
- Department of Virology 1, National Institute of Infectious Diseases, Tokyo, Japan
Ebola and Marburg hemorrhagic fevers (EHF and MHF) are caused by the Filoviridae family, Ebolavirus and Marburgvirus (ebolavirus and marburgvirus), respectively. These severe diseases have high mortality rates in humans. Although EHF and MHF are endemic to sub-Saharan Africa. A novel filovirus, Lloviu virus, which is genetically distinct from ebolavirus and marburgvirus, was recently discovered in Spain where filoviral hemorrhagic fever had never been reported. The virulence of this virus has not been determined. Ebolavirus and marburgvirus are classified as biosafety level-4 (BSL-4) pathogens and Category A agents, for which the US government requires preparedness in case of bioterrorism. Therefore, preventive measures against these viral hemorrhagic fevers should be prepared, not only in disease-endemic regions, but also in disease-free countries. Diagnostics, vaccines, and therapeutics need to be developed, and therefore the establishment of animal models for EHF and MHF is invaluable. Several animal models have been developed for EHF and MHF using non-human primates (NHPs) and rodents, which are crucial to understand pathophysiology and to develop diagnostics, vaccines, and therapeutics. Rhesus and cynomolgus macaques are representative models of filovirus infection as they exhibit remarkably similar symptoms to those observed in humans. However, the NHP models have practical and ethical problems that limit their experimental use. Furthermore, there are no inbred and genetically manipulated strains of NHP. Rodent models such as mouse, guinea pig, and hamster, have also been developed. However, these rodent models require adaptation of the virus to produce lethal disease and do not mirror all symptoms of human filovirus infection. This review article provides an outline of the clinical features of EHF and MHF in animals, including humans, and discusses how the animal models have been developed to study pathophysiology, vaccines, and therapeutics.
Introduction
The Family Filoviridae includes three accepted genera, Ebolavirus (ebolavirus), Marburgvirus (marburgvirus), and Cuevavirus (Figure 1) (Kuhn et al., 2011, 2013). Filoviruses are classified as biosafety level 4 (BSL-4) agents because they cause severe hemorrhagic fevers in humans and non-human primates (NHPs) with high case-fatality rates, ranging between 23 and 90% (Sanchez et al., 2007). Each of the Marburgvirus and Cuevavirus genera consists of a single species, Marburg marburgvirus and Lloviu cuevavirus, respectively. The genus Marburgvirus has two subspecies: Marburg virus (MARV) and Ravn virus (RAVV). The genus Ebolavirus is divided into five distinct species, Zaire ebolavirus (Ebola virus, EBOV), Sudan ebolavirus (Sudan virus, SUDV), Tai Forest ebolavirus (Tai Forest virus, TAFV), Bundibugyo ebolavirus (Bundibugyo virus, BDBV), and Reston ebolavirus (Reston virus, RESTV; Kuhn et al., 2013). EBOV is highly virulent to humans and NHPs with a mortality rate of up to 90% in African epidemics. The case fatality rate of SUDV and BDBV is ~50 and 25%, respectively; the only person known to have been infected with TAFV survived. RESTV has been known to cause symptomatic disease in NHPs but not in humans. Lloviu virus belonging to the genus Cuevavirus was identified in the absence of replicating isolates during an investigation of die-off bats in Spain and the virulence for humans and NHPs has not been assessed (Negredo et al., 2011).

Figure 1. Phylogenetic analysis of filovirus based on nucleotide sequence. The phylogenetic tree based on complete viral genome sequences was constructed by using the neighbor-joining method. Numbers at branch points indicate bootstrap values (1000 replicates). The GenBank accession numbers of Tai Forest virus (TAFV), Bundibugyo virus (BDBV), Ebola virus (EBOV), Reston virus (RESTV), Sudan virus (SUDV), Lloviu virus (LLOV), Marburg virus (MARV), and Ravn virus (RAVV) are FJ217162, FJ217161, AF086833, AB050936, AY729654, JF828358, DQ217792, and EF446131, respectively.
Although there has been an increasing frequency of filovirus outbreaks reported from endemic regions of Africa and Asia in recent years, there are no licensed vaccines or effective therapeutics for filovirus hemorrhagic fever.
The primary source of patients with filovirus hemorrhagic fever was mainly linked to exposure to animal carcasses found in the forest or to the putative bat reservoir, resulting in subsequent transmission through direct person-to-person contact (Leroy et al., 2004, 2009). Filoviruses enter the body via direct contact with infectious blood and/or body fluids. After an incubation period of 2–21 days, non-specific initial symptoms such as fever, chills, fatigue, headache, and myalgia appear. About 5–7 days after onset, a maculopapular rash usually develops on the face, buttocks, trunk, and/or arms and later generalizes over the entire body. As disease progresses, systemic (prostration, lethargy), gastrointestinal (anorexia, vomiting, abdominal pain, diarrhea), respiratory (chest pain, breath shortness, cough, nasal discharge), vascular (conjunctival injection, postural hypotension, edema), and neurological (headache, confusion, coma) manifestations are observed. Some patients develop multiple foci of mucosal hemorrhage, which is especially evident in conjunctiva and gingiva together with bleeding from venipuncture sites. Hemorrhagic symptoms observed during the peak of the illness include petechiae, ecchymoses, epistaxis, mucosal hemorrhages, and/or visceral hemorrhagic effusions. In fatal cases, patients die with hypovolemic shock and multiple organ failure between Day 6 and 16.
Animal models of filovirus infection have been developed in mice, guinea pigs, hamsters, and NHPs (Connolly et al., 1999; Bente et al., 2009; Bradfute et al., 2012; Wahl-Jensen et al., 2012). The development of animal models that accurately reflect human disease is critical to understanding the pathogenesis of Ebola and Marburg hemorrhagic fevers (EHF and MHF, respectively), because filoviral outbreaks in humans are sporadic and there is limited clinical data and access to human tissue. Since the wild-type virus replicates to high titers in NHPs and the virus causes symptoms, including hemorrhage and shock, which are similar to those of patients with EHF and MHF, NHP models may be the most useful to evaluate the efficacy of candidate vaccines and treatment measures. However, small animal models are also needed for preliminary evaluation of vaccines and therapeutic interventions against filovirus diseases, because of the ethical and handling issues related to using NHPs.
Here, we summarize and discuss the animal models developed for the study of hemorrhagic fever caused by filoviruses.
Mouse Models
In contrast to the development of the NHP and guinea pig models, as described in later sections, development of a mouse model of filovirus infection has been unsuccessful due to the fact that adult immunocompetent mice were resistant to wild-type filovirus infection. The intraperitoneal or intracerebral inoculation of newborn mice and 4-day-old suckling mice with non-mouse adapted EBOV was shown to cause lethal infections, but 8-day-old or older mice did not show any symptoms (Johnson et al., 1995; Bray, 2001). Serial passage of wild-type EBOV in suckling mice was needed for adaptation, in which the virus acquired lethal virulence to adult immunocompetent mice (Bray, 2001). Intraperitoneal inoculation of mouse-adapted EBOV with a 1–100 plaque forming unit (pfu) dose (30–3000 times the median lethal dose) caused lethal infection to 5-week-old BALB/c, C57BL/6 and ICR (CD-1) mice, but subcutaneous inoculation of the virus at a dose of 106 pfu did not cause symptomatic illness in 3-week-old adult mice (Bray, 2001). This phenomenon is not observed in NHP and guinea pig models, which are susceptible to wild-type EBOV infection through any route of inoculation. CD8+ T cells and perforin, but not B cells and CD4+ T cells, are required for resistance to subcutaneous inoculation of EBOV (Gupta et al., 2005). It is supposed that the presence of regional lymph nodes and/or Langerhans cells in the skin contributes to protection from filoviral subcutaneous infection via activation of CD8+ cells, however, there are no reports to prove this hypothesis.
It has been shown that mouse-adapted filovirus is fatal over a broad range of ages in BALB/c mice. Infected mice became acutely ill with symptoms of ruffled fur, reduced activity, and weight loss on Day 3–4 post-infection and died on Days 5–7, although these lengths differed depending on the challenge dose (Bray et al., 1999; Warfield et al., 2009). Virus titer in the liver and spleen exceeded 107 pfu/g within 3 days after infection and then reached a maximum of over 109 pfu/g at Day 5 post-inoculation. These titers exceeded the peak viral concentrations in the liver and spleen of infected guinea pigs (about 106 pfu/g) and NHPs (about 107 pfu/g; Bray et al., 1999). The virus is generally undetectable in serum on Day 1, but by Day 3 the viremia level peaks at approximately 107 pfu/ml, which was comparable to that in NHPs and exceeds that in guinea pigs (104−5 pfu/ml; Bray et al., 1999).
As seen in the NHP model, the systemic viral spread results in extensive infection and necrosis of the liver, spleen, and other organs (Bray et al., 1999; Warfield et al., 2009). In liver from mice infected with mouse-adapted EBOV or RAVV, viral replication was observed in hepatocytes, Kupffer cells, and sinusoidal endothelial lining cells. Histological lesions were observed by Day 4 after inoculation, including coalescing, foci of hepatocellular vacuolar change, degeneration, and necrosis of hepatocytes. In the spleen, viral antigen was detected on Day 2 after infection, at which point coagulopathy, such as disseminated intravascular coagulation (DIC) accompanied by prolongation of prothrombin time (PT) and activated partial thromboplastin time (aPTT), was not observed in the moribund mice (Bray et al., 2001; Warfield et al., 2009).
Mouse-adapted EBOV initially infects macrophages and other mononuclear phagocytes at the site of invasion and in regional lymph nodes. The major target cells of infection are as the same as those in humans, NHPs, and guinea pigs (Davis et al., 1997; Connolly et al., 1999; Zaki and Goldsmith, 1999; Gibb et al., 2001). Viral replication in mononuclear phagocytes in the lymph node, spleen, and thymus and an increase in the number of virus-infected Kupffer cells in the liver were observed by Day 3 after infection. Most mononuclear phagocytes throughout the body appear to be infected and the mice died by Days 5–6.
The adaptation of EBOV to adult mice resulted in 8 amino acid changes in both the coding and non-coding regions of the virus genome compared to the original wild-typed virus (Ebihara et al., 2006). Nucleotide substitutions leading to amino acid changes were found in VP35, VP24, NP, and L viral proteins. VP24 and VP35 are known as type I interferon (IFN) antagonists and interfere with type I IFN-mediated antiviral response in vitro (Bowen et al., 1980; Basler et al., 2000; Bente et al., 2009; Halfmann et al., 2011). VP24 functions as an IFN antagonist by binding karyopherin α and blocking nuclear accumulation of signal transducer and activator of transcription 1 (STAT1; Reid et al., 2007). VP35 is also implicated in blocking type I IFN responses by inhibiting phosphorylation of interferon regulatory factor (IRF) 3 and 7 by the Tank binding kinase-1 and I-Kappa-B kinase epsilon, and sequestering the viral RNA from detection by RIG-I like receptor (Ramanan et al., 2011, 2012). It is considered that there is a significant relationship between filoviral virulence and the ability of the virus to evade the type I IFN-induced antiviral response (van der Groen et al., 1979). The mutations in NP and VP24 genes were found to be critical for acquisition of EBOV virulence in adult mice, but not VP35 mutations (Ebihara et al., 2006). NP is tightly coupled with viral RNA and forms the nucleocapsid complex together with L, VP30, and VP35. Although it is unclear how NP is involved in the IFN response, NP is thought to confer evasion from the IFN-stimulated antiviral responses directly or indirectly in infected mice.
The mutations identified for adaptation of marburgvirus to mice differed from those required for that of ebolavirus. The amino acid mutations were found in VP40, VP35, NP, and VP30 in mouse-adapted RAVV compared to those of the wild-type virus derived from a patient (Warfield et al., 2009). It is still unclear what is the role and necessity of each of the mutations in mouse-adapted RAVV. Further experiments are required to clarify which mutations are critical for adaptation to mice.
Adult mice treated with antibodies against IFN-α/β became susceptible to infections with non-adapted EBOV or SUDV infected via the intraperitoneal route, and to mouse-adapted virus infected via the subcutaneous route (Bray, 2001). Furthermore, non-adapted EBOV, SUDV, MARV, or RAVV caused illness in KO mice lacking type I IFN receptors or the STAT1 protein (Bray, 2001). These results suggest that inhibition of type I IFN response against filovirus infections is critical for pathogenesis in mouse models.
Mice infected with the mouse-adapted filovirus are different from humans and NHPs infected with the original filoviruses in terms of a lack of severe coagulation disorder and fibrin deposition. Mouse models are useful tools for studying basic aspects of replication, pathogenesis, and immune responses and also serve as an irreplaceable platform for evaluating the efficacy of the wide range of the candidate vaccines and therapeutic agents.
