 Yorihiro Nishimura* Hiroyuki Shimizu
Yorihiro Nishimura* Hiroyuki Shimizu
- Department of Virology II, National Institute of Infectious Diseases, Musashimurayama-shi, Tokyo, Japan
Human enterovirus species A (HEV-A) is one of the four species of HEV in the genus Enterovirus in the family Picornaviridae. Among HEV-A, coxsackievirus A16 (CVA16) and enterovirus 71 (EV71) are the major causative agents of hand, foot, and mouth disease (HFMD). Some other types of HEV-A are commonly associated with herpangina. Although HFMD and herpangina due to HEV-A are common febrile diseases among infants and children, EV71 can cause various neurological diseases, such as aseptic meningitis and fatal encephalitis. Recently, two human transmembrane proteins, P-selectin glycoprotein ligand-1 (PSGL-1) and scavenger receptor class B, member 2 (SCARB2), were identified as functional receptors for EV71 and CVA16. In in vitro infection experiments using the prototype HEV-A strains, PSGL-1 and SCARB2 could be responsible for the specific receptors for EV71 and CVA16. However, the involvement of both receptors in the in vitro and in vivo infections of clinical isolates of HEV-A has not been clarified yet. To elucidate a diverse array of the clinical outcome of HEV-A-associated diseases, the identification and characterization of HEV-A receptors may provide useful information in understanding the HEV-A pathogenesis at a molecular level.
Introduction
The genus Enterovirus within family Picornaviridae, non-enveloped viruses with a single-stranded RNA genome of positive polarity, is comprised of more than 100 serotypes (Pallansch and Roos, 2007). Human enteroviruses (HEVs) are presently classified into four species, HEV species A, B, C, and D (HEV-A, B, C, and D). At present, coxsackievirus A2 (CVA2), CVA3, CVA4, CVA5, CVA6, CVA7, CVA8, CVA10, CVA12, CVA14, CVA16, enterovirus 71 (EV71), EV76, EV89, EV90, EV91, and EV92 have been classified as HEV-A (Oberste et al., 2004, 2005, 2008). In addition, four types of simian enteroviruses are classified as HEV-A (Oberste et al., 2007). Although most enterovirus infections are asymptomatic, particular clinical manifestations are associated with specific types of enteroviruses (Pallansch and Roos, 2007). CVA2, CVA4, CVA5, CVA6, and CVA10 are commonly associated with herpangina. EV71 and CVA16 are major causative agents of hand, foot, and mouth disease (HFMD), a common febrile disease occurring mainly in young children, characterized by skin rash involving palms and soles, and ulcers on oral mucosa. Recently, HFMD outbreaks mainly due to CVA6 have been reported (Fujimoto et al., 2012). Clinical manifestations of HFMD caused by EV71, CVA6, and CVA16 are usually mild and self-limited. However, EV71 infection causes a diverse range of neurological diseases, such as aseptic meningitis, acute flaccid paralysis, brainstem encephalitis, and neurogenic pulmonary edema, and may result in long-term neurological sequelae, mainly in infants and young children (Alexander et al., 1994; McMinn, 2002; Modlin, 2007).
Recently, two human transmembrane proteins, P-selectin glycoprotein ligand-1 (PSGL-1; Nishimura et al., 2009) and scavenger receptor class B, member 2 (SCARB2; Yamayoshi et al., 2009), were identified as functional receptors for EV71 and CVA16 (Patel and Bergelson, 2009). In addition, annexin II (Yang et al., 2011), sialic acid (SA; Yang et al., 2009), and dendritic cell (DC)-specific ICAM3-grabbing non-integrin (DC-SIGN; Lin et al., 2009b) were found to be cellular factors involved in the early stages of EV71 infection. This review summarizes our current understanding of the EV71/CVA16 receptors and their role in HEV-A infection.
P-Selectin Glycoprotein Ligand-1
Patients with severe EV71-associated encephalitis and neurological pulmonary edema showed a significant depletion of T cells and high levels of proinflammatory cytokines (Lin et al., 2003; Wang et al., 2003), suggesting the possible involvement of lymphocytes in EV71 infection and the immunopathogenesis. Therefore, we generated a cDNA library from Jurkat T cells and used it for expression cloning to identify a receptor on lymphocytes that specifically binds to EV71. Finally we identified PSGL-1 as a functional EV71 receptor on Jurkat T cells (Nishimura et al., 2009).
