
- Department of Molecular Microbiology and Immunology, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA
Kaposi’s sarcoma-associated herpesvirus (KSHV), a member of the herpesvirus family, has evolved to establish a long-term, latent infection of cells such that while they carry the viral genome gene expression is highly restricted. Latency is a state of cryptic viral infection associated with genomic persistence in their host and this hallmark of KSHV infection leads to several clinical–epidemiological diseases such as KS, a plasmablastic variant of multicentric Castleman’s disease, and primary effusion lymphoma upon immune suppression of infected hosts. In order to sustain efficient life-long persistency as well as their life cycle, KSHV dedicates a large portion of its genome to encode immunomodulatory proteins that antagonize its host’s immune system. In this review, we will describe our current knowledge of the immune evasion strategies employed by KSHV at distinct stages of its viral life cycle to control the host’s immune system.
Introduction
Herpesviruses have co-evolved with their hosts for more than one hundred million years. During this coevolution, hosts have come to equip themselves with elaborate immune system to defend themselves from invading viruses and other pathogens. In order to establish infection and maintain latency, herpesviruses have acquired a number of genes that cause selective suppression of normal immune system functions and allow for an apathogenic, persistent infection.
Kaposi’s sarcoma-associated herpesvirus [KSHV; also known as human herpesvirus 8 (HHV8)] is a large double-stranded, DNA oncogenic virus belonging to the gammaherpesvirus subfamily. Its genome consists of a long unique region (LUR) encoding over 87 open reading frames (ORFs), at least 17 microRNAs, and a variable number of GC-rich 801-bp large terminal repeat (TR) elements. Like all other herpesviruses, KSHV can adopt one of two lifestyles known as latency and lytic replication. In latency, the viral genome is retained as a circular episome in the nucleus, expressing only a minimal number of genes without producing progeny virions. Lytic replication can be induced from the latency state by a single master-switch viral gene, which encodes a transcription factor known as RTA. The switch to lytic replication from latency leads to the expression of the entire viral genome as well as the production of progeny virions. Interestingly, KSHV dedicates almost a quarter of its genome to immunomodulatory genes, most of which seem to be pirated from the host and are cellular orthologs. Through these proteins, KSHV has developed elaborate mechanisms for outsmarting and adapting to the host’s immune responses, ultimately establishing a life-long, persistent infection within an immunocompetent host after primary infection.
Herein, we summarize recent advances in our understanding of how KSHV utilizes immunomodulatory genes to subvert the antiviral immune responses of its host (Figure 1 and Table 1).
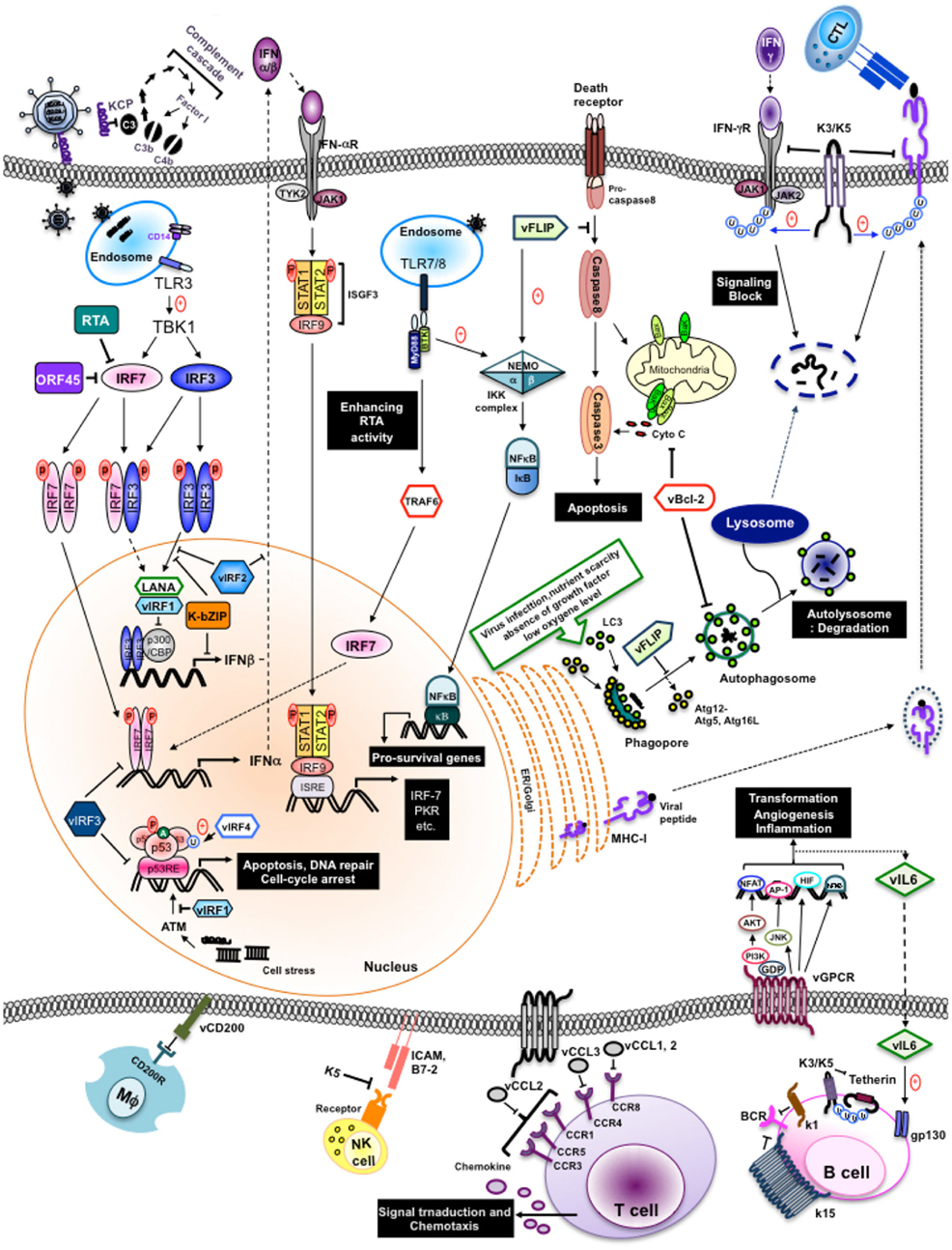
Figure 1. Evasion of immune responses by individual KSHV products. Individual KSHV products are shown to circumvent immune response, both innate and adapt, during their life cycle.
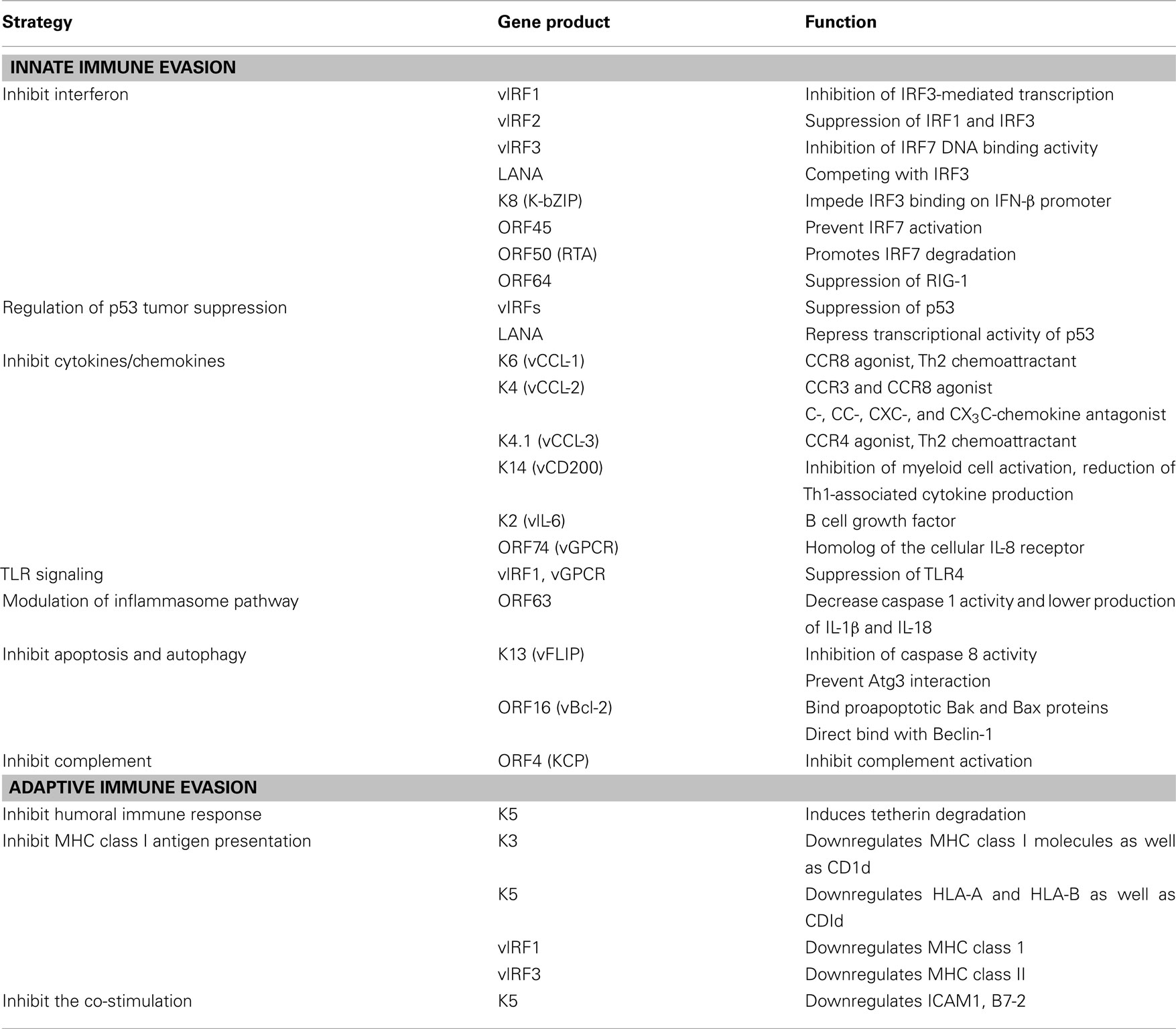
Table 1. Anti-immune strategies of KSHV.
Host Immune System Overview
The immune system provides numerous mechanisms of protection against invading pathogens like viruses. These immune responses include both broad spectrum, innate responses, and highly specific, adaptive responses. The innate immune system is our first line of defense against invading organisms while the adaptive immune system acts as a second line of defense that also affords protection against re-exposure to the same pathogen. In order to overcome innate responses, KSHV proteins deregulate interferon responses, the complement cascade, inflammatory cytokine production, natural killer (NK) cell immunity, Toll-like receptors (TLRs), apoptosis, and autophagy pathways. Furthermore, KSHV employs several strategies to block the two adaptive arms of the immune system: the humoral immune response arm (production of antibodies by B cells) and the cellular immune response arm (activities carried out by cytotoxic CD4+ and CD8+ T cells).
Innate Immunity
Interferon
Interferon (IFN) plays an important role as a primary defense mechanism against viral infection by inhibiting viral replication, suppressing cell growth, promoting apoptosis, upregulating antigen presentation, and modulating several signal transduction pathways. To date, three distinct classes of IFN designated as types I to III have been identified, classified according to their receptor complex and amino acid sequence (Pestka et al., 2004; Sadler and Williams, 2008). Type II IFN consists of a single gene, IFN-γ, that can interact with IFN-γ receptor (IFNγR) complexes and induce broad immune responses to pathogens other than viruses. Type III IFNs (namely, IFN-λ1, -λ2, and -λ3, also known as IL-28/29), and type I IFNs (IFN-α/β) mainly regulate antiviral responses. Virus-activated IFN-regulatory factors (IRFs) undergo phosphorylation, dimerization, and nuclear translocation, ultimately turning on positive regulators of the transcription of type I IFN and type III IFN genes (Sadler and Williams, 2008; Trinchieri, 2010).
