
- 1 Department of Medicine, University of California San Diego School of Medicine, La Jolla, CA, USA
- 2 Veterans Administration Medical Center, La Jolla, CA, USA
Salmonella strains cause three main types of diseases in people: gastroenteritis, enteric (typhoid) fever, and non-typhoid extra-intestinal disease with bacteremia. Genetic analysis indicates that each clinical syndrome requires distinct sets of virulence genes, and Salmonella isolates differ in their constellation of virulence traits. The spv locus is strongly associated with strains that cause non-typhoid bacteremia, but are not present in typhoid strains. The spv region contains three genes required for the virulence phenotype in mice: the positive transcriptional regulator spvR and two structural genes spvB and spvC. SpvB and SpvC are translocated into the host cell by the Salmonella pathogenicity island-2 type-three secretion system. SpvB prevents actin polymerization by ADP-ribosylation of actin monomers, while SpvC has phosphothreonine lyase activity and has been shown to inhibit MAP kinase signaling. The exact mechanisms by which SpvB and SpvC act in concert to enhance virulence are still unclear. SpvB exhibits a cytotoxic effect on host cells and is required for delayed cell death by apoptosis following intracellular infection. Strains isolated from systemic infections of immune compromised patients, particularly HIV patients, usually carry the spv locus, strongly suggesting that CD4 T cells are required to control disease due to Salmonella that are spv positive. This association is not seen with typhoid fever, indicating that the pathogenesis and immunology of typhoid have fundamental differences from the syndrome of non-typhoid bacteremia.
Salmonella enterica strains are facultative intracellular pathogens that can produce both localized and disseminated, systemic disease in humans and a variety of other vertebrates (Guiney et al., 1994, 1995; Fierer and Guiney, 2001; Coburn et al., 2007). Non-typhoid Salmonella strains are endemic in domestic agricultural animal populations throughout the world, and provide a huge reservoir for human infection. An extensive body of work has shown that all S. enterica strains share common, core features of pathogenesis. This analysis has also shown that the ability to produce the distinctive clinical syndromes of typhoid fever, enteritis, or non-typhoid disseminated infection is due to the presence of specific sets of genes that are frequently located in pathogenicity islands and on phage or virulence plasmids (Fierer and Guiney, 2001).
Salmonella Type III Protein Secretion Systems in Pathogenesis
Common features of Salmonella pathogenesis include the ability to induce intestinal epithelial cells to take up the organism into a Salmonella-containing vacuole (SCV), and the ability to manipulate the intracellular trafficking of the vacuole to promote survival and replication of the pathogen. These core virulence processes depend on the function of two distinct type-three secretion systems (abbreviated TTSS or T3SS) that transfer multiple effector proteins into the host cell cytoplasm. Following ingestion, Salmonella attach to intestinal epithelial cells and induce uptake of the bacteria into specialized membrane-bound vesicles termed SCV (reviewed in Galan, 2001; Ly and Casanova, 2007). This invasion process requires the TTSS encoded in the Salmonella pathogenicity island-1 (SPI-1) locus. The SPI-1 TTSS transfers a number of effector proteins into host epithelial cells, acting on Rho family GTPases and actin to produce physiological and structural changes in the cytoskeleton leading to bacterial uptake. The Rho family GTPases lead to activation of pro-inflammatory signaling pathways involving MAP kinases and NF-κB. Inside the SCV, Salmonella strains express a second TTSS encoded by the SPI-2 locus (Knodler and Steele-Mortimer, 2003; Waterman and Holden, 2003; Abrahams and Hensel, 2006). A variety of SPI-2 effectors regulate the trafficking of the SCV and interactions with the endosomal vesicular sorting process. These effectors position the SCV in a perinuclear position closely associated with the Golgi apparatus (Abrahams et al., 2006; Deiwick et al., 2006), stimulate the accumulation of actin filaments around the SCV (Poh et al., 2007), and also induce the formation of long, filamentous vesicular structures called Sifs (Brumell et al., 2002). Salmonella are able to replicate inside intestinal epithelial cells and induce apoptosis after 18–24 h (Kim et al., 1998; Paesold et al., 2002). In polarized intestinal monolayers, Salmonella can also pass through the epithelial cells and exit on the basolateral side before monolayer disruption occurs. Mucosal macrophages ingest Salmonella, but a massive influx of neutrophils quickly ensues in animals that develop clinical enteritis. Salmonella can also pass through the intestinal mucosa and disseminate by an SPI-1 independent process requiring CD18 positive cells (Vazquez-Torres et al., 1999).
The SPI-2 TTSS and a subset of effectors have been shown to be required to produce systemic disease (Knodler and Steele-Mortimer, 2003; Waterman and Holden, 2003; Abrahams and Hensel, 2006). A large body of experimental evidence indicates that extra-intestinal Salmonella infection occurs inside tissue macrophages (Fields et al., 1986; Vassiloyanakopoulos et al., 1998). Both clinical experience in humans and experimental studies in mice indicate that extracellular antibiotics such as the aminoglycosides are not effective against systemic Salmonella infections, demonstrating that Salmonella can grow within cells and spread from cell-to-cell without significant exposure to the extracellular space (Fierer et al., 1990; Vassiloyanakopoulos et al., 1998). SPI-2 TTSS effectors appear to act to promote intracellular infection by increasing the ability to Salmonella to resist the antimicrobial activity of macrophages and to grow within the cells. In macrophages, Salmonella appear to inhibit the recruitment of the NADPH oxidase to the phagosome, and this inhibition requires the SPI-2 TTSS function (Vazquez-Torres et al., 2000; Gallois et al., 2001). In support of the interaction between the SPI-2 virulence system and oxidative killing, gp91 phox−/− knockout mice lacking oxidase activity are fully susceptible to infection with SPI-2 mutants, while SPI-2 mutants are severely attenuated in mice with an intact NADPH oxidase (Vazquez-Torres et al., 2000). Unfortunately, the specific SPI-2 effectors involved in downregulating the NADPH oxidase have not been identified.
The spv Virulence Locus in Salmonella
Non-typhoid Salmonella strains associated with extra-intestinal infections in humans and animals carry an additional locus termed spv (Guiney et al., 1994, 1995; Fierer and Guiney, 2001). The spv genes are located within a highly homologous region contained on virulence plasmids found in the subspecies 1 lineage of S. enterica, and were named for the designation Salmonella plasmid virulence (Figure 1). The spv locus enhances mouse virulence by several orders of magnitude in LD50, depending on the Salmonella serovar and the mouse strain. The virulence phenotype is seen in both ItyS and ItyR mice, also referred to as Nramp1 (Slc11a1) mutant and wild-type mice respectively. Furthermore the spv effect has also been documented experimentally in calves and pigs and by molecular epidemiology in humans (Fierer et al., 1992; Libby et al., 1997). Subsequent work showed that the spv genes are located in the chromosome in certain other S. enterica lineages (Boyd and Hartl, 1998; Libby et al., 2002). spv-carrying serovars associated with human disease include Typhimurium, Enteritidis, Choleraesuis, Dublin, and Arizona. Certain host-adapted animal pathogens, such as Gallinarum/Pullorum and Abortusovis, also contain the spv locus. The plasmid spvABCD genes are arranged in an operon positively regulated by the upstream spvR gene, as shown in Figure 1 (Fang et al., 1991; Krause et al., 1991, 1992). The spvD gene is missing in the chromosomal locus as found in serovar Arizona (Libby et al., 2002). The SpvR protein is a positive transcriptional regulator of the LysR family and binds to inverted repeat recognition sequences upstream of its own promoter and the spvA promoter (Krause et al., 1991, 1995; Grob and Guiney, 1996; Grob et al., 1997). Transcription initiation at both promoters requires the alternative sigma factor RpoS (“stationary phase sigma factor”; Fang et al., 1992; Chen et al., 1995). Expression of the spv operon is induced by the intracellular environment of host cells and is dependent on both SpvR and RpoS (Fierer et al., 1993; Chen et al., 1996). Genetic analysis demonstrates that the spvR and spvBC genes are required for the virulence phenotype of the spv locus, while mutations in spvA and spvD do not have a reproducible virulence phenotype in mice (Roudier et al., 1992).
