
- Department of Ocean Sciences, University of California, Santa Cruz, Santa Cruz, CA, USA
Unicellular nitrogen-fixing cyanobacteria are important components of marine phytoplankton. Although non-nitrogen-fixing marine phytoplankton generally exhibit high gene sequence and genomic diversity, gene sequences of natural populations and isolated strains of Crocosphaera watsonii, one of the two most abundant open ocean unicellular cyanobacteria groups, have been shown to be 98–100% identical. The low sequence diversity in Crocosphaera is a dramatic contrast to sympatric species of Prochlorococcus and Synechococcus, and raises the question of how genome differences can explain observed phenotypic diversity among Crocosphaera strains. Here we show, through whole genome comparisons of two phenotypically different strains, that there are strain-specific sequences in each genome, and numerous genome rearrangements, despite exceptionally low sequence diversity in shared genomic regions. Some of the strain-specific sequences encode functions that explain observed phenotypic differences, such as exopolysaccharide biosynthesis. The pattern of strain-specific sequences distributed throughout the genomes, along with rearrangements in shared sequences is evidence of significant genetic mobility that may be attributed to the hundreds of transposase genes found in both strains. Furthermore, such genetic mobility appears to be the main mechanism of strain divergence in Crocosphaera which do not accumulate DNA microheterogeneity over the vast majority of their genomes. The strain-specific sequences found in this study provide tools for future physiological studies, as well as genetic markers to help determine the relative abundance of phenotypes in natural populations.
Introduction
Marine phytoplankton, which are dominated by cyanobacteria in most of the world’s open oceans, are important in global marine biogeochemical cycles and account for half of global carbon fixation (Waterbury et al., 1986; Goericke and Welschmeyer, 1993; Liu et al., 1997; Partensky et al., 1999; Scanlan and West, 2002). The immense genetic diversity of phytoplankton communities has been revealed through rRNA sequences and genomic sequencing of cultivated species, as well as large scale environmental sequencing efforts (Rocap et al., 2002, 2003; Ernst et al., 2003; Venter et al., 2004; Rusch et al., 2007; Partensky and Garczarek, 2010). As direct descendents of ancient phototrophs with deeply rooted phylogenies, it is not surprising that cyanobacteria typically show a large amount of genomic sequence heterogeneity, even among closely related species (Zhao and Qin, 2007; Dufresne et al., 2008). However, the genome diversity among Crocosphaera strains and populations is an intriguing deviation from that observed trend.
In oligotrophic regions, phytoplankton production is often limited by nutrients, especially nitrogen (N), and in those areas, nitrogen (N2) fixation provides an important source of new N that supports primary productivity (Karl et al., 1997, 2002; Bonnet et al., 2009; Kitajima et al., 2009; Shiozaki et al., 2010). The major marine N2-fixing cyanobacterial taxa can be categorized into three groups based on life-style and morphology: (1) symbiotic, including Richelia spp., and Calothrix spp. (2) free-living and filamentous, like Trichodesmium spp., and (3) free-living and unicellular, such as Crocosphaera spp. The two most abundant taxa of unicellular diazotrophs, as defined by nitrogenase gene (nifH) phylogeny, are the uncultivated Group A (i.e., UCYN-A) and Group B, which is represented in culture by a number of Crocosphaera watsonii strains. Previously, free-living unicellular diazotrophs were thought to be relatively minor contributors to total marine N2 fixation (Capone et al., 1997). However, more recent studies have reported high abundances using qPCR and direct cell counts (Zehr et al., 2001; Falcon et al., 2004; Church et al., 2005a, 2008; Langlois et al., 2008; Moisander et al., 2008, 2010), and measured high rates of in situ unicellular cyanobacterial N2 fixation (Zehr et al., 2001; Falcon et al., 2004; Montoya et al., 2004; Kitajima et al., 2009; Moisander et al., 2010), demonstrating that unicellular diazotrophs are often significant contributors of new N in the global ocean.
Crocosphaera strains have been isolated from the Atlantic and Pacific Oceans between 28°S and 24°N latitudes. All isolates are strains of the species C. watsonii. These strains have important phenotypic differences with ecological implications, such as the possible absence of phosphorus scavenging genes in some strains (Dyhrman and Haley, 2006), and differences in cell size, temperature growth optima, exopolysaccharide (EPS) production, and N2 fixation rates (Webb et al., 2009). It is likely that these differences affect the way each phenotype interacts with the surrounding environment. For example, EPS production may alter cell sinking rates, and has also been shown to have cell-protective properties in some cyanobacteria (Pereira et al., 2009). The two strains described in this study have contrasting phenotypes. C. watsonii WH8501, isolated from the South Atlantic in 1984, has a smaller cell size (2–4 μm), narrower temperature range, and does not produce EPS. C. watsonii WH0003, isolated from the North Pacific in 2000, has larger cells (4.5–5.5 μm), produces large amounts of EPS, and has per-cell N2 fixation rates approximately five times higher than the WH8501 strain (Webb et al., 2009).
Despite differences in phenotype, a high degree of genetic similarity has been observed among Crocosphaera strains. For example, when comparing a 950 bp fragment of the typically variable 16S–23S rRNA ITS region, no strain was found to vary at more than five of six total variable single base positions (Webb et al., 2009). Another study examined sequences of five functional genes in seven Crocosphaera strains and two large-insert environmental clones (BACs) and found that all strains and BACs shared > 99% nucleotide identity for all gene fragments, suggesting that there is remarkably little DNA mutation accumulation among strains in this genus (Zehr et al., 2007). A metagenomic study supported this finding when similar levels of sequence identity were observed between environmental sequences and the C. watsonii WH8501 genome (Hewson et al., 2009). Such observations of Crocosphaera genetic conservation are a striking contrast to non-N2-fixing marine cyanobacteria genera (Prochlorococcus and Synechococcus) that exhibit a large degree of genomic sequence divergence (Scanlan et al., 2009; Partensky and Garczarek, 2010). Genome-wide analyses of those genera have shown nucleotide sequences of orthologous genes often differ by 20–50% even when comparing very closely related species (Zhao and Qin, 2007; Dufresne et al., 2008). The average nucleotide identity of orthologous genes in pair-wise whole genome comparisons of cultivated Synechococcus and Prochlorococcus species (both within and between genera) was between 50 and 78%, even in comparisons between species with > 96% 16S rRNA identity (Coleman et al., 2006; Zhao and Qin, 2007; Dufresne et al., 2008). In addition, large scale environmental sequencing showed that this degree of sequence variation is also present in natural populations (Rusch et al., 2007).
Observed phenotypic variation among C. watsonii strains could be explained by genomic rearrangements such as that reported from alignment of environmental BAC sequences to the WH8501 draft genome (Zehr et al., 2007). That study also observed transposase genes (the genes responsible for genetic movement in transposons) near rearrangements, hinting at a mechanism for genetic mobility. More evidence that transposase genes may be important in Crocosphaera spp., was provided shortly after the release of the C. watsonii WH8501 draft genome, when the unusually high abundance of transposase genes was recognized, and evidence was found for positive evolutionary selection in a subset of those genes (Mes and Doeleman, 2006). A more recent study showed that this was not a culture-based phenomenon by observing expression of some of those transposase genes in natural Crocosphaera populations (Hewson et al., 2009).
Transposons are highly abundant mobile genetic elements that mediate genome shuffling within and among all domains of life (Mahillon et al., 1999; Lander et al., 2001; Feschotte et al., 2002; Waterston et al., 2002; Goodchild et al., 2004; Filee et al., 2007; Touchon and Rocha, 2007; Aziz et al., 2010). The abundance of transposable elements in genomes has been correlated with both genome size and the frequency of horizontal gene transfer (HGT), and insertion sequence (IS) elements have been observed in prokaryotes at frequencies from zero to over 300 per genome, with proteobacteria and cyanobacteria species containing some of the highest numbers (Kaneko et al., 2007; Touchon and Rocha, 2007; Frangeul et al., 2008; Stucken et al., 2010). However, many cyanobacteria species do not have any recognized transposases in their genomes (e.g., Prochlorococcus), and a study which examined a small number of cyanobacterial genomes found very low numbers (median = 1 per genome) even in the genomes which had transposases (Touchon and Rocha, 2007). More recently, researchers found high abundances of transposases in the deep oceans, suggesting they play an important role in microbial communities in a variety of marine environments (Konstantinidis et al., 2009).
The aim of this study was to compare the genomes of two Crocosphaera strains (C. watsonii WH8501 and C. watsonii WH0003, referred to hereafter as WH8501 and WH0003 respectively) in order to answer the following questions: (1) Is the lack of DNA sequence divergence found in previous studies generalized across the entire genome? and (2) are there strain-specific regions in each genome that can explain the phenotypic differences between strains?
Materials and Methods
WH0003 Genomic DNA Amplification and 454 Sequencing
A non-axenic culture of C. watsonii WH0003 was grown in nitrogen-free SO medium (Waterbury et al., 1986, 1988) in polycarbonate tissue culture flasks with a 0.2 μm pore-size vent cap (Corning Inc., Corning, NY, USA) at 26°C under a 12:12 h light/dark cycle. Because the cells cannot be directly separated from their EPS matrix, DNA could not be extracted from cultured cells using standard methods. Instead, an aliquot of densely grown cells was subjected to 60 s of bead beating on a Mini-Beadbeater-96 (Biospec Products, Bartlesville, OK, USA) with a mixture of 0.5 and 0.1 mm beads to physically separate a portion of the cells from their EPS. After bead beating, the resulting mixture of cells and EPS was passed through a 10 μm swinex filter prior to being sorted using the Influx Mariner flow cytometer and cell sorter (Cytopeia Corp, Seattle, WA, USA). The flow rate was adjusted to allow approximately 2,000 events per second during sorting. Replicates of 5,000 cells each were sorted into 1.5 ml microcentrifuge tubes containing 150 μL of TE buffer, and stored at −80°C.
