 Song Yang1 Janet B. Matsen1 Michael Konopka1 Abigail Green-Saxena2 Justin Clubb1 Martin Sadilek3 Victoria J. Orphan4 David Beck1,5 Marina G. Kalyuzhnaya6*
Song Yang1 Janet B. Matsen1 Michael Konopka1 Abigail Green-Saxena2 Justin Clubb1 Martin Sadilek3 Victoria J. Orphan4 David Beck1,5 Marina G. Kalyuzhnaya6*- 1Department of Chemical Engineering, University of Washington, Seattle, WA, USA
- 2Division of Biology, California Institute of Technology, Pasadena, CA, USA
- 3Department of Chemistry, University of Washington, Seattle, WA, USA
- 4Division of Geological and Planetary Sciences; California Institute of Technology, Pasadena, CA, USA
- 5eScience Institute, University of Washington, Seattle, WA, USA
- 6Department of Microbiology, University of Washington, Seattle, WA, USA
In this work we use metabolomics and 13C-labeling data to refine central metabolic pathways for methane utilization in Methylosinus trichosporium OB3b, a model alphaproteobacterial methanotrophic bacterium. We demonstrate here that similar to non-methane utilizing methylotrophic alphaproteobacteria the core metabolism of the microbe is represented by several tightly connected metabolic cycles, such as the serine pathway, the ethylmalonyl-CoA (EMC) pathway, and the citric acid (TCA) cycle. Both in silico estimations and stable isotope labeling experiments combined with single cell (NanoSIMS) and bulk biomass analyses indicate that a significantly larger portion of the cell carbon (over 60%) is derived from CO2 in this methanotroph. Our13 C-labeling studies revealed an unusual topology of the assimilatory network in which phosph(enol) pyruvate/pyruvate interconversions are key metabolic switches. A set of additional pathways for carbon fixation are identified and discussed.
Introduction
Microbial oxidation of methane is one of the key elements of the global carbon cycle. The ability to oxidize methane has been demonstrated in two classes of Proteobacteria, Alpha, and Gamma. Methylosinus trichosporium OB3b was first described by Whittenbury et al. (1970) and has served as a model system for the investigation of methane utilization in obligate alphaproteobacterial methanotrophs for decades (Lawrence and Quayle, 1970; Strom et al., 1974; Cornish et al., 1984; Jollie and Lipscomb, 1991; Park et al., 1991, 1992; DiSpirito et al., 1998; Lontoh and Semrau, 1998; Gilbert et al., 2000; Trotsenko and Murrell, 2008). While the key metabolic pathways for carbon assimilation in M. trichosporium OB3b have been predicted (Strom et al., 1974), several fundamental questions have never been answered, such as how cells regenerate glyoxylate (Anthony, 1982), what is the role of the TCA cycle in methanotrophic metabolism is (Patel et al., 1979; Shishkina and Trotsenko, 1982), and why CO2 supplementation has a significant positive effect on cell growth (Park et al., 1991, 1992). A draft genome of M. trichosporium OB3b has recently been generated (Stein et al., 2010), providing a genetic framework for characterization of the methanotrophy. In this work, models of carbon assimilation pathways in M. trichosporium OB3b predicted by biochemical characterization (Strom et al., 1974; Trotsenko and Murrell, 2008) and global gene expression analysis (Matsen et al., 2013) are further tested by metabolomic and 13C-labeled studies.
Results and Discussion
Growth Parameters and Incorporation from Methane and CO2 via 13C-Labeling Experiments
A strong dependence of the Methylosinus trichosporium OB3b growth upon CO2 supplementation has been previously demonstrated (Park et al., 1991, 1992). Similarly, a lag-period (up to 24 h) was observed in methane supplemented cultures inoculated at low cell density (OD600 < 0.05) at ambient concentrations of CO2. The initial lag-period was shortened by the addition of CO2 (5% of head space). Furthermore, the specific growth rate was also increased to 0.057 ± 0.002 h−1 (versus μ = 0.038 ± 0.004 h−1, in cultures grown without additional CO2). CO2 supplementation did not have any significant effect on the final cell density (data not shown).
It has been speculated that the positive effect of CO2 is a result of the high demand of the serine pathway for CO2 (Park et al., 1991, 1992). However, addition of extra CO2 does not affect the growth of non-methane utilizing methylotrophs such as Methylobacterium extorquens AM1 with the serine pathway for carbon utilization (Yanfen Fu, Mary E. Lidstrom personal communication). Several putative carboxylating systems were identified in the M. trichosporium OB3b genome: phosphoenolpyruvate carboxylase (Ppc); two propionyl-CoA carboxylases; crotonyl-CoA reductase (ccr); NAD(P)-dependent malic enzyme (mae), acetyl-CoA carboxylase (accABD), phosphoribosylaminoimidazole carboxylase, pyruvate carboxylase (pcx), and a putative 2-oxoacid ferredoxin oxidoreductase. To investigate the fate of CO2 in OB3b, we estimated the C1-carbon incorporation derived from methane (at the level of methylene-tetrahydrofolate, MeH4F) and/or bicarbonate into biomass using stable isotope labeling experiments and bulk biomass analyses. For these experiments, cells of M. trichosporium OB3b were grown on 13CH4 (20%) with or without external CO2 supplementation (5% atmosphere). A Cameca nanoSIMS 50 L was used to analyze the 13C/12C ratio of individual cells from either methane only or methane + carbon dioxide treatment. Individual M. trichosporium OB3b cells from the 13CH4 treatment showed substantial 13C enrichment (13C/12C ratio of 0.351 ± 0.231), albeit with significant cell-to-cell variation (Table 1). Cells grown on 13CH4 in the presence of added 12CO2 showed less cell-to-cell variation. The amount of 13C-enriched biomass derived from methane dropped dramatically in the presence of elevated CO2 (13C/12C ratio of 0.134 ± 0.046), reflecting the reincorporation of 13CO2 derived from 13CH4 when 12CO2 is not elevated (Crowther et al., 2008; Table 1). Analysis of bulk cell pellets from M. trichosporium OB3b cultures incubated under the same conditions (13CH4 with and without elevated 12CO2) also showed a similar trend with significantly lower 13C enrichment in biomass from cultures incubated with additional CO2 relative to treatments with only methane (data not shown). Overall, 13C-labeling data suggest significant assimilation of CO2 relative to CH4. Assuming that in the presence of 5% 12CO2, no reincorporation of 13CH4-derived 13CO2 occurred, our results suggest that at least 62% of the assimilated carbon is from CO2, significantly greater than the 50% observed for M. extorquens AM1, a non-methane utilizing serine cycle methylotroph (Peyraud et al., 2011).
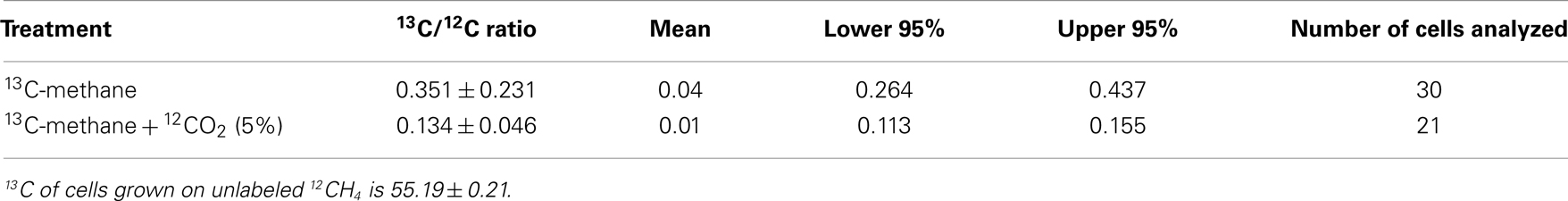
Table 1. 13Carbon enrichment of Methylosinus trichosporiu m OB3b cells grown on 13C-labeled methane with or without additional supplementation with unlabeled carbon dioxide.
Assimilation: In silico and Metabolomics Studies
The functional organization of the M. trichosporium OB3b metabolic network operating during growth on methane has been discussed in an accompanying manuscript by Matsen et al. (2013). A summary of the central metabolism is shown in Figure 1. The chemical composition of M. trichosporium OB3b (RNA, DNA, protein, polyhydroxybutyrate (PHB), fatty acids, intracellular metabolites) was determined experimentally or taken from available literature (Weaver et al., 1975; Williams, 1988; Guckert et al., 1991; Sun and Wood, 1996; Lloyd et al., 1999; Doronina et al., 2008). Proteins comprise a significant portion of the cell dry weight (55%). The specific amino acid composition of the cell proteins was estimated from RNA-seq data, and the data were used to estimate contribution of C2-C6 intermediates into biomass (shown in Figure 1, Table A1 in Appendix).