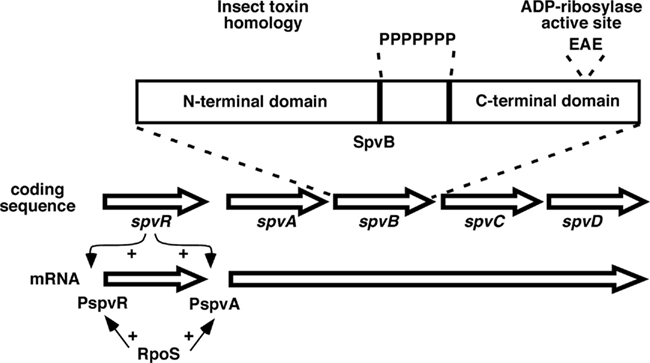
Figure 1. Map of the spv region found on virulence plasmids in subspecies 1 strains, not drawn to scale. The spvR gene is transcribed separately from the spvABCD genes. SpvR activates transcription at both promoters in concert with the RpoS sigma factor. The expansion shows the general structure of the SpvB protein, with N- and C-terminal domains separated by a run of proline residues (P). EAE denotes residues at the active site for the ADP-ribosylation activity.
Biochemical activities for SpvB and SpvC have been identified (Lesnick et al., 2001; Li et al., 2007). As shown in Figure 1, the SpvB protein has two distinct domains separated by a run of seven proline residues (nine in Dublin; Guiney and Lesnick, 2005). The N-terminal domain belongs to a large family of bacterial proteins identified by genome sequencing. Early work found members of this family in certain insect toxins from Enterobacteriaceae that infect insects, but the regions of homology do not involve the toxin activity (Lesnick and Guiney, 2001). At present, the functions of this widely distributed protein family are unknown. In contrast, the C-terminal domain of SpvB contains ADP-ribosyltransferase activity that covalently modifies G-actin monomers and prevents their polymerization into F-actin filaments (Lesnick et al., 2001; Hochmann et al., 2006). Since F-actin is continuously formed and depolymerized in the cell, the activity of SpvB in the host cell cytoplasm leads to loss of the F-actin cytoskeleton (Lesnick et al., 2001; Browne et al., 2002). The C-terminal domain is both necessary and sufficient to depolymerize cellular F-actin, and the N-terminal domain is not required (Lesnick et al., 2001). A site-specific mutation at the active site (see Figure 1) was used to show that the actin ADP-ribosylating activity of SpvB is required for the virulence phenotype in mice and for actin depolymerization in infected macrophages (Lesnick et al., 2001). SpvB is required for Salmonella proliferation in a subset of monocyte-derived human macrophages, and is required for the late apoptosis seen in host cells during Salmonella infection (Libby et al., 2000; Paesold et al., 2002). Purified, enzymatically active SpvB protein does not enter intact eukaryotic cells (Lesnick et al., 2001). Instead, SpvB is translocated from Salmonella in the SCV into the host cell cytoplasm by a process dependent on SPI-2 (Browne et al., 2002, 2008). Recently, SpvC and the related Shigella effector OspF have been shown to have phosphothreoninelyase activity that irreversibly inactivates host cell MAP kinases by removal of phosphate and modification of the target threonine (Li et al., 2007). The role of this activity in the virulence phenotype of SpvC has yet to be determined. SpvC is also translocated by the SPI-2 TTSS (Mazurkiewicz et al., 2008). Although SpvB and SpvC have distinct and seemingly unrelated biochemical actions, both are required to produce the spv virulence phenotype (Roudier et al., 1992). Cloned spvB can complement a non-polar spvB mutant, but spvB without spvC does not have a detectable virulence phenotype (Guiney and Fierer, unpublished). These results strongly suggest that SpvB and SpvC act at different stages to affect a common pathway or process in the host cell required for resistance to infection.
Host Cell Death Induced by Salmonella
Salmonella strains produce host cell death during infection of cell cultures by several different mechanisms. Salmonella grown under conditions to express the SPI-1 TTSS activate the NLRC4 inflammasome in macrophages by a mechanism that depends on the SPI-1 TTSS rod component PrgJ and flagellin subunits (Franchi et al., 2006; Miao et al., 2006, 2010). The inflammasome activates caspase-1 leading to processing and release of IL-1β and IL-18, and cell death by a process termed pyroptosis (Fink and Cookson, 2007). The macrophages lyse, and bacteria are released to the extracellular space. The significance of this process for Salmonella pathogenesis is unknown. Recent studies show that caspase-1 apparently has a protective role for the host during systemic Salmonella infection, suggesting that caspase-1 activation by Salmonella would be detrimental to the organism in disseminated disease (Lara-Tejero et al., 2006; Raupach et al., 2006). However, these studies do not distinguish the roles of caspase-1 in the release of cytokines versus the induction of pyroptosis. Furthermore, SPI-1 TTSS mutants (including SipB mutants) have normal virulence for systemic disease in mice, indicating that SipB-mediated macrophage pyroptosis may not have a significant role in systemic infection (Guiney, 2005). The SPI-2 TTSS, which is essential for systemic virulence, does not appear to translocate flagellin nor activate the inflammasome (Miao et al., 2010). In contrast, human intestinal epithelial cells do not undergo pyroptosis even though Salmonella invasion involves the SPI-1 TTSS and SipB (Kim et al., 1998; Paesold et al., 2002). Instead, Salmonella strains proliferate inside epithelial cells and induce delayed cell death (16–24 h after infection) by a process that depends on both the SPI-2 TTSS and the spv locus (Paesold et al., 2002). Salmonella-induced epithelial cell death has features of classical apoptosis including cell surface exposure of phosphatidylserine, mitochondrial membrane depolarization, caspase-3 activation, cytokeritin-3 cleavage, and DNA fragmentation between nucleosomes (Kim et al., 1998; Paesold et al., 2002). A similar process occurs in macrophages infected with Salmonella that are grown under conditions that do not induce SPI-1 expression, or when SPI-1 mutants are used (Libby et al., 2000; Browne et al., 2002; Hsu et al., 2004). An important role for this delayed process of macrophage apoptosis during systemic Salmonella infection in vivo is supported by (1) histologic evidence of apoptosis in liver macrophages (Richter-Dahlfors et al., 1997), and (2) correspondence between the bacterial factors (such as SpvB) required for apoptosis in cell culture and for virulence in vivo (Libby et al., 2000; Browne et al., 2002; Hsu et al., 2004).