After freezing, cells were thawed and pelleted at 14,000 rpm (21,000 × g) for approximately 40 min and the supernatant was discarded. Cells were resuspended in 7 μL of GenomiPhi V2 sample buffer (Amersham Biosciences, Piscataway, NJ, USA), lysed by adding 2 μL of lysis buffer (400 mM KOH + 10 mM EDTA) and incubating at 65°C for 3 min. The lysis was terminated by adding 2 μL of neutralization buffer (600 mM Tris HCl, pH 7.5, 400 mM HCl) and placing the samples on ice. The resulting whole cell lysis was used directly in a 21 μL reaction by adding 8.5 μL of reaction buffer, and 1.5 μL of GenomiPhi V2 enzyme mix (Amersham Biosciences, Piscataway, NJ, USA). Amplification was carried out in a thermal cycler at 30°C for 105 min, terminated at 65°C for 10 min, followed by temporary storage at 4°C, and long-term storage at −20°C. Prior to 454 sequencing, amplified genomic DNA was quantified using Pico Green (Invitrogen Corporation, Carlsbad, CA, USA).
Shotgun library construction was carried out at the UCSC Genome Sequencing Center1 using sorted cell amplified DNA and sequenced on the Genome Sequencer FLX instrument using Titanium Series protocols according to the manufacturer’s specifications (454 Life Sciences, Branford, CT, USA).
Sequence Assembly and Analysis and ORF Identification
The 1/2 chip 454 sequencing run produced 540,451 reads, with an average length of 418 bp for a total of 225,977,489 bp (∼37× coverage of the genome). All reads were assembled using Version 2.0.00 of the Newbler GS De Novo Assembler program (454 Life Sciences, Branford, CT, USA). The assembly was run via command line interface using the “-nrm,” “-consed,” and “-large” flags. All other parameters used were the default values, as described in the manufacturer’s publication, “Genome Sequencer Data Analysis Software Manual.”
The assembly resulted in 1390 contigs, ranging in length from 500 to 46,275 bp with a total length of 6,130,298 bp. Each contig was compared to the C. watsonii WH8501 draft genome (GenBank GI #67858163) using nucleotide BLAST (Altschul et al., 1990), and contigs with over 100 bp sequence alignments to WH8501 were assigned to the WH0003 draft genome. There were 899 such contigs with a total length of 5,465,610 bp. The remaining 491 contigs that showed less similarity, or no similarity, to WH8501 were compared to a database of all prokaryotic proteins using BLASTx (translated nucleotide query vs. protein DB), and divided according to the taxonomy of their best BLAST alignments. There were 227 contigs (totaling 424,894 bp) which were most similar to known cyanobacterial sequences. Those contigs were labeled as “probable” WH0003 genome sequence. The remaining 260 contigs (237,640 bp) showed no homology to known cyanobacteria, and were discarded from further analysis.
Features in the WH0003 draft genome sequence were identified and annotated using RAST (Aziz et al., 2008). The concatenated proxy genome was the input sequence, and 5,693 features were predicted. Those features were annotated as follows; 3 rRNA sequences (in a single operon), 39 tRNA sequences (71–87 bp each), and 5,651 open reading frames (ORFs). In order to correct for the fact that the genome was artificially concatenated, 618 ORFs which were predicted by RAST to read across the ends of contigs were broken at the contig ends, and manually re-annotated using BLAST results of the resulting broken ORFs. This process produced 762 non-broken ORFs (from the original 618 contig-spanning sequences), resulting in a total of 5,795 ORFs in the WH0003 draft genome (4,553,866 bp or 83.3% coding sequence). For the WH8501 genome, the existing GenBank locations and functional annotations for all 5,958 ORFS were used, except for 1,211 ORFs that were identified as transposase genes and subsequently re-annotated with their corresponding IS family assignments (see Materials and Methods and Table S1 in Supplementary Material).
The WH0003 genome sequences and annotations are publicly available in GenBank2. The Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession AESD00000000. The version described in this paper is the first version, AESD01000000. The 899 contigs confidently assigned to the WH0003 have accession numbers AESD01000001–AESD01000899, and the additional 227 contigs that are “probable” WH0003 sequences have accession numbers AESD01000900–AESD01001126.
Transposase Annotation
The observation of highly repetitive ORFs in the WH8501 genome, most of which were annotated as hypothetical proteins, led to a reassessment of the functions of those ORFs. Using a nucleotide sequence identity cutoff of 98%, ORFs were placed into isoform groups, and the number of copies of each isoform was tabulated (Table S2 in Supplementary Material). Amino acid sequences for a representative of each isoform were used as query sequences in a protein BLAST (BLASTp) against all prokaryotic proteins. Sequences were annotated as transposase genes using the conservative criteria of a total identity (tID = percent identity × percent of the ORF length aligned) of more than 50% to known transposases. Those ORFs, as well as any originally annotated transposase genes not represented in the isoform groups, were assigned to IS families according to sequence similarity to known families using the BLAST tool on the ISfinder website3 with default parameters (Siguier et al., 2006). All of the re-annotated ORFs are listed in Table S1 in Supplementary Material. A similar analysis was carried out on the WH0003 genome. ORFs initially annotated as hypothetical or unknown proteins were compared (BLASTp) to all prokaryotic proteins. Sequences with alignments to known transposases were annotated based on tID as follows; >50% tID: annotated as “transposase,” 35–50% tID: annotated as “similar to transposase,” 10–35% tID: annotated as “possible transposase,” ORFs in all of those categories, as well as those annotated as transposases by the RAST automated annotation were assigned to IS families according to sequence similarity to known families using the ISfinder BLAST tool (see text footnote 3; Siguier et al., 2006).
Genome Comparisons
To assist in visualization of genome-wide comparison between the two strains, proxy genome sequences were created by concatenating the draft genome contigs into a single sequence. For C. watsonii WH8501, all 323 contigs were placed in the same order in which they are listed in GenBank, roughly in order of descending contig length. The WH0003 contigs were ordered according to the location of their best BLAST alignment to the WH8501 proxy genome. The resulting two proxy genomes were aligned and visualized using the WebACT4 version of the Artemis Comparison Tool (Carver et al., 2005).
Nucleotide sequences for intergenic spaces (IGSs) over 50 bp and all ORFs from each strain were used as query sequences in BLASTn comparisons against the proxy genome of the other strain. The percent identity of the best BLAST alignment for each sequence (for sequences with alignments ≥ 50 bp) were used to determined shared and strain-specific genome features as discussed in those sections below. For the taxonomic analysis of the WH0003 ORFs least similar to WH8501, sequences were placed into three bins based on tID of the best BLAST alignment (35–50% tID, 20–35% tID, and <20% tID). Translated amino acid sequences for all sequences in each bin were compared to the NCBI nr protein database using BLASTp. The results of those BLAST comparisons were used to construct the taxonomic distributions (and likely origins) of the ORFs using the MEGAN program (Huson et al., 2007).
Microarray Gene Expression
Methods for growth, RNA extraction, and microarray design and hybridization of whole genome expression experiments were described in Shi et al. (2010). Briefly, C. watsonii WH8501 cultures were grown under a 12:12 h light/dark cycle, and RNA was extracted at eight time points (four in dark and four in light). The RNA samples from each time point were hybridized to an oligonucleotide array designed from the WH8501 draft genome (NimbleGen design ID 2007-03-14_EW_C_watsonii). A total of 320 oligonucleotide probes representing transposase genes in three IS families and one putative transposase family (average of 80 probes per family) were included on the array, which enabled the analysis described in this manuscript. For each gene, the overall mean expression for all eight time points was calculated, and the relative expression for each time point was calculated relative to that mean.
Results and Discussion
Broad Genome Comparison
The genomes of the two Crocosphaera strains (WH8501 and WH0003) were similar in size, %G + C, and number of predicted ORFs. Genomic DNA from the WH0003 strain was sequenced, assembled and analyzed (see Materials and Methods), resulting in a draft genome of 5.5 Mb in 899 contigs. There were an additional 227 contigs (0.4 Mb) that had no similarity to WH8501, but were identified as “probable” WH0003 genome sequence, based on similarity to other cyanobacterial sequences (Table 1). The total length of WH0003 contigs (5.9 Mb) was similar in size to the previously sequenced 6.2 Mb genome of the WH8501 strain. The WH8501 genome is composed of fewer (323) and longer contigs than that of WH0003, despite the fact that the sequence data was over 35× coverage of the WH0003 genome. This may have been due to the difference between sequencing methods, since other pyrosequencing projects have had similar difficulties assembling genomes without paired-end data (Goldberg et al., 2006; Hofreuter et al., 2006; Rothberg and Leamon, 2008; Tripp et al., 2010). The %G + C of the two genomes was very similar (37.1% for WH8501 and 37.7% for WH0003) and both genomes had just under 6,000 predicted ORFs (Table 1). Genome sequences of other cyanobacteria have much higher variability in GC content within a single genus. For example, total %G + C ranges from 31 to 51% in completed Prochlorococcus genomes and 52–66% in Synechococcus genomes (Partensky and Garczarek, 2010). Genome size also varies more within these groups than in the two Crocosphaera strains (<6% variation), with completed genome sizes (as listed in NCBI completed genomes5) ranging from 2.2 to 3.4 Mb (up to 50% variation) in Synechococcus and 1.6–2.7 Mb (up to 56% variation) in Prochlorococcus.
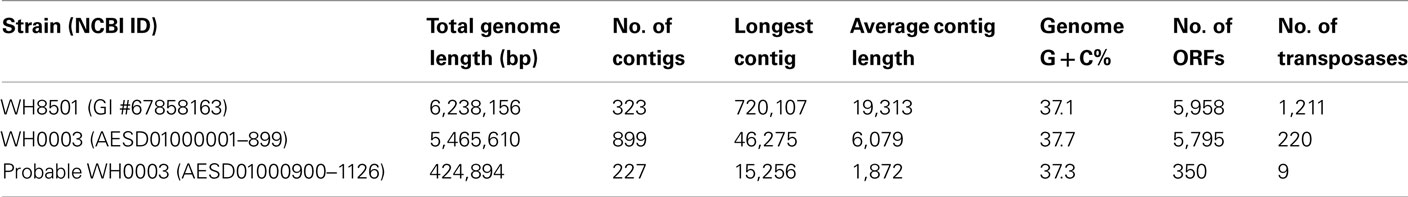
Table 1. Genome assembly information and annotation summary.
