




95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 02 March 2012
Sec. Virology
Volume 3 - 2012 | https://doi.org/10.3389/fmicb.2012.00073
This article is part of the Research Topic Research on Kaposi's sarcoma-associated herpesvirus: past, present, and future View all 17 articles
Soon after the discovery of Kaposi sarcoma (KS)-associated herpesvirus (KSHV), it was appreciated that this virus was associated with most cases of multicentric Castleman disease (MCD) arising in patients infected with human immunodeficiency virus. It has subsequently been recognized that KSHV–MCD is a distinct entity from other forms of MCD. Like MCD that is unrelated to KSHV, the clinical presentation of KSHV–MCD is dominated by systemic inflammatory symptoms including fevers, cachexia, and laboratory abnormalities including cytopenias, hypoalbuminemia, hyponatremia, and elevated C-reactive protein. Pathologically KSHV–MCD is characterized by polyclonal, IgM-lambda restricted plasmacytoid cells in the intrafollicular areas of affected lymph nodes. A portion of these cells are infected with KSHV and a sizable subset of these cells express KSHV lytic genes including a viral homolog of interleukin-6 (vIL-6). Patients with KSHV–MCD generally have elevated KSHV viral loads in their peripheral blood. Production of vIL-6 and induction of human (h) IL-6 both contribute to symptoms, perhaps in combination with overproduction of IL-10 and other cytokines. Until recently, the prognosis of patients with KSHV–MCD was poor. Recent therapeutic advances targeting KSHV-infected B cells with the anti-CD20 monoclonal antibody rituximab and utilizing KSHV enzymes to target KSHV-infected cells have substantially improved patient outcomes. Recently another KSHV-associated condition, the KSHV inflammatory cytokine syndrome (KICS) has been described. Its clinical manifestations resemble those of KSHV–MCD but lymphadenopathy is not prominent and the pathologic nodal changes of KSHV–MCD are absent. Patients with KICS exhibit elevated KSHV viral loads and elevation of vIL-6, homolog of human interleukin-6 and IL-10 comparable to those seen in KSHV–MCD; the cellular origin of these is a matter of investigation. KICS may contribute to the inflammatory symptoms seen in some patients with severe KS or primary effusion lymphoma. Additional research is needed to better define the clinical spectrum of KICS and its relationship to KSHV–MCD. In additional, research is needed to better understand the pathogenesis and epidemiology of both KICS and KSHV–MCD, as well as the optimal therapy for both of these disorders.
Kaposi sarcoma (KS)-associated herpesvirus (KSHV) is a gamma herpesvirus that is established as the causative agent of a several human illnesses, including KS, primary effusion lymphoma (PEL), and, in the setting of human immunodeficiency virus (HIV) infection, almost all cases of multicentric Castleman disease (MCD; Chang et al., 1994; Cesarman et al., 1995; Soulier et al., 1995). In KS, the predominant cell type is the spindle cell, which expresses endothelial markers, while the cell of origin in PEL and KSHV–MCD is of B-cell lineage. KS and KSHV–MCD are polyclonal in nature, while PEL is considered to be monoclonal. Like other herpesviruses, KSHV infection can exhibits both latent and lytic phases, distinguished by their viral gene expression patterns (Moore and Chang, 1998; Staskus et al., 1999; Sun et al., 1999). KSHV can establish latency in various cell types including endothelial cells and B-lymphocytes (Monini et al., 1999; Wu et al., 2006; Della Bella et al., 2008). Gene expression in latent phase is extremely limited (Jenner et al., 2001), with expressed genes primarily being directed to enhance cell survival, inhibit apoptosis, and evade the host immune response (Moore and Chang, 1998; Wen and Damania, 2010). Following activation of the lytic switch gene, RTA, encoded by open reading frame (ORF) 50, the virus enters the lytic phase, wherein the full complement of viral encoded genes is expressed and the host cellular machinery is redirected to the manufacture and assembly of progeny virions. The triggers of lytic activation in vivo remain unclear. It is speculated that intermittent lytic cycles may play a role in maintaining viral persistence in the host (Gregory et al., 2009).
The KSHV genome is notable for its molecular piracy of cellular genes (Moore et al., 1996; Ganem, 2010). These appear to promote viral survival, and there is evidence that they contribute to the pathogenesis of KSHV-associated diseases through interactions with the human cellular machinery and immune system. These gene mimics include viral homologs of interferon regulatory factors (IRF), ORF K9/vIRF-1, and ORF K11.5/vIRF-2; a viral homolog of human interleukin-6 (hIL-6) called viral IL-6 (vIL-6; encoded by ORF K2); and a constitutively active G-protein coupled receptor (GPCR) with pro-angiogenic and immunomodulatory functions encoded by ORF74 (Aoki et al., 2000; Jenner et al., 2001; Yarchoan, 2006). While vIL-6 is commonly referred to as a lytic gene, in some circumstances it may be expressed in the absence of full lytic activation (Chatterjee et al., 2002). KSHV also encodes at least 12 small non-coding microRNAs (miRNAs), whose functions are still to be comprehensively determined (Ganem and Ziegelbauer, 2008; Umbach and Cullen, 2010). There is evidence that these also interact with host proteins in ways that favor viral survival and may also induce expression of other host inflammatory proteins including IL-10 (Abend et al., 2010; Lei et al., 2010; Lu et al., 2010; Qin et al., 2010).
Viral IL-6 exhibits a number of important differences from its human counterpart. Their sequence homology is approximately 25% (Moore et al., 1996; Neipel et al., 1997). vIL-6 has a signaling potency 100- to 1000-fold lower that that seen with hIL-6 in in vitro models (Aoki et al., 2000). However, unlike hIL-6, vIL-6 is able to signal by forming a tetrameric complex with the IL-6 receptor superfamily gp130 component, which is widely expressed in human tissues, without requiring binding to the classical gp80 IL-6 receptor (Heinrich et al., 1998; Boulanger et al., 2004; Meads and Medveczky, 2004; Hu and Nicholas, 2006). As a result, vIL-6 may be able to initiate signaling in a wider variety of cell types than does hIL-6 (Osborne et al., 1999; Meads and Medveczky, 2004). In addition, there is in vitro evidence of vIL-6 autocrine action in lytically infected cells, though the function and significance of this activity is as yet uncertain (Chen et al., 2009a,b).