Guinea Pig Models
Guinea pigs are susceptible to several arenaviruses, Lassa fever virus, Junin virus, and Guanarito virus and used as animal models for human viral hemorrhagic fevers caused by these viruses (Bowen et al., 1977; Jahrling et al., 1982; Kenyon et al., 1990; Hall et al., 1996). However, infection of guinea pigs with wild-type filovirus usually causes only a transient febrile illness (Simpson et al., 1968; Robin et al., 1971; Bowen et al., 1977). Filoviruses need to be serially passaged in guinea pigs to acquire the ability to cause lethal infection in guinea pigs (Simpson et al., 1968; Robin et al., 1971; Ryabchikova et al., 1996). Guinea pigs inoculated with guinea pig (GP)-adapted virus showed similar symptoms such as fever, anorexia, and dehydration, to those reported in humans and NHPs infected with wild-type filovirus (Simpson et al., 1968; Robin et al., 1971; Connolly et al., 1999). GP-adapted EBOV-infected guinea pigs showed fibrin deposition coincident with decreases in platelet count during the late stage of infection (Connolly et al., 1999). GP-adapted EBOV replicated to high titers in the spleen, liver, adrenal gland, and lung, resulting in viremia in guinea pig models, although the peak titers were less than those demonstrated in NHPs. Viremia in guinea pigs developed within 2 days after inoculation and increased during the course of the disease, reaching a peak on Day 7 (>104 pfu/ml; Connolly et al., 1999; Subbotina et al., 2010). The guinea pigs infected with the GP-adapted filovirus died on 7–9 days after infection (Simpson et al., 1968; Robin et al., 1971; Connolly et al., 1999; Subbotina et al., 2010).
Histopathological changes in the liver of guinea pigs infected with GP-adapted filoviruses included replication of the viruses in Kupffer cells, multifocal necrosis of hepatocytes, and congestion and destruction of the sinusoid wall, which were also similar to those reported in humans and NHPs infected with wild-type filoviruses (Korb and Slenczka, 1971; Connolly et al., 1999; Ryabchikova et al., 2003). However, infiltrations of inflammatory cells in the liver and other organs were mild or absent (Connolly et al., 1999; Ryabchikova et al., 2003). Lymphoid necrosis was observed in the spleen and lymph nodes of guinea pig models.
Neutrophilia and lymphopenia became detectable in the guinea pig model as early as 2 days after infection and the severity continued to increase over the course of infection (Connolly et al., 1999; Subbotina et al., 2010). However, lymphocyte bystander apoptosis, an important feature in NHPs and mice, was not prominent in guinea pigs (Bray et al., 1999; Connolly et al., 1999; Bradfute et al., 2007). Thrombocytopenia was marked during the late stages of the disease when guinea pigs became moribund and platelets fell from a mean of ~500,000 to <50,000/μl. Fibrin deposition was a late event, beginning only modestly in the liver and spleen on Day 4, with increases in distribution and amount on Days 7–9, coincident with decreases in platelet counts.
Comparative sequence analysis of the complete genomes of the GP-adapted EBOV and wild-type virus showed 8 nucleotide differences, which led to 5 amino acid substitutions; single amino acid mutations in NP and L and 3 mutations in VP24 (Volchkov et al., 2000). Using a reverse genetics approach, it was shown that VP24 had a critical role in the pathogenesis and the amino acid changes in VP24 were essential to achieve EBOV virulence in guinea pigs. VP24 was demonstrated to antagonize IFN signaling by binding host karyopherin α proteins and prevent transport of the tyrosine phosphorylated transcription factor STAT-1 to the nucleus (Reid et al., 2007; Mateo et al., 2010; Zhang et al., 2012). One of the substitutions in VP24 of GP-adapted EBOV was located in the proximal domain, which was recently shown to be involved in karyopherin binding and required for efficient control of the IFN response (Mateo et al., 2010). However, the mutations associated with EBOV adaptation to the guinea pigs did not affect the ability of VP24 to inhibit IFN signaling (Mateo et al., 2011). VP24 participates in the assembly and/or proper formation of viral nucleocapsids. The lack of virulence of wild-type virus in guinea pigs was associated with an inability of the virus to replicate in and/or be released from hepatocytes and macrophages efficiently (Mateo et al., 2011). Wild-type VP24 is somehow incapable of participating in assembly of viral nucleocapsids in guinea pigs. Mutations in VP24 for adaption to guinea pigs recovered the ability of EBOV to replicate in both macrophages and hepatocytes and to facilitate the systemic spread of the virus.
Syrian Golden Hamster Models
The pathogenesis of rodent-adapted filoviruses differs in some aspects from those of humans and NHPs infected with wild-typed virus (Table 1). Fever and cutaneous rash, which are major clinical signs of EHF and MHF in humans and NHPs, are absent in mice infected with mouse-adapted virus. Fever appears in guinea pigs infected with GP-adapted virus, but rash does not develop in these animals. Mice infected with mouse-adapted virus do not consistently display coagulation abnormalities. Compared to mice, guinea pigs infected with GP-adapted virus develop coagulation defects, including a drop in platelet counts and an increase in coagulation time, but coagulopathy (i.e., DIC) are not as marked as that observed in NHPs. Furthermore, lymphocyte apoptosis observed in humans, NHPs, and mice was not determined in the guinea pig model. Because of these differences in the rodent models, some vaccines (e.g., irradiated virion) and therapeutics (e.g., passive immunization with antiserum) that were effective in rodents challenged with adapted virus fail to protect NHPs challenged with wild-type virus (Wahl-Jensen et al., 2012).
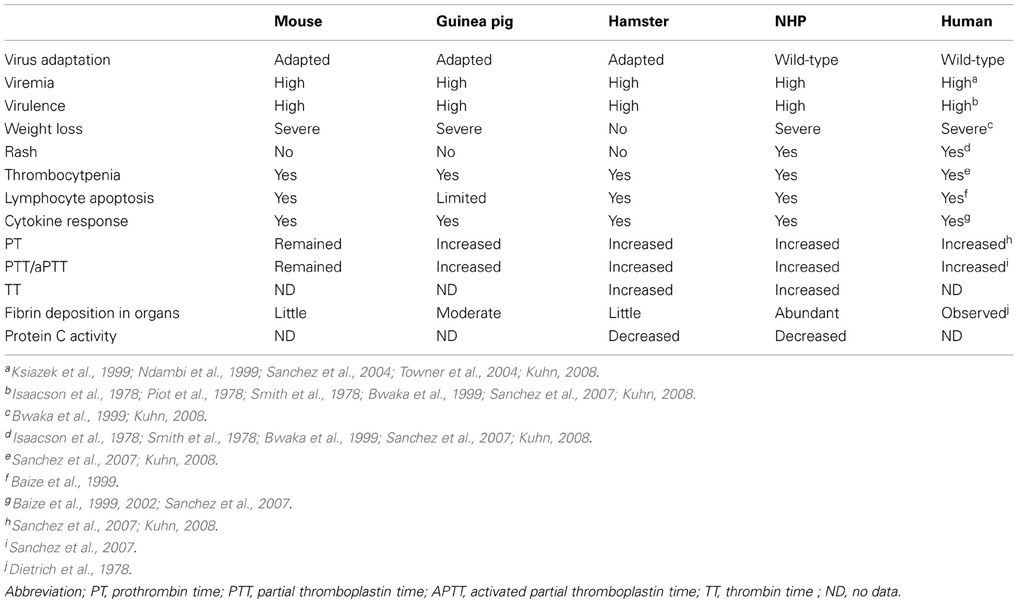
Table 1. Comparison of pathological features of different animal models of filovirus infection.
Moreover, in the guinea pig model, the lack of available reagents and tools, such as quantitative reverse-transcription polymerase chain reaction (qRT-PCR) and ELISA for cytokine profiling, makes the guinea pig model less desirable. Therefore, the development of other rodent models that better recapitulate EHF and MHF in humans was desired for more relevant pathogenesis studies and high throughput screening of prophylactic and post-exposure treatment prior to their testing in NHPs.
Syrian golden hamster (Mesocricetus auratus) is broadly used in animal models for human infectious diseases (Zivcec et al., 2011). Suckling hamsters were susceptible to wild-type MARV but disease was evident in only 40–80% of the animals following either intracerebral or intraperitoneal inoculation (Zlotnik, 1971). The symptoms and the mortality which was up to 90% were observed by inoculation with ninth passage materials in 5–6-week-old hamsters. While the pathological changes in the hamsters are similar to those observed in other animal models including patients, encephalitis, which is not observed in other animals, were constantly demonstrated in all suckling hamsters, irrespective of route of inoculation, and in adult hamsters, when the virus was inoculated intracerebrally.
The Syrian golden hamster model was developed for EHF based on infection with mouse-adapted EBOV. Six-week-old hamsters infected intraperitoneally with 103 focus forming unit (ffu) of mouse-adapted EBOV started to show clinical signs of disease, including ruffled fur and decreased activity, by Day 3 after infection and succumbed to disease within 4–5 days after infection. When inoculated subcutaneously, mouse-adapted virus failed to produce lethal infection in hamsters in the same way as the mouse model. Mouse-adapted EBOV-infected hamsters showed severe coagulopathy with prolonged PT, aPTT, and thrombin time (TT), in the late stage of infection (Ebihara et al., 2013). Other factors, including increased fibrinogen, decreased protein C activity, thrombocytopenia, and coagulation disorder were observed in the hamster model. The target organs were the mesenteric lymph node, spleen, liver, and adrenal gland. In the mesenteric lymph node, the target cells were the macrophages and dendritic cells (DCs). The viral antigens were found in macrophages, marginal reticular-like cells, and DC-like cells in the spleen, and Kupffer cells and hepatocytes in the liver. Histopathological changes, including inflammatory cell infiltration, cellular necrosis, and apoptosis, were mainly noted in the lymphoid organs (spleen and mesenteric lymph node) and liver. These pathological changes were similar to those demonstrated in NHPs and other rodent models (Baskerville et al., 1978; Fisher-Hoch et al., 1992; Jahrling et al., 1996; Ryabchikova et al., 1996, 1999; Bray et al., 1999; Connolly et al., 1999; Warfield et al., 2009). Fibrin deposits in liver sinuses, which are a hallmark of EBOV infection in humans and NHPs, were detected to a lesser extent in the hamster model (Ebihara et al., 2013). Suppression of type I IFN that enhances viral replication in target cells and contributes to lethal disease was observed (Ebihara et al., 2013).
It has been demonstrated that the mouse-adapted EBOV-based Syrian golden hamster model shows the most similar clinical and pathological features, including coagulation abnormalities, to those observed in humans and NHPs infected with wild-type EBOV.
Non-Human Primate Models
Although guinea pigs, mice, and hamsters models have been developed to study EHF and MHF as stated above, the rodent models are not ideal because mice and guinea pigs, except a novel hamster model, do not entirely exhibit coagulation disorders that are associated with human and NHPs filovirus infections (Table 1). Additionally the bystander death of large numbers of uninfected lymphocytes due to apoptosis that are hallmark features in filovirus-infected humans and NHPs is not present in infected guinea pigs. In mouse models, the bystander lymphocyte apoptosis was reported, but the process and morphology of lymphocyte apoptosis was different from those of filovirus-infecting humans and NHPs (Bradfute et al., 2007).
In NHP models, apoptosis was the primary reason for lymphocyte death, but the lymphocyte death in mouse models appeared to occur by apoptosis and apoptosis-like programmed cell death. Furthermore, NHPs are lethally infected with non-adapted filovirus isolates resulting in pathophysiology similar to that demonstrated in humans, although rodent models required serial passages of the virus for adaptation to produce lethal disease. Because of the aforementioned disadvantages and differences in the disease pathology, NHPs remain the most useful and reasonable model of EHF and MHF despite practical and ethical considerations leading to the restriction of experiments.
Marburgvirus Infection in NHPs
The first documented outbreak of MHF was associated with wild-caught African green monkeys (Chlorocebus aethiops) in Uganda and imported to Marburg and Frankfurt, Germany, and to Belgrade, Serbia Montenegro, former Yugoslavia, in 1967 (Martini, 1971). Since the first outbreak of MHF originated from the wild-caught African green monkeys, this species was an obvious choice for an animal model of MHF. At that time, rhesus macaques (Macaca mulatta) were found to be equally susceptible to infection and showed symptoms after inoculation with MARV (Hass and Maass, 1971). Cynomolgus macaques (Macaca fascicularis) were also well characterized as an MHF model (Hensley et al., 2011). After an incubation period of 2–6 days, the monkeys showed febrile illness, anorexia, diarrhea, skin rash, and hemorrhagic manifestations by any routes of MARV-inoculation (Simpson et al., 1968; Simpson, 1969; Murphy et al., 1971; Geisbert et al., 2007; Alves et al., 2010; Hensley et al., 2011). Death occurred by 6–13 days post-infection after a sudden decrease in body temperature and the mortality rate was almost 100%. It was shown that reducing the virus inoculum led to delayed onset of the disease and longer time to death without reduction in mortality rate (Hass and Maass, 1971). In the macaques, petechial rashes on the forehead, chest, axillae, and groins were prominent and resembled the rashes that appeared in patients with MHF, but intriguingly the rashes were not seen in African green monkeys (Simpson, 1969).