P-selectin glycoprotein ligand-1 is a sialomucin leukocyte membrane protein expressed as a homodimer of disulfide-linked subunits and it can bind to three different selectins (P, E, and L; Sako et al., 1993; Laszik et al., 1996; Somers et al., 2000). The tissue distribution of PSGL-1 is restricted to myeloid, lymphoid, and dendritic lineages, and platelets. PSGL-1 is also expressed on DCs of lymph nodes and macrophages in the intestinal mucosa (Laszik et al., 1996), which could be the primary sites of EV71 replication. PSGL-1 plays critical roles in the tethering and rolling of leukocytes for the recruitment of cells from blood vessels to the sites of acute inflammation upon stimulation by infection.
We found that some representative EV71 strains bind to PSGL-1 but other strains did not (Nishimura et al., 2009). According to their PSGL-1 binding capability, we classified the EV71 isolates as PSGL-1 binding strains (EV71-PB) and PSGL-1-non-binding strains (EV71-non-PB). The replication of EV71-PB in Jurkat T cells was inhibited by anti-PSGL-1 monoclonal antibody (KPL1), indicating that EV71-PB replicated in Jurkat cells in a PSGL-1-dependent manner. On the other hand, EV71 replicated in non-leukocyte cells (such as RD cells) expressing little or no PSGL-1, and the replication was not affected by KPL1. Therefore we conclude that EV71 does not use PSGL-1 as the major cellular receptor on RD cells and other receptor(s), including SCARB2 or annexin II, may be responsible for EV71 infection in non-leukocyte cells expressing little or no PSGL-1.
Post-translational modifications of the N-terminal region of PSGL-1 contribute the efficient binding to selectins and chemokines (Pouyani and Seed, 1995; Sako et al., 1995; Wilkins et al., 1995; Liu et al., 1998; Hirata et al., 2004). In this region, there are a potential O-glycosylation residue (T57) and three potential tyrosine sulfation sites (Y46, Y48, and Y51). We demonstrated that tyrosine sulfation, not O-glycosylation, of the N-terminal region of PSGL-1 facilitates its binding to EV71-PB and viral replication in Jurkat T cells (Nishimura et al., 2010).
Coxsackievirus A16 is genetically and antigenically related to EV71 and is a major causative agent of HFMD as well as EV71 (Oberste et al., 2004). The inoculation of L-PSGL-1.1 cells, mouse L929 cells stably expressing human PSGL-1, with the prototype CVA16-G-10 strain induced faint cytopathic effects (CPE) (Nishimura et al., 2009). The replication of CVA16-G-10 was partially inhibited by KPL1 in L-PSGL-1.1 cells. This result indicated that the prototype CVA16 strain may use human PSGL-1 and another unidentified receptor(s) to infect L-PSGL-1.1 cells. CVA16-G-10 replication in Jurkat cells was not apparently inhibited by KPL1 (Nishimura et al., 2009), but significantly inhibited by a sulfation inhibitor, sodium chlorate (Nishimura et al., 2010). Therefore some sulfated molecules other than PSGL-1 might be involved in the replication of CVA16 in Jurkat cells. CVA16-G-10 would use unidentified receptor(s) to infect Jurkat T cells (Nishimura et al., 2009; Patel and Bergelson, 2009).
To investigate the PSGL-1-dependent replication phenotype of HEV-A, we tested the replication of 10 prototype HEV-A strains in L-PSGL-1.1 cells in the presence or absence of KPL1 (Figure 1A). On day 6 post-inoculation, there was no significant replication of CVA4, CVA5, CVA6, or CVA8 in L-bsd cells (blasticidin-resistant control L929 cells) or L-PSGL-1.1 cells. Although higher viral titers were found for CVA2 and CVA7 in L-PSGL-1.1 cells compared with those in L-bsd cells, replication was not affected by KPL1. These results suggest that CVA2 and CVA7 may infect to L-PSGL-1.1 cells in an alternative pathway via PSGL-1 or glycosylated PSGL-1, without the interaction between EV71-PB and the N-terminal region of PSGL-1 recognized by KPL1. We could not demonstrate any PSGL-1-dependent replication of the CVA3, CVA10, CVA12, and CVA14 strains in L-PSGL-1.1 cells, because they replicated even in PSGL-1 negative L-bsd cells as previously reported for certain HEV-A field isolates (Nadkarni and Deshpande, 2003; Yamayoshi et al., 2009).
