18 is proteolytically processed to LL-37 by the azurophilic granule protein proteinase-3 (Sorensen et al., 2001). hCAP-18/LL-37 is also synthesized by mucosal epithelial cells, and is readily detected in cervicovaginal secretions (mean LL-37 concentration of 10 μg/ml) and seminal plasma (mean hCAP-18 concentration of 86 μg/ml; Malm et al., 2000; Tjabringa et al., 2005). Gc infection increases the levels of hCAP18/LL-37 by two- to four-fold in cervical and urethral washes (Porter et al., 2005; Tjabringa et al., 2005). These concentrations of LL-37 would be sufficient to exert antibacterial activity on Gc, since the mean inhibitory concentration of LL-37 for Gc is 6 μg/ml (Shafer et al., 1998). The antigonococcal mechanism of action of LL-37 remains enigmatic and may be related to its ability to form pores that disrupt the integrity of bacterial membranes (Brogden, 2005).
Although Gc are susceptible to cathepsin G and LL-37 in vitro, the ability of some percentage of Gc to survive PMN exposure suggests that the bacteria have evolved mechanisms to counter these antimicrobial components. These mechanisms involve direct modulation of PMN granule release, changes to the Gc surface to resist non-oxidative antimicrobial components, and active export of these components (Figure 4B).
Modulation of PMN granule release
Both pili and porin have been reported to reduce PMN granule fusion with the plasma membrane or phagosomes. When added to primary PMNs, purified porins inhibit azurophilic and specific granule exocytosis (Bjerknes et al., 1995; Lorenzen et al., 2000). “Type 1,” piliated Gc was also reported to inhibit azurophilic granule exocytosis relative to “type 4,” non-piliated Gc, but additional surface structures expressed on type 1 bacteria may have mediated this result (Densen and Mandell, 1978). More detailed studies with isogenic Gc strains are necessary to determine whether and how Gc surface structures influence granule mobilization.
Modifications to the Gc surface
Gc LOS is thought to mask proteins which are degraded by cathepsin G, since truncation or loss of LOS results in increased binding of cathepsin G and increased susceptibility to cathepsin G-mediated killing (Shafer, 1988). Two modifications of LOS impact bacterial interactions with host cells and host defenses: phosphoethanolamine (PEA) substitution on lipid A or the oligosaccharide, and sialylation of the terminal Galβ1-4GlcNAc epitopes of the oligosaccharide (Mandrell et al., 1990; Plested et al., 1999). PEA addition to the heptose group on the beta chain of the core oligosaccharide enhances Gc serum resistance but does not affect susceptibility to antimicrobial peptides (Lewis et al., 2009). In contrast, PEA addition to lipid A by the LptA enzyme increases resistance to both normal human serum and cationic antimicrobial peptides indicating that structural changes in LOS contribute to the ability of gonococci to resist the bactericidal action of these innate immune components (Lewis et al., 2009). In the related bacterium N. meningitidis, expression of lptA is positively regulated by the misR/misS two-component regulatory system (Newcombe et al., 2005; Tzeng et al., 2008). The roles of MisR/MisS and LptA in Gc pathogenesis remain to be examined. The gonococcal α-2,3-sialyltransferase Lst transfers sialyl groups from host-derived CMP-N-acetylneuraminic acid to the terminal galactose residue on the oligosaccharide of LOS (Gilbert et al., 1996). Sialylation contributes to Gc resistance to normal human serum as well as PMN-derived oxygen-independent antimicrobial factors (Shafer et al., 1986; Parsons et al., 1992). Importantly, sialylated Gc are more resistant to PMNs in vitro, and sialylation contributes to Gc survival in the murine female genital tract (Kim et al., 1992; Rest and Frangipane, 1992; Gill et al., 1996; Wu and Jerse, 2006). In addition to LOS, changes in other surface components may contribute to Gc resistance to non-oxidative antimicrobial factors. For instance, loss of Opa expression enhances Gc resistance to serine proteases (Blake et al., 1981; Cole et al., 2010), and N. meningitidis lacking pili (due to insertional mutagenesis of the pilMNOPQ operon) are more resistant to the model antimicrobial peptide polymyxin B (Tzeng et al., 2005).
Gc export of antimicrobial components
The multiple transferable resistance (mtr) locus is a key determinant of Gc resistance to antimicrobial agents (Shafer et al., 1998). Mtr, a member of the resistance-nodulation-cell division (RND) family of efflux pumps, is encoded by a three gene operon designated mtrCDE. MtrC spans the periplasm to link the inner membrane protein MtrD, the multidrug efflux transporter, with outer membrane protein MtrE, the channel for export of antimicrobials to the extracellular environment (Hagman et al., 1995). MtrCDE uses the proton motive force to export a variety of compounds from the Gc cytoplasm, including antibiotics, detergents, and antimicrobial peptides (Hagman et al., 1995; Veal et al., 2002). mtrCDE is negatively regulated by the MtrR transcriptional repressor (Hagman and Shafer, 1995) and positively regulated by the MtrA transcriptional activator (Rouquette et al., 1999). Mutations in mtrR and mtrA that modulate expression of MtrCDE affect Gc resistance to antimicrobial peptides (Hagman et al., 1995; Hagman and Shafer, 1995). MtrCDE expression promotes Gc survival in the murine female genital tract (Jerse et al., 2003) and enhances resistance to murine antimicrobial peptides (Warner et al., 2008), but its role in defense of Gc against PMNs is unclear. Gc also uses the FarAB efflux pump system to confer resistance to long-chain fatty acids, independent of Mtr activity (Lee and Shafer, 1999). The Far system is composed of the FarA membrane-spanning linker, the FarB cytoplasmic membrane transporter, and MtrE. Far expression is believed to be important for survival of isolates at the rectal mucosal surface, which is rich in diet-derived fatty acids, and does not contribute to Gc survival in the murine genital tract (Jerse et al., 2003). How the Far system contributes to defense of Gc against PMNs, which may release fatty acids (Huang et al., 2010), remains to be explored.