A marked lymphocytosis was observed at the beginning of the illness (Simpson et al., 1968; Simpson, 1969; Gonchar et al., 1991; Spiridonov et al., 1992; Johnson et al., 1996; Geisbert et al., 2007). Thrombocytopenia and leukocytosis due to increased neutrophilia were prominent on 5–6 days after infection (Hensley et al., 2011). Changes in coagulation systems, such as a decrease in circulating levels of protein C, an increase in levels of circulating D-dimer and fibrin deposition in tissues were noted at late stages of the disease (Geisbert et al., 2007; Hensley et al., 2011). The pathological changes in liver including multifocal necrosis of the parenchyma cells, and lymphocyte apoptosis in lymphoid tissues were prominent (Geisbert et al., 2000). Monocyte/macrophages and DCs in the lymphoid tissues as well as Kupffer cells and sinusoids lining cells in the liver were the primary target cells for infections with MARV (Hensley et al., 2011). The infection then progressed to parenchymal cells in the liver, adrenal gland, and high endothelial venules in lymphoid tissues. Finally, the infection spread to endothelial cells in a variety of organ tissues (Hensley et al., 2011). The virus or viral antigen was detected in liver, lymph nodes, spleen, adrenal grand, kidney, and blood in infected cynomolgus macaques. Onset of viremia occurred on Day 3, and in cynomolgus macaques and African green monkeys the maximum titer was 107−8 pfu/ml on Day 8 after infection (Hass and Maass, 1971; Hensley et al., 2011).
Under experimental conditions, the possibility of aerosol transmission of MARV was shown in macaque models, although such a transmission route has not been described in human outbreaks (Pokhodiaev et al., 1991; Alves et al., 2010).
Ebolavirus Infection in NHPs
African green monkeys, cynomolgus macaques, rhesus macaques, and hamadryas baboons (Papio hamadryas) have been employed as a model of EBOV infection (Baskerville et al., 1978, 1985; Bowen et al., 1978, 1980; Ellis et al., 1978; Fisher-Hoch et al., 1985, 1992; Johnson et al., 1995; Jaax et al., 1996; Jahrling et al., 1996; Davis et al., 1997; Ryabchikova et al., 1999; Ignatiev et al., 2000; Geisbert et al., 2003b,e). The monkeys infected with EBOV became febrile 3 days after infection with temperatures above 40°C. Pyrexia usually persisted throughout the course of the disease, which usually ended in a decrease in temperature followed by death, which occurred within 5–8 days after infection (Baskerville et al., 1978; Bowen et al., 1978, 1980; Ellis et al., 1978; Fisher-Hoch et al., 1985; Luchko et al., 1995). By Day 4, anorexia developed with a loss of drinking ability, causing severe weight loss and dehydration. Some monkeys that survived until Day 5 had diarrhea, rectal bleeding, and/or intermittent melena. Petechial skin rashes appeared on the forehead, fore and hind limbs, and chest 4–5 days post-infection in macaques, but on Day 7 in baboons (Bowen et al., 1978; Ellis et al., 1978; Luchko et al., 1995; Ignatiev et al., 2000; Geisbert et al., 2003b). African green monkeys did not develop the cutaneous rash as demonstrated in monkeys infected with MARV (Simpson, 1969; Baskerville et al., 1978). Viremia became detectable within 3 days after infection with the maximum virus titer at the level of 106.5−7 pfu/ml on Day 4–5 (Bowen et al., 1978; Fisher-Hoch et al., 1992; Jahrling et al., 1996; Geisbert et al., 2003b). The virus was positive in liver, spleen and lung on Day 4 and also appeared to have lower affinity for kidney, adrenal, lung, testis, lymph node, and pancreas (Baskerville et al., 1978, 1985; Bowen et al., 1978; Geisbert et al., 2003b). Mean virus titers in these organs increased progressively and reached the highest level of 105.5−8.6 pfu/g on Day 6 (Geisbert et al., 2003b).
Total blood cell counts revealed marked neutrophilia and lymphopenia in the monkeys. Neutrophils and immature neutrophils increased remarkably by Day 4 (Fisher-Hoch et al., 1985; Geisbert et al., 2003b; Ebihara et al., 2011). Coincident with this process, severe lymphopenia due to lymphocyte apoptosis developed by Day 3 (Fisher-Hoch et al., 1985; Geisbert et al., 2003b). Extensive lymphocyte apoptosis, both in the vasculature and in lymphoid tissue, appears to be critical to the pathogenesis of EHF. Especially within the CD8+ subset, the NK cell population dropped dramatically in the early stage of infection (Geisbert et al., 2003b). Lymphocytes were not productively infected and the apoptosis was not associated with direct viral infection (Geisbert et al., 2000). However, the mechanism underlying such apoptosis is unclear. Another characteristic feature was the abnormality of platelet function preceding thrombocytopenia (Fisher-Hoch et al., 1985; Geisbert et al., 2003b). Thrombocytopenia developed between 3 and 4 days and abnormalities in coagulation parameters, including prolonged PT, aPTT, and TT appeared (Geisbert et al., 2003b; Ebihara et al., 2011). Examination of coagulation parameters revealed that decreased protein C coagulation inhibitor activity due to excessive consumption triggered severe coagulopathy as indicated by prolonged coagulation times and decreased fibrinogen levels (Ebihara et al., 2011).
The NHP model has been proven to be valuable in providing new information regarding filoviral pathogenesis. EBOV spreads from the initial infection site via monocytes/macrophages and DCs to regional lymph nodes, likely via lymphatics, and to liver and spleen through the blood stream. Tissue macrophages, including Kupffer cells, DCs, and fibroblastic reticular cells become infected with EBOV at this stage. EBOV activates DCs by upregulating expression of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), which is expressed on DCs and mediates their cytotoxic activity (Geisbert et al., 2003b). Such overexpression of TRAIL is enhanced by overexpression of IFN-α in NHPs infected with EBOV and triggers lymphocyte apoptosis. Monocytes/macrophages infected with EBOV release various soluble factors including proinflammatory cytokines to recruit additional target cells to areas of infection. As disease progresses, increased levels of oxygen free radicals (e.g., nitric oxide) released from virus-infecting macrophages at the inflammatory sites trigger apoptosis of NK cells. The lymphocyte apoptosis caused by TRAIL and nitric oxide interferes with the innate immune response, resulting in escape of EBOV infection from mounting an adaptive response. Coagulation abnormalities are not the direct result of EBOV replication-induced cytolysis of endothelial cells, but are likely triggered by immune-mediated mechanisms (Geisbert et al., 2003d,e). Extensive viral replication leads to increased levels of additional proinflammatory cytokines, notably IL-6, which triggers the coagulation irregularities. This is probably through upregulation of tissue factor expression/release from virus-infected monocytes/macrophages. Tissue factor works as the primary cellular inhibitor of coagulation protease cascades. Activation of the coagulation cascade induces the fibrinogenic and fibrinolytic pathways and finally leads to DIC, hemorrhagic shock, thrombosis-related organ failure and death (Arai et al., 2000).
Monkey species-specific disease features of the pathogenesis of EBOV infection were observed, not only in the development of cutaneous rash but also in the impairment of the clotting systems of African green monkeys and baboons infected with EBOV. In African green monkeys, fibrin thrombosis was generalized in all visceral organs, while in baboons hemorrhages were prominent in visceral organs, most notably in the liver and spleen (Ryabchikova et al., 1999; Ignatiev et al., 2000). Genetic differences, even among the same animal species, and the origin of a species may influence disease presentation and progression.
The dose and species of challenge virus affects the progression of disease. Intramuscular inoculation of cynomolgus macaques with 103 pfu of EBOV produced a 100% lethal infection, with deaths occurring 6–7 days post-infection (Geisbert et al., 2002). When the challenge dose was lowered to 10 pfu, uniform lethality was still achieved, but deaths occurred 9–12 days post-infection (Geisbert et al., 2003c). Viremia was demonstrated as early as 24 h after subcutaneous infection of rhesus macaques with a high infectious dose (105 pfu) of EBOV. In rhesus and cynomolgus macaques infected with 103 pfu of EBOV, viremia is first detected by Day 3 after infection. SUDV causes slower disease progression than EBOV and it has been reported that some monkeys infected with SUDV did not die and recovered from the illness (Ellis et al., 1978; Bowen et al., 1980; Fisher-Hoch et al., 1992). One of four rhesus macaques infected with SUDV died on Day 12 but the other macaques survived and remained normal thereafter (Ellis et al., 1978). The dead macaque had small numbers of virus particles in the liver but no virus particles were found in the kidney, spleen, heart, lung, and brain. The liver, lung, and spleen from EBOV infected macaques, which were moribund and killed on Day 6 contained large numbers of virions. The apparent limitation of viral replication in the liver of SUDV-infected host and the contrasting widespread involvement of liver and other organs such as the spleen and lung of EBOV-infected host are similar in patients and macaques. RESTV, which is considered not to be virulent to humans, is clearly less pathogenic than EBOV and SUDV in African green monkeys and cynomolgus macaques (Fisher-Hoch et al., 1992; Jahrling et al., 1996). Only 5 of 16 monkeys infected with EBOV or SUDV survived, whereas 11 of 15 monkeys infected with RESTV survived (Fisher-Hoch et al., 1992). Viremia, clinical signs (temperature rise, anorexia, depression, or evidence of disturbed hemostasis), serum chemistry changes (elevated aspartate aminotransferase and lactate dehydrogenase activities) and pathological changes (necrosis of hepatocytes, adrenalcytes, and lymphoid elements of the spleen and prominent fibrin thrombi and fibrin precipitation) in RESTV infected monkeys developed slower and/or milder than those observed in monkeys infected with EBOV and/or SUDV.
Most human cases are thought to occur by direct contact with blood and/or body secretions from patients or animal cadavers. Aerosol transmission among humans has not been reported. However, evidence of intercage transmission of RESTV was observed in the 1989–1990 epizootic cases of RESTV in the Hazleton facility in Reston, Virginia, and demonstration of high concentrations of ebolavirus in nasal secretions and alveoli in experimental infection implicated the potency of aerosol transmission of ebolavirus (Baskerville et al., 1978, 1985; Bowen et al., 1978; Jahrling et al., 1990, 1996; Dalgard et al., 1992; Jaax et al., 1995; Miranda et al., 1999, 2002). Furthermore, the rhesus macaques experimentally challenged with aerosolized EBOV developed the same disease as macaques infected parenterally (Johnson et al., 1995). Regardless of the route of infection (intramuscular, subcutaneous, conjunctival, and aerosol injections), NHPs are highly susceptible to EBOV infection (Baskerville et al., 1978; Bowen et al., 1978, 1980; Ellis et al., 1978; Baskerville et al., 1985; Fisher-Hoch et al., 1985, 1992; Johnson et al., 1995; Jaax et al., 1996; Jahrling et al., 1996; Davis et al., 1997; Ryabchikova et al., 1999; Ignatiev et al., 2000; Geisbert et al., 2003b,e).
Vaccines
Inactivated Whole Virion
The development of filovirus vaccines has been performed based on inactivated whole virion preparations. About half of the rhesus macaques or African green monkeys treated were protected against homologous MARV challenge, when formalin- or gamma-inactivated whole MARV virions were used as vaccine candidates (Ignat'ev et al., 1991; Ignatyev et al., 1996). Vaccination with formalin-inactivated EBOV virions protected 4 of 5 hamadryas baboons (Mikhailov et al., 1994), while other studies suggested that inactivated virus did not induce sufficient immunity to protect baboons against a lethal challenge (Chupurnov et al., 1995). Furthermore, vaccination with gamma-irradiated EBOV virions alone or in a form of liposomes containing lipid A failed to protect cynomolgus macaques against lethal infection (Geisbert et al., 2002). Overall, these vaccine candidates based on inactivated virions did not confer sufficient protection in NHP models. Furthermore, these vaccines are unlikely to be used in humans due to safety risk of incomplete inactivation. However, these results promoted the development of an alternative vaccine platform, such as DNA-based vaccines, recombinant viral vector, or virus-like particles as described below and Table 2.