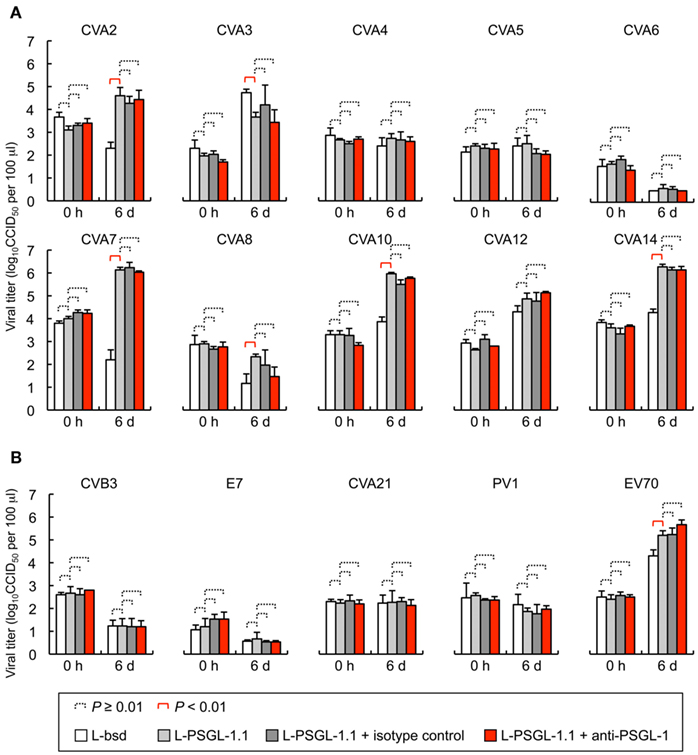
Figure 1. Human enteroviruses replication in L-PSGL-1.1 cells. (A) Replication of the HEV-A strains (Table 1) in L-PSGL-1.1 cells in the presence or absence of anti-PSGL-1 mAb (KPL1) or an isotype control. Cells were inoculated with viruses at 10 CCID50/cell for 1 h, washed, and incubated in the medium, as described previously (Nishimura et al., 2009). Cells were incubated at 34°C. For mAb inhibition, the cells were pretreated with 10 μg/ml mAb for 1 h, washed, and maintained in the medium with 10 μg/ml mAb. At the indicated time [just after infection (0 h) and 6 days postinfection (6 d)], the infected cells and supernatants were freeze-thawed and viral titers were determined by CCID50 titration using RD cells. The titers are expressed as the mean and error bars indicate SD of triplicate analyses. The mean viral titers were compared using Student’s t-test. P values < 0.01 were considered statistically significant. (B) Viral replication of HEV-B, C, and D in L-PSGL-1.1 cells. Replication of two HEV-B [CVB3-Nancy and echovirus 7 (E7)-Wallace] and two HEV-C strains [CVA21-Coe and poliovirus 1 (PV1)-Sabin 1], and one HEV-D (EV70-J670/71) strains in L-PSGL-1.1 cells in the presence or absence of KPL1 or an isotype control. The titers are expressed as the mean and error bars indicate SD of triplicate analyses. The mean viral titers were compared using Student’s t-test. P values < 0.01 were considered statistically significant.
Coxsackievirus A7 and CVA14 infection induced CPE in L-PSGL-1.1 cells, but not in L-bsd cells (Table 1). On the other hand, CVA7 and CVA14 induced CPE in L-Empty cells (puromycin-resistant control L929 cells; Table 1; Yamayoshi et al., 2009). The difference in the CPE induction by some HEV-A strains might be due to the maintenance or cultivation conditions of the mouse L929-derived cells regardless of the receptor expression of PSGL-1 or SCARB2. Some strains of HEV-A are able to infect mouse L929 cells regardless of expression of PSGL-1 or SCARB2 (Nadkarni and Deshpande, 2003; Yamayoshi et al., 2009). It is therefore impossible to determine receptor usage of HEV-A by simply investigating the susceptibility of mouse L929 cells expressing the putative cellular receptor. Receptor usage of HEV-A should be determined carefully by showing several lines of evidence such as acquisition of susceptibility by expressing a putative receptor in non-susceptible cells, loss of susceptibility by knocking down of the receptor in susceptible cells, and direct binding of the virus to the receptor, etc.
