
- 1Australian Institute of Marine Science, Townsville, QLD, Australia
- 2Northern Australian Marine Research Alliance, Arafura Timor Research Facility Darwin, Brinkin, NT, Australia
- 3Research Institute for the Environment and Livelihoods, Charles Darwin University, Casuarina, NT, Australia
- 4Australian Centre for Ecogenomics, University of Queensland, Brisbane, QLD, Australia
- 5Centre for Microbial Innovation, School of Biological Sciences, The University of Auckland, Auckland, New Zealand
- 6Department of Fisheries, Government of Western Australia, North Beach, WA, Australia
- 7Marine Ecology Research Centre, School of Environment, Science and Engineering, Southern Cross University, Lismore, NSW, Australia
Symbioses in marine sponges involve diverse consortia of microorganisms that contribute to the health and ecology of their hosts. The microbial communities of 13 taxonomically diverse Great Barrier Reef (GBR) sponge species were assessed by DGGE and 16S rRNA gene sequencing to determine intra and inter species variation in bacterial symbiont composition. Microbial profiling revealed communities that were largely conserved within different individuals of each species with intra species similarity ranging from 65–100%. 16S rRNA gene sequencing revealed that the communities were dominated by Proteobacteria, Chloroflexi, Acidobacteria, Actinobacteria, Nitrospira, and Cyanobacteria. Sponge-associated microbes were also highly host-specific with no operational taxonomic units (OTUs) common to all species and the most ubiquitous OTU found in only 5 of the 13 sponge species. In total, 91% of the OTUs were restricted to a single sponge species. However, GBR sponge microbes were more closely related to other sponge-derived bacteria than they were to environmental communities with sequences falling within 50 of the 173 previously defined sponge-(or sponge-coral) specific sequence clusters (SC). These SC spanned the Acidobacteria, Actinobacteria, Proteobacteria, Bacteroidetes, Chloroflexi, Cyanobacteria, Gemmatimonadetes, Nitrospira, and the Planctomycetes-Verrucomicrobia-Chlamydiae superphylum. The number of sequences assigned to these sponge-specific clusters across all species ranged from 0 to 92%. No relationship between host phylogeny and symbiont communities were observed across the different sponge orders, although the highest level of similarity was detected in two closely related Xestospongia species. This study identifies the core microbial inhabitants in a range of GBR sponges thereby providing the basis for future studies on sponge symbiotic function and research aiming to predict how sponge holobionts will respond to environmental perturbation.
Introduction
Associations between sponges and bacteria have existed for 600 million years making them one of the most ancient of all symbioses between microbes and metazoa (Wilkinson, 1984). Most sponges host diverse and abundant communities of microorganisms (Hentschel et al., 2006; Taylor et al., 2007; Webster et al., 2010), which contribute to host health, ecology and evolution [reviewed in (Taylor et al., 2007) and (Webster and Taylor, 2012)]. The importance of the relationship between sponges and their associated microbial communities is supported by the fact that microorganisms can contribute to more than 35% of the sponge biomass (Vacelet, 1975) and undertake diverse functional roles including nutrition, cycling of metabolites and host defense [reviewed in (Hentschel et al., 2012; Webster and Taylor, 2012)]. At least 32 bacterial phyla and candidate phyla have so far been found in sponges, either via cultivation or molecular characterization (Taylor et al., 2007; Schmitt et al., 2012b; Webster and Taylor, 2012). Some of these may be transient members of the sponge microbiota, potentially being filtered from the seawater as food. However, the “core” taxa, thought to represent the stable sponge inhabitants or true symbionts, include the Acidobacteria, Actinobacteria, Chloroflexi, Cyanobacteria, Gemmatimonadetes, Nitrospira, Proteobacteria, (especially Alpha, Delta, Gamma classes) and the candidate phylum “Poribacteria” (Taylor et al., 2007; Schmitt et al., 2012b).
The existence of sponge-specific microorganisms was first reported over a decade ago based on the finding that distantly related sponges from geographically separated regions shared microbes that had not been recovered from any other source, including the surrounding seawater (Hentschel et al., 2002; Taylor et al., 2007). Clusters of sponge-specific sequences were defined if groups of at least three rRNA gene sequences derived from more than two sponge species shared higher similarity to each other compared to sequences from other environments (Hentschel et al., 2002). Recently, the concept of sponge-specific microbes was comprehensively explored by performing phylogenetic analyses of all publicly available 16S and 18S rRNA gene sequences that originated from sponges. In total, 27% of all sponge-derived sequences fell into monophyletic, sponge-specific sequence clusters (SC) within the Bacteria, Archaea, and Fungi (Simister et al., 2012a) and additional sequences fell within clusters containing both sponge and coral derived sequences (SCC). Within the Bacteria, Chloroflexi, Cyanobacteria, “Poribacteria”, Betaproteobacteria, and Acidobacteria contained the highest abundance of these SCs. However, deep sequencing of diverse marine environments including seawater, sediments, hydrothermal vents, salt marshes, microbial mats, and corals has recently demonstrated that the rare biosphere may be a reservoir for some previously designated “sponge-specific” microbes (Webster et al., 2010; Taylor et al., 2012).
It is well established that particular sponge species can host stable microbial populations that are different to the communities in other species (Wilkinson et al., 1981; Taylor et al., 2004, 2005; Webster et al., 2004, 2010; Holmes and Blanch, 2007; Erwin and Thacker, 2008b; Turque et al., 2008; Lee et al., 2009; Erwin et al., 2011, 2012a,b; Schmitt et al., 2012b) and some molecular evidence also supports the potential for host-symbiont coevolution (Erpenbeck et al., 2002; Thacker and Starnes, 2003; Fan et al., 2012). Throughout the sponge literature and within this manuscript, the term “symbiosis” is used in its loosest possible definition, referring to the stable host-microbe association rather than implying any symbiotic function to the relationship. To test whether geographic or host-specific subpopulations of sponge microbes exist, Schmitt and colleagues performed 454 amplicon sequencing on 32 sponge species collected from eight locations around the world (Schmitt et al., 2012b). Whilst tropical sponge species shared more similarity in their microbial communities than they did with sub-tropical species, no other geographic or host phylogeny patterns were detected (Schmitt et al., 2012b). Interestingly, only a small “core” bacterial community was present in all 32 sponge species with the majority of bacterial operational taxonomic units (OTUs) occurring in only a single sponge species. Whilst the different sponge species were found to contain unique microbial populations, most of these sponge-associated bacteria were more closely related to other sponge-derived bacteria than they were to microbes from other environmental origins. These findings suggest that exploration of further sponge species may reveal additional novel sequences which would enhance our understanding of these sponge-specific microbial communities.
