 Christelle Brégnard
Christelle Brégnard Monsef Benkirane
Monsef Benkirane Nadine Laguette
Nadine Laguette- Laboratoire de Virologie Moléculaire, Institut de Génétique Humaine, CNRS UPR1142, Montpellier, France
Viruses have been long known to perturb cell cycle regulators and key players of the DNA damage response to benefit their life cycles. In the case of the human immunodeficiency virus (HIV), the viral auxiliary protein Vpr activates the structure-specific endonuclease SLX4 complex to promote escape from innate immune sensing and, as a side effect, induces replication stress in cycling cells and subsequent cell cycle arrest at the G2/M transition. This novel pathway subverted by HIV to prevent accumulation of viral reverse transcription by-products adds up to facilitating effects of major cellular exonucleases that degrade pathological DNA species. Within this review we discuss the impact of this finding on our understanding of the interplay between HIV replication and nucleic acid metabolism and its implications for cancer-related chronic inflammation.
Introduction
Efficient human immunodeficiency virus (HIV) replication in target cells relies on its ability to use cellular resources and to overthrow host defense mechanisms. Indeed, viral fitness is defined by both the availability of cellular dependency factors and the ability to escape cellular blocks. One of the most challenging steps of HIV life cycle is the delivery of its single stranded RNA (ssRNA) genome and its conversion into double stranded DNA (dsDNA) in the host cell without inducing innate immune responses. Indeed, cellular “sensors” specialized in the recognition of foreign or pathological nucleic acids are present in different compartments through which viruses enter target cell. These nucleic acid sensors belong to the pattern recognition receptors (PRRs) family that recognize pathogen associated molecular patterns (PAMPs) and subsequently trigger a signaling cascade that culminates in the production of pro-inflammatory cytokines, including antiviral interferon (IFN; for review Kawai and Akira, 2006). Once recognition established, a signaling cascade is triggered, endowing an antiviral state and a cellular response aiming to clear the infection. In the course of reverse transcription of the HIV ssRNA genome into dsDNA, several intermediate, and sometime abortive, nucleic acid species are generated, including DNA:RNA hybrids, DNA flap structures and dsDNA. Exposure of these in the cytoplasm engages various sensors. Those include Toll like receptor 7 (TLR7) that detects viral RNA in endosomes (Beignon et al., 2005), Gamma-interferon-inducible protein 16 (IFI16) that recognizes virus-derived DNA in the cytoplasm of lymphoid quiescent CD4 T cells (Monroe et al., 2013) and the cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS; Gao et al., 2013).
To avoid recognition of reverse-transcription intermediates, viral genomes are protected within the capsid core. If uncoating is correctly orchestrated, this ensures delivery of fully reverse-transcribed viral genomes into the nucleus that are subsequently integrated into the host cell genome, a pre-requisite for the establishment of productive infection. However, as reviewed in Yan and Hasan within this issue, while few viruses efficiently undergo these steps, abortive infection events also occur, leading to accumulation of “junk” nucleic acid species in the host cell cytoplasm that may be detected and thereby have adverse effects on the infection. The frailty of viral reverse-transcription is highlighted by the plethora of antiviral factors that target this specific step. Those include three of the prototypical restriction factors. Although their mechanism of action being beyond the scope of this review, it is worthy to mention that Tripartite Motif 5 alpha (TRIM5α) causes untimely uncoating, leading to premature exposure of virus-derived nucleic acid species in the host cell cytoplasm, apolipoprotein B mRNA-editing, enzyme-catalytic, polypeptide-like 3G (APOBEC3G) induces hypermutations in the viral genome generating non-functional unstable genomes and SAM domain and HD domain 1 (SAMHD1) deprives the viral reverse transcriptase of the deoxynucleoside building blocks required for its action (Malim and Bieniasz, 2012 and this review series).