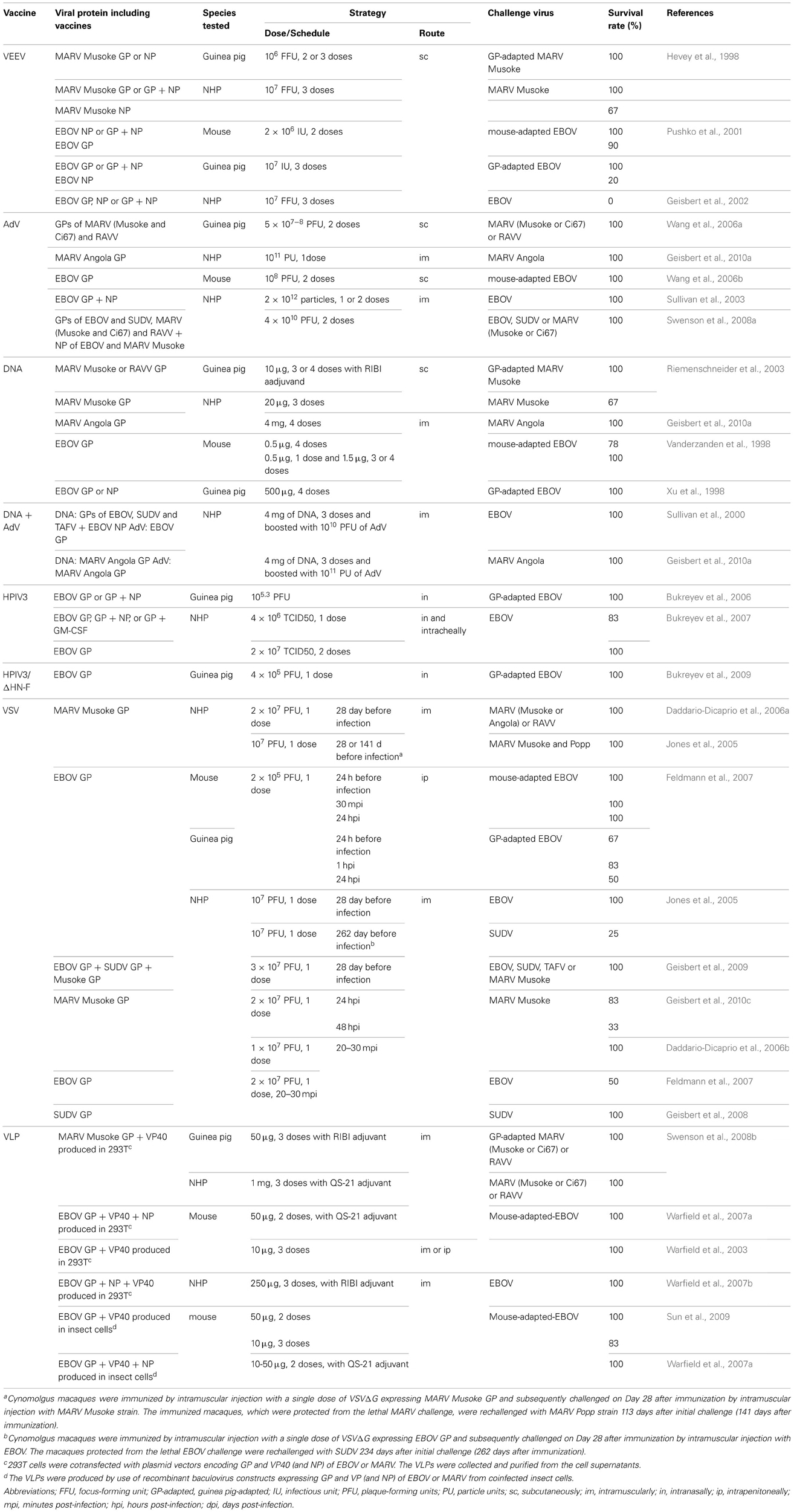
Table 2. Efficacy of vaccines in animal models of filovirus infection.
Venezuelan Equine Encephalitis Virus Replicon
Venezuelan equine encephalitis virus (VEEV) replicons that express either GP or NP of MARV Musoke protected guinea pigs from viremia and death caused by GP-adapted MARV challenge (Hevey et al., 1998). Cynomolgus macaques administrated with MARV Musoke GP-expressing VEEV replicons alone or in combination with NP were also protected from lethal infection with the homologous Musoke strain, but not from heterologous RAVV (Hevey et al., 1998; Falzarano et al., 2011). additionally, vaccination with NP alone prevented death but not disease onset in two of three monkeys and allowed all animals to become viremic. For EBOV, EBOV GP-expressing VEEV replicons, alone or in combination with EBOV NP-expressing VEEV replicons, protected mice, and guinea pigs from lethal infection, whereas immunization with EBOV NP-expressing VEEV replicons alone protected mice but not guinea pigs (Pushko et al., 2001). Furthermore, vaccination with recombinant VEEV, expressing EBOV GP, NP, or both GP and NP, failed to protect cynomolgus macaques from a lethal EBOV infection (Geisbert et al., 2002). One recent study produced different results, whereby a VEEV-based vaccine was fully protective in cynomolgus macaques against EBOV, SUDV, and MARV (Friedrich et al., 2012). The results obtained from these studies are inconsistent, suggesting that VEEV-based vaccine may be promising although further research is needed.
Adenovirus-Based Vaccines
Adenovirus (AdV) vectors commonly used are based on serotype 5 (AdV5). A single infection of the recombinant MARV Angola GP-expressing AdV5 resulted in complete protection of cynomolgus macaques from illness and death by challenge with homologous virus (Geisbert et al., 2010a). Vaccination with a mixture of EBOV GP—expressing AdV5 and EBOV NP—expressing AdV5 have demonstrated 100% protection in cynomolgus macaques against homologous virus challenge (Sullivan et al., 2003). However, the genome insert size in this first generation AdV vector was restricted to as little as a single filovirus GP gene. The second generation AdV vector has the advantage of being able to express multiple antigens in a single construct over the first generation vector. The second generation bivalent AdV vector expressing GPs of EBOV and SUDV led to efficient induction of antibodies specific to EBOV and SUDV (Wang et al., 2006b). A trivalent AdV vector expressing MARV GPs of Ci67, Musoke and RAVV efficiently led to MARV-specific antibodies in mice and guinea pigs and showed complete protection of guinea pigs against MARV and RAVV infections (Wang et al., 2006a). Additionally, vaccination of cynomolgus macaques with second generation AdV vectors, which expressed multiple filovirus GPs of EBOV, SUDV, Ci67, Musoke, and RAVV, induced 100% protection against challenge with EBOV and SUDV and two different strains of MARV (Ci67 and Musoke; Swenson et al., 2008a). Although the AdV-based vaccines showed efficacy, the vaccines have a major obstacle: the prevalence of pre-existing immunity to AdV that may substantially limit their immunogenicity and clinical utility. It is estimated that the prevalence of antibody to AdV5 is up to 60% in the general human population and up to 85% in Africa (Schulick et al., 1997; Piedra et al., 1998). Indeed, macaques pre-immunized against AdV5 and vaccinated with EBOV GP-expressing AdV5 were not protected from lethal challenge with EBOV infection (Geisbert et al., 2010b). AdV serotype 26 and 35 segregated genetically from AdV5 exhibit lower seroprevalence in humans (Vogels et al., 2003; Abbink et al., 2007; Mast et al., 2010). Therefore, AdV serotype 26 and 35 vectors with expression of EBOV or SUDV GPs have been generated and the protective efficacy examined by using the NHP model, but these vectors failed to protect cynomolgus macaques against lethal EBOV challenge (Geisbert et al., 2011).
DNA
The plasmid coding the DNA of GP from MARV Musoke or RAVV demonstrated efficacy in protection of guinea pigs and cynomolgus macaques against lethal infection of each homologous strain (Riemenschneider et al., 2003). All of the guinea pigs vaccinated three or four times with DNA vaccines were aviremic and appeared healthy. In cynomolgus macaques, four of six monkeys immunized with 3 doses of DNA vaccine encoding Musoke GP were protected from homologous challenge with MARV Musoke (Riemenschneider et al., 2003). In a report of DNA vaccines encoding GP of MARV Angola strain, the 4 vaccination doses resulted in protection of all four vaccinated monkeys, but three of the four monkeys showed symptoms and/or lymphopenia (Geisbert et al., 2010a). A combination vaccine regimen (3 times injection with DNA and boost with recombinant Angola GP-expressing AdV vector) protected the monkeys from lethal infections but two of the four monkeys showed rash or lymphopenia. A single inoculation with AdV vaccine induced optimal immune responses to eliminate symptoms and death by itself. These data suggest that DNA vaccines do not optimally control MARV infection (Geisbert et al., 2010a). However, three-plasmid DNA vaccines encoding EBOV GP, SUDV GP, and EBOV NP were evaluated in a phase I trial as safe and immunogenic in humans (Martin et al., 2006). The EBOV DNA vaccine also protected mice and guinea pigs against a lethal challenge (Vanderzanden et al., 1998; Xu et al., 1998; Martin et al., 2006). In one study, cynomolgus macaques, which received 3 injections of DNA vectors encoding EBOV GP, SUDV GP, TAFV GP, and EBOV NP, were boosted with recombinant EBOV GP-expressing AdV (Sullivan et al., 2000). All four monkeys vaccinated survived and showed no symptoms of EBOV infection. This prime-boost strategy provided a sufficient immune response to clear the virus efficiently.
Human Parainfluenza Virus
In an outbreak of RESTV in the Hazleton facility in Reston, Virginia, aerosol transmission between NHPs may have occurred (Jahrling et al., 1990; Dalgard et al., 1992; Miranda et al., 1999, 2002). To address the assumed aerosol transmission of filovirus, a vaccine that induces a strong immune response in the respiratory tract was developed. Human parainfluenza virus type 3 (HPIV3), a common respiratory virus, was modified as a form of vaccine vector and used for development of a vaccine against EBOV. The HPIV3 vectors, which express EBOV GP or EBOV GP together with NP, protected guinea pigs and rhesus macaques against EBOV challenge (Bukreyev et al., 2006, 2007). In guinea pigs, a single intranasal inoculation with HPIV3 expressing EBOV GP or both GP and NP showed complete protection against signs of illness and death (Bukreyev et al., 2006). The rhesus macaques were immunized with a single dose of EBOV GP-expressing HPIV3, or EBOV GP and NP-expressing HPIV3, through a combined intranasal and intratracheal inoculation. Five of six monkeys immunized with the HPIV3 based vaccine survived and four of six monkeys did not show any clinical illness (Bukreyev et al., 2007). Two doses of intranasal immunizations showed greater efficacy, including complete protection of all three rhesus macaques against clinical illness and death. However, HPIV3 may not be effective as a vaccine vectors in humans, since HPIV3 is a common childhood pathogen and the majority of the population have pre-existing immunity to HPIV3. To overcome the problem of pre-existing immunity, a chimeric HPIV3, where both HPIV3 surface proteins, HN and F, were deleted and replaced with EBOV GP was developed (Bukreyev et al., 2009). A single immunization with the vaccine completely protected guinea pigs against a lethal infection. It was shown that the HPIV3 based vaccine, which expressed EBOV GP, was immunogenic equally among HPIV3-naïve and HPIV3 antibody-positive subjects and effective when vaccinated twice. However, pre-existing HPIV3-specific immunity in rhesus macaques reduced the replicative capacity of the HPIV3-based vaccine in the respiratory tract (Bukreyev et al., 2010). Nevertheless, this study indicated that the vaccination induced an appropriate antibody response.
Vesicular Stomatitis Virus
A vaccine to resolve the problem of pre-existing immunity utilized the recombinant vesicular stomatitis virus (VSV) vector, which expresses filovirus GP. VSV is mainly a veterinary pathogen and human infection with VSV is rare and not associated with disease in humans. A single intramuscular vaccination of cynomolgus macaques with recombinant VSV with expression of MARV Musoke GP elicited complete protection against a high dose (103 pfu) intramuscular challenge with both homologous Musoke strain and heterologous Popp strain, Angola strain, and RAVV (Jones et al., 2005; Daddario-Dicaprio et al., 2006a). For EBOV, a single immunization of cynomolgus macaques with recombinant VSV vector, which expresses EBOV GP, also elicited complete protection against EBOV challenge (Jones et al., 2005). The surviving macaques from lethal EBOV infection were re-challenged with heterologous SUDV, but the cross-protection was not observed (Jones et al., 2005). Administration of the EBOV GP-expressing VSV vaccine through the oral or intranasal route completely protected cynomolgus macaques from EBOV challenge (Qiu et al., 2009). A blended vaccine consisting of equal amounts of 3 different VSV vectors, which expresses GP of each of EBOV, SUDV, and MARV, generated complete protection of cynomolgus macaques against challenges with EBOV, TAFV, and MARV (Geisbert et al., 2009). Macaques vaccinated with the blended vaccine followed by challenge with SUDV showed mild clinical sign of illness including fever, lymphopenia, and mild anorexia, and the macaques recovered from illness. Importantly, none of the macaques vaccinated with the blended vaccine succumbed to a filovirus challenge. The efficacy of the recombinant VSV vaccine has been evaluated as a post-exposure prophylaxis for filovirus infections. Administration of recombinant VSV with MARV Musoke GP expression to rhesus macaques shortly after a homologous high-dose MARV challenge resulted in complete protection of all subjects from clinical illness and death (Daddario-Dicaprio et al., 2006b). Furthermore, administration of recombinant MARV Musoke GP-expressing VSV at 24 and 48 h following infection resulted in protection of 83 and 33% of rhesus macaques, respectively (Geisbert et al., 2010c). When recombinant EBOV GP-expressing VSV were administrated to mice 24 h prior to challenge, and 1 and 24 h post-challenge, all treated mice survived (Feldmann et al., 2007). In guinea pigs treated with EBOV GP-expressing VSV at 24 h prior to challenge, and 1 or 24 h post-challenge, the survival rates were 67, 83, and 50%, respectively. It was also demonstrated that post-exposure vaccination with the recombinant VSV GP vectors for EBOV and SUDV in rhesus macaques was effective against challenge with homologous viruses, although the protection rate was dependent on the species of ebolavirus. The survival rates of the EBOV- or SUDV-infected monkeys were 50 and 100%, respectively (Feldmann et al., 2007; Geisbert et al., 2008).