Shared Genome Features
Most coding and non-coding regions of each genome were nearly identical between the two Crocosphaera strains. Nucleotide BLAST comparisons of coding sequences (ORFs) and non-coding IGSs between the genomes revealed that over 80% of each genome was >98% identical to the other strain at the DNA sequence level (Figure 1). Below the highest category of sequence identity, the number of sequences in each bin dropped rapidly, and for all BLAST alignments over 50 bp, there were very few (∼5%) sequences between 92 and 97% identical, and none was less than 78% identical (Figure 1). The finding that most of the two Crocosphaera strain genomes are nearly identical is consistent with previous results that reported little to no sequence variation among a number of genetic markers targeting functional genes of cultivated strains and natural populations (Zehr et al., 2007). Surveys of the C. watsonii nitrogenase gene nifH also showed very little, if any, sequence variation in either the Atlantic (Langlois et al., 2005) or the Pacific Oceans (Church et al., 2005a,b). Additionally, 16S–23S rRNA ITS sequences for 10 Crocosphaera strains varied at fewer than five single base positions in a 950 bp amplicon, and some strains shared 100% identity in this region that is typically variable (Webb et al., 2009). This level of genetic conservation is particularly notable in phenotypically distinct Crocosphaera strains that have been isolated from multiple natural populations, over several decades and from multiple ocean basins.
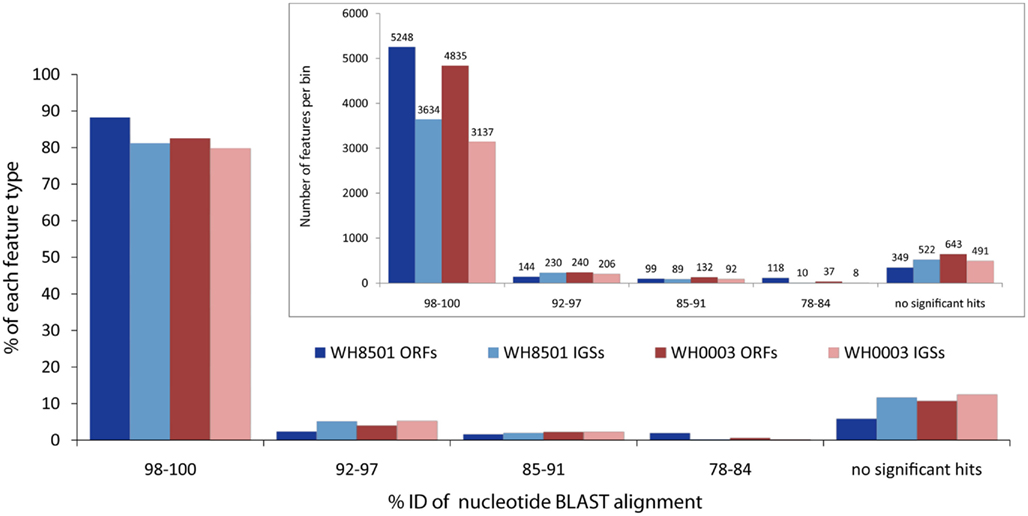
Figure 1. Nucleotide BLAST similarity of open reading frames (ORFs) and intergenic spaces (IGSs) of the two C. watsonii genomes to the opposite strain. Each feature was binned according to the percent identity of the top BLAST alignment. E-values above 0.001 and alignments shorter than 50 bp were considered not significant. Bars in inset figure show total number of features in each bin, and the main figure bars represent the percent of the total number each feature type.
The genome conservation observed in the two Crocosphaera genomes described in this study and the previously described environmental sequences (Zehr et al., 2007) could be explained using three possible mechanisms. First, the species could have a very effective DNA replication and error correction system, similar to Deinococcus radiodurans (Slade et al., 2009). Second, the Crocosphaera population could have undergone a relatively recent global selective sweep as a result of a single strain gaining a trait that conferred a significant advantage over all other strains in the species. The third possibility is that a recent bottleneck could have severely reduced the population and resulted in loss of most genetic variation followed by a global re-distribution of the species throughout tropical waters. However, with the limited sequence data sets currently available, it is not possible to establish which of these three best explains the current observations. Future studies of genomic sequence variation within and between additional strains, and among natural Crocosphaera populations would help answer this question.
The high degree of nucleotide sequence conservation in Crocosphaera strains also contrasts with sequence divergence observed in the sympatric, non-N2-fixing cyanobacteria Prochlorococcus and Synechococcus (Rocap et al., 2002; Ernst et al., 2003; Brown and Fuhrman, 2005; Rusch et al., 2007; Partensky and Garczarek, 2010). Pair-wise, whole genome comparisons of Synechococcus and Prochlorococcus species as well as metagenomic sequencing of environmental samples showed a much higher degree of variation in cultivated strains and in natural populations (Coleman et al., 2006; Rusch et al., 2007; Zhao and Qin, 2007; Dufresne et al., 2008). However, such sequence divergence was not seen in comparisons of environmental samples to the WH8501 genome (Zehr et al., 2007; Hewson et al., 2009), nor in the comparison between the Crocosphaera strains described in this manuscript, which suggests that genome sequence diversity among Crocosphaera strains is much different than in Synechococcus and Prochlorococcus taxa.
Strain-Specific Genome Features
While much of the genome of C. watsonii is highly conserved at the nucleotide sequence level, genetic variation between strains mostly is present as genome rearrangements, insertions and deletions. Alignments of Prochlorococcus genomes showed that strain-specific genetic material is often localized to large islands of variation (10–90 kb each) that do not have homology to other strains (Coleman et al., 2006). The sequences in each Crocosphaera genome that were strain-specific (10–15% of coding and non-coding sequences, see Figure 1) were further analyzed to determine their genomic locations and whether they were localized to similar islands of variation. Proxy genome sequences were constructed by concatenating the contigs from each strain into a single sequence (see Materials and Methods). The BLAST percent identity of each ORF and IGS from one strain were plotted at the position of the best BLAST match on the proxy genome of the other strain to illustrate sequence similarity across the genomes as well as regions where there was little or no sequence identity (Figure 2). The vast majority of the nearly identical coding and non-coding sequences were spread across both proxy genomes, and regions without similarity (i.e., strain-specific regions) occurred mostly in small fragments across both genomes, rather than grouped into large islands. Because the proxy genomes were both constructed from many contigs, it is possible that some larger stretches of strain-specific sequence were not properly arranged, so their full lengths would be unknown, but that cannot be assessed without closed genomes. However, the vast majority of WH0003 contigs contained some regions that were shared with high nucleotide identity to WH8501; and yet, even on those contigs, the strain-specific regions were consistently small and numerous. This suggests that it is not an artifact of the genome status, but that a multitude of insertions, deletions and genomic rearrangements have occurred since the strains diverged.
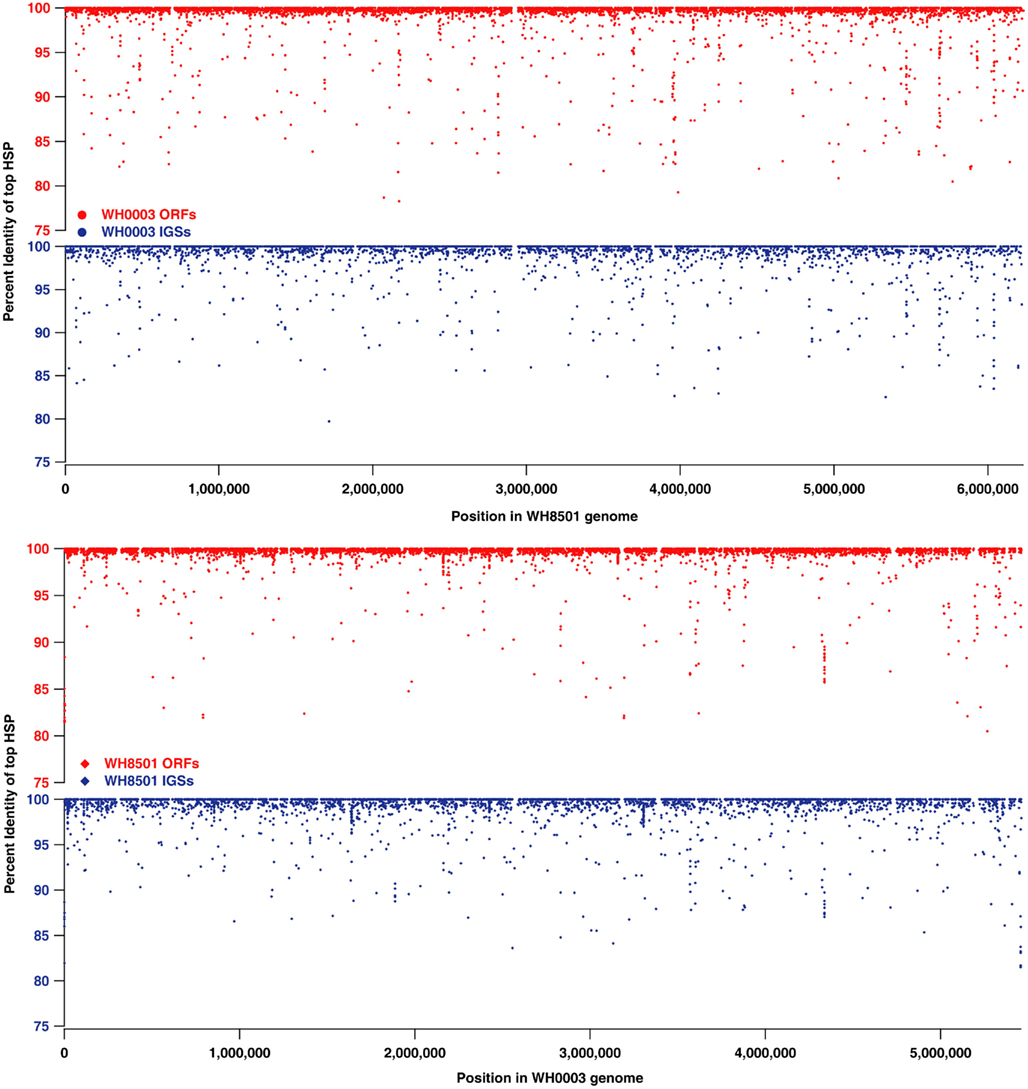
Figure 2. Comparison of nucleotide sequence identity of open reading frames (ORFs, in red) and intergenic spaces (IGSs, in blue) between C. watsonii genomes. Each point represents a single sequence with the x-coordinate as the subject position of top BLAST hit (i.e., highest scoring pair or “HSP”) in the proxy genome sequence, and the y-coordinate as the percent identity of the top BLAST alignment when compared to the alternate strain genome.