In the absence of HIV infection or other causes of immunodeficiency, most infection with KSHV is clinically silent, even when intermittent lytic activation cycles occur. However, as T cell immunity declines, KSHV-infected patients become increasingly likely to develop KSHV-associated tumors, particularly KS. It is not unusual for patients to manifest more than one KSHV-associated tumor. Each of these tumors has also been reported in other immunosuppressed and older individuals (Teruya-Feldstein et al., 1998). In KS tissue, KSHV is found predominantly in latent phase as assessed by immunohistochemical and genetic analysis of tumor tissue, while there is limited expression of KSHV viral genes, especially vIL-6, in PEL cells (Teruya-Feldstein et al., 1998; Jones et al., 1999; Staskus et al., 1999; Parravicini et al., 2000). By contrast, and unusually among herpesvirus-associated tumors, KSHV–MCD is associated with lytically active KSHV in a substantial proportion of KSHV-infected cells (Oksenhendler et al., 1996; Staskus et al., 1999; Parravicini et al., 2000). This lytic activation, and particularly the production of vIL-6, is thought to underlie its pathogenesis (Oksenhendler et al., 2000; Aoki et al., 2001a; Polizzotto et al., 2011).
There are several forms of Castleman disease, which historically have been distinguished by clinical findings (based on whether involved nodes are singular or multiple), pathologic findings, and more recently etiology (idiopathic or KSHV-associated; Cronin and Warnke, 2009). The idiopathic forms, as originally described, include a hyaline vascular form (usually unicentric) and a plasmacytoid form (usually multicentric). The KSHV-associated form is characteristically multicentric, with distinct pathologic features mirroring its unique etiology, as described below (Oksenhendler et al., 1996). Nearly all Castleman disease arising in the setting of HIV infection is KSHV-associated MCD (KSHV–MCD), while KSHV–MCD also rarely occurs in non-HIV infected individuals, particularly the immunocompromised. KSHV–MCD is considered to be rare, although its incidence is not well defined. By contrast to KS, there is evidence that MCD has become more common since the advent of combination anti-retroviral therapy (cART; Powles et al., 2009).
KSHV-MCD is characterized by intermittent flares of inflammatory symptoms, including fevers, fatigue, and cachexia, and edema, together with lymphadenopathy and/or splenomegaly (Oksenhendler et al., 1996; Stebbing et al., 2008). Gastrointestinal and respiratory symptoms are common. Flares are often severe and can be fatal. Common laboratory abnormalities include anemia, thrombocytopenia, hypoalbuminemia, hyponatremia, and elevated inflammatory markers such as C-reactive protein (CRP; Oksenhendler et al., 2000; Oksenhendler, 2009). The clinical course waxes and wanes, but until recently, has generally been fatal within 2 years of diagnosis, with patients succumbing to the severe inflammatory syndrome, concurrent infections, or progressing to lymphoma (Oksenhendler et al., 2000; Stebbing et al., 2008). The differential diagnosis of fever and adenopathy, even with other laboratory abnormalities, in the HIV infected individuals is broad. As a result, KSHV–MCD may be difficult to diagnose and is often missed. Also, while there are few reports of KSHV–MCD in Africa, there is recent evidence that it may be substantially underdiagnosed in this region (Ayers et al., 2011).
The diagnosis of KSHV–MCD is made pathologically by a lymph node biopsy demonstrating hypocellular germinal centers with a vascularized core and by a proliferation of polyclonal, IgM λ-restricted plasmacytoid cells in the interfollicular area (Oksenhendler et al., 1996; Du et al., 2001; Chadburn et al., 2008). A proportion of these cells are KSHV-infected, as demonstrated by expression of the KSHV latency associated nuclear antigen (LANA-1), and of these a subset express KSHV lytic genes, especially vIL-6 (Du et al., 2001; Figure 1). Areas of KS spindle cell proliferation in affected nodes are also not uncommon (Naresh et al., 2008). However, the majority of the cellular burden within affected nodes is comprised of uninfected plasmacytoid B-lymphocytes (Chadburn et al., 2008; Naresh et al., 2009).
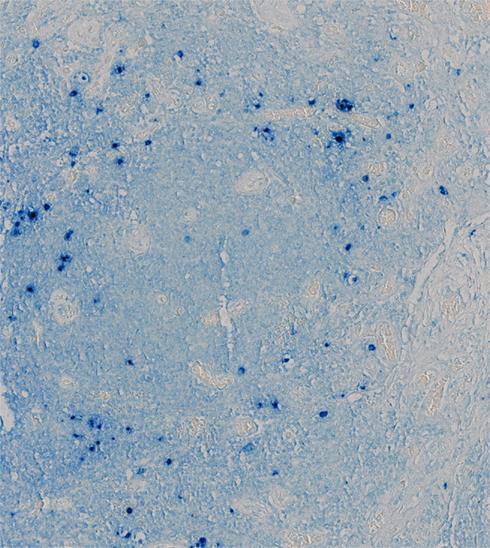
Figure 1. Lymph Node from a Patient with KSHV–MCD Stained for vIL-6. Germinal center of a lymph node from a patient with KSHV–MCD, stained with a monoclonal antibody to vIL-6 and an alkaline phosphatase substrate. Cells expressing vIL-6 stain in blue; these cells are located primarily in the mantle region around the germinal center. The tissue was studied under a protocol approved by the Institutional Review Board of the National Cancer Institute, and the patient gave written informed consent.
As noted, the symptom complex of KSHV–MCD is associated with inflammatory dysregulation attributable to KSHV lytic activation. Both production of vIL-6 and induction of host production of hIL-6 have separately been postulated to contribute to symptoms, perhaps in combination with enhanced production of human interleukin-10 (IL-10) and other human cytokines (Oksenhendler et al., 2000; Aoki et al., 2001a,b; Bower et al., 2009). Viral and human IL-6 over-expression in mouse models can each produce phenotypes resembling that of KSHV–MCD (Brandt et al., 1990; Aoki et al., 2001b). Symptomatic episodes (“flares”) are also associated with high KSHV viral loads (Oksenhendler et al., 2000; Polizzotto et al., 2011). Histopathologic evaluation of nodes involved by KSHV–MCD suggests that the cellular origins of vIL-6 and hIL-6 are largely distinct: involved nodes show vIL-6 production within the periphery of affected germinal center, in the lytic subset of the relatively small number of KSHV-infected cells. By contrast, the larger number of KSHV-uninfected cells in the core of affected nodes can produce hIL-6 but not vIL-6 (Chadburn et al., 2008).