Escape from innate immune sensing is therefore paramount to the establishment of productive viral infections. In recent years, several lines of evidence have shown that HIV has evolved highly specialized mechanisms to elude cellular blocks. For example, blocks imposed by restriction factors are mostly overcome through the use of viral accessory proteins (Vpx, Vpr, Nef, Vif, Vpu). Accessory proteins, initially qualified as such because unrequired for in vitro replication in permissive cells, are encoded by lentiviral genomes in addition to the essential structural and enzymatic proteins required for mature viral particles production (Gag, Pol, Env, Tat, and Rev). Additional mechanisms deployed by viruses to avoid innate immune sensing include a direct action on the IFN signaling cascade: inhibition of IFN synthesis, IFN receptor decoy and inhibition of IFN signaling (for review Stetson and Medzhitov, 2006; Broz and Monack, 2013). HIV also takes advantage of pre-existing cellular processes. Importantly, while cellular nucleic acid sensors recognize virus-derived nucleic acids and thereby detect incoming virions, they also play crucial roles in cellular metabolism and are usually constitutively expressed. They may therefore detect the presence of nucleic acid species resulting from DNA damage-associated repair mechanism or endogenous retroelement life cycle. Thus, cellular processes co-exist to prevent accumulation of abnormal self-nucleic acids, thereby preventing auto-initiation of pro-inflammatory responses (for review Yan and Hasan). These include major cellular exonucleases that have also been shown to positively impact of HIV life cycle: ribonuclease H2 (RNaseH2) and three prime repair exonuclease 1 (TREX1) (Yan et al., 2010; Genovesio et al., 2011). Importantly, these proteins involved in nucleic acid metabolism belong to a family of genes that, when mutated, lead to the Aicardi-Gouttière syndrome (AGS). This rare autosomal recessive genetic encephalopathy is characterized by neurological dysfunctions, intracranial calcifications, brain atrophy, psychomotor retardation and increased plasma levels of IFN that lead to chronic inflammation (Lebon et al., 1988). We recently established that the SLX4 structure-specific endonuclease regulator complex also acts as a facilitator of HIV infection (Laguette et al., 2014). This finding bears substantial similarities with what was shown for the TREX1 exonuclease. Indeed, similar to TREX1, the SLX4 complex is involved in nucleic acid metabolism and plays crucial roles in the repair of DNA lesions. In addition, the core component of this complex is the SLX4 molecular scaffold that assembles structure-specific endonuclease modules. Biallelic mutations in SLX4 are involved in the onset of Fanconi anemia (FA), a cancer predisposition syndrome characterized by congenital malformations, hypersensitivity to DNA interstrand cross-linking agents and progressive bone marrow failure (Sasaki and Tonomura, 1973). In addition, FA patients experience heightened pro-inflammatory cytokines levels (Whitney et al., 1996; Rathbun et al., 1997; Dufour et al., 2003; Briot et al., 2008). The latter is a feature shared with AGS patients and supports a potential link between proteins involved in DNA damage response and the development of inflammatory responses. These recent findings also shed a new light on the implication on proteins involved in the maintenance of genomic stability and the HIV life cycle.
Cross-Talk between Cell Cycle Regulation Machinery and Viral Infections
Viruses have been long known to keep a privileged relationship with cell cycle regulatory mechanisms. Indeed, an estimated 20% of human cancers arise from infection with DNA or RNA viruses. Malignancy frequently results from side-tracking of cell cycle regulatory elements. A stricking example is virus-driven oncogenesis that results from subversion of the boundaries between the DNA replication step (S), segregation of sister chromatids (mitosis) and gap phases (G1 and G2). This is frequently achieved through viral non-structural proteins that modulate cell cycle regulators. Transforming viruses essentially subvert the G1/S boundary, thereby pushing cells into proliferation. In the case of the retrovirus HTLV-I, that encodes several potential oncogenes, the well-studied Tax protein is necessary and sufficient to initiate cellular transformation, while the HBz protein is required for its maintenance (Gatza et al., 2003). Similarly, HBx from the DNA virus HBV has the ability to transform immortalized cell lines and to provoke liver cancer in mice (Kim et al., 1991). Small DNA tumor viruses often encode potent oncoproteins that can cause cellular transformation in vitro [for example E6/E7 from HPV – for review (Howley and Livingston, 2009)]. In contrast certain viruses such as EBV require the concerted action of several proteins to achieve cellular transformation (Kutok and Wang, 2006).