Regulation of apoptosis in macrophages is complex, with competing pathways leading to pro- and anti-apoptotic factors that govern the ultimate outcome. In general, primary macrophages exposed to bacterial products and/or pro-inflammatory, death-inducing cytokines such as TNF become activated rather than undergo apoptosis, due to the induction of factors that favor cell survival (Park et al., 2002; Hsu et al., 2004). Clearly, macrophages have to survive at sites of infection in order to function as immune effector cells. However, certain bacterial pathogens have evolved specific virulence mechanisms that block the pro-survival pathways of macrophages, tipping the balance of signals in favor of apoptosis and resulting in the death of the macrophage. Figure 2 shows a schematic representation of the key pathways leading to the induction of apoptosis during infection of macrophages with Salmonella. For clarity, many factors and co-factors in the individual pathways are not shown. The data in support for this scheme were obtained primarily through the use of macrophages from specific knockout and transgenic mice in combination with specific bacterial mutants. As seen in Figure 2, apoptosis of wild-type macrophages in this system requires the SPI-2 TTSS but not the SPI-1 TTSS (SipB). Therefore, the process of apoptotic cell death clearly differs from pyroptosis dependent on SipB as described above (Fink and Cookson, 2007). Salmonella infection triggers activation of TLR4 by LPS. Although Salmonella produces ligands for other TLRs such as TLR2, TLR5, and TLR9, TLR4 has been shown to be required for apoptosis induced by Salmonella, so we have not shown the other TLRs in this scheme. TLR4 ligation triggers activation of the adapter proteins MyD88 and TRIF (also known as lps2). As seen in Figure 2, the TRIF pathway is a major activator of apoptosis, through a key intermediate kinase, PKR (also known as dsRNA responsive protein kinase because of its involvement in TLR3 signaling). Macrophages deficient in TRIF or PKR show marked reduction in Salmonella-induced apoptosis. PKR activates interferon-response factor-3 (IRF-3) and also phosphorylates eukaryotic translation initiation factor 2α (eIF2α), inhibiting protein synthesis and therefore blocking the expression of anti-apoptotic factors. Macrophages lacking IRF-3, and transgenic knock-in macrophages expressing a mutant eIF2α that is not phosphorylated by PKR both show reduced apoptosis after Salmonella infection (data in reference Hsu et al., 2004). These results clearly establish the TRIF pathway as a major transducer of pro-apoptotic signaling during Salmonella infection.
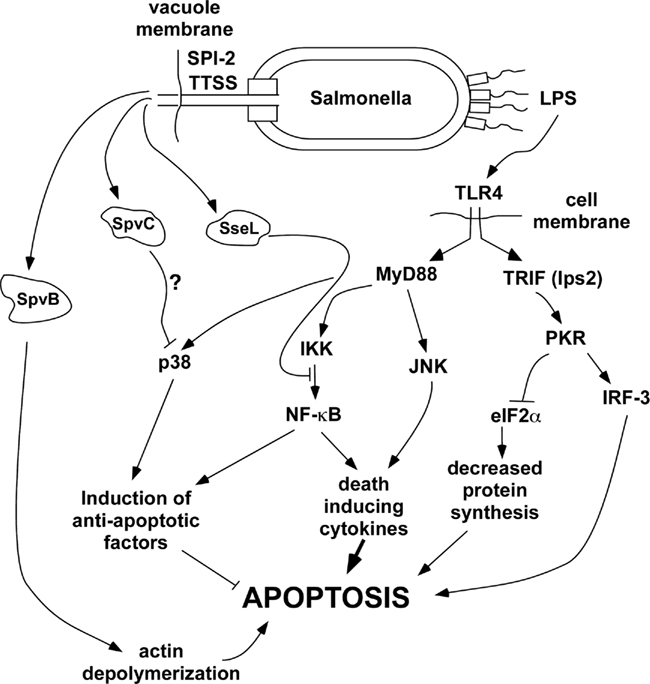
Figure 2. General scheme of pathways affecting delayed apoptosis in Salmonella-infected host cells. Roles for each of the host factors were demonstrated using knockout mice or inhibitors. SpvB is known to be required and presumably acts through actin depolymerization, but the exact mechanism connecting SpvB to the induction of apoptosis remains to be clarified. Although SpvC and SseL have been shown to inhibit p38 and NF-κB respectively, the roles of these effectors in apoptosis is hypothetical at present.
Ligation of TLR4 also activates pathways dependent on the MyD88 adapter protein, leading to activation of MAP kinases and NF-κB. The MAP kinase isoform JNK acts with NF-κB to induce pro-inflammatory, death-inducing cytokines such as TNF. However, NF-κB and the MAP kinase p38 have critical roles in the induction of anti-apoptotic factors leading to macrophage survival after exposure to LPS. Macrophages treated with a p38 inhibitor, or that have knockouts in MyD88 or IKKα undergo apoptosis after exposure to LPS alone (Park et al., 2002; Hsu et al., 2004). However, it is important to note that normal macrophages do not undergo apoptosis after treatment with LPS because the anti-apoptotic signaling dependent on p38 and NF-κB predominates.
Roles of Salmonella Virulence Factors in Host Cell Apoptosis
The delayed apoptosis of macrophages after Salmonella infection requires PhoP (Valle and Guiney, 2005) and the SPI-2 TTSS, but not SPI-1 (Hsu et al., 2004). PhoP is a master regulatory factor that controls the expression of a large number of genes during intracellular infection. We found that PhoP is required independent of SPI-2 for Salmonella to induce death by a novel mechanism in THP-1 human macrophage-like cells (Valle and Guiney, 2005). The SPI-2 TTSS translocates effector proteins across the membrane of the SCV into the host cell cytoplasm. Without the intervention of specific Salmonella effector proteins, the p38 and NF-κB anti-apoptotic pathways dominate, and cells infected with Salmonella SPI-2 mutants do not undergo delayed apoptosis (Hsu et al., 2004). Two major SPI-2 TTSS-secreted effectors that induce apoptosis have been identified: SpvB and SseL. The spv locus is required for the induction of apoptosis in human macrophages (Libby et al., 2000), and the ADP-ribosylating activity of SpvB is required for caspase-3 activation during human macrophage infection. However, the SpvB mutant does not reduce apoptosis to the level seen with an SPI-2 mutant that abolishes the TTSS function. This result suggests that other SPI-2 secreted effectors may also contribute to apoptosis. One of these candidates is SseL, shown to be involved in macrophage cytotoxicity (Rytkonen et al., 2007). NF-κB activation is significantly increased after infection of macrophages with an SseL mutant, and this activity can be decreased to wild-type levels by complementation with a low copy plasmid expressing SseL. These results show that the action of SseL is to decrease NF-κB signaling, thereby decreasing the anti-apoptotic and pro-inflammatory effects of this pathway. SseL deubiquitinates IκBα, the major regulator of the classical NF-κB activation pathway. In the NF-κB pathway, activation of IKK leads to phosphorylation of the inhibitor IκBα, targeting the inhibitor for ubiquitination and degradation by the proteosome. However, the deubiquitinating activity of SseL blocks degradation of IκBα and maintains the inhibition of NF-κB, preventing nuclear translocation and induction of NF-κB-dependent genes. As a result of the action of SseL in the host cell, both pro-inflammatory and anti-apoptotic pathways dependent on NF-κB are inhibited, allowing the pro-apoptotic effects through TRIF and PKR (see Figure 2) to dominate. Therefore, these results provide a mechanistic explanation for the cytotoxic effect of SseL previously reported (Rytkonen et al., 2007). The production of the pro-inflammatory cytokines TNF and IL-12 are decreased during infection of mice with wild-type serovar Typhimurium compared to the SseL mutant. These results support the idea that SseL may also act in concert with other effectors to decrease activation of innate immunity.
