
- Friedrich Loeffler Institute of Medical Microbiology, University of Greifswald, Greifswald, Germany
The Gram-negative facultative intracellular rod Burkholderia pseudomallei causes melioidosis, an infectious disease with a wide range of clinical presentations. Among the observed visceral abscesses, the liver is commonly affected. However, neither this organotropism of B. pseudomallei nor local hepatic defense mechanisms have been thoroughly investigated so far. Own previous studies using electron microscopy of the murine liver after systemic infection of mice indicated that hepatocytes might be capable of killing B. pseudomallei. Therefore, the aim of this study was to further elucidate the interaction of B. pseudomallei with these cells and to analyze the role of hepatocytes in anti-B. pseudomallei host defense. In vitro studies using the human hepatocyte cell line HepG2 revealed that B. pseudomallei can invade these cells. Subsequently, B. pseudomallei is able to escape from the vacuole, to replicate within the cytosol of HepG2 cells involving its type 3 and type 6 secretion systems, and to induce actin tail formation. Furthermore, stimulation of HepG2 cells showed that IFNγ can restrict growth of B. pseudomallei in the early and late phase of infection whereas the combination of IFNγ, IL-1β, and TNFα is required for the maximal antibacterial activity. This anti-B. pseudomallei defense of HepG2 cells did not seem to be mediated by inducible nitric oxide synthase-derived nitric oxide or NADPH oxidase-derived superoxide. In summary, this is the first study describing B. pseudomallei intracellular life cycle characteristics in hepatocytes and showing that IFNγ-mediated, but nitric oxide- and reactive oxygen species-independent, effector mechanisms are important in anti-B. pseudomallei host defense of hepatocytes.
Introduction
The Gram-negative saprophyte Burkholderia pseudomallei is the causative agent of melioidosis, an emerging infectious disease of humans and animals in certain areas of the tropics and subtropics. B. pseudomallei is an intracellular pathogen that can invade a variety of host cells (Jones et al., 1996). After invasion, B. pseudomallei can escape from the endocytotic vesicle of murine macrophage cells into the host cytosol depending on a functional type 3 secretion system-3 (T3SS-3; Stevens et al., 2002; Burtnick et al., 2008; Muangsombut et al., 2008; Gong et al., 2011). Within the cytosol bacteria can multiply and induce the BimA-dependent formation of actin tails, facilitating intracellular motility as well as spreading of B. pseudomallei into neighboring cells (Kespichayawattana et al., 2000; Breitbach et al., 2003; Stevens et al., 2005). A recent paper proposed that B. pseudomallei-induced cell fusion and the formation of multinucleated giant cells represent the primary path for intercellular spread and plaque formation of this pathogen (French et al., 2011).
Several reports have shown that interferon γ (IFNγ) is essential for the early control of B. pseudomallei infection in mice (Santanirand et al., 1999; Breitbach et al., 2006). In vitro experiments also demonstrated a pivotal role for IFNγ to eliminate intracellular B. pseudomallei in macrophages (Miyagi et al., 1997; Utaisincharoen et al., 2001). We recently demonstrated that the downstream effector molecule of IFNγ, nitric oxide (NO), has a dual role among resistant and susceptible mouse strains after B. pseudomallei infection. NO had rather detrimental effects in innate resistant C57BL/6 mice in a murine model of melioidosis and was not involved in killing activity of C57BL/6 macrophages (Breitbach et al., 2006, 2011). In contrast, NO contributed to complete resistance in innate susceptible BALB/c mice and to growth restriction of B. pseudomallei in macrophages from those mice (Breitbach et al., 2011). In a previous study we revealed that NADPH oxidase-mediated mechanisms contribute to early resistance in bone marrow-derived macrophages and C57BL/6 mice (Breitbach et al., 2006).
The liver plays an essential role in the innate immune response, providing the first line of defense against microbes crossing the intestinal barrier (Crispe, 2009). Kupffer cells are important for the rapid clearance of microorganisms from the systemic circulation, and can facilitate the generation of a local inflammatory response leading to recruitment of inflammatory cells. Furthermore, hepatocytes can also secrete inflammatory cytokines and chemokines in response to cytokine activation and/or bacterial invasion (Rowell et al., 1997; Santos et al., 2005). However, to date little is known about the antimicrobial responses of hepatocytes. Only few studies indicate that IFNγ can restrict growth of Listeria monocytogenes and Salmonella typhimurium in murine hepatocytes (Gregory and Wing, 1993; Lajarin et al., 1996).
In previous studies we and others revealed that the organotropism of B. pseudomallei for the spleen and liver in melioidosis patients can be mimicked by infection of mice (Hoppe et al., 1999; Santanirand et al., 1999; Liu et al., 2002). Electron microscopic investigations of the murine liver demonstrated that B. pseudomallei-containing phagosomes in hepatocytes fuse with lysosomes, leading to bacterial degradation (Hoppe et al., 1999). Therefore, the present study aimed to establish an in vitro hepatocyte infection model with human polarized HepG2 cells to study host defense mechanisms against B. pseudomallei. We investigated whether B. pseudomallei is able to invade and survive within hepatocytes and whether bacterial replication is dependent on the B. pseudomallei type 3 or type 6 secretion systems. Finally, we addressed the role of cytokines in enhancing anti-B. pseudomallei activity in HepG2 cells and a possible contribution of nitric oxide and reactive oxygen species in the bactericidal activity against B. pseudomallei.
Materials and Methods
Materials
Cytochalasin D (CytoD), latrunculin B (LatB), jasplakinolide (Jasp), and nocodazole (Noco) were obtained from Enzo Life Sciences (Lörrach, Germany). Catalase–polyethylene glycol (CAT–PEG), superoxide dismutase–polyethylene glycol (SOD–PEG), N-acetyl-cysteine (NAC), aminoguanidine (AG), and colchicine (Col) were from Sigma Aldrich (Taufkirchen, Germany). hIFNγ was purchased from Miltenyi Biotec GmbH (Bergisch Gladbach, Germany), and both mIL-1β and mTNFα were from Roche (Mannheim, Germany). Apocynin (Apo) was obtained from Calbiochem (Darmstadt, Germany).
Bacterial Strains
Burkholderia pseudomallei wild-type strain E8 comprises a soil isolate from the surrounding area of Ubon Ratchathani in northeast Thailand (Wuthiekanun et al., 1996) and was used throughout the study. T3SS-3 mutant ΔBPSS1539 (16:48) and T6SS-1 mutant ΔBPSS1509 (5:45) were generated by Tn5-OT182 mutagenesis of B. pseudomallei E8 as previously described (Pilatz et al., 2006). Bacteria were grown on Columbia agar at 37°C for 24 h and adjusted to the desired concentration in Dulbecco’s phosphate-buffered saline (D-PBS; Invitrogen, Darmstadt, Germany) or the respective cell culture medium.
Cell Culture and Infection of HepG2 Cells
Human hepatocellular carcinoma HepG2 cells were obtained from the DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany). Cells were cultured in RPMI 1640 medium (Biochrom AG, Berlin, Germany) supplemented with 10% fetal calf serum (PAA Laboratories GmbH, Cölbe, Germany) at 37°C in a humidified atmosphere containing 95% air and 5% CO2.
Twenty-four hours prior to infection, cells were seeded in 48 well plates (1.5 × 105 cells per well), grown to 80% confluence, and infected with B. pseudomallei strain E8 at the indicated multiplicity of infection (MOI). For invasion assays well plates were additionally centrifuged for 4 min at 120 × g. After infection for 30 min cells were washed twice with D-PBS and incubated in kanamycin (250 μg/ml) containing medium to eliminate remaining extracellular bacteria. To minimize re-infection and extracellular replication the culture medium was replaced by fresh medium containing 125 μg/ml kanamycin 6 h after infection. At indicated time points (time zero was taken 25 min after incubation under antibiotic-containing medium) the number of intracellular colony forming units (CFU) was determined. Consequently, cells were washed twice with D-PBS and subsequently lysed using 150 μl D-PBS containing 0.5% Tergitol TMN (Fluka, Buchs, Switzerland) and 1% bovine serum albumin (BSA) per well. After 15 min of incubation appropriate dilutions of lysates were plated on Mueller–Hinton agar and incubated at 37°C for 48 h.