Over 8500 sponge species have been described globally (van Soest et al., 2012) with an estimated 2500 species occurring in Australian waters, although many of these remain to be formally described (Hooper, 2009). Baseline data on the composition and stability of symbiotic microbial communities is lacking for most sponge species and this knowledge gap makes it difficult to determine the role of microorganisms in sponge morbidity and mortality events. Sponge disease and mass mortalities have increased over recent years (Webster, 2007; Maldonado et al., 2010; Angermeier et al., 2011, 2012), including numerous reports of diseases that affect Great Barrier Reef (GBR) sponge species (Webster et al., 2002; Webster, 2007; Luter et al., 2010a,b). The study of sponge disease (including the identification of causative agents) is hampered by the high bacterial diversity within sponges combined with inadequate knowledge of the inherent microbial communities for most species. Understanding how sponges are likely to respond to a rapidly changing environment has also been a recent research focus (Webster et al., 2008; López-Legentil et al., 2010; Simister et al., 2012b), but scientists require a much better understanding of the diversity and specificity of microbes before assessments of environmental change can be validated.
Enhanced understanding of the ecological and evolutionary implications of sponge-bacterial symbioses gained over the past decade has prompted considerable new research in this field (Webster and Taylor, 2012), however, some regions such as the GBR remain largely understudied. The GBR is home to over 1500 sponge species although the microbial associates of the vast majority of species are yet to be explored. Here we surveyed the taxonomic composition of microbial associates in 13 of the most abundant and ecologically important GBR sponge species spanning five orders within the Porifera. By profiling replicate individuals per sponge species we are also able to address questions related to intra and inter species specificity.
Materials and Methods
Sample Collection
Sponge species were selected to encompass a wide range of genera covering five taxonomic orders: (1) Dictyoceratida included Luffariella variabilis, Coscinoderma matthewsi, Carteriospongia foliascens, and Ircinia sp. (2) Haplosclerida included Xestospongia testudinaria, Xestospongia exigua, and Haliclona sp. (3) Poescilosclerida included Coelocarteria singaporensis, Paramyxilla sp., and Hamigera sp. (4) Halichondrida included Stylissa sp. and Cymbastella coralliophila and (5) Spirophorida included Cinachyra sp. Triplicate individuals per species were collected on SCUBA from Orpheus Island (18°33.617′S; 146°29.077′E) at a depth range of 5–10 m in July 2010. Samples were photographed, immediately frozen in liquid nitrogen and stored at −80°C until subsequent molecular analysis.
DNA Extraction, DGGE and 16S rRNA Gene Sequence Analysis
DNA was extracted from all sponge samples using the Power Plant DNA Isolation kit, MoBio Laboratories (Carlsbad, CA) according to the manufacturer's protocol. The 16S rRNA gene from each sponge sample was amplified by PCR with primers 1055f and 1406r (Ferris et al., 1996) with the reverse primer incorporating the GC clamp (Muyzer et al., 1993). PCR reactions were performed as described by Ferris et al. (1996). Products from triplicate PCR reactions were combined and 15 μl applied to duplicate 8% w/v polyacrylamide (37.5:1) gels containing a 50–70% denaturing gradient of formamide and urea. Gels were electrophoresed at 60°C for 17 h in 1 × TAE buffer at 50 V using the Ingeny D-Code system. Gels were stained with 1 × Sybr Gold for 30 min, visualized under UV illumination and photographed. Representative bands from each species were excised and sequenced using the forward primer at Macrogen Inc., the PRISM Ready Reaction Kit (PE Applied Biosystems) and the ABI 310 and 373 automated sequencers.
For a more comprehensive phylogenetic comparison between the different sponge species, the 16S rRNA gene from triplicate samples of each sponge species was amplified by PCR with primers 63f and 1387r (Marchesi et al., 1998). PCR products were combined for all sponge replicates per species and ligated into the TOPO TA cloning vector (Invitrogen). Ligations were sent to Macrogen Inc for transformation and sequencing of up to 96 clones for each library. All sequences were submitted to Genbank under accession numbers JX455220—JX455743.
Data Analysis (DGGE)
16S rRNA gene fingerprints of the DGGE gels were manually assessed and a matrix was constructed using the presence (1) or absence (0) of a band in each sample. All analyses of the DGGE data were conducted using the Primer + PERMANOVA software package.
To determine if the microbial communities were significantly different between host species, Permutational Multivariate Analysis of Variance (PERMANOVA) was conducted using a Euclidean distance matrix and 9999 permutations (Anderson et al., 2008). The analysis consisted of one fixed factor (host species) for all samples. PERMANOVA pair-wise comparisons were subsequently made amongst the levels of each significant factor (using the Euclidean distance matrix and 9999 permutations).
Canonical analysis of principal coordinates (CAP) was then used to display differences in the microbial community structure among the different sponge species (Anderson et al., 2008) and DGGE bands with a correlation greater than 0.5 were overlaid on the plots as vectors. Cluster analysis was performed using Group Average and Bray–Curtis similarity.
Data Analysis (16S rRNA Gene Sequences)
Clone sequence quality was checked manually using Sequencher (Genesearch, Brisbane). Chimeric sequences were identified using the programs CHECK_CHIMERA (Maidak et al., 1999) and Bellerophon (Huber et al., 2004). Sequences were imported into the ARB software package (http://www.arb-home.de) (Ludwig et al., 1998), automatically aligned using WebAligner and manually edited. Phylogenetic trees were calculated with almost complete 16S rRNA (1400 bp) sequences for all close relatives of target sequences using the neighbor-joining and Maximum Parsimony methods in ARB. Partial sequences were subsequently imported to the tree without changing branch topology using the ARB parsimony-interactive method. Taxonomic bar charts were constructed using the phylogenetic assignments produced in ARB and plotted using Sigmaplot (V7.101, SPSS Inc.).
Distance matrices were generated in MOTHUR V1.8.1 (DeSantis et al., 2006) using a Jukes Cantor correction and OTU assignment, diversity estimates and OTU heatmaps were subsequently produced in MOTHUR using a distance of 0.03 (Schloss et al., 2009). All published, sponge-derived sequences within a manually curated SILVA reference database were used to assign clone sequences to previously defined sponge-specific and sponge/coral-specific SC (Simister et al., 2012a). In brief, a BLAST search against the curated SILVA database was performed for each clone sequence and the 10 best hits were aligned so that sequence similarity could be determined. A 75% sequence similarity threshold was applied to assign the clone sequences to SC or SCCs.