In addition to these well-recognized KSHV-associated diseases, our group has recently described what appears to be a novel inflammatory symptom complex associated with KSHV infection (Uldrick et al., 2010a). In six patients with HIV and KSHV co-infection, but without any pathological evidence of MCD, we observed an inflammatory syndrome resembling that seen in MCD. These patients presented with abnormalities including fevers, sweats, fatigue, wasting, cytopenias, hypoalbuminemia, and hyponatremia, associated in some cases with lymphadenopathy or effusions (Table 1). Notably, four of the six patients also had severe KS. Each patient underwent a least one tissue biopsy to rule out KSHV–MCD as a cause of symptoms. None had pathologic evidence of KSHV–MCD (or KSHV-associated lymphoma), although abnormal KSHV-infected mononuclear cells were sometimes detected in biopsy specimens. None developed KSHV–MCD on follow-up, and one patient who died had no evidence of KSHV–MCD or lymphoma at autopsy (Uldrick et al., 2010a).
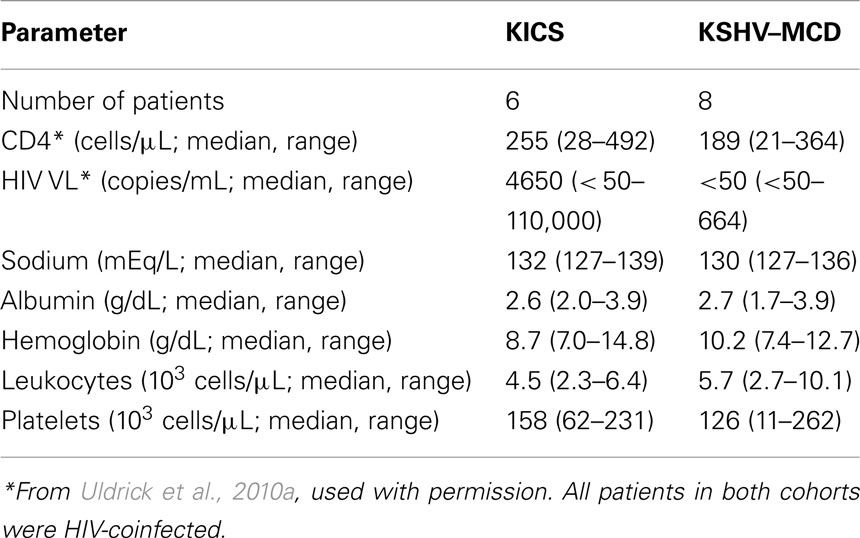
Table 1. Key clinical and laboratory findings in patients with KICS and KSHV–MICD.
We hypothesized that while these symptoms did not appear to be caused by KSHV–MCD, they might nonetheless similarly result from KSHV lytic activation and consequent cytokine production. On investigation, this cohort indeed exhibited KSHV viral burdens and cytokines profiles similar those seen with KSHV lytic activity in MCD flares, including substantial elevation of vIL-6 and hIL-6 (Figure 2; Uldrick et al., 2010a). Patients also exhibited markedly elevated KSHV viral loads in peripheral blood mononuclear cells. While the number of patients is small, the large differences in the immunologic and virologic milieu in these patients compared to KS patients without inflammatory symptoms was statistically significant (Figure 2). Our findings in this small cohort provide evidence that patients with lytically active KSHV may manifest systemic clinical symptoms even in the absence of the pathological changes diagnostic of MCD. These features are distinguishable from the chronic immune activation of HIV infection by the presence both of markedly elevated KSHV VLs in peripheral blood and by the detection of vIL-6 in KSHV inflammatory cytokine syndrome (KICS). As seen in Figure 2, neither of these features are evident in patients with KSHV and HIV co-infection who have KS but do not have systemic inflammatory symptoms. While in the original series all patients were HIV infected, we have subsequently observed this syndrome in patients without HIV.
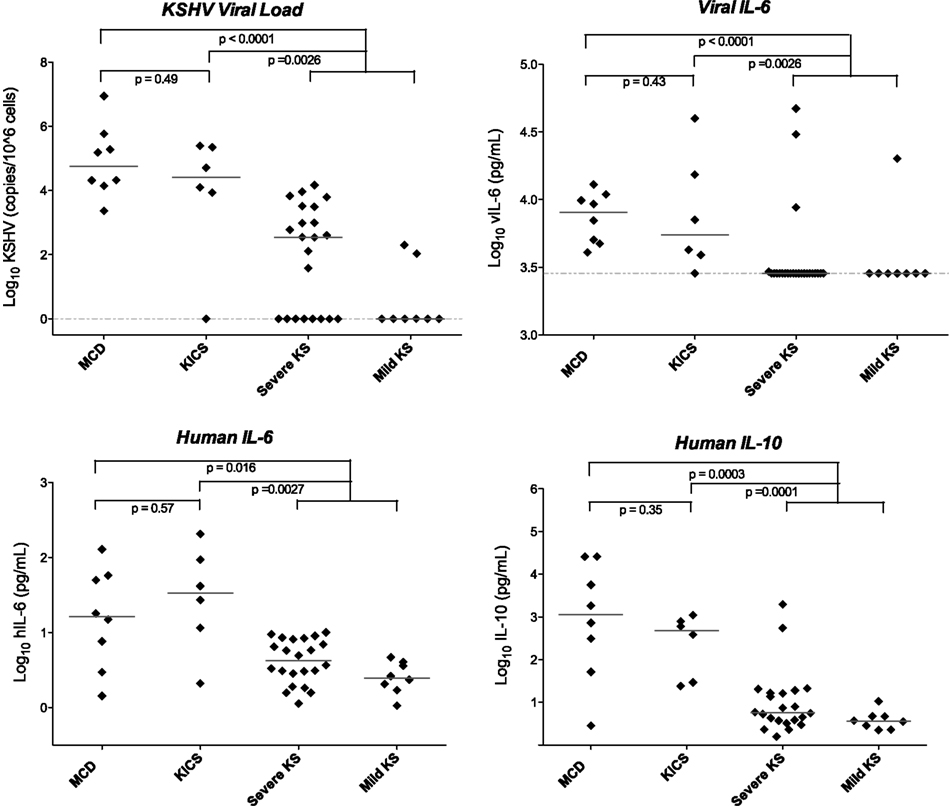
Figure 2. Viral and cytokine elevations in patients with KICS, KSHV–MCD, and KS. Statistically significant differences in KSHV viral load, viral, and human cytokines were seen in KICS patients compared with controls with KS alone, at levels comparable to those seen in KSHV–MCD. Horizontal lines indicate median value, and the dashed line indicates the lower limit of detection. Values are log transformed; viral and human IL-6 and IL-10 are picograms per milliliter; KSHV VL is copies/106 cells. Note that several of the patients with severe KS and elevated vIL-6 had laboratory abnormalities suggestive of KICS, but did not meet the operative case definition for this series. Modified from Uldrick et al. (2010a), with permission.