Discussion
Despite the prevalence of gonorrhea in the human population and the abundance of PMNs during acute gonorrheal disease, we are just beginning to understand the molecular mechanisms underlying Gc interactions with and resistance to PMNs. There are three overarching questions which remain currently in the field. First, how does a subset of Gc survive PMN challenge? As we have described, Gc possesses gene products which protect against oxidative and non-oxidative components that are made by PMNs. Many of these gene products are necessary for in vitro protection against isolated antimicrobial components and some provide a selective advantage in vivo. However, in many cases, it has not been investigated whether these gene products also confer a survival advantage in the context of PMN challenge. Second, how does Gc persist over time inside PMNs, as is seen in PMNs isolated from gonorrheal exudates? Although virulence-associated Gc surface structures such as Opa proteins, pili, porin, and LOS have been highly investigated for their biochemistry and impact on Gc-epithelial interactions, their effects on Gc survival inside PMNs remain enigmatic. How complement or immunoglobulin opsonization affects Gc phagocytosis by and survival inside PMNs also needs to be examined. Finally, how and why does Gc stimulate PMN recruitment? That is, what is the benefit of recruiting professional antimicrobial cells to the site of Gc infection? Given the long history of Gc in the human population, Gc could have evolved mechanisms for inhibiting PMN recruitment; instead, Gc LOS and lipoproteins are strong initiators of the host innate immune response (Massari et al., 2002; Pridmore et al., 2003; Zughaier et al., 2004). The answer to this question remains enigmatic, but may be revealed once we have a better understanding of how Gc manipulates PMNs in vitro and in vivo.
Our current knowledge of Gc interactions with PMNs demonstrates the impressive ability of Gc to survive PMN challenge. Although we are just beginning to piece together the roles of many Gc surface structures and gene products in Gc survival after PMN exposure, we now have model systems in hand that will allow these issues to be directly addressed. We are optimistic that continuing to investigate the mechanisms used by Gc to defend against PMN antimicrobial responses will shed light on how Gc has remained a fixture in the human population for all of recorded history (Wain, 1947; Morton, 1977). This research also has the potential to reveal novel human and Gc targets that can be exploited for new therapeutics to treat the ever-growing threat of highly antibiotic-resistant gonorrhea.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Jeffrey Tessier for obtaining the clinical sample for the image in Figure 1. We thank Louise Ball, Joanna Goldberg, and the two reviewers for helpful comments on the manuscript. This work was supported in part by NIH R00 TW008042 (Alison K. Criss) and T32 AI007046 (M. Brittany Johnson).
References
Achard, M. E., Hamilton, A. J., Dankowski, T., Heras, B., Schembri, M. S., Edwards, J. L., Jennings, M. P., and Mcewan, A. G. (2009). A periplasmic thioredoxin-like protein plays a role in defense against oxidative stress in Neisseria gonorrhoeae. Infect. Immun. 77, 4934–4939.
Anonymous. (2001). Global Prevalence and Incidence of Selected Curable Sexually Transmitted Infections: Overview and Estimates. Geneva: World Health Organization.
Apicella, M. A., Ketterer, M., Lee, F. K., Zhou, D., Rice, P. A., and Blake, M. S. (1996). The pathogenesis of gonococcal urethritis in men: confocal and immunoelectron microscopic analysis of urethral exudates from men infected with Neisseria gonorrhoeae. J. Infect. Dis. 173, 636–646.
Archibald, F. S., and Duong, M. N. (1984). Manganese acquisition by Lactobacillus plantarum. J. Bacteriol. 158, 1–8.
Archibald, F. S., and Duong, M. N. (1986). Superoxide dismutase and oxygen toxicity defenses in the genus Neisseria. Infect. Immun. 51, 631–641.
Bauer, F. J., Rudel, T., Stein, M., and Meyer, T. F. (1999). Mutagenesis of the Neisseria gonorrhoeae porin reduces invasion in epithelial cells and enhances phagocyte responsiveness. Mol. Microbiol. 31, 903–913.
Bjerknes, R., Guttormsen, H. K., Solberg, C. O., and Wetzler, L. M. (1995). Neisserial porins inhibit human neutrophil actin polymerization, degranulation, opsonin receptor expression, and phagocytosis but prime the neutrophils to increase their oxidative burst. Infect. Immun. 63, 160–167.
Blake, M. S., Gotschlich, E. C., and Swanson, J. (1981). Effects of proteolytic enzymes on the outer membrane proteins of Neisseria gonorrhoeae. Infect. Immun. 33, 212–222.
Blake, M. S., and Swanson, J. (1978). Studies on gonococcus infection. XVI. Purification of Neisseria gonorrhoeae immunoglobulin A1 protease. Infect. Immun. 22, 350–358.
Borregaard, N., Sorensen, O. E., and Theilgaard-Monch, K. (2007). Neutrophil granules: a library of innate immunity proteins. Trends Immunol. 28, 340–345.
Britigan, B. E., Klapper, D., Svendsen, T., and Cohen, M. S. (1988). Phagocyte-derived lactate stimulates oxygen consumption by Neisseria gonorrhoeae. An unrecognized aspect of the oxygen metabolism of phagocytosis. J. Clin. Invest. 81, 318–324.
Brogden, K. A. (2005). Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250.