Results
A microbial profiling approach revealed that GBR sponge-associated microbial communities are highly conserved within each species (Figures 1, 2), but differ significantly between host species (p = 0.0001, Table 1, Figures 1, 2). Ordination analysis of the DGGE data showed clear clustering of replicate samples for all GBR sponge species (Figure 1). In particular, microbial communities in triplicate individuals of C. coralliophila, and Cinachyra sp. were identical within each species as were two of the three C. singaporensis replicates (Figure 2). L. variabilis had the greatest intra-species variation but individuals still shared >65% similarity in microbial community composition (Figure 2). Microorganisms that contributed most to the discrimination (correlation greater than 0.5, Figure 1) between host sponges were affiliated with the Archaea and the bacterial phyla Proteobacteria, Chloroflexi and Cyanobacteria with BLAST analysis showing that most sequences had highest similarity to microbes previously retrieved from sponges and corals (Table 2).
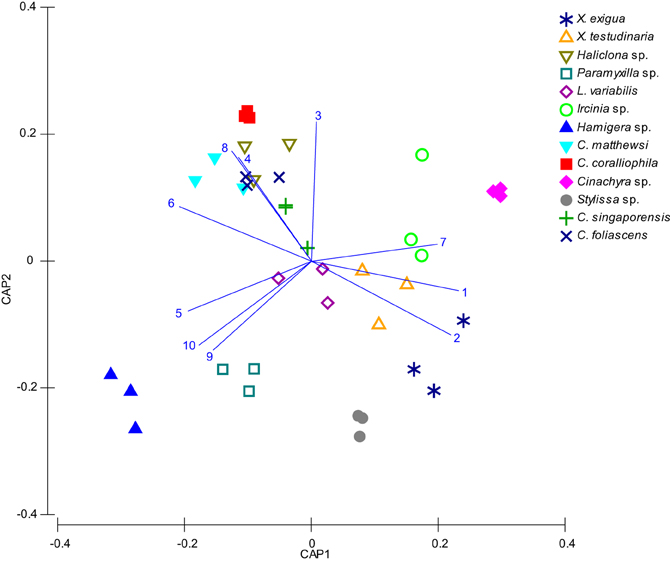
Figure 1. CAP Analysis of microbial communities (based on DGGE banding pattern data) for different sponge species which were all sampled in triplicate (depicted by each shape). DGGE bands with a correlation greater than 0.5 have been overlaid on the plots as vectors and the sequence identity of these are reported in Table 2.
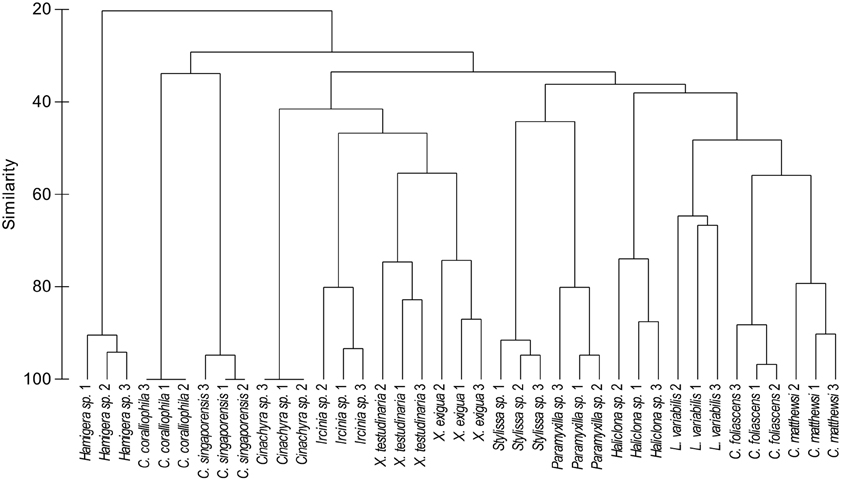
Figure 2. Cluster analysis (based on DGGE banding pattern data) for different sponge species using Group Average and Bray Curtis similarity.
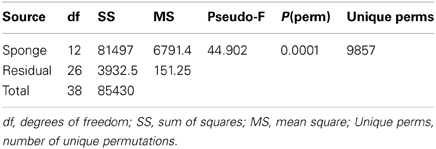
Table 1. Summary PERMANOVA statistics (as per Anderson et al., 2008) of the DGGE derived symbiont assemblages reveal that microbial communities differ significantly between host sponge species (p = 0.0001).
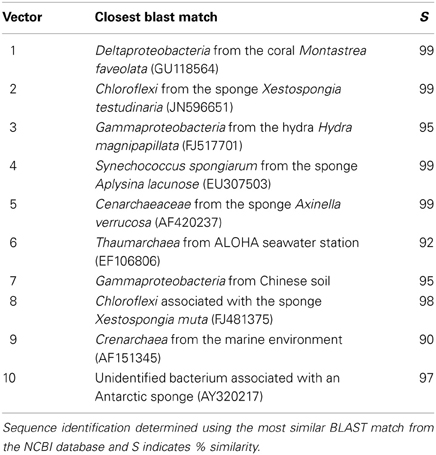
Table 2. Microbial sequences detected by DGGE that discriminate between sponge hosts (correlation >0.5).
Pairwise comparisons revealed interesting patterns in the microbial assemblages amongst the sampled sponges (Table 3). For example, L. variabilis had a “generalist” microbial community that did not differ significantly from many of the other sampled sponges, whereas the microbial communities in Hamigera sp., Cinachyra sp., C. coralliophila, and C. singaporensis were significantly different to most other sponge species (Table 3). Cluster analysis confirmed that the microbial community in Hamigera sp. shared only 20% similarity to the other species (Figure 2). The highest similarity detected between host species (55%) occurred in the two Xestospongia species (Figure 2). Despite the similarity between X. testudinaria and X. exigua, no discernable clustering according to higher host taxonomy was evident. Three of the four Dictyoceratid species (L. variabilis, C. matthewsi, and C. foliascens) grouped together but this grouping shared less than 50% similarity in their microbial communities (Figure 2).

Table 3. Significant PERMANOVA pairwise comparisons of the microbial communities amongst the different sponge species.