Proposed Model for the Virulence Effects of SpvB and SpvC in Salmonella Infection
Early in the analysis of the spv genes, genetic studies indicated that both SpvB and SpvC are required for the virulence phenotype encoded by the spv region (Roudier et al., 1992). A model to account for this finding is proposed in Figure 3. In this model, SpvB and SpvC act by different biochemical mechanisms, but they affect the same cellular pathways involved in the pathogenesis of intracellular Salmonella infection. Both SpvB and SpvC are translocated into the host cell cytoplasm by the SPI-2 TTSS (Browne et al., 2008; Mazurkiewicz et al., 2008). Actin depolymerization and induction of apoptosis are major effects of SpvB during intracellular Salmonella infection (Libby et al., 2000; Lesnick et al., 2001; Browne et al., 2002). Recent studies have shown that the NADPH oxidase associates with actin filaments during the organization and assembly of the oxidase on the membrane (elBenna et al., 1994; Allen et al., 1999; Zhan et al., 2004; Tamura et al., 2006), and wild-type Salmonella decreases NADPH oxidase recruitment to the phagosome by an SPI-2-dependent mechanism (Vazquez-Torres et al., 2000; Gallois et al., 2001). Therefore, SpvB-mediated actin depolymerization may decrease oxidase assembly and recruitment to the phagosome, and therefore could decrease oxidative killing of Salmonella. The second effect of SpvB-mediated actin depolymerization is to induce apoptosis (Libby et al., 2000; Lesnick et al., 2001). The phosphothreonine lyase activity of SpvC acts to decrease MAP kinase signaling, and inhibition of p38 has been demonstrated. As proposed in Figure 3, SpvC inhibition of the p38 MAP kinase isoform would block the synthesis of anti-apoptotic factors and therefore trigger apoptosis, in concert with the effects of SpvB. Macrophage apoptosis is one potential common pathway for the actions of SpvB and SpvC. In addition, SpvC may block the pro-inflammatory signaling function of the MAP kinase isoforms, leading to lower levels of key macrophage-activating cytokines such as TNF. Salmonella infections are known to be more severe in TNF-deficient mice (Vazquez-Torres et al., 2001). TNF has been shown promote localization of the NADPH oxidase to the phagosome of Salmonella-infected macrophages (Vazquez-Torres et al., 2001). Furthermore, the MAP kinase p38 has a direct effect in promoting oxidase assembly through phosphorylation of the p47 phox subunit (Laroux et al., 2005). Therefore, SpvC could act in concert with SpvB to block the recruitment of a functional NADPH oxidase to the phagosome. The SpvB and SpvC effects may be enhanced by the chromosomal sseL gene product, which further promotes apoptosis and decreases macrophage activation through blockade of NF-κB activity.
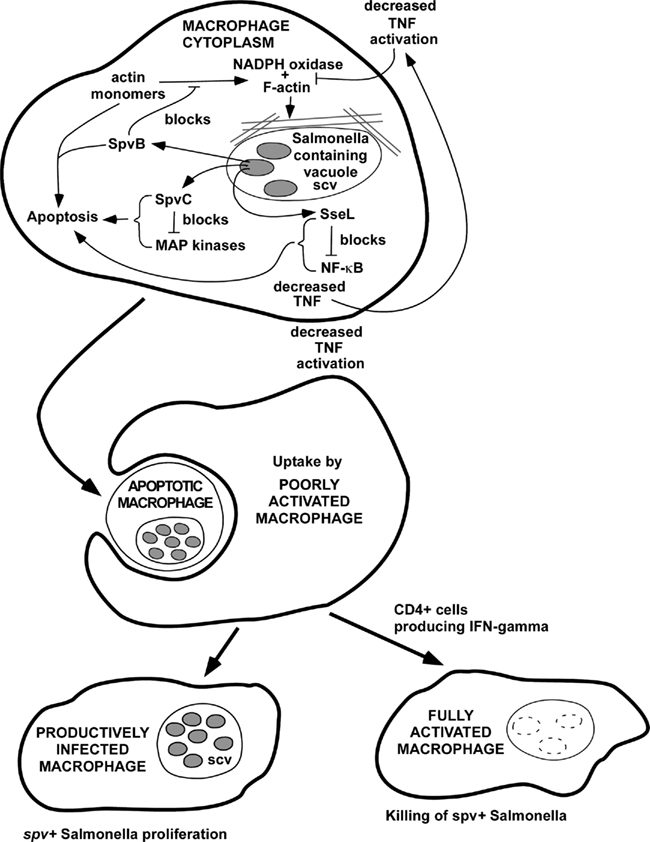
Figure 3. Hypothetical model for the combined effects of SpvB, SpvC, and SseL to promote Salmonella virulence. In this model, SpvB, SpvC, and SseL could act by different biochemical mechanisms to inhibit NADPH oxidase recruitment to the phagosome and also to promote host cell apoptosis. The overall effect of host cell apoptosis may to be promote cell-to-cell spread of the infection, since extracellular antibiotics such as gentamicin, and phagocytosis of extracellular bacteria by neutrophils, are not able to terminate infections with Salmonella strains that express spv genes. This model would provide an explanation for the experimental and clinical evidence that CD4 T cells and IFN-gamma are required to control infections due to spv+ Salmonella, likely involving macrophage activation and killing of intracellular bacteria.
The virulence effect of decreasing NADPH oxidase activity is readily apparent, and infections in phox knockout mice have shown that oxidative killing is a major host defense mechanism against Salmonella (Mastroeni et al., 2000). The virulence effect of inducing apoptosis in infected macrophages has not been established. Host cell apoptosis has been described with many infectious agents (Guiney, 2005). In some systems, pathogen virulence factors appear to inhibit apoptosis, while in other systems, key virulence factors promote apoptosis. For Salmonella, we propose that induction of macrophage apoptosis late in the intracellular infection cycle is a specific virulence mechanism that facilitates cell-to-cell spread of the bacteria (Guiney, 2005). Apoptosis induces cell surface changes that stimulate receptor-mediated uptake of apoptotic cells by surrounding macrophages, thereby transferring infection to these naïve macrophages. Therefore, the induction of apoptosis by Salmonella may represent a specific mechanism promoting cell-to-cell spread and subsequent rounds of macrophage infection. If Salmonella produced necrosis or pyroptosis during infection, free bacteria would be released into the extracellular space. The inability of extracellular antibiotics such as gentamicin to control Salmonella infection is strong evidence in favor of the importance of a cell-to-cell spread mechanism (Fierer et al., 1990). The model proposes that this intracellular infection cycle of spv+ Salmonella can only be terminated by the induction of specific CD4+ T cell immunity.
The ability of Salmonella to cause sustained intracellular infection has been a well-established paradigm in pathogenesis, and this principle has been confirmed by numerous experimental studies indicating that CD4+ T cell immunity is required for control of the infection (reviewed in Mittrucker and Kaufmann, 2000; Mastroeni and Menager, 2003). This concept is strongly supported by the key clinical observation in humans that the CD4+ cell depletion seen in HIV infection and other immune deficiency conditions strongly predisposes to persistent disseminated non-typhoid Salmonella disease (Gilks et al., 1990; Arthur et al., 2001).
Role of the spv Loci in Human Infections with Salmonella
The role of the spv locus in the mouse model of salmonellosis is well established, but its role in human infection is less appreciated. S. enterica strains cause three distinct illnesses in humans. The first and most common disease is gastroenteritis; it is estimated that more than one million cases occur annually in the USA alone (Mead et al., 1999). The other two less common syndromes are typhoid or enteric fever (Tauxe, 1997), and Salmonella bacteremia. Of the greater than 2000 serovars of S. enterica that are identified using the Kauffman–White typing scheme, about 20 serovars, mostly in O antigen groups B, C, D, and E (Jones et al., 2008), cause the vast majority of cases of gastroenteritis, as these strains are widely distributed amongst domestic food animals and so enter the food chain (Foley and Lynne, 2008). Bacteremia rarely accompanies gastroenteritis, and when it does, it is usually transient (Goldberg and Rubin, 1988). Of the most commonly isolated serovars from stool cultures only Typhimurium and Enteritidis can carry virulence plasmids that encode the spv operon, indicating that spv genes are not required to cause gastroenteritis in people.