Remarkably, KSHV encodes four viral IRFs (vIRF1–4) that show homology with cellular IRFs in one cluster region of its genome, and employs them as negative regulators that subvert host IFN-mediated immune responses (Takaoka et al., 2008). They are all expressed during lytic reactivation, but vIRF1 and vIRF3 [also know as latency-associated nuclear antigen (LANA2)] have also been detected in latently infected cells. This suggests that although vIRFs may function redundantly, they may act independently depending on the cell type and the stage of the viral lifecycle to thereby elicit disparate and distinct mechanisms of IFN evasion (Cunningham et al., 2003; Lee et al., 2009a). A number of previous studies have shown that vIRFs associate with and regulate cellular IRFs, other transcriptional factors and cofactors rather than directly interacting with DNA to inhibit IFN-induced gene transcripts (Takaoka et al., 2008; Tamura et al., 2008; Lee et al., 2009a, 2010).
vIRF1 (K9). Viral IRF1 was the first vIRF found to effectively repress cellular IFN responses (Gao et al., 1997; Zimring et al., 1998). vIRF1 does not compete with IRF1 for DNA binding although it inhibits IRF1 transcriptional activity (Zimring et al., 1998). Alternatively, vIRF1 binds to transcriptional cofactor p300 and interferes with CBP/p300–IRF3 complex formation along with p300 histone acetyltransferase (HAT) activity, thus preventing IRF3-mediated transcriptional activation (Li et al., 2000; Lin et al., 2001).
vIRF2 (K11/K11.1). Full length vIRF2, translated from two spliced exons (K11 and K11.1), accelerates IRF3 degradation and inhibits IRF3 transactivation, thereby repressing IRF3-mediated IFN-β transcriptional activity (Areste et al., 2009). In addition, vIRF2 inhibits IFN-α/β driven signaling as well as signaling induced by IFN-λ (Fuld et al., 2006). The underlying mechanism, however, has yet to be defined. In a recent study, vIRF2 reduced the activation of the IFN-induced interferon-response element (ISRE) promoter through the deregulation of IFN-stimulated gene factor-3 (ISGF-3), (Mutocheluh et al., 2011). It is suggested that vIRF2 possesses pleiotropic activity of inhibiting early type I IFN (IFN enhanceosome-dependent) and delayed type I IFN (ISGF-dependent) responses. Previous studies have shown that the first exon of vIRF2 (K11.1) interacts with cellular IRF1, IRF2, IRF8, RelA, and p300 (Burysek and Pitha, 2001). Furthermore, vIRF2 prevents PKR kinase activity to overcome IFN-α/β-mediated antiviral effects (Burysek and Pitha, 2001).
vIRF3 (K10.5). Viral IRF3 interaction with cellular IRF7 suppresses IRF7 DNA binding activity and, therefore, inhibits IFN-mediated immunity through the inhibition of IFN-α production (Joo et al., 2007). Remarkably, a putative double α-helix motif of vIRF3 (residues 240–280) that has been shown to be responsible for the interaction of vIRF3 with IRF7 is also sufficient to bind to IRF5 (Wies et al., 2009). As a result of this interaction, vIRF3 inhibits IRF5-mediatd ISRE and IFN-β promoter activity (Wies et al., 2009). It was recently shown that vIRF3 is required for the survival of primary effusion lymphoma (PEL) cells. RNA interference (RNAi) knockdown of vIRF3 in PEL cells reduced cell proliferation by releasing IRF5 from p21 promoter transcription complexes.
While it remains to be discovered whether vIRF4, the most recently identified member of the vIRF family, affects IFN-mediated innate immunity, the downregulation of the IFN-regulatory pathway is a common characteristic of the vIRFs, whose functions have been well studied, as IRF3 and IRF7 are key initiation factors of the host immune surveillance program against viral infection. Accordingly, in addition to vIRFs, other viral proteins restrain the type I and II IFN signaling pathways. The LANA protein, expressed during viral latency, competes with IRF3 for binding to the IFN-β promoter, thus preventing the expression of the CREB-binding protein as well as inhibiting IFN-β transcription (Cloutier and Flamand, 2010). Three immediately early gene products, K8, ORF45, and RTA, are also involved in the inhibition of IFN pathway in distinctive manners. The K8 (K-bZIP) precludes efficient IFN-β gene expression by impeding IRF3 binding to the IFN-β promoter through a direct interaction with the IFN-β promoter (Lefort et al., 2007). ORF45 interacts with IRF7, preventing its phosphorylation and subsequent translocation into the nucleus, hence impairing this amplification loop in type I IFN production (Zhu et al., 2002). RTA antagonizes type I IFN-mediated antiviral response, enhancing the ability of RTA-associated ubiquitin ligases (RAUL) to degrade IRF7 and IRF3 (Yu et al., 2005; Yu and Hayward, 2010). Much less is known about the state of type I IFN signaling in infected cells. Bisson et al. (2009) demonstrated that lytically infected cells display profound defects in IFN-mediated STAT activation. More recent studies have demonstrated that an early protein RIF (the product of ORF10) forms complexes with Jak1, Tyk2, STAT2, and both IFNAR subunits, resulting in the inhibition of the kinase activities of both Jak1 and Tyk2 (Bisson et al., 2009). In addition, KSHV ORF64, a tegument protein with deubiquitinase (DUB) activity, suppresses retinoic acid-inducible gene I-mediated IFN signaling by reducing the ubiquitination of RIG-1 upon KSHV infection (Inn et al., 2011). In summary, KSHV dedicates almost 10% of its gene production to modulating host IFN responses, further indicating the importance of IFN-mediated innate immunity as a primary defense mechanism against viral infection. Recently, HIV-1 Vpu led to the discovery of the interferon-induced transmembrane protein, tetherin (BST-2, CD317), as a novel component of host innate defense against enveloped viruses. Thus far, six viral proteins have been reported to counteract tetherin: HIV-1 Vpu, HIV-2, SIV Env, SIV Nef, Ebola glycoprotein (GP), and KSHV K5 (Neil et al., 2008; Van Damme et al., 2008; Jia et al., 2009; Kaletsky et al., 2009; Le Tortorec and Neil, 2009; Mansouri et al., 2009; Zhang et al., 2009). KSHV K5 induces the proteasomal degradation of tetherin during primary infection and upon reactivation from latency in endothelial cells. Furthermore, tetherin reduces KSHV release upon inhibition of K5 expression by small interfering RNA (siRNA), suggesting that tetherin is part of the IFN-induced innate immune response against KSHV (Mansouri et al., 2009; Pardieu et al., 2010).
p53 tumor suppression regulation
p53 (also known as tumor protein 53, TP53) is a transcriptional factor often described as the “guardian of the genome” because of its major roles in cell cycle arrest and in inducing apoptosis in response to a myriad of cellular stresses such as DNA damage and viral infection (Vogelstein et al., 2000; Collot-Teixeira et al., 2004). Therefore, viruses openly employ products that overcome the action of p53 to circumvent its growth-suppressive and proapoptotic actions (Coscoy, 2007; Liang et al., 2008; Lee et al., 2009a).
p53 is activated in a specific manner by post-translational modifications that lead to either cell cycle arrest, a program that induces cell senescence, or cellular apoptosis. Activation of the p53 network by stressors including virus replication and DNA damage is marked by two major events. First, the half-life of the p53 protein is increased drastically, leading to a quick accumulation of p53 in stressed cells (Collot-Teixeira et al., 2004). Second, a conformational change activates p53 as a transcriptional regulator in these cells, stimulating the phosphorylation and acetylation of p53 to increase the levels of activated p53. Activated p53 then binds to its regulatory region, which activates the expression of several target genes leading to diverse cellular responses, such as apoptosis, cell cycle arrest, or DNA repair (Harris and Levine, 2005). When p53 is no longer needed, it is ubiquitinated by murine double minute 2 (MDM2) and translocates from the nucleus to be degraded (Michael and Oren, 2003; Bond et al., 2005). In order to hinder p53-mediated irreversible cell cycle arrest and apoptosis, viruses need to tightly regulate the expression and/or functions of p53.
Viral IRFs. Notably, mounting studies show that vIRFs have remarkable functional redundancy in inhibiting p53-mediated cell cycle arrest and apoptosis. vIRF1 suppresses its acetylation, impeding the transcriptional activation of p53, resulting in a decrease in p53 target gene expression and transcription (Nakamura et al., 2001; Seo et al., 2001). Moreover, vIRF1 blocks ATM kinase activity, thereby reducing p53 phosphorylation and increasing p53 ubiquitination (Shin et al., 2006). Like vIRF1, vIRF3, and vIRF4 also interfere with p53 signaling. The B cell-specific, latently expressed vIRF3 inhibits p53-mediated transcriptional activity and apoptosis through its interaction with p53, although the way in which it targets the p53 pathway is not clear (Rivas et al., 2001). vIRF4 targets two major components, HAUSP and MDM2, of the p53 pathway to effectively reducing p53 and suppressing p53-mediated apoptosis. Furthermore, vIRF4’s actions with HAUSP and MDM2 are functionally and genetically separable and the vIRF4–MDM2 interaction has a more significant role in downregulating p53 than the vIRF4–HAUSP interaction (Lee et al., 2009b, 2011).
LANA (ORF73). Similar to vIRF3, the LANA also acts as negative regulator of the p53 pathway. LANA physically interacts with p53 to repress its transcriptional and translational activity (Friborg et al., 1999; Si and Robertson, 2006; Chen et al., 2010). Ultimately, LANA inhibits the ability of p53 to induce cell death, thereby, promoting chromosomal instability (Friborg et al., 1999; Si and Robertson, 2006). Initially, the modulation of p53 transcriptional activity by LANA was reported to be neither through p53 degradation nor inhibition of its DNA binding ability. However, a later report indicated that expression of LANA leads to decreased p53 protein levels due to LANA enhanced ubiquitination and degradation of p53 (Suzuki et al., 2010).
Cytokine regulation
Viral infection stimulates the production of cytokines and chemokines, secreted proteins that enable either the positive or negative regulation of the immune responses. They have crucial roles in inducing the migration of immune cells to areas of infection, immune regulation, antiviral defense, as well as the capacity of targets cells to support viral replication. Therefore, it is not surprising to find that many viruses benefit from an antagonism of cytokine activity by encoding proteins homologous to cellular cytokines (virokines), chemokine receptors (viroceptors), and/or secreting chemokine-binding proteins. Alternatively, viruses also utilize viral proteins to neutralize cytokine activities and may use cytokine pathways to induce cell proliferation, cell migration, or control homeostasis to enhance viral replication.