16S rRNA gene libraries revealed diverse bacterial assemblages with clear taxonomic differences in the microbial communities inhabiting each sponge species (Figure 3). Paramyxilla sp., C. matthewsi, Stylissa sp., Haliclona sp., and Hamigera sp. were dominated by Gammaproteobacteria; Cinachyra sp. and X. exigua were dominated by Chloroflexi with a large proportion of Actinobacteria; L. variabilis and X. testudinaria were dominated by Acidobacteria and C. coralliophila, C. singaporensis, and C. foliascens contained a large abundance of Cyanobacteria (although C. foliascens was actually dominated by Bacteroidetes). Ircinia sp. had a more diverse and even distribution of bacterial phyla and classes within the Proteobacteria (Figure 3). No patterns according to host taxonomy were observed in the microbial community composition.
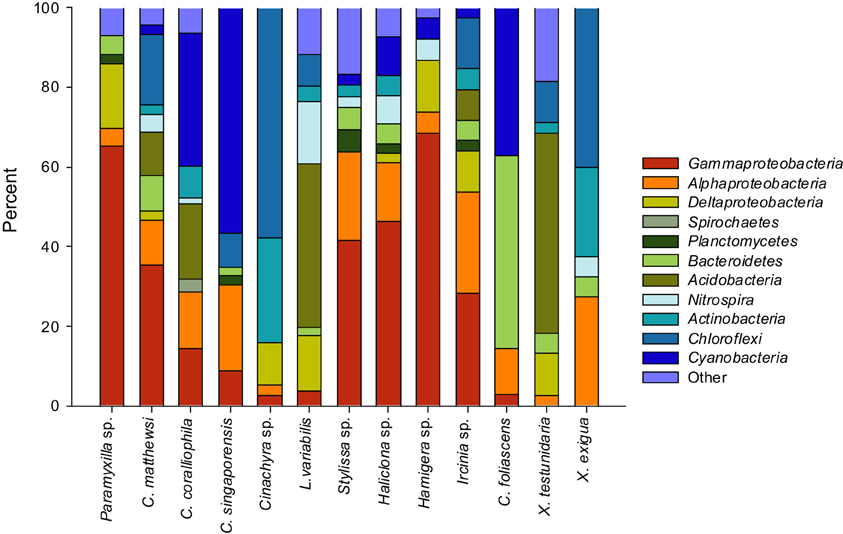
Figure 3. Bar charts showing the relative abundance of each bacterial phyla (and class for the Proteobacteria) within each host species.
Whilst it was not possible to accurately estimate total sponge microbial diversity due to the low sequencing depth, comparison of diversity estimates within the GBR sponges (Table 4) indicated that X. testudinaria, C. matthewsi and L. variabilis hosted higher bacterial diversity, whereas Cinachyra sp., C. foliascens, Hamigera sp., and C. singaporensis had lower microbial diversity. Whilst two of the three species with highest bacterial diversity (C. matthewsi and L. variabilis) resided within the Dictyoceratida, the low microbial diversity species encompassed three different taxonomic orders. No observed OTUs (97% sequence similarity) were common to all sponge species (Figure 4) and the most ubiquitous OTU was shared between only five sponge species (Stylissa sp., Haliclona sp., Ircinia sp., C. coraliophilla, and Hamigera sp.). In total, 91% of the observed OTUs were detected in a single sponge species, 6% occurred across two species, 2% across three species and only 1% occurred in four or more species. These results indicate that the vast majority of dominant microbes (i.e., those detected at low sequencing depth) are species-specific (Figure 4).
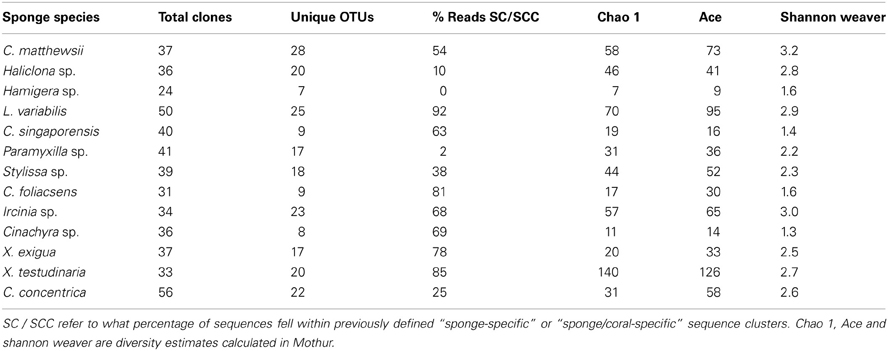
Table 4. Diversity indices calculated from sequences of 16S rRNA genes using a 97% sequence similarity threshold.
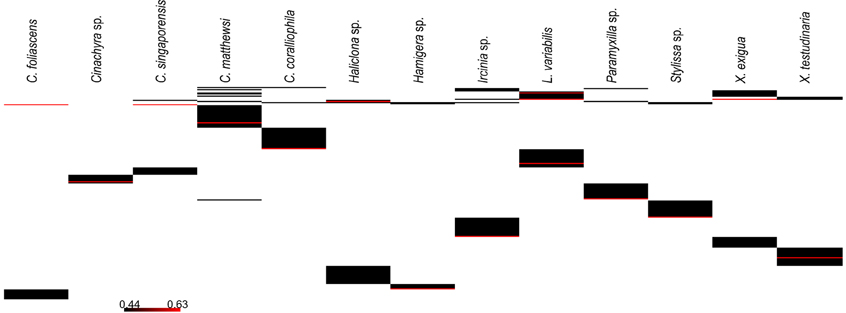
Figure 4. OTU heatmaps generated with a distance of 0.03 for clone libraries from each sponge species. Whilst there are a small number of shared OTUs, the vast majority are species-specific.
Bacterial sequences retrieved from the 13 GBR sponge species fell within 50 of the 173 previously defined SC and SCCs. These SC and SCCs encompassed nine phyla including Acidobacteria, Actinobacteria, Proteobacteria, Bacteroidetes, Chloroflexi, Cyanobacteria, Gemmatimonadetes, Nitrospira, and the Planctomycetes-Verrucomicrobia-Chlamydiae superphylum (Figure 5). The number of sequences falling within SC/SCCs varied greatly between sponges, ranging from 0% in Hamigera sp. to 92% of the total sequences in L. variabilis (Table 4). No SC/SCCs were represented in more than 4 of the 13 sponge species with the most ubiquitous clusters being SC122 (Deltaproteobacteria) and SCC 17 (Nitrospira) which occurred in four different sponge species (Figure 5).