Brooks, G. F., Israel, K. S., and Petersen, B. H. (1976). Bactericidal and opsonic activity against Neisseria gonorrhoeae in sera from patients with disseminated gonococcal infection. J. Infect. Dis. 134, 450–462.
Casey, S. G., Shafer, W. M., and Spitznagel, J. K. (1985). Anaerobiosis increases resistance of Neisseria gonorrhoeae to O2-independent antimicrobial proteins from human polymorphonuclear granulocytes. Infect. Immun. 47, 401–407.
Casey, S. G., Shafer, W. M., and Spitznagel, J. K. (1986). Neisseria gonorrhoeae survive intraleukocytic oxygen-independent antimicrobial capacities of anaerobic and aerobic granulocytes in the presence of pyocin lethal for extracellular gonococci. Infect. Immun. 52, 384–389.
Casey, S. G., Veale, D. R., and Smith, H. (1979). Demonstration of intracellular growth of gonococci in human phagocytes using spectinomycin to kill extracellular organisms. J. Gen. Microbiol. 113, 395–398.
Casey, S. G., Veale, D. R., and Smith, H. (1980). Intracellular survival of Neisseria gonorrhoeae in human urethral exudate. FEMS Microbiol. Lett. 8, 97–100.
Cohen, M. S., and Cannon, J. G. (1999). Human experimentation with Neisseria gonorrhoeae: progress and goals. J. Infect. Dis. 179(Suppl. 2), S375–S379.
Cole, J. G., Fulcher, N. B., and Jerse, A. E. (2010). Opacity proteins increase Neisseria gonorrhoeae fitness in the female genital tract due to a factor under ovarian control. Infect. Immun. 78, 1629–1641.
Collins, S. J., Gallo, R. C., and Gallagher, R. E. (1977). Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature 270, 347–349.
Connell, T. D., Shaffer, D., and Cannon, J. G. (1990). Characterization of the repertoire of hypervariable regions in the Protein II (opa) gene family of Neisseria gonorrhoeae. Mol. Microbiol. 4, 439–449.
Criss, A. K., Katz, B. Z., and Seifert, H. S. (2009). Resistance of Neisseria gonorrhoeae to non-oxidative killing by adherent human polymorphonuclear leucocytes. Cell. Microbiol. 11, 1074–1087.
Criss, A. K., and Seifert, H. S. (2008). Neisseria gonorrhoeae suppresses the oxidative burst of human polymorphonuclear leukocytes. Cell. Microbiol. 10, 2257–2270.
Davidsen, T., Bjoras, M., Seeberg, E. C., and Tonjum, T. (2005). Antimutator role of DNA glycosylase MutY in pathogenic Neisseria species. J. Bacteriol. 187, 2801–2809.
Dempsey, J. A., Litaker, W., Madhure, A., Snodgrass, T. L., and Cannon, J. G. (1991). Physical map of the chromosome of Neisseria gonorrhoeae FA1090 with locations of genetic markers, including opa and pil genes. J. Bacteriol. 173, 5476–5486.
Densen, P., and Mandell, G. L. (1978). Gonococcal interactions with polymorphonuclear neutrophils: importance of the phagosome for bactericidal activity. J. Clin. Invest. 62, 1161–1171.
Dilworth, J. A., Hendley, J. O., and Mandell, G. L. (1975). Attachment and ingestion of gonococci human neutrophils. Infect. Immun. 11, 512–516.
Edwards, J. L., and Apicella, M. A. (2004). The molecular mechanisms used by Neisseria gonorrhoeae to initiate infection differ between men and women. Clin. Microbiol. Rev. 17, 965–981, table of contents.
Edwards, J. L., Brown, E. J., Uk-Nham, S., Cannon, J. G., Blake, M. S., and Apicella, M. A. (2002). A co-operative interaction between Neisseria gonorrhoeae and complement receptor 3 mediates infection of primary cervical epithelial cells. Cell. Microbiol. 4, 571–584.
Eschenbach, D. A., Davick, P. R., Williams, B. L., Klebanoff, S. J., Young-Smith, K., Critchlow, C. M., and Holmes, K. K. (1989). Prevalence of hydrogen peroxide-producing Lactobacillus species in normal women and women with bacterial vaginosis. J. Clin. Microbiol. 27, 251–256.
Estabrook, M. M., Zhou, D., and Apicella, M. A. (1998). Nonopsonic phagocytosis of group C Neisseria meningitidis by human neutrophils. Infect. Immun. 66, 1028–1036.
Fang, F. C. (2004). Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2, 820–832.
Farrell, C. F., and Rest, R. F. (1990). Up-regulation of human neutrophil receptors for Neisseria gonorrhoeae expressing PII outer membrane proteins. Infect. Immun. 58, 2777–2784.
Farzadegan, H., and Roth, I. L. (1975). Scanning electron microscopy and freeze-etching of gonorrhoeal urethral exudate. Br. J. Vener. Dis. 51, 83–91.
Fischer, S. H., and Rest, R. F. (1988). Gonococci possessing only certain P.II outer membrane proteins interact with human neutrophils. Infect. Immun. 56, 1574–1579.
Frangipane, J. V., and Rest, R. F. (1992). Anaerobic growth of gonococci does not alter their Opa-mediated interactions with human neutrophils. Infect. Immun. 60, 1793–1799.
Gilbert, M., Watson, D. C., Cunningham, A. M., Jennings, M. P., Young, N. M., and Wakarchuk, W. W. (1996). Cloning of the lipooligosaccharide alpha-2,3-sialyltransferase from the bacterial pathogens Neisseria meningitidis and Neisseria gonorrhoeae. J. Biol. Chem. 271, 28271–28276.