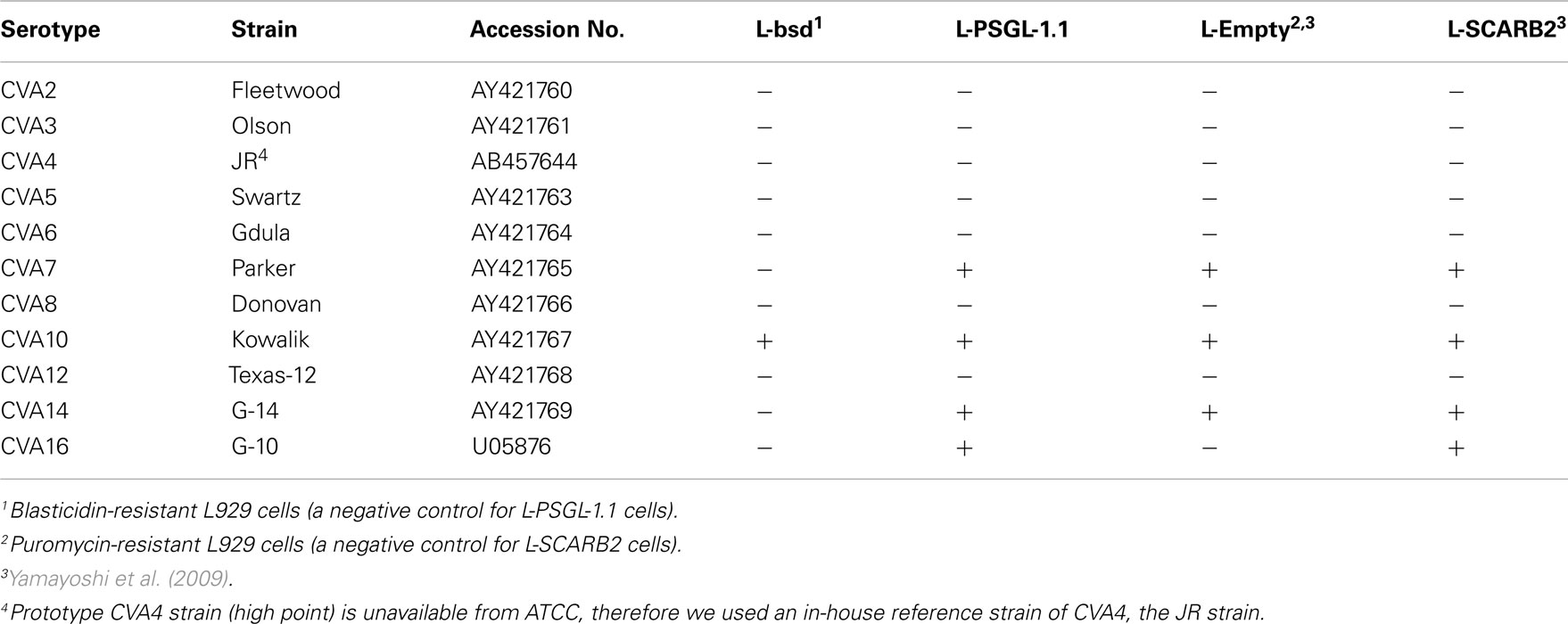
Table 1. Induction of CPE by the HEV-A strains.
L-PSGL-1.1 cells did not support PSGL-1-dependent replication of the HEV-B and HEV-C strains (Figure 1B); however, the prototype EV70 strain (HEV-D) replicated in L-PSGL-1.1 cells more efficiently than in L-bsd cells. Although EV70 replication was not affected by KPL1 (Figure 1B), we cannot exclude the possibility that EV70 utilizes α2,3-linked SA, which could be a receptor for EV70 (Nokhbeh et al., 2005).
Recently we reported that that four out of five EV71-PB strains replicated poorly in L-PSGL-1.1 cells (Miyamura et al., 2011). We found that EV71 variants, which were propagated once in L-PSGL-1.1 cells, have several possible adaptive mutations, including a putative amino acid determinant of the adaptive phenotype in L-PSGL-1.1 cells at VP2-149 (Miyamura et al., 2011). The results suggest that adaptive mutations, along with a PB phenotype, may facilitate efficient PSGL-1-dependent replication of the EV71 variants in L-PSGL-1.1 cells. It is possible that HEV-A strains other than EV71 also require adaptive mutations for efficient replication in L-PSGL-1.1 cells.