Based on this original cohort, we have proposed naming this putative clinical syndrome KICS. The assessment of this small retrospective cohort of patients suggest that clinical manifestations of KICS are protean, and none are specific to the syndrome. Rather, they likely reflect common endpoints of proinflammatory cytokine deregulation in the setting of KSHV lytic replication and pathologic expression of KSHV encoded genes such as vIL-6. These symptoms may also be observed in other inflammatory states, and at times can be confused clinically with sepsis. KICS is therefore a diagnosis of exclusion, requiring pathologic exclusion of MCD; careful evaluation to exclude alternate explanations for the clinical manifestations, such as serious intercurrent infection; demonstration of systemic inflammation; and demonstration of KSHV viral activity. Based on the initial case series, we have proposed a working case definition for KICS (Table 2), incorporating clinical, laboratory, and radiographic manifestations together with evidence of KSHV activation and systemic inflammation, and a requirement that KSHV–MCD be excluded on pathological examination. A clinical protocol to study this syndrome prospectively is underway at the National Cancer Institute, National Institutes of Health, based on this working definition (NCT01419561).

Table 2. Proposed working case definition of KSHV inflammatory cytokine syndrome.
The precise relationship of KICS to KSHV–MCD is unclear, and it is possible that KICS is a heterogeneous condition. Pathological evaluation and perhaps functional imaging with 18-[F]-fluorodeoxyglucose positron emission tomography (FDG-PET) may shed additional light on this question, and perhaps assist in identifying lymph nodes to biopsy that may reveal KSHV–MCD. We have preliminary evidence to show that in KSHV–MCD, pathologically involved nodes are detectable by FDG-PET, and that PET findings are distinct from those of suppressed HIV or intercurrent lymphoma (Polizzotto et al., 2010). Comparison of PET findings in KICS and KSHV–MCD may also provide a non-invasive means to assess disease activity and shed light on syndrome pathogenesis, including the possible cellular sites of disease activity. It is also possible that some patients with KICS may represent a prodrome to KSHV–MCD or an unusual or limited presentation of KSHV–MCD, and may eventually evolve into frank KSHV–MCD. Alternatively, certain patients may have another cellular source apart from lymphocytes responsible for the excess vIL-6 cytokine production, perhaps extensive KS spindle cells or KSHV-infected monocytes or macrophages. Notably, while several patients with KICS in the original cohort had severe KS, several other patients in this cohort with severe KS had less severe laboratory abnormalities suggestive of KICS in conjunction with detectable vIL-6 levels, although these abnormalities were not sufficiently severe for the patients to be identified as KICS based on the clinical criteria used for the initial case series. Similarly, PEL patients can exhibit severe inflammatory symptoms with disease activity, and may exhibit elevated KSHV viral loads (Uldrick et al., 2011b). Taken together, these observations raise the possibility that KSHV lytic activation and associated inflammatory clinical manifestations may be more common than appreciated in patients with other KSHV-associated diseases. It is possible that significant clinical overlap exists between KICS, KS, and PEL.
Among patients with KSHV and HIV co-infection, KICS may be an important cause of morbidity and mortality. KICS was associated with 50% mortality in our small series (which likely captured relatively severe cases). We also hypothesize that KS may be relatively refractory to therapy in patients with KICS because the cytokine excess can help drive KS spindle cell proliferation. In a small number of patients our group has seen with KICS and KS that was difficult to treat, strategies to reduce KSHV burden and intercurrent inflammation (including rituximab with cytotoxic chemotherapy) appear to have led to improvements in KS. One can thus hypothesize that KICS may in some cases be associated with the worsening of KS that can be observed after starting cART [KS immune reconstitution syndrome (IRIS)], although KICS has not been formally studied in this setting. Notably, some investigators report elevated hIL-6 levels in IRIS patients without KS, though KSHV and viral cytokines have not, to our knowledge, been assessed in relation to systemic symptoms (Stone et al., 2001).
There is no standard therapy for KSHV–MCD, and most articles describing therapy have consisted of case reports or very small series. Some of the approaches that have shown activity include interferon-alpha, surgical resection of the spleen, cytotoxic chemotherapy (either single agents or combinations), ganciclovir, glucocorticoids, and rituximab (Boulanger et al., 2004; Waterston and Bower, 2004; Dispenzieri and Gertz, 2005; Neuville et al., 2005; Bower et al., 2007; Mylona et al., 2008; Bower, 2010). Our group has explored selective targeting of KSHV–MCD plasmablasts based on their expression of KSHV lytic genes that phosphorylate certain drugs (Davis et al., 2007). One KSHV lytic gene, ORF36, encodes a phosphotransferase that activates ganciclovir to a cytotoxic triphosphate moiety, and another, ORF21, encodes a thymidine kinase that phosphorylates zidovudine (AZT), again to a cytotoxic moiety (Cannon et al., 1999; Gustafson et al., 2000). Members of our group have shown that PEL cells in which KSHV was lytically activated produced increased amounts of triphosphate moieties of AZT and ganciclovir and that AZT and ganciclovir had synergistic toxicity, at doses attainable in patients, against these PEL lines with activated KSHV (Davis et al., 2007). No comparable cytotoxicity was seen in non-KSHV-infected Burkitt lymphoma cell lines. Based on these in vitro results, we explored the combination of high dose zidovudine and valganciclovir, an oral pro-drug of ganciclovir, in 14 patients with HIV infection and KSHV–MCD (Uldrick et al., 2011a). This regimen delivered symptomatic control and normalization of correlates of lytic KSHV activity (including KSHV viral load, vIL-6, and hIL-6) in a high proportion of MCD patients. Responses were evaluated based on newly established criteria (NCI–HAMB criteria) for improvement in clinical symptoms, laboratory parameters (including hemoglobin, platelets, CRP, albumin, and sodium), and radiographic parameters (Uldrick et al., 2011a). Overall, 12 patients (86%) attained major clinical responses by these criteria, with seven patients (50%) attaining clinical complete responses, three (21%) attaining symptom free disease, and two (14%) attaining partial clinical response. In addition, seven patients (50%) attained major biochemical responses, with three (21%) attaining a complete response, four (29%) attaining a partial response. Finally, five patients (36%) attained a major radiographic responses including four (29%) with complete radiographic responses. Relapses were relatively common with this regimen, and the progression-free survival was 6 months. However, patients overall did well, and with 43 months of median follow-up, the overall survival was 86%.