The WH0003 ORF sequences that were least similar to the WH8501 genome were compared to public sequence databases and were generally most similar to closely related cyanobacteria. Using a combination of sequence identity and length of BLAST alignment, the total percent identity [(% ID) multiplied by (% of sequence length aligned)] to the WH8501 genome was calculated for WH0003 ORF sequences. There were 930 ORFs in theWH0003 genome with less than 50% tID to the WH8501 genome. A combined tree of presumed taxonomy (Figure 3) showed that about 15% of these ORFs (142 of the 930 total), with all tID categories proportionately represented, could not be assigned or had no BLAST hits at the MEGAN “MinScore” value of 35 (Huson et al., 2007). Nearly all assigned bacterial ORFs (672 of 762) were most similar to known cyanobacteria. Most (34 of 49) of the ORFs taxonomically identified as C. watsonii WH8501 were from the two tID categories most similar to the WH8501 genome, and most of the remaining sequences (379 ORFs) were assigned to various Cyanothece spp. (a closely related unicellular N2-fixing cyanobacterium). Only ORFs from the least similar tID category were assigned to any species other than Crocosphaera and Cyanothece, and most of those were assigned to other cyanobacterial genera (Figure 3). This suggested that most of the WH0003 strain-specific ORFs were either horizontally transferred from cyanobacteria, or were ancestral cyanobacterial genes that have been lost from the WH8501 genome. The 11 ORFs that were taxonomically similar to non-cyanobacterial taxa could have been acquired via HGT, or could be genes that do not have homologs in genomes of other cyanobacteria sequenced to-date. Because there are many transposase genes in the Crocosphaera genomes and the abundance of insertion sequences in genomes is positively correlated with the extent of HGT (Touchon and Rocha, 2007), it is not surprising that these genomes show some evidence of HGT.
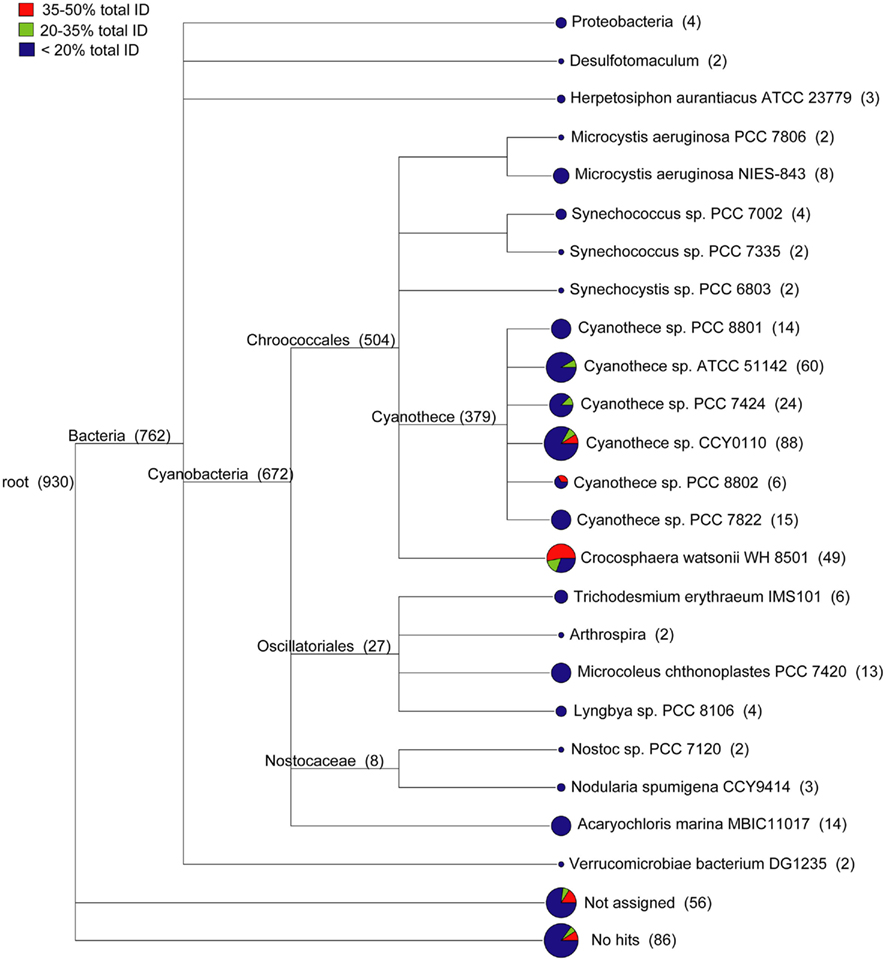
Figure 3. Taxonomic distribution of 930 ORFs in the WH0003 genome that show little or no sequence similarity to the WH8501 genome. WH0003 sequences were binned according to the level of sequence similarity to the WH8501 genome across the entire ORF. The number of sequences in each bin were as follows: 751 in the <20% bin; 75 in the 20–35% bin; and 104 in the 35–50% bin. Pie charts show the relative contributions of the three bins to each branch.
Most of the strain-specific ORFs in both genomes did not have annotated functions, were transposases, or were redundant with the functions of shared genes, leaving a relatively small number of gene functions that could be correlated with phenotypic divergence. There were 351 ORFs in WH8501 that had no BLAST similarity (i.e., no alignments >50 bp) to WH0003 (Table S3 in Supplementary Material), half of which (176 ORFs) were transposases (also noted with an ** in Table S1 in Supplementary Material) and ∼100 more were annotated as hypothetical or unknown function. The majority of these genes showed diel expression patterns (listed in right column of Table S3 in Supplementary Material) in a previous microarray study (Shi et al., 2010). The functions of the 71 ORFs with annotated functions (aside from transposases) are listed in the top two sections of Table S3 in Supplementary Material. Most of those had an identical or nearly identical function annotated in the WH0003 genome, suggesting that the function is not missing from the WH0003 genome, but is being performed by an homologous gene. There were only nine WH8501 ORFs with functions not found in the WH0003 genome (listed at the top of Table S3 in Supplementary Material). In contrast, the WH0003 genome had a larger number (609) of ORFs without BLAST similarity to the WH8501 genome (Table S4 in Supplementary Material). The majority (370) of those ORFs had no assigned function, and only 24 were transposases. The functions of the remaining 215 ORFs with non-transposase functions are listed in the top section of Table S4 in Supplementary Material. A significant portion (57) of those 215 ORFs were annotated with functions that had no homologs in the WH8501 genome (using annotated gene descriptions). The observation that the WH8501 genome contains a much smaller number of ORFs without functional homologs than the WH003 genome may be an indication that WH8501 has lost genetic functionality with the accumulation of the highly abundant transposase genes throughout its genome. Based on that observation, as well the larger total number of strain-specific ORFs, it seems likely that the WH0003 strain has a number of genetic capabilities that are not present in the WH8501 strain, and which may help explain the phenotypic differences between the strains.
Examination of the two longest WH0003 strain-specific regions showed that one is probably involved in DNA processing, and the other is involved in EPS biosynthesis, which is distinctive of that strain’s phenotype. The largest region unique to the WH0003 genome was 28.5 kb long and was dominated by ORFs annotated as hypothetical or unknown (Table S5 in Supplementary Material). Five of the seven functionally annotated ORFs were related to DNA replication or transcription, and the function of one other ORF (bmgA) is a mobilization protein that plays a role in HGT. These predicted ORF functions suggest that the region could provide an aspect of DNA processing not carried out by the WH8501 strain. The second largest (∼25 kb) strain-specific region of the WH0003 genome contained 23 ORFs, a number of which had annotated functions related to polysaccharide biosynthesis and export (Table 2). Alignment of the two genomes using the flanking shared sequences (6.6 kb from the beginning of the upstream contig and 2.3 kb to the end of the downstream contig) showed that the entire 25 kb region has been replaced by a single transposase gene in the WH8501 genome (Figure 4). The %G + C of the 25 kb region is 37.2%, which is very close to the average for the entire WH0003 genome, and most of the highest quality blast alignments for ORFs in the region are to Cyanothece spp., a closely related cyanobacterial species. Thus, this region does not appear to be a horizontally transferred addition to the WH0003 genome, but rather a deletion from the WH8501 genome. It is also notable that six of the 15 functionally annotated ORFs in this region had functions that are not found in the WH8501 genome (indicated by an asterisk in Table 2), and eight of the 15 had functions (based on annotation or COG similarity) related to polysaccharide synthesis and export (shown in bold in Table 2 and with arrowheads in Figure 4). Those included three of the five genes proposed as the core of the cyanobacterial EPS pathway (Pereira et al., 2009); specifically, the wzx gene (CWATWH0003_3507), the wza gene (CWATWH0003_3516), and the wzc gene (CWATWH0003_3517). There are no ORFs with sequence homology, or even conserved domain similarity, to any of these three genes in the WH8501 genome. The other two genes in the EPS pathway (wzb and wzy) have homologs in both strains, which further supports the supposition that WH8501 once had the ability to produce EPS, but lost that functionality through one or more genomic deletion events. Because this region of the WH0003 genome appears to be important in the EPS production that is characteristic of its phenotype, it would be a prime target for physiological studies focused on EPS production, and also as a possible phenotypic marker in future studies of cultivated strains and natural populations.