In contrast to gastroenteritis, enteric fever is caused by two serovars: Typhi and Paratyphi A (Goldberg and Rubin, 1988). These organisms are host adapted to humans and are spread fecal-orally via contaminated food or water. Enteric fever is a sub-acute to chronic disease characterized by a transient rash, fever, weight loss, cough, and headache, but rarely diarrhea. Clinically, the infection localizes in the RES, as hepatosplenomegaly and abnormal liver function tests are common. Bacteremia is present in the first week of the illness but is either transient or very low grade in the following weeks. However, the organism can still be recovered from the bone marrow, another indication of its RES localization (Hoffman et al., 1984). Neither Typhi nor Paratyphi A possesses spv genes. Furthermore, typhoid fever does not have an increased incidence or altered pathogenesis in HIV patients, indicating that CD4+ T cell immunity is not crucial for the human host response to typhoid.
In contrast, non-typhoid Salmonella bacteremias are usually due to strains that encode the spv locus (Guiney et al., 1995), implying that spv products are required for bacteremia. The evidence for this is indirect, since it would be unethical to try to compare the dissemination potential of isogenic strains of Salmonella in humans. Therefore, the evidence for this association is obtained from molecular epidemiological studies of patients with Salmonella bacteremia to determine if there is a relationship between the clinical syndrome and infections with spv+ Salmonella. Salmonella bacteremia is distinct from enteric fever, in most cases presenting as high fever, usually without a rash or localizing signs or symptoms unless it has already caused a metastatic infection, e.g., infection of a bone or joint. Diarrhea is uncommon, and often there is no history of recent intestinal symptoms. The diagnosis of Salmonella bacteremia is established by positive blood cultures for non-typhoid S. enterica. In older immunocompetent patients S. enterica bacteremia in the absence of localizing signs or symptoms suggests the presence of a mycotic aneurysm (Cohen et al., 1978). With rare exceptions the aneurysms are infected with serovars known to carry virulence plasmids (Mendelowitz et al., 1979).
The syndrome of non-typhoid Salmonella bacteremia was first recognized as a manifestation of human infection with serovar Choleraesuis, a pig-adapted pathogen. In a study of 329 culture-proven cases of Choleraesuis in the northeastern US, only 26 had isolated gastroenteritis, while 239 isolates were from blood, and another 88 were from extra-intestinal sites of infection, such as CSF and abscesses (Saphra and Winter, 1957). Since all strains of Choleraesuis carry a virulence plasmid with the spv operon, this establishes an association between spv genes and the extra-intestinal infection syndrome, even though those particular isolates were not tested to see if they were spv+. This study and others (Maccready et al., 1957) showed that Choleraesuis can cause invasive infections in patients with no known immunological defects. In most developed countries S. Choleraesuis is now an uncommon human isolate, probably because of improvements in animal husbandry that have greatly reduced the risk of human infection.
The other spv bearing serovars are relatively less virulent in humans than Choleraesuis, mostly infecting patients with immunodeficiencies. More recently serovar Dublin has emerged as a cause of Salmonella bacteremia (Fierer, 1983; Fang and Fierer, 1991). Like Choleraesuis, Dublin infections present primarily as bacteremia without gastroenteritis (Fang and Fierer, 1991). Most infections with this bovine-adapted serovar are acquired from drinking unpasteurized milk or other dairy products (Headrick et al., 1998). Patients with T cell immune defects are over-represented in case series of Dublin infection (Werner et al., 1979; Fang and Fierer, 1991), suggesting that the functions of spv genes are largely controlled by a normal CD4 T cell response to infection. The most frequent serovars causing Salmonella bacteremia are Typhimurium and Enteritidis. Since they are also the most common causes of gastroenteritis one might assume that their prominence in the former reflects their prominence in the latter syndrome. However, if this was the whole explanation, one would expect stool and blood isolates to have the same prevalence of spv genes, but this is not the case (Fierer et al., 1992). In epidemiologically unrelated isolates from different states in the United States, 76% of the blood isolates carried spv genes, about twice the percentage of fecal isolates. Since clinical information was not available for these cases, it is not certain that all the blood isolates were from patients with the syndrome of Salmonella bacteremia, which may explain why nearly 25% of blood isolates did not have spv genes. Heithoff et al. (2008) examined S. Typhimurium isolates from Utah and they found that 10/10 blood isolates had the spv locus, while only 19/29 fecal isolates had spv. Others have examined the proportion of isolates of different serovars that cause bacteremia and Dublin and Choleraesuis stand out because they have such a high ratio of blood to fecal isolates (Blaser and Feldman, 1981; Jones et al., 2008).
The AIDS epidemic revealed the importance of CD4 T cells in preventing Salmonella bacteremia, a syndrome that was often the presenting manifestation of AIDS (Celum et al., 1987; Gruenewald et al., 1994). In those publications that included information about the serovars involved, they were nearly always strains that are known to carry virulence plasmids (Blaser and Feldman, 1981; Celum et al., 1987; Gruenewald et al., 1994). One exception was S. enterica serovar Arizona, an infection of patients who had AIDS and ingested folk remedies made from rattlesnakes (Noskin and Clarke, 1990). Subsequently, these isolates were shown to have the spv operon inserted in the chromosome (Libby et al., 2002).
Non-typhoid Salmonella bacteremia is a very common problem in sub-Saharan Africa, in both adults and children. Very few studies have looked for the presence of spv genes in the African blood culture isolates, but one large study of S. Typhimurium blood culture isolates from Malawi and two regions of Kenya found that all of them had virulence plasmids (Kingsley et al., 2009). Disturbingly, many of the isolates carried a pSLT virulence plasmid that had a large transposon insert encoding multiple antibiotic resistance genes. This genetic association has also been observed in S. Typhimurium isolates from the Asturia region of Spain, potentially linking selection for virulence with that for antibiotic resistance. Recently, analysis of 24 blood culture isolates from AIDS patients in Uganda, comprising serovars Typhimurium, Typhimurium var Copenhagen, and Enteritidis, has found that all contained the spvB gene (unpublished results; Fierer and Guiney). Thus, it appears likely that the spv operon is necessary for the syndrome of Salmonella bacteremia in AIDS, regardless of the serovar involved. To establish this, one would need to examine isolates for people in the same communities with gastroenteritis and show that a lower percentage had the virulence plasmid.
Summary
The spv genes comprise an accessory virulence locus in Salmonella located on virulence plasmids in the subspecies I lineage and in the chromosome of certain other subspecies. Regulation of expression of the spv operon occurs through the SpvR transcription activator and the RpoS sigma factor, and responds physiologically to the stationary phase of growth and the intracellular environment of host cells. SpvB and SpvC have been identified as essential effector proteins for the spv virulence phenotype and their biochemical activities have been characterized. Both SpvB and SpvC are translocated by the TTSS encoded by SPI-2. SpvB is an ADP-ribosyltransferase that modifies G-actin and prevents polymerization to F-actin, thereby disrupting the actin cytoskeleton. SpvC has phosphothreoninelyase activity and inhibits MAP kinases. The mechanisms of the concerted virulence effects of SpvB and SpvC remain to be determined. SpvB has been shown to be required for apoptosis in human macrophages, and may act together with SpvC and other Salmonella effectors.