Virus-Like Particle
Virus-like particle (VLPs), which mimic authentic virions structurally but do not contain infectious genetic material, are non-infectious and safer than replicating vaccines. The efficiency of a MARV vaccine consisting of VLPs with MARV Musoke GP and VP40 was assessed in guinea pig and cynomolgus macaque models (Swenson et al., 2008b). The guinea pigs and monkeys immunized three times with MARV-Musoke VLPs with RIBI or QS-21 adjuvant were challenged with Musoke strain, Ci67 strain, or RAVV. All guinea pigs and eight monkeys were protected from death and clinical illness following the lethal challenge, except for a single monkey. The monkey challenged with RAVV, which is the most genetically distinct strain of marburgvirus, developed minor signs of disease without detectable viremia. For ebolavirus, mice vaccinated with EBOV VLP in the presence or absence of adjuvant were protected from lethal EBOV infection in a dose-dependent manner (Warfield et al., 2003, 2007a; Sun et al., 2009). Furthermore, the efficacy of the EBOV VLP, which consists of EBOV GP, NP, and VP40 was evaluated in cynomolgus macaques (Warfield et al., 2007b). All five monkeys that received three injections of the EBOV VLPs with RIBI adjuvant were completely protected against EBOV challenge.
There are some other vaccine candidates, including an EBOV lacking VP30 (which encodes the essential transcription factor), an Fc portion of a human IgG fused to EBOV-GP, a bean yellow dwarf virus-derived replicon system, and a cytomegalovirus-based vaccine encoding an EBOV NP CTL epitope (Halfmann et al., 2009; Konduru et al., 2011; Phoolcharoen et al., 2011; Tsuda et al., 2011a). However, the immunogenic efficacy of these vaccines has only been confirmed in the rodent models and further studies are needed to evaluate the protective efficacy and safety in NHPs.
Treatments
Recombinant Nematode Anticoagulant Protein C2
Coagulation abnormalities are one of the most prominent hallmarks of filovirus infection. It has been suggested that tissue factor plays an important role in triggering the hemorrhagic complications in NHPs infected with filoviruses (Geisbert et al., 2003d). Overexpression of tissue factor that performs as the primary cellular inhibitor of the coagulation protease cascades is one of the causes of DIC and thrombosis-related organ failure. The effect of blocking the pathway leading from the complex of activated factor VII and tissue factor to thrombin was examined in filovirus infection. Recombinant nematode anticoagulant protein c2 (rNAPc2), which directly inhibits factor VII and tissue factor, provided partial post-exposure protection to rhesus macaques infected with filovirus (Geisbert et al., 2003a, 2009). In rNAPc2-treated rhesus macaques, the mean survival time (11.7 days) was longer than that in untreated control monkeys (8.3 days) and 33% of EBOV-infected macaques survived. In MARV Angola-infected rhesus macaques treated with rNAPc2, 1 of 6 (17%) monkeys survived and the mean survival time for the five dead monkeys was significantly prolonged compared with that of the untreated control monkeys. rNAPc2 demonstrated a clear improvement in terms of survival rate and an increase in mean survival time in a normally 100% lethal model of filovirus infection.
Recombinant Human Activated Protein C
Activated protein C (APC) is generated from the protein C, which is a vitamin K-dependent plasma protein and inactivates factors V and VIII to down-regulate thrombin generation. It has been reported that circulating levels of protein C were rapidly and significantly reduced in cynomolgus macaques and rhesus macaques during EBOV infections, because the protein C might be produced in the liver, which is a main target of filovirus infection (Geisbert et al., 2003a). In rhesus macaque models, administration of recombinant human APC (rhAPC) at 30–60 min after challenge and continuing for 7 days, protected 2 of 11 (18%) monkeys against lethal EBOV infection (Hensley et al., 2007). The mean survival time in the rhAPC-treated monkeys was prolonged compared with the untreated monkeys (Hensley et al., 2007).
Phosphorodiamidate Morpholino Oligomer
Phosphorodiamidate morpholino oligomers (PMOs) inhibit targeted gene translation by steric blockage of ribosomal assembly. A combination of EBOV-specific PMOs targeting sequences of viral mRNAs for the VP24, VP35, and RNA polymerase L protected rodents in both pre- and post-exposure therapeutic regimens (Warfield et al., 2006). In rhesus macaque models, treatment with a combination of the PMOs of VP24, VP35, and L from 2 days prior to EBOV challenge through Day 9 of the infection protected 3 of 4 (75%) rhesus macaques against lethal infection (Warfield et al., 2006). Furthermore, it was demonstrated that the antiviral potency of PMOs could be enhanced by chemical modification, either by conjugating PMOs with peptides or by introducing positively charge to the PMOs (PMOplus™, Avi BioPharma, Inc.; Swenson et al., 2009). Subsequently, PMOplus targeting EBOV VP24 and VP35 or MARV Musoke VP24 and NP showed significant protection of mice and guinea pigs against lethal challenge with EBOV and MARV Musoke, respectively (Warren et al., 2010). AVI-6002 PMOplus against both EBOV VP24 and VP35, and AVI-6003 PMOplus against MARV VP24 and NP, were developed and tested for treatment efficacy using NHP models. These PMOs, delivered 30–60 min post-exposure, protected 62.5% of rhesus macaques against lethal EBOV infection and 100% of cynomolgus macaques against MARV Musoke infection (Warren et al., 2010). AVI-6002 and AVI-6003 are currently in phase I clinical trials.
RNA Interference
RNA interference (RNAi) inhibits gene expression to the extent that their function is abrogated through a highly regulated enzyme-mediated process. It was demonstrated that small-interfering RNA (siRNA) down-regulated various MARV mRNA transcripts, resulting in a significant decrease in viral protein production and subsequent viral release in vitro (Fowler et al., 2005). Furthermore, siRNA targeting the EBOV RNA polymerase L protein formulated in stable nucleic acid-lipid particles (SNALPs) completely protected guinea pigs when administered shortly after a lethal EBOV infection (Geisbert et al., 2006). In rhesus macaques, a combination of siRNA targeting the EBOV L, VP24, and VP35 were formulated in SNALPs and administrated to the monkeys. Two of three monkeys, which were treated four times with siRNA at 30 min, 1, 3, and 5 days after challenge, survived lethal infection. Furthermore, all four monkeys treated seven times at 30 min, 1–6 days after challenge survived (Geisbert et al., 2010d).
Therapeutic Efficacy in the Mouse Model and in vitro
In the mouse model, administration of recombinant mannose-binding lectin and hexaamminecobalt (III) chloride showed efficacy in protecting against EBOV infections (Michelow et al., 2011). Mannose-binding lectin targets diverse microorganisms for phagocytosis and complement-mediated lysis by binding specific surface glycans. Hexaamminecobalt (III) chloride is a complex of a cobalt (III) ion surrounded by six ammonia ligands in a full octahedral coordination. Furthermore, by high-throughput screening, some compounds such as FGI-103, FGI-106, and NSC 62914 (a reactive oxygen species scavenger), were identified to have high antiviral activity against filoviruses (Aman et al., 2009; Warren et al., 2010). Some other substances, for example inhibitors of heat-shock protein 90 and Niemann-Pick C1, showed antiviral activity in vitro (Smith et al., 2010; Cote et al., 2011). As mentioned above (Table 3), several candidates are discussed as therapeutic agents for Ebola and Marburg HFs, but no licensed therapeutics are yet available (Friedrich et al., 2012).
Table 3. Efficacy of post-exposure treatment in animal models of filovirus infection.
Conclusions
Significant progress has been made in developing animal models, including mice, guinea pigs, hamsters and NHPs, for EHF and MHF. The NHPs are the most feasible model, because they are the only animals that are lethally infected with non-adapted virus isolates and the pathophysiology is close to that demonstrated in patients. The rodent models need serial passages of original filoviruses in rodents for acquiring lethal infection capacity and they have limited value, because the disease course in rodents differs from that demonstrated in humans and NHPs. However, the rodent models are the first choice for preliminary studies to explore vaccines and therapeutic agents, because of their ease to handling. The newly developed Golden hamster model will also be used for studies on pathogenesis and evaluation of efficacy of candidate vaccines and therapeutics because they show manifestations similar to those of patients and NHPs, including severity of coagulopathy that is lacking in mouse and guinea pig models. Among the candidate vaccines so far developed, recombinant VSV-based vaccines against EHF and MHF are confirmed to be effective in mouse, guinea pig, Golden hamster, and NHP models, and are the only platform with the potential to prevent lethal infection, especially via both vaccine and post-exposure treatment (Jones et al., 2005; Daddario-Dicaprio et al., 2006b; Feldmann et al., 2007; Geisbert et al., 2008, 2009, 2010c; Qiu et al., 2009; Tsuda et al., 2011b). Furthermore, the VSV have been used as a treatment following a recent laboratory exposure (Tuffs, 2009). Further research is needed to develop vaccines with sufficient long-term efficacy by single-dose vaccination, because expensive and time-consuming vaccinations may pose difficulties due to logistical and financial problems in developing countries, where EHF and MHF are endemic. Neither licensed vaccines nor therapeutic agents are available so far. The development of vaccines and therapeutic testing using the animal models has only recently begun to progress. We hope that further research facilitates progress toward elucidating the disease pathophysiology and developing prophylactic and therapeutic measures against EHF and MHF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbink, P., Lemckert, A. A., Ewald, B. A., Lynch, D. M., Denholtz, M., Smits, S., et al. (2007). Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J. Virol. 81, 4654–4663. doi: 10.1128/JVI.02696-06
Alves, D. A., Glynn, A. R., Steele, K. E., Lackemeyer, M. G., Garza, N. L., Buck, J. G., et al. (2010). Aerosol exposure to the angola strain of marburg virus causes lethal viral hemorrhagic Fever in cynomolgus macaques. Vet. Pathol. 47, 831–851. doi: 10.1177/0300985810378597
Aman, M. J., Kinch, M. S., Warfield, K., Warren, T., Yunus, A., Enterlein, S., et al. (2009). Development of a broad-spectrum antiviral with activity against Ebola virus. Antiviral Res. 83, 245–251. doi: 10.1016/j.antiviral.2009.06.001
Arai, A., Hirano, H., Ueta, Y., Hamada, T., Mita, T., and Shirahata, A. (2000). Detection of mononuclear cells as the source of the increased tissue factor mRNA in the liver from lipopolysaccharide-treated rats. Thromb. Res. 97, 153–162. doi: 10.1016/S0049-3848(99)00147-4
Baize, S., Leroy, E. M., Georges, A. J., Georges-Courbot, M. C., Capron, M., Bedjabaga, I., et al. (2002). Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 128, 163–168. doi: 10.1046/j.1365-2249.2002.01800.x
Baize, S., Leroy, E. M., Georges-Courbot, M. C., Capron, M., Lansoud-Soukate, J., Debre, P., et al. (1999). Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med. 5, 423–426. doi: 10.1038/7422
Baskerville, A., Bowen, E. T., Platt, G. S., McArdell, L. B., and Simpson, D. I. (1978). The pathology of experimental Ebola virus infection in monkeys. J. Pathol. 125, 131–138. doi: 10.1002/path.1711250303
Baskerville, A., Fisher-Hoch, S. P., Neild, G. H., and Dowsett, A. B. (1985). Ultrastructural pathology of experimental Ebola haemorrhagic fever virus infection. J. Pathol. 147, 199–209. doi: 10.1002/path.1711470308
Basler, C. F., Wang, X., Muhlberger, E., Volchkov, V., Paragas, J., Klenk, H. D., et al. (2000). The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl. Acad. Sci. U.S.A. 97, 12289–12294. doi: 10.1073/pnas.220398297
Bente, D., Gren, J., Strong, J. E., and Feldmann, H. (2009). Disease modeling for Ebola and Marburg viruses. Dis. Model. Mech. 2, 12–17. doi: 10.1242/dmm.000471
Bowen, E. T., Lloyd, G., Harris, W. J., Platt, G. S., Baskerville, A., and Vella, E. E. (1977). Viral haemorrhagic fever in southern Sudan and northern Zaire. Preliminary studies on the aetiological agent. Lancet 1, 571–573. doi: 10.1016/S0140-6736(77)92001-3
Bowen, E. T., Platt, G. S., Lloyd, G., Raymond, R. T., and Simpson, D. I. (1980). A comparative study of strains of Ebola virus isolated from southern Sudan and northern Zaire in 1976. J. Med. Virol. 6, 129–138. doi: 10.1002/jmv.1890060205
Bowen, E. T., Platt, G. S., Simpson, D. I., McArdell, L. B., and Raymond, R. T. (1978). Ebola haemorrhagic fever: experimental infection of monkeys. Trans. R. Soc. Trop. Med. Hyg. 72, 188–191. doi: 10.1016/0035-9203(78)90058-5
Bradfute, S. B., Braun, D. R., Shamblin, J. D., Geisbert, J. B., Paragas, J., Garrison, A., et al. (2007). Lymphocyte death in a mouse model of Ebola virus infection. J. Infect. Dis. 196(Suppl. 2), S296–S304. doi: 10.1086/520602
Bradfute, S. B., Warfield, K. L., and Bray, M. (2012). Mouse models for filovirus infections. Viruses 4, 1477–1508. doi: 10.3390/v4091477
Bray, M. (2001). The role of the Type I interferon response in the resistance of mice to filovirus infection. J. Gen. Virol. 82, 1365–1373.