Several recent studies have reported that KSHV–MCD can respond to therapy with the anti-CD20 monoclonal antibody rituximab (Corbellino et al., 2001; Marcelin et al., 2003; Ocio et al., 2005; Bower et al., 2007). It is postulated that in this setting, it acts by targeting the B-lymphocyte KSHV reservoirs, though it should be noted that the level of expression (weak or absent) of CD20 on lytic KSHV-infected B-lymphocytes themselves is contentious. Alternatively, it may target IL-6 production by other B cells that are stimulated by the KSHV-infected plasmablasts. However, different investigators have reported variable results with this agent, and two studies noted that its use was associated with a worsening of KS (Neuville et al., 2005; Bower et al., 2007). Because of this concern, and because many patients with MCD either have KS or are at risk of KS, we have explored the combination of rituximab and liposomal doxorubicin (Doxil) in KSHV–MCD (R/Dox). Liposomal doxorubicin has a high affinity for the pathogenic spindle cell in KS lesions, and, when available, is currently the standard of care for first line therapy of KS (Northfelt et al., 1998; Cianfrocca et al., 2010). It was also possible that it could help target the KSHV-infected MCD plasmablasts. In an ongoing study, we have treated 12 patients with this R/Dox regimen (Uldrick et al., 2010b). R/Dox was highly effective even in heavily pretreated patients. All achieved a clinical CR by NCI–HAMB criteria (after median two cycles therapy, range 1–5); nine (75) achieved a biochemical CR, one (8%) biochemical PR, and two (17%) biochemical SD; while best radiographic response (nodes and spleen) was CR in six (50%) and PR in six (50%). Toxicity was not marked, comprising predominantly infusion reactions and neutropenia.
Until recently, the expected survival of most patients with KSHV–MCD was 2 years or less (Oksenhendler et al., 2000). With the recent advances in therapy, along with the use of highly active anti-retroviral therapy (HAART) for those patients with HIV-associated KSHV–MCD, the overall prognosis of patients with KSHV–MCD has improved markedly, and a number of patients can now attain long remissions. Nonetheless, patients with flares of KSHV–MCD can rapidly become critically ill, with a clinical presentation similar to patients with sepsis, and may require careful monitoring, sometimes in an intensive care unit, during the initial treatment. Our experience is that some patients may benefit from short-term glucocorticoids during initial KSHV–MCD treatment, although patients should be probably weaned as soon as possible as glucocorticoids may mask intercurrent infections and can exacerbate KS (Gill et al., 1989). It should be noted, however, that there is no standard therapy for KSHV–MCD and it may be worthwhile to consider a variety of approaches, depending on disease severity and other parameters. Our group has been exploring the use of consolidation therapy after patients achieve remissions with several months of either interferon-alpha or high dose zidovudine with valganciclovir. Additional studies will be needed to explore new therapies, to establish the best therapy for KSHV–MCD, and perhaps through a combination of treatments, to establish the best way to optimize therapy for different patients with the goal of durable long-term remission.
Because of the small number of patients with KSHV–MCD, there is little commercial interest in developing drugs specifically for this disease, and it is unlikely that a therapeutic antibody to vIL-6 will be developed in the near future. However, an antibody to the IL-6 receptor is in clinical use for rheumatoid arthritis, while a different antibody against IL-6 is being evaluated in the setting of MCD not associated with KSHV. Because hIL-6 is markedly elevated and believed to contribute to disease pathogenesis in KSHV–MCD, antibodies targeting IL-6 signaling may have activity, and we are currently conducting a clinical study of tocilizumab, an anti-IL-6 receptor antibody in patients with symptomatic KSHV–MCD (NCT01441063).
Kaposi sarcoma-associated herpesvirus inflammatory cytokine syndrome is a newly described syndrome, and there is little clinical experience to guide therapy. In those cases where KICS accompanies KS or PEL, it is reasonable to hypothesize that treatment of the underlying tumor may help reduce the KSHV-associated cytokines. However, it should be noted that in the original series describing this syndrome KS was often quite difficult to treat, perhaps because of stimulation by the IL-6 or other cytokines. It may be worthwhile to consider treatment approaches to KICS similar to those used for KSHV–MCD. These may include, for example, antiviral drugs such as ganciclovir with activity against KSHV, the use of high doses of zidovudine or ganciclovir that can be activated by KSHV to toxic moieties as described above, the use of rituximab to kill B cells that may harbor KSHV or be producing cytokines, or the use of liposomal doxorubicin to kill KS spindle cells. In the original series of six patients of KICS, one patient received high dose zidovudine and valganciclovir, one patient received valganciclovir alone, and two received liposomal doxorubicin alone (Uldrick et al., 2010a). The patient who received zidovudine/valganciclovir demonstrated an initial improvement in laboratory parameters, though his clinical course was later complicated significantly by intercurrent illness, while one patient who received valganciclovir monotherapy had a transient clinical response. The two patients who received liposomal doxorubicin (both with KS requiring therapy) also had transient clinical responses.
Soon after the discovery of KSHV in 1994, it became apparent that KSHV-associated PEL and KSHV–MCD were separate disease entities. Since that time, we have learned much about the pathogenesis of these diseases, and for KSHV–MCD in particular, therapeutic advances have led to substantial improvement in survival. At the same time, many questions remain concerning KSHV–MCD and its relationship to other KSHV-induced diseases. More recently, KICS has been described as a new syndrome caused by KSHV; but there is much to be learned about this entity. What cells are primarily responsible for the cytokine overproduction in KICS, and what pathophysiologic mechanisms are responsible for its overproduction? What is the spectrum of disease, and what is its incidence and prevalence in different populations? How is it best managed? It is not unreasonable to hope that answers to many of these questions will be forthcoming in the next decade and that this research will help lead to improved tools to diagnose and treat these entities.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Intramural Research Program on the NIH, National Cancer Institute. The authors thank the patients who volunteered for the protocols and their families. The authors also acknowledge the help of Dr. Stefania Pittaluga, Dr. Denise Whitby, Dr. Richard Little, Dr. Victoria Wang, all of the NCI, the research nurses of HAMB, NCI, and the nurses, physicians, and medical staff of the Medical Oncology Branch of the NCI, and the medical staff of the NIH Clinical Center.
The authors wrote this paper as part of their duties as US Government employees and no Copyright is claimed by them. The work is in the public domain.