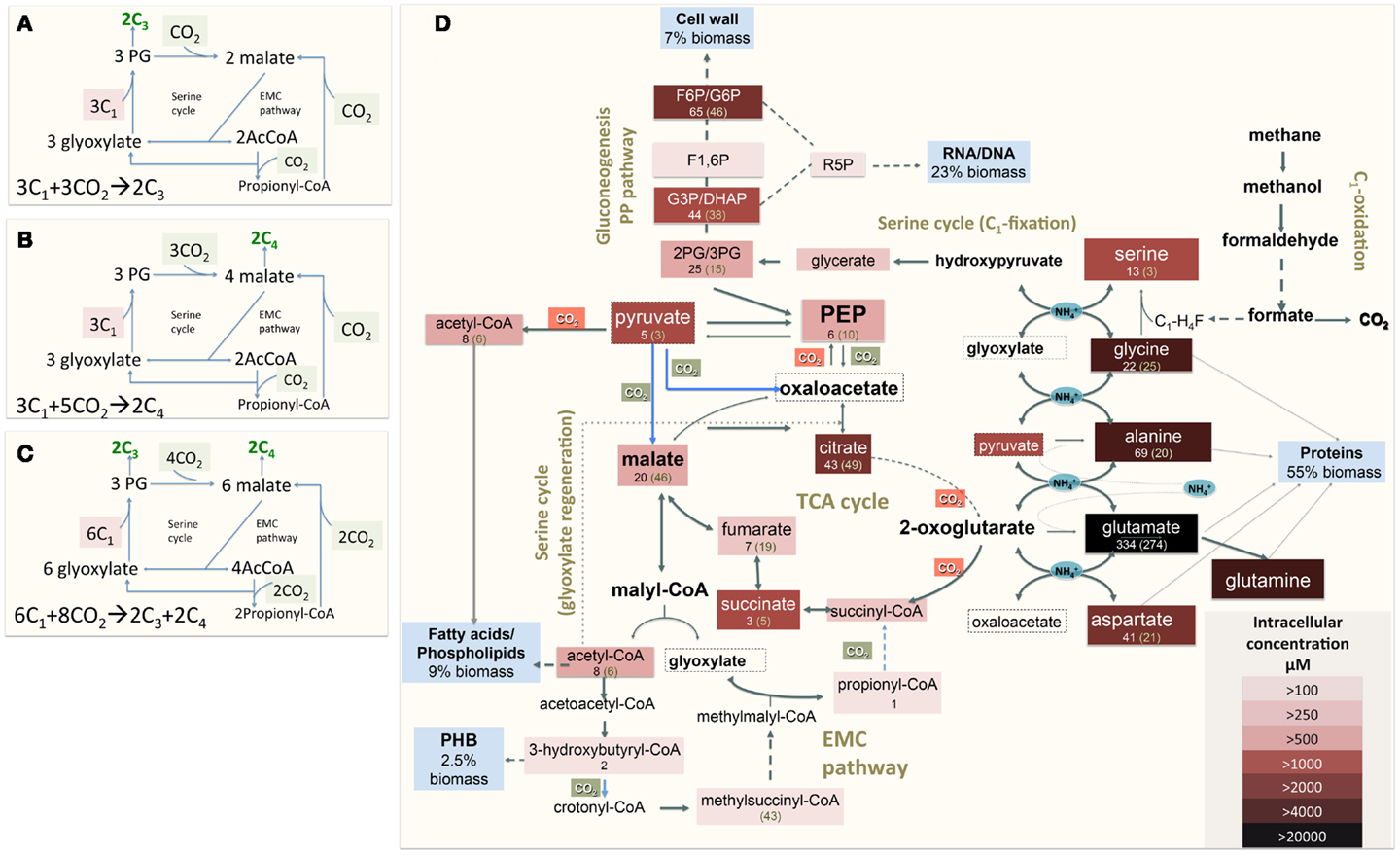
Figure 1. (A) Schematic representation of the serine cycle with the EMC pathway for carbon assimilation [adapted from (Peyraud et al., 2009, 2011)]. (B) Schematic representation of the serine cycle with the EMC pathway for carbon assimilation (adapted from Anthony, 2011); (C) Schematic representation of the serine cycle with the EMC pathway for carbon assimilation in Methylosinus trichosporium OB3b (see description in the text). (D) Central metabolism of Methylosinus trichosporium OB3b grown on methane as deduced from the genome sequences, transcriptomic, and metabolomic studies. Metabolites boxed were measured. Box color is an indicator of the metabolite pool, with darker colors indicating a larger pool. Numbers under a metabolite names are rates of 13C-carbon incorporation: the first number is the rate of CH4-derived carbon; number in parentheses- is the rate of CO2-derived carbon incorporation. PHB-polyhydroxybutyrate; F1, 6P, Fructose-1, 6-bisphosphate; F6P/G6P, fructose-6-phosphate/glucose-6-phosphate; GAP/DHAP, glyceraldehyde-3-phosphate/dihydroxyacetone phosphate; 2PG/3PG, 2-phosphoglycerate/3-phosphoglycerate.
As background information for physiological pathways, 27 targeted intermediates involved in the serine cycle, EMC pathway, TCA cycle, and gluconeogenesis were quantified (Table 2). Among those, the lowest concentration was observed for propionyl-CoA (108.6 μM) and the highest concentration was 24 mM for glutamate (Table 2). Relatively high concentrations of pyruvate (1 mM) and its corresponding amino acid, alanine (4.5 mM) were detected.
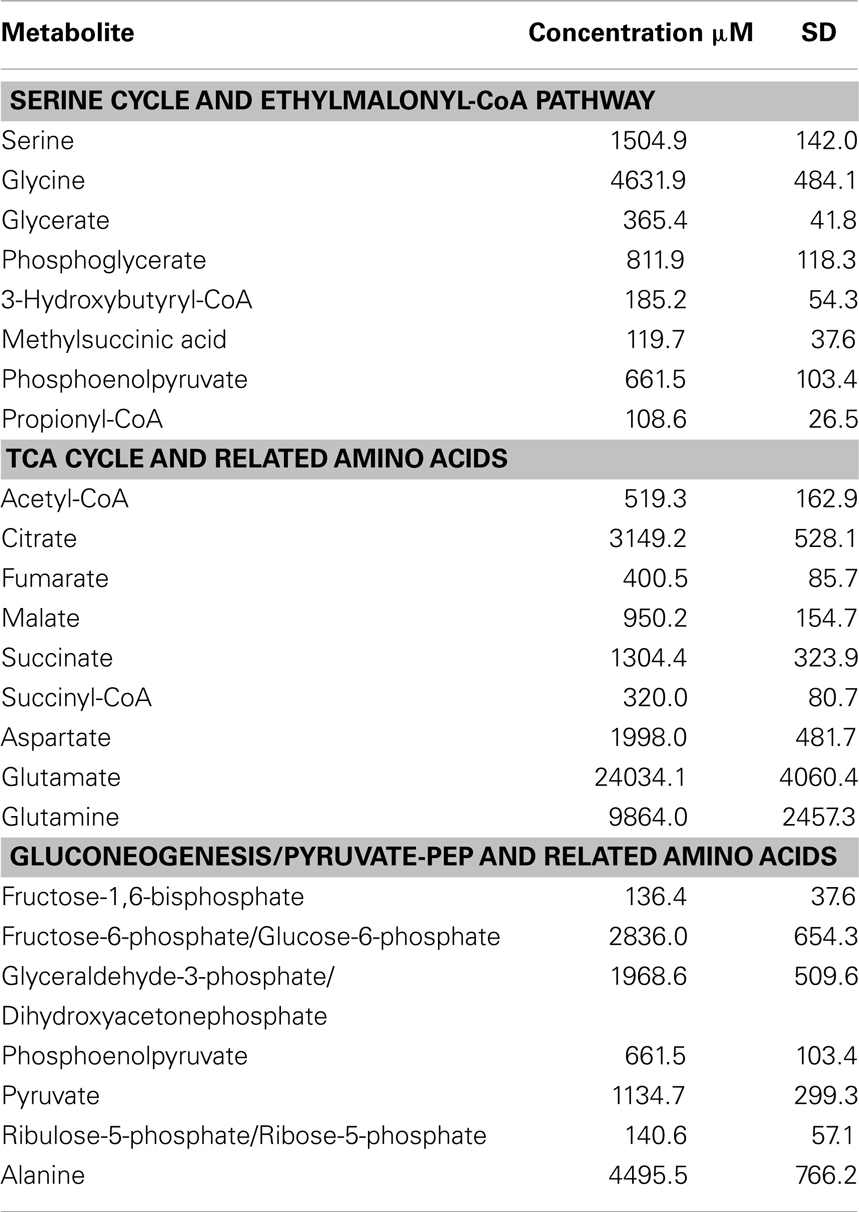
Table 2. Intracellular pool of key metabolites in M. trichosporiu m OB3b.
Metabolome Analysis: 13C-Labeling
To address some of the hypotheses generated by gene expression and in silico studies and to probe metabolic pathways for methane and CO2 assimilation in M. trichosporium OB3b, we monitored the dynamic 13C-incorporation of intermediates through two different 13C-tracing experiments: switching from 12CH4 to 13CH4 (switch experiment) and spiking 13CO2 into cell cultures grown on 12CH4 (spike experiment) (Figures 2–4, Figure A1 in Appendix). The data generated were compared with predicted pathways for carbon assimilation (summarized in Figure 1, see also Matsen et al., 2013). All key intermediates of central pathways could be divided into three groups: (1) efficiently labeled with methane-derived carbon, such as serine, alanine, G6P/F6P, citric acid (Figures 2A–F); (2) more efficiently labeled with CO2-derived carbon, such as malate, fumarate, and succinate (Figures 2G–I); and (3) similarly labeled by both CO2 and CH4 carbon, such as glutamate (Figure 2J).