Gill, M. J., Mcquillen, D. P., Van Putten, J. P., Wetzler, L. M., Bramley, J., Crooke, H., Parsons, N. J., Cole, J. A., and Smith, H. (1996). Functional characterization of a sialyltransferase-deficient mutant of Neisseria gonorrhoeae. Infect. Immun. 64, 3374–3378.
Gray-Owen, S. D., and Blumberg, R. S. (2006). CEACAM1: contact-dependent control of immunity. Nat. Rev. Immunol. 6, 433–446.
Griffiss, J. M., Jarvis, G. A., O’Brien, J. P., Eads, M. M., and Schneider, H. (1991). Lysis of Neisseria gonorrhoeae initiated by binding of normal human IgM to a hexosamine-containing lipooligosaccharide epitope(s) is augmented by strain-specific, properdin-binding-dependent alternative complement pathway activation. J. Immunol. 147, 298–305.
Groves, E., Dart, A. E., Covarelli, V., and Caron, E. (2008). Molecular mechanisms of phagocytic uptake in mammalian cells. Cell. Mol. Life Sci. 65, 1957–1976.
Gu, A., Zhang, Z., Zhang, N., Tsark, W., and Shively, J. E. (2010). Generation of human CEACAM1 transgenic mice and binding of Neisseria Opa protein to their neutrophils. PLoS ONE 5, e10067.
Gulati, S., Cox, A., Lewis, L. A., Michael, F. S., Li, J., Boden, R., Ram, S., and Rice, P. A. (2005). Enhanced factor H binding to sialylated gonococci is restricted to the sialylated lacto-N-neotetraose lipooligosaccharide species: implications for serum resistance and evidence for a bifunctional lipooligosaccharide sialyltransferase in gonococci. Infect. Immun. 73, 7390–7397.
Hagman, K. E., Pan, W., Spratt, B. G., Balthazar, J. T., Judd, R. C., and Shafer, W. M. (1995). Resistance of Neisseria gonorrhoeae to antimicrobial hydrophobic agents is modulated by the mtrRCDE efflux system. Microbiology 141(Pt 3), 611–622.
Hagman, K. E., and Shafer, W. M. (1995). Transcriptional control of the mtr efflux system of Neisseria gonorrhoeae. J. Bacteriol. 177, 4162–4165.
Haines, K. A., Yeh, L., Blake, M. S., Cristello, P., Korchak, H., and Weissmann, G. (1988). Protein I, a translocatable ion channel from Neisseria gonorrhoeae, selectively inhibits exocytosis from human neutrophils without inhibiting O2-generation. J. Biol. Chem. 263, 945–951.
Hassett, D. J., Charniga, L., and Cohen, M. S. (1990). recA and catalase in H2O2-mediated toxicity in Neisseria gonorrhoeae. J. Bacteriol. 172, 7293–7296.
Hedges, S. R., Sibley, D. A., Mayo, M. S., Hook, E. W., III and Russell, M. W. (1998). Cytokine and antibody responses in women infected with Neisseria gonorrhoeae: effects of concomitant infections. J. Infect. Dis. 178, 742–751.
Huang, C. B., George, B., and Ebersole, J. L. (2010). Antimicrobial activity of n-6, n-7 and n-9 fatty acids and their esters for oral microorganisms. Arch. Oral Biol. 55, 555–560.
James, J. F., and Swanson, J. (1978). Studies on gonococcus infection. XIII. Occurrence of color/opacity colonial variants in clinical cultures. Infect. Immun. 19, 332–340.
Jerse, A. E. (1999). Experimental gonococcal genital tract infection and opacity protein expression in estradiol-treated mice. Infect. Immun. 67, 5699–5708.
Jerse, A. E., Cohen, M. S., Drown, P. M., Whicker, L. G., Isbey, S. F., Seifert, H. S., and Cannon, J. G. (1994). Multiple gonococcal opacity proteins are expressed during experimental urethral infection in the male. J. Exp. Med. 179, 911–920.
Jerse, A. E., Sharma, N. D., Simms, A. N., Crow, E. T., Snyder, L. A., and Shafer, W. M. (2003). A gonococcal efflux pump system enhances bacterial survival in a female mouse model of genital tract infection. Infect. Immun. 71, 5576–5582.
Johansson, L., Rytkonen, A., Bergman, P., Albiger, B., Kallstrom, H., Hokfelt, T., Agerberth, B., Cattaneo, R., and Jonsson, A. B. (2003). CD46 in meningococcal disease. Science 301, 373–375.
Johnson, S. R., Steiner, B. M., Cruce, D. D., Perkins, G. H., and Arko, R. J. (1993). Characterization of a catalase-deficient strain of Neisseria gonorrhoeae: evidence for the significance of catalase in the biology of N. gonorrhoeae. Infect. Immun. 61, 1232–1238.
Kim, J. J., Zhou, D., Mandrell, R. E., and Griffiss, J. M. (1992). Effect of exogenous sialylation of the lipooligosaccharide of Neisseria gonorrhoeae on opsonophagocytosis. Infect. Immun. 60, 4439–4442.
King, G., James, J. F., and Swanson, J. (1978). Studies on gonococcus infection. XI. Comparison of in vivo and vitro association of Neisseria gonorrhoeae with human neutrophils. J. Infect. Dis. 137, 38–43.
King, G. J., and Swanson, J. (1978). Studies on gonococcus infection. XV. Identification of surface proteins of Neisseria gonorrhoeae correlated with leukocyte association. Infect. Immun. 21, 575–584.
Lacy, P., and Eitzen, G. (2008). Control of granule exocytosis in neutrophils. Front. Biosci. 13, 5559–5570.