Bray, M., Davis, K., Geisbert, T., Schmaljohn, C., and Huggins, J. (1999). A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J. Infect. Dis. 179(Suppl. 1), S248–S258. doi: 10.1086/514292
Bray, M., Hatfill, S., Hensley, L., and Huggins, J. W. (2001). Haematological, biochemical and coagulation changes in mice, guinea-pigs and monkeys infected with a mouse-adapted variant of Ebola Zaire virus. J. Comp. Pathol. 125, 243–253. doi: 10.1053/jcpa.2001.0503
Bukreyev, A., Marzi, A., Feldmann, F., Zhang, L., Yang, L., Ward, J. M., et al. (2009). Chimeric human parainfluenza virus bearing the Ebola virus glycoprotein as the sole surface protein is immunogenic and highly protective against Ebola virus challenge. Virology 383, 348–361. doi: 10.1016/j.virol.2008.09.030
Bukreyev, A., Rollin, P. E., Tate, M. K., Yang, L., Zaki, S. R., Shieh, W. J., et al. (2007). Successful topical respiratory tract immunization of primates against Ebola virus. J. Virol. 81, 6379–6388. doi: 10.1128/JVI.00105-07
Bukreyev, A., Yang, L., Zaki, S. R., Shieh, W. J., Rollin, P. E., Murphy, B. R., et al. (2006). A single intranasal inoculation with a paramyxovirus-vectored vaccine protects guinea pigs against a lethal-dose Ebola virus challenge. J. Virol. 80, 2267–2279. doi: 10.1128/JVI.80.5.2267-2279.2006
Bukreyev, A. A., Dinapoli, J. M., Yang, L., Murphy, B. R., and Collins, P. L. (2010). Mucosal parainfluenza virus-vectored vaccine against Ebola virus replicates in the respiratory tract of vector-immune monkeys and is immunogenic. Virology 399, 290–298. doi: 10.1016/j.virol.2010.01.015
Bwaka, M. A., Bonnet, M. J., Calain, P., Colebunders, R., De Roo, A., Guimard, Y., et al. (1999). Ebola hemorrhagic fever in Kikwit, Democratic Republic of the Congo: clinical observations in 103 patients. J. Infect. Dis. 179(Suppl. 1), S1–S7. doi: 10.1086/514308
Chupurnov, A. A., Chernukhin, I. V., Ternovoi, V. A., Kudoiarova, N. M., Makhova, N. M., Azaev, M., et al. (1995). Attempts to develop a vaccine against Ebola fever. Vopr. Virusol. 40, 257–260.
Connolly, B. M., Steele, K. E., Davis, K. J., Geisbert, T. W., Kell, W. M., Jaax, N. K., et al. (1999). Pathogenesis of experimental Ebola virus infection in guinea pigs. J. Infect. Dis. 179(Suppl. 1), S203–S217. doi: 10.1086/514305
Cote, M., Misasi, J., Ren, T., Bruchez, A., Lee, K., Filone, C. M., et al. (2011). Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477, 344–348. doi: 10.1038/nature10380
Daddario-Dicaprio, K. M., Geisbert, T. W., Geisbert, J. B., Stroher, U., Hensley, L. E., Grolla, A., et al. (2006a). Cross-protection against Marburg virus strains by using a live, attenuated recombinant vaccine. J. Virol. 80, 9659–9666. doi: 10.1128/JVI.00959-06
Daddario-Dicaprio, K. M., Geisbert, T. W., Stroher, U., Geisbert, J. B., Grolla, A., Fritz, E. A., et al. (2006b). Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: an efficacy assessment. Lancet 367, 1399–1404. doi: 10.1016/S0140-6736(06)68546-2
Dalgard, D. W., Hardy, R. J., Pearson, S. L., Pucak, G. J., Quander, R. V., Zack, P. M., et al. (1992). Combined simian hemorrhagic fever and Ebola virus infection in cynomolgus monkeys. Lab. Anim. Sci. 42, 152–157.
Davis, K. J., Anderson, A. O., Geisbert, T. W., Steele, K. E., Geisbert, J. B., Vogel, P., et al. (1997). Pathology of experimental Ebola virus infection in African green monkeys. Involvement of fibroblastic reticular cells. Arch. Pathol. Lab. Med. 121, 805–819.
Dietrich, M., Schumacher, H. H., Peters, D., and Knobloch, J. (1978). “Human pathology of Ebola (Maridi) virus infection in the Sudan,” in Ebola Virus Haemorrhagic Fever, ed S. R. Pattyn (New York, NY: Elsevier, North-Holland Biomedical Press), 37–41.
Ebihara, H., Rockx, B., Marzi, A., Feldmann, F., Haddock, E., Brining, D., et al. (2011). Host response dynamics following lethal infection of rhesus macaques with Zaire ebolavirus. J. Infect. Dis. 204(Suppl. 3), S991–999. doi: 10.1093/infdis/jir336
Ebihara, H., Takada, A., Kobasa, D., Jones, S., Neumann, G., Theriault, S., et al. (2006). Molecular determinants of Ebola virus virulence in mice. PLoS Pathog. 2:e73. doi: 10.1371/journal.ppat.0020073
Ebihara, H., Zivcec, M., Gardner, D., Falzarano, D., Lacasse, R., Rosenke, R., et al. (2013). A syrian golden hamster model recapitulating ebola hemorrhagic fever. J. Infect. Dis. 207, 306–318. doi: 10.1093/infdis/jis626
Ellis, D. S., Bowen, E. T., Simpson, D. I., and Stamford, S. (1978). Ebola virus: a comparison, at ultrastructural level, of the behaviour of the Sudan and Zaire strains in monkeys. Br. J. Exp. Pathol. 59, 584–593.
Falzarano, D., Geisbert, T. W., and Feldmann, H. (2011). Progress in filovirus vaccine development: evaluating the potential for clinical use. Expert Rev. Vaccines 10, 63–77. doi: 10.1586/erv.10.152
Feldmann, H., Jones, S. M., Daddario-Dicaprio, K. M., Geisbert, J. B., Stroher, U., Grolla, A., et al. (2007). Effective post-exposure treatment of Ebola infection. PLoS Pathog. 3:e2. doi: 10.1371/journal.ppat.0030002
Fisher-Hoch, S. P., Brammer, T. L., Trappier, S. G., Hutwagner, L. C., Farrar, B. B., Ruo, S. L., et al. (1992). Pathogenic potential of filoviruses: role of geographic origin of primate host and virus strain. J. Infect. Dis. 166, 753–763. doi: 10.1093/infdis/166.4.753
Fisher-Hoch, S. P., Platt, G. S., Neild, G. H., Southee, T., Baskerville, A., Raymond, R. T., et al. (1985). Pathophysiology of shock and hemorrhage in a fulminating viral infection (Ebola). J. Infect. Dis. 152, 887–894. doi: 10.1093/infdis/152.5.887
Fowler, T., Bamberg, S., Moller, P., Klenk, H. D., Meyer, T. F., Becker, S., et al. (2005). Inhibition of Marburg virus protein expression and viral release by RNA interference. J. Gen. Virol. 86, 1181–1188. doi: 10.1099/vir.0.80622-0
Friedrich, B. M., Trefry, J. C., Biggins, J. E., Hensley, L. E., Honko, A. N., Smith, D. R., et al. (2012). Potential vaccines and post-exposure treatments for filovirus infections. Viruses 4, 1619–1650. doi: 10.3390/v4091619
Geisbert, T. W., Bailey, M., Geisbert, J. B., Asiedu, C., Roederer, M., Grazia-Pau, M., et al. (2010a). Vector choice determines immunogenicity and potency of genetic vaccines against Angola Marburg virus in nonhuman primates. J. Virol. 84, 10386–10394. doi: 10.1128/JVI.00594-10
Geisbert, T. W., Bausch, D. G., and Feldmann, H. (2010b). Prospects for immunisation against Marburg and Ebola viruses. Rev. Med. Virol. 20, 344–357.
Geisbert, T. W., Hensley, L. E., Geisbert, J. B., Leung, A., Johnson, J. C., Grolla, A., et al. (2010c). Postexposure treatment of Marburg virus infection. Emerging Infect. Dis. 16, 1119–1122. doi: 10.3201/eid1607.100159
Geisbert, T. W., Lee, A. C., Robbins, M., Geisbert, J. B., Honko, A. N., Sood, V., et al. (2010d). Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: a proof-of-concept study. Lancet 375, 1896–1905. doi: 10.1016/S0140-6736(10)60357-1
Geisbert, T. W., Bailey, M., Hensley, L., Asiedu, C., Geisbert, J., Stanley, D., et al. (2011). Recombinant adenovirus serotype 26 (Ad26) and Ad35 vaccine vectors bypass immunity to Ad5 and protect nonhuman primates against ebolavirus challenge. J. Virol. 85, 4222–4233. doi: 10.1128/JVI.02407-10
Geisbert, T. W., Daddario-Dicaprio, K. M., Geisbert, J. B., Young, H. A., Formenty, P., Fritz, E. A., et al. (2007). Marburg virus Angola infection of rhesus macaques: pathogenesis and treatment with recombinant nematode anticoagulant protein c2. J. Infect. Dis. 196(Suppl. 2), S372–S381. doi: 10.1086/520608
Geisbert, T. W., Daddario-Dicaprio, K. M., Williams, K. J., Geisbert, J. B., Leung, A., Feldmann, F., et al. (2008). Recombinant vesicular stomatitis virus vector mediates postexposure protection against Sudan Ebola hemorrhagic fever in nonhuman primates. J. Virol. 82, 5664–5668. doi: 10.1128/JVI.00456-08
Geisbert, T. W., Geisbert, J. B., Leung, A., Daddario-Dicaprio, K. M., Hensley, L. E., Grolla, A., et al. (2009). Single-injection vaccine protects nonhuman primates against infection with marburg virus and three species of ebola virus. J. Virol. 83, 7296–7304. doi: 10.1128/JVI.00561-09
Geisbert, T. W., Hensley, L. E., Gibb, T. R., Steele, K. E., Jaax, N. K., and Jahrling, P. B. (2000). Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab. Invest. 80, 171–186. doi: 10.1038/labinvest.3780021
Geisbert, T. W., Hensley, L. E., Jahrling, P. B., Larsen, T., Geisbert, J. B., Paragas, J., et al. (2003a). Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet 362, 1953–1958. doi: 10.1016/S0140-6736(03)15012-X
Geisbert, T. W., Hensley, L. E., Larsen, T., Young, H. A., Reed, D. S., Geisbert, J. B., et al. (2003b). Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 163, 2347–2370.
Geisbert, T. W., Jahrling, P. B., Larsen, T., Davis, K. J., and Hensley, L. E. (2003c). Filovirus Pathogenesis in Nonhuman Primates. Wymondham, Norfolk: Taylor and Francis.
Geisbert, T. W., Young, H. A., Jahrling, P. B., Davis, K. J., Kagan, E., and Hensley, L. E. (2003d). Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis. 188, 1618–1629. doi: 10.1086/379724
Geisbert, T. W., Young, H. A., Jahrling, P. B., Davis, K. J., Larsen, T., Kagan, E., et al. (2003e). Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am. J. Pathol. 163, 2371–2382.
Geisbert, T. W., Hensley, L. E., Kagan, E., Yu, E. Z., Geisbert, J. B., Daddario-Dicaprio, K., et al. (2006). Postexposure protection of guinea pigs against a lethal ebola virus challenge is conferred by RNA interference. J. Infect. Dis. 193, 1650–1657. doi: 10.1086/504267
Geisbert, T. W., Pushko, P., Anderson, K., Smith, J., Davis, K. J., and Jahrling, P. B. (2002). Evaluation in nonhuman primates of vaccines against Ebola virus. Emerging Infect. Dis. 8, 503–507. doi: 10.3201/eid0805.010284
Gibb, T. R., Norwood, D. A. Jr., Woollen, N., and Henchal, E. A. (2001). Development and evaluation of a fluorogenic 5′ nuclease assay to detect and differentiate between Ebola virus subtypes Zaire and Sudan. J. Clin. Microbiol. 39, 4125–4130. doi: 10.1128/JCM.39.11.4125-4130.2001
Gonchar, N. I., Pshenichnov, V. A., Pokhodiaev, V. A., Lopatov, K. L., and Firsova, I. V. (1991). The sensitivity of different experimental animals to the Marburg virus. Vopr. Virusol. 36, 435–437.
Gupta, M., Greer, P., Mahanty, S., Shieh, W. J., Zaki, S. R., Ahmed, R., et al. (2005). CD8-mediated protection against Ebola virus infection is perforin dependent. J. Immunol. 174, 4198–4202.
Halfmann, P., Ebihara, H., Marzi, A., Hatta, Y., Watanabe, S., Suresh, M., et al. (2009). Replication-deficient ebolavirus as a vaccine candidate. J. Virol. 83, 3810–3815. doi: 10.1128/JVI.00074-09
Halfmann, P., Neumann, G., and Kawaoka, Y. (2011). The Ebolavirus VP24 protein blocks phosphorylation of p38 mitogen-activated protein kinase. J. Infect. Dis. 204(Suppl. 3), S953–S956. doi: 10.1093/infdis/jir325
Hall, W. C., Geisbert, T. W., Huggins, J. W., and Jahrling, P. B. (1996). Experimental infection of guinea pigs with Venezuelan hemorrhagic fever virus (Guanarito): a model of human disease. Am. J. Trop. Med. Hyg. 55, 81–88.