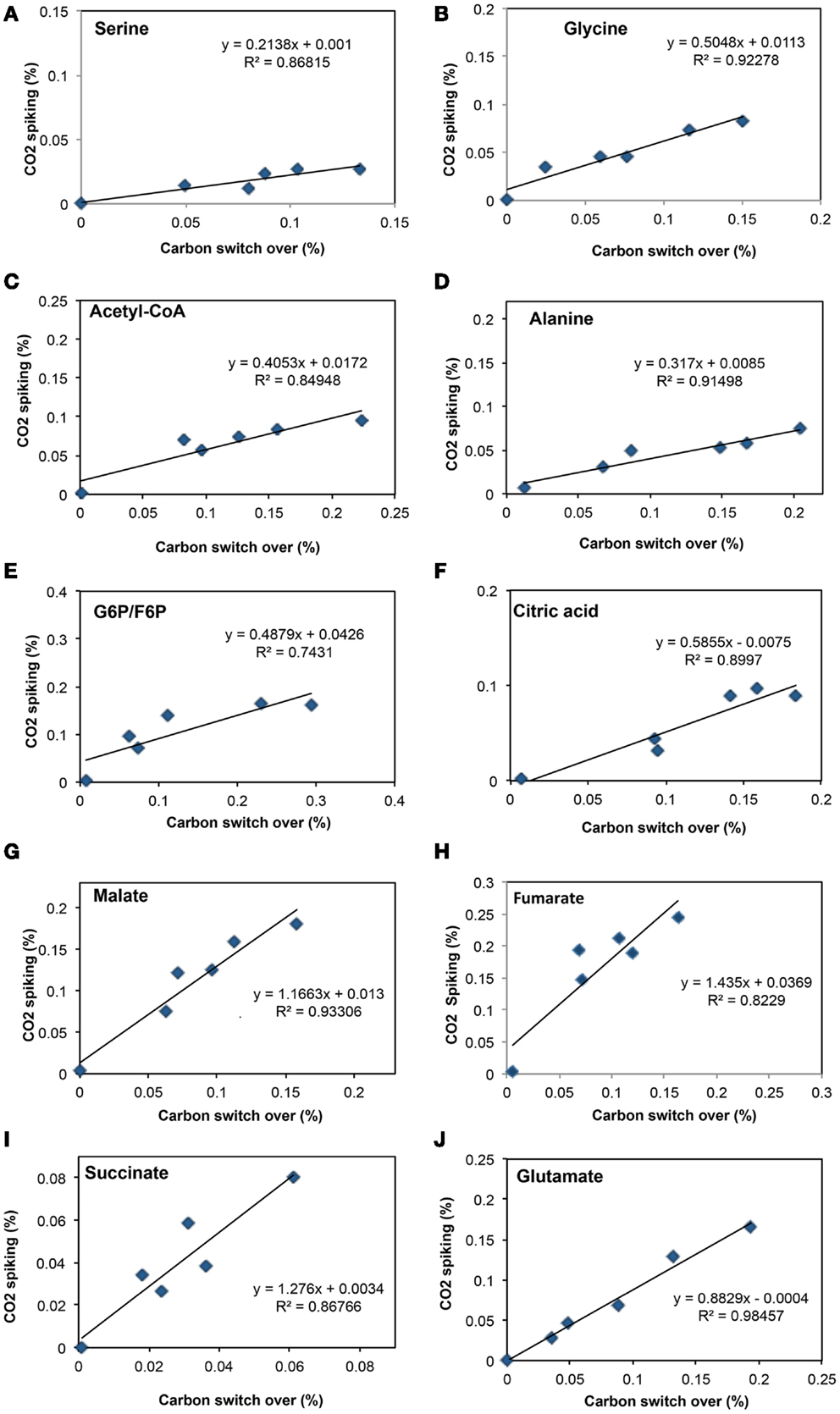
Figure 2. Comparison of total 13C-incorporation (%) between switch from 12CH4 to 13CH4 versus that of 13CO2 spiking in a time course (0, 1, 2, 5, 10, 20 min). In each panel, the total 13C-incorporation under carbon switchover and CO2 spiking conditions are plotted against each other for each time point. The six points corresponding to 0, 1, 2, 4, 10, and 20 min were best fit and the slope of the resulting line used to estimate the relative rate of incorporation into serine (A), glycine (B), acetyl-CoA (C), alanine (D), glucose-6-phosphate/fructose-6-phosphate (E), citric acid (F), malate (G), fumarate (H), succinate (I), and glutamate (J).
As was expected during the switch from 12CH4 to 13CH4, singly labeled serine was generated quickly, demonstrating that any unlabeled glycine rapidly reacted with labeled methylene H4F (Figure 3). Doubly labeled serine appeared later, followed by triply labeled serine. About 25% of the total serine pool (as determined by normalization to total pool of serine) was labeled after 20 min. The low 13C-incorporation suggested a slow uptake that was consistent with the methane consumption rate (in M. trichosporium OB3b). The labeling distribution of glycine (herein an indicator of glyoxylate) is shown in Figures 3B and 4A. Around 25% of the glycine (an indicator of glyoxylate) was labeled in 20 min in the switch experiment. Ten percent of the singly label glycine occurred after 10 min in the 13CO2 spiking experiments (Figure 4A), which is also consistent with regeneration of glyoxylate via the EMC pathway (Figure 4).
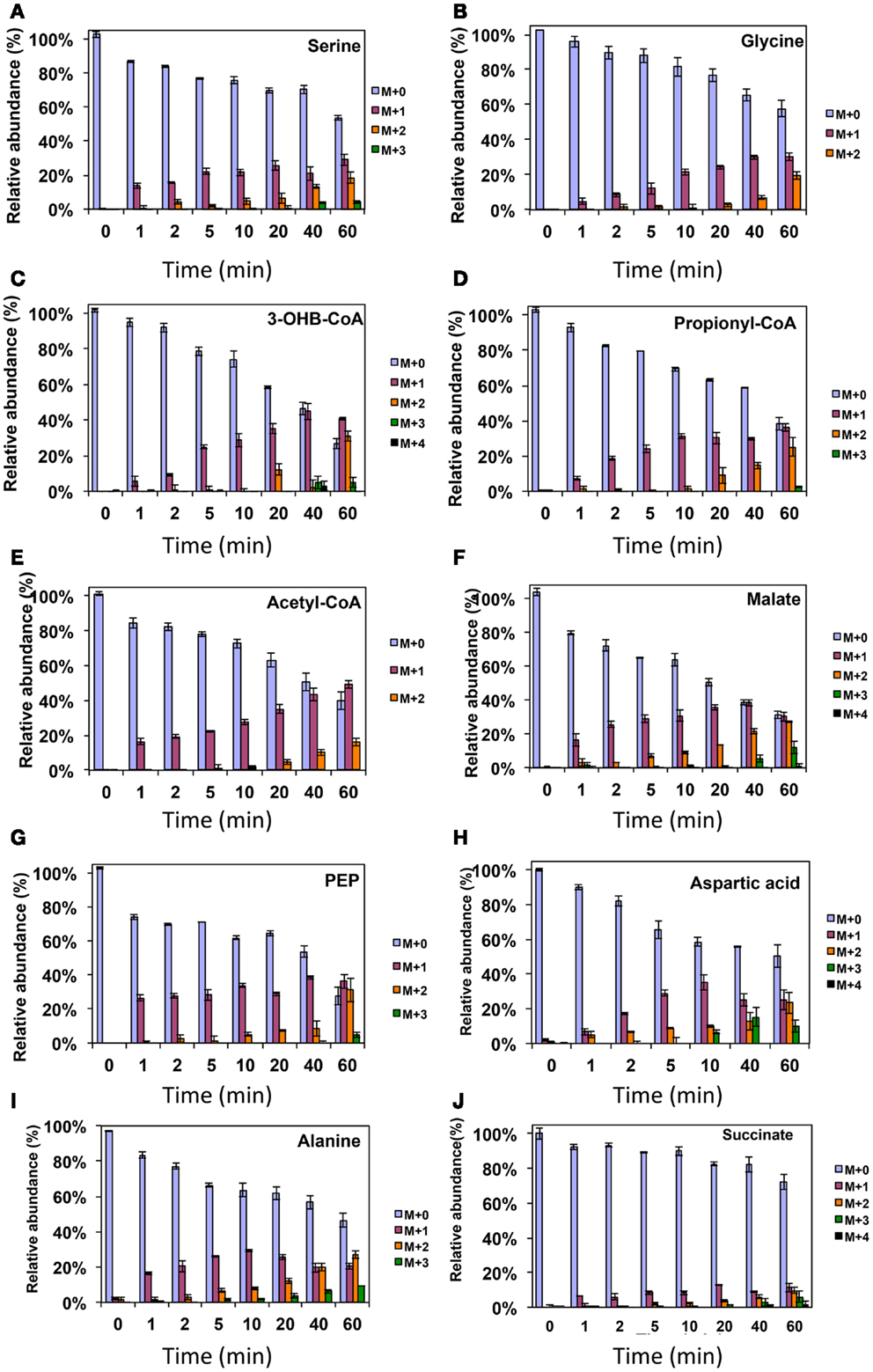
Figure 3. 13C-incorporation during the switch from 12CH4 to 13CH4 in Methylosinus trichosporium OB3b. (A)12C/13C-isotopomer distributions of serine (A), glycine (B), 3-hydroxybutyric acid (C), propionyl-CoA (D), acetyl-CoA (E), malate (F), phosphoenolpyruvate (G), aspartic acid (H), alanine (I), and succinate (J). Additional data are shown in Figure A1 in Appendix. M + 0 represented non-labeled compound, M + 1 represented compound with one 13C-label, M + 2 represented compound with two 13C-labels and so on.