Activation of HepG2 cells was performed using 500 ng/ml IFNγ, 50–200 U/ml IL-1β, or 10 ng/ml TNFα 24 h prior to infection. For in vitro inhibition of inducible nitric oxide synthase (iNOS), NADPH oxidase, or ROS generation, HepG2 cells were treated by adding 2 mM aminoguanidine (24 h), 500 μM apocynin, or 20 U/ml superoxide dismutase, 200 U/ml catalase, and 200 μM N-acetyl-cysteine or corresponding vehicle into the culture medium 1 h (unless otherwise indicated) prior to infection and during the incubation with kanamycin-containing medium. For in vitro inhibition of the actin cytoskeleton or the microtubules, HepG2 were incubated for 1 h with 1 μM cytochalasin D, 0.1 μM latrunculin B, 0.5 μM jasplakinolide, 10 μM nocodazole, 5 μM colchicine, or corresponding vehicle, followed by infection with B. pseudomallei. All inhibitors and vehicles were kept in the infection medium throughout the experiment.
Immunofluorescence Staining
Twenty-four hours prior to infection, HepG2 cells were seeded on collagen type I coated cover slips in 24 well plates (2 × 105 cells per well) and infected with B. pseudomallei strain E8 at a MOI of 400 by centrifugation of the well plates for 4 min at 120 × g. After infection for 30 min cells were washed twice with D-PBS and incubated in kanamycin (250 μg/ml) containing medium to eliminate remaining extracellular bacteria. At indicated time points HepG2 cells were washed with PBS, incubated for 10 min in ice-cold methanol, and washed three times with IF buffer [0.2% (w/v) BSA, 0.05% (w/v) saponin, 0.1% (w/v) sodium azide in PBS, pH 7.4]. To block non-specific antibody binding, cells were incubated for up to 1 h in IF buffer followed by an overnight incubation at 4°C in a humidity chamber with monoclonal mouse anti-B. pseudomallei 3015 γ2b (1:2000; Pilatz et al., 2006) and polyclonal rabbit anti-β-actin (1:100; Cell Signaling, Frankfurt am Main, Germany) or monoclonal mouse anti-lysosomal-associated membrane protein-1 (anti-LAMP-1) γ1 (1:400; BD Biosciences, Heidelberg, Germany) antibodies. After a wash with IF buffer, the immunoreacted primary antibodies were visualized with green fluorescent Alexa Fluor 488 anti-mouse IgG2b (1:800; Invitrogen, Darmstadt, Germany) and red fluorescent Cy3-conjugated goat anti-rabbit IgG (1:400; Dianova, Hamburg, Germany) or red fluorescent Alexa Fluor 568 anti-mouse IgG1 (1:800; Invitrogen) by incubation for 1 h at room temperature in the dark. After another wash with IF buffer, slices were covered with Fluorprep (bioMérieux, Nürtingen, Germany) and observed by fluorescent microscopy with a BZ-9000 microscope (Keyence Corporation, Neu-Isenburg, Germany).
LDH Assay
To quantify the extent of cell damage after infection, release of lactate dehydrogenase (LDH) in cell culture supernatants was determined. HepG2 cells were seeded in 96 well plates (3.75 × 104 cells per well) and infected at the indicated MOI with B. pseudomallei for 30 min. Cells were washed twice with D-PBS, and 100 μl of medium containing 250 μg/ml of kanamycin was added to each well to eliminate extracellular bacteria. At the indicated time points, cell culture supernatant was collected, and LDH activity was detected by using the CytoTox-One homogeneous membrane integrity assay (Promega, Mannheim, Germany) according to the manufacturer’s instructions. Briefly, 50 μl of supernatant was added to the kit reagent and incubated for 10 min. After addition of stopping solution, the fluorescence intensity was measured using the microplate reader Infinite M200 PRO (Tecan, Crailsheim, Germany) at excitation wavelength of 560 nm and emission wavelength of 590 nm.
Real-Time Cell Analysis
HepG2 cells were seeded in 96 well E-plates (3 × 104 cells per well), grown for 24 h, and then infected at the indicated MOI with B. pseudomallei for 30 min followed by incubation in kanamycin (250 μg/ml) containing medium. Cellular events were monitored in real-time using the xCELLigence system according to the manufacturer’s instructions (Roche, Mannheim, Germany). The system measures electrical impedance across gold microelectrodes integrated into the bottom of tissue culture E-plates providing real-time, quantitative information about the biological status of the cells, including cell number, viability, morphology, and degree of cell adhesion. In the absence of cells on the electrode surface, the electrical impedance describes only the background. Changes in impedance are dependent on either the number of cells attached to the electrodes or the dimensional change of attached cells on the electrode surface. The xCELLigence system detects changes in impedance and calculates them as dimensionless parameter termed Cell Index: CI = (Zi − Z0)/15 where Zi is the impedance at an individual time point of the experiment and Z0 describes the background measurement at the beginning of the experiment. To evaluate the impact of B. pseudomallei on HepG2 cells, the normalized cell index (NCIti) was used. Consequently, all selected wells were set on impedance value of 1 at a given time point (time of infection), and all further values were calculated as the cell index at a given time point (CIti) divided by the Cell Index at the normalization time point (CInml_time).
Data Presentation and Statistical Analysis
Figures and statistical analysis were performed using GraphPad Prism, version 4.0. Student’s t-test was used to detect statistically significant differences in the intracellular bacterial numbers. p-Values of <0.05 were considered to be statistically significant.
Results
Burkholderia pseudomallei is Able to Invade and Replicate within HepG2 Cells
The invasion and intracellular replication of B. pseudomallei in hepatocytes or hepatocyte cell lines has not been investigated so far. Therefore, we first investigated the invasion and the course of the intracellular bacterial burden in human hepatocellular carcinoma HepG2 cells after infection with B. pseudomallei wild-type strain E8 at a MOI of 29 and 599, respectively. As shown in Figure 1A, B. pseudomallei was able to enter HepG2 cells. Within the first 3 h after infection, a strong reduction in Burkholderia counts could be observed which indicates that effective bactericidal defense mechanisms are activated in HepG2 cells in the early phase of infection. Six hours after infection the intracellular bacterial burden increased, suggesting that B. pseudomallei is able to multiply within cells. After 24 h 10-fold more bacteria were found in Burkholderia-infected HepG2 cells compared to the beginning.