Viral cytokine (vIL-6). KSHV K2, an early lytic gene, encodes a viral interleukin, vIL-6, which shows homology to cellular IL-6 (Swanton et al., 1997). vIL-6 directly binds to and activates gp130 without the need of the gp80 α-subunit of the IL-6 receptor for IL-6 signal transduction (Molden et al., 1997; Hoischen et al., 2000; Chow et al., 2001; Chatterjee et al., 2002; Chen et al., 2009). As a functional consequence, vIL-6 mimics a number of IL-6 activities, including stimulation of IL-6 dependent proliferation of B cells, activation of the JAK/STAT pathway and stimulation of cells by intracellular signaling (Molden et al., 1997; Burger et al., 1998; Meads and Medveczky, 2004). Furthermore, vIL-6 plays important roles as an angiogenic factor through the induction of vascular endothelial growth factor (VEGF; Aoki et al., 1999), the promotion of Th2-cell development by increasing CCL2 expression, as well as blocking IFN response (Chatterjee et al., 2002; Diehl and Rincon, 2002). Moreover, secreted vIL-6 can be detected from latently infected KSHV-positive B cells and contributes to the survival and growth of PEL cells (Moore et al., 1996; Foussat et al., 1999; Mori et al., 2000; Chen et al., 2009). Hence, it is indicated that vIL-6 has distinctive functions in latency, where it promotes the growth and survival of infected cells for the maintenance of the latent virus within host cells, and lytic replication, where it protects virally infected cells from undergoing growth arrest and cell death.
Viral chemokines (vCCL-1, vCCL-2, vCCL-3). KSHV contains three virally encoded chemokines: vCCL-1 (K6/vMIP-I), vCCL-2 (K4/vMIP-II), and vCCL-3 (K4.1/vMIP-III), (Lee et al., 2010). vCCL-1, vCCL-2, and vCCL-3 can activate signal transduction through CCR3/CCR8, CCR8, and CCR4, respectively. Interestingly, they appear to be chemoattractants that specifically target Th2 lymphocytes as these chemokine receptors are preferentially expressed on Th2 cells, and not Th1 cells (Choi and Nicholas, 2008). It is indicated that KSHV CCLs act as Th2-cell chemokine-receptor agonists and can polarize the adaptive immune response toward a predominantly Th2-type response at sites of KSHV infection, potentially reducing the efficacy of host antiviral responses. Likewise, vCCL-2 acts as an antagonist by inhibiting a broad range of CC and CXC receptors expressed by Th1 cells and effectively blocks the RANTES-induced arrest of monocytes while promoting the arrest of eosinophils (Chen et al., 1998; Weber et al., 2001; Choi and Nicholas, 2008). Aside from their immune modulatory functions, vCCLs may further contribute to KSHV-associated disease by enhancing angiogenic responses through the induction of VEGF (Masood et al., 1997; Nakamura et al., 1997; Dittmer et al., 1998).
vGPCR (ORF74). The KSHV-encoded vGPCR (ORF74) is a seven-transmembrane, IL-8 receptor homolog that possesses promiscuous chemokine-binding activity (Swanton et al., 1997; Bais et al., 2003). Moreover, it has been shown that vGPCR constitutively activates mitogen-activated protein kinase (MAPK) cascades, p38 MAPK (Sodhi et al., 2000), Akt/protein kinase B (Cannon, 2007), mTOR (Montaner, 2007), as well as some small GTPases, like RhoA and Rac1 (Bottero et al., 2010). Consequently, certain transcription factors, including AP-1, NFAT, NF-κB, HIF-α, and cyclic AMP response element binding protein (CREB) have been shown to be activated by vGPCR expression, which in turn, lead to the expression of growth factors, proinflammatory cytokines, as well as angiogenic factors (Sodhi et al., 2000; Cannon et al., 2003; Cannon and Cesarman, 2004; Montaner et al., 2004). Expression of vGPCR in transgenic mice causes KS-like lesions characterized by increased vascularization and the recruitment of inflammatory cells (Bais et al., 2003; Montaner et al., 2003). Among the multiple intracellular pathways stimulated by vGPCR mentioned above, the PI3K/Akt/mTOR pathway has recently drawn great attention for its contribution to KS development. Notably, vGPCR expression was detected in only a very few cells within the tumor mass and other tissues, indicating that there is a paracrine effect, perhaps due to the enhanced activation of PI3Kγ (Martin et al., 2011). The fact that expression of vGPCR is restricted to the lytic phase of KSHV replication has raised arguments against its role in pathogenesis and further examination will be required.
vCD200 (K14). Another negative regulator of inflammatory signaling is K14, a surface glycoprotein (vOX2) expressed during the early lytic phase. It shows significant homology with OX2, also named CD200, a member of the immunoglobulin superfamily that is broadly distributed on the surface of cells (Chung et al., 2002). It was initially thought that vCD200 promotes the secretion of proinflammatory cytokines upon stimulation of monocytes, macrophages and DCs through an undefined receptor, to promote cytokine-mediated proliferation of KSHV-infected cells (Chung et al., 2002). However, vCD200 was later shown to down-modulate proinflammatory cytokines via a direct interaction with cellular CD200R, inhibiting myeloid cell activation and reducing Th1-cell-associated cytokine production (Foster-Cuevas et al., 2004; Rezaee et al., 2005; Shiratori et al., 2005). Furthermore, vCD200 has also been shown to suppress neutrophil-mediated inflammation in mice (Rezaee et al., 2005), suggestive of vCD200-mediated immunosuppressive activities in KSHV infection. Overall, mounting data has led to the conclusion that vCD200 can cause immune dysfunction associated with persistent infection, but the immunomodulatory role of vCD200 is still controversial. Recently, Salata et al. (2009) reported that vCD200 acts as a proinflammatory stimulus in the early stage of viral infection (or reactivation), while contributing to the downregulation of macrophage immune response activities, from phagocytosis to antigen presentation, in later stages.
TLR signaling
Toll-like receptors are transmembrane proteins expressed by various immune and non-immune cells of the innate immune system that recognize invading infectious agents and initiate signaling pathways, culminating in the increased expression of immune and inflammatory genes. To date, 11 human TLRs and 13 murine TLRs have been identified. Each TLR contains 21–25 leucine-rich repeat regions that specifically recognize a variety of pathogen associated molecular patterns (PAMPs), (Akira et al., 2006). TLRs are expressed in distinct cellular compartments: TLR1, TLR2, TLR4, TLR5, and TLR6 are expressed on the cell surface, whereas TLR3, TLR7, TLR8, and TLR9 are expressed in intracellular vesicles such as the endosome and ER (O’Neill and Bowie, 2010). Accumulated evidence in relation to TLR signal transduction has demonstrated that TLR3, TLR4, TLR7, TLR8, and TLR9 are involved in the recognition of viruses by way of binding to RNA, DNA, or viral glycoproteins (Akira and Takeda, 2004; Kawai and Akira, 2006). A recent growing body of information is beginning to shed light on TLR recognition of KSHV: KSHV infection of human monocytes enhance TLR3 expression and induction of cytokines including IFN-β1, CCL2, and CXCL10 (West and Damania, 2008). Infection of endothelial cells with KSHV causes suppression of TLR4 through the activation of the extracellular signal-regulated kinase (ERK) MAPK pathway via viral gene expression-independent mechanisms and viral gene expression dependent mechanisms by way of vIRF1 and vGPCR (Lagos et al., 2008). In addition, stimulation of TLR7 and TLR8 reactivates KSHV in latently infected B lymphocytes (Gregory et al., 2009). A later study has shown that KSHV infection of plasmacytoid dendritic cells (pDCs) activates the TLR9 signaling pathway, leading to the upregulation of CD83 and CD86, and secretion of IFN-α (West et al., 2011). Overall, there is evidence that KSHV has developed different ways to escape TLR-mediated detection throughout their lifecycle to limit KSHV lytic replication and facilitate the establishment of latency.
Modulation of inflammasome pathway
Inflammasomes are emerging as key regulators of host responses against infectious agents. When a pathogen invades sterile tissues or elicits cellular damage, it can cause the activation of NLRs (nucleotide binding and oligomerization, leucine-rich repeat) that then trigger inflammasomes, molecular platforms composed of oligomers of specific NLRs, pro-caspase 1, and an adaptor protein called ASC (apoptotic-associated speck-like; Martinon et al., 2002). These cytosolic multiprotein complexes activate cysteine protease caspase 1, leading to the processing and secretion of the proinflammatory cytokines, IL-1β, IL-18, and IL-33 (Masters et al., 2009; Schroder and Tschopp, 2010; Lamkanfi and Dixit, 2011). In addition, production of IL-1β and IL-18 is associated with a caspase 1-mediated inflammatory cell death program known as pyrotosis (Ting et al., 2008; Miao et al., 2010). Therefore, it is not surprising that viruses have evolved a variety of strategies to interfere with inflammasome activation and downstream signaling cascades (Lamkanfi and Dixit, 2011).
Recently, Gregory et al. found that KSHV ORF63 shows sequence similarity to a NLR family member, NLRP1, and hinders the interaction between NLRP1 and pro-caspase 1 through a direct interaction with NLRP1 oligomerization domains. Thus, ORF63 reduces caspase 1 activity and lowers production of IL-1β and IL-18. Interestingly, ORF63 also interacts with two additional NLR family members, NOD2 and NLRP3, and may thus broadly inhibit NLR inflammasome responses (Gregory et al., 2011). Taken together, KSHV deploys ORF63 to prevent inflammasome assembly and further enhance viral virulence.
Apoptosis and autophagy
A common cellular defense against pathogenic invasion is cell death by two programmed cell death (PCD) mechanisms: apoptosis (“self-killing”) and autophagy (“self-eating”), (Maiuri et al., 2007). These two PCDs are characterized by distinctive morphological and biochemical changes and both are highly conserved and tightly regulated processes that are essential for cell homeostasis, disease, and development. Apoptosis is the best-described cellular suicide mechanism against invading pathogens that involves a cascade of internal proteolytic digestion that result in the rapid demolition of cellular infrastructure (Danial and Korsmeyer, 2004; Green, 2005). By contrast, autophagy has been historically recognized as a cellular survival program that is involved in homeostasis by removing damaged or superfluous organelles through lysosomal degradation and recycling of cytoplasmic material delivered to autophagosomes (Maiuri et al., 2007; Kim et al., 2010; Liang and Jung, 2010). Recently, a growing body of evidence suggests that apoptosis and autophagy are not mutually exclusive pathways even though these two pathways seem to act independently. The functional relationship between apoptosis and autophagy is complex, with autophagy serving as a cell survival pathway that suppresses apoptosis that can also lead to cellular demise called autophagic cell death (type II PCD) in other cellular settings. Currently, a number of pathways that link the apoptotic and autophagic machineries and polarize the cellular response to favor one over the other have been deciphered at the molecular level. For instance, several apoptosis signaling molecules including TNF-α, TRAIL, FADD, and p53, also mediated autophagy. Likewise, Atg5, an autophagy effector, triggers apoptosis through its interaction with FADD (Mills et al., 2004; Feng et al., 2005; Levine, 2005; Lum et al., 2005; Pyo et al., 2005; Thorburn et al., 2005), while the inhibition of the class I PI3K, AKT, and mTOR pathways suppress both apoptosis and autophagy (Arico et al., 2001). Hence, it is not surprising that KSHV employs some gene products that target the crosstalk between apoptotic and autophagic signaling to evade and subvert this part of the overall host surveillance mechanism directed at blocking viral replication and dissemination.