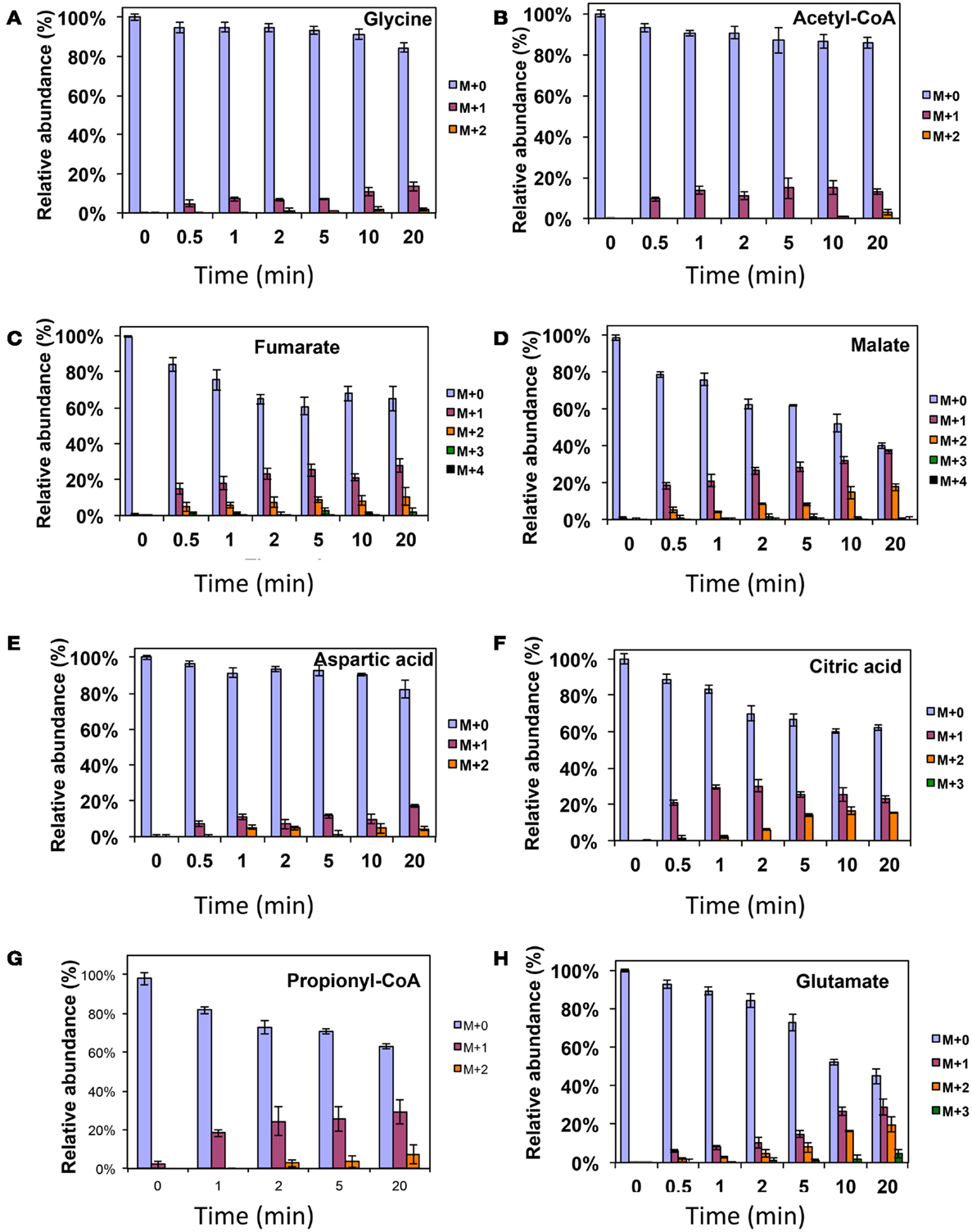
Figure 4. 13C-incorporation during 12CO2 spiking in Methylosinus trichosporium OB3b grown on 12CH4. 12C/13C-isotopomer distributions of glycine (A), acetyl-CoA (B), fumarate (C), malate (D), aspartic acid (E), citric acid (F), propionyl-CoA (G), glutamate (H). Additional data are shown in Figure A2 in Appendix. M + 0 represented non-labeled compound, M + 1 represented compound with one 13C-label, M + 2 represented compound with two 13C-labels and so on.
In addition, two metabolites involved in the EMC pathway [3-OHB-CoA (entry) and propionyl-CoA (exit)] were monitored during CH4 spike (Figures 3C,D). Both compounds were singly labeled quickly. These could be generated from one singly labeled acetyl-CoA and one unlabeled acetyl-CoA (Figure 3E). The low incorporation for the second carbon can be explained by the additional synthesis of unlabeled acetyl-CoA via the EMC pathway itself. 13CO2-carbon was also quickly incorporated into propionyl-CoA (Figure 4G). These data also provide metabolic proof of EMC operation in the strain.
About 40% of the total malate pool was labeled throughout the course of the 13CH4-experiments (Figure 3F). Both methane and CO2 (generated from 13CH4-oxidation) can contribute to the label. However, the rate of CO2 incorporation into malate and fumarate was high too (Figures 2G,H). The possible routes for the CO2 incorporation into malate labeling are through the serine cycle, EMC pathway and carboxylation of pyruvate (Figure 1). As shown in Figure 4D, singly labeled malate increased to around 20% after 0.5 min and remained at a similarly high percentage throughout the experiment, adding support to the hypothesis that the carboxylation via the EMC pathway and pyruvate contributes significantly to the pool of malate in addition to PEP-carboxylation. The labeling pattern of fumarate favors the hypothesis of the EMC pathway is a major source of malate. However, it should be noted that the 13CO2-fumarate labeling may be explained by the reversible reaction catalyzed by fumC. On the other hand, the rate of CO2 incorporation into aspartate (herein an indicator of oxaloacetate) was slow compared to malate (Figures 4D,E). The rates of C1-incorporation (13CH4-derived carbon) into malate and aspartate are almost identical in the switch over experiment (Figures 3F,H). Thus, the much slower rate of CO2 incorporation into aspartate at the early time points could not be attributed only to the large pool of the compound. The labeling patterns of malate and aspartate during 13CO2-spikes indicate that at least one of the compounds (most likely malate) comes from alternative sources. Doubly labeled malate was observed throughout the experiment (Figure 4D). The reversible conversions of malyl-CoA to malate and malate to fumarate may result in fumarate scrambling and contribute to doubly labeled malate. However, 13CO2 is incorporated into fumarate and succinate more efficiently than C1-labeled carbon (Figures 2H,I). Together the presented data indicate that the ECM pathway may serve as a significant source of intracellular malate.
According to current pathway prediction, both acetyl-CoA and glyoxylate are generated from malyl-CoA. The labeling rate of these two compounds was similar during the switch over experiments (Figures 3B,E). The incorporation of CO2-labeling into intracellular acetyl-CoA was slow compared to the 13C-carbon label that originated from methane. The labeling pattern of acetyl-CoA is similar to other intermediates of the serine cycle generated upstream from phosphoenolpyruvate, including all intermediates of gluconeogenesis/PPP, and pyruvate/alanine (Figures 3C,I; Figures A1D and A2A-D in Appendix). As a rule, these intermediates were labeled with methane-derived carbon faster than with CO2 (Figure 2). This suggests that significant flux of carbon assimilated as MeH4F is incorporated into biomass via gluconeogenesis/PPP and that pyruvate contributes to at least 1/3 of the intracellular pool of acetyl-CoA. Such conversion seems to be essential in the strain, which is known to have high demand for acetyl-CoA as a key metabolite for membrane and PHB biosynthesis.
Singly and doubly 13C1-labeled citric acid and glutamate were generated quickly, followed by triply and quadruple labeled variants (Figures 3 and 4). About 32% of the total pool of citric acid was labeled throughout the experiments demonstrating significant flux through the forward TCA cycle. During 13CO2 experiments, singly labeled citric acid appeared quickly at the early time points most likely as a result of CO2 incorporation into oxaloacetate (Figure 4F). The concentration of doubly labeled citric acid eventually increased to 15%, yet triply labeled citric acid (the limit of detection of citric acid was less than 0.2 pmol on column) was not clearly detected within 20 min. For glutamate, singly and doubly labeled patterns rose slowly due to its high concentration (Figure 4H). However, 4.7% of the glutamate pool (567 ± 268 μM) was triply labeled after 20 min. 13C-labeling data indicate that cells could produce α-ketoglutarate from succinyl-CoA. The genome analysis suggests one system that may perform this function, namely related to 2-oxoacid ferredoxin oxidoreductase (METTOv1_1080004). This gene is expressed (Matsen et al., 2013). The presence of a partially reversible TCA has never been predicted for methanotrophic bacteria and represents an interesting subject for follow-up studies.