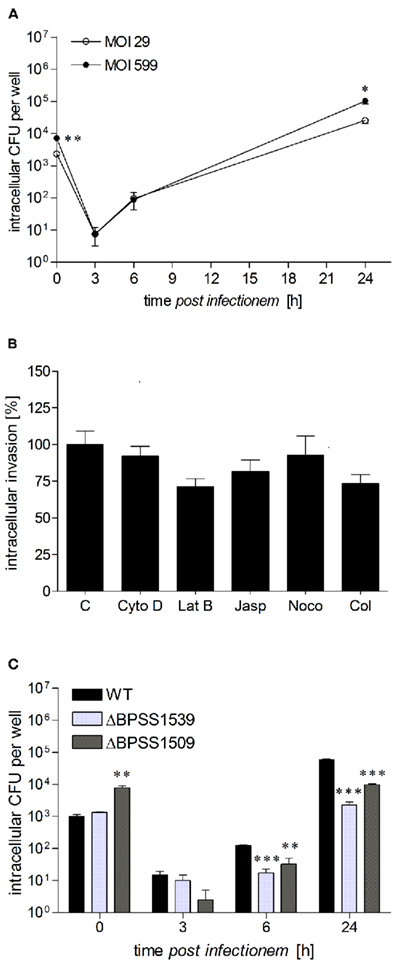
Figure 1. Burkholderia pseudomallei can multiply in human hepatocytes. (A) HepG2 cells were infected with B. pseudomallei E8 at multiplicities of infection of 29:1 (opened circles) and 599:1 (closed circles), and cells were lysed at different time points after infection to determine the number of viable intracellular bacteria by plating lysates on Mueller–Hinton agar. (B) Invasion of B. pseudomallei in HepG2 cells at a MOI of ~400:1 in the presence of inhibitors of actin filaments, including cytochalasin D (CytoD, 1 μM), latrunculin B (LatB, 0.1 μM), and jasplakinolide (Jasp, 0.5 μM) as well as inhibitors of microtubules, including nocodazole (Noco, 10 μM) and colchicine (Col, 5 μM). Invasion of B. pseudomallei in the absence of an inhibitor was set to 100%. (C) HepG2 cells were infected with either B. pseudomallei wild-type (WT), T3SS-3 mutant ΔBPSS1539, or T6SS-1 mutant ΔBPSS1509 using a MOI of ~40:1, and intracellular replication was monitored as above. Data are presented as mean with SEM of triplicate determinations. One representative experiment out of three independent experiments is shown. Statistical analyses were performed using a Student’s t-test (*p < 0.05, **p < 0.01, ***p < 0.001).
To provide some information about the mechanism used by B. pseudomallei to enter HepG2 cells, we analyzed the role of the host actin cytoskeleton as well as microtubules by using well defined inhibitors. As shown in Figure 1B, there was a tendency of actin cytoskeleton inhibitors (1 μM cytochalasin D, 0.1 μM latrunculin B, 0.5 μM jasplakinolide) as well as microtubule inhibitors (10 μM nocodazole, 5 μM colchicine) to reduce invasion of B. pseudomallei, as determined by enumeration of intracellular bacteria, although statistical significance was not reached. Therefore, our results indicate that the host actin cytoskeleton may contribute to B. pseudomallei uptake in HepG2 cells, but other invasion mechanisms may also play an important role.
As B. pseudomallei type 3 and type 6 secretion systems (T3SS, T6SS) are important for invasion of murine macrophages or human epithelial cells as well as for intracellular survival (Stevens et al., 2002, 2003; Burtnick et al., 2011), we analyzed whether they are also involved in the replication of B. pseudomallei in hepatocytes. Consequently, HepG2 cells were infected with B. pseudomallei wild-type strain E8, as well as a type 3 (ΔBPSS1539) and type 6 (ΔBPSS1509) secretion mutant, which have previously been shown to be attenuated in vivo (Pilatz et al., 2006). Surprisingly, mutant ΔBPSS1509, but not ΔBPSS1539 revealed a significantly higher invasion in HepG2 cells compared to the wild-type strain (Figure 1C). However, 24 h postinfection intracellular CFU counts were at least one log lower in cells infected with both mutant strains than in those infected with the wild-type strain. These results suggest that B. pseudomallei can infect and replicate in human hepatocytes and that the T3SS and T6SS contribute to optimal replication.
Burkholderia pseudomallei Can Escape from the Vacuole and Induce Formation of Actin Tails in HepG2 Cells
To further characterize the hepatocyte–pathogen-interaction we examined the intracellular localization of B. pseudomallei wild-type, T3SS, and T6SS mutant strains in HepG2 cells relative to LAMP-1 containing vacuoles as well as actin tail formation. Immunofluorescence microscopy revealed that 8 h after infection the wild-type was rarely seen co-localized with regions of LAMP-1 staining (Figure 2A), however a part of the B. pseudomallei population was able to induce actin tails (data not shown). Sixteen hours after infection the majority of bacteria displayed long actin tails (Figure 2B). In contrast, T3SS mutant ΔBPSS1539 was almost exclusively observed in association with LAMP-1, suggesting that the mutant is unable to escape from endocytotic vesicles 8 h after infection (Figure 2A). In accordance with our recent study using HeLa cells (Pilatz et al., 2006) this mutant was still able to multiply within the phagosomes of HepG2 cells, leading to large vesicles filled with bacteria. As B. pseudomallei was trapped within the phagosomes, no actin tail formation could be observed at this time point (data not shown). However, 16 h after infection some of these huge vacuoles seemed to become leaky and B. pseudomallei ΔBPSS1539 could be detected within the cytosol in association with actin tails (Figure 2B). As the wild-type the T6SS mutant ΔBPSS1509 was rarely seen associated with LAMP-1 8 h after infection, but actin tails could not be detected at this time point (data not shown). However, 16 h after infection the mutant was able to induce actin tails, but to a lesser degree compared to the wild-type (data not shown).
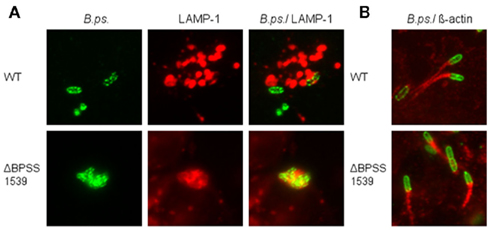
Figure 2. Burkholderia pseudomallei is able to escape from the endocytotic vesicle and to form actin tails in hepatocytes. HepG2 cells were infected with either B. pseudomallei wild-type (WT) or T3SS-3 mutant ΔBPSS1539 using a MOI of ~400:1 and analyzed by immunofluorescence microscopy (magnification ×1000). (A) Representative micrographs showing the association of intracellular B. pseudomallei with LAMP-1-containing vacuoles 8 h after infection. Bacteria were stained green with mouse anti-B. pseudomallei EPS (3015 γ2b) and Alexa Fluor 488 anti-mouse IgG2b antibodies. LAMP-1 was stained red with mouse anti-LAMP-1 γ1 and Alexa Fluor 568 anti-mouse IgG1 antibodies. (B) Representative micrographs showing the formation of actin tails by B. pseudomallei 16 h after infection. Bacteria were stained green with mouse anti-B. pseudomallei EPS (3015 γ2b) and Alexa Fluor 488 anti-mouse IgG2b antibodies. Actin tails were stained red with rabbit anti-β-actin and Cy3-conjugated goat anti-rabbit IgG antibodies.
Burkholderia pseudomallei Infection Induces Cell Damage of HepG2 Cells
To determine whether B. pseudomallei affects the viability of hepatocytes, cytosolic LDH activity was measured in cell culture supernatants of infected and control HepG2 cells at 24 h postinfection as an indicator of cytotoxicity. Figure 3A demonstrates that infection of HepG2 cells with B. pseudomallei wild-type strain E8 induced only a small MOI-dependent release of LDH compared to uninfected cells. However, this slight cytotoxicity was not found to be mediated by activation of apoptotic caspases 3 and 9 using Western blot analysis (data not shown).