Abend, J. R., Uldrick, T., and Ziegelbauer, J. M. (2010). Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi’s sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J. Virol. 84, 12139–12151.
Aoki, Y., Jones, K. D., and Tosato, G. (2000). Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. J. Hematother. Stem Cell Res. 9, 137–145.
Aoki, Y., Tosato, G., Fonville, T. W., and Pittaluga, S. (2001a). Serum viral interleukin-6 in AIDS-related multicentric Castleman disease. Blood 97, 2526.
Aoki, Y., Yarchoan, R., Wyvill, K., Okamoto, S., Little, R. F., and Tosato, G. (2001b). Detection of viral interleukin-6 in Kaposi sarcoma-associated herpesvirus-linked disorders. Blood 97, 2173.
Ayers, L. W., Lukande, R., NDung’u, J. R., Chumba, D. K., and Turnwine, L. K. (2011). “Multicentric Castleman’s disease (MCD) and other related KSHV lymphoproliferations in East Africa,” in International Conference on Malignancies in AIDS and Other Acquired Immunodeficiencies, Bethesda, MD.
Boulanger, M. J., Chow, D., Brevnova, E., Martick, M., Sandford, G., Nicholas, J., and Garcia, K. C. (2004). Molecular mechanisms for viral mimicry of a human cytokine: activation of gp130 by HHV-8 interleukin-6. J. Mol. Biol. 335, 641–654.
Bower, M., Powles, T., Williams, S., Davis, T. N., Atkins, M., Montoto, S., Orkin, C., Webb, A., Fisher, M., Nelson, M., Gazzard, B., Stebbing, J., and Kelleher, P. (2007). Brief communication: rituximab in HIV-associated multicentric Castleman disease. Ann. Intern. Med. 147, 836–839.
Bower, M., Veraitch, O., Szydlo, R., Charles, P., Kelleher, P., Gazzard, B., Nelson, M., and Stebbing, J. (2009). Cytokine changes during rituximab therapy in HIV-associated multicentric Castleman disease. Blood 113, 4521–4524.
Brandt, S. J., Bodine, D. M., Dunbar, C. E., and Nienhuis, A. W. (1990). Dysregulated interleukin 6 expression produces a syndrome resembling Castleman’s disease in mice. J. Clin. Invest. 86, 592–599.
Cannon, J. S., Hamzeh, F., Moore, S., Nicholas, J., and Ambinder, R. F. (1999). Human herpesvirus 8-encoded thymidine kinase and phosphotransferase homologues confer sensitivity to ganciclovir. J. Virol. 73, 4786–4793.
Cesarman, E., Chang, Y., Moore, P. S., Said, J. W., and Knowles, D. M. (1995). Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332, 1186–1191.
Chadburn, A., Hyjek, E. M., Tam, W., Liu, Y., Rengifo, T., Cesarman, E., and Knowles, D. M. (2008). Immunophenotypic analysis of the Kaposi sarcoma herpesvirus (KSHV; HHV-8)-infected B cells in HIV+ multicentric Castleman disease (MCD). Histopathology 53, 513–524.
Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J., Knowles, D. M., and Moore, P. S. (1994). Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865–1869.
Chatterjee, M., Osborne, J., Bestetti, G., Chang, Y., and Moore, P. S. (2002). Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science 298, 1432–1435.
Chen, D., Choi, Y. B., Sandford, G., and Nicholas, J. (2009a). Determinants of secretion and intracellular localization of human herpesvirus 8 interleukin-6. J. Virol. 83, 6874–6882.
Chen, D., Sandford, G., and Nicholas, J. (2009b). Intracellular signaling mechanisms and activities of human herpesvirus 8 interleukin-6. J. Virol. 83, 722–733.
Cianfrocca, M., Lee, S., Von Roenn, J., Tulpule, A., Dezube, B. J., Aboulafia, D. M., Ambinder, R. F., Lee, J. Y., Krown, S. E., and Sparano, J. A. (2010). Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma: evidence of symptom palliation from chemotherapy. Cancer 116, 3969–3977.
Corbellino, M., Bestetti, G., Scalamogna, C., Calattini, S., Galazzi, M., Meroni, L., Manganaro, D., Fasan, M., Moroni, M., and Galli, M. (2001). Long-term remission of Kaposi sarcoma-associated herpesvirus-related multicentric Castleman disease with anti-CD20 monoclonal antibody therapy. Blood 98, 3473.
Cronin, D. M., and Warnke, R. A. (2009). Castleman disease: an update on classification and the spectrum of associated lesions. Adv. Anat. Pathol. 16, 236–246.
Davis, D. A., Singer, K. E., Reynolds, I. P., Haque, M., and Yarchoan, R. (2007). Hypoxia enhances the phosphorylation and cytotoxicity of ganciclovir and zidovudine in Kaposi’s sarcoma-associated herpesvirus infected cells. Cancer Res. 67, 7003–7010.
Della Bella, S., Taddeo, A., Calabrò, M. L., Brambilla, L., Bellinvia, M., Bergamo, E., Clerici, M., and Villa, M. L. (2008). Peripheral blood endothelial progenitors as potential reservoirs of Kaposi’s sarcoma-associated herpesvirus. PLoS ONE 3, e1520. doi: 10.1371/journal.pone.0001520
Dispenzieri, A., and Gertz, M. A. (2005). Treatment of Castleman’s disease. Curr. Treat. Options Oncol. 6, 255–266.
Du, M. Q., Liu, H., Diss, T. C., Ye, H., Hamoudi, R. A., Dupin, N., Meignin, V., Oksenhendler, E., Boshoff, C., and Isaacson, P. G. (2001). Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in Castleman disease and associated lymphoproliferative disorders. Blood 97, 2130–2136.
Ganem, D. (2010). KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 120, 939–949.
Ganem, D., and Ziegelbauer, J. (2008). MicroRNAs of Kaposi’s sarcoma-associated herpes virus. Semin. Cancer Biol. 18, 437–440.
Gill, P. S., Loureiro, C., Bernstein-Singer, M., Rarick, M. U., Sattler, F., and Levine, A. M. (1989). Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann. Intern. Med. 110, 937–940.
Gregory, S. M., West, J. A., Dillon, P. J., Hilscher, C., Dittmer, D. P., and Damania, B. (2009). Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. U.S.A. 106, 11725–11730.
Gustafson, E. A., Schinazi, R. F., and Fingeroth, J. D. (2000). Human herpesvirus 8 open reading frame 21 is a thymidine and thymidylate kinase of narrow substrate specificity that efficiently phosphorylates zidovudine but not ganciclovir. J. Virol. 74, 684.