Conclusion
In this work we used 13C-labeling to test and refine the previous genome/transriptome based reconstruction of the central metabolic pathways in Methylosinus trichosporium OB3b grown on methane (Matsen et al., 2013). While some metabolic functions correlate well with previous enzymatic and genetic studies, several novel functions are newly predicted. The major outcomes of our work are listed below:
1. M. trichosporium OB3b uses the EMC variant of the serine cycle for carbon assimilation. Both in silico and in vivo labeling data indicate that a significant fraction of biomass comes from C4-metabolites (after carboxylation step). Thus the overall balance of the pathway is shifted to a higher CO2-assimilation mode, whereby eight molecules of CO2 are consumed per six molecules of MeH4F (Figure 1C). The data also suggest that a significant fraction of methane oxidized to CO2 is incorporated back via the EMC pathway. Thus this methanotroph differs from facultative methanol utilizers with respect to overall CO2 assimilation. Type II methanotrophs may represent a better system for C1-based commercial production of chemicals than serine cycle methylotrophs, as the methanotrophs will reincorporate more CO2 per unit of substrate oxidized.
2. Both genomic and trancriptomic data predict a variety of reactions at the PEP-pyruvate-oxaloacetate node. Metabolomic data indicate that the metabolic interconversions play an important role in the distribution of carbon flux between the major metabolic pathways. A significant fraction of PEP is converted to pyruvate, which serves as a precursor for alanine and is also used as an acceptor in two anaplerotic CO2-fixation reactions: pyruvate carboxytransferase and malic enzyme. 13C-labeling data strongly suggest that a part of the intracellular pool of the acetyl-CoA comes from pyruvate most likely via pyruvate dehydrogenase. The contribution of pyruvate to acetyl-CoA has never been discussed before, as it was assumed that the serine cycle refills the cellular needs for this intermediate.
3. A significant fraction of intracellular malate comes from the EMC pathway. The data suggest that the serine cycle is split into two functional branches that have different metabolic control. The first, labeled as “C1-fixation” part (Figure 1), includes all steps from glycine to PEP. This branch of the assimilatory pathway contributes to 53% of biomass. Most of the C1-carbon incorporated at first step of the branch is directed to gluconeogenesis (RNA/DNA, cell wall biosynthesis) and amino acid (serine, cysteine, tryptophan tyrosine, and phenylalanine) biosynthesis. The second part of the serine cycle (named “glyoxylate regeneration” in Figure 1) overlaps with the EMC pathway. Our data suggest the EMC pathway contributes significantly to replenish the intermediates (mostly malate, acetyl-CoA, and glyoxylate) of this part of the assimilation.
Experimental Procedures
Strain and Cultivation Conditions
Methylosinus trichosporium strain OB3b was kindly provided by Dr. Lisa Stein. The culture was grown in 250 mL glass bottles on modified NMS that contained the following contents (Whittenbury et al., 1970): 1 g KNO3, 1 g MgSO4⋅7H2O, 0.134 g CaCl2⋅2H2O, 0.25 g KH2PO4, 0.7 g Na2HPO4⋅12H2O, and 2 mL of trace elements solution. The trace elements solution contained 0.5 g Na2-EDTA, 1.0 g FeSO4⋅7H2O, 0.75 g Fe-EDTA, 0.8 g ZnSO4⋅7H2O, 0.005 g MnCl2⋅4H2O, 0.03 g H3BO3, 0.05 g CoCl2⋅6H2O, 0.4 g Cu-EDTA, 0.6 g CuCl2⋅2H2O, 0.002 g NiCl2⋅6H2O, and 0.05 g Na2MoO4⋅2H2O in 1 L of water. The bottles were sealed with rubber stoppers and aluminum caps. 50 mL of methane were added to the 200 mL headspace. If necessary, 10 mL of CO2 were added to the headspace. Bottles were shaken at 250 RPM at 30°C for 1–4 days.
Growth Parameters and Methane Consumption Rate Measurements
Methane consumption rates and cell density (OD600) were measured in triplicate as cultures grew. Methane measurements were collected on a Shimadzu Gas Chromatograph GC-14A, using FID detection with helium as the carrier gas. Concentrations were deduced from standard curves. OD600 was measured on a Beckman DU® 640B spectrophotometer in plastic 1.5 mL cuvettes with a 1 cm path length.
13CO2 and 13CH4 Labeling Experiments for Single Cells and Bulk Biomass: EA-IRMS and NanoSIMS Analyses
Experiments to determine the relative contributions of methylene H4F (produced from methane) and CO2 to labeled biomass were performed by modifying a previous method (Crowther et al., 2008). Cells were grown in NMS medium supplemented with either 20% 13CH4, 20% 12CH4, 20% 13CH4 and 5% 12CO2, or 20% 12CH4 and 5% 13CO2. After initial incubations at the described gas mixtures, cell cultures (OD600 = 0.4) were diluted (1:50) with fresh medium and transferred into new vials containing the same gases. For bulk and single cell NanoSIMS measurements, cells were harvested at OD600 = 0.4.
Bulk δ13C Isotopic Analysis via Elemental Analysis Coupled to Isotopic Ratio Mass Spectrometry
For bulk elemental analysis coupled to isotopic ratio mass spectrometry (EA-IRMS) measurements, harvested cell pellets were initially frozen at −80°C, then lyophilized prior to δ13C analysis. As these were isotopically enriched samples, cell material was initially diluted in unlabeled glucose in order to lower the δ13C values to an acceptable range for the instrument. Specifically, 2 mg of dry cell mass was re-suspended in a glucose solution (1.455 M final concentration, Mallinckrodt Chemicals) and diluted to create a 3 point standard curve. Samples were then aliquoted into tin capsules, dried under desiccant, and sealed. The δ13C values of these samples were measured using an ECS 4010 Elemental Analyzer (Costech, Valencia, CA, USA) connected to a Finnigan ThermoQuest DeltaplusXL IRMS and using CO2 as reference gas. δ13C values were calculated using the ISODAT 2.0 software (Thermo-Fisher Scientific, Bremen, Germany) and are reported in permil (‰) relative to the VPDB standard. Blank tin capsules along with urea and acetanilide standards of known δ13C value were analyzed after every 15 samples as well as at the beginning and end of each run in order to track instrument accuracy and precision.
As the cell cultures were diluted in unlabeled glucose, the raw δ13C values represented that of cellular carbon + glucose, and a back calculation was necessary in order to determine the δ13C of the cellular carbon. Three-point pure glucose standard curves were run in order to generate a linear regression of μmoles carbon versus peak area. The resulting equation was then used to determine the total μmoles carbon (glucose + cells) added to each sample run, in order to calculate μmoles of cellular carbon added. This value was then used to determine the δ13C of the cellular carbon.
δ13C Isotopic Analysis via Nanoscale Secondary Ion Mass Spectrometry
For bulk and single cell Nanoscale Secondary Ion Mass Spectrometry (NanoSIMS) measurements, cells were harvested at OD600 = 0.4 and fixed in 3% formaldehyde. Fixed cells were then washed in sterile buffer and deposited onto indium tin oxide (ITO) coated glass slides. Samples were analyzed using a CAMECA NanoSIMS 50 L housed at Caltech, using a mass resolving power approximately 5,000. A primary Cs+ ion beam (2.5 pA) was used to raster over target cells, and ion images ranging from 5 to 20 μm were collected at 256 × 256 pixel resolution with a dwell time of 14,000 ct/pixel. Several masses were collected in parallel including: 12C−, 13C−, 14N12C−, and 14N13C− using electron multiplier detectors. Resulting ion images were processed using L’Image software (http://www.dtm.ciw.edu/users/nittler/limage_manual.pdf). In order to compare bulk (average δ13C over entire rastor) and single cell δ13C values, NanoSIMS targets included both aggregated cells as well as less densely populated areas of the slide containing single cells. δ13C values for single cells were calculated by creating “regions of interest” around individual cells in L’Image, using a 0.636 micron diameter circle which matched the 12C14N count space for an individual cell.
CO2 incorporation was determined by assuming that the 13C in the cells incubated with 5% 12CO2 was solely from direct 13CH4 incorporation, while in the sample without external 12CO2, incorporation of 12CO2 was minimal. Instead, CO2 incorporation in the latter samples was assumed to be from 13CO2 generated from 13CH4. These assumptions have been shown to be valid for the serine cycle methanol-utilizer, M. extorquens AM1 (Crowther et al., 2008). It was not possible to carry out experiments with M. trichosporium OB3b in the absence of any external CO2 to confirm these assumptions directly, since this strain grows poorly under these conditions. Therefore the calculation represents a minimum, and may be higher.
In silico Amino Acid Usage Estimation
The amino acid usage data were generated from the codon usage as follows: nucleotide sequences for each predicted ORF were split into consecutive codons. The start and stop codons were ignored. The frequency of each codon was tabulated for each ORF. The amino acid usage table for each predicted protein product was multiplied by the mean number of reads mapped from replicates one and two. For each amino acid, the aggregate sum across predicted protein products was then normalized by the aggregate sum of all amino acids. Those fractions were also weighted by the neutral amino acid mass to predict the percent of the total pool by weight. The contribution of each amino acid into biomass was re-calculated using the estimation that proteins comprise 55% of biomass. The data are shown in Table A1 in Appendix.