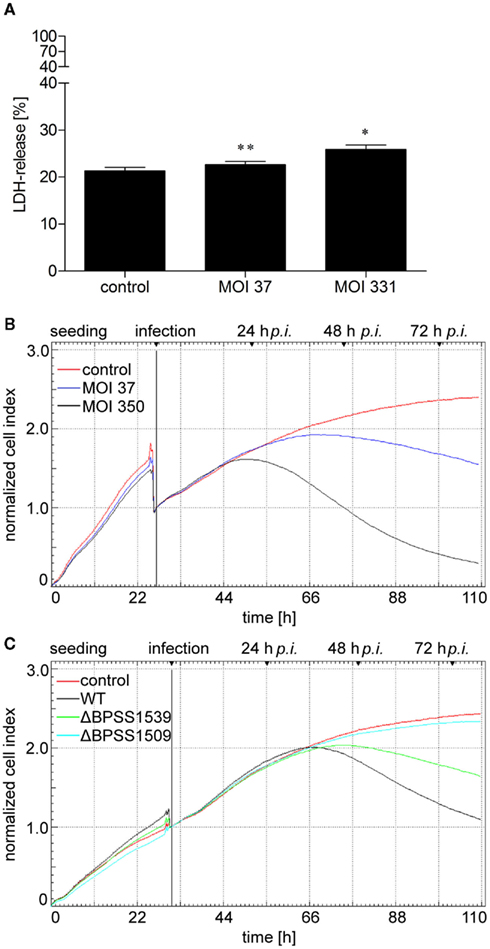
Figure 3. Burkholderia pseudomallei infection leads to cell damage of hepatocytes. (A) HepG2 cells were infected with B. pseudomallei E8 at multiplicities of infection of 37:1 and 331:1, respectively, and lactate dehydrogenase (LDH) release was determined 24 h after infection. Values are expressed as percentages relative to the LDH activity in supernatants from the 100% lysis control. Data are presented as mean with SEM of triplicate determinations. One representative experiment out of three independent experiments is shown. Statistical analyses were performed using a Student’s t-test (*p < 0.05, **p < 0.01). (B,C) HepG2 cells were seeded on 96 well E-plates and continuously monitored using the xCELLigence system. At the indicated time point, cells were infected with (B) B. pseudomallei at MOI of 37:1 and 350:1, respectively, or (C) B. pseudomallei wild-type (WT), T3SS-3 mutant ΔBPSS1539, or T6SS-1 mutant ΔBPSS1509 at MOI of ~400:1, and cell response was monitored every 30 min by impedance measurement. One representative experiment out of three independent experiments is shown.
Since B. pseudomallei infection led to detachment of hepatocytes after 24 h in a MOI-dependent manner as determined by microscopic analyses (data not shown), we aimed to quantify these cellular damages by performing electrical impedance measurements providing real-time information about the biological status of hepatocytes such as viability, morphology, and degree of cell adhesion. Such an experimental approach has recently been described for meningococcal infection in human brain microvascular endothelial cells (Slanina et al., 2011). HepG2 cells were grown for 24 h leading to an increase of the Normalized Cell Index (Figure 3B). Infection with B. pseudomallei wild-type strain E8 at MOI of 37 or 350 resulted in a transient decrease of the corresponding Cell Index caused by the cell culture medium exchange. Due to the proliferation and spreading of hepatocytes, non-infected cells led to enhanced electrical impedance within 110 h. In contrast, infection of HepG2 cells with B. pseudomallei at a MOI of 37 resulted in a loss of electrical impedance 36 h after infection, whereas infection at a MOI of 350 led to a loss of electrical impedance already 24 h postinfection (Figure 3B), correlating with the low LDH release after 24 h (Figure 3A). To exclude that the impedance was influenced by bacterial overload of the electrodes, electrical impedance was investigated in response to bacterial growth over the same period. However, no effect on the Cell Index could be observed when bacteria alone were grown in the wells (data not shown).
To determine whether reduced intracellular growth of T3SS and T6SS mutants correlates with an increased viability of hepatocytes, we measured the electrical impedance after infection of HepG2 cells with B. pseudomallei wild-type as well as the T3SS (ΔBPSS1539) and T6SS (ΔBPSS1509) mutants at a MOI of ~400. Figure 3C illustrates that the electrical impedance of HepG2 cells after infection with the T6SS mutant ΔBPSS1509 did not differ significantly from the one of uninfected cells whereas infection with the wild-type strain resulted in a strong loss of electrical impedance. The decrease of the Normalized Cell Index observed in HepG2 cells infected with the T3SS mutant ΔBPSS1539 was significantly delayed compared to wild-type infected cells. Our data indicate that B. pseudomallei induces cell damage of HepG2 cells depending on a functional T3SS and T6SS.
Interferon γ Increases Antibacterial Activity of HepG2 Cells Against B. pseudomallei
IFNγ is crucial for in vivo resistance against B. pseudomallei (Santanirand et al., 1999; Breitbach et al., 2006) and has been shown to restrict replication of B. pseudomallei in macrophages (Miyagi et al., 1997; Utaisincharoen et al., 2001). We therefore compared the intracellular growth of B. pseudomallei between unstimulated or IFNγ-stimulated (500 ng/ml) HepG2 cells. Although invasion of B. pseudomallei into HepG2 cells was comparable, already 3 and 6 h after infection intracellular bacterial load was reduced in IFNγ-activated cells, which was most prominent after 24 h (Figure 4A). These data indicate that IFNγ is able to increase the antibacterial capacity of HepG2 cells against intracellular B. pseudomallei in the early and late phase of infection.
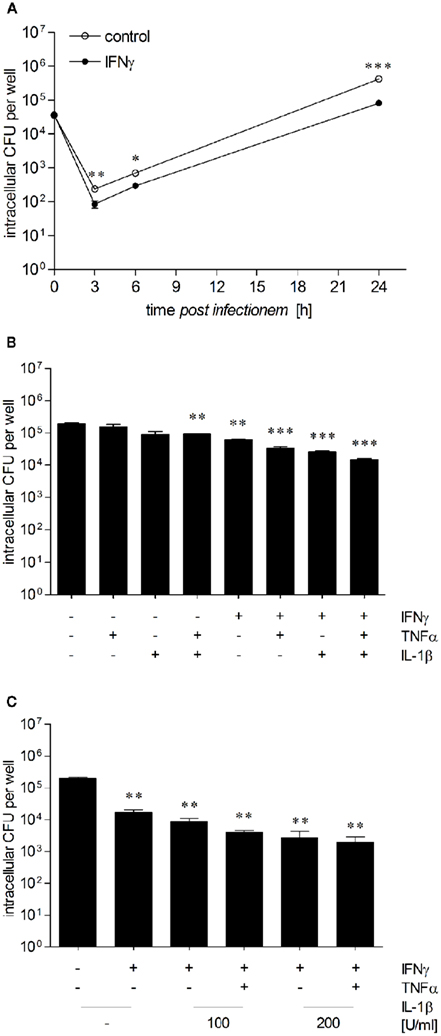
Figure 4. Proinflammatory cytokines increase anti-B. pseudomallei activity of hepatocytes. (A) Intracellular survival of B. pseudomallei E8 in unstimulated (opened circles) and IFNγ-stimulated (500 ng/ml; closed circles) HepG2 cells infected with an MOI of 36:1. (B) Intracellular survival of B. pseudomallei E8 24 h after infection (MOI 37:1) of HepG2 cells pretreated with IFNγ (500 ng/ml), IL-1β (50 U/ml), or TNFα (10 ng/ml) alone or in combination. (C) Intracellular survival of B. pseudomallei E8 24 h after infection (MOI 36:1) of HepG2 cells activated with IFNγ alone (500 ng/ml) or in combination with IL-1β (100–200 U/ml) and TNFα (10 ng/ml). Data are presented as mean with SEM of triplicate determinations. One representative experiment out of three independent experiments is shown. Statistical analyses were performed using a Student’s t-test (*p < 0.05, **p < 0.01, ***p < 0.001).