Heinrich, P. C., Behrmann, I., Muller-Newen, G., Schaper, F., and Graeve, L. (1998). Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 334, 297–314.
Hu, F., and Nicholas, J. (2006). Signal transduction by human herpesvirus 8 viral interleukin-6 (vIL-6) is modulated by the nonsignaling gp80 subunit of the IL-6 receptor complex and is distinct from signaling induced by human IL-6. J. Virol. 80, 10874–10878.
Jenner, R. G., Albà, M. M., Boshoff, C., and Kellam, P. (2001). Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 75, 891–902.
Jones, K. D., Aoki, Y., Chang, Y., Moore, P. S., Yarchoan, R., and Tosato, G. (1999). Involvement of interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi’s sarcoma herpesvirus-associated infected primary effusion lymphoma cells. Blood 94, 2871–2879.
Lei, X., Bai, Z., Ye, F., Xie, J., Kim, C. G., Huang, Y., and Gao, S. J. (2010). Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 12, 193–199.
Lu, C. C., Li, Z., Chu, C. Y., Feng, J., Feng, J., Sun, R., and Rana, T. M. (2010). MicroRNAs encoded by Kaposi’s sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep. 11, 784–790.
Marcelin, A. G., Aaron, L., Mateus, C., Gyan, E., Gorin, I., Viard, J. P., Calvez, V., and Dupin, N. (2003). Rituximab therapy for HIV-associated Castleman disease. Blood 102, 2786–2788.
Meads, M. B., and Medveczky, P. G. (2004). Kaposi’s sarcoma-associated herpesvirus-encoded viral interleukin-6 is secreted and modified differently than human interleukin-6: evidence for a unique autocrine signaling mechanism. J. Biol. Chem. 279, 51793–51803.
Monini, P., Colombini, S., Sturzl, M., Goletti, D., Cafaro, A., Sgadari, C., Butto, S., Franco, M., Leone, P., and Fais, S. (1999). Reactivation and persistence of human herpesvirus-8 infection in B cells and monocytes by Th-1 cytokines increased in Kaposi’s sarcoma. Blood 93, 4044.
Moore, P. S., Boshoff, C., Weiss, R. A., and Chang, Y. (1996). Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 274, 1739–1744.
Moore, P. S., and Chang, Y. (1998). Kaposi’s sarcoma-associated herpesvirus-encoded oncogenes and oncogenesis. J. Natl. Cancer Inst. Monogr. 23, 65–71.
Mylona, E., Baraboutis, I. G., Georgiou, O., Rondogianni, D., Lekakis, L. J., Papastamopoulos, V., Apostolidis, I., and Skoutelis, A. T. (2008). Solid variant of primary effusion lymphoma in successfully treated HIV infection: a case report. Int. J. STD AIDS 19, 570–572.
Naresh, K. N., Rice, A. J., and Bower, M. (2008). Lymph nodes involved by multicentric Castleman disease among HIV-positive individuals are often involved by Kaposi sarcoma. Am. J. Surg. Pathol. 32, 1006–1012.
Naresh, K. N., Trivedi, P., Horncastle, D., and Bower, M. (2009). CD20 expression in the HHV-8-infected lymphoid cells in multicentric Castleman disease. Histopathology 55, 358–359.
Neipel, F., Albrecht, J. C., Ensser, A., Huang, Y. Q., Li, J. J., Friedman-Kien, A. E., and Fleckenstein, B. (1997). Human herpesvirus 8 encodes a homolog of interleukin-6. J. Virol. 71, 839.
Neuville, S., Agbalika, F., Rabian, C., Briere, J., and Molina, J. M. (2005). Failure of rituximab in human immunodeficiency virus-associated multicentric Castleman disease. Am. J. Hematol. 79, 337–339.
Northfelt, D. W., Dezube, B. J., Thommes, J. A., Miller, B. J., Fischl, M. A., Friedman-Kien, A., Kaplan, L. D., Du Mond, C., Mamelok, R. D., and Henry, D. H. (1998). Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi’s sarcoma: results of a randomized phase III clinical trial. J. Clin. Oncol. 16, 2445–2451.
Ocio, E. M., Sanchez-Guijo, F. M., Diez-Campelo, M., Castilla, C., Blanco, O. J., Caballero, D., and San Miguel, J. F. (2005). Efficacy of rituximab in an aggressive form of multicentric Castleman disease associated with immune phenomena. Am. J. Hematol. 78, 302–305.
Oksenhendler, E., Carcelain, G., Aoki, Y., Boulanger, E., Maillard, A., Clauvel, J. P., and Agbalika, F. (2000). High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric Castleman disease in HIV-infected patients. Blood 96, 2069–2073.
Oksenhendler, E., Duarte, M., Soulier, J., Cacoub, P., Welker, Y., Cadranel, J., Cazals-Hatem, D., Autran, B., Clauvel, J. P., and Raphael, M. (1996). Multicentric Castleman’s disease in HIV infection: a clinical and pathological study of 20 patients. AIDS 10, 61.
Osborne, J., Moore, P. S., and Chang, Y. (1999). KSHV-encoded viral IL-6 activates multiple human IL-6 signaling pathways. Hum. Immunol. 60, 921–927.
Parravicini, C., Chandran, B., Corbellino, M., Berti, E., Paulli, M., Moore, P. S., and Chang, Y. (2000). Differential viral protein expression in Kaposi’s sarcoma-associated herpesvirus-infected diseases: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Am. J. Pathol. 156, 743–749.
Polizzotto, M. N., Millo, C., Uldrick, T. S., Aleman, K., Whatley, M., Wyvill, K. M., O’Mahony, D., Marshall, V., Whitby, D., Steinberg, S. M., Little, R. F., and Yarchoan, R. (2010). “[18F]-fluoro-D-deoxyglucose-positron emission tomography findings in Kaposi sarcoma-associated herpesvirus associated multicentric Castleman disease in people with HIV: correlation with clinical, inflammatory and virologic parameters,” in Conference on Retroviruses and Opportunistic Infections, Boston, MA.
Polizzotto, M. N., Uldrick, T. S., Wang, V., Aleman, K., Wyvill, K. M., Marshall, V., Pittaluga, S., O’Mahony, D., Whitby, D., Tosato, G., Steinberg, S. M., Little, R. F., and Yarchoan, R. (2011). “Distinct human and viral interleukin-6 profiles and other viral and immunologic abnormalities in KSHV-associated multicentric Castleman disease: relationship with disease activity and individual disease manifestations,” in American Society of Hematology Annual Meeting, San Diego, CA, A1573.