Metabolomics: Chemicals, Sampling, and Metabolite Extraction
Acyl-CoAs, amino acids, organic acids, and sugar phosphates used as standards were of analytical grade and obtained from Sigma (St. Louis, MO, USA). Absolute ethanol (EMD Chemicals; Gibbstown, NJ, USA) was used in the metabolite extraction. All solvents used for liquid chromatography were LC-MS grade (Fisher Scientific; Fair Lawn, NJ, USA). Millipore purified water was used in the preparation of standard and sample solutions. For GC-MS sample derivatization, pyridine was purchased from EMD Chemicals (Gibbstown, NJ, USA), the trimethylsilylation (TMS) reagent [N,O–Bis (trimethylsilyl) trifluoroacetamide (BSTFA) + trimethylchlorosilane (TMCS), 99:1] and methoxyamine hydrochloride were both obtained from Sigma (St. Louis, MO, USA).
Samples (3 mL) of mid-exponential cultures (OD600 = 0.35 ± 0.02) were rapidly harvested by vacuum filtration using S-Pak™ membrane filters (0.22 μm, 47 mm) (Millipore; Billerica, MA, USA) and washed with 3 mL of fresh medium. The filter was immediately transferred to a petri dish located on the surface of a Cool Beans Chill Bucket™ (ISC Bioexpress; Kaysville, UT, USA) at −5°C. To collect cells, the following three sequential rinse solutions were applied: (i) 0.5 mL of 25 mM ice cold HEPES buffer (pH 5.2), (ii) 0.5 mL of −20°C ethanol solution (75/25, v/v, ethanol/aqueous 25 mM HEPES buffer, pH 5.2), and (iii) 1.5 mL of −20°C ethanol. The resulting solution was transferred to a precooled tube and stored in a −80°C freezer until it was ready for subsequent extractions.
Extraction of metabolites from M. trichosporium OB3b samples were carried out as previously published for M. extorquens AM1 (Yang et al., 2009). Briefly, the samples were incubated in a 100°C water bath for 3 min. The extracted cell suspension was cooled on ice for 5 min, then cell debris was removed by centrifugation at 5,000 RPM (4,300 × g) for 5 min. The cell-free metabolite extract was centrifuged at 14,000 RPM (20,800 × g) for 8 min. The supernatant was dried in a vacuum centrifuge (CentriVap® Concentrator System; Labconco, MO, USA) and stored at −80°C. For LC-MS/MS analysis, each dried sample was dissolved in 50 μL of purified water. For GC-MS analysis, each sample was further derivatized in two steps. First, keto group were methoximated by adding 50 μL of methoxyamine solution (25 mg/ml methoxyamine hydrochloride in pyridine) and incubated at 60°C for 30 min. Second, trimethylsilylation was performed by adding 50 μl of a TMS reagent (BSTFA/TMCS, 99:1) and incubated at 30°C for 90 min.
Metabolite Measurement and Absolute Quantification
LC-MS/MS experiments were carried out on a Waters© (Milford, MA, USA) LC-MS system consisting of a 1,525 μ binary HPLC pump with a 2,777°C autosampler coupled to a Quattro Micro™ API triple-quadrupole mass spectrometer (Micromass®; Manchester, UK). The HILIC method employing gradient elution was carried out using the previously described column (Luna NH2, 250 mm × 2 mm, 5 μm; Phenomenex®; Torrance, CA, USA) and nearly identical conditions as described below. Gradient elution was carried out with 20 mM ammonium acetate +0.35% NH4OH (28%) in water (v/v)/acetonitrile (95:5, v/v) with pH 9.7 (mobile phase A), and acetonitrile (mobile phase B). The linear gradients used were 85–0% B for 15 min, 0% B for 11 min, 0–85% B for 1 min, and 85% B for 15 min. The total run time was 42 min at 0.15 mL/min. The injection volume was 10 μL. The eluent from each LC column was directed into the ion source of a mass spectrometer. The multiple reaction monitoring (MRM) experiments were carried out as previously described (Yang et al., 2010). The dwell time for each MRM transition was 0.08 s. All peaks were integrated using MassLynx™ Applications Manager (version 4.1) software.
GC-MS experiments were performed using an Agilent 7890/5975C GC-MS (Agilent Corp; Santa Clara, CA, USA). The column was HP-5MS (30 m × 0.25 mm × 0.25 μm film; Restek; Bellefonte, PA, USA). Ultra high purity helium was used as a carrier gas at a constant flow rate of 1 mL/min, 1 μL of a given sample was injected in split-less mode through an Agilent 7890 auto sampler. The inlet temperature was set to 250°C. The temperature began at 60°C, was held for 0.25 min, and then increased at 8°C/min to 280°C where it held for 10 min. The ion source temperature was set to 250°C. Mass spectra were collected from m/z 50–500 at 3 spectra/s with a 7.4 min solvent delay. The peaks were evaluated using Agilent data analysis software.
The absolute intracellular metabolite quantification was determined using an isotope ratio-based approach as previously published (Yang et al., 2010). Briefly, 13C-labeled cell extracts produced from the continuous cultivation of M. extorquens AM1 served as 13C-labeled internal standards (13C-labeled IS). After fast filtration of M. trichosporium OB3b, a fixed amount of 13C-labeled IS was added to the petri dish prior to the cell storage. After the metabolites were extracted, they were analyzed by LC-MS/MS and GC-MS as previously described. The calibration curve was developed by adding a fixed amount of 13C-labeled IS to different dilutions of primary stock solutions of 12C-standard mixtures. Calibration plots were obtained by plotting the ratio of 12C-standard to 13C-labeled IS versus the 12C-standard concentration.
Dynamic 13C-Incorporation
For the 13CH4 labeling experiment, M. trichosporium OB3b grown on 12C-methane to mid-exponential phase (corresponding to 25% of the added methane consumed) were rapidly transferred to a fresh flask and supplemented with the same percentage of 13CH4. At the defined time points (0, 1, 2, 5, 10, 20, 40, and 60 min), the cell culture was harvested, and metabolites were analyzed as previously described. The mass isotopomer distributions were corrected for the natural isotope contribution by using a matrix-based method (Yang et al., 2012) and calculated as the relative abundances of the different possible mass isotopomers of a metabolite.
For the 13CO2 tracing experiment, mid-exponential cultures were spiked with 13CO2, resulting in 5% 13CO2 in the headspace. These were also harvested at 0, 0.5, 1, 2, 5, 10, and 20 min, and metabolites were analyzed as previously described.
Calculation of 13C-Incorporation Rate
Total 13C-incorporation of each metabolite was obtained by normalizing to its total carbon number. Relative isotopic abundance (Mi) for a metabolite in which i 13C atoms were incorporated was calculated by the Eq. 1:
Where mi represented the isotopic abundance for a metabolite in which i13C atoms were incorporated and n represented the maximum number of 13C atoms incorporated.
Total 13C-incorporation of a metabolite with N carbon atoms was obtained by normalizing to its total carbon number according to the following Eq. 2:
13C-incorporation rate was calculated from the initial slope of all 13C-isotopologues versus time. Comparison of 13C-incorporation was obtained by plotting 13C-incorporation (%) during a switchover from 12CH4 to 13CH4 versus 13CO2 spiking in a time course. The correlation coefficients (R2) between 13CO2 vs. 13CH4 were calculated using linear regression.
Cell Volume Determination
An average OB3b cell volume was determined based on confocal images of OB3b cells stained with the lipid dye FM 1–43 (Invitrogen™). After growing colonies on agar plates, OB3b cells were re-suspended in NMS1 media as described and stained for 1 h at 20°C with FM 1–43 (5 μg/mL). The cells were then washed with NMS1 media, seeded on a poly lysine coated slide, and imaged on a Zeiss Axiovert 200 M microscope with an LSM 510 META confocal attachment with a 100× oil immersion objective (NA = 1.30). Image spacing in the Z-axis was 0.5 μm. Complete images of non-mobile cells were analyzed using ImageJ with the Volumest plug-in to determine the average volume (3.79 fL/cell; n = 60). The concentration of cells per mL was estimated using a CyFlow® (Partec) flow cytometer with true volumetric absolute counting (TVAC). An average M. trichosporium OB3b cell number per 1 mL per OD = 1 is 8.6 × 107 ± 2.6 × 106.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are very grateful to Dr. Mary E. Lidstrom, Dr. Ivan Berg and Dr. Ludmila Chistoserdova for insightful suggestions on the manuscript. This work was supported by the DOE (DE-SC0005154).
References
Anthony, C. (2011). How half a century of research was required to understand bacterial growth on C1 and C2 compounds; the story of the serine cycle and the ethylmalonyl-CoA pathway. Sci. Prog. 94, 109–137.
Cornish, A., Nicholls, K. M., Scott, D., Hunter, B. K., Aston, W. J., Higgins, I. J., et al. (1984). In vivo super 13C NMR investigations of methanol oxidation by the obligate methanotroph Methylosinus trichosporium OB3b. J. Gen. Microbiol. 130, 2565–2575.