Proinflammatory Cytokines Enhance Antibacterial Activity of IFNγ-Activated HepG2 Cells
IFNγ, IL-1β, and TNFα have been detected in livers of BALB/c and C57BL/6 mice after B. pseudomallei infection (Ulett et al., 2000a,b), and numerous studies have shown that early cytokine responses are important for resistance to intracellular pathogens (Ehlers et al., 1992; Autenrieth et al., 1994; Bohn et al., 1994; Santanirand et al., 1999). To examine the impact of these inflammatory cytokines in controlling intracellular B. pseudomallei growth in hepatocytes, we analyzed the intracellular survival kinetics of B. pseudomallei in HepG2 cells in the presence of several cytokines. Therefore, HepG2 cells were stimulated with IFNγ (500 ng/ml), IL-1β (50–200 U/ml), TNFα (10 ng/ml), or a combination of these cytokines for 24 h followed by infection with B. pseudomallei. As shown in Figure 4B, IL-1β or TNFα alone had no or only a marginal effect on intracellular bacterial replication, whereas the combination of both cytokines caused a small, but significant growth inhibition of B. pseudomallei. In contrast, IFNγ alone reduced bacterial multiplication. This inhibitory effect was enhanced, when IFNγ was combined with either IL-1β or TNFα. The combination of all three cytokines produced a maintained decrease in intracellular bacterial growth compared to that observed in unstimulated controls (Figure 4C), suggesting that IFNγ, IL-1β, and TNFα are essential for the maximal activation of hepatocytes.
Inducible Nitric Oxide Synthase is not Essential to Eliminate B. pseudomallei
Previous studies provided evidence for a crucial role of NO in growth inhibition of B. pseudomallei in BALB/c macrophages, whereas NO did not play any role in killing activity of C57BL/6 macrophages (Breitbach et al., 2006, 2011). Thus, we investigated the role of iNOS-generated NO with respect to intracellular elimination of B. pseudomallei in HepG2 cells. First, we determined the amount of NO released from untreated and IFNγ-treated HepG2 cells 24 h after infection with B. pseudomallei. However, we could neither detect NO in cell culture supernatants nor iNOS protein expression in lysates of infected HepG2 cells (data not shown). To assess the contribution of the NO pathway in the antibacterial effect of activated HepG2 cells, we used the iNOS inhibitor aminoguanidine (2 mM). However, after treatment with the iNOS inhibitor and infection of resting or IFNγ-stimulated HepG2 cells, we could not observe any significant difference in intracellular killing of B. pseudomallei (Figure 5). This suggests that iNOS-derived NO is not necessary to eliminate B. pseudomallei in hepatocytes.
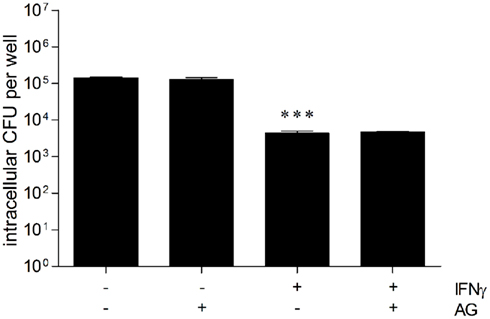
Figure 5. Inducible nitric oxide synthase (iNOS) is not necessary to eliminate B. pseudomallei in hepatocytes. Intracellular replication of B. pseudomallei E8 24 h after infection (MOI 40:1) of resting and IFNγ-activated (500 ng/ml) HepG2 cells in the presence of iNOS inhibitor aminoguanidine (AG, 2 mM). Data are presented as mean with SEM of triplicate determinations. One representative experiment out of three independent experiments is shown. Statistical analyses were performed using a Student’s t-test (***p < 0.001).
NADPH Oxidase is not Involved in the Control of B. pseudomallei Infection
In a recent study we demonstrated that NADPH oxidase-mediated mechanisms contribute to early resistance in C57BL/6 mice in vivo and in BMM (Breitbach et al., 2006). Consequently, we investigated the role of NADPH oxidase in the intracellular B. pseudomallei elimination of hepatocytes. For this reason unstimulated and IFNγ-stimulated HepG2 cells were pretreated with apocynin (500 μM), a potent and selective inhibitor of NADPH oxidase, followed by infection with B. pseudomallei. In spite of a significantly higher invasion of apocynin-inhibited cells (data not shown), our experiments indicated that bacterial growth of B. pseudomallei was not impaired by apocynin (Figure 6), suggesting that, in contrast to primary murine macrophages, NADPH oxidase does not contribute to antibacterial activity of human hepatocyte cells.
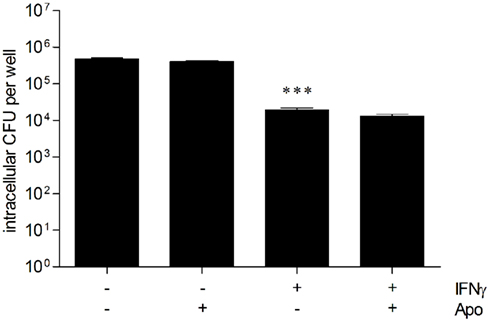
Figure 6. NADPH oxidase is not important for the control of B. pseudomallei hepatocyte infection. Intracellular survival of B. pseudomallei E8 24 h after infection (MOI 31:1) of resting and IFNγ-activated (500 ng/ml) HepG2 cells in the presence of NADPH oxidase inhibitor apocynin (Apo, 500 μM). Data are presented as mean with SEM of triplicate determinations. One representative experiment out of three independent experiments is shown. Statistical analyses were performed using a Student’s t-test (***p < 0.001).
Scavengers/Inhibitors of Reactive Oxygen Species Do not Inhibit the Antibacterial Activity
To further study the involvement of reactive oxygen species as effector molecules of the antibacterial activity of hepatocytes, unstimulated, and IFNγ-stimulated HepG2 cells were incubated with the free radical scavengers catalase (CAT, 200 U/ml), superoxide dismutase (SOD, 20 U/ml), and/or N-acetyl-cysteine (NAC, 200 μM) prior to infection with B. pseudomallei. Figure 7 shows that treatment of IFNγ-stimulated HepG2 cells with catalase and SOD slightly, but not significantly reversed the antibacterial activity. The effect of both antioxidant enzymes added at the same time was not further enhanced by addition of NAC. However, treatment of resting HepG2 cells did not increase the number of CFU recovered, indicating that basal, ROS-mediated antibacterial mechanisms do not exist in hepatocytes. Our results imply that reactive oxygen species do not play an important role in the control of B. pseudomallei replication.
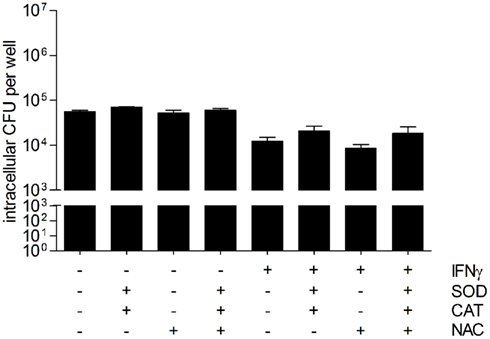
Figure 7. Scavengers/inhibitors of reactive oxygen intermediates do not inhibit the antibacterial activity of hepatocytes. Intracellular survival of B. pseudomallei E8 24 h after infection (MOI 42:1) of unstimulated and IFNγ-stimulated (500 ng/ml) HepG2 cells in the presence of antioxidants superoxide dismutase (SOD, 20 U/ml), catalase (CAT, 200 U/ml), and N-acetyl-cysteine (NAC, 200 μM). Data are presented as mean with SEM of triplicate determinations. One representative experiment out of three independent experiments is shown. Statistical analyses were performed using a Student’s t-test.
Discussion
The liver consists of both hepatocytes and different types of non-parenchymal cells including endothelial sinusoidal cells, Kupffer cells, and other immune cells. Although hepatocytes comprise 60% of cells in the liver and 80% of the hepatic volume (Malarkey et al., 2005), their role in antimicrobial defense is poorly understood. As the liver is among the most commonly affected organs in melioidosis, the present study was designed to examine the contribution of hepatocytes in host defense against B. pseudomallei.