Lammel, C. J., Sweet, R. L., Rice, P. A., Knapp, J. S., Schoolnik, G. K., Heilbron, D. C., and Brooks, G. F. (1985). Antibody-antigen specificity in the immune response to infection with Neisseria gonorrhoeae. J. Infect. Dis. 152, 990–1001.
Le Cabec, V., Calafat, J., and Borregaard, N. (1997). Sorting of the specific granule protein, NGAL, during granulocytic maturation of HL-60 cells. Blood 89, 2113–2121.
LeCuyer, B. E., Criss, A. K., and Seifert, H. S. (2010) Genetic characterization of the nucleotide excision repair system of Neisseria gonorrhoeae. J. Bacteriol. 192, 665–673.
Lee, E. H., and Shafer, W. M. (1999). The farAB-encoded efflux pump mediates resistance of gonococci to long-chained antibacterial fatty acids. Mol. Microbiol. 33, 839–845.
Lewis, L. A., Choudhury, B., Balthazar, J. T., Martin, L. E., Ram, S., Rice, P. A., Stephens, D. S., Carlson, R., and Shafer, W. M. (2009). Phosphoethanolamine substitution of lipid A and resistance of Neisseria gonorrhoeae to cationic antimicrobial peptides and complement-mediated killing by normal human serum. Infect. Immun. 77, 1112–1120.
Lorenzen, D. R., Gunther, D., Pandit, J., Rudel, T., Brandt, E., and Meyer, T. F. (2000). Neisseria gonorrhoeae porin modifies the oxidative burst of human professional phagocytes. Infect. Immun. 68, 6215–6222.
Malm, J., Sorensen, O., Persson, T., Frohm-Nilsson, M., Johansson, B., Bjartell, A., Lilja, H., Stahle-Backdahl, M., Borregaard, N., and Egesten, A. (2000). The human cationic antimicrobial protein (hCAP-18) is expressed in the epithelium of human epididymis, is present in seminal plasma at high concentrations, and is attached to spermatozoa. Infect. Immun. 68, 4297–4302.
Mandrell, R. E., Lesse, A. J., Sugai, J. V., Shero, M., Griffiss, J. M., Cole, J. A., Parsons, N. J., Smith, H., Morse, S. A., and Apicella, M. A. (1990). In vitro and in vivo modification of Neisseria gonorrhoeae lipooligosaccharide epitope structure by sialylation. J. Exp. Med. 171, 1649–1664.
Massari, P., Henneke, P., Ho, Y., Latz, E., Golenbock, D. T., and Wetzler, L. M. (2002). Cutting edge: immune stimulation by neisserial porins is toll-like receptor 2 and MyD88 dependent. J. Immunol. 168, 1533–1537.
McCaw, S. E., Liao, E. H., and Gray-Owen, S. D. (2004). Engulfment of Neisseria gonorrhoeae: revealing distinct processes of bacterial entry by individual carcinoembryonic antigen-related cellular adhesion molecule family receptors. Infect. Immun. 72, 2742–2752.
McMillan, A., Mcneillage, G., and Young, H. (1979). Antibodies to Neisseria gonorrhoeae: a study of the urethral exudates of 232 men. J. Infect. Dis. 140, 89–95.
Merz, A. J., and So, M. (2000). Interactions of pathogenic neisseriae with epithelial cell membranes. Annu. Rev. Cell Dev. Biol. 16, 423–457.
Murphy, G. L., Connell, T. D., Barritt, D. S., Koomey, M., and Cannon, J. G. (1989). Phase variation of gonococcal protein II: regulation of gene expression by slipped-strand mispairing of a repetitive DNA sequence. Cell 56, 539–547.
Newburger, P. E., Chovaniec, M. E., Greenberger, J. S., and Cohen, H. J. (1979). Functional changes in human leukemic cell line HL-60. A model for myeloid differentiation. J. Cell Biol. 82, 315–322.
Newcombe, J., Jeynes, J. C., Mendoza, E., Hinds, J., Marsden, G. L., Stabler, R. A., Marti, M., and Mcfadden, J. J. (2005). Phenotypic and transcriptional characterization of the meningococcal PhoPQ system, a magnesium-sensing two-component regulatory system that controls genes involved in remodeling the meningococcal cell surface. J. Bacteriol. 187, 4967–4975.
Ofek, I., Beachey, E. H., and Bisno, A. L. (1974). Resistance of Neisseria gonorrhoeae to phagocytosis: relationship to colonial morphology and surface pili. J. Infect. Dis. 129, 310–316.
O’Hanlon, D. E., Lanier, B. R., Moench, T. R., and Cone, R. A. (2010). Cervicovaginal fluid and semen block the microbicidal activity of hydrogen peroxide produced by vaginal lactobacilli. BMC Infect. Dis. 10, 120.
Ovcinnikov, N. M., and Delektorskij, V. V. (1971). Electron microscope studies of gonococci in the urethral secretions of patients with gonorrhoea. Br. J. Vener. Dis. 47, 419–439.
Pantelic, M., Chen, I., Parker, J., Zhang, P., Grunert, F., and Chen, T. (2004). Retinoic acid treated HL60 cells express CEACAM1 (CD66a) and phagocytose Neisseria gonorrhoeae. FEMS Immunol. Med. Microbiol. 42, 261–266.
Papayannopoulos, V., and Zychlinsky, A. (2009). NETs: a new strategy for using old weapons. Trends Immunol. 30, 513–521.
Parsons, N. J., Curry, A., Fox, A. J., Jones, D. M., Cole, J. A., and Smith, H. (1992). The serum resistance of gonococci in the majority of urethral exudates is due to sialylated lipopolysaccharide seen as a surface coat. FEMS Microbiol. Lett. 69, 295–299.