Crowther, G. J., Kosály, G., and Lidstrom, M. E. (2008). Formate as the main branch point for methylotrophic metabolism in Methylobacterium extorquens AM1. J. Bacteriol. 190, 5057–5062.
DiSpirito, A. A., Zahn, J. A., Graham, D. W., Kim, H. J., Larive, C. K., Derrick, T. S., et al. (1998). Copper-binding compounds from Methylosinus trichosporium OB3b. J. Bacteriol. 180, 3606–3613.
Doronina, N. V., Ezhov, V. A., and Trotsenko, Y. uA. (2008). Growth of Methylosinus trichosporium OB3b on methane and poly-beta-hydroxybutyrate biosynthesis. Appl. Biochem. Microbiol. 44, 182–185.
Gilbert, B., McDonald, I., Finch, R., Stafford, G., Nielsen, A., and Murrell, J. (2000). Molecular analysis of the pmo (particulate methane monooxygenase) operon from two type II methano- trophs. Appl. Environ. Microbiol. 66, 966–975.
Guckert, J. B., Ringelberg, D. B., White, D. C., Hanson, R. S., and Bratina, B. J. (1991). Membrane fatty acids as phenotypic markers in the polyphasic taxonomy of methylotrophs within the proteobacteria. J. Gen. Microbiol. 137, 2631–2641.
Jollie, D. R., and Lipscomb, J. D. (1991). Formate dehydrogenase from Methylosinus trichosporium OB3b. Purification and spectroscopic characterization of the cofactors. J. Biol. Chem. 266, 21853–21863.
Lawrence, A. J., and Quayle, J. R. (1970). Alternative carbon assimilation pathways in methane-utilizing bacteria. J. Gen. Microbiol. 63, 371–374.
Lloyd, J. S., Finch, R., Dalton, H., and Murrell, J. C. (1999). Homologous expression of soluble methane monooxygenase genes in Methylosinus trichosporium OB3b. Microbiology 145, 461–470.
Lontoh, S., and Semrau, J. D. (1998). Methane and trichloroethylene degradation by Methylosinus trichosporium OB3b expressing particulate methane monooxygenase. Appl. Environ. Microbiol. 64, 1106–1114.
Matsen, J. B., Yang, S., Stein, L. Y., Beck, D., and Kalyuzhnaya, M. G. (2013). Global molecular analyses of methane methabolism in methanotrophic Alphaproteobacterium, Methylosinus trichosporium OB3b. Part I. Transcripomic study. Front. Microbiol. 4:40. doi:10.3389/fmicb.2013.00040.
Neidhardt, F. C., Curtiss, R. III, Ingraham, J. L., Lin, E. C. C., Low, K. B., Magasanik, B., et al. (1996). Escherichia Coli and Salmonella Typhimurium: Cellular and Molecular Biology, 2nd Edn. Washington: ASM Press.
Park, S., Hanna, L. M., Taylor, R. T., and Droege, M. W. (1991). Batch cultivation of Methylosinus trichosporium OB3b. I: production of soluble methane monooxygenase. Biotechnol. Bioeng. 38, 423–433.
Park, S., Shah, N. N., Taylor, R. T., and Droege, M. W. (1992). Batch cultivation of Methylosinus trichosporium OB3b: II. production of particulate methane monooxygenase. Biotechnol. Bioeng. 40, 151–157.
Patel, R. N., Hoare, S. L., and Hoare, D. S. (1979). (14C) acetate assimilation by obligate methylotrophs, Pseudomonas methanica and Methylosinus trichosporium. Antonie Van Leeuwenhoek 45, 499–511.
Peyraud, R., Kiefer, P., Christen, P., Massou, S., Portais, J. C., and Vorholt, J. A. (2009). Demonstration of the ethylmalonyl-CoA pathway by using 13C metabolomics. Proc. Natl. Acad. Sci. U.S.A. 106, 4846–4851.
Peyraud, R., Schneider, K., Kiefer, P., Massou, S., Vorholt, J., and Portais, J. P. (2011). Genome-scale reconstruction and system level investigation of the metabolic network of Methylobacterium extorquens AM1. BMC Syst. Biol. 5:189. doi:10.1186/1752-0509-5-189
Shishkina, V. N., and Trotsenko, Y. A. (1982). Multiple enzymatic lesions in obligate methanotrophic bacteria. FEMS Microbiol. Lett. 13, 237–242.
Stein, L. Y., Yoon, S., Semrau, J. D., Dispirito, A. A., Crombie, A., Murrell, J. C., et al. (2010). Genome sequence of the obligate methanotroph Methylosinus trichosporium strain OB3b. J. Bacteriol. 192, 6497–6498.
Strom, T., Ference, T., and Quayle, J. R. (1974). The carbon assimilation pathways of Methylococcus capsulatus, Pseudomonas methanica, and Methylosinus trichosporium (OB3B) during growth on methane. Biochem. J. 144, 465–476.
Sun, A. K., and Wood, T. K. (1996). Trichloroethylene degradation and mineralization by pseudomonads and Methylosinus trichosporium OB3b. Appl. Microbiol. Biotechnol. 45, 248–256.
Trotsenko, Y. A., and Murrell, J. C. (2008). Metabolic aspects of aerobic obligate methanotrophy. Adv. Appl. Microbiol. 63, 183–229.
Weaver, T. L., Patrick, M. A., and Dugan, P. R. (1975). Whole-cell and membrane lipids o the methanotrophic bacterium Methylosinus trichosporium. J. Bacteriol. 124, 602–605.
Whittenbury, R., Phillips, K. C., and Wilkinson, J. F. (1970). Enrichment, isolation and some properties of methane-utilizing bacteria. J. Gen. Microbiol. 61, 205–218.
Williams, A. M. (1988). The Biochemistry and Physiology of Poly-Beta-Hydroxybutyrate Metabolism in Methylosinus trichosporium OB3b. Ph.D. thesis, Cranfield Institute of Technology, Biotechnology Centre, Cranfield.
Yang, S., Nadeau, J. S., Humston-Fulmer, E. M., Hoggard, J. C., Lidstrom, M. E., and Synovec, R. E. (2012). Gas chromatography-mass spectrometry with chemometric analysis for determining 12C and 13C labeled contributions in metabolomics and 13C flux analysis. J. Chromatogr. A 1240, 156–164.
Yang, S., Sadilek, M., and Lidstrom, M. E. (2010). Streamlined pentafluorophenyl propyl column liquid chromatography-tandem quadrupole mass spectrometry and global (13)C-labeled internal standards improve performance for quantitative metabolomics in bacteria. J. Chromatogr. A 1217, 7401–7410.
Yang, S., Sadilek, M., Synovec, R. E., and Lidstrom, M. E. (2009). Liquid chromatography-tandem quadrupole mass spectrometry and comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry measurement of targeted metabolites of Methylobacterium extorquens AM1 grown on two different carbon sources. J. Chromatogr. A 1216, 3280–3289.
Appendix
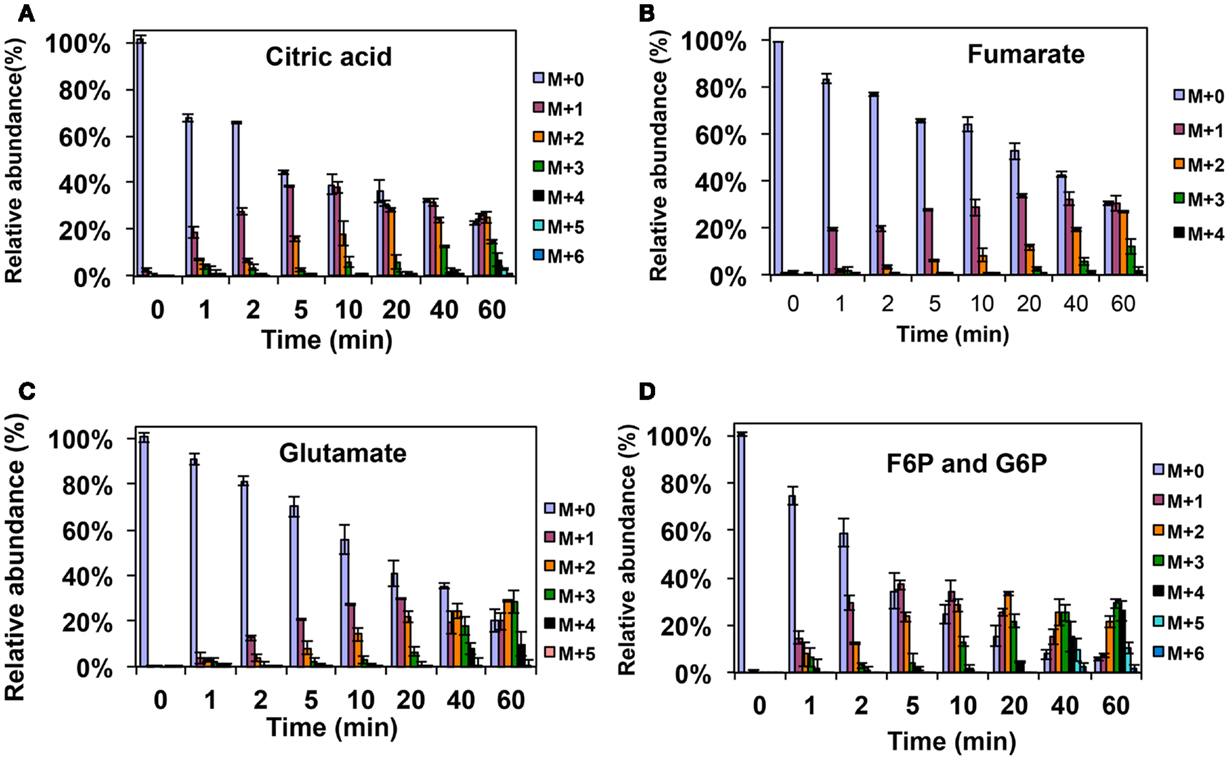
Figure A1. 13C-incorporation during the switch from 12CH4 to 13CH4 in Methylosinus trichosporium OB3b. (A)12C/13C-isotopomer distributions of citric acid (A), fumarate (B), glutamate (C), and fructose-6-phosphate/glucose-6-phosphate (D).