To our knowledge, this is the first study showing that B. pseudomallei can infect and replicate within HepG2 cells (Figure 1A). The intracellular multiplication in hepatocytes is in accordance with the well-known ability of B. pseudomallei to proliferate inside several cell types, including phagocytic and non-phagocytic cells (Jones et al., 1996; Harley et al., 1998; Kespichayawattana et al., 2000). In addition, our results are consistent with other recently published data describing that intracellular L. monocytogenes (Gregory and Wing, 1993; Gregory et al., 1993; Wood et al., 1993; Szalay et al., 1995), S. typhimurium (Lajarin et al., 1996), and Brucella abortus (Delpino et al., 2010) are able to invade and multiply within primary hepatocytes and hepatoma cells, respectively.
Several in vitro studies have demonstrated that B. pseudomallei T3SS-3 facilitates bacterial invasion of non-phagocytic cells (Stevens et al., 2003), contributes to escape from endosomes, intracellular survival, and replication (Stevens et al., 2002), and is necessary for the induction of caspase-1-dependent cell death in phagocytic cells (Sun et al., 2005). It has been reported that BsaU seems to be essential for control of needle length in T3SS-3 secretion apparatus (Sun and Gan, 2010) and important for the secretion of the T3SS-3 translocator protein BipD as well as the effector protein BopE (Hii et al., 2008). We report here that the intracellular growth of B. pseudomallei in hepatocytes is partially dependent on a functional T3SS, since mutant ΔBPSS1539 (BsaU) exhibited reduced intracellular replication (Figure 1C) as well as decreased cell damage (Figure 3C) in HepG2 cells. In agreement with our previous study using HeLa cells (Pilatz et al., 2006), we found here that the transposon mutant of BsaU is unable to escape from the endocytotic vesicle after invasion, but is still able to proliferate within the vacuole (Figure 2A). We previously described that mutant ΔBPSS1509, encoded within the B. pseudomallei T6SS-1 cluster, displays no intracellular growth defect in J774A.1 macrophages and HeLa cells, but is required for virulence in mice (Pilatz et al., 2006). In contrast, the present study indicates that mutant ΔBPSS1509 reveals delayed and reduced induction of actin polymerization (data not shown) as well as decreased intracellular replication (Figure 1C) and reduced cell damage (Figure 3C) in hepatocytes compared to the wild-type. These results are in line with recent reports showing that different T6SS-1 mutants of B. pseudomallei exhibit reduced intracellular growth and cytotoxicity in RAW264.7 macrophages (Burtnick et al., 2011; Chen et al., 2011). Thus, our results suggest that BPSS1539 encoded by T3SS-3 and BPSS1509 encoded by T6SS-1 are potential virulence determinants that enable intracellular growth of B. pseudomallei and induce cell damage in hepatocytes. However, these factors are unlikely the main players responsible for intracellular survival in these cells, since HepG2 cells were not able to eliminate the corresponding mutants.
Hepatocytes have been shown to express a variety of cytokines (Rowell et al., 1997; Stonans et al., 1999) and to respond to bacterial or viral infections with enhanced secretion of inflammatory cytokines and chemokines (Rowell et al., 1997; Santos et al., 2005). Since it has been shown that numerous cytokines can modulate resistance to intracellular bacterial infections, primarily by stimulating antimicrobial activities (Ehlers et al., 1992; Autenrieth et al., 1994; Bohn et al., 1994; Santanirand et al., 1999), we investigated the impact of cytokine-activated hepatocytes in intracellular B. pseudomallei clearance. IFNγ was previously shown to be a major factor in primary host defense to B. pseudomallei (Santanirand et al., 1999; Breitbach et al., 2006). In addition, several reports described that both primary murine hepatocytes and hepatocyte cell lines are efficiently activated by IFNγ to kill intracellular L. monocytogenes or S. typhimurium (Gregory and Wing, 1993; Lajarin et al., 1996). This study, demonstrating that IFNγ is capable to enhance the anti-Burkholderia activity of HepG2 cells (Figure 4A) argues for a potential role of activated hepatoma cells in host defense against this pathogen. Although IL-1β and TNFα alone failed to significantly activate antibacterial activity of HepG2 cells, the combination of both cytokines together with IFNγ caused the most prominent growth inhibition in hepatoma cells (Figures 4B,C). These results are in line with several reports indicating that murine hepatocytes and hepatoma cell lines stimulated with a combination of IFNγ, TNFα, IL-6, IL-1β, or LPS exhibited the strongest antibacterial activity against L. monocytogenes (Szalay et al., 1995; Haponsaph and Czuprynski, 1996) and S. typhimurium (Lajarin et al., 1996, 1999). Therefore, our findings suggest that inflammatory cytokines, particularly IFNγ, are important for resistance of hepatocytes against B. pseudomallei.
However, the mechanism responsible for the anti-B. pseudomallei activity of cytokine-activated hepatoma cells remains unclear. In a previous study we could show that iNOS-generated NO contributes to resistance in innate susceptible BALB/c mice in a murine model of melioidosis as demonstrated by enhanced mortality rates and higher bacterial loads in liver and spleen of aminoguanidine-treated mice compared to untreated control mice (Breitbach et al., 2011). Moreover, inhibition of iNOS in IFNγ-stimulated and B. pseudomallei-infected BALB/c-derived macrophage cell lines revealed that reactive nitrogen species are involved in restricting the intracellular growth of B. pseudomallei (Miyagi et al., 1997; Utaisincharoen et al., 2001, 2003). Our present data indicate, however, that reactive nitrogen species do not play any role in controlling B. pseudomallei growth in hepatocytes since inhibition of NO production by aminoguanidine did not reverse the antibacterial activity of HepG2 cells (Figure 5). These results are in good accordance with reports demonstrating that elimination of L. monocytogenes (Gregory et al., 1993; Szalay et al., 1995; Haponsaph and Czuprynski, 1996) and S. typhimurium (Lajarin et al., 1996) from activated hepatocytes is not mediated by nitric oxide.
Besides the production of NO, the generation of ROS is another potential mechanism in the control of bacterial pathogens (Inoue et al., 1995; Vazquez-Torres et al., 2000; Breitbach et al., 2006). In a previous study we demonstrated that NADPH oxidase is able to restrict intracellular replication of B. pseudomallei in primary murine macrophages (Breitbach et al., 2006). But the present findings suggest that neither NADPH oxidase (Figure 6) nor other ROS-generating enzymes (Figure 7) are involved in the anti-B. pseudomallei activity of activated hepatoma cells. In contrast, some reports provided evidence of ROS-dependent mechanisms in both primary murine hepatocytes and hepatocyte cell lines against intracellular L. monocytogenes (Gregory and Wing, 1993) or S. typhimurium (Lajarin et al., 1996) since antibacterial activity was inhibited by the presence of scavengers or inhibitors of reactive oxygen species. In this context, Lajarin et al. (1999) indicated that cyclooxygenases (COX) are a potential source of ROS in S. typhimurium infected murine hepatocyte cell lines, and thus contribute to antibacterial host defense. However, several studies revealed that COX are involved in intracellular replication and survival of different pathogens (Uchiya and Nikai, 2004; Sadikot et al., 2007; Serezani et al., 2007).
In summary, the present study describes the in vitro invasion and intracellular replication of B. pseudomallei in human hepatocyte HepG2 cells involving type 3 and type 6 secretion systems. We provide evidence that IFNγ contributes to growth restriction of B. pseudomallei whereas the combination with IL-1β and TNFα is necessary for the maximal antibacterial activity of hepatocytes. As the IFNγ-mediated effects are independent of NO and ROS production, we assume that other defense mechanisms are important to inhibit intracellular multiplication of B. pseudomallei. Thus, further investigations are required to elucidate the NO- and ROS-independent mechanisms responsible for restricting intracellular growth of B. pseudomallei in cytokine-activated hepatocytes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Claudia Weber and Eylin Topfstedt for excellent technical assistance.