vFLIP (K13). The K13 (ORF71) latent viral gene encodes viral FLICE (Fas-associated death-domain like IL-1 β-converting enzyme) inhibitory protein (vFLIP), a truncated homolog of cellular FLIP (cFLIP), (Dittmer et al., 1998; Fakhari and Dittmer, 2002). The vFLIP gene is expressed in KS and PEL cells from a polycistronic mRNA encompassing the latency locus. As such, vFLIP can inhibit extrinsic death-receptor-mediated apoptosis pathway by preventing the activation of caspases, including caspase 8 (Djerbi et al., 1999), and additionally associates with the IKK complex and heat shock protein 90 (HSP90) to induce the expression of anti-apoptotic proteins via the activation of NF-κB (Chaudhary et al., 1999; Liu et al., 2002; Field et al., 2003; Bubman et al., 2007). The induction of NF-κB activity has been linked to at least two aspects of KSHV-infected pathogenesis: viral latency and oncogenesis. On the other hand, NF-κB activation by vFLIP is crucial for its inhibition of lytic replication via the AP-1 pathway (Zhao et al., 2007; Ye et al., 2008). vFLIP likely contributes to a proinflammatory microenvironment given that its expression was found to induce NF-κB-regulated cytokine expression and secretion (Grossmann et al., 2006; Sun et al., 2006). Taken together, vFLIP appears to directly contribute to KSHV pathogenesis with functions in viral infected cell survival, transformation, inflammatory activation and latency control. Lately, vFLIP has not only been recognized to possess anti-apoptotic activities, but additionally serves as an anti-autophagy protein through its prevention of Atg3 binding to and processing of LC3, a protein involved in the elongation step of autophagy (Lee et al., 2009c).
vBcl-2 (ORF16). KSHV ORF16 encodes the viral Bcl-2 protein that generally shares 15–20% amino acid identity with human cellular homologs and inhibits apoptosis induced by virus infection and the pro-death protein BAX (Cheng et al., 1997; Sarid et al., 1997). Furthermore, recent evidence shows that vBcl-2 suppresses the cellular autophagy pathway through its direct interaction with the autophagy protein Beclin-1 (Pattingre et al., 2005; Liang et al., 2006). Although vBcl-2 possesses low degrees of homology with most regions of cellular Bcl-2 except the Bcl-2 homology (BH) 1 and BH2 domains, vBcl-2 shares three-dimensional structural conservation with Bcl-2 family members such that they all contain a central hydrophobic cleft called the BH3-peptide binding groove (Cheng et al., 1997; Huang et al., 2002; Loh et al., 2005). Interestingly, the BH3 binding cleft of vBcl-2 binds with high affinity and specificity with BH3-containing molecules (BAX, BAK, BIM, PUMA, BID, and Noxa) as well as selective binding to Beclin-1. It is thus possible that vBcl-2 may direct apoptosis and autophagy through a coordinated control of the two pathways. vBcl-2 is transcribed during lytic viral infection (Sarid et al., 1997; Paulose-Murphy et al., 2001) so inhibition of apoptosis and autophagy by vBcl-2 may function to prolong the life-span of KSHV-infected cells, which in turn enhances viral replication and establishment of latency. In summary, KSHV has evolved to acquire viral versions of Bcl-2 and FLIP to avoid elimination by the host’s apoptotic and autophagy-mediated immune responses.
Complement
The complement system has long been considered to be a part of the innate immune defense system that also bridges innate and adaptive immune responses, activated spontaneously in response to pathogen surface components or antibodies to help clear pathogens from an organism (Blue et al., 2004; Morgan et al., 2005). The complement system is comprised of 30 different proteins that are either circulating in the serum or attached to the cell surface, and they shared a common structural motif known as the short consensus repeat (SCR). They orchestrate three distinct cascades, termed the classic, alternative, and mannose-binding lectin pathways, which contribute to four major functions: lysis of infectious organisms, activation of inflammation, opsonization, and immune clearance. All three cascades converge at C3 convertase assembly, which can either initiate the opsonization of the foreign body or continue to activate C5 and thus propagate the cascade (Dunkelberger and Song, 2010).
KCP (ORF4)
It is not surprising that KSHV encodes its own inhibitor of the complement system in ORF4, designated as KSHV complement control protein (KCP). Three lytic protein isoforms are produced by alternative splicing (Spiller et al., 2003b). All three isoforms retain four SCR domains and a transmembrane region, and can regulate complement by accelerating the decay of the classical C3 convertase and acting as cofactors for the inactivation of C3b and C4b, components of the C3 and C5 convertases (Spiller et al., 2003a, 2006; Mark et al., 2008). KCPs are expressed and incorporated onto the surface of the virions during lytic infection, and have been detected at the surface of de novo infected endothelial cells after induction of lytic replication as well as at the surface of KSHV-infected cells from patients with PEL (Spiller et al., 2003b, 2006). Moreover, other γ-herpesviruses also encode KCP homologs indicating that an anti-complement defense system is essential during KSHV infection (Fodor et al., 1995; Kapadia et al., 1999; Okroj et al., 2009).
Adaptive immunity
B cell-dependent immune response
B lymphocytes are the major cell type involved in the creation of antibodies, also known as humoral immunity. Antibodies (e.g., IgG, IgM, and IgA) are produced by plasma cells, have been stimulated by CD4+ Th cells, which activate B cells through a signaling mechanism involving binding of CD40 on the B cell surface to CD40 ligand. In lymph nodes, naïve B cells recognize cognate antigen by their surface antibodies, become activated, switch from IgM to IgG production (class-switch), increase their immunoglobulin specificity and affinity, and differentiate into plasma cells or memory B cells as the cell continues to divide in the presence of cytokines. As a general host defense mechanism, antibodies can directly neutralize viruses by sterically hindering the receptor–virus ligand interaction or by inducing conformational changes in viral receptor ligands. Other indirect effects caused by antibodies include the recruitment or activation of the innate immune effector system such as antibody-dependent cell cytoxicity (ADCC), engulfment of antibody-coated (opsonized) viruses by phagocytes, and complement activation. B cells are the likely cellular reservoir of KSHV infection and KSHV seems to influence several aspects of B cell biology through the modulation of humoral immune responses.
Mounting evidence shows that a B cell terminal differentiation factor, X-box binding protein 1 (XBP-1), can effectively initiate KSHV reactivation by activating the RTA promoter, thus providing a link between B cell development and KSHV pathogenesis (Wilson et al., 2007; Yu et al., 2007; Dalton-Griffin et al., 2009). Interestingly, PEL cells predominantly express the inactive form of XBP-1, XBP-1u, but when the active form, XBP-1s, is induced, the KSHV lytic cycle is activated (Reimold et al., 1996). This raises the possibility that in KSHV-infected B cells, latency is maintained until plasma cell differentiation occurs.
T cell-dependent immune response
Unlike B cells, T cells recognize antigenic determinants associated with self MHC molecules on the surface of antigen-presenting cells (APCs) instead of soluble antigens. Classically, during viral infection, the recognition of viral peptides presented by MHC class I molecules on cytotoxic T lymphocytes (CTLs) is a key event in the elimination of cells producing abnormal or foreign proteins, specifically during a virus infection. CTLs thus play a critical role in the control of a viral infection, especially as a long-term immune surveillance effector that can more quickly react against the same virus after a primary infection (Micheletti et al., 2002). CTL evasion is hence a prerequisite for the replication of persistent viruses particularly in the case of herpesviruses, which must establish a persistent, latent infection, and must then reactivate in immunologically primed hosts to shed infectious virions. All herpesviruses implement strategies that target key stages of the MHC class I antigen presentation pathway with the goal of preventing the presentation of viral peptide to CTLs (for review see Alcami and Koszinowski, 2000; Ambagala et al., 2005). For instance, KSHV encodes two well-known inhibitors of MHC class I cell surface molecules that effectively suppress CTL response to virus infected cells.
Inhibition of MHC class I antigen presentation. The K3 and K5 proteins, also known as modulator of immune recognition (MIR) 1 and MIR2, consist of an N-terminal RING-CH domain harboring ubiquitin E3 ligases activity and two transmembrane domains responsible for substrate recognition (Lehner et al., 2005). In contrast to other viral inhibitors of MHC class I, K3 and K5 do not affect assembly or transportation of MHC complexes to the cell surface. Instead, they interact with MHC class I molecules through transmembrane interactions and trigger endocytosis and proteasomal degradation of MHC class I molecules without affecting their assembly or transport by ubiquitinating its cytoplasmic tail (Coscoy et al., 2001). Interestingly, K3 downregulates the expression of both canonical and non-canonical MHC class I molecules in humans (HLA-A, -B, -C, and -E), whereas K5 primarily downregulates HLA-A and -B alleles due to substrate specificity arising from TM interactions (Coscoy and Ganem, 2000; Ishido et al., 2000; Sanchez et al., 2002; Wang et al., 2004). Additionally, vIRF1 has been implicated to interact with p300 to prevent basal transcription of MHC class I molecules and thereby downregulate MHC class I molecules on the cell surface of infected cells (Lagos et al., 2007). Notably, from the viral perspective, the virus must thwart both CTLs and NK cells because the downregulation of only MHC class I molecules renders infected cells sensitive to NK cell-mediated lysis. NK cells recognize infected cells in an antigen-independent manner and destroy such cells via cytotoxic activities. Furthermore, they can rapidly produce large amounts of IFN-γ, which induces diverse antiviral immune responses upon receptor activation, including the augmentation of antigen presentation and activation/polarization of CD4+ T cells and macrophages (Goodbourn et al., 2000). Hence, K3 and K5 also target gamma interferon receptor 1 (IFN-γR1) by inducing its ubiquitination, endocytosis, and degradation, ultimately resulting in the inhibition of IFN-γR1-mediated activation of transcription factors (Li et al., 2007). Very recently, Schmidt et al. reported that vIRF3 also contributes to viral immunoevasion by downregulating of IFN-γ and class II transactivator (CIITA), thus MHC class II expression (Schmidt et al., 2011). Overall, it is suggested that the downregulation of surface MHC class I molecules and IFN- γR1 by K3- and K5-mediated ubiquitination and lysosomal degradation inhibits primary host immunity against viral infection.
Inhibition of co-stimulation. The generation of a robust adaptive immune response requires the engagement of T cells with professional APCs such as DCs, macrophages, and B cells. To sufficiently activate T cells, costimulatory signals such as the interaction between CD28 and its ligands, B7-1 and B7-2 on APC surfaces (Sperling and Bluestone, 1996; Chambers and Allison, 1999), and between intercellular adhesion molecules 1 (ICAM1) and lymphocyte function-associated antigen 1 (LFA1; Dustin and Springer, 1989) are essential. In addition to lowering the level of MHC class I molecules, K5 also downregulates other components of the immune synapse on the B cell surface such as ICAM (CD54) and B7-2 (CD86), two co-activating molecules for NK cell activation, by inducing their endocytosis and degradation (Ishido et al., 2000; Coscoy and Ganem, 2001).
Taken together, K3 seems to specifically target MHC class I molecules, whereas K5 additionally targets multiple receptors including MHC class I, CD86, and ICAM, with the overall effect of preventing detection by CTL and NK cells (Nathan and Lehner, 2009; Manes et al., 2010). Even in a single gene overexpression system, the resultant downregulation is sufficient to confer resistance to immune cells in culture, and therefore may protect KSHV-infected cells undergoing lytic replication in vivo. The contribution of K3 and K5 to immune evasion in the context of viral genome during latency, however, is less clear. Their influence appears to be most important in the early stages of KSHV infection, when diminished detection by T cells can result in a reduced antiviral cytokine responses and an impaired production of CTLs, thereby allowing the virus to establish a persistent infection.