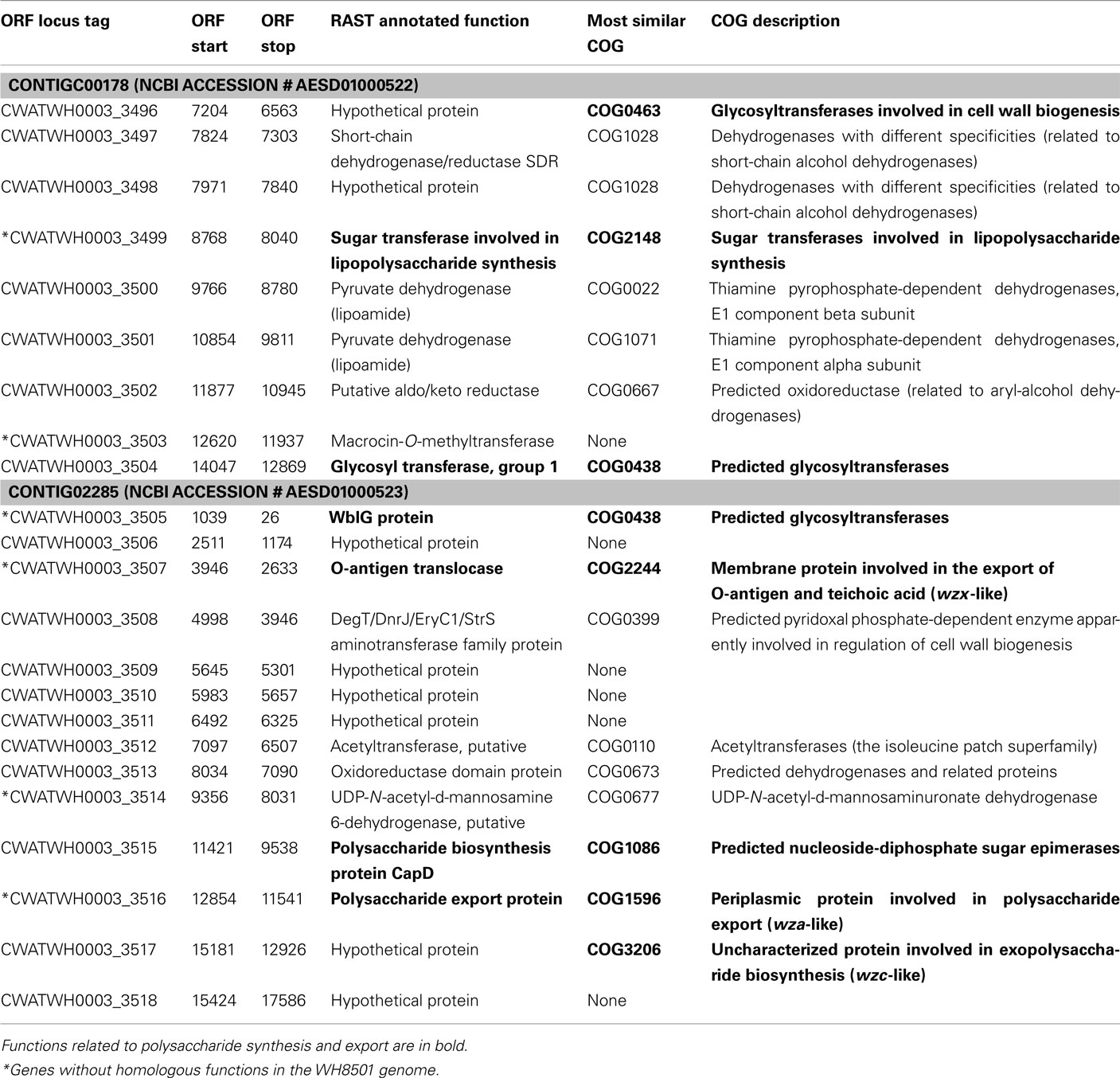
Table 2. ORFs within WH0003 strain-specific genome region.
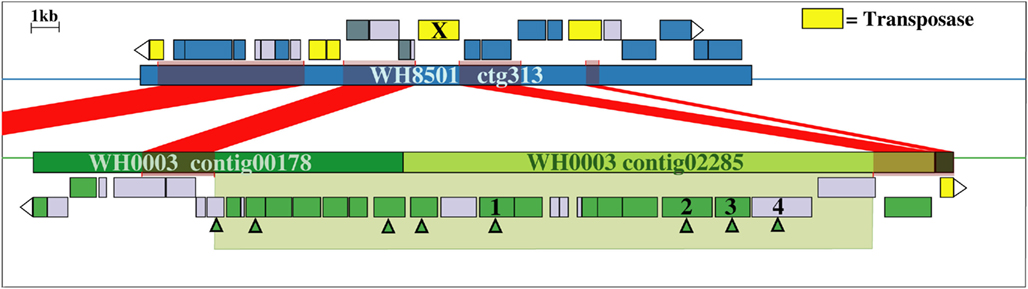
Figure 4. Alignment of WH8501 contig (top) to WH0003 contigs (bottom), showing a 25 kb region of the WH0003 genome (within large green shaded box) that has been replaced by a single transposase gene in the WH8501 genome (marked with an X). Red connecting bars and shading indicate regions of sequence homology. Hypothetical genes are in light gray, transposase genes are yellow and ORFs with other annotated functions are in blue (WH8501) or green (WH0003). ORFs with functions related to polysaccharide synthesis or export are marked with green arrowheads. Descriptions of the numbered genes are listed below. See Table 2 for annotated functions and COG similarities of the 25 contiguous, WH0003-specific ORFs.(1) CWATWH0003_3507: “O-antigen translocase,” similar to wzx, (2) CWATWH0003_3515: “polysaccharide biosynthesis protein CapD,” (3) CWATWH0003_3516: “polysaccharide export protein,” similar to wza, and (4) CWATWH0003_3517: “uncharacterized protein involved in exopolysaccharide biosynthesis,” similar to wzc.
Transposase Gene Comparisons
In contrast to the genome similarities between Crocosphaera strains, the number of transposase genes per genome showed a six-fold difference with over 1,200 transposases identified in the WH8501 genome, and just over 200 identified in the WH0003 genome. Some of the highest numbers of transposases previously found in cyanobacterial genomes were 362 and 469 in two Microcystis aeruginosa strains (Kaneko et al., 2007; Frangeul et al., 2008), and 260 in T. erythraeum IMS101(Stucken et al., 2010). The number of transposase genes in the WH0003 genome (220) was similar in magnitude to those species, but the WH8501 genome contained significantly more than previously reported cyanobacterial genomes (Kaneko et al., 2007; Frangeul et al., 2008). In fact, the 1,211 genes annotated as transposases (see Materials and Methods) constituted more than 20% of the predicted ORFs in the WH8501 genome, and was far higher than the average of 40 transposases per genome computed for 630 transposase-containing bacterial genomes (Aziz et al., 2010).
To further characterize the transposases in the genomes of both Crocosphaera strains, they were assigned to IS families based on sequence similarity using IS finder (Siguier et al., 2006). The resulting IS family distributions showed that WH8501 had many more genes in most families, and the relative proportions of families were quite different between strains (Table 3). The most numerous IS families in the WH8501 genome contained many identical copies of the same sequence, suggesting that their abundance is a result of widespread replication of those genes (see Materials and Methods and Table S4 in Supplementary Material). For instance, of the 294 ORFs assigned to the IS5 family 283 are isoforms of the same sequence, and for the IS1380 family, 119 of 120 ORFs are isoforms of the same sequence. Such replication may partly explain the disparity in transposase abundance between the genomes, as these genes were not highly replicated in the WH0003 genome (Table 3). However, it was not clear why transposases in the same IS families, and even with similar sequences have not undergone similar levels of replication in the WH0003 strain. Because homologous recombination can be enhanced between multi-copy IS elements (Touchon and Rocha, 2007), it is likely that WH8501 has undergone more genomic recombination compared to WH0003, but that is difficult to assess without finished genome sequences. The differing patterns of IS family abundance and replication between the genomes suggests that there are strain-distinct mechanisms of regulating IS element activity.
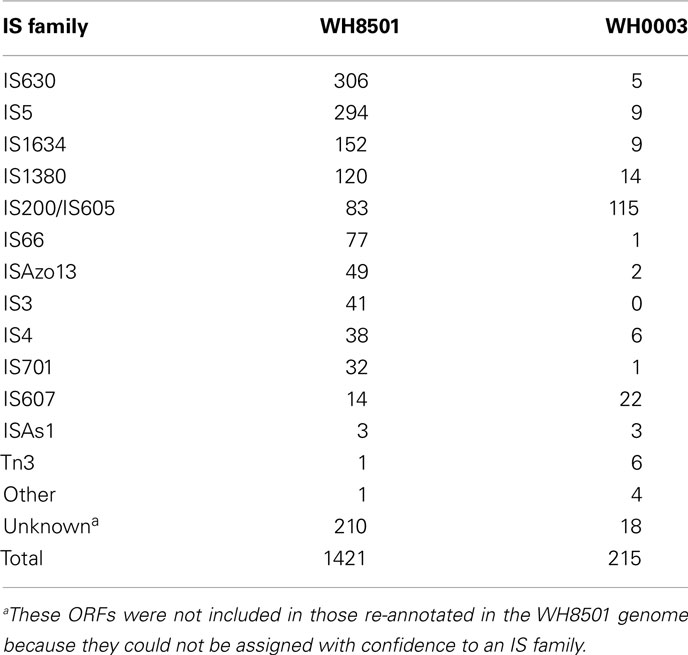
Table 3. Transposase IS family distribution in both genomes.
The large number of transposase genes in WH8501 may have resulted from genome assembly error or from the strain being maintained in culture for a relatively long time. At the time of genome sequencing, WH8501 had been continuously cultivated for 20 years, but WH0003 was in culture for less than half that time (∼9 years) when its genome was sequenced. However, recent metatranscriptome data has shown that some of the transposases found in the WH8501 genome were actively transcribed in natural Crocosphaera populations (Hewson et al., 2009). In addition, microarray expression data showed that transposase genes in four IS families in the WH8501 genome are up- and down-regulated on a daily cycle in culture. The similarity to the pattern observed for the dnaA gene (Figure 5), which encodes for a DNA replication initiation protein (Messer, 2002; Zakrzewska-Czerwinska et al., 2007) suggests that transposase expression may be coordinated with DNA replication as has been observed in other organisms (Ton-Hoang et al., 2010). While more work is required to investigate the full range and activity of transposase genes in these strains, the available data suggest that transposase genes are actively expressed in culture and natural populations, and some exhibit a diel expression pattern.