Abbreviations
AG, aminoguanidine hemisulfate; Apocynin, 4-hydroxy-3-methoxyacetophenone; BMM, bone marrow-derived macrophages; CAT, catalase; CFU, colony forming units; COX, cyclooxygenase; EPS, exopolysaccharide; IFNγ, interferon γ; IL-1β, interleukin-1β; iNOS, inducible nitric oxide synthase; LAMP-1, lysosomal-associated membrane protein-1; LDH, lactate dehydrogenase; LPS, lipopolysaccharide; MOI, multiplicity of infection; NAC, N-acetyl-cysteine; NADPH oxidase, nicotinamide adenine dinucleotide phosphate oxidase; NO, nitric oxide; ROS, reactive oxygen species; SOD, superoxide dismutase; T3SS, type three secretion system; T6SS, type six secretion system; TNFα, tumor necrosis factor.
References
Autenrieth, I. B., Beer, M., Bohn, E., Kaufmann, S. H., and Heesemann, J. (1994). Immune responses to Yersinia enterocolitica in susceptible BALB/c and resistant C57BL/6 mice: an essential role for gamma interferon. Infect. Immun. 62, 2590–2599.
Bohn, E., Heesemann, J., Ehlers, S., and Autenrieth, I. B. (1994). Early gamma interferon mRNA expression is associated with resistance of mice against Yersinia enterocolitica. Infect. Immun. 62, 3027–3032.
Breitbach, K., Klocke, S., Tschernig, T., Van Rooijen, N., Baumann, U., and Steinmetz, I. (2006). Role of inducible nitric oxide synthase and NADPH oxidase in early control of Burkholderia pseudomallei infection in mice. Infect. Immun. 74, 6300–6309.
Breitbach, K., Rottner, K., Klocke, S., Rohde, M., Jenzora, A., Wehland, J., and Steinmetz, I. (2003). Actin-based motility of Burkholderia pseudomallei involves the Arp 2/3 complex, but not N-WASP and Ena/VASP proteins. Cell. Microbiol. 5, 385–393.
Breitbach, K., Wongprompitak, P., and Steinmetz, I. (2011). Distinct roles for nitric oxide in resistant C57BL/6 and susceptible BALB/c mice to control Burkholderia pseudomallei infection. BMC Immunol. 12, 20. doi:10.1186/1471-2172-12-20
Burtnick, M. N., Brett, P. J., Harding, S. V., Ngugi, S. A., Ribot, W. J., Chantratita, N., Scorpio, A., Milne, T. S., Dean, R. E., Fritz, D. L., Peacock, S. J., Prior, J. L., Atkins, T. P., and Deshazer, D. (2011). The cluster 1 type VI secretion system is a major virulence determinant in Burkholderia pseudomallei. Infect. Immun. 79, 1512–1525.
Burtnick, M. N., Brett, P. J., Nair, V., Warawa, J. M., Woods, D. E., and Gherardini, F. C. (2008). Burkholderia pseudomallei type III secretion system mutants exhibit delayed vacuolar escape phenotypes in RAW 264.7 murine macrophages. Infect. Immun. 76, 2991–3000.
Chen, Y., Wong, J., Sun, G. W., Liu, Y., Tan, G. Y., and Gan, Y. H. (2011). The regulation of type VI secretion system during Burkholderia pseudomallei infection. Infect. Immun. 79, 3064–3073.
Delpino, M. V., Barrionuevo, P., Scian, R., Fossati, C. A., and Baldi, P. C. (2010). Brucella-infected hepatocytes mediate potentially tissue-damaging immune responses. J. Hepatol. 53, 145–154.
Ehlers, S., Mielke, M. E., Blankenstein, T., and Hahn, H. (1992). Kinetic analysis of cytokine gene expression in the livers of naive and immune mice infected with Listeria monocytogenes. The immediate early phase in innate resistance and acquired immunity. J. Immunol. 149, 3016–3022.
French, C. T., Toesca, I. J., Wu, T. H., Teslaa, T., Beaty, S. M., Wong, W., Liu, M., Schroder, I., Chiou, P. Y., Teitell, M. A., and Miller, J. F. (2011). Dissection of the Burkholderia intracellular life cycle using a photothermal nanoblade. Proc. Natl. Acad. Sci. U.S.A. 108, 12095–12100.
Gong, L., Cullinane, M., Treerat, P., Ramm, G., Prescott, M., Adler, B., Boyce, J. D., and Devenish, R. J. (2011). The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS ONE 6, e17852. doi:10.1371/journal.pone.0017852
Gregory, S. H., and Wing, E. J. (1993). IFN-gamma inhibits the replication of Listeria monocytogenes in hepatocytes. J. Immunol. 151, 1401–1409.
Gregory, S. H., Wing, E. J., Hoffman, R. A., and Simmons, R. L. (1993). Reactive nitrogen intermediates suppress the primary immunologic response to Listeria. J. Immunol. 150, 2901–2909.
Haponsaph, R., and Czuprynski, C. J. (1996). Inhibition of the multiplication of Listeria monocytogenes in a murine hepatocyte cell line (ATCC TIB73) by IFN-gamma and TNF-alpha. Microb. Pathog. 20, 287–295.
Harley, V. S., Dance, D. A., Drasar, B. S., and Tovey, G. (1998). Effects of Burkholderia pseudomallei and other Burkholderia species on eukaryotic cells in tissue culture. Microbios 96, 71–93.
Hii, C. S., Sun, G. W., Goh, J. W., Lu, J., Stevens, M. P., and Gan, Y. H. (2008). Interleukin-8 induction by Burkholderia pseudomallei can occur without Toll-like receptor signaling but requires a functional type III secretion system. J. Infect. Dis. 197, 1537–1547.
Hoppe, I., Brenneke, B., Rohde, M., Kreft, A., Haussler, S., Reganzerowski, A., and Steinmetz, I. (1999). Characterization of a murine model of melioidosis: comparison of different strains of mice. Infect. Immun. 67, 2891–2900.
Inoue, S., Itagaki, S., and Amano, F. (1995). Intracellular killing of Listeria monocytogenes in the J774.1 macrophage-like cell line and the lipopolysaccharide (LPS)-resistant mutant LPS1916 cell line defective in the generation of reactive oxygen intermediates after LPS treatment. Infect. Immun. 63, 1876–1886.
Jones, A. L., Beveridge, T. J., and Woods, D. E. (1996). Intracellular survival of Burkholderia pseudomallei. Infect. Immun. 64, 782–790.
Kespichayawattana, W., Rattanachetkul, S., Wanun, T., Utaisincharoen, P., and Sirisinha, S. (2000). Burkholderia pseudomallei induces cell fusion and actin-associated membrane protrusion: a possible mechanism for cell-to-cell spreading. Infect. Immun. 68, 5377–5384.
Lajarin, F., Rubio, G., Galvez, J., and Garcia-Penarrubia, P. (1996). Adhesion, invasion and intracellular replication of Salmonella typhimurium in a murine hepatocyte cell line. Effect of cytokines and LPS on antibacterial activity of hepatocytes. Microb. Pathog. 21, 319–329.
Lajarin, F., Rubio, G., Lorenzo, N., Gamiz, P., Hernandez-Caselles, T., and Garcia-Penarrubia, P. (1999). Implication of reactive oxygen species in the antibacterial activity against Salmonella typhimurium of hepatocyte cell lines. Free Radic. Biol. Med. 27, 1008–1018.
Liu, B., Koo, G. C., Yap, E. H., Chua, K. L., and Gan, Y. H. (2002). Model of differential susceptibility to mucosal Burkholderia pseudomallei infection. Infect. Immun. 70, 504–511.