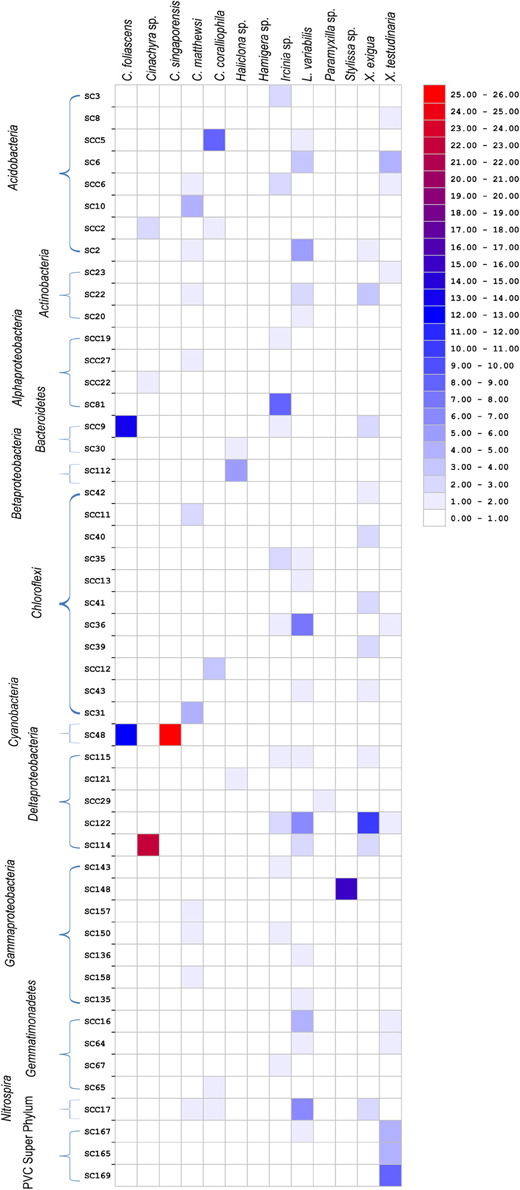
Figure 5. Heatmap showing the distribution of clone sequences phylogenetically assigned to previously described “sponge-specific” 16S rRNA gene sequence clusters (Simister et al., 2012a) among the 13 different sponge species. Clusters with an SC prefix contain sequences previously reported only from sponges; SCC prefix signifies clusters containing only sponge- and coral-derived sequences. Units are the percentage of total sequences from each sponge species that fall within SC/SCC.
For the five species dominated by Gammaproteobacteria; Paramyxilla sp., Haliclona sp. and Hamigera sp. contained no sequences residing within Gammaproteobacteria SC/SCCs whereas the Gammaproteobacteria in C. matthewsi were spread across three different SCs and all of the Gammaproteobacteria within Sylissa sp. clustered within a single SC. Whilst C. foliascens, C. singaporensis and C. coralliophila all contained abundant Cyanobacteria, only the first two species contained sequences within Cyanobacteria SC. Both Xestospongia species contained abundant sequences within SC/SCCs from multiple bacterial phyla. As illustrated in Figure 3, C. matthewsi, Ircinia sp. and L. variabilis had diverse bacterial assemblages and this was also evident by the relatively broad spread of SC/SCCs across taxa (Figure 5).
Discussion
The microbial communities of taxonomically diverse GBR sponges were dominated by phyla previously reported to associate with marine sponges. The microbial populations were highly conserved within different individuals of each species yet varied significantly between species. This is consistent with patterns for other GBR sponges (Luter et al., 2010b, 2012; Webster et al., 2011; Fan et al., 2012) and in species from other broad geographic locations (Schmitt et al., 2012b; Webster and Taylor, 2012). Whilst BLAST analysis did not reveal common indicator species between the DGGE and the clone library analyses, both datasets were dominated by sequences that had sponge and coral derived bacteria as their closest relatives. Further highlighting the specific nature of GBR sponge-bacterial populations was the finding that all species (with the exception of Hamigera sp.) contained clone sequences that fell within previously defined clusters of sponge-specific bacterial sequences (Simister et al., 2012a).
The dominance of Proteobacteria, Chloroflexi, Acidobacteria, Actinobacteria, Nitrospira, and Cyanobacteria across the GBR sponge species accords with the dominant microbial groups described from other studies of sponge-associated microbial assemblages (Taylor et al., 2007; Webster and Taylor, 2012). Two of the GBR sponge species from this study (C. coralliophila and Stylissa sp.) were recently assessed using metagenomic shotgun sequencing (Fan et al., 2012). Whilst the different sequencing approaches were generally consistent, some methodological bias was evident with metagenomic sequencing detecting an abundance of Chloroflexi in C. coralliophila and a dominance of Archaea in Stylissa sp. which were not detected by DGGE or clone sequencing. The community composition in X. testudinaria from the present study showed high similarity with the microbial community described for this species in Indonesia (Montalvo and Hill, 2011). However, Cyanobacteria, which are well studied associates of Caribbean Xestospongia spp., were not detected in either of the GBR Xestospongia species (Steindler et al., 2005; Erwin and Thacker, 2007, 2008a). Episodes of both cyclic and fatal bleaching have dramatically effected populations of Xestospongia muta in the Caribbean (López-Legentil et al., 2008; McMurray et al., 2011), whereas there is currently no evidence of bleaching within GBR Xestospongia species. The absence of photosynthetic cyanobacteria in GBR species not only distinguishes these geographically separated congeners, but raises questions of GBR populations being less vulnerable to bleaching impacts associated with a changing climate. Results from the present study indicate that C. singaporensis, C. foliascens, and C. coralliophila are likely to be phototrophic species due to the high representation of Cyanobacteria. This is an important finding for studies that may want to distinguish differential responses of heterotrophs or phototrophs to environmental perturbation.
The Chloroflexi were present in over 50% of the surveyed GBR sponge species and were particularly abundant in Cinachyra sp., X. exigua, and C. matthewsi. Another recent investigation compared the presence of Chloroflexi in sponge species containing either high (HMA) or low (LMA) microbial abundance (Schmitt et al., 2011). Clear differences were observed between HMA and LMA species including a greater abundance, diversity and sponge-specificity of Chloroflexi in HMA sponges. In contrast, LMA species tended to be highly variable and contain Chloroflexi sequences related to seawater-derived microorganisms. These findings indicate a functional importance for Chloroflexi in HMA sponges and whilst we would predict that Cinachyra sp., X. exigua, and C. matthewsi from the GBR are HMA species based on sequence analysis of Chloroflexi, further microscopy analysis would be required to determine the microbial abundance within each of our GBR sponges.