Viral microRNAs
MicroRNAs (miRNAs) are a copious class of evolutionarily conserved small non-coding RNAs that are thought to modulate gene expression post-transcriptionally by targeting mRNAs for degradation or translational repression. miRNAs are 21–23 nucleotide long RNAs that lack protein coding capacity and primarily regulate gene expression by binding to the 3′ untranslated region (3′UTR) of target mRNAs (Bartel, 2004). The genomic regions encoding miRNAs are generally transcribed into primary miRNAs (pri-miRNAs) by RNA polymerase II. The nuclease Drosha cleaves pri-miRNA into precursor miRNAs (pre-miRNAs), which are further processed by Dicer to produce mature miRNAs. miRNAs are loaded into a protein complex known as the RNA-induced silencing complex (RISC) to inhibit the translation of target mRNAs (Bartel, 2004). Through various methods of microRNA target identification, miRNA have been found to regulate diverse biological processes including cell cycle, development, differentiation, and metabolism. Given the propensity of viruses to co-opt cellular systems and activities for their benefit, it is perhaps not surprising that several viruses have now been shown to reshape the cellular environment by reprogramming the host’s RNAi machinery. Indeed, most herpesviruses encode miRNAs and KSHV in particular encodes 12 pre-miRNAs genes (miR-K1–12) located in the latency locus (Ganem and Ziegelbauer, 2008; Cullen, 2009; Lei et al., 2010a; Ziegelbauer, 2011). These pre-miRNAs are ultimately processed into mature miRNAs. Interestingly, in some cases, mature miRNAs can undergo RNA editing along with variations at the 5′ end of the mature miRNA, leading to at least five different mature miRNAs arising from one pre-miRNA (Pfeffer et al., 2004; Lin et al., 2010; Umbach and Cullen, 2010). Very recently, Gottwein et al. revealed that KSHV miRNAs directly target numerous cellular mRNAs, including many involved in pathways relevant to KSHV pathogenesis and replication (Table 2). Notably, around 60% of these miRNAs are also target EBV miRNAs in PEL cells (Gottwein et al., 2011).
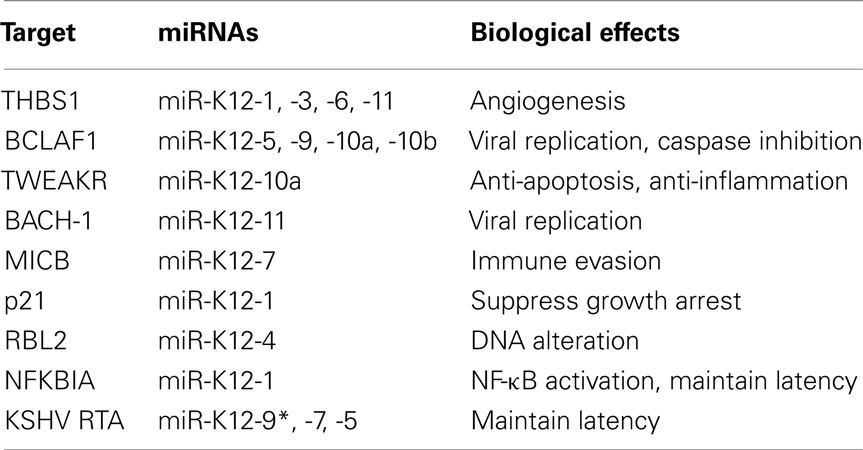
Table 2. Summary of KSHV miRNAs.
Targeting of cellular mRNAs
As with other viral mechanisms of host interaction, viral miRNAs display redundancy in that multiple miRNAs target and repress an individual cellular gene. In a gene expression profile using microarray, Samols et al. identified a total of 81 genes whose expression were significantly changed in cells stably expressing KSHV-encoded miRNAs. Among them, thrombospondin 1 (THBS1), a major regulator of cell adhesion, migration, and angiogenesis showed a significant decrease by several KSHV miRNAs including miR-K12-1, miR-K12-3-3p, miR-K-12-6-3p, and miR-K-12-11. This consequently led to reduced TGF-β activity (Samols et al., 2007). Hence, it is indicated that these KSHV miRNAs potentially contribute to KS angiogenesis by repressing the expression of an angiogenesis inhibitor, THBS1. Another microarray study showed that at least four miRNAs (miR-K-12-5, -9, -10a, and -10b) target BCLAF1 (BCL-2 associated factor). Intriguingly, these miRNAs sensitized latently infected cells to stimuli that induce lytic reactivation (Ziegelbauer et al., 2009). This is a different phenotype from that proposed for the effect of KSHV miRNAs on its own viral proteins, RTA (Lei et al., 2010b; Lu et al., 2010). Thus, it is suggested that KSHV employs its miRNAs to fine-tune gene regulation to either stabilize or destabilize latency. miR-K12-10a, it causes the miR-K12-10a mediated knockdown of TWEAKR (Tumor necrosis factor-like weak inducer of apoptosis receptor) reduces the level of apoptosis and interleukin-8 (IL-8), (Abend et al., 2010). Remarkably, the seed sequence of KSHV miR-K12-11 is identical to that of cellular miR-155. These two miRNAs consequently function as orthologs that downregulate the same target genes including BACH-1(Gottwein et al., 2007; Skalsky et al., 2007). Suppressing BACH-1 by miR-K12-11 increases expression of an amino acid transport protein, xCT, which enables KSHV propagation by cell fusion (Qin et al., 2010). Like HCMV and EBV, KSHV miR-K12-7 has been shown to inhibit MICB (MHC class I polypeptide-related sequence B) expression, thereby preventing NK cell-mediated cell killing during KSHV infection (Nachmani et al., 2009). To investigate the role of KSHV miRNAs in viral latency, Lei et al. generated a mutant KSHV lacking its 14 miRNAs. Interestingly, KSHV miR-K12-1 targets the mRNA of NF-κB inhibitor IκBα, thus promoting viral latency by activating NF-κB (Lei et al., 2010b). miR-K12-1 was also found to inhibit the cellular dependent kinase inhibitor p21 through a different mechanism (Gottwein and Cullen, 2010). KSHV miR-K12-4-5P epigenetically regulates KSHV lytic replication by inhibiting a repressor of DNA methyl transferases, Rbl2 (retinoblastoma-like protein2), to increase the mRNA levels of DNA methyl transferase 1, 3a, and 3b (Iorio et al., 2007). In summary, KSHV miRNAs manipulate host survival pathways to regulate viral latency and lytic replication.
Targeting of viral mRNAs
Currently, a validated viral target of KSHV miRNAs is RTA, a well-known viral latent-lytic switch protein. In order to control the expression of viral lytic genes, KSHV miR-K12-9* directly targets RTA (ORF50, lytic immediately early transcription factor) to prevent viral reactivation (Lu et al., 2010). Another viral miRNA, miR-K12-5, also antagonizes KSHV reactivation by consistently inhibiting RTA expression (Lei et al., 2010b). Taken together, miRNAs are essential for controlling KSHV replication and latency by either directly targeting the expression of key viral lytic genes or indirectly targeting cellular regulatory genes.
Concluding Remarks
As we have seen, viruses have learned to manipulate host immune control mechanisms to facilitate their propagation by using captured host genes or evolving genes to target specific immune pathways. Thus, viral genomes can be regarded as repositories of important information of immune processes, offering us a viral view of the host immune system. Hence, studies that add to our growing knowledge of viral immunomodulatory proteins might help us uncover new human genes that control immunity. Their characterization will increase our understanding of not only viral pathogenesis, but also normal immune mechanisms. Furthermore, mechanisms used by viral proteins suggest strategies of immune modulation that might have therapeutic potential. With regards to the discovery of potential therapeutic reagents through virus research, our recent study sets a notable precedent: two peptides derived from the KSHV vIRF4 can additively inhibit HAUSP, leading to p53-mediated tumor regression (Lee et al., 2011). Despite this significant advance, it is only the tip of the iceberg. Although numerous immune evasion mechanisms employed by KSHV seem to be well understood, it is too early to translate the knowledge we have obtained from basic science research into developing more effective clinical management and therapies due to the absence of tractable tissues culture or in vivo models for KSHV. Time after time, there is more to be learned from viruses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was partly supported by CA82057, CA91819, CA31363, CA115284, CA147868, CA148616, DE019085, AI073099, AI083025, Hastings Foundation, Fletcher Jones Foundation, National Agenda Project grant from Korea Research Council of Fundamental Science and Technology and KRIBB Initiative program (Grant no. KGM0821113), and the GRL Program (K20815000001) from the National Research Foundation of Korea. We thank Stacy Lee for manuscript preparation. Finally, we thank all of Jae U. Jung’s lab members for their discussions.
References
Abend, J. R., Uldrick, T., and Ziegelbauer, J. M. (2010). Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi’s sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J. Virol. 84, 12139–12151.
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801.
Alcami, A., and Koszinowski, U. H. (2000). Viral mechanisms of immune evasion. Trends Microbiol. 8, 410–418.
Ambagala, A. P., Solheim, J. C., and Srikumaran, S. (2005). Viral interference with MHC class I antigen presentation pathway: the battle continues. Vet. Immunol. Immunopathol. 107, 1–15.
Aoki, Y., Jaffe, E. S., Chang, Y., Jones, K., Teruya-Feldstein, J., Moore, P. S., and Tosato, G. (1999). Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood 93, 4034–4043.
Areste, C., Mutocheluh, M., and Blackbourn, D. J. (2009). Identification of caspase-mediated decay of interferon regulatory factor-3, exploited by a Kaposi sarcoma-associated herpesvirus immunoregulatory protein. J. Biol. Chem. 284, 23272–23285.
Arico, S., Petiot, A., Bauvy, C., Dubbelhuis, P. F., Meijer, A. J., Codogno, P., and Ogier-Denis, E. (2001). The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 276, 35243–35246.
Bais, C., Van Geelen, A., Eroles, P., Mutlu, A., Chiozzini, C., Dias, S., Silverstein, R. L., Rafii, S., and Mesri, E. A. (2003). Kaposi’s sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/KDR. Cancer Cell 3, 131–143.
Bisson, S. A., Page, A. L., and Ganem, D. (2009). A Kaposi’s sarcoma-associated herpesvirus protein that forms inhibitory complexes with type I interferon receptor subunits, Jak and STAT proteins, and blocks interferon-mediated signal transduction. J. Virol. 83, 5056–5066.
Blue, C. E., Spiller, O. B., and Blackbourn, D. J. (2004). The relevance of complement to virus biology. Virology 319, 176–184.
Bond, G. L., Hu, W., and Levine, A. J. (2005). MDM2 is a central node in the p53 pathway: 12 years and counting. Curr. Cancer Drug Targets 5, 3–8.
Bottero, V., Kerur, N., Sadagopan, S., Patel, K., Sharma-Walia, N., and Chandran, B. (2010). Phosphorylation and polyubiquitination of transforming growth factor beta-activated kinase 1 are necessary for activation of NF-kappaB by the Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor. J. Virol. 85, 1980–1993.
Bubman, D., Guasparri, I., and Cesarman, E. (2007). Deregulation of c-Myc in primary effusion lymphoma by Kaposi’s sarcoma herpesvirus latency-associated nuclear antigen. Oncogene 26, 4979–4986.
Burger, R., Neipel, F., Fleckenstein, B., Savino, R., Ciliberto, G., Kalden, J. R., and Gramatzki, M. (1998). Human herpesvirus type 8 interleukin-6 homologue is functionally active on human myeloma cells. Blood 91, 1858–1863.
Burysek, L., and Pitha, P. M. (2001). Latently expressed human herpesvirus 8-encoded interferon regulatory factor 2 inhibits double-stranded RNA-activated protein kinase. J. Virol. 75, 2345–2352.
Cannon, M. (2007). The KSHV and other human herpesviral G protein-coupled receptors. Curr. Top. Microbiol. Immunol. 312, 137–156.
Cannon, M., Philpott, N. J., and Cesarman, E. (2003). The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor has broad signaling effects in primary effusion lymphoma cells. J. Virol. 77, 57–67.