Evidence from experimental models and human epidemiologic data indicate that the spv genes promote the virulence of non-typhoid Salmonella serovars to cause extra-intestinal disease. The spv genes appear to subvert the innate immune mechanisms in the host that are able to terminate the common intestinal infections due to non-typhoid Salmonella manifested as self-limited gastroenteritis. Resistance to spv-expressing Salmonella causing disseminated infection appears to require acquired immunity mediated by CD4+ T cells as demonstrated experimentally in mice and by the high incidence of spv+ non-typhoid Salmonella bacteremia in HIV infection. Analysis of the distribution of the spv locus in Salmonella serovars has underscored a key difference between the pathogenesis of typhoid fever and non-typhoid Salmonella bacteremia in people. The typhoid serovars Typhi and Paratyphi lack the spv genes, and are not associated with increased infections in patients deficient in CD4+ T cells. Therefore, the mechanisms of pathogenesis and host immunity are likely to have important differences between typhoid and non-typhoid serovars of Salmonella.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
Work by the authors was supported in part by NIH grants AI032178 and AI077661.
References
Abrahams, G. L., and Hensel, M. (2006). Manipulating cellular transport and immune responses: dynamic interactions between intracellular Salmonella enterica and its host cells. Cell. Microbiol. 8, 728–737.
Abrahams, G. L., Muller, P., and Hensel, M. (2006). Functional dissection of SseF, a type III effector protein involved in positioning the Salmonella-containing vacuole. Traffic 7, 950–965.
Allen, L. A., DeLeo, F. R., Gallois, A., Toyoshima, S., Suzuki, K., and Nauseef, W. M. (1999). Transient association of the nicotinamide adenine dinucleotide phosphate oxidase subunits p47phox and p67phox with phagosomes in neutrophils from patients with X-linked chronic granulomatous disease. Blood 93, 3521–3530.
Arthur, G., Nduba, V. N., Kariuki, S. M., Kimari, J., Bhatt, S. M., and Gilks, C. F. (2001). Trends in bloodstream infections among human immunodeficiency virus-infected adults admitted to a hospital in Nairobi, Kenya, during the last decade. Clin. Infect. Dis. 33, 248–256.
Blaser, M. J., and Feldman, R. A. (1981). From the centers for disease control. Salmonella bacteremia: reports to the Centers for Disease Control, 1968–1979. J. Infect. Dis. 143, 743–746.
Boyd, E. F., and Hartl, D. L. (1998). Salmonella virulence plasmid. Modular acquisition of the spv virulence region by an F-plasmid in Salmonella enterica subspecies I and insertion into the chromosome of subspecies II, IIIa, IV and VII isolates. Genetics 149, 1183–1190.
Browne, S. H., Hasegawa, P., Okamoto, S., Fierer, J., and Guiney, D. G. (2008). Identification of Salmonella SPI-2 secretion system components required for SpvB-mediated cytotoxicity in macrophages and virulence in mice. FEMS Immunol. Med. Microbiol. 52, 194–201.
Browne, S. H., Lesnick, M. L., and Guiney, D. G. (2002). Genetic requirements for Salmonella-induced cytopathology in human monocyte-derived macrophages. Infect. Immun. 70, 7126–7135.
Brumell, J. H., Goosney, D. L., and Finlay, B. B. (2002). SifA, a type III secreted effector of Salmonella typhimurium, directs Salmonella-induced filament (Sif) formation along microtubules. Traffic 3, 407–415.
Celum, C. L., Chaisson, R. E., Rutherford, G. W., Barnhart, J. L., and Echenberg, D. F. (1987). Incidence of salmonellosis in patients with AIDS. J. Infect. Dis. 156, 998–1002.
Chen, C. Y., Buchmeier, N. A., Libby, S., Fang, F. C., Krause, M., and Guiney, D. G. (1995). Central regulatory role for the RpoS sigma factor in expression of Salmonella dublin plasmid virulence genes. J. Bacteriol. 177, 5303–5309.
Chen, C. Y., Eckmann, L., Libby, S. J., Fang, F. C., Okamoto, S., Kagnoff, M. F., Fierer, J., and Guiney, D. G. (1996). Expression of Salmonella typhimurium rpoS and rpoS-dependent genes in the intracellular environment of eukaryotic cells. Infect. Immun. 64, 4739–4743.
Coburn, B., Grassl, G. A., and Finlay, B. B. (2007). Salmonella, the host and disease: a brief review. Immunol. Cell Biol. 85, 112–118.
Cohen, P. S., O’Brien, T. F., Schoenbaum, S. C., and Medeiros, A. A. (1978). The risk of endothelial infection in adults with Salmonella bacteremia. Ann. Intern. Med. 89, 931–932.
Deiwick, J., Salcedo, S. P., Boucrot, E., Gilliland, S. M., Henry, T., Petermann, N., Waterman, S. R., Gorvel, J. P., Holden, D. W., and Méresse, S. (2006). The translocated Salmonella effector proteins SseF and SseG interact and are required to establish an intracellular replication niche. Infect. Immun. 74, 6965–6972.
elBenna, J., Ruedi, J. M., and Babior, B. M. (1994). Cytosolic guanine nucleotide-binding protein Rac2 operates in vivo as a component of the neutrophil respiratory burst oxidase. Transfer of Rac2 and the cytosolic oxidase components p47phox and p67phox to the submembranous actin cytoskeleton during oxidase activation. J. Biol. Chem. 269, 6729–6734.
Fang, F. C., Krause, M., Roudier, C., Fierer, J., and Guiney, D. G. (1991). Growth regulation of a Salmonella plasmid gene essential for virulence. J. Bacteriol. 173, 6783–6789.
Fang, F. C., Libby, S. J., Buchmeier, N. A., Loewen, P. C., Switala, J., Harwood, J., and Guiney, D. G. (1992). The alternative sigma factor katF (rpoS) regulates Salmonella virulence. Proc. Natl. Acad. Sci. U.S.A. 89, 11978–11982.
Fields, P. I., Swanson, R. V., Haidaris, C. G., and Heffron, F. (1986). Mutants of Salmonella typhimurium that cannot survive within the macrophage are a virulent. Proc. Natl. Acad. Sci. U.S.A. 83, 5189–5193.
Fierer, J. (1983). Invasive Salmonella dublin infections associated with drinking raw milk. West. J. Med. 138, 665–669.
Fierer, J., Eckmann, L., Fang, F., Pfeifer, C., Finlay, B. B., and Guiney, D. (1993). Expression of the Salmonella virulence plasmid gene spvB in cultured macrophages and nonphagocytic cells. Infect. Immun. 61, 5231–5236.
Fierer, J., and Guiney, D. G. (2001). Diverse virulence traits underlying different clinical outcomes of Salmonella infection. J. Clin. Invest. 107, 775–780.
Fierer, J., Hatlen, L., Lin, J. P., Estrella, D., Mihalko, P., and Yau-Young, A. (1990). Successful treatment using gentamicin liposomes of Salmonella dublin infections in mice. Antimicrob. Agents Chemother. 34, 343–348.
Fierer, J., Krause, M., Tauxe, R., and Guiney, D. (1992). Salmonella typhimurium bacteremia: association with the virulence plasmid. J. Infect. Dis. 166, 639–642.
Fink, S. L., and Cookson, B. T. (2007). Pyroptosis and host cell death responses during Salmonella infection. Cell. Microbiol. 9, 2562–2570.
Foley, S. L., and Lynne, A. M. (2008). Food animal-associated Salmonella challenges: pathogenicity and antimicrobial resistance. J. Anim. Sci. 86, E173–E187.