Scavenger Receptor Class B, Member 2
Yamayoshi et al. (2009) identified SCARB2 (also known as lysosomal integral membrane protein II, or CD36b like-2) as an EV71 receptor on RD cells, widely used for isolation of EV71 from clinical specimens. They transfected EV71-non-susceptible mouse L929 cells with the genomic DNA of RD cells and selected two cell clones that were susceptible for EV71 infection. By a transcriptome analysis, SCARB2 was identified as an EV71 receptor on RD cells.
Scavenger receptor class B, member 2 is a heavily N-glycosylated type III transmembrane protein consists from 478 amino acids and belongs to the CD36 family of scavenger receptor proteins (Fujita et al., 1992; Calvo et al., 1995). SCARB2 has a N-terminal transmembrane domain, a ∼400 amino acid lumeral domain, a C-terminal transmembrane domain, and a C-terminal cytoplasmic tail of ∼20 amino acids (Fujita et al., 1992). SCARB2 involves in an enlargement of early endosomes and late endosomes/lysosomes and an impairment of endocytic membrane out of the enlarged compartments (Kuronita et al., 2002). SCARB2 deficiency caused ureteric pelvic junction obstruction, deafness, and peripheral neuropathy in mice (Gamp et al., 2003). SCARB2 is expressed ubiquitously in human tissues (Eskelinen et al., 2003); therefore, it might be involved in systemic EV71 infections (Yamayoshi et al., 2009).
Human SCARB2 has 10 potential N-glycosylation sites (Fujita et al., 1992). But the carbohydrate chains of human SCARB2 are not essential for the interaction between EV71 and human SCARB2 (Yamayoshi and Koike, 2011). Experiments using a series of chimeric proteins between human and mouse SCARB2 identified that the amino acids 142–204 of human SCARB2 (encoded by human SCARB2 exon 4) are responsible for EV71 binding and infection (Yamayoshi and Koike, 2011).
Mouse L929 cells expressing human SCARB2 in the presence of puromycin (L-SCARB2 cells) permitted the replication of all EV71 strains tested, including the non-PB strains (Yamayoshi et al., 2009). CVA16 induced CPE in L-SCARB2 cells, whereas CVA2, CVA3, CVA4, CVA5, CVA6, CVA8, and CVA12 did not. CVA16 grew efficiently in L-SCARB2, whereas CVA2, CVA3, CVA4, CVA5, CVA6, CVA8, and CVA12 did not (Table 1). Yamayoshi et al. (2009) concluded that CVA16 also infect L-SCARB2 cells in a SCARB2-dependent manner and that infection with most other HEV-A is not dependent upon SCARB2. CVA7, CVA10, and CVA14 induced CPE in both L-Empty cells and L-SCARB2 cells (Yamayoshi et al., 2009). They could not determine whether the CPE induced by these viruses were due to human SCARB2–mediated infection.
Annexin II
Yang et al. (2011) identified annexin II as an EV71 VP1-binding protein on RD cells. Using a recombinant VP1 protein of EV71 fused with a calmodulin-binding peptide, they tried to identify VP1-binding proteins from the total cellular proteins of RD cells. A virus-overlay protein-binding assay followed by a mass spectrometry analysis identified annexin II as a VP1-binding protein.
Annexin II is a member of the annexin family – the multifunctional phospholipid-binding proteins. Annexin II on the surface of endothelial cells acts as a profibrinolytic coreceptor for both plasminogen and tissue plasminogen activator facilitating the generation of plasmin (Kim and Hajjar, 2002). The interaction to annexin II was specific to EV71; CVA16 did not bind to annexin II in the virus-overlay protein-binding assay (Yang et al., 2011).