No relationship between host phylogeny and bacterial communities were observed within the Dictyoceratida, Haplosclerida, Poescilosclerida, Halichondrida, and Pirophorida from the GBR. This finding is consistent with a 454 amplicon pyrosequencing study that surveyed sponge microbes across 32 different host species encompassing nine sponge orders (Schmitt et al., 2012b). Species within the same orders did not contain more similar microbial communities than species in different orders nor was there any clear correlation to host phylogeny when three species each within the genera Aplysina, Hyrtios, and Ircinia were compared (Schmitt et al., 2012b). Cospeciation of sponges and their symbionts would be most likely to occur if the microbial associates were transmitted strictly vertically (from adult to gametes/larvae). Research over the past decade has revealed that many sponges use both vertical (Usher et al., 2001; Oren et al., 2005; Enticknap et al., 2006; Schmitt et al., 2007; Sharp et al., 2007) and horizontal (Taylor et al., 2007; Schmitt et al., 2008; Webster et al., 2010) transmission strategies to maintain their complex and diverse microbial communities. The lack of correlation between host phylogeny and bacterial composition in the GBR sponges also suggests a strategy of symbiont acquisition that incorporates both vertical and horizontal transmission.
It has been hypothesized that abundant microbes common to all samples within a given habitat must be essential for the function of that community (Shade and Handelsman, 2012). Hence, the identification of core microbiomes (microorganisms that are shared between two or more samples) in complex microbial habitats can enhance our understanding of systems ecology. Across the 13 GBR sponge species investigated here, the core microbiome within each species was high yet the microbiome shared between species was low. However, it should be noted that additional sequencing for each species may reveal further sequences common to multiple samples. No OTUs were present in more than five different sponge species and 91% of the total OTUs were species-specific. Schmitt and colleagues (2012b) also identified a minimal core bacterial community in 32 different sponge species. In another study of five Mediterranean sponge species, 72% of the detected OTUs were species-specific, 26% were common to two or more species and only 2% were shared amongst all five species (Schmitt et al., 2012a). The study of three sympatric Mediterranean Ircinia sp. also revealed host species-specific assemblages and identified one proportion of the OTUs that were shared between the two most phylogenetically related species and a second component that was shared between the two species sharing the same cryptic habitat (Erwin et al., 2012a). These findings indicate that factors relevant to each host species can contribute to structuring the distinct microbial communities. Whilst next generation sequencing would further elucidate the host-specific nature of GBR sponge microbial communities, our results contribute to this growing body of evidence for species-specific microbial associations in sponges.
The vast majority of GBR sponges surveyed in the current study had high proportions of sequences that were more similar to other sponge-derived sequences than they were to sequences from the environment or other sources. These findings are consistent with our understanding of sponge-specific SC (Hentschel et al., 2002; Taylor et al., 2007; Simister et al., 2012a) and further our knowledge of their distribution and geographic range. In the global sponge microbial survey, each of the 32 species was also found to host bacteria that were more closely related to each other than they were to non-sponge derived bacterial species (Schmitt et al., 2012b). However, a recent survey of the rare microbial biosphere from ~650 marine samples collected from diverse habitats detected 77 of the 173 known sponge-specific SC in environmental (non-sponge) samples, although these were generally found at extremely low abundances (Taylor et al., 2012). To date, little is known about the functional roles of these “sponge-specific” bacteria although single celled genomics of a sponge-specific Poribacteria indicated potential symbiotic functions including autotrophic carbon fixation and vitamin B12 production (Siegl et al., 2011). Interestingly, sponge species from the present study which contained no sequences within sponge-specific clusters still maintained highly conserved bacterial populations, indicating that species without these “sponge-specific” sequences are still capable of structuring their communities and are not just reflecting the microbial composition of the surrounding seawater. For example, Hamigera sp. had no sequences within SC/SCCs and only 2% of Paramyxilla sp. fell within a SC yet the replicate individuals within each species shared 90 and 80% similarity respectively. Similarly, the highest number of sequences falling within SC/SCCs occurred in L. variabilis (92% of total sequences), yet this species had the lowest intra-species similarity based on microbial profiling (65%).
Whilst the number of microbes shared between different GBR sponge species was small, the core microbiome within each species was large, with the vast majority of microbes conserved in all replicate individuals per species. This indicates that these microbes are likely to be functionally important to the ecology of each species. Metagenomic sequence analysis recently highlighted how core symbiont functions can be provided in different sponge species by functionally equivalent microbes and analogous enzymes or biosynthetic pathways (Fan et al., 2012). Therefore, a “core microbiome” at the phylogenetic level does not need to exist across species. As indicated by Shade and Handelsman (2012), identifying the “core” microorganisms in any habitat is essential for defining the healthy community and subsequently predicting how the community will respond to perturbation. Given the increasing incidence of sponge disease and health declines associated with climate change, an enhanced understanding of the species-specific symbionts in GBR sponges will be a valuable resource.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anderson, M. J., Gorley, R. N., and Clarke, K. R. (2008). “PERMANOVA+ for PRIMER: Guide to software and statistical methods,” in PRIMER-E, (Plymouth).
Angermeier, H., Glöckner, V., Pawlik, J. R., Lindquist, N. L., and Hentschel, U. (2012). Sponge white patch disease affecting the Caribbean sponge Amphimedon compressa. Dis. Aquat. Organ. 99, 95–102.
Angermeier, H., Kamke, J., Abdelmohsen, U. R., Krohne, G., Pawlik, J. R., Lindquist, N. L., et al. (2011). The pathology of sponge orange band disease affecting the Caribbean barrel sponge Xestospongia muta. FEMS Microbiol. Ecol. 75, 218–230.
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072.
Enticknap, J. J., Kelly, M., Peraud, O., and Hill, R. T. (2006). Characterization of a culturable alphaproteobacterial symbiont common to many marine sponges and evidence for vertical transmission via sponge larvae. Appl. Environ. Microbiol. 72, 3724–3732.
Erpenbeck, D., Breeuwer, J. A. J., van der Velde, H. C., and van Soest, R. W. M. (2002). Unravelling host and symbiont phylogenies of halichondrid sponges (Demospongiae, Porifera) using a mitochondrial marker. Mar. Biol. 141, 377–386.