Cannon, M. L., and Cesarman, E. (2004). The KSHV G protein-coupled receptor signals via multiple pathways to induce transcription factor activation in primary effusion lymphoma cells. Oncogene 23, 514–523.
Chambers, C. A., and Allison, J. P. (1999). Costimulatory regulation of T cell function. Curr. Opin. Cell Biol. 11, 203–210.
Chatterjee, M., Osborne, J., Bestetti, G., Chang, Y., and Moore, P. S. (2002). Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science 298, 1432–1435.
Chaudhary, P. M., Jasmin, A., Eby, M. T., and Hood, L. (1999). Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 18, 5738–5746.
Chen, D., Sandford, G., and Nicholas, J. (2009). Intracellular signaling mechanisms and activities of human herpesvirus 8 interleukin-6. J. Virol. 83, 722–733.
Chen, S., Bacon, K. B., Li, L., Garcia, G. E., Xia, Y., Lo, D., Thompson, D. A., Siani, M. A., Yamamoto, T., Harrison, J. K., and Feng, L. (1998). In vivo inhibition of CC and CX3C chemokine-induced leukocyte infiltration and attenuation of glomerulonephritis in Wistar-Kyoto (WKY) rats by vMIP-II. J. Exp. Med. 188, 193–198.
Chen, W., Hilton, I. B., Staudt, M. R., Burd, C. E., and Dittmer, D. P. (2010). Distinct p53, p53:LANA, and LANA complexes in Kaposi’s sarcoma – associated herpesvirus lymphomas. J. Virol. 84, 3898–3908.
Cheng, E. H., Nicholas, J., Bellows, D. S., Hayward, G. S., Guo, H. G., Reitz, M. S., and Hardwick, J. M. (1997). A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. U.S.A. 94, 690–694.
Choi, Y. B., and Nicholas, J. (2008). Autocrine and paracrine promotion of cell survival and virus replication by human herpesvirus 8 chemokines. J. Virol. 82, 6501–6513.
Chow, D., He, X., Snow, A. L., Rose-John, S., and Garcia, K. C. (2001). Structure of an extracellular gp130 cytokine receptor signaling complex. Science 291, 2150–2155.
Chung, Y. H., Means, R. E., Choi, J. K., Lee, B. S., and Jung, J. U. (2002). Kaposi’s sarcoma-associated herpesvirus OX2 glycoprotein activates myeloid-lineage cells to induce inflammatory cytokine production. J. Virol. 76, 4688–4698.
Cloutier, N., and Flamand, L. (2010). Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen inhibits interferon (IFN) beta expression by competing with IFN regulatory (factor)-3 for binding to IFNB promoter. J. Biol. Chem. 285, 7208–7221.
Collot-Teixeira, S., Bass, J., Denis, F., and Ranger-Rogez, S. (2004). Human tumor suppressor p53 and DNA viruses. Rev. Med. Virol. 14, 301–319.
Coscoy, L. (2007). Immune evasion by Kaposi’s sarcoma-associated herpesvirus. Nat. Rev. Immunol. 7, 391–401.
Coscoy, L., and Ganem, D. (2000). Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. U.S.A. 97, 8051–8056.
Coscoy, L., and Ganem, D. (2001). A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J. Clin. Invest. 107, 1599–1606.
Coscoy, L., Sanchez, D. J., and Ganem, D. (2001). A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J. Cell Biol. 155, 1265–1273.
Cullen, B. R. (2009). Viral and cellular messenger RNA targets of viral microRNAs. Nature 457, 421–425.
Cunningham, C., Barnard, S., Blackbourn, D. J., and Davison, A. J. (2003). Transcription mapping of human herpesvirus 8 genes encoding viral interferon regulatory factors. J. Gen. Virol. 84, 1471–1483.
Dalton-Griffin, L., Wilson, S. J., and Kellam, P. (2009). X-box binding protein 1 contributes to induction of the Kaposi’s sarcoma-associated herpesvirus lytic cycle under hypoxic conditions. J. Virol. 83, 7202–7209.
Diehl, S., and Rincon, M. (2002). The two faces of IL-6 on Th1/Th2 differentiation. Mol. Immunol. 39, 531–536.
Dittmer, D., Lagunoff, M., Renne, R., Staskus, K., Haase, A., and Ganem, D. (1998). A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 72, 8309–8315.
Djerbi, M., Screpanti, V., Catrina, A. I., Bogen, B., Biberfeld, P., and Grandien, A. (1999). The inhibitor of death receptor signaling, FLICE-inhibitory protein defines a new class of tumor progression factors. J. Exp. Med. 190, 1025–1032.
Dunkelberger, J. R., and Song, W. C. (2010). Complement and its role in innate and adaptive immune responses. Cell Res 20, 34–50.
Dustin, M. L., and Springer, T. A. (1989). T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature 341, 619–624.
Fakhari, F. D., and Dittmer, D. P. (2002). Charting latency transcripts in Kaposi’s sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76, 6213–6223.
Feng, Z., Zhang, H., Levine, A. J., and Jin, S. (2005). The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. U.S.A. 102, 8204–8209.
Field, N., Low, W., Daniels, M., Howell, S., Daviet, L., Boshoff, C., and Collins, M. (2003). KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell. Sci. 116, 3721–3728.
Fodor, W. L., Rollins, S. A., Bianco-Caron, S., Rother, R. P., Guilmette, E. R., Burton, W. V., Albrecht, J. C., Fleckenstein, B., and Squinto, S. P. (1995). The complement control protein homolog of herpesvirus saimiri regulates serum complement by inhibiting C3 convertase activity. J. Virol. 69, 3889–3892.
Foster-Cuevas, M., Wright, G. J., Puklavec, M. J., Brown, M. H., and Barclay, A. N. (2004). Human herpesvirus 8 K14 protein mimics CD200 in down-regulating macrophage activation through CD200 receptor. J. Virol. 78, 7667–7676.
Foussat, A., Wijdenes, J., Bouchet, L., Gaidano, G., Neipel, F., Balabanian, K., Galanaud, P., Couderc, J., and Emilie, D. (1999). Human interleukin-6 is in vivo an autocrine growth factor for human herpesvirus-8-infected malignant B lymphocytes. Eur. Cytokine Netw. 10, 501–508.
Friborg, J. Jr., Kong, W., Hottiger, M. O., and Nabel, G. J. (1999). p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402, 889–894.
Fuld, S., Cunningham, C., Klucher, K., Davison, A. J., and Blackbourn, D. J. (2006). Inhibition of interferon signaling by the Kaposi’s sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. J. Virol. 80, 3092–3097.
Ganem, D., and Ziegelbauer, J. (2008). MicroRNAs of Kaposi’s sarcoma-associated herpes virus. Semin. Cancer Biol. 18, 437–440.
Gao, S. J., Boshoff, C., Jayachandra, S., Weiss, R. A., Chang, Y., and Moore, P. S. (1997). KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene 15, 1979–1985.
Goodbourn, S., Didcock, L., and Randall, R. E. (2000). Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol. 81, 2341–2364.
Gottwein, E., Corcoran, D. L., Mukherjee, N., Skalsky, R. L., Hafner, M., Nusbaum, J. D., Shamulailatpam, P., Love, C. L., Dave, S. S., Tuschl, T., Ohler, U., and Cullen, B. R. (2011). Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 10, 515–526.
Gottwein, E., and Cullen, B. R. (2010). A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J. Virol. 84, 5229–5237.
Gottwein, E., Mukherjee, N., Sachse, C., Frenzel, C., Majoros, W. H., Chi, J. T., Braich, R., Manoharan, M., Soutschek, J., Ohler, U., and Cullen, B. R. (2007). A viral microRNA functions as an orthologue of cellular miR-155. Nature 450, 1096–1099.
Gregory, S. M., Davis, B. K., West, J. A., Taxman, D. J., Matsuzawa, S., Reed, J. C., Ting, J. P., and Damania, B. (2011). Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331, 330–334.
Gregory, S. M., West, J. A., Dillon, P. J., Hilscher, C., Dittmer, D. P., and Damania, B. (2009). Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. U.S.A. 106, 11725–11730.
Grossmann, C., Podgrabinska, S., Skobe, M., and Ganem, D. (2006). Activation of NF-kappaB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J. Virol. 80, 7179–7185.
Harris, S. L., and Levine, A. J. (2005). The p53 pathway: positive and negative feedback loops. Oncogene 24, 2899–2908.
Hoischen, S. H., Vollmer, P., Marz, P., Ozbek, S., Gotze, K. S., Peschel, C., Jostock, T., Geib, T., Mullberg, J., Mechtersheimer, S., Fischer, M., Grötzinger, J., Galle, P. R., and Rose-John, S. (2000). Human herpes virus 8 interleukin-6 homologue triggers gp130 on neuronal and hematopoietic cells. Eur. J. Biochem. 267, 3604–3612.
Huang, Q., Petros, A. M., Virgin, H. W., Fesik, S. W., and Olejniczak, E. T. (2002). Solution structure of a Bcl-2 homolog from Kaposi sarcoma virus. Proc. Natl. Acad. Sci. U.S.A. 99, 3428–3433.
Inn, K. S., Lee, S. H., Rathbun, J. Y., Wong, L. Y., Toth, Z., Machida, K., Ou, J. H., and Jung, J. U. (2011). Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 85, 10899–10904.
Iorio, M. V., Visone, R., Di Leva, G., Donati, V., Petrocca, F., Casalini, P., Taccioli, C., Volinia, S., Liu, C. G., Alder, H., Calin, G. A., Ménard, S., and Croce, C. M. (2007). MicroRNA signatures in human ovarian cancer. Cancer Res. 67, 8699–8707.
Ishido, S., Wang, C., Lee, B. S., Cohen, G. B., and Jung, J. U. (2000). Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74, 5300–5309.
Jia, B., Serra-Moreno, R., Neidermyer, W., Rahmberg, A., Mackey, J., Fofana, I. B., Johnson, W. E., Westmoreland, S., and Evans, D. T. (2009). Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 5, e1000429. doi:10.1371/journal.ppat.1000429
Joo, C. H., Shin, Y. C., Gack, M., Wu, L., Levy, D., and Jung, J. U. (2007). Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 81, 8282–8292.
Kaletsky, R. L., Francica, J. R., Agrawal-Gamse, C., and Bates, P. (2009). Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 106, 2886–2891.
Kapadia, S. B., Molina, H., van Berkel, V., Speck, S. H., and Virgin, H. W. T. (1999). Murine gammaherpesvirus 68 encodes a functional regulator of complement activation. J. Virol. 73, 7658–7670.
Kawai, T., and Akira, S. (2006). Innate immune recognition of viral infection. Nat. Immunol. 7, 131–137.
Kim, H. J., Lee, S., and Jung, J. U. (2010). When autophagy meets viruses: a double-edged sword with functions in defense and offense. Semin. Immunopathol. 32, 323–341.
Lagos, D., Trotter, M. W., Vart, R. J., Wang, H. W., Matthews, N. C., Hansen, A., Flore, O., Gotch, F., and Boshoff, C. (2007). Kaposi sarcoma herpesvirus-encoded vFLIP and vIRF1 regulate antigen presentation in lymphatic endothelial cells. Blood 109, 1550–1558.
Lagos, D., Vart, R. J., Gratrix, F., Westrop, S. J., Emuss, V., Wong, P. P., Robey, R., Imami, N., Bower, M., Gotch, F., and Boshoff, C. (2008). Toll-like receptor 4 mediates innate immunity to Kaposi sarcoma herpesvirus. Cell Host Microbe 4, 470–483.