Malarkey, D. E., Johnson, K., Ryan, L., Boorman, G., and Maronpot, R. R. (2005). New insights into functional aspects of liver morphology. Toxicol. Pathol. 33, 27–34.
Miyagi, K., Kawakami, K., and Saito, A. (1997). Role of reactive nitrogen and oxygen intermediates in gamma interferon-stimulated murine macrophage bactericidal activity against Burkholderia pseudomallei. Infect. Immun. 65, 4108–4113.
Muangsombut, V., Suparak, S., Pumirat, P., Damnin, S., Vattanaviboon, P., Thongboonkerd, V., and Korbsrisate, S. (2008). Inactivation of Burkholderia pseudomallei bsaQ results in decreased invasion efficiency and delayed escape of bacteria from endocytic vesicles. Arch. Microbiol. 190, 623–631.
Pilatz, S., Breitbach, K., Hein, N., Fehlhaber, B., Schulze, J., Brenneke, B., Eberl, L., and Steinmetz, I. (2006). Identification of Burkholderia pseudomallei genes required for the intracellular life cycle and in vivo virulence. Infect. Immun. 74, 3576–3586.
Rowell, D. L., Eckmann, L., Dwinell, M. B., Carpenter, S. P., Raucy, J. L., Yang, S. K., and Kagnoff, M. F. (1997). Human hepatocytes express an array of proinflammatory cytokines after agonist stimulation or bacterial invasion. Am. J. Physiol. 273, G322–G332.
Sadikot, R. T., Zeng, H., Azim, A. C., Joo, M., Dey, S. K., Breyer, R. M., Peebles, R. S., Blackwell, T. S., and Christman, J. W. (2007). Bacterial clearance of Pseudomonas aeruginosa is enhanced by the inhibition of COX-2. Eur. J. Immunol. 37, 1001–1009.
Santanirand, P., Harley, V. S., Dance, D. A., Drasar, B. S., and Bancroft, G. J. (1999). Obligatory role of gamma interferon for host survival in a murine model of infection with Burkholderia pseudomallei. Infect. Immun. 67, 3593–3600.
Santos, S. A., Andrade, D. R., and Andrade Junior, D. R. (2005). Rat hepatocyte invasion by Listeria monocytogenes and analysis of TNF-alpha role in apoptosis. Rev. Inst. Med. Trop. Sao Paulo 47, 73–80.
Serezani, C. H., Chung, J., Ballinger, M. N., Moore, B. B., Aronoff, D. M., and Peters-Golden, M. (2007). Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am. J. Respir. Cell Mol. Biol. 37, 562–570.
Slanina, H., Konig, A., Claus, H., Frosch, M., and Schubert-Unkmeir, A. (2011). Real-time impedance analysis of host cell response to meningococcal infection. J. Microbiol. Methods 84, 101–108.
Stevens, M. P., Friebel, A., Taylor, L. A., Wood, M. W., Brown, P. J., Hardt, W. D., and Galyov, E. E. (2003). A Burkholderia pseudomallei type III secreted protein, BopE, facilitates bacterial invasion of epithelial cells and exhibits guanine nucleotide exchange factor activity. J. Bacteriol. 185, 4992–4996.
Stevens, M. P., Stevens, J. M., Jeng, R. L., Taylor, L. A., Wood, M. W., Hawes, P., Monaghan, P., Welch, M. D., and Galyov, E. E. (2005). Identification of a bacterial factor required for actin-based motility of Burkholderia pseudomallei. Mol. Microbiol. 56, 40–53.
Stevens, M. P., Wood, M. W., Taylor, L. A., Monaghan, P., Hawes, P., Jones, P. W., Wallis, T. S., and Galyov, E. E. (2002). An Inv/Mxi-Spa-like type III protein secretion system in Burkholderia pseudomallei modulates intracellular behaviour of the pathogen. Mol. Microbiol. 46, 649–659.
Stonans, I., Stonane, E., Russwurm, S., Deigner, H. P., Bohm, K. J., Wiederhold, M., Jager, L., and Reinhart, K. (1999). HepG2 human hepatoma cells express multiple cytokine genes. Cytokine 11, 151–156.
Sun, G. W., and Gan, Y. H. (2010). Unraveling type III secretion systems in the highly versatile Burkholderia pseudomallei. Trends Microbiol. 18, 561–568.
Sun, G. W., Lu, J., Pervaiz, S., Cao, W. P., and Gan, Y. H. (2005). Caspase-1 dependent macrophage death induced by Burkholderia pseudomallei. Cell. Microbiol. 7, 1447–1458.
Szalay, G., Hess, J., and Kaufmann, S. H. (1995). Restricted replication of Listeria monocytogenes in a gamma interferon-activated murine hepatocyte line. Infect. Immun. 63, 3187–3195.
Uchiya, K., and Nikai, T. (2004). Salmonella enterica serovar typhimurium infection induces cyclooxygenase 2 expression in macrophages: involvement of Salmonella pathogenicity island 2. Infect. Immun. 72, 6860–6869.
Ulett, G. C., Ketheesan, N., and Hirst, R. G. (2000a). Cytokine gene expression in innately susceptible BALB/c mice and relatively resistant C57BL/6 mice during infection with virulent Burkholderia pseudomallei. Infect. Immun. 68, 2034–2042.
Ulett, G. C., Ketheesan, N., and Hirst, R. G. (2000b). Proinflammatory cytokine mRNA responses in experimental Burkholderia pseudomallei infection in mice. Acta Trop. 74, 229–234.
Utaisincharoen, P., Anuntagool, N., Limposuwan, K., Chaisuriya, P., and Sirisinha, S. (2003). Involvement of beta interferon in enhancing inducible nitric oxide synthase production and antimicrobial activity of Burkholderia pseudomallei-infected macrophages. Infect. Immun. 71, 3053–3057.
Utaisincharoen, P., Tangthawornchaikul, N., Kespichayawattana, W., Chaisuriya, P., and Sirisinha, S. (2001). Burkholderia pseudomallei interferes with inducible nitric oxide synthase (iNOS) production: a possible mechanism of evading macrophage killing. Microbiol. Immunol. 45, 307–313.
Vazquez-Torres, A., Jones-Carson, J., Mastroeni, P., Ischiropoulos, H., and Fang, F. C. (2000). Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. I. Effects on microbial killing by activated peritoneal macrophages in vitro. J. Exp. Med. 192, 227–236.
Wood, S., Maroushek, N., and Czuprynski, C. J. (1993). Multiplication of Listeria monocytogenes in a murine hepatocyte cell line. Infect. Immun. 61, 3068–3072.
Keywords: Burkholderia, cytoskeleton, hepatocytes, interferon γ, iNOS, NADPH oxidase, secretion system
Citation: Bast A, Schmidt IHE, Brauner P, Brix B Breitbach K and Steinmetz I (2012) Defense Mechanisms of Hepatocytes Against Burkholderia Pseudomallei. Front. Microbio. 2:277. doi: 10.3389/fmicb.2011.00277
Received: 26 July 2011;
Accepted: 24 December 2011;
Published online: 10 January 2012.
Edited by:
Alfredo G. Torres, University of Texas Medical Branch, USAReviewed by:
Jose A. Bengoechea, Fundacion Caubet – CIMERA Illes Balears, SpainYufeng Yao, Shanghai Jiao Tong University School of Medicine, China
Copyright: © 2012 Bast, Schmidt, Brauner, Brix, Breitbach and Steinmetz. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ivo Steinmetz, Friedrich Loeffler Institut für Medizinische Mikrobiologie, Universitätsmedizin Greifswald KdöR, Martin-Luther-Str. 6, 17475 Greifswald, Germany. e-mail: steinmetz.ivo@uni-greifswald.de