Franchi, L., Amer, A., Body-Malapel, M., Kanneganti, T. D., Ozoren, N., Jagirdar, R., Inohara, N., Vandenabeele, P., Bertin, J., Coyle, A., Grant, E. P., and Núñez, G. (2006). Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in Salmonella-infected macrophages. Nat. Immunol. 7, 576–582.
Galan, J. E. (2001). Salmonella interactions with host cells: type III secretion at work. Annu. Rev. Cell Dev. Biol. 17, 53–86.
Gallois, A., Klein, J. R., Allen, L. A., Jones, B. D., and Nauseef, W. M. (2001). Salmonella pathogenicity island 2-encoded type III secretion system mediates exclusion of NADPH oxidase assembly from the phagosomal membrane. J. Immunol. 166, 5741–5748.
Gilks, C. F., Brindle, R. J., Newnham, R. S., Watkins, W. M., Waiyaki, P. G., Were, J. B. O., Otieno, L. S., Simani, P. M., Bhatt, S. M., Lule, G. N., Okelo, G. B. A., Brindle, R. J., Newnham, R. S., Gilks, C. F., and Warrell, D. A. (1990). Life-threatening bacteraemia in HIV-1 seropositive adults admitted to hospital in Nairobi, Kenya. Lancet 336, 545–549.
Goldberg, M. B., and Rubin, R. H. (1988). The spectrum of Salmonella infection. Infect. Dis. Clin. North Am. 2, 571–598.
Grob, P., and Guiney, D. G. (1996). In vitro binding of the Salmonella dublin virulence plasmid regulatory protein SpvR to the promoter regions of spvA and spvR. J. Bacteriol. 178, 1813–1820.
Grob, P., Kahn, D., and Guiney, D. G. (1997). Mutational characterization of promoter regions recognized by the Salmonella dublin virulence plasmid regulatory protein SpvR. J. Bacteriol. 179, 5398–5406.
Gruenewald, R., Blum, S., and Chan, J. (1994). Relationship between human immunodeficiency virus infection and salmonellosis in 20- to 59-year-old residents of New York City. Clin. Infect. Dis. 18, 358–363.
Guiney, D. G. (2005). The role of host cell death in Salmonella infections. Curr. Top. Microbiol. Immunol. 289, 131–150.
Guiney, D. G., Fang, F. C., Krause, M., and Libby, S. (1994). Plasmid-mediated virulence genes in non-typhoid Salmonella serovars. FEMS Microbiol. Lett. 124, 1–9.
Guiney, D. G., Fang, F. C., Krause, M., Libby, S., Buchmeier, N. A., and Fierer, J. (1995). Biology and clinical significance of virulence plasmids in Salmonella serovars. Clin. Infect. Dis. 21(Suppl. 2), S146–S151.
Guiney, D. G., and Lesnick, M. (2005). Targeting of the actin cytoskeleton during infection by Salmonella strains. Clin. Immunol. 114, 248–255.
Headrick, M. L., Korangy, S., Bean, N. H., Angulo, F. J., Altekruse, S. F., Potter, M. E., and Klontz, K. C. (1998). The epidemiology of raw milk-associated foodborne disease outbreaks reported in the United States, 1973 through 1992. Am. J. Public Health 88, 1219–1221.
Heithoff, D. M., Shimp, W. R., Lau, P. W., Badie, G., Enioutina, E. Y., Daynes, R. A., Byrne, B. A., House, J. K., and Mahan, M. J. (2008). Human Salmonella clinical isolates distinct from those of animal origin. Appl. Environ. Microbiol. 74, 1757–1766.
Hochmann, H., Pust, S., von Figura, G., Aktories, K., and Barth, H. (2006). Salmonella enterica SpvB ADP-ribosylates actin at position arginine-177-characterization of the catalytic domain within the SpvB protein and a comparison to binary clostridial actin-ADP-ribosylating toxins. Biochemistry 45, 1271–1277.
Hoffman, S. L., Punjabi, N. H., Rockhill, R. C., Sutomo, A., Rivai, A. R., and Pulungsih, S. P. (1984). Duodenal string-capsule culture compared with bone-marrow, blood, and rectal-swab cultures for diagnosing typhoid and paratyphoid fever. J. Infect. Dis. 149, 157–161.
Hsu, L. C., Park, J. M., Zhang, K., Luo, J. L., Maeda, S., Kaufman, R. J., Eckmann, L., Guiney, D. G., and Karin, M. (2004). The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature 428, 341–345.
Jones, T. F., Ingram, L. A., Cieslak, P. R., Vugia, D. J., Tobin-D’Angelo, M., Hurd, S., Medus, C., Cronquist, A., and Angulo, F. J. (2008). Salmonellosis outcomes differ substantially by serotype. J. Infect. Dis. 198, 109–114.
Kim, J. M., Eckmann, L., Savidge, T. C., Lowe, D. C., Witthoft, T., and Kagnoff, M. F. (1998). Apoptosis of human intestinal epithelial cells after bacterial invasion. J. Clin. Invest. 102, 1815–1823.
Kingsley, R. A., Msefula, C. L., Thomson, N. R., Kariuki, S., Holt, K. E., Gordon, M. A., Harris, D., Clarke, L., Whitehead, S., Sangal, V., Marsh, K., Achtman, M., Molyneux, M. E., Cormican, M., Parkhill, J., MacLennan, C. A., Heyderman, R. S., and Dougan, G. (2009). Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res. 19, 2279–2287.
Knodler, L. A., and Steele-Mortimer, O. (2003). Taking possession: biogenesis of the Salmonella-containing vacuole. Traffic 4, 587–599.
Krause, M., Fang, F. C., el-Gedaily, A., Libby, S., and Guiney, D. G. (1995). Mutational analysis of SpvR binding to DNA in the regulation of the Salmonella plasmid virulence operon. Plasmid 34, 37–47.
Krause, M., Fang, F. C., and Guiney, D. G. (1992). Regulation of plasmid virulence gene expression in Salmonella dublin involves an unusual operon structure. J. Bacteriol. 174, 4482–4489.
Krause, M., Roudier, C., Fierer, J., Harwood, J., and Guiney, D. (1991). Molecular analysis of the virulence locus of the Salmonella dublin plasmid pSDL2. Mol. Microbiol. 5, 307–316.
Lara-Tejero, M., Sutterwala, F. S., Ogura, Y., Grant, E. P., Bertin, J., Coyle, A. J., Flavell, R. A., and Galán, J. E. (2006). Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J. Exp. Med. 203, 1407–1412.
Laroux, F. S., Romero, X., Wetzler, L., Engel, P., and Terhorst, C. (2005). Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of gram-negative bacteria. J. Immunol. 175, 5596–5600.
Lesnick, M. L., and Guiney, D. G. (2001). The best defense is a good offense – Salmonella deploys an ADP-ribosylating toxin. Trends Microbiol. 9, 2–4; discussion 4–5.
Lesnick, M. L., Reiner, N. E., Fierer, J., and Guiney, D. G. (2001). The Salmonella spvB virulence gene encodes an enzyme that ADP-ribosylates actin and destabilizes the cytoskeleton of eukaryotic cells. Mol. Microbiol. 39, 1464–1470.
Li, H., Xu, H., Zhou, Y., Zhang, J., Long, C., Li, S., Chen, S., Zhou, J. M., and Shao, F. (2007). The phosphothreonine lyase activity of a bacterial type III effector family. Science 315, 1000–1003.
Libby, S. J., Adams, L. G., Ficht, T. A., Allen, C., Whitford, H. A., Buchmeier, N. A., Bossie, S., and Guiney, D. G. (1997). The spv genes on the Salmonella dublin virulence plasmid are required for severe enteritis and systemic infection in the natural host. Infect. Immun. 65, 1786–1792.