Lamkanfi, M., and Dixit, V. M. (2011). Modulation of inflammasome pathways by bacterial and viral pathogens. J. Immunol. 187, 597–602.
Le Tortorec, A., and Neil, S. J. (2009). Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J. Virol. 83, 11966–11978.
Lee, H. R., Choi, W. C., Lee, S., Hwang, J., Hwang, E., Guchhait, K., Haas, J., Toth, Z., Jeon, Y. H., Oh, T. K., Kim, M. H., and Jung, J. U. (2011). Bilateral inhibition of HAUSP deubiquitinase by a viral interferon regulatory factor protein. Nat. Struct. Mol. Biol. 18, 1336–1344.
Lee, H. R., Kim, M. H., Lee, J. S., Liang, C., and Jung, J. U. (2009a). Viral interferon regulatory factors. J. Interferon Cytokine Res. 29, 621–627.
Lee, H. R., Toth, Z., Shin, Y. C., Lee, J. S., Chang, H., Gu, W., Oh, T. K., Kim, M. H., and Jung, J. U. (2009b). Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 4 targets MDM2 to deregulate the p53 tumor suppressor pathway. J. Virol. 83, 6739–6747.
Lee, J. S., Li, Q., Lee, J. Y., Lee, S. H., Jeong, J. H., Lee, H. R., Chang, H., Zhou, F. C., Gao, S. J., Liang, C., and Jung, J. U. (2009c). FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 11, 1355–1362.
Lee, H. R., Lee, S., Chaudhary, P. M., Gill, P., and Jung, J. U. (2010). Immune evasion by Kaposi’s sarcoma-associated herpesvirus. Future Microbiol. 5, 1349–1365.
Lefort, S., Soucy-Faulkner, A., Grandvaux, N., and Flamand, L. (2007). Binding of Kaposi’s sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J. Virol. 81, 10950–10960.
Lehner, P. J., Hoer, S., Dodd, R., and Duncan, L. M. (2005). Downregulation of cell surface receptors by the K3 family of viral and cellular ubiquitin E3 ligases. Immunol. Rev. 207, 112–125.
Lei, X., Bai, Z., Ye, F., Huang, Y., and Gao, S. J. (2010a). Regulation of herpesvirus lifecycle by viral microRNAs. Virulence 1, 433–435.
Lei, X., Bai, Z., Ye, F., Xie, J., Kim, C. G., Huang, Y., and Gao, S. J. (2010b). Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 12, 193–199.
Levine, B. (2005). Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120, 159–162.
Li, M., Damania, B., Alvarez, X., Ogryzko, V., Ozato, K., and Jung, J. U. (2000). Inhibition of p300 histone acetyltransferase by viral interferon regulatory factor. Mol. Cell. Biol. 20, 8254–8263.
Li, Q., Means, R., Lang, S., and Jung, J. U. (2007). Downregulation of gamma interferon receptor 1 by Kaposi’s sarcoma-associated herpesvirus K3 and K5. J. Virol. 81, 2117–2127.
Liang, C., Feng, P., Ku, B., Dotan, I., Canaani, D., Oh, B. H., and Jung, J. U. (2006). Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 8, 688–699.
Liang, C., and Jung, J. U. (2010). Autophagy genes as tumor suppressors. Curr. Opin. Cell Biol. 22, 226–233.
Liang, C., Lee, J. S., and Jung, J. U. (2008). Immune evasion in Kaposi’s sarcoma-associated herpes virus associated oncogenesis. Semin. Cancer Biol. 18, 423–436.
Lin, R., Genin, P., Mamane, Y., Sgarbanti, M., Battistini, A., Harrington, W. J. Jr., Barber, G. N., and Hiscott, J. (2001). HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 20, 800–811.
Lin, Y. T., Kincaid, R. P., Arasappan, D., Dowd, S. E., Hunicke-Smith, S. P., and Sullivan, C. S. (2010). Small RNA profiling reveals antisense transcription throughout the KSHV genome and novel small RNAs. RNA 16, 1540–1558.
Liu, L., Eby, M. T., Rathore, N., Sinha, S. K., Kumar, A., and Chaudhary, P. M. (2002). The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J. Biol. Chem. 277, 13745–13751.
Loh, J., Huang, Q., Petros, A. M., Nettesheim, D., van Dyk, L. F., Labrada, L., Speck, S. H., Levine, B., Olejniczak, E. T., and Virgin, H. W. T. (2005). A surface groove essential for viral Bcl-2 function during chronic infection in vivo. PLoS Pathog. 1, e10. doi:10.1371/journal.ppat.0010010
Lu, F., Stedman, W., Yousef, M., Renne, R., and Lieberman, P. M. (2010). Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J. Virol. 84, 2697–2706.
Lum, J. J., DeBerardinis, R. J., and Thompson, C. B. (2005). Autophagy in metazoans: cell survival in the land of plenty. Nat. Rev. Mol. Cell Biol. 6, 439–448.
Maiuri, M. C., Zalckvar, E., Kimchi, A., and Kroemer, G. (2007). Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8, 741–752.
Manes, T. D., Hoer, S., Muller, W. A., Lehner, P. J., and Pober, J. S. (2010). Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins block distinct steps in transendothelial migration of effector memory CD4+ T cells by targeting different endothelial proteins. J. Immunol. 184, 5186–5192.
Mansouri, M., Viswanathan, K., Douglas, J. L., Hines, J., Gustin, J., Moses, A. V., and Fruh, K. (2009). Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 83, 9672–9681.
Mark, L., Proctor, D. G., Blackbourn, D. J., Blom, A. M., and Spiller, O. B. (2008). Separation of decay-accelerating and cofactor functional activities of Kaposi’s sarcoma-associated herpesvirus complement control protein using monoclonal antibodies. Immunology 123, 228–238.
Martin, D., Galisteo, R., Molinolo, A. A., Wetzker, R., Hirsch, E., and Gutkind, J. S. (2011). PI3Kgamma mediates Kaposi’s sarcoma-associated herpesvirus vGPCR-induced sarcomagenesis. Cancer Cell 19, 805–813.
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426.
Masood, R., Cai, J., Zheng, T., Smith, D. L., Naidu, Y., and Gill, P. S. (1997). Vascular endothelial growth factor/vascular permeability factor is an autocrine growth factor for AIDS-Kaposi sarcoma. Proc. Natl. Acad. Sci. U.S.A. 94, 979–984.
Masters, S. L., Simon, A., Aksentijevich, I., and Kastner, D. L. (2009). Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu. Rev. Immunol. 27, 621–668.
Meads, M. B., and Medveczky, P. G. (2004). Kaposi’s sarcoma-associated herpesvirus-encoded viral interleukin-6 is secreted and modified differently than human interleukin-6: evidence for a unique autocrine signaling mechanism. J. Biol. Chem. 279, 51793–51803.
Miao, E. A., Leaf, I. A., Treuting, P. M., Mao, D. P., Dors, M., Sarkar, A., Warren, S. E., Wewers, M. D., and Aderem, A. (2010). Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142.
Michael, D., and Oren, M. (2003). The p53-Mdm2 module and the ubiquitin system. Semin. Cancer Biol. 13, 49–58.
Micheletti, F., Monini, P., Fortini, C., Rimessi, P., Bazzaro, M., Andreoni, M., Giuliani, M., Traniello, S., Ensoli, B., and Gavioli, R. (2002). Identification of cytotoxic T lymphocyte epitopes of human herpesvirus 8. Immunology 106, 395–403.
Mills, K. R., Reginato, M., Debnath, J., Queenan, B., and Brugge, J. S. (2004). Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc. Natl. Acad. Sci. U.S.A. 101, 3438–3443.
Molden, J., Chang, Y., You, Y., Moore, P. S., and Goldsmith, M. A. (1997). A Kaposi’s sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J. Biol. Chem. 272, 19625–19631.
Montaner, S. (2007). Akt/TSC/mTOR activation by the KSHV G protein-coupled receptor: emerging insights into the molecular oncogenesis and treatment of Kaposi’s sarcoma. Cell Cycle 6, 438–443.
Montaner, S., Sodhi, A., Molinolo, A., Bugge, T. H., Sawai, E. T., He, Y., Li, Y., Ray, P. E., and Gutkind, J. S. (2003). Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi’s sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell 3, 23–36.
Montaner, S., Sodhi, A., Servitja, J. M., Ramsdell, A. K., Barac, A., Sawai, E. T., and Gutkind, J. S. (2004). The small GTPase Rac1 links the Kaposi sarcoma-associated herpesvirus vGPCR to cytokine secretion and paracrine neoplasia. Blood 104, 2903–2911.
Moore, P. S., Boshoff, C., Weiss, R. A., and Chang, Y. (1996). Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 274, 1739–1744.
Morgan, B. P., Marchbank, K. J., Longhi, M. P., Harris, C. L., and Gallimore, A. M. (2005). Complement: central to innate immunity and bridging to adaptive responses. Immunol. Lett. 97, 171–179.
Mori, Y., Nishimoto, N., Ohno, M., Inagi, R., Dhepakson, P., Amou, K., Yoshizaki, K., and Yamanishi, K. (2000). Human herpesvirus 8-encoded interleukin-6 homologue (viral IL-6) induces endogenous human IL-6 secretion. J. Med. Virol. 61, 332–335.
Mutocheluh, M., Hindle, L., Areste, C., Chanas, S. A., Butler, L. M., Lowry, K., Shah, K., Evans, D. J., and Blackbourn, D. J. (2011). Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor-2 inhibits type 1 interferon signalling by targeting interferon-stimulated gene factor-3. J. Gen. Virol. 92, 2394–2398.
Nachmani, D., Stern-Ginossar, N., Sarid, R., and Mandelboim, O. (2009). Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 5, 376–385.
Nakamura, H., Li, M., Zarycki, J., and Jung, J. U. (2001). Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J. Virol. 75, 7572–7582.
Nakamura, S., Murakami-Mori, K., Rao, N., Weich, H. A., and Rajeev, B. (1997). Vascular endothelial growth factor is a potent angiogenic factor in AIDS-associated Kaposi’s sarcoma-derived spindle cells. J. Immunol. 158, 4992–5001.
Nathan, J. A., and Lehner, P. J. (2009). The trafficking and regulation of membrane receptors by the RING-CH ubiquitin E3 ligases. Exp. Cell Res. 315, 1593–1600.
Neil, S. J., Zang, T., and Bieniasz, P. D. (2008). Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–430.
Okroj, M., Mark, L., Stokowska, A., Wong, S. W., Rose, N., Blackbourn, D. J., Villoutreix, B. O., Spiller, O. B., and Blom, A. M. (2009). Characterization of the complement inhibitory function of rhesus rhadinovirus complement control protein (RCP). J. Biol. Chem. 284, 505–514.
O’Neill, L. A., and Bowie, A. G. (2010). Sensing and signaling in antiviral innate immunity. Curr. Biol. 20, R328–R333.
Pardieu, C., Vigan, R., Wilson, S. J., Calvi, A., Zang, T., Bieniasz, P., Kellam, P., Towers, G. J., and Neil, S. J. (2010). The RING-CH ligase K5 antagonizes restriction of KSHV and HIV-1 particle release by mediating ubiquitin-dependent endosomal degradation of tetherin. PLoS Pathog. 6, e1000843. doi:10.1371/journal.ppat.1000843
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., Packer, M., Schneider, M. D., and Levine, B. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939.
Paulose-Murphy, M., Ha, N. K., Xiang, C., Chen, Y., Gillim, L., Yarchoan, R., Meltzer, P., Bittner, M., Trent, J., and Zeichner, S. (2001). Transcription program of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus). J. Virol. 75, 4843–4853.