Sialic Acid
Sialic acid is usually found as terminal monosaccharides on the glycan chains of glycolipids and glycoproteins (Varki and Varki, 2007). Coxsackievirus A24 variant (CVA24v) uses SA-containing glycoconjugates as attachment receptors on corneal cells (Nilsson et al., 2008). Yang et al. (2009) hypothesized that SA would be important for EV71 infection, as the transmission route of EV71 and CVA24v is fecal-oral and/or droplet-aerosol route. EV71 infection to DLD-1 intestinal cells was inhibited by an O-glycan synthesis inhibitor, but not by an N-glycan synthesis inhibitor. Sialidase treatment decreased EV71 replication in DLD-1 cells. Furthermore, DLD-1 cells co-cultured with SA-linked galactose significantly reduced the EV71 infection. Thus Yang et al. (2009) concluded that SA-linked glycans are EV71 receptors on DLD-1 cells. Recently, Neu5Acα2,3Gal disaccharides on PSGL-1 were reported as a candidate receptor of CVA24v (Mistry et al., 2011). It is unknown whether other enteroviruses, including HEV-A, recognize SA-containing glycans as the entry receptors.
Dendritic Cell-Specific ICAM3-Grabbing Non-Integrin
Dendritic cells play crucial roles in antiviral immunity by functioning as professional antigen-presenting cells to prime T cells and by secreting cytokines to modulate immune responses. In a mouse model of EV71 infection, DCs from the brains of EV71-infected, but not of uninfected, mice expressed viral antigen and primed T cells efficiently (Lin et al., 2009a). Lin et al. (2009b) reported that EV71 infection enhances mouse DCs to elicit protective immune response and also found that EV71 infects human immature DCs and that viral entry is partially inhibited by anti-DC-SIGN antibody. However, the direct interaction between EV71 and DC-SIGN is still unclear. It is essential to characterize the role of DC-SIGN and other receptors for EV71 in DCs for understanding the host immunological responses and immunopathogenesis of HEV-A including EV71.
Conclusion
Identification of PSGL-1 and SCARB2 as the cellular receptors for EV71 and CVA16 has advanced our understanding of the early stages of HEV-A infections at the molecular level. However, further experiments using clinical HEV-A isolates are necessary to clarify the general role of PSGL-1 and SCARB2 in HEV-A infection and their pathogenesis. Most of the prototype (laboratory-adapted) HEV-A strains other than EV71 and CVA16 may use unidentified receptor(s) to infect susceptible human cells such as RD cells. Characterization of the identified and unidentified HEV-A receptors is essential to understand the mechanism of HEV-A infection and development of a diverse array of the clinical outcomes of HEV-A-associated diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Junko Wada for excellent technical assistance. This work was supported by a Grant-in-Aid for Research on Emerging and Re-emerging Infectious Diseases and JSPS KAKENHI [Grant-in-Aid for Scientific Research (B), 22390092]. Yorihiro Nishimura and Hiroyuki Shimizu were supported in part by a Grant-in-Aid for the Promotion of Polio Eradication, from the Ministry of Health, Labour and Welfare, Japan.
References
Alexander, J. P. Jr., Baden, L., Pallansch, M. A., and Anderson, L. J. (1994). Enterovirus 71 infections and neurologic disease – United States, 1977–1991. J. Infect. Dis. 169, 905–908.
Calvo, D., Dopazo, J., and Vega, M. A. (1995). The CD36, CLA-1 (CD36L1), and LIMPII (CD36L2) gene family: cellular distribution, chromosomal location, and genetic evolution. Genomics 25, 100–106.
Eskelinen, E. L., Tanaka, Y., and Saftig, P. (2003). At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 13, 137–145.
Fujimoto, T., Iizuka, S., Enomoto, M., Abe, K., Yamashita, K., Hanaoka, N., Okabe, N., Yoshida, H., Yasui, Y., Kobayashi, M., Fujii, Y., Tanaka, H., Yamamoto, M., and Shimizu, H. (2012). Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg. Infect. Dis. 18, 337–339.
Fujita, H., Takata, Y., Kono, A., Tanaka, Y., Takahashi, T., Himeno, M., and Kato, K. (1992). Isolation and sequencing of a cDNA clone encoding the 85 kDa human lysosomal sialoglycoprotein (hLGP85) in human metastatic pancreas islet tumor cells. Biochem. Biophys. Res. Commun. 184, 604–611.
Gamp, A. C., Tanaka, Y., Lüllmann-Rauch, R., Wittke, D., D’Hooge, R., De Deyn, P. P., Moser, T., Maier, H., Hartmann, D., Reiss, K., Illert, A. L., von Figura, K., and Saftig, P. (2003). LIMP-2/LGP85 deficiency causes ureteric pelvic junction obstruction, deafness and peripheral neuropathy in mice. Hum. Mol. Genet. 12, 631–646.