Hass, R., and Maass, G. (1971). “Experimental infection of monkeys with the marburg virus,” in Marburg Virus Disease, eds G. A. Martini and R. Siegert (New York, NY: Springer, Berlin Heidelberg), 136–143.
Hensley, L. E., Alves, D. A., Geisbert, J. B., Fritz, E. A., Reed, C., Larsen, T., et al. (2011). Pathogenesis of Marburg hemorrhagic fever in cynomolgus macaques. J. Infect. Dis. 204(Suppl. 3), S1021–S1031. doi: 10.1093/infdis/jir339
Hensley, L. E., Stevens, E. L., Yan, S. B., Geisbert, J. B., Macias, W. L., Larsen, T., et al. (2007). Recombinant human activated protein C for the postexposure treatment of Ebola hemorrhagic fever. J. Infect. Dis. 196(Suppl. 2), S390–S399. doi: 10.1086/520598
Hevey, M., Negley, D., Pushko, P., Smith, J., and Schmaljohn, A. (1998). Marburg virus vaccines based upon alphavirus replicons protect guinea pigs and nonhuman primates. Virology 251, 28–37. doi: 10.1006/viro.1998.9367
Ignat'ev, G. M., Agafonov, A. P., Strel'tsova, M. A., Kuz'min, V. A., Mainagasheva, G. I., Spirin, G. V., et al. (1991). A comparative study of the immunological indices in guinea pigs administered an inactivated Marburg virus. Vopr. Virusol. 36, 421–423.
Ignatiev, G. M., Dadaeva, A. A., Luchko, S. V., and Chepurnov, A. A. (2000). Immune and pathophysiological processes in baboons experimentally infected with Ebola virus adapted to guinea pigs. Immunol. Lett. 71, 131–140. doi: 10.1016/S0165-2478(99)00169-8
Ignatyev, G. M., Agafonov, A. P., Streltsova, M. A., and Kashentseva, E. A. (1996). Inactivated Marburg virus elicits a nonprotective immune response in Rhesus monkeys. J. Biotechnol. 44, 111–118. doi: 10.1016/0168-1656(95)00104-2
Isaacson, M., Sureau, P., Courteille, G., and Pattyn, S. R. (1978). “Clinical aspets of Ebola virus disease at the Ngaliema hospital, Kinshasa, Zaire, 1976,” in Ebola Virus Haemorrhagic Fever, ed S. R. Pattyn (New York, NY: Elsevier, North-Holland Biomedical Press), 15–20.
Jaax, N., Jahrling, P., Geisbert, T., Geisbert, J., Steele, K., McKee, K., et al. (1995). Transmission of Ebola virus (Zaire strain) to uninfected control monkeys in a biocontainment laboratory. Lancet 346, 1669–1671. doi: 10.1016/S0140-6736(95)92841-3
Jaax, N. K., Davis, K. J., Geisbert, T. J., Vogel, P., Jaax, G. P., Topper, M., et al. (1996). Lethal experimental infection of rhesus monkeys with Ebola-Zaire (Mayinga) virus by the oral and conjunctival route of exposure. Arch. Pathol. Lab. Med. 120, 140–155.
Jahrling, P. B., Geisbert, T. W., Dalgard, D. W., Johnson, E. D., Ksiazek, T. G., Hall, W. C., et al. (1990). Preliminary report: isolation of Ebola virus from monkeys imported to USA. Lancet 335, 502–505. doi: 10.1016/0140-6736(90)90737-P
Jahrling, P. B., Geisbert, T. W., Jaax, N. K., Hanes, M. A., Ksiazek, T. G., and Peters, C. J. (1996). Experimental infection of cynomolgus macaques with Ebola-Reston filoviruses from the 1989–1990 U.S. epizootic. Arch. Virol. Suppl. 11, 115–134. doi: 10.1007/978-3-7091-7482-1_11
Jahrling, P. B., Smith, S., Hesse, R. A., and Rhoderick, J. B. (1982). Pathogenesis of Lassa virus infection in guinea pigs. Infect. Immun. 37, 771–778.
Johnson, E., Jaax, N., White, J., and Jahrling, P. (1995). Lethal experimental infections of rhesus monkeys by aerosolized Ebola virus. Int. J. Exp. Pathol. 76, 227–236.
Johnson, E. D., Johnson, B. K., Silverstein, D., Tukei, P., Geisbert, T. W., Sanchez, A. N., et al. (1996). Characterization of a new Marburg virus isolated from a 1987 fatal case in Kenya. Arch. Virol. Suppl. 11, 101–114. doi: 10.1007/978-3-7091-7482-1_10
Jones, S. M., Feldmann, H., Stroher, U., Geisbert, J. B., Fernando, L., Grolla, A., et al. (2005). Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med. 11, 786–790. doi: 10.1038/nm1258
Kenyon, R. H., Condie, R. M., Jahrling, P. B., and Peters, C. J. (1990). Protection of guinea pigs against experimental Argentine hemorrhagic fever by purified human IgG: importance of elimination of infected cells. Microb. Pathog. 9, 219–226. doi: 10.1016/0882-4010(90)90010-N
Konduru, K., Bradfute, S. B., Jacques, J., Manangeeswaran, M., Nakamura, S., Morshed, S., et al. (2011). Ebola virus glycoprotein Fc fusion protein confers protection against lethal challenge in vaccinated mice. Vaccine 29, 2968–2977. doi: 10.1016/j.vaccine.2011.01.113
Korb, G., and Slenczka, W. (1971). “Histologic findings in livers and spleens of guinea pigs after inoculation by the marburg virus,” in Marburg Virus Disease, eds G. A. Martini and R. Siegert (New York, NY: Springer, Berlin Heidelberg), 123–128.
Ksiazek, T. G., Rollin, P. E., Williams, A. J., Bressler, D. S., Martin, M. L., Swanepoel, R., et al. (1999). Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J. Infect. Dis. 179(Suppl. 1), S177–S187. doi: 10.1086/514321
Kuhn, J. H. (2008). Filoviruses: A Compendium of 40 Years of Epidemiological, Clinical, and Laboratory Studies. New York, NY: Springer Wien.
Kuhn, J. H., Bao, Y., Bavari, S., Becker, S., Bradfute, S., Brister, J. R., et al. (2013). Virus nomenclature below the species level: a standardized nomenclature for natural variants of viruses assigned to the family Filoviridae. Arch. Virol. 158, 301–311. doi: 10.1007/s00705-012-1454-0
Kuhn, J. H., Dodd, L. E., Wahl-Jensen, V., Radoshitzky, S. R., Bavari, S., and Jahrling, P. B. (2011). Evaluation of perceived threat differences posed by filovirus variants. Biosecur. Bioterror. 9, 361–371. doi: 10.1089/bsp.2011.0051
Leroy, E. M., Epelboin, A., Mondonge, V., Pourrut, X., Gonzalez, J. P., Muyembe-Tamfum, J. J., et al. (2009). Human Ebola outbreak resulting from direct exposure to fruit bats in Luebo, Democratic Republic of Congo, 2007. Vector Borne Zoonotic Dis. 9, 723–728. doi: 10.1089/vbz.2008.0167
Leroy, E. M., Rouquet, P., Formenty, P., Souquiere, S., Kilbourne, A., Froment, J. M., et al. (2004). Multiple Ebola virus transmission events and rapid decline of central African wildlife. Science 303, 387–390. doi: 10.1126/science.1092528
Luchko, S. V., Dadaeva, A. A., Ustinova, E. N., Sizikova, L. P., Riabchikova, E. I., and Sandakhchiev, L. S. (1995). Experimental study of Ebola hemorrhagic fever in baboon models. Biull. Eksp. Biol. Med. 120, 302–304. doi: 10.1007/BF02445023
Martin, J. E., Sullivan, N. J., Enama, M. E., Gordon, I. J., Roederer, M., Koup, R. A., et al. (2006). A DNA vaccine for Ebola virus is safe and immunogenic in a phase I clinical trial. Clin. Vaccine Immunol. 13, 1267–1277. doi: 10.1128/CVI.00162-06
Martini, G. A. (1971). “Marburg virus disease. Clinical symptome,” in Marburg Virus Disease, eds G. A. Martini and R. Siegert (New York, NY: Springer, Berlin Heidelberg), 1–9.
Mast, T. C., Kierstead, L., Gupta, S. B., Nikas, A. A., Kallas, E. G., Novitsky, V., et al. (2010). International epidemiology of human pre-existing adenovirus (Ad) type-5, type-6, type-26 and type-36 neutralizing antibodies: correlates of high Ad5 titers and implications for potential HIV vaccine trials. Vaccine 28, 950–957. doi: 10.1016/j.vaccine.2009.10.145
Mateo, M., Carbonnelle, C., Reynard, O., Kolesnikova, L., Nemirov, K., Page, A., et al. (2011). VP24 is a molecular determinant of Ebola virus virulence in guinea pigs. J. Infect. Dis. 204(Suppl. 3), S1011–S1020. doi: 10.1093/infdis/jir338
Mateo, M., Reid, S. P., Leung, L. W., Basler, C. F., and Volchkov, V. E. (2010). Ebolavirus VP24 binding to karyopherins is required for inhibition of interferon signaling. J. Virol. 84, 1169–1175. doi: 10.1128/JVI.01372-09
Michelow, I. C., Lear, C., Scully, C., Prugar, L. I., Longley, C. B., Yantosca, L. M., et al. (2011). High-dose mannose-binding lectin therapy for Ebola virus infection. J. Infect. Dis. 203, 175–179. doi: 10.1093/infdis/jiq025
Mikhailov, V. V., Borisevich, I. V., Chernikova, N. K., Potryvaeva, N. V., and Krasnianskii, V. P. (1994). The evaluation in hamadryas baboons of the possibility for the specific prevention of Ebola fever. Vopr. Virusol. 39, 82–84.
Miranda, M. E., Ksiazek, T. G., Retuya, T. J., Khan, A. S., Sanchez, A., Fulhorst, C. F., et al. (1999). Epidemiology of Ebola (subtype Reston) virus in the Philippines, 1996. J. Infect. Dis. 179(Suppl. 1), S115–S119. doi: 10.1086/514314
Miranda, M. E., Yoshikawa, Y., Manalo, D. L., Calaor, A. B., Miranda, N. L., Cho, F., et al. (2002). Chronological and spatial analysis of the 1996 Ebola Reston virus outbreak in a monkey breeding facility in the Philippines. Exp. Anim. 51, 173–179. doi: 10.1538/expanim.51.173
Murphy, F. A., Simpson, D. I., Whitfield, S. G., Zlotnik, I., and Carter, G. B. (1971). Marburg virus infection in monkeys. Ultrastructural studies. Lab. Invest. 24, 279–291.
Ndambi, R., Akamituna, P., Bonnet, M. J., Tukadila, A. M., Muyembe-Tamfum, J. J., and Colebunders, R. (1999). Epidemiologic and clinical aspects of the Ebola virus epidemic in Mosango, Democratic Republic of the Congo, 1995. J. Infect. Dis. 179(Suppl. 1), S8–S10. doi: 10.1086/514297
Negredo, A., Palacios, G., Vazquez-Moron, S., Gonzalez, F., Dopazo, H., Molero, F., et al. (2011). Discovery of an ebolavirus-like filovirus in europe. PLoS Pathog. 7:e1002304. doi: 10.1371/journal.ppat.1002304
Phoolcharoen, W., Bhoo, S. H., Lai, H., Ma, J., Arntzen, C. J., Chen, Q., et al. (2011). Expression of an immunogenic Ebola immune complex in Nicotiana benthamiana. Plant Biotechnol. J. 9, 807–816. doi: 10.1111/j.1467-7652.2011.00593.x
Piedra, P. A., Poveda, G. A., Ramsey, B., McCoy, K., and Hiatt, P. W. (1998). Incidence and prevalence of neutralizing antibodies to the common adenoviruses in children with cystic fibrosis: implication for gene therapy with adenovirus vectors. Pediatrics 101, 1013–1019. doi: 10.1542/peds.101.6.1013
Piot, P., Sureau, P., Breman, G., Heymann, D., Kintoki, V., Masamba, M., et al. (1978). “Clinical aspects of Ebola virus infection in Tambuku area, Zaire, 1976,” in Ebola Virus Haemorrhagic Fever, ed S. R. Pattyn (New York, NY: Elsevier, North-Holland Biomedical Press), 7–14.
Pokhodiaev, V. A., Gonchar, N. I., and Pshenichnov, V. A. (1991). An experimental study of the contact transmission of the Marburg virus. Vopr. Virusol. 36, 506–508.