Importantly, manipulation of the cell cycle or of cell cycle regulators is not solely confined to transforming viruses. Indeed, several DNA and RNA viruses are able to cause cell cycle arrest at the G2/M transition, including HIV (Davy and Doorbar, 2007). The molecular mechanisms underlying virus-induced G2/M arrest vary widely and have been extensively studied. Nonetheless, the understanding of the biological end-point of G2/M arrest remains poor despite suggestions that G2/M arrest may decrease propensity to secrete IFN (Lee and Rozee, 1970), increase RNA production rates (Lee and Rozee, 1970), and overall boost early step of the HIV life cycle (Groschel and Bushman, 2005). Subversion of the host cell cycle by HIV-1 relies on the highly conserved viral protein regulatory (Vpr) protein that causes a potent G2/M arrest in most cycling eukaryotic cells (Di Marzio et al., 1995; He et al., 1995; Jowett et al., 1995; Re et al., 1995; Rogel et al., 1995).
From a mechanistic stand point, Vpr-induced G2/M arrest is a well-documented phenotype. To understand how G2/M arrest is achieved, it is necessary to recapitulate the mechanism underlying this cell cycle check-point (for more details see Guenzel et al, this issue). Indeed, in healthy cells, the G2/M transition is controlled by Cyclin-dependent kinase1:CyclinB1 (CDK1:CCNB1). As cells progress through G2, CDK1:CCNB1 is progressively activated and once the G2/M boundary crossed, the complex is inactivated. The G2/M check-point serves as a quality-control step during the replication of the cellular genome that ensures the transmission to daughter cells of a complete unaltered set of chromosomes. Thus, when genotoxic stress is incurred, entry into mitosis is prevented to provide an opportunity to repair genomic lesions (Stark and Taylor, 2006). This is achieved through preventing CDK1:CCNB1 activation (Sanchez et al., 1997). This response is regulated through a signaling cascade that involves detection of the DNA lesion by the key DNA damage response regulators ataxia-telangiectasia-mutated kinase (ATM), ATM and Rad3-related kinase (ATR), and DNA-dependent protein kinase [DNA-PK; for review (Smith et al., 2010; Sirbu and Cortez, 2013)]. When damaged DNA or unreplicated regions of the genome are detected, these kinases activate downstream CHK1 or CHK2 that in turn inactivate CDK1:CCNB1 (Lopez-Girona et al., 1999).
Similar to what is observed following genotoxic stress, Vpr expression activates ATR, ATM, and the downstream CHK1/CHK2 kinases (Roshal et al., 2003; Munoz et al., 2009), thereby inactivating CDK1:CCNB1. In agreement, treatment of Vpr-expressing cells with caffeine, which inhibits ATR and ATM, relieves the cell cycle block (Poon et al., 1997; Shostak et al., 1999). Intriguingly, although mobilization onto sub-regions of the chromatin of breast cancer susceptibility protein 1 (BRCA1) and γH2ax have been reported upon Vpr expression, it remains unclear whether actual DNA breaks occur in the presence of Vpr and whether these lesions would be the trigger for cell cycle arrest. Rather, the prevailing view is that Vpr mediates ATR-dependent replication stress. Importantly, since the only consensual cellular partner of Vpr for the induction of G2/M arrest is the VPRBP-DDB1-CUL4 E3-ligase complex (Belzile et al., 2007; DeHart et al., 2007; Hrecka et al., 2007; Le Rouzic et al., 2007; Tan et al., 2007; Wen et al., 2007), it was assumed that Vpr would provoke the proteasomal degradation of a cell cycle regulatory element governing the G2/M transition. We recently identified the SLX4 complex as being the Vpr partner required for G2/M arrest (Laguette et al., 2014). Indeed, the structure specific endonucleases ERCC4XPF-ERCC1 and MUS81-EME1 together with the SLX4FANCP scaffold protein co-purified with Vpr as well as the poorly characterized TSPYL1 and C20orf94SLX4IP subunits. Vpr binds to the C-terminus of SLX4, inducing the recruitment of VPRBP and kinase-active PLK1. This leads to VPRBP-induced ubiquitination of MUS81 and hyperphosphorylation of EME1, the consequence of which being activation of SLX4-associated MUS81-EME1. Vpr-induced untimely activation of SLX4-bound MUS81-EME1 results in replication stress, ultimately leading to G2/M cell cycle arrest (Laguette et al., 2014).