Powles, T., Stebbing, J., Bazeos, A., Hatzimichael, E., Mandalia, S., Nelson, M., Gazzard, B., and Bower, M. (2009). The role of immune suppression and HHV-8 in the increasing incidence of HIV-associated multicentric Castleman’s disease. Ann. Oncol. 20, 775–779.
Qin, Z., Kearney, P., Plaisance, K., and Parsons, C. H. (2010). Kaposi’s sarcoma-associated herpesvirus (KSHV)-encoded microRNA specifically induce IL-6 and IL-10 secretion by macrophages and monocytes. J. Leukoc. Biol. 87, 25–34.
Soulier, J., Grollet, L., Oksenhendler, E., Cacoub, P., Cazals-Hatem, D., Babinet, P., d’Agay, M. F., Clauvel, J. P., Raphael, M., Degos, L., and Sigaux, F. (1995). Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86, 1276–1280.
Staskus, K. A., Sun, R., Miller, G., Racz, P., Jaslowski, A., Metroka, C., Brett-Smith, H., and Haase, A. T. (1999). Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. J. Virol. 73, 4181–4187.
Stebbing, J., Pantanowitz, L., Dayyani, F., Sullivan, R. J., Bower, M., and Dezube, B. J. (2008). HIV-associated multicentric Castleman’s disease. Am. J. Hematol. 83, 498–503.
Stone, S. F., Price, P., Brochier, J., and French, M. A. (2001). Plasma bioavailable interleukin-6 is elevated in human immunodeficiency virus-infected patients who experience herpesvirus-associated immune restoration disease after start of highly active antiretroviral therapy. J. Infect. Dis. 184, 1073–1077.
Sun, R., Lin, S. F., Staskus, K., Gradoville, L., Grogan, E., Haase, A., and Miller, G. (1999). Kinetics of Kaposi’s sarcoma-associated herpesvirus gene expression. J. Virol. 73, 2232.
Teruya-Feldstein, J., Zauber, P., Setsuda, J. E., Berman, E. L., Sorbara, L., Raffeld, M., Tosato, G., and Jaffe, E. S. (1998). Expression of human herpesvirus-8 oncogene and cytokine homologues in an HIV-seronegative patient with multicentric Castleman’s disease and primary effusion lymphoma. Lab. Invest. 78, 1637–1642.
Uldrick, T. S., Polizzotto, M. N., Aleman, K., O’Mahony, D., Wyvill, K. M., Wang, V., Marshall, V., Pittaluga, S., Steinberg, S. M., Tosato, G., Whitby, D., Little, R. F., and Yarchoan, R. (2011a). High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood 117, 6977–6986.
Uldrick, T. S., Polizzotto, M. N., Wyvill, K. M., Aleman, K., Marshall, V., Little, R. F., Filie, A., Raffeld, M., Steinberg, S. M., Pittaluga, S., Whitby, D., and Yarchoan, R. (2011b). “Patients with Kaposi sarcoma herpesvirus (KSHV)-associated aggressive lymphomas have markedly elevated interleukin-10, abnormalities in other inflammatory cytokines, and high circulating KSHV viral load,” in International Conference on Malignancies in AIDS and Other Acquired Immunodeficiencies, Bethesda, MD.
Uldrick, T. S., Wang, V., O’Mahony, D., Aleman, K., Wyvill, K. M., Marshall, V., Steinberg, S. M., Pittaluga, S., Maric, I., Whitby, D., Tosato, G., Little, R. F., and Yarchoan, R. (2010a). An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without multicentric Castleman disease. Clin. Infect. Dis. 51, 350–358.
Uldrick, T. S., Polizzotto, M. N., O’Mahony, D., Aleman, K., Wyvill, K. M., Steinberg, S. M., Pittaluga, S., Marshall, V., Whitby, D., Tosato, G., Little, R. F., and Yarchoan, R. (2010b). Rituximab combined with liposomal doxorubicin (R-Dox) in HIV-infected patients with severe Kaposi sarcoma-associated herpes virus (KSHV) associated multicentric Castleman disease (MCD). Infect. Agents Cancer 5(Suppl. 1), A72.
Umbach, J. L., and Cullen, B. R. (2010). In-depth analysis of Kaposi’s sarcoma-associated herpesvirus microRNA expression provides insights into the mammalian microRNA-processing machinery. J. Virol. 84, 695–703.
Waterston, A., and Bower, M. (2004). Fifty years of multicentric Castleman’s disease. Acta Oncol. 43, 698–704.
Wen, K. W., and Damania, B. (2010). Kaposi sarcoma-associated herpesvirus (KSHV): molecular biology and oncogenesis. Cancer Lett. 289, 140–150.
Wu, W., Vieira, J., Fiore, N., Banerjee, P., Sieburg, M., Rochford, R., Harrington, W., and Feuer, G. (2006). KSHV/HHV-8 infection of human hematopoietic progenitor (CD34+) cells: persistence of infection during hematopoiesis in vitro and in vivo. Blood 108, 141–151.
Keywords: Kaposi sarcoma-associated herpesvirus, human herpesvirus 8, multicentric Castleman disease, KSHV inflammatory cytokine syndrome, interleukin-6, human immunodeficiency virus
Citation: Polizzotto MN, Uldrick TS, Hu D and Yarchoan R (2012) Clinical manifestations of Kaposi sarcoma herpesvirus lytic activation: multicentric Castleman disease (KSHV–MCD) and the KSHV inflammatory cytokine syndrome. Front. Microbio. 3:73. doi: 10.3389/fmicb.2012.00073
Received: 30 December 2011; Paper pending published: 19 January 2012;
Accepted: 12 February 2012; Published online: 02 March 2012.
Edited by:
Keiji Ueda, Osaka University Graduate School of Medicine, JapanReviewed by:
Keiji Ueda, Osaka University Graduate School of Medicine, JapanCopyright: © 2012 This work is authored by Mark N. Polizzotto, Thomas S. Uldrick, Duosha Hu and Robert Yarchoan on behalf of the U.S. Government and, as regards Drs. Polizzotto, Uldrick, Hu and Yarchoan and the US government, is not subject to copyright protection in the United States. Foreign and other copyrights may apply. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Yarchoan, HIV/AIDS Malignancy Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Room 6N106, Building 10, Bethesda, MD 20892-1868, USA. e-mail:robert.yarchoan@mail.nih.gov
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.