Plested, J. S., Makepeace, K., Jennings, M. P., Gidney, M. A., Lacelle, S., Brisson, J., Cox, A. D., Martin, A., Bird, A. G., Tang, C. M., Mackinnon, F. M., Richards, J. C., and Moxon, E. R. (1999). Conservation and accessibility of an inner core lipopolysaccharide epitope of Neisseria meningitidis. Infect. Immun. 67, 5417–5426.
Porter, E., Yang, H., Yavagal, S., Preza, G. C., Murillo, O., Lima, H., Greene, S., Mahoozi, L., Klein-Patel, M., Diamond, G., Gulati, S., Ganz, T., Rice, P. A., and Quayle, A. J. (2005). Distinct defensin profiles in Neisseria gonorrhoeae and Chlamydia trachomatis urethritis reveal novel epithelial cell–neutrophil interactions. Infect. Immun. 73, 4823–4833.
Potter, A. J., Kidd, S. P., Edwards, J. L., Falsetta, M. L., Apicella, M. A., Jennings, M. P., and Mcewan, A. G. (2009). Thioredoxin reductase is essential for protection of Neisseria gonorrhoeae against killing by nitric oxide and for bacterial growth during interaction with cervical epithelial cells. J. Infect. Dis. 199, 227–235.
Pridmore, A. C., Jarvis, G. A., John, C. M., Jack, D. L., Dower, S. K., and Read, R. C. (2003). Activation of toll-like receptor 2 (TLR2) and TLR4/MD2 by Neisseria is independent of capsule and lipooligosaccharide (LOS) sialylation but varies widely among LOS from different strains. Infect. Immun. 71, 3901–3908.
Qu, X. D., Harwig, S. S., Oren, A. M., Shafer, W. M., and Lehrer, R. I. (1996). Susceptibility of Neisseria gonorrhoeae to protegrins. Infect. Immun. 64, 1240–1245.
Ram, S., Cullinane, M., Blom, A. M., Gulati, S., Mcquillen, D. P., Monks, B. G., O’Connell, C., Boden, R., Elkins, C., Pangburn, M. K., Dahlback, B., and Rice, P. A. (2001). Binding of C4b-binding protein to porin: a molecular mechanism of serum resistance of Neisseria gonorrhoeae. J. Exp. Med. 193, 281–295.
Ram, S., Lewis, L. A., and Rice, P. A. (2010). Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin. Microbiol. Rev. 23, 740–780.
Ram, S., Mcquillen, D. P., Gulati, S., Elkins, C., Pangburn, M. K., and Rice, P. A. (1998a). Binding of complement factor H to loop 5 of porin protein 1A: a molecular mechanism of serum resistance of nonsialylated Neisseria gonorrhoeae. J. Exp. Med. 188, 671–680.
Ram, S., Sharma, A. K., Simpson, S. D., Gulati, S., Mcquillen, D. P., Pangburn, M. K., and Rice, P. A. (1998b). A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J. Exp. Med. 187, 743–752.
Ramsey, K. H., Schneider, H., Cross, A. S., Boslego, J. W., Hoover, D. L., Staley, T. L., Kuschner, R. A., and Deal, C. D. (1995). Inflammatory cytokines produced in response to experimental human gonorrhea. J. Infect. Dis. 172, 186–191.
Rest, R. F. (1979). Killing of Neisseria gonorrhoeae by human polymorphonuclear neutrophil granule extracts. Infect. Immun. 25, 574–579.
Rest, R. F., Fischer, S. H., Ingham, Z. Z., and Jones, J. F. (1982). Interactions of Neisseria gonorrhoeae with human neutrophils: effects of serum and gonococcal opacity on phagocyte killing and chemiluminescence. Infect. Immun. 36, 737–744.
Rest, R. F., and Frangipane, J. V. (1992). Growth of Neisseria gonorrhoeae in CMP-N-acetylneuraminic acid inhibits nonopsonic (opacity-associated outer membrane protein-mediated) interactions with human neutrophils. Infect. Immun. 60, 989–997.
Rest, R. F., and Pretzer, E. (1981). Degradation of gonococcal outer membrane proteins by human neutrophil lysosomal proteases. Infect. Immun. 34, 62–68.
Rice, P. A., and Kasper, D. L. (1982). Characterization of serum resistance of Neisseria gonorrhoeae that disseminate. Roles of blocking antibody and gonococcal outer membrane proteins. J. Clin. Invest. 70, 157–167.
Rock, J. P., and Rest, R. F. (1988). Rapid damage to membranes of Neisseria gonorrhoeae caused by human neutrophil granule extracts. J. Gen. Microbiol. 134, 509–519.
Roos, D., Van Bruggen, R., and Meischl, C. (2003). Oxidative killing of microbes by neutrophils. Microbes Infect. 5, 1307–1315.
Rouquette, C., Harmon, J. B., and Shafer, W. M. (1999). Induction of the mtrCDE-encoded efflux pump system of Neisseria gonorrhoeae requires MtrA, an AraC-like protein. Mol. Microbiol. 33, 651–658.
Sadarangani, M., Pollard, A. J., and Gray-Owen, S. D. (2011). Opa proteins and CEACAMs: pathways of immune engagement for pathogenic Neisseria. FEMS Microbiol. Rev. 35, 498–514.
Saigh, J. H., Sanders, C. C., and Sanders, W. E. Jr. (1978). Inhibition of Neisseria gonorrhoeae by aerobic and facultatively anaerobic components of the endocervical flora: evidence for a protective effect against infection. Infect. Immun. 19, 704–710.