Viral Protein Regulatory and SLX4 Complex Regulation
Vpr and the Fanconi Anemia Pathway
SLX4, also as known as FANCP, together with the fifteen additional identified FA or FA-like proteins, is involved in the FA DNA repair pathway. This pathway has been extensively described in reviews (Garner and Smogorzewska, 2011; Su and Huang, 2011; Constantinou, 2012). Briefly, FANCM binds chromatin at damage sites and recruits the E3 ubiquitin ligase activity-containing FA core complex (Kim et al., 2008). The FA core complex monoubiquitinates FANCD2–FANCI and stabilizes them at sites of damage (Garcia-Higuera et al., 2001). FANCD2–FANCI subsequently activate DNA repair proteins, including the SLX4 complex (Yamamoto et al., 2011). The latter is involved in the repair of double strand breaks, interstrand cross-links (ICL), and collapsed/damaged replication forks by homologous recombination (HR; Munoz et al., 2009; Svendsen et al., 2009; Kim et al., 2011; Stoepker et al., 2011). HR allows accurate repair by using the sister chromatid as a template and leads to the formation of four-way DNA structures, Holliday junctions (HJ), that must be removed prior to chromosome segregation. It is important to note that in somatic cells, the favored pathway to remove HJ relies on non-endonucleolytic dissolution by Bloom (BLM)-related helicases, a process that prevents sister chromatid exchanges (Wu and Hickson, 2003). Sister chromatid exchanges are particularly disfavored in somatic cells because they may engender loss of heterozygosity thereby predisposing cells to cancer (Matos et al., 2011, 2013; Gallo-Fernandez et al., 2012; Dehe et al., 2013; Saugar et al., 2013; Szakal and Branzei, 2013). However, in certain cases, for example in the absence of BLM or when the levels of damage incurred are above those that can be salvaged through dissolution, structure-specific endonucleases activities associated with the SLX4 complex may be mobilized (Schwartz and Heyer, 2011; Garner et al., 2013). In vitro studies have shown that SLX4–SLX1 (Fekairi et al., 2009; Munoz et al., 2009; Svendsen et al., 2009) and MUS81-EME1 have 5′ and 3′ endonuclease activities, respectively (Boddy et al., 2001; Doe et al., 2002; Ciccia et al., 2003; Gaillard et al., 2003). However, SLX4-associated resolvase activity requires interaction of SLX1 and MUS81-EME1 with the SLX4 scaffold (Castor et al., 2013; Garner et al., 2013; Wyatt et al., 2013).
Because activation of MUS81-EME1 during S phase may cause pathological processing of healthy replication forks (Dehe et al., 2013; Matos et al., 2013; Saugar et al., 2013) and replication stress (Blais et al., 2004; Matos et al., 2013; Szakal and Branzei, 2013), under physiological conditions and when DNA damage is incurred, acquisition of MUS81-EME1 endonuclease activity is under tight regulatory circuits. Those ensure that MUS81-EME1 activity is mostly confined to late G2-early M, when bulk DNA synthesis has been completed (Dehe et al., 2013; Saugar et al., 2013). The molecular mechanism underlying Mus81-Mms4EME1 regulation has been extensively studied, and is achieved through phosphorylation of EMEMms4 by PLK1Cdc5 in budding yeast and in mammalian cells (Matos et al., 2011; Gallo-Fernandez et al., 2012; Saugar et al., 2013) or Cdc2CDK1 in fission yeast (Dehe et al., 2013). Importantly, work in mammalian cells has recently shown that MUS81-EME1 is regulated through phosphorylation of EME1 within the SLX4 complex (Castor et al., 2013; Garner et al., 2013; Wyatt et al., 2013).