Erwin, P. M., López-Legentil, S., González-Pech, R., and Turon, X. (2012a). A specific mix of generalists: bacterial symbionts in Mediterranean Ircinia spp. FEMS Microbiol. Ecol. 79, 619–637.
Erwin, P. M., Pita, L., Lopez-Legentil, S., and Turon, X. (2012b). Stability of sponge-associated bacteria over large seasonal shifts in temperature and irradiance. Appl. Environ. Microbiol. 78, 7358–7368.
Erwin, P. M., Olson, J. B., and Thacker, R. W. (2011). Phylogenetic diversity, host-specificity and community profiling of sponge-associated bacteria in the northern Gulf of Mexico. PLoS ONE 6:e26806. doi: 10.1371/journal.pone.0026806
Erwin, P. M., and Thacker, R. W. (2007). Incidence and identity of photosynthetic symbionts in Caribbean coral reef sponge assemblages. J. Mar. Biol. Assoc. U.K. 87, 1687–1692.
Erwin, P. M., and Thacker, R. W. (2008a). Cryptic diversity of the symbiotic cyanobacterium Synechcoccus spongiarum among sponge hosts. Mol. Ecol. 17, 2937–2947.
Erwin, P. M., and Thacker, R. W. (2008b). Phototrophic nutrition and symbiont diversity of two Caribbean sponge-cyanobacteria symbioses. Mar. Ecol. Prog. Ser. 362, 139–147.
Fan, L., Reynolds, D., Liu, M., Stark, M., Kjelleberg, S., Webster, N. S., et al. (2012). Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. U.S.A. 109, E1878–E1887.
Ferris, M. J., Muyzer, G., and Ward, D. M. (1996). Denaturing gradient gel electrophoresis profiles of 16S rRNA-defined populations inhabiting a hot spring microbial mat community. Appl. Environ. Microbiol. 62, 340–346.
Hentschel, U., Hopke, J., Horn, M., Friedrich, A. B., Wagner, M., Hacker, J., et al. (2002). Molecular evidence for a uniform microbial community in sponges from different oceans. Appl. Environ. Microbiol. 68, 4431–4440.
Hentschel, U., Piel, J., Degnan, S. M., and Taylor, M. W. (2012). Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 10, 641–654.
Hentschel, U., Usher, K. M., and Taylor, M. W. (2006). Marine sponges as microbial fermenters. FEMS Microbiol. Ecol. 55, 167–177.
Holmes, B., and Blanch, H. (2007). Genus-specific associations of marine sponges with group I crenarchaeotes. Mar. Biol. 150, 759–772.
Hooper, J. N. A. (2009). “Sponges,” in The Great Barrier Reef: Biology, Environment and Management, eds P. Hutchings, M. J. Kingsford, and O. Hoegh-Guldberg (VIC, Australia: CSIRO Publishing), 171–187.
Huber, T., Faulkner, G., and Hugenholtz, P. (2004). Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20, 2317–2319.
Lee, O. O., Wong, Y. H., and Qian, P. Y. (2009). Inter- and intraspecific variations of bacterial communities associated with marine sponges from San Juan Island, Washington. Appl. Environ. Microbiol. 75, 3513–3521.
López-Legentil, S., Erwin, P. E., Pawlik, J. R., and Song, B. (2010). Effects of sponge bleaching on ammonia-oxidizing Archaea: distribution and relative expression of ammonia monoxygenase genes associated with the giant barrel sponge Xestospongia muta. Microb. Ecol. 60, 561–571.
López-Legentil, S., Song, B., McMurray, S. E., and Pawlik, J. R. (2008). Bleaching and stress in coral reef ecosystems: hsp70 expression by the giant barrel sponge Xestospongia muta. Mol. Ecol. 17, 1840–1849.
Ludwig, W., Strunk, O., Klugbauer, S., Klugbauer, N., Weizenegger, M., Neumaier, J., et al. (1998). Bacterial phylogeny based on comparative sequence analysis. Electrophoresis 19, 554–568.
Luter, H. M., Whalan, S., and Webster, N. S. (2010a). Exploring the role of microorganisms in the disease-like syndrome affecting the sponge Ianthella basta. Appl. Environ. Microbiol. 76, 5736–5744.
Luter, H. M., Whalan, S., and Webster, N. S. (2010b). Prevalence of tissue necrosis and brown spot lesions in a common marine sponge. Mar. Freshwater Res. 61, 481–484.
Luter, H. M., Whalan, S., and Webster, N. S. (2012). Thermal and sedimentation stress are unlikely causes of brown spot syndrome in the coral reef sponge, Ianthella basta PLoS ONE 7:e39779. doi: 10.1371/journal.pone.0039779
Maidak, B. L., Cole, J. R., Parker, C. T. Jr., Garrity, G. M., Larsen, N., Li, B., et al. (1999). A new version of the RDP (Ribosomal Database Project). Nucleic Acids Res. 27, 171–173.
Maldonado, M., Sánchez-Tocino, L., and Navarro, C. (2010). Recurrent disease outbreaks in corneous demosponges of the genus Ircinia: epidemic incidence and defense mechanisms. Mar. Biol. 157, 1577–1590.
Marchesi, J. R., Sato, T., Weightman, A. J., Martin, T. A., Fry, J. C., Hiom, S. J., et al. (1998). Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA [published erratum appears in Appl Environ Microbiol 1998 Jun;64, 2333]. Appl. Environ. Microbiol. 64, 795–799.
McMurray, S. E., Blum, J. E., Leichter, J. J., and Pawlik, J. R. (2011). Bleaching of the giant barrel sponge Xestospongia muta in the Florida Keys. Limnol. Oceanogr. 56, 2243–2250.
Montalvo, N. F., and Hill, R. T. (2011). Sponge-associated bacteria are strictly maintained in two closely-related but geographically distant sponge hosts. Appl. Environ. Microbiol. 77, 7207–7216.
Muyzer, G., de Waal, E. C., and Uitterlinden, A. G. (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59, 695–700.
Oren, M., Steindler, L., and Ilan, M. (2005). Transmission, plasticity and the molecular identification of cyanobacterial symbionts in the Red Sea sponge Diacarnus erythraenus. Mar. Biol. 148, 35–41.
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541.