Pushko, P., Bray, M., Ludwig, G. V., Parker, M., Schmaljohn, A., Sanchez, A., et al. (2001). Recombinant RNA replicons derived from attenuated Venezuelan equine encephalitis virus protect guinea pigs and mice from Ebola hemorrhagic fever virus. Vaccine 19, 142–153. doi: 10.1016/S0264-410X(00)00113-4
Qiu, X., Fernando, L., Alimonti, J. B., Melito, P. L., Feldmann, F., Dick, D., et al. (2009). Mucosal immunization of cynomolgus macaques with the VSVDeltaG/ZEBOVGP vaccine stimulates strong ebola GP-specific immune responses. PLoS ONE 4:e5547. doi: 10.1371/journal.pone.0005547
Ramanan, P., Edwards, M. R., Shabman, R. S., Leung, D. W., Endlich-Frazier, A. C., Borek, D. M., et al. (2012). Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc. Natl. Acad. Sci. U.S.A. 109, 20661–20666. doi: 10.1073/pnas.1213559109
Ramanan, P., Shabman, R. S., Brown, C. S., Amarasinghe, G. K., Basler, C. F., and Leung, D. W. (2011). Filoviral immune evasion mechanisms. Viruses 3, 1634–1649. doi: 10.3390/v3091634
Reid, S. P., Valmas, C., Martinez, O., Sanchez, F. M., and Basler, C. F. (2007). Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J. Virol. 81, 13469–13477. doi: 10.1128/JVI.01097-07
Riemenschneider, J., Garrison, A., Geisbert, J., Jahrling, P., Hevey, M., Negley, D., et al. (2003). Comparison of individual and combination DNA vaccines for B. anthracis, Ebola virus, Marburg virus and Venezuelan equine encephalitis virus. Vaccine 21, 4071–4080. doi: 10.1016/S0264-410X(03)00362-1
Robin, Y., Bres, P., and Camain, R. (1971). “Passage of marburg virus in guinea pigs,” in Marburg Virus Disease, eds G. A. Martini and R. Siegert (New York, NY: Springer, Berlin Heidelberg), 117–122.
Ryabchikova, E., Kolesnikova, L., Smolina, M., Tkachev, V., Pereboeva, L., Baranova, S., et al. (1996). Ebola virus infection in guinea pigs: presumable role of granulomatous inflammation in pathogenesis. Arch. Virol. 141, 909–921. doi: 10.1007/BF01718165
Ryabchikova, E., Smolina, M. A. G., and Rassadkin, J. (2003). Ebola virus infection in the guinea pig. Wymondham, Norfolk: Taylor and Francis.
Ryabchikova, E. I., Kolesnikova, L. V., and Luchko, S. V. (1999). An analysis of features of pathogenesis in two animal models of Ebola virus infection. J. Infect. Dis. 179(Suppl. 1), S199–S202. doi: 10.1086/514293
Sanchez, A., Geisbert, T. W., and Felsmann, H. (2007). Marburg and Ebola Viruses. Philadelphia, PA: Lippincott Williams and Wilkins.
Sanchez, A., Lukwiya, M., Bausch, D., Mahanty, S., Sanchez, A. J., Wagoner, K. D., et al. (2004). Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: cellular responses, virus load, and nitric oxide levels. J. Virol. 78, 10370–10377. doi: 10.1128/JVI.78.19.10370-10377.2004
Schulick, A. H., Vassalli, G., Dunn, P. F., Dong, G., Rade, J. J., Zamarron, C., et al. (1997). Established immunity precludes adenovirus-mediated gene transfer in rat carotid arteries. Potential for immunosuppression and vector engineering to overcome barriers of immunity. J. Clin. Invest. 99, 209–219. doi: 10.1172/JCI119149
Simpson, D. I. (1969). Marburg agent disease: in monkeys. Trans. R. Soc. Trop. Med. Hyg. 63, 303–309. doi: 10.1016/0035-9203(69)90002-9
Simpson, D. I., Zlotnik, I., and Rutter, D. A. (1968). Vervet monkey disease. Experiment infection of guinea pigs and monkeys with the causative agent. Br. J. Exp. Pathol. 49, 458–464.
Smith, D. H., Francis, F., and Simpson, D. I. H. (1978). “African haemorrhagic fever in the Southern Sudan, 1976: the clinical manifestations,” in Ebola Virus Haemorrhagic Fever, ed S. R. Pattyn (New York, NY: Elsevier, North-Holland Biomedical Press), 21–26.
Smith, D. R., McCarthy, S., Chrovian, A., Olinger, G., Stossel, A., Geisbert, T. W., et al. (2010). Inhibition of heat-shock protein 90 reduces Ebola virus replication. Antiviral Res. 87, 187–194. doi: 10.1016/j.antiviral.2010.04.015
Spiridonov, V. A., Bazhutin, N. B., Belanov, E. F., Voitenko, A. V., Zolin, V. V., Krivenchuk, N. A., et al. (1992). Changes in the blood serum aminotransferase activity in the experimental infection of Cercopithecus aethiops monkeys with the Marburg virus. Vopr. Virusol. 37, 156–157.
Subbotina, E., Dadaeva, A., Kachko, A., and Chepurnov, A. (2010). Genetic factors of Ebola virus virulence in guinea pigs. Virus Res. 153, 121–133. doi: 10.1016/j.virusres.2010.07.015
Sullivan, N. J., Geisbert, T. W., Geisbert, J. B., Xu, L., Yang, Z. Y., Roederer, M., et al. (2003). Accelerated vaccination for Ebola virus haemorrhagic fever in non-human primates. Nature 424, 681–684. doi: 10.1038/nature01876
Sullivan, N. J., Sanchez, A., Rollin, P. E., Yang, Z. Y., and Nabel, G. J. (2000). Development of a preventive vaccine for Ebola virus infection in primates. Nature 408, 605–609. doi: 10.1038/35046108
Sun, Y., Carrion, R. Jr., Ye, L., Wen, Z., Ro, Y. T., et al. (2009). Protection against lethal challenge by Ebola virus-like particles produced in insect cells. Virology 383, 12–21. doi: 10.1016/j.virol.2008.09.020
Swenson, D. L., Wang, D., Luo, M., Warfield, K. L., Woraratanadharm, J., Holman, D. H., et al. (2008a). Vaccine to confer to nonhuman primates complete protection against multistrain Ebola and Marburg virus infections. Clin. Vaccine Immunol. 15, 460–467. doi: 10.1128/CVI.00431-07
Swenson, D. L., Warfield, K. L., Larsen, T., Alves, D. A., Coberley, S. S., and Bavari, S. (2008b). Monovalent virus-like particle vaccine protects guinea pigs and nonhuman primates against infection with multiple Marburg viruses. Expert Rev. Vaccines 7, 417–429.
Swenson, D. L., Warfield, K. L., Warren, T. K., Lovejoy, C., Hassinger, J. N., Ruthel, G., et al. (2009). Chemical modifications of antisense morpholino oligomers enhance their efficacy against Ebola virus infection. Antimicrob. Agents Chemother. 53, 2089–2099. doi: 10.1128/AAC.00936-08
Towner, J. S., Rollin, P. E., Bausch, D. G., Sanchez, A., Crary, S. M., Vincent, M., et al. (2004). Rapid diagnosis of Ebola hemorrhagic fever by reverse transcription-PCR in an outbreak setting and assessment of patient viral load as a predictor of outcome. J. Virol. 78, 4330–4341. doi: 10.1128/JVI.78.8.4330-4341.2004
Tsuda, Y., Caposio, P., Parkins, C. J., Botto, S., Messaoudi, I., Cicin-Sain, L., et al. (2011a). A replicating cytomegalovirus-based vaccine encoding a single Ebola virus nucleoprotein CTL epitope confers protection against Ebola virus. PLoS Negl. Trop. Dis. 5:e1275. doi: 10.1371/journal.pntd.0001275
Tsuda, Y., Safronetz, D., Brown, K., Lacasse, R., Marzi, A., Ebihara, H., et al. (2011b). Protective efficacy of a bivalent recombinant vesicular stomatitis virus vaccine in the Syrian hamster model of lethal Ebola virus infection. J. Infect. Dis. 204(Suppl. 3), S1090–S1097. doi: 10.1093/infdis/jir379
Tuffs, A. (2009). Experimental vaccine may have saved Hamburg scientist from Ebola fever. BMJ 338:b1223. doi: 10.1136/bmj.b1223
van der Groen, G., Jacob, W., and Pattyn, S. R. (1979). Ebola virus virulence for newborn mice. J. Med. Virol. 4, 239–240. doi: 10.1002/jmv.1890040309
Vanderzanden, L., Bray, M., Fuller, D., Roberts, T., Custer, D., Spik, K., et al. (1998). DNA vaccines expressing either the GP or NP genes of Ebola virus protect mice from lethal challenge. Virology 246, 134–144. doi: 10.1006/viro.1998.9176
Vogels, R., Zuijdgeest, D., van Rijnsoever, R., Hartkoorn, E., Damen, I., de Bethune, M. P., et al. (2003). Replication-deficient human adenovirus type 35 vectors for gene transfer and vaccination: efficient human cell infection and bypass of preexisting adenovirus immunity. J. Virol. 77, 8263–8271. doi: 10.1128/JVI.77.15.8263-8271.2003
Volchkov, V. E., Chepurnov, A. A., Volchkova, V. A., Ternovoj, V. A., and Klenk, H. D. (2000). Molecular characterization of guinea pig-adapted variants of Ebola virus. Virology 277, 147–155. doi: 10.1006/viro.2000.0572
Wahl-Jensen, V., Bollinger, L., Safronetz, D., de Kok-Mercado, F., Scott, D. P., and Ebihara, H. (2012). Use of the Syrian hamster as a new model of ebola virus disease and other viral hemorrhagic fevers. Viruses 4, 3754–3784. doi: 10.3390/v4123754
Wang, D., Hevey, M., Juompan, L. Y., Trubey, C. M., Raja, N. U., Deitz, S. B., et al. (2006a). Complex adenovirus-vectored vaccine protects guinea pigs from three strains of Marburg virus challenges. Virology 353, 324–332. doi: 10.1016/j.virol.2006.05.033
Wang, D., Raja, N. U., Trubey, C. M., Juompan, L. Y., Luo, M., Woraratanadharm, J., et al. (2006b). Development of a cAdVax-based bivalent ebola virus vaccine that induces immune responses against both the Sudan and Zaire species of Ebola virus. J. Virol. 80, 2738–2746. doi: 10.1128/JVI.80.6.2738-2746.2006
Warfield, K. L., Bosio, C. M., Welcher, B. C., Deal, E. M., Mohamadzadeh, M., Schmaljohn, A., et al. (2003). Ebola virus-like particles protect from lethal Ebola virus infection. Proc. Natl. Acad. Sci. U.S.A. 100, 15889–15894. doi: 10.1073/pnas.2237038100
Warfield, K. L., Bradfute, S. B., Wells, J., Lofts, L., Cooper, M. T., Alves, D. A., et al. (2009). Development and characterization of a mouse model for Marburg hemorrhagic fever. J. Virol. 83, 6404–6415. doi: 10.1128/JVI.00126-09
Warfield, K. L., Posten, N. A., Swenson, D. L., Olinger, G. G., Esposito, D., Gillette, W. K., et al. (2007a). Filovirus-like particles produced in insect cells: immunogenicity and protection in rodents. J. Infect. Dis. 196(Suppl. 2), S421–429.
Warfield, K. L., Swenson, D. L., Olinger, G. G., Kalina, W. V., Aman, M. J., and Bavari, S. (2007b). Ebola virus-like particle-based vaccine protects nonhuman primates against lethal Ebola virus challenge. J. Infect. Dis. 196(Suppl. 2), S430–S437.
Warfield, K. L., Swenson, D. L., Olinger, G. G., Nichols, D. K., Pratt, W. D., Blouch, R., et al. (2006). Gene-specific countermeasures against Ebola virus based on antisense phosphorodiamidate morpholino oligomers. PLoS Pathog. 2:e1. doi: 10.1371/journal.ppat.0020001
Warren, T. K., Warfield, K. L., Wells, J., Swenson, D. L., Donner, K. S., van Tongeren, S. A., et al. (2010). Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat. Med. 16, 991–994. doi: 10.1038/nm.2202
Xu, L., Sanchez, A., Yang, Z., Zaki, S. R., Nabel, E. G., Nichol, S. T., et al. (1998). Immunization for Ebola virus infection. Nat. Med. 4, 37–42. doi: 10.1038/nm0198-037
Zaki, S. R., and Goldsmith, C. S. (1999). Pathologic features of filovirus infections in humans. Curr. Top. Microbiol. Immunol. 235, 97–116. doi: 10.1007/978-3-642-59949-1_7
Zhang, A. P., Bornholdt, Z. A., Liu, T., Abelson, D. M., Lee, D. E., Li, S., et al. (2012). The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog. 8:e1002550. doi: 10.1371/journal.ppat.1002550
Keywords: Ebola virus, Marburg virus, filovirus, animal models, viral hemorrhagic fever
Citation: Nakayama E and Saijo M (2013) Animal models for Ebola and Marburg virus infections. Front. Microbiol. 4:267. doi: 10.3389/fmicb.2013.00267
Received: 09 June 2013; Paper pending published: 03 July 2013;
Accepted: 19 August 2013; Published online: 05 September 2013.
Edited by:
Akio Adachi, The University of Tokushima Graduate School, JapanReviewed by:
Stefan Pöhlmann, German Primate Center, GermanyTakeo Ohsugi, Kumamoto University, Japan
Copyright © 2013 Nakayama and Saijo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masayuki Saijo, Department of Virology 1, National Institute of Infectious Diseases, Toyama 1-23-1, Shinjuku-ku, Tokyo 162-8640, Japan e-mail: msaijo@nih.go.jp