Libby, S. J., Lesnick, M., Hasegawa, P., Kurth, M., Belcher, C., Fierer, J., and Guiney, D. G. (2002). Characterization of the spv locus in Salmonella enterica serovar Arizona. Infect. Immun. 70, 3290–3294.
Libby, S. J., Lesnick, M., Hasegawa, P., Weidenhammer, E., and Guiney, D. G. (2000). The Salmonella virulence plasmid spv genes are required for cytopathology in human monocyte-derived macrophages. Cell. Microbiol. 2, 49–58.
Ly, K. T., and Casanova, J. E. (2007). Mechanisms of Salmonella entry into host cells. Cell. Microbiol. 9, 2103–2111.
Maccready, R. A., Reardon, J. P., and Saphra, I. (1957). Salmonellosis in Massachusetts; a sixteen-year experience. N. Engl. J. Med. 256, 1121–1128.
Mastroeni, P., and Menager, N. (2003). Development of acquired immunity to Salmonella. J. Med. Microbiol. 52, 453–459.
Mastroeni, P., Vazquez-Torres, A., Fang, F. C., Xu, Y., Khan, S., Hormaeche, C. E., and Dougan, G. (2000). Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. II. Effects on microbial proliferation and host survival in vivo. J. Exp. Med. 192, 237–248.
Mazurkiewicz, P., Thomas, J., Thompson, J. A., Liu, M., Arbibe, L., Sansonetti, P., and Holden, D. W. (2008). SpvC is a Salmonella effector with phosphothreoninelyase activity on host mitogen-activated protein kinases. Mol. Microbiol. 67, 1371–1383.
Mead, P. S., Slutsker, L., Dietz, V., McCaig, L. F., Bresee, J. S., Shapiro, C., Griffin, P. M., and Tauxe, R. V. (1999). Food-related illness and death in the United States. Emerg. Infect. Dis. 5, 607–625.
Mendelowitz, D. S., Ramstedt, R., Yao, J. S., and Bergan, J. J. (1979). Abdominal aortic salmonellosis. Surgery 85, 514–519.
Miao, E. A., Alpuche-Aranda, C. M., Dors, M., Clark, A. E., Bader, M. W., Miller, S. I., and Aderem, A. (2006). Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7, 569–575.
Miao, E. A., Mao, D. P., Yudkovsky, N., Bonneau, R., Lorang, C. G., Warren, S. E., Leaf, I. A., and Aderem, A. (2010). Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 107, 3076–3080.
Mittrucker, H. W., and Kaufmann, S. H. (2000). Immune response to infection with Salmonella typhimurium in mice. J. Leukoc. Biol. 67, 457–463.
Noskin, G. A., and Clarke, J. T. (1990). Salmonella arizonae bacteremia as the presenting manifestation of human immunodeficiency virus infection following rattlesnake meat ingestion. Rev. Infect. Dis. 12, 514–517.
Paesold, G., Guiney, D. G., Eckmann, L., and Kagnoff, M. F. (2002). Genes in the Salmonella pathogenicity island 2 and the Salmonella virulence plasmid are essential for Salmonella-induced apoptosis in intestinal epithelial cells. Cell. Microbiol. 4, 771–781.
Park, J. M., Greten, F. R., Li, Z. W., and Karin, M. (2002). Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 297, 2048–2051.
Poh, J., Odendall, C., Spanos, A., Boyle, C., Liu, M., Freemont, P., and Holden, D. W. (2007). SteC is a Salmonella kinase required for SPI-2-dependent F-actin remodelling. Cell. Microbiol. 10, 20–30.
Raupach, B., Peuschel, S. K., Monack, D. M., and Zychlinsky, A. (2006). Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 74, 4922–4926.
Richter-Dahlfors, A., Buchan, A. M., and Finlay, B. B. (1997). Murine salmonellosis studied by confocal microscopy: Salmonella typhimurium resides intracellularly inside macrophages and exerts a cytotoxic effect on phagocytes in vivo. J. Exp. Med. 186, 569–580.
Roudier, C., Fierer, J., and Guiney, D. G. (1992). Characterization of translation termination mutations in the spv operon of the Salmonella virulence plasmid pSDL2. J. Bacteriol. 174, 6418–6423.
Rytkonen, A., Poh, J., Garmendia, J., Boyle, C., Thompson, A., Liu, M., Freemont, P., Hinton, J. C., and Holden, D. W. (2007). SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc. Natl. Acad. Sci. U.S.A. 104, 3502–3507.
Saphra, I., and Winter, J. W. (1957). Clinical manifestations of salmonellosis in man; an evaluation of 7779 human infections identified at the New York Salmonella Center. N. Engl. J. Med. 256, 1128–1134.
Tamura, M., Itoh, K., Akita, H., Takano, K., and Oku, S. (2006). Identification of an actin-binding site in p47phox an organizer protein of NADPH oxidase. FEBS Lett. 580, 261–267.
Tauxe, R. V. (1997). Emerging foodborne diseases: an evolving public health challenge. Emerging Infect. Dis. 3, 425–434.
Valle, E., and Guiney, D. G. (2005). Characterization of Salmonella-induced cell death in human macrophage-like THP-1 cells. Infect. Immun. 73, 2835–2840.
Vassiloyanakopoulos, A. P., Okamoto, S., and Fierer, J. (1998). The crucial role of polymorphonuclear leukocytes in resistance to Salmonella dublin infections in genetically susceptible and resistant mice. Proc. Natl. Acad. Sci. U.S.A. 95, 7676–7681.
Vazquez-Torres, A., Fantuzzi, G., Edwards, C. K. III, Dinarello, C. A., and Fang, F. C. (2001). Defective localization of the NADPH phagocyte oxidase to Salmonella-containing phagosomes in tumor necrosis factor p55 receptor-deficient macrophages. Proc. Natl. Acad. Sci. U.S.A. 98, 2561–2565.
Vazquez-Torres, A., Jones-Carson, J., Baumler, A. J., Falkow, S., Valdivia, R., Brown, W., Le, M., Berggren, R., Parks, W. T., and Fang, F. C. (1999). Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature 401, 804–808.
Vazquez-Torres, A., Xu, Y., Jones-Carson, J., Holden, D. W., Lucia, S. M., Dinauer, M. C., Mastroeni, P., and Fang, F. C. (2000). Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science 287, 1655–1658.
Waterman, S. R., and Holden, D. W. (2003). Functions and effectors of the Salmonella pathogenicity island 2 type III secretion system. Cell. Microbiol. 5, 501–511.
Werner, S. B., Humphrey, G. L., and Kamei, I. (1979). Association between raw milk and human Salmonella dublin infection. Br. Med. J. 2, 238–241.
Keywords: Salmonella, spv, virulence, non-typhoid bacteremia, apoptosis, ADP-ribosylation, phosphothreonine lyase, CD4
Citation: Guiney DG and Fierer J (2011) The role of the spv genes in Salmonella pathogenesis. Front. Microbio. 2:129. doi: 10.3389/fmicb.2011.00129
Received: 16 March 2011;
Accepted: 25 May 2011;
Published online: 14 June 2011.
Edited by:
John S. Gunn, The Ohio State University, USAReviewed by:
Corrie Detweiler, University of Colorado Boulder, USAAndres Vazquez-Torres, University of Colorado Medical School, USA
Copyright: © 2011 Guiney and Fierer. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Donald G. Guiney, Department of Medicine 0640, University of California San Diego School of Medicine, 9500 Gilman Drive, La Jolla, CA 92093-0640, USA. e-mail: dguiney@ucsd.edu