Sarafian, S. K., Tam, M. R., and Morse, S. A. (1983). Gonococcal protein I-specific opsonic IgG in normal human serum. J. Infect. Dis. 148, 1025–1032.
Schook, P. O., Stohl, E. A., Criss, A. K., and Seifert, H. S. (2011). The DNA-binding activity of the Neisseria gonorrhoeae LexA orthologue NG1427 is modulated by oxidation. Mol. Microbiol. 79, 846–860.
Schwalbe, R. S., Sparling, P. F., and Cannon, J. G. (1985). Variation of Neisseria gonorrhoeae protein II among isolates from an outbreak caused by a single gonococcal strain. Infect. Immun. 49, 250–252.
Seib, K. L., Simons, M. P., Wu, H. J., Mcewan, A. G., Nauseef, W. M., Apicella, M. A., and Jennings, M. P. (2005). Investigation of oxidative stress defenses of Neisseria gonorrhoeae by using a human polymorphonuclear leukocyte survival assay. Infect. Immun. 73, 5269–5272.
Seib, K. L., Wu, H. J., Kidd, S. P., Apicella, M. A., Jennings, M. P., and Mcewan, A. G. (2006). Defenses against oxidative stress in Neisseria gonorrhoeae: a system tailored for a challenging environment. Microbiol. Mol. Biol. Rev. 70, 344–361.
Shafer, W. M. (1988). Lipopolysaccharide masking of gonococcal outer-membrane proteins modulates binding of bacterial cathepsin G to gonococci. J. Gen. Microbiol. 134, 539–545.
Shafer, W. M., and Morse, S. A. (1987). Cleavage of the protein III and major iron-regulated protein of Neisseria gonorrhoeae by lysosomal cathepsin G. J. Gen. Microbiol. 133, 155–162.
Shafer, W. M., Onunka, V. C., Jannoun, M., and Huthwaite, L. W. (1990). Molecular mechanism for the antigonococcal action of lysosomal cathepsin G. Mol. Microbiol. 4, 1269–1277.
Shafer, W. M., Onunka, V. C., and Martin, L. E. (1986). Antigonococcal activity of human neutrophil cathepsin G. Infect. Immun. 54, 184–188.
Shafer, W. M., Qu, X., Waring, A. J., and Lehrer, R. I. (1998). Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. U.S.A. 95, 1829–1833.
Siegel, M., Olsen, D., Critchlow, C., and Buchanan, T. M. (1982). Gonococcal pili: safety and immunogenicity in humans and antibody function in vitro. J. Infect. Dis. 145, 300–310.
Simons, M. P., Nauseef, W. M., and Apicella, M. A. (2005). Interactions of Neisseria gonorrhoeae with adherent polymorphonuclear leukocytes. Infect. Immun. 73, 1971–1977.
Simons, M. P., Nauseef, W. M., Griffith, T. S., and Apicella, M. A. (2006). Neisseria gonorrhoeae delays the onset of apoptosis in polymorphonuclear leukocytes. Cell. Microbiol. 8, 1780–1790.
Skaar, E. P., Tobiason, D. M., Quick, J., Judd, R. C., Weissbach, H., Etienne, F., Brot, N., and Seifert, H. S. (2002). The outer membrane localization of the Neisseria gonorrhoeae MsrA/B is involved in survival against reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 99, 10108–10113.
Soler-Garcia, A. A., and Jerse, A. E. (2004). A Neisseria gonorrhoeae catalase mutant is more sensitive to hydrogen peroxide and paraquat, an inducer of toxic oxygen radicals. Microb. Pathog. 37, 55–63.
Soler-Garcia, A. A., and Jerse, A. E. (2007). Neisseria gonorrhoeae catalase is not required for experimental genital tract infection despite the induction of a localized neutrophil response. Infect. Immun. 75, 2225–2233.
Sorensen, O. E., Follin, P., Johnsen, A. H., Calafat, J., Tjabringa, G. S., Hiemstra, P. S., and Borregaard, N. (2001). Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 97, 3951–3959.
St Amant, D. C., Valentin-Bon, I. E., and Jerse, A. E. (2002). Inhibition of Neisseria gonorrhoeae by Lactobacillus species that are commonly isolated from the female genital tract. Infect. Immun. 70, 7169–7171.
Stohl, E. A., Criss, A. K., and Seifert, H. S. (2005). The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol. Microbiol. 58, 520–532.
Stohl, E. A., and Seifert, H. S. (2006). Neisseria gonorrhoeae DNA recombination and repair enzymes protect against oxidative damage caused by hydrogen peroxide. J. Bacteriol. 188, 7645–7651.
Swanson, J., Barrera, O., Sola, J., and Boslego, J. (1988). Expression of outer membrane protein II by gonococci in experimental gonorrhea. J. Exp. Med. 168, 2121–2129.
Tapsall, J. W. (2009). Neisseria gonorrhoeae and emerging resistance to extended spectrum cephalosporins. Curr. Opin. Infect. Dis. 22, 87–91.
Tjabringa, G. S., Vos, J. B., Olthuis, D., Ninaber, D. K., Rabe, K. F., Schalkwijk, J., Hiemstra, P. S., and Zeeuwen, P. L. (2005). Host defense effector molecules in mucosal secretions. FEMS Immunol. Med. Microbiol. 45, 151–158.
Tramont, E. C., Ciak, J., Boslego, J., Mcchesney, D. G., Brinton, C. C., and Zollinger, W. (1980). Antigenic specificity of antibodies in vaginal secretions during infection with Neisseria gonorrhoeae. J. Infect. Dis. 142, 23–31.