Schmitt, S., Angermeier, H., Schiller, R., Lindquist, N., and Hentschel, U. (2008). Molecular microbial diversity survey of sponge reproductive stages and mechanistic insights into vertical transmission of microbial symbionts. Appl. Environ. Microbiol. 74, 7694–7708.
Schmitt, S., Deines, P., Behnam, F., Wagner, M., and Taylor, M. W. (2011). Chloroflexi bacteria are more diverse, abundant, and similar in high than in low microbial abundance sponges. FEMS Microbiol. Ecol. 78, 497–510.
Schmitt, S., Hentschel, U., and Taylor, M. W. (2012a). Deep sequencing reveals diversity and community structure of complex microbiota in five Mediterranean sponges. Hydrobiologia 687, 341–351.
Schmitt, S., Tsai, P., Bell, J., Fromont, J., Ilan, M., Lindquist, N., et al. (2012b). Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J. 6, 564–576.
Schmitt, S., Weisz, J. B., Lindquist, N., and Hentschel, U. (2007). Vertical transmission of a phylogenetically complex microbial consortium in the viviparous sponge Ircinia felix. Appl. Environ. Microbiol. 73, 2067–2078.
Shade, A., and Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12.
Sharp, K. H., Eam, B., Faulkner, D. J., and Haygood, M. G. (2007). Vertical transmission of diverse microbes in the tropical sponge Corticium sp. Appl. Environ. Microbiol. 73, 622–629.
Siegl, A., Kamke, J., Hochmuth, T., Piel, J., Richter, M., Liang, C., et al. (2011). Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 5, 61–70.
Simister, R. L., Deines, P., Botté, E. S., Webster, N. S., and Taylor, M. W. (2012a). Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Environ. Microbiol. 14, 517–524.
Simister, R. L., Taylor, M. W., Tsai, P., Fan, L., Bruxner, T., Crowe, M., et al. (2012b). Thermal stress responses in the bacterial biosphere of the Great Barrier Reef sponge, Rhopaloeides odorabile. Environ. Microbiol. 14, 3232–3246.
Steindler, L., Huchon, D., Avni, A., and Ilan, M. (2005). 16S rRNA phylogeny of sponge-associated Cyanobacteria. Appl. Environ. Microbiol. 71, 4127–4131.
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347.
Taylor, M. W., Schupp, P. J., Dahllof, I., Kjelleberg, S., and Steinberg, P. D. (2004). Host specificity in marine sponge-associated bacteria, and potential implications for marine microbial diversity. Environ. Microbiol. 6, 121–130.
Taylor, M. W., Schupp, P. J., de Nys, R., Kjelleberg, S., and Steinberg, P. D. (2005). Biogeography of bacteria associated with the marine sponge Cymbastela concentrica. Environ. Microbiol. 7, 419–433.
Taylor, M. W., Tsai, P., Simister, R. L., Deines, P., Botte, E., Ericson, G., et al. (2012). ‘Sponge-specific’ bacteria are widespread (but rare) in diverse marine environments. ISME J. doi: 10.1038/ismej.2012.111. [Epub ahead of print].
Thacker, R. W., and Starnes, S. (2003). Host specificity of the symbiotic cyanobacterium Oscillatoria spongeliae in marine sponges, Dysidea spp. Mar. Biol. 142, 643–648.
Turque, A. S., Cardoso, A. M., Silveira, C. B., Vieira, R. P., Freitas, F. A. D., Albano, R. M., et al. (2008). Bacterial communities of the marine songes Hymeniacidon heliophila and Polymastia janeirensis and their environment in Rio de Janeiro, Brazil. Mar. Biol. 155, 135–146.
Usher, K. M., Kuo, J., Fromont, J., and Sutton, D. C. (2001). Vertical transmission of cyanobacterial symbionts in the marine sponge Chondrilla australiensis (Demospongiae). Hydrobiologia 461, 9–13.
Vacelet, J. (1975). Etude en microscopie electronique de l'association entre bacteries et spongiaires du genre Verongia (Dictyoceratida). J. Microsc. Biol. Cell 23, 271–288.
van Soest, R. W. M., Boury-Esnault, N., Vacelet, J., Dohrmann, M., Erpenbeck, D., De Voogd, N. J., et al. (2012). Global diversity of sponges (Porifera). PLoS ONE 7:e35105. doi: 10.1371/journal.pone.0035105
Webster, N. S., Cobb, R. E., and Negri, A. P. (2008). Temperature thresholds for bacterial symbiosis with a sponge. ISME J. 2, 830–842.
Webster, N. S., Cobb, R. E., Soo, R., Anthony, S. L., Battershill, C. N., Whalan, S., et al. (2011). Bacterial community dynamics in the marine sponge Rhopaloeides odorabile under in situ and ex situ cultivation. Mar. Biotech. 13, 296–304.
Webster, N. S., Negri, A. P., Munro, M. M., and Battershill, C. N. (2004). Diverse microbial communities inhabit Antarctic sponges. Environ. Microbiol. 6, 288–300.
Webster, N. S., Negri, A. P., Webb, R. I., and Hill, R. T. (2002). A spongin-boring alpha proteobacterium is the etiological agent of disease in the Great Barrier Reef sponge, Rhopaloeides odorabile. Mar. Ecol. Prog. Ser. 232, 305–309.
Webster, N. S., and Taylor, M. W. (2012). Marine sponges and their microbial symbionts: love and other relationships. Environ. Microbiol. 14, 335–346.
Webster, N. S., Taylor, M. W., Behnam, F., Lücker, S., Rattei, T., Whalan, S., et al. (2010). Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 12, 2070–2082.
Keywords: sponge, microorganism, symbiont, diversity, Great Barrier Reef
Citation: Webster NS, Luter HM, Soo RM, Botté ES, Simister RL, Abdo D and Whalan S (2013) Same, same but different: symbiotic bacterial associations in GBR sponges. Front. Microbio. 3:444. doi:10.3389/fmicb.2012.00444
Received: 17 October 2012; Paper pending published: 02 November 2012;
Accepted: 31 December 2012; Published online: 18 January 2013.
Edited by:
Karla B. Heidelberg, University of Southern California, USAReviewed by:
Susannah G. Tringe, DOE Joint Genome Institute, USASpencer V. Nyholm, University of Connecticut, USA
Copyright © 2013 Webster, Luter, Soo, Botté, Simister, Abdo and Whalan. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: N. S. Webster, Australian Institute of Marine Science, PMB 3, Townsville Mail Centre, QLD 4810, Australia. e-mail: n.webster@aims.gov.au