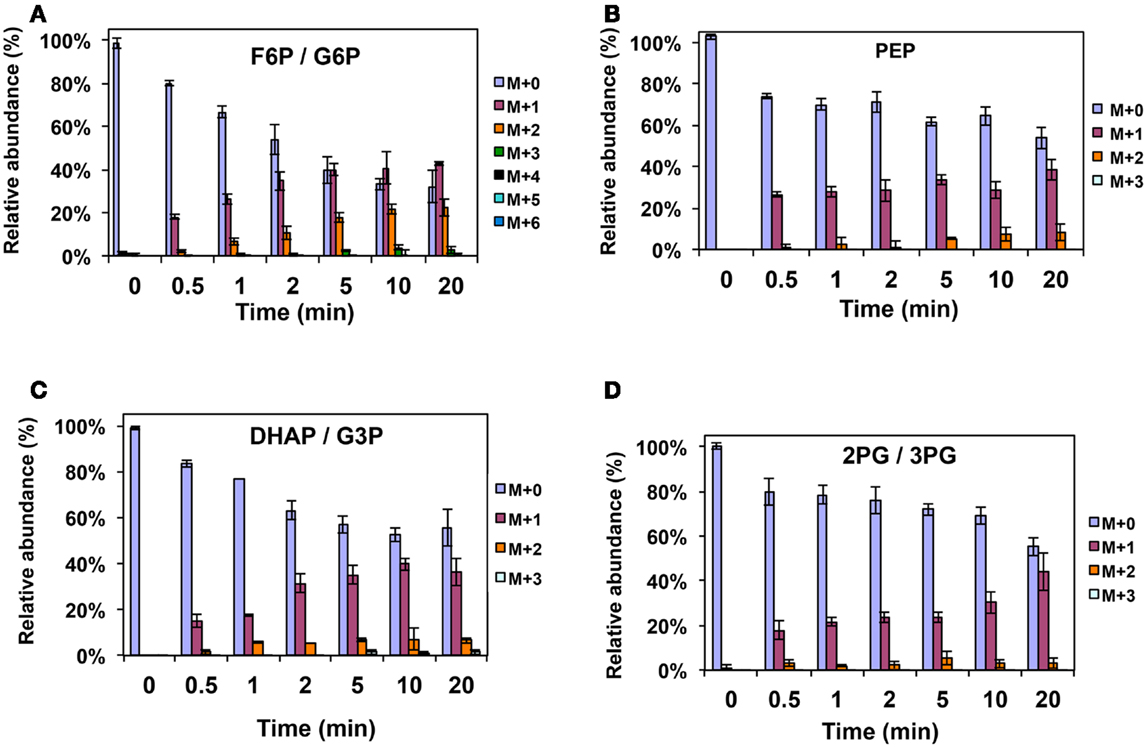
Figure A2. 13C-incorporation during 12CO2 spiking in Methylosinus trichosporium OB3b grown on 12CH4. 12C/13C-isotopomer distributions of fructose-6-phosphate/glucose-6-phosphate (A), phosphoenolpyruvate (B), dihydroxyacetone phosphate/glyceraldehyde 3-phosphate (C), 2-phosphoglycerate/3-phosphoglycerate (D).
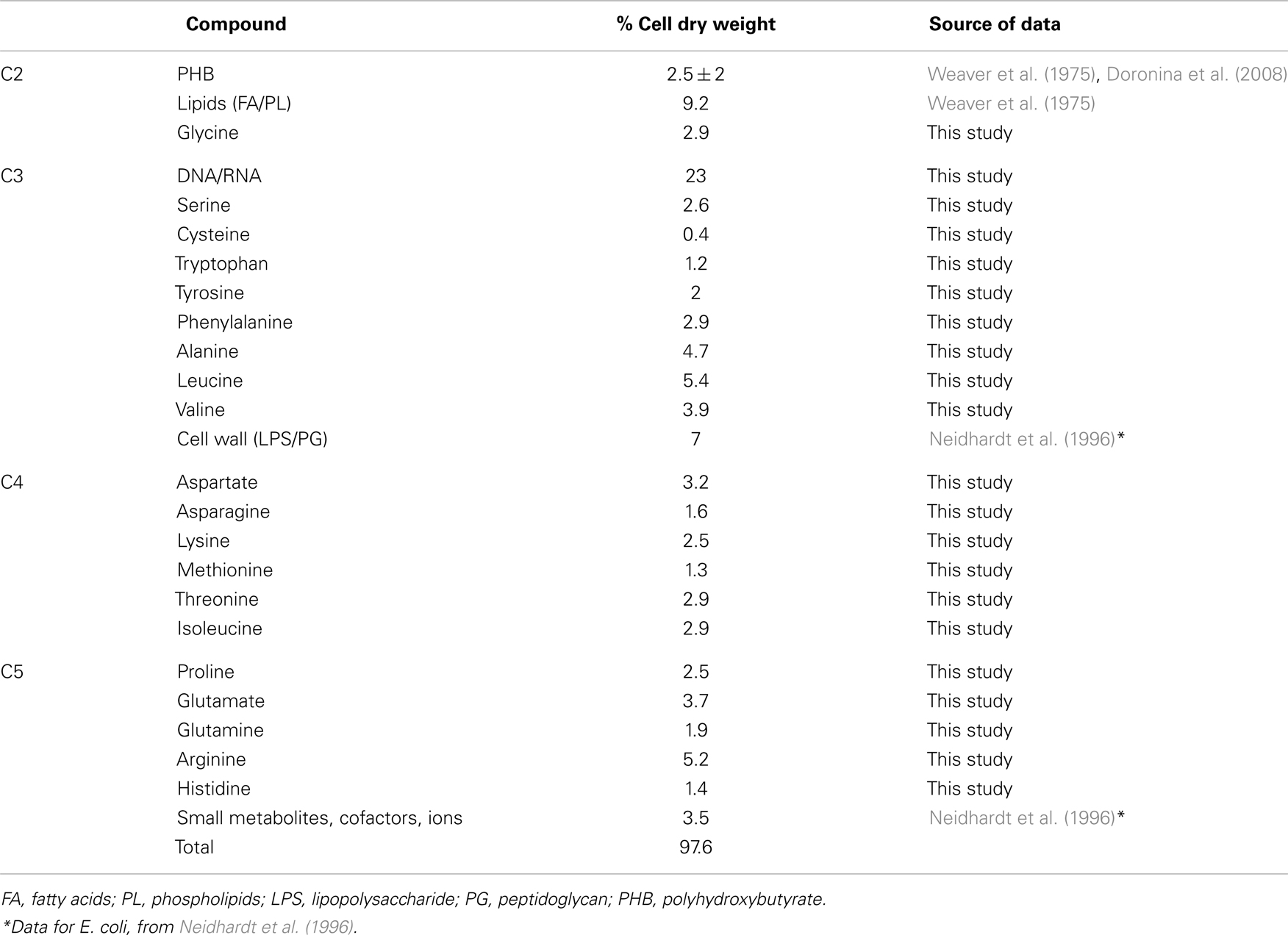
Table A1. Macromolecule and amino acid composition of cells grown on methane.
Keywords: methanotrophic proteobacteria, serine cycle, ethylmalonyl-CoA pathway in methanotrophs, metabolic fluxes labeling
Citation: Yang S, Matsen JB, Konopka M, Green-Saxena A, Clubb J, Sadilek M, Orphan VJ, Beck D and Kalyuzhnaya MG (2013) Global molecular analyses of methane metabolism in methanotrophic Alphaproteobacterium, Methylosinus trichosporium OB3b. Part II. metabolomics and 13C-labeling study. Front. Microbiol. 4:70. doi: 10.3389/fmicb.2013.00070
Received: 01 January 2013; Paper pending published: 28 January 2013;
Accepted: 12 March 2013; Published online: 03 April 2013.
Edited by:
Amy V. Callaghan, University of Oklahoma, USAReviewed by:
Alan A. DiSpirito, Ohio State University, USAAmy V. Callaghan, University of Oklahoma, USA
Copyright: © 2013 Yang, Matsen, Konopka, Green-Saxena, Clubb, Sadilek, Orphan, Beck and Kalyuzhnaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Marina G. Kalyuzhnaya, Department of Microbiology, University of Washington, Benjamin Hall IRB RM 455, 616 NE Northlake Place, Seattle, WA 98105, USA. e-mail: mkalyuzh@uw.edu