Tseng, H. J., Mcewan, A. G., Apicella, M. A., and Jennings, M. P. (2003). OxyR acts as a repressor of catalase expression in Neisseria gonorrhoeae. Infect. Immun. 71, 550–556.
Tseng, H. J., Srikhanta, Y., Mcewan, A. G., and Jennings, M. P. (2001). Accumulation of manganese in Neisseria gonorrhoeae correlates with resistance to oxidative killing by superoxide anion and is independent of superoxide dismutase activity. Mol. Microbiol. 40, 1175–1186.
Turner, S., Reid, E., Smith, H., and Cole, J. (2003). A novel cytochrome c peroxidase from Neisseria gonorrhoeae: a lipoprotein from a Gram-negative bacterium. Biochem. J. 373, 865–873.
Tzeng, Y. L., Ambrose, K. D., Zughaier, S., Zhou, X., Miller, Y. K., Shafer, W. M., and Stephens, D. S. (2005). Cationic antimicrobial peptide resistance in Neisseria meningitidis. J. Bacteriol. 187, 5387–5396.
Tzeng, Y. L., Kahler, C. M., Zhang, X., and Stephens, D. S. (2008). MisR/MisS two-component regulon in Neisseria meningitidis. Infect. Immun. 76, 704–716.
Veal, W. L., Nicholas, R. A., and Shafer, W. M. (2002). Overexpression of the MtrC-MtrD-MtrE efflux pump due to an mtrR mutation is required for chromosomally mediated penicillin resistance in Neisseria gonorrhoeae. J. Bacteriol. 184, 5619–5624.
Veale, D. R., Finch, H., and Smith, H. (1976). Penetration of penicillin into human phagocytes containing Neisseria gonorrhoeae: intracellular survival and growth at optimum concentrations of antibiotic. J. Gen. Microbiol. 96, 353–363.
Veale, D. R., Goldner, M., Penn, C. W., Ward, J., and Smith, H. (1979). The intracellular survival and growth of gonococci in human phagocytes. J. Gen. Microbiol. 113, 383–393.
Veale, D. R., Sen, D., Penn, C. W., Finch, H., Smith, H., and Witt, K. (1977). Interactions of Neisseria gonorrhoeae with guinea pig defence mechanisms in subcutaneously implanted chambers. FEMS Microbiol. Lett. 1, 3–6.
Virji, M. (2009). Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat. Rev. Microbiol. 7, 274–286.
Virji, M., and Heckels, J. E. (1986). The effect of protein II and pili on the interaction of Neisseria gonorrhoeae with human polymorphonuclear leucocytes. J. Gen. Microbiol. 132, 503–512.
Warner, D. M., Shafer, W. M., and Jerse, A. E. (2008). Clinically relevant mutations that cause derepression of the Neisseria gonorrhoeae MtrC-MtrD-MtrE Efflux pump system confer different levels of antimicrobial resistance and in vivo fitness. Mol. Microbiol. 70, 462–478.
Witt, K., Veale, D. R., Finch, H., Penn, C. W., Sen, D., and Smith, H. (1976). Resistance of Neisseria gonorrhoeae grown in vivo to ingestion and digestion by phagocytes of human blood. J. Gen. Microbiol. 96, 341–350.
Workowski, K. A., and Berman, S. (2010). Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm. Rep. 59, 1–110.
Wu, H., and Jerse, A. E. (2006). Alpha-2,3-sialyltransferase enhances Neisseria gonorrhoeae survival during experimental murine genital tract infection. Infect. Immun. 74, 4094–4103.
Wu, H., Soler-Garcia, A. A., and Jerse, A. E. (2009). A strain-specific catalase mutation and mutation of the metal-binding transporter gene mntC attenuate Neisseria gonorrhoeae in vivo but not by increasing susceptibility to oxidative killing by phagocytes. Infect. Immun. 77, 1091–1102.
Wu, H. J., Seib, K. L., Edwards, J. L., Apicella, M. A., Mcewan, A. G., and Jennings, M. P. (2005). Azurin of pathogenic Neisseria spp. is involved in defense against hydrogen peroxide and survival within cervical epithelial cells. Infect. Immun. 73, 8444–8448.
Wu, H. J., Seib, K. L., Srikhanta, Y. N., Kidd, S. P., Edwards, J. L., Maguire, T. L., Grimmond, S. M., Apicella, M. A., Mcewan, A. G., and Jennings, M. P. (2006). PerR controls Mn-dependent resistance to oxidative stress in Neisseria gonorrhoeae. Mol. Microbiol. 60, 401–416.
Zheng, H. Y., Alcorn, T. M., and Cohen, M. S. (1994). Effects of H2O2-producing lactobacilli on Neisseria gonorrhoeae growth and catalase activity. J. Infect. Dis. 170, 1209–1215.
Keywords: Neisseria gonorrhoeae, virulence factors, neutrophils, polymorphonuclear leukocytes, phagocytosis, reactive oxygen species, antimicrobial peptides, neutrophil proteases
Citation: Johnson MB and Criss AK (2011) Resistance of Neisseria gonorrhoeae to neutrophils. Front. Microbio. 2:77. doi:10.3389/fmicb.2011.00077
Received: 21 February 2011; Paper pending published: 16 March 2011;
Accepted: 31 March 2011; Published online: 13 April 2011.
Edited by:
Cynthia N. Cornelissen, Virginia Commonwealth University School of Medicine, USAReviewed by:
William Shafer, Emory University School of Medicine, USARick Rest, Drexel University College of Medicine, USA
Copyright: © 2011 Johnson and Criss. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Alison K. Criss, Department of Microbiology, University of Virginia Health Sciences Center, Box 800734, Charlottesville, VA 22908-0734, USA. e-mail: akc2r@virginia.edu