Hirata, T., Furukawa, Y., Yang, B. G., Hieshima, K., Fukuda, M., Kannagi, R., Yoshie, O., and Miyasaka, M. (2004). Human P-selectin glycoprotein ligand-1 (PSGL-1) interacts with the skin-associated chemokine CCL27 via sulfated tyrosines at the PSGL-1 amino terminus. J. Biol. Chem. 279, 51775–51782.
Kim, J., and Hajjar, K. A. (2002). Annexin II: a plasminogen-plasminogen activator co-receptor. Front. Biosci. 7, d341–d348.
Kuronita, T., Eskelinen, E. L., Fujita, H., Saftig, P., Himeno, M., and Tanaka, Y. (2002). A role for the lysosomal membrane protein LGP85 in the biogenesis and maintenance of endosomal and lysosomal morphology. J. Cell Sci. 115, 4117–4131.
Laszik, Z., Jansen, P. J., Cummings, R. D., Tedder, T. F., McEver, R. P., and Moore, K. L. (1996). P-selectin glycoprotein ligand-1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood 88, 3010–3021.
Lin, T. Y., Hsia, S. H., Huang, Y. C., Wu, C. T., and Chang, L. Y. (2003). Proinflammatory cytokine reactions in enterovirus 71 infections of the central nervous system. Clin. Infect. Dis. 36, 269–274.
Lin, Y. W., Chang, K. C., Kao, C. M., Chang, S. P., Tung, Y. Y., and Chen, S. H. (2009a). Lymphocyte and antibody responses reduce enterovirus 71 lethality in mice by decreasing tissue viral loads. J. Virol. 83, 6477–6483.
Lin, Y. W., Wang, S. W., Tung, Y. Y., and Chen, S. H. (2009b). Enterovirus 71 infection of human dendritic cells. Exp. Biol. Med. (Maywood) 234, 1166–1173.
Liu, W. J., Ramachandran, V., Kang, J., Kishimoto, T. K., Cummings, R. D., and McEver, R. P. (1998). Identification of N-terminal residues on P-selectin glycoprotein ligand-1 required for binding to P-selectin. J. Biol. Chem. 273, 7078–7087.
McMinn, P. C. (2002). An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol. Rev. 26, 91–107.
Mistry, N., Inoue, H., Jamshidi, F., Storm, R. J., Oberste, M. S., and Arnberg, N. (2011). Coxsackievirus A24 variant uses sialic acid-containing O-linked glycoconjugates as cellular receptors on human ocular cells. J. Virol. 85, 11283–11290.
Miyamura, K., Nishimura, Y., Abo, M., Wakita, T., and Shimizu, H. (2011). Adaptive mutations in the genomes of enterovirus 71 strains following infection of mouse cells expressing human P-selectin glycoprotein ligand-1. J. Gen. Virol. 92, 287–291.
Nadkarni, S. S., and Deshpande, J. M. (2003). Recombinant murine L20B cell line supports multiplication of group A coxsackieviruses. J. Med. Virol. 70, 81–85.
Nilsson, E. C., Jamshidi, F., Johansson, S. M. C., Oberste, M. S., and Arnberg, N. (2008). Sialic acid is a cellular receptor for coxsackievirus A24 variant, an emerging virus with pandemic potential. J. Virol. 82, 3061–3068.
Nishimura, Y., Shimojima, M., Tano, Y., Miyamura, T., Wakita, T., and Shimizu, H. (2009). Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat. Med. 15, 794–797.
Nishimura, Y., Wakita, T., and Shimizu, H. (2010). Tyrosine sulfation of the amino terminus of PSGL-1 is critical for enterovirus 71 infection. PLoS Pathog. 6, e1001174. doi:10.1371/journal.ppat.1001174
Nokhbeh, M. R., Hazra, S., Alexander, D. A., Khan, A., McAllister, M., Suuronen, E. J., Griffith, M., and Dimock, K. (2005). Enterovirus 70 binds to different glycoconjugates containing α2,3-linked sialic acid on different cell lines. J. Virol. 79, 7087–7094.
Oberste, M. S., Jiang, X., Maher, K., Nix, W. A., and Jiang, B. (2008). The complete genome sequences for three simian enteroviruses isolated from captive primates. Arch. Virol. 153, 2117–2122.