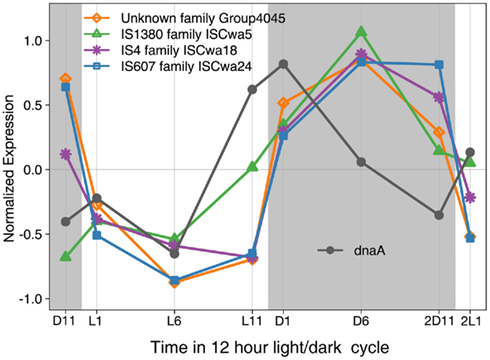
Figure 5. Expression of four IS family genes and dnaA over 26 h time period with a 12 h light (L in white)/dark (D in gray) cycle. Expression values for each gene were normalized to average expression for that gene over the entire 26 h time course, with negative expression values indicating down regulation, and positive values indicating up regulation.
Conclusion
The whole genome comparison of two Crocosphaera strains revealed that, although the strains have divergent phenotypes, the vast majority of the two genomes are essentially identical at the nucleotide level, and only a small fraction of ORFs in each genome are strain-specific. ORFs in one of the two largest contiguous strain-specific regions in the WH0003 genome are likely to play a role in EPS biosynthesis, and therefore likely to be important in establishing phenotypic characteristics. Many of the strain-specific ORFs did not have annotated functions, and future discovery of their functions may help further explain the physiological differences between strains. Strain-specific sequences will also be useful for studying genetic and phenotypic variability in natural populations. Both genomes contained an unusually large number of transposase genes, but the WH8501 strain harbored roughly six times the number of these genes compared to the WH0003 strain, and the IS family patterns of the strains were quite different. Overall, these observations support the conclusion that Crocosphaera spp., maintain an unusually high degree of genomic sequence conservation, without accumulating significant nucleotide level mutations, and strains diverge through genomic insertions, deletions and rearrangements.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge funding from NSF grant EF0424599 for the Center for Microbial Oceanography: Research and Education (CMORE) and from the Gordon and Betty Moore Foundation Marine Microbiology Initiative (Jonathan P. Zehr). We would also like to thank Brandon Carter and the MEGAMER facility for assistance with flow cytometry and cell sorting and John Waterbury for providing Crocosphaera isolates. Pia Moisander, Rachel Foster, Jason Hilton, Kendra Turk, and Mary Hogan provided valuable scientific input and technical assistance. We are also grateful to Eric Webb for critical insight and discussion, and Sara Pereira for generous assistance in EPS pathway gene identification.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/aquatic_microbiology/abstract/16402
Table S1. WH8501 Re-annotated Transposase ORFs.
Table S2. WH8501 Transposase Isoform Counts.
Table S3. WH8501 strain-specific ORFs.
Table S4. WH0003 strain-specific ORFs.
Table S5. WH0003 ORFs in longest strain-specific region.
Footnotes
References
Altschul, S. F., Gish, W., Miller, W., Meyers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Aziz, R., Bartels, D., Best, A., Dejongh, M., Disz, T., Edwards, R., Formsma, K., Gerdes, S., Glass, E., Kubal, M., Meyer, F., Olsen, G., Olson, R., Osterman, A., Overbeek, R., Mcneil, L., Paarmann, D., Paczian, T., Parrello, B., Pusch, G., Reich, C., Stevens, R., Vassieva, O., Vonstein, V., Wilke, A., and Zagnitko, O. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9, 75. doi:10.1186/1471-2164-9-75
Aziz, R. K., Breitbart, M., and Edwards, R. A. (2010). Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Res. 38, 4207–4217.
Bonnet, S., Biegala, I. C., Dutrieux, P., Slemons, L. O., and Capone, D. G. (2009). Nitrogen fixation in the western equatorial Pacific: rates, diazotrophic cyanobacterial size class distribution, and biogeochemical significance. Global Biogeochem. Cycles 23, GB3012.
Brown, M. V., and Fuhrman, J. A. (2005). Marine bacterial microdiversity as revealed by internal transcribed spacer analysis. Aquat. Microb. Ecol. 41, 15–23.
Capone, D. G., Zehr, J. P., Paerl, H. W., Bergman, B., and Carpenter, E. J. (1997). Trichodesmium: a globally significant marine cyanobacterium. Science 276, 1221–1229.
Carver, T. J., Rutherford, K. M., Berriman, M., Rajandream, M.-A., Barrell, B. G., and Parkhill, J. (2005). ACT: the Artemis comparison tool. Bioinformatics 21, 3422–3423.
Church, M. J., Bjorkman, K. M., Karl, D. M., Saito, M. A., and Zehr, J. P. (2008). Regional distributions of nitrogen-fixing bacteria in the Pacific Ocean. Limnol. Oceanogr. 53, 63–77.
Church, M. J., Jenkins, B. D., Karl, D. M., and Zehr, J. P. (2005a). Vertical distributions of nitrogen-fixing phylotypes at Stn ALOHA in the oligotrophic North Pacific Ocean. Aquat. Microb. Ecol. 38, 3–14.
Church, M. J., Short, C. M., Jenkins, B. D., Karl, D. M., and Zehr, J. P. (2005b). Temporal patterns of nitrogenase gene (nifH) expression in the oligotrophic North Pacific Ocean. Appl. Environ. Microbiol. 71, 5362–5370.
Coleman, M. L., Sullivan, M. B., Martiny, A. C., Steglich, C., Barry, K., Delong, E. F., and Chisholm, S. W. (2006). Genomic islands and the ecology and evolution of Prochlorococcus. Science 311, 1768–1770.
Dufresne, A., Ostrowski, M., Scanlan, D., Garczarek, L., Mazard, S., Palenik, B., Paulsen, I., De Marsac, N., Wincker, P., Dossat, C., Ferriera, S., Johnson, J., Post, A., Hess, W., and Partensky, F. (2008). Unraveling the genomic mosaic of a ubiquitous genus of marine cyanobacteria. Genome Biol. 9, R90.
Dyhrman, S. T., and Haley, S. T. (2006). Phosphorus scavenging in the unicellular marine diazotroph Crocosphaera watsonii. Appl. Environ. Microbiol. 72, 1452–1458.
Ernst, A., Becker, S., Wollenzien, U. I. A., and Postius, C. (2003). Ecosystem-dependent adaptive radiations of picocyanobacteria inferred from 16S rRNA and ITS-1 sequence analysis. Microbiology 149, 217–228.
Falcon, L. I., Carpenter, E. J., Cipriano, F., Bergman, B., and Capone, D. G. (2004). N2 Fixation by unicellular bacterioplankton from the Atlantic and Pacific Oceans: phylogeny and in situ rates. Appl. Environ. Microbiol. 70, 765–770.
Feschotte, C., Jiang, N., and Wessler, S. R. (2002). Plant transposable elements: where genetics meets genomics. Nat. Rev. Genet. 3, 329–341.
Filee, J., Siguier, P., and Chandler, M. (2007). Insertion sequence diversity in Archaea. Microbiol. Mol. Biol. Rev. 71, 121–157.
Frangeul, L., Quillardet, P., Castets, A.-M., Humbert, J.-F., Matthijs, H., Cortez, D., Tolonen, A., Zhang, C.-C., Gribaldo, S., Kehr, J.-C., Zilliges, Y., Ziemert, N., Becker, S., Talla, E., Latifi, A., Billault, A., Lepelletier, A., Dittmann, E., Bouchier, C., and Tandeau De Marsac, N. (2008). Highly plastic genome of Microcystis aeruginosa PCC 7806, a ubiquitous toxic freshwater cyanobacterium. BMC Genomics 9, 274. doi:10.1186/1471-2164-9-274
Goericke, R., and Welschmeyer, N. A. (1993). The marine prochlorophyte Prochlorococcus contributes significantly to phytoplankton biomass and primary production in the Sargasso Sea. Deep Sea Res. Part 1 Oceanogr. Res. Pap. 40, 2283–2294.
Goldberg, S. M. D., Johnson, J., Busam, D., Feldblyum, T., Ferriera, S., Friedman, R., Halpern, A., Khouri, H., Kravitz, S. A., Lauro, F. M., Li, K., Rogers, Y.-H., Strausberg, R., Sutton, G., Tallon, L., Thomas, T., Venter, E., Frazier, M., and Venter, J. C. (2006). A Sanger/pyrosequencing hybrid approach for the generation of high-quality draft assemblies of marine microbial genomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11240–11245.
Goodchild, A., Raftery, M., Saunders, N. F. W., Guilhaus, M., and Cavicchioli, R. (2004). Biology of the cold adapted Archaeon, Methanococcoides burtonii determined by proteomics using liquid chromatography-tandem mass spectrometry. J. Proteome Res. 3, 1164–1176.
Hewson, I., Poretsky, R. S., Beinart, R. A., White, A. E., Shi, T., Bench, S. R., Moisander, P. H., Paerl, R. W., Tripp, H. J., Montoya, J. P., Moran, M. A., and Zehr, J. P. (2009). In situ transcriptomic analysis of the globally important keystone N2-fixing taxon Crocosphaera watsonii. ISME J. 3, 618–631.
Hofreuter, D., Tsai, J., Watson, R. O., Novik, V., Altman, B., Benitez, M., Clark, C., Perbost, C., Jarvie, T., Du, L., and Galan, J. E. (2006). Unique features of a highly pathogenic Campylobacter jejuni strain. Infect. Immun. 74, 4694–4707.
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386.