Pestka, S., Krause, C. D., and Walter, M. R. (2004). Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202, 8–32.
Pfeffer, S., Zavolan, M., Grasser, F. A., Chien, M., Russo, J. J., Ju, J., John, B., Enright, A. J., Marks, D., Sander, C., and Tuschl, T. (2004). Identification of virus-encoded microRNAs. Science 304, 734–736.
Pyo, J. O., Jang, M. H., Kwon, Y. K., Lee, H. J., Jun, J. I., Woo, H. N., Cho, D. H., Choi, B., Lee, H., Kim, J. H., Mizushima, N., Oshumi, Y., and Jung, Y. K. (2005). Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 280, 20722–20729.
Qin, Z., Freitas, E., Sullivan, R., Mohan, S., Bacelieri, R., Branch, D., Romano, M., Kearney, P., Oates, J., Plaisance, K., Renne, R., Kaleeba, J., and Parsons, C. (2010). Upregulation of xCT by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in an environment of oxidative stress. PLoS Pathog. 6, e1000742. doi:10.1371/journal.ppat.1000742
Reimold, A. M., Ponath, P. D., Li, Y. S., Hardy, R. R., David, C. S., Strominger, J. L., and Glimcher, L. H. (1996). Transcription factor B cell lineage-specific activator protein regulates the gene for human X-box binding protein 1. J. Exp. Med. 183, 393–401.
Rezaee, S. A., Gracie, J. A., McInnes, I. B., and Blackbourn, D. J. (2005). Inhibition of neutrophil function by the Kaposi’s sarcoma-associated herpesvirus vOX2 protein. AIDS 19, 1907–1910.
Rivas, C., Thlick, A. E., Parravicini, C., Moore, P. S., and Chang, Y. (2001). Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 75, 429–438.
Sadler, A. J., and Williams, B. R. (2008). Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8, 559–568.
Salata, C., Curtarello, M., Calistri, A., Sartori, E., Sette, P., de Bernard, M., Parolin, C., and Palu, G. (2009). vOX2 glycoprotein of human herpesvirus 8 modulates human primary macrophages activity. J. Cell. Physiol. 219, 698–706.
Samols, M. A., Skalsky, R. L., Maldonado, A. M., Riva, A., Lopez, M. C., Baker, H. V., and Renne, R. (2007). Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 3, e65. doi:10.1371/journal.ppat.0030065
Sanchez, D. J., Coscoy, L., and Ganem, D. (2002). Functional organization of MIR2, a novel viral regulator of selective endocytosis. J. Biol. Chem. 277, 6124–6130.
Sarid, R., Sato, T., Bohenzky, R. A., Russo, J. J., and Chang, Y. (1997). Kaposi’s sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nat. Med. 3, 293–298.
Schmidt, K., Wies, E., and Neipel, F. (2011). Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 3 inhibits gamma interferon and major histocompatibility complex class II expression. J. Virol. 85, 4530–4537.
Seo, T., Park, J., Lee, D., Hwang, S. G., and Choe, J. (2001). Viral interferon regulatory factor 1 of Kaposi’s sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J. Virol. 75, 6193–6198.
Shin, Y. C., Nakamura, H., Liang, X., Feng, P., Chang, H., Kowalik, T. F., and Jung, J. U. (2006). Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 80, 2257–2266.
Shiratori, I., Yamaguchi, M., Suzukawa, M., Yamamoto, K., Lanier, L. L., Saito, T., and Arase, H. (2005). Down-regulation of basophil function by human CD200 and human herpesvirus-8 CD200. J. Immunol. 175, 4441–4449.
Si, H., and Robertson, E. S. (2006). Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J. Virol. 80, 697–709.
Skalsky, R. L., Samols, M. A., Plaisance, K. B., Boss, I. W., Riva, A., Lopez, M. C., Baker, H. V., and Renne, R. (2007). Kaposi’s sarcoma-associated herpesvirus encodes an ortholog of miR-155. J. Virol. 81, 12836–12845.
Sodhi, A., Montaner, S., Patel, V., Zohar, M., Bais, C., Mesri, E. A., and Gutkind, J. S. (2000). The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 60, 4873–4880.
Sperling, A. I., and Bluestone, J. A. (1996). The complexities of T-cell co-stimulation: CD28 and beyond. Immunol. Rev. 153, 155–182.
Spiller, O. B., Blackbourn, D. J., Mark, L., Proctor, D. G., and Blom, A. M. (2003a). Functional activity of the complement regulator encoded by Kaposi’s sarcoma-associated herpesvirus. J. Biol. Chem. 278, 9283–9289.
Spiller, O. B., Robinson, M., O’Donnell, E., Milligan, S., Morgan, B. P., Davison, A. J., and Blackbourn, D. J. (2003b). Complement regulation by Kaposi’s sarcoma-associated herpesvirus ORF4 protein. J. Virol. 77, 592–599.
Spiller, O. B., Mark, L., Blue, C. E., Proctor, D. G., Aitken, J. A., Blom, A. M., and Blackbourn, D. J. (2006). Dissecting the regions of virion-associated Kaposi’s sarcoma-associated herpesvirus complement control protein required for complement regulation and cell binding. J. Virol. 80, 4068–4078.
Sun, Q., Matta, H., Lu, G., and Chaudhary, P. M. (2006). Induction of IL-8 expression by human herpesvirus 8 encoded vFLIP K13 via NF-kappaB activation. Oncogene 25, 2717–2726.
Suzuki, T., Isobe, T., Kitagawa, M., and Ueda, K. (2010). Kaposi’s sarcoma-associated herpesvirus-encoded LANA positively affects on ubiquitylation of p53. Biochem. Biophys. Res. Commun. 403, 194–197.
Swanton, C., Mann, D. J., Fleckenstein, B., Neipel, F., Peters, G., and Jones, N. (1997). Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature 390, 184–187.
Takaoka, A., Tamura, T., and Taniguchi, T. (2008). Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer Sci. 99, 467–478.
Tamura, T., Yanai, H., Savitsky, D., and Taniguchi, T. (2008). The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584.
Thorburn, J., Moore, F., Rao, A., Barclay, W. W., Thomas, L. R., Grant, K. W., Cramer, S. D., and Thorburn, A. (2005). Selective inactivation of a Fas-associated death domain protein (FADD)-dependent apoptosis and autophagy pathway in immortal epithelial cells. Mol. Biol. Cell 16, 1189–1199.
Ting, J. P., Willingham, S. B., and Bergstralh, D. T. (2008). NLRs at the intersection of cell death and immunity. Nat. Rev. Immunol. 8, 372–379.
Umbach, J. L., and Cullen, B. R. (2010). In-depth analysis of Kaposi’s sarcoma-associated herpesvirus microRNA expression provides insights into the mammalian microRNA-processing machinery. J. Virol. 84, 695–703.
Van Damme, N., Goff, D., Katsura, C., Jorgenson, R. L., Mitchell, R., Johnson, M. C., Stephens, E. B., and Guatelli, J. (2008). The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3, 245–252.
Wang, X., Lybarger, L., Connors, R., Harris, M. R., and Hansen, T. H. (2004). Model for the interaction of gammaherpesvirus 68 RING-CH finger protein mK3 with major histocompatibility complex class I and the peptide-loading complex. J. Virol. 78, 8673–8686.
Weber, K. S., Grone, H. J., Rocken, M., Klier, C., Gu, S., Wank, R., Proudfoot, A. E., Nelson, P. J., and Weber, C. (2001). Selective recruitment of Th2-type cells and evasion from a cytotoxic immune response mediated by viral macrophage inhibitory protein-II. Eur. J. Immunol. 31, 2458–2466.
West, J., and Damania, B. (2008). Upregulation of the TLR3 pathway by Kaposi’s sarcoma-associated herpesvirus during primary infection. J. Virol. 82, 5440–5449.
West, J. A., Gregory, S. M., Sivaraman, V., Su, L., and Damania, B. (2011). Activation of plasmacytoid dendritic cells by Kaposi’s sarcoma-associated herpesvirus. J. Virol. 85, 895–904.
Wies, E., Hahn, A. S., Schmidt, K., Viebahn, C., Rohland, N., Lux, A., Schellhorn, T., Holzer, A., Jung, J. U., and Neipel, F. (2009). The Kaposi’s sarcoma-associated herpesvirus-encoded vIRF-3 inhibits cellular IRF-5. J. Biol. Chem. 284, 8525–8538.
Wilson, S. J., Tsao, E. H., Webb, B. L., Ye, H., Dalton-Griffin, L., Tsantoulas, C., Gale, C. V., Du, M. Q., Whitehouse, A., and Kellam, P. (2007). X box binding protein XBP-1s transactivates the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J. Virol. 81, 13578–13586.
Ye, F. C., Zhou, F. C., Xie, J. P., Kang, T., Greene, W., Kuhne, K., Lei, X. F., Li, Q. H., and Gao, S. J. (2008). Kaposi’s sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-kappaB-mediated suppression of the AP-1 pathway: a novel mechanism of virus control of latency. J. Virol. 82, 4235–4249.
Yu, F., Feng, J., Harada, J. N., Chanda, S. K., Kenney, S. C., and Sun, R. (2007). B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi’s sarcoma-associated herpesvirus. FEBS Lett. 581, 3485–3488.
Yu, Y., and Hayward, G. S. (2010). The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33, 863–877.
Yu, Y., Wang, S. E., and Hayward, G. S. (2005). The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22, 59–70.
Zhang, F., Wilson, S. J., Landford, W. C., Virgen, B., Gregory, D., Johnson, M. C., Munch, J., Kirchhoff, F., Bieniasz, P. D., and Hatziioannou, T. (2009). Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6, 54–67.
Zhao, J., Punj, V., Matta, H., Mazzacurati, L., Schamus, S., Yang, Y., Yang, T., Hong, Y., and Chaudhary, P. M. (2007). K13 blocks KSHV lytic replication and deregulates vIL6 and hIL6 expression: a model of lytic replication induced clonal selection in viral oncogenesis. PLoS ONE 2, e1067. doi:10.1371/journal.pone.0001067
Zhu, F. X., King, S. M., Smith, E. J., Levy, D. E., and Yuan, Y. (2002). A Kaposi’s sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. U.S.A. 99, 5573–5578.
Ziegelbauer, J. M. (2011). Functions of Kaposi’s sarcoma-associated herpesvirus microRNAs. Biochim. Biophys. Acta 1809, 623–630.
Ziegelbauer, J. M., Sullivan, C. S., and Ganem, D. (2009). Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat. Genet. 41, 130–134.
Keywords: KSHV, modulation of immune system, viral evasion strategies
Citation: Lee H-R, Brulois K, Wong L and Jung JU (2012) Modulation of immune system by Kaposi’s sarcoma-associated herpesvirus: lessons from viral evasion strategies. Front. Microbio. 3:44. doi: 10.3389/fmicb.2012.00044
Received: 18 December 2011; Paper pending published: 07 January 2012;
Accepted: 27 January 2012; Published online: 05 March 2012.
Edited by:
Keiji Ueda, Osaka University Graduate School of Medicine, JapanReviewed by:
Keiji Ueda, Osaka University Graduate School of Medicine, JapanFanxiu Zhu, Florida State University, USA
Copyright: © 2012 Lee, Brulois, Wong and Jung. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Hye-Ra Lee, Department of Molecular Microbiology and Immunology, University of Southern California, Harlyne J. Norris Cancer Research Tower, Room 5517, 1450 Biggy Street, Los Angeles, CA 90033, USA. e-mail: hyelee@usc.edu