Oberste, M. S., Maher, K., Michele, S. M., Belliot, G., Uddin, M., and Pallansch, M. A. (2005). Enteroviruses 76, 89, 90 and 91 represent a novel group within the species human enterovirus A. J. Gen. Virol. 86, 445–451.
Oberste, M. S., Maher, K., and Pallansch, M. A. (2007). Complete genome sequences for nine simian enteroviruses. J. Gen. Virol. 88, 3360–3372.
Oberste, M. S., Peñaranda, S., Maher, K., and Pallansch, M. A. (2004). Complete genome sequences of all members of the species Human enterovirus A. J. Gen. Virol. 85, 1597–1607.
Pallansch, M., and Roos, R. (2007). “Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses,” in Fields Virology, 5th Edn, eds D. M. Knipe and P. M. Howley (Philadelphia, PA: Lippincott Williams and Wilkins), 839–893.
Patel, K. P., and Bergelson, J. M. (2009). Receptors identified for hand, foot and mouth virus. Nat. Med. 15, 728–729.
Pouyani, T., and Seed, B. (1995). PSGL-1 recognition of P-selectin is controlled by a tyrosine sulfation consensus at the PSGL-1 amino terminus. Cell 83, 333–343.
Sako, D., Chang, X. J., Barone, K. M., Vachino, G., White, H. M., Shaw, G., Veldman, G. M., Bean, K. M., Ahern, T. J., Furie, B., Cumming, D. A., and Larsen, G. R. (1993). Expression cloning of a functional glycoprotein ligand for P-selectin. Cell 75, 1179–1186.
Sako, D., Comess, K. M., Barone, K. M., Camphausen, R. T., Cumming, D. A., and Shaw, G. D. (1995). A sulfated peptide segment at the amino terminus of PSGL-1 is critical for P-selectin binding. Cell 83, 323–331.
Somers, W. S., Tang, J., Shaw, G. D., and Camphausen, R. T. (2000). Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLeX and PSGL-1. Cell 103, 467–479.
Varki, N. M., and Varki, A. (2007). Diversity in cell surface sialic acid presentations: implications for biology and disease. Lab. Invest. 87, 851–857.
Wang, S. M., Lei, H. Y., Huang, K. J., Wu, J. M., Wang, J. R., Yu, C. K., Su, I. J., and Liu, C. C. (2003). Pathogenesis of enterovirus 71 brainstem encephalitis in pediatric patients: roles of cytokines and cellular immune activation in patients with pulmonary edema. J. Infect. Dis. 188, 564–570.
Wilkins, P. P., Moore, K. L., McEver, R. P., and Cummings, R. D. (1995). Tyrosine sulfation of P-selectin glycoprotein ligand-1 is required for high affinity binding to P-selectin. J. Biol. Chem. 270, 22677–22680.
Yamayoshi, S., and Koike, S. (2011). Identification of a human SCARB2 region that is important for enterovirus 71 binding and infection. J. Virol. 85, 4937–4946.
Yamayoshi, S., Yamashita, Y., Li, J., Hanagata, N., Minowa, T., Takemura, T., and Koike, S. (2009). Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat. Med. 15, 798–801.
Yang, B., Chuang, H., and Yang, K. D. (2009). Sialylated glycans as receptor and inhibitor of enterovirus 71 infection to DLD-1 intestinal cells. Virol. J. 6, 141.
Keywords: human enterovirus species A, enterovirus 71, receptor, PSGL-1, SCARB2
Citation: Nishimura Y and Shimizu H (2012) Cellular receptors for human enterovirus species A. Front. Microbio. 3:105. doi: 10.3389/fmicb.2012.00105
Received: 30 November 2011; Accepted: 02 March 2012;
Published online: 27 March 2012.
Edited by:
Kazutaka Terahara, National Institute of Infectious Diseases, JapanReviewed by:
Kazutaka Terahara, National Institute of Infectious Diseases, JapanSatoshi Koike, Tokyo Metropolitan Institute of Medical Science, Japan
Copyright: © 2012 Nishimura and Shimizu. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Yorihiro Nishimura, Department of Virology II, National Institute of Infectious Diseases, 4-7-1 Gakuen, Musashimurayama-shi, Tokyo 208-0011, Japan. e-mail: ynishi@nih.go.jp