Kaneko, T., Nakajima, N., Okamoto, S., Suzuki, I., Tanabe, Y., Tamaoki, M., Nakamura, Y., Kasai, F., Watanabe, A., Kawashima, K., Kishida, Y., Ono, A., Shimizu, Y., Takahashi, C., Minami, C., Fujishiro, T., Kohara, M., Katoh, M., Nakazaki, N., Nakayama, S., Yamada, M., Tabata, S., and Watanabe, M. M. (2007). Complete genomic structure of the bloom-forming toxic cyanobacterium Microcystis aeruginosa NIES-843. DNA Res. 14, 247–256.
Karl, D., Letelier, R., Tupas, L., Dore, J., Christian, J., and Hebel, D. (1997). The role of nitrogen fixation in biogeochemical cycling in the subtropical North Pacific Ocean. Nature 388, 533–538.
Karl, D., Michaels, A., Bergman, B., Capone, D., Carpenter, E., Letelier, R., Lipschultz, F., Paerl, H., Sigman, D., and Stal, L. (2002). Dinitrogen fixation in the world’s oceans. Biogeochemistry 57/58, 47–98.
Kitajima, S., Furuya, K., Hashihama, F., Takeda, S., and Kanda, J. (2009). Latitudinal distribution of diazotrophs and their nitrogen fixation in the tropical and subtropical western North Pacific. Limnol. Oceanogr. 54, 537–547.
Konstantinidis, K. T., Braff, J., Karl, D. M., and Delong, E. F. (2009). Comparative metagenomic analysis of a microbial community residing at a depth of 4,000 meters at station ALOHA in the North Pacific Subtropical Gyre. Appl. Environ. Microbiol. 75, 5345–5355.
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., Fitzhugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland, J., Kann, L., Lehoczky, J., Levine, R., Mcewan, P., Mckernan, K., Meldrim, J., Mesirov, J. P., Miranda, C., Morris, W., Naylor, J., Raymond, C., Rosetti, M., Santos, R., Sheridan, A., Sougnez, C., Stange-Thomann, N., Stojanovic, N., Subramanian, A., Wyman, D., Rogers, J., Sulston, J., Ainscough, R., Beck, S., Bentley, D., Burton, J., Clee, C., Carter, N., Coulson, A., Deadman, R., Deloukas, P., Dunham, A., Dunham, I., Durbin, R., French, L., Grafham, D., Gregory, S., Hubbard, T., Humphray, S., Hunt, A., Jones, M., Lloyd, C., Mcmurray, A., Matthews, L., Mercer, S., Milne, S., Mullikin, J. C., Mungall, A., Plumb, R., Ross, M., Shownkeen, R., Sims, S., Waterston, R. H., Wilson, R. K., Hillier, L. W., Mcpherson, J. D., Marra, M. A., Mardis, E. R., Fulton, L. A., Chinwalla, A. T., Pepin, K. H., Gish, W. R., Chissoe, S. L., Wendl, M. C., Delehaunty, K. D., Miner, T. L., Delehaunty, A., Kramer, J. B., Cook, L. L., Fulton, R. S., Johnson, D. L., Minx, P. J., Clifton, S. W., Hawkins, T., Branscomb, E., Predki, P., Richardson, P., Wenning, S., Slezak, T., Doggett, N., Cheng, J.-F., Olsen, A., Lucas, S., Elkin, C., Uberbacher, E., Frazier, M., Gibbs, R. A., Muzny, D. M., Scherer, S. E., Bouck, J. B., Sodergren, E. J., Worley, K. C., Rives, C. M., Gorrell, J. H., Metzker, M. L., Naylor, S. L., Kucherlapati, R. S., Nelson, D. L., Weinstock, G. M., Sakaki, Y., Fujiyama, A., Hattori, M., Yada, T., Toyoda, A., Itoh, T., Kawagoe, C., Watanabe, H., Totoki, Y., Taylor, T., Weissenbach, J., Heilig, R., Saurin, W., Artiguenave, F., Brottier, P., Bruls, T., Pelletier, E., Robert, C., Wincker, P., Smith, D. R., Doucette-Stamm, L., Rubenfield, M., Weinstock, K., Lee, H. M., Dubois, J., Rosenthal, A., Platzer, M., Nyakatura, G., Taudien, S., Rump, A., Yang, H., Yu, J., Wang, J., Huang, G., Gu, J., Hood, L., Rowen, L., Madan, A., Qin, S., Davis, R. W., Federspiel, N. A., Abola, A. P., Proctor, M. J., Myers, R. M., Schmutz, J., Dickson, M., Grimwood, J., Cox, D. R., Olson, M. V., Kaul, R., Raymond, C., Shimizu, N., Kawasaki, K., Minoshima, S., Evans, G. A., Athanasiou, M., Schultz, R., Roe, B. A., Chen, F., Pan, H., Ramser, J., Lehrach, H., Reinhardt, R., McCombie, W. R., de la Bastide, M., Dedhia, N., Blöcker, H., Hornischer, K., Nordsiek, G., Agarwala, R., Aravind, L., Bailey, J. A., Bateman, A., Batzoglou, S., Birney, E., Bork, P., Brown, D. G., Burge, C. B., Cerutti, L., Chen, H. C., Church, D., Clamp, M., Copley, R. R., Doerks, T., Eddy, S. R., Eichler, E. E., Furey, T. S., Galagan, J., Gilbert, J. G., Harmon, C., Hayashizaki, Y., Haussler, D., Hermjakob, H., Hokamp, K., Jang, W., Johnson, L. S., Jones, T. A., Kasif, S., Kaspryzk, A., Kennedy, S., Kent, W. J., Kitts, P., Koonin, E. V., Korf, I., Kulp, D., Lancet, D., Lowe, T. M., McLysaght, A., Mikkelsen, T., Moran, J. V., Mulder, N., Pollara, V. J., Ponting, C. P., Schuler, G., Schultz, J., Slater, G., Smit, A. F., Stupka, E., Szustakowski, J., Thierry-Mieg, D., Thierry-Mieg, J., Wagner, L., Wallis, J., Wheeler, R., Williams, A., Wolf, Y. I., Wolfe, K. H., Yang, S. P., Yeh, R. F., Collins, F., Guyer, M. S., Peterson, J., Felsenfeld, A., Wetterstrand, K. A., Patrinos, A., Morgan, M. J., de Jong, P., Catanese, J. J., Osoegawa, K., Shizuya, H., Choi, S., Chen, Y. J., and International Human Genome Sequencing Consortium. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921.
Langlois, R. J., Hummer, D., and Laroche, J. (2008). Abundances and distributions of the dominant nifH phylotypes in the Northern Atlantic Ocean. Appl. Environ. Microbiol. 74, 1922–1931.
Langlois, R. J., Laroche, J., and Raab, P. A. (2005). Diazotrophic diversity and distribution in the tropical and subtropical Atlantic ocean. Appl. Environ. Microbiol. 71, 7910–7919.
Liu, H., Nolla, H. A., and Campbell, L. (1997). Prochlorococcus growth rate and contribution to primary production in the equatorial and subtropical North Pacific Ocean. Aquat. Microb. Ecol. 12, 39–47.
Mahillon, J., Léonard, C., and Chandler, M. (1999). IS elements as constituents of bacterial genomes. Res. Microbiol. 150, 675–687.
Mes, T. H. M., and Doeleman, M. (2006). Positive selection on transposase genes of insertion sequences in the Crocosphaera watsonii genome. J. Bacteriol. 188, 7176–7185.
Messer, W. (2002). The bacterial replication initiator DnaA. DnaA and oriC, the bacterial mode to initiate DNA replication. FEMS Microbiol. Rev. 26, 355–374.
Moisander, P. H., Beinart, R. A., Hewson, I., White, A. E., Johnson, K. S., Carlson, C. A., Montoya, J. P., and Zehr, J. P. (2010). Unicellular cyanobacterial distributions broaden the oceanic N2 fixation domain. Science 327, 1512–1514.
Moisander, P. H., Beinart, R. A., Voss, M., and Zehr, J. P. (2008). Diversity and abundance of diazotrophic microorganisms in the South China Sea during intermonsoon. ISME J. 2, 954–967.
Montoya, J. P., Holl, C. M., Zehr, J. P., Hansen, A., Villareal, T. A., and Capone, D. G. (2004). High rates of N2 fixation by unicellular diazotrophs in the oligotrophic Pacific Ocean. Nature 430, 1027–1031.
Partensky, F., Hess, W. R., and Vaulot, D. (1999). Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol. Mol. Biol. Rev. 63, 106–127.
Partensky, F. D. R., and Garczarek, L. (2010). Prochlorococcus: advantages and limits of minimalism. Ann. Rev. Mar. Sci. 2, 305–331.
Pereira, S., Zille, A., Micheletti, E., Moradas-Ferreira, P., Philippis, R. D., and Tamagnini, P. (2009). Complexity of cyanobacterial exopolysaccharides: composition, structures, inducing factors and putative genes involved in their biosynthesis and assembly. FEMS Microbiol. Rev. 33, 917–941.
Rocap, G., Distel, D. L., Waterbury, J. B., and Chisholm, S. W. (2002). Resolution of Prochlorococcus and Synechococcus ecotypes by using 16S-23S ribosomal DNA internal transcribed spacer sequences. Appl. Environ. Microbiol. 68, 1180–1191.
Rocap, G., Larimer, F. W., Lamerdin, J., Malfatti, S., Chain, P., Ahlgren, N. A., Arellano, A., Coleman, M., Hauser, L., Hess, W. R., Johnson, Z. I., Land, M., Lindell, D., Post, A. F., Regala, W., Shah, M., Shaw, S. L., Steglich, C., Sullivan, M. B., Ting, C. S., Tolonen, A., Webb, E. A., Zinser, E. R., and Chisholm, S. W. (2003). Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature 424, 1042–1047.
Rothberg, J. M., and Leamon, J. H. (2008). The development and impact of 454 sequencing. Nat. Biotechnol. 26, 1117–1124.
Rusch, D. B., Halpern, A. L., Sutton, G., Heidelberg, K. B., Williamson, S., Yooseph, S., Wu, D., Eisen, J. A., Hoffman, J. M., Remington, K., Beeson, K., Tran, B., Smith, H., Baden-Tillson, H., Stewart, C., Thorpe, J., Freeman, J., Andrews-Pfannkoch, C., Venter, J. E., Li, K., Kravitz, S., Heidelberg, J. F., Utterback, T., Rogers, Y.-H., Falcã3n, L. I., Souza, V., Bonilla-Rosso, G. N., Eguiarte, L. E., Karl, D. M., Sathyendranath, S., Platt, T., Bermingham, E., Gallardo, V., Tamayo-Castillo, G., Ferrari, M. R., Strausberg, R. L., Nealson, K., Friedman, R., Frazier, M., and Venter, J. C. (2007). The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biol. 5, e77. doi:10.1371/journal.pbio.0050077
Scanlan, D. J., Ostrowski, M., Mazard, S., Dufresne, A., Garczarek, L., Hess, W. R., Post, A. F., Hagemann, M., Paulsen, I., and Partensky, F. (2009). Ecological genomics of marine picocyanobacteria. Microbiol. Mol. Biol. Rev. 73, 249–299.
Scanlan, D. J., and West, N. J. (2002). Molecular ecology of the marine cyanobacterial genera Prochlorococcus and Synechococcus. FEMS Microbiol. Ecol. 40, 1–12.
Shi, T., Ilikchyan, I., Rabouille, S., and Zehr, J. P. (2010). Genome-wide analysis of diel gene expression in the unicellular N2-fixing cyanobacterium Crocosphaera watsonii WH 8501. ISME J. 4, 621–632.
Shiozaki, T., Furuya, K., Kodama, T., Kitajima, S., Takeda, S., Takemura, T., and Kanda, J. (2010). New estimation of N2 fixation in the western and central Pacific Ocean and its marginal seas. Global Biogeochem. Cycles 24, GB1015.
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36.
Slade, D., Lindner, A. B., Paul, G., and Radman, M. (2009). Recombination and replication in DNA repair of heavily irradiated Deinococcus radiodurans. Cell 136, 1044–1055.
Stucken, K., John, U., Cembella, A., Murillo, A. A., Soto-Liebe, K., Fuentes-Valdes, J. J., Friedel, M., Plominsky, A. M., Vasquez, M., and Glockner, G. (2010). The smallest known genomes of multicellular and toxic cyanobacteria: comparison, minimal gene sets for linked traits and the evolutionary implications. PLoS ONE 5, e9235. doi:10.1371/journal.pone.0009235
Ton-Hoang, B., Pasternak, C., Siguier, P., Guynet, C., Hickman, A. B., Dyda, F., Sommer, S., and Chandler, M. (2010). Single-stranded DNA transposition is coupled to host replication. Cell 142, 398–408.
Touchon, M., and Rocha, E. P. C. (2007). Causes of insertion sequences abundance in prokaryotic genomes. Mol. Biol. Evol. 24, 969–981.
Tripp, H. J., Bench, S. R., Turk, K. A., Foster, R. A., Desany, B. A., Niazi, F., Affourtit, J. P., and Zehr, J. P. (2010). Metabolic streamlining in an open-ocean nitrogen-fixing cyanobacterium. Nature 464, 90–94.
Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., Wu, D., Paulsen, I., Nelson, K. E., Nelson, W., Fouts, D. E., Levy, S., Knap, A. H., Lomas, M. W., Nealson, K., White, O., Peterson, J., Hoffman, J., Parsons, R., Baden-Tillson, H., Pfannkoch, C., Rogers, Y.-H., and Smith, H. O. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74.
Waterbury, J. B., Watson, S. W., Valois, F. W., and Franks, D. G. (1986). Biological and ecological characterization of the marine unicellular cyanobacterium Synechococcus. Can. Bull. Fish. Aquat. Sci. 71–120.
Waterbury, J. B., Willey, J. M., Packer, L., and Alexander, N. G. (1988). Isolation and growth of marine planktonic cyanobacteria. Methods Enzymol. 167, 100–105.
Waterston, R. H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J. F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., Antonarakis, S. E., Attwood, J., Baertsch, R., Bailey, J., Barlow, K., Beck, S., Berry, E., Birren, B., Bloom, T., Bork, P., Botcherby, M., Bray, N., Brent, M. R., Brown, D. G., Brown, S. D., Bult, C., Burton, J., Butler, J., Campbell, R. D., Carninci, P., Cawley, S., Chiaromonte, F., Chinwalla, A. T., Church, D. M., Clamp, M., Clee, C., Collins, F. S., Cook, L. L., Copley, R. R., Coulson, A., Couronne, O., Cuff, J., Curwen, V., Cutts, T., Daly, M., David, R., Davies, J., Delehaunty, K. D., Deri, J., Dermitzakis, E. T., Dewey, C., Dickens, N. J., Diekhans, M., Dodge, S., Dubchak, I., Dunn, D. M., Eddy, S. R., Elnitski, L., Emes, R. D., Eswara, P., Eyras, E., Felsenfeld, A., Fewell, G. A., Flicek, P., Foley, K., Frankel, W. N., Fulton, L. A., Fulton, R. S., Furey, T. S., Gage, D., Gibbs, R. A., Glusman, G., Gnerre, S., Goldman, N., Goodstadt, L., Grafham, D., Graves, T. A., Green, E. D., Gregory, S., Guigó, R., Guyer, M., Hardison, R. C., Haussler, D., Hayashizaki, Y., Hillier, L. W., Hinrichs, A., Hlavina, W., Holzer, T., Hsu, F., Hua, A., Hubbard, T., Hunt, A., Jackson, I., Jaffe, D. B., Johnson, L. S., Jones, M., Jones, T. A., Joy, A., Kamal, M., Karlsson, E. K., Karolchik, D., Kasprzyk, A., Kawai, J., Keibler, E., Kells, C., Kent, W. J., Kirby, A., Kolbe, D. L., Korf, I., Kucherlapati, R. S., Kulbokas, E. J., Kulp, D., Landers, T., Leger, J. P., Leonard, S., Letunic, I., Levine, R., Li, J., Li, M., Lloyd, C., Lucas, S., Ma, B., Maglott, D. R., Mardis, E. R., Matthews, L., Mauceli, E., Mayer, J. H., McCarthy, M., McCombie, W. R., McLaren, S., McLay, K., McPherson, J. D., Meldrim, J., Meredith, B., Mesirov, J. P., Miller, W., Miner, T. L., Mongin, E., Montgomery, K. T., Morgan, M., Mott, R., Mullikin, J. C., Muzny, D. M., Nash, W. E., Nelson, J. O., Nhan, M. N., Nicol, R., Ning, Z., Nusbaum, C., O’Connor, M. J., Okazaki, Y., Oliver, K., Overton-Larty, E., Pachter, L., Parra, G., Pepin, K. H., Peterson, J., Pevzner, P., Plumb, R., Pohl, C. S., Poliakov, A., Ponce, T. C., Ponting, C. P., Potter, S., Quail, M., Reymond, A., Roe, B. A., Roskin, K. M., Rubin, E. M., Rust, A. G., Santos, R., Sapojnikov, V., Schultz, B., Schultz, J., Schwartz, M. S., Schwartz, S., Scott, C., Seaman, S., Searle, S., Sharpe, T., Sheridan, A., Shownkeen, R., Sims, S., Singer, J. B., Slater, G., Smit, A., Smith, D. R., Spencer, B., Stabenau, A., Stange-Thomann, N., Sugnet, C., Suyama, M., Tesler, G., Thompson, J., Torrents, D., Trevaskis, E., Tromp, J., Ucla, C., Ureta-Vidal, A., Vinson, J. P., Von Niederhausern, A. C., Wade, C. M., Wall, M., Weber, R. J., Weiss, R. B., Wendl, M. C., West, A. P., Wetterstrand, K., Wheeler, R., Whelan, S., Wierzbowski, J., Willey, D., Williams, S., Wilson, R. K., Winter, E., Worley, K. C., Wyman, D., Yang, S., Yang, S. P., Zdobnov, E. M., Zody, M. C., and Lander, E. S. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562.
Webb, E. A., Ehrenreich, I. M., Brown, S. L., Valois, F. W., and Waterbury, J. B. (2009). Phenotypic and genotypic characterization of multiple strains of the diazotrophic cyanobacterium, Crocosphaera watsonii, isolated from the open ocean. Environ. Microbiol. 11, 338–348.
Zakrzewska-Czerwinska, J., Jakimowicz, D., Zawilak-Pawlik, A., and Messer, W. (2007). Regulation of the initiation of chromosomal replication in bacteria. FEMS Microbiol. Rev. 31, 378–387.
Zehr, J. P., Bench, S. R., Mondragon, E. A., Mccarren, J., and Delong, E. F. (2007). Low genomic diversity in tropical oceanic N2-fixing cyanobacteria. Proc. Natl. Acad. Sci. U.S.A. 104, 17807–17812.
Zehr, J. P., Waterbury, J. B., Turner, P. J., Montoya, J. P., Omoregie, E., Steward, G. F., Hansen, A., and Karl, D. M. (2001). Unicellular cyanobacteria fix N2 in the subtropical North Pacific Ocean. Nature 412, 635–638.
Keywords: comparative genomics, Crocosphaera, exopolysaccharide biosynthesis, genome conservation, mobile genetic elements, nitrogen fixation
Citation: Bench SR, Ilikchyan IN, Tripp HJ and Zehr JP (2011) Two strains of Crocosphaera watsonii with highly conserved genomes are distinguished by strain-specific features. Front. Microbio. 2:261. doi: 10.3389/fmicb.2011.00261
Received: 22 September 2011;
Paper pending published: 23 October 2011;
Accepted: 06 December 2011;
Published online: 29 December 2011.
Edited by:
Julie LaRoche, Leibniz Institute of Marine Sciences, GermanyReviewed by:
Katherine McMahon, University of Wisconsin-Madison, USAMartin W. Hahn, Austrian Academy of Sciences, Austria
Copyright: © 2011 Bench, Ilikchyan, Tripp and Zehr. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jonathan P. Zehr, Department of Ocean Sciences, University of California, Santa Cruz, 1156 High Street, Santa Cruz, CA 95064, USA. e-mail: zehrj@ucsc.edu