Importantly, interaction of Vpr with the SLX4 scaffold protein induces recruitment of VPRBP and kinase-active PLK1, thereby activating the MUS81-EME1 endonuclease module independently of the cell cycle stage (Figure 1). This results in replication stress as visualized by accumulation of FANCD2 on sub-regions of the chromatin that likely mark the sites of abnormal processing of replication intermediates (Naim and Rosselli, 2009). Thus, as supported by previous work, Vpr causes cell cycle arrest through a S phase-dependent mechanism (Li et al., 2010), which is congruent with activation of the ATR pathway in Vpr expressing cells (Roshal et al., 2003; Lobrich and Jeggo, 2007). Interestingly, SLX4 has also been identified as an ATR substrate (Matsuoka et al., 2007; Mu et al., 2007) and in yeast, phosphorylation of Eme1 requires Rad53ATR activation (Dehe et al., 2013). Thus, aberrant processing of stalled replication forks by Vpr-activated SLX4-associated MUS81-EME1 would cause replication stress, ATR-CHK1 pathway activation, resulting in inhibition of CDC25C. This signaling cascade will ultimately lead to inability of CDC25C to activate CCNB1:CDK1 and thus result in G2/M arrest (Figure 1).
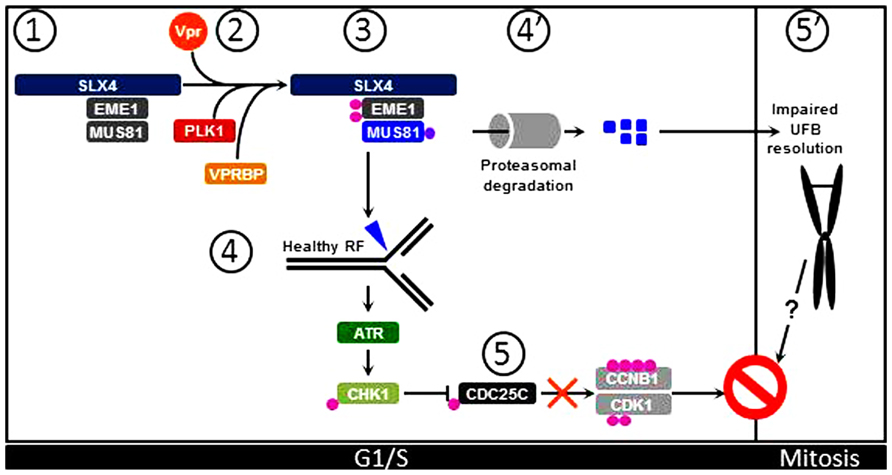
FIGURE 1. Vpr induces G2/M arrest through activation of the SLX4 complex. (1) Under physiological conditions, inactive MUS81-EME1 interact with the SLX4 scaffold. (2) Upon Vpr expression, PLK1 and VPRBP are recruited to SLX4. (3) PLK1 phosphorylates EME1 while VPRBP causes ubiquitination of MUS81. (4) These posttranslational modifications contribute to activation of SLX4-bound MUS81-EME1 that can process healthy replication forks (RF) in cycling cells. (5) This leads to activation of the ATR signaling pathway and subsequent activation of CHK1. Activated CHK1 provokes inhibitory phosphorylation of CDC25C, leading to inhibition of CCNB1:CDK1 and cell cycle arrest at the G2/M transition. (4′) In addition, ubiquitinated MUS81 molecules are degraded by the proteasome machinery, leading to decreased steady-state levels of MUS81. (5′) Consequently, UFBs are not processed and persist in Mitosis. This possibly contributes to overall G2/M arrest in Vpr expressing cells.
Vpr and Genomic Instability
The MUS81-EME1 endonuclease module plays an important role in the removal of ultrafine DNA bridges (UFBs). These DNA structures that arise from regions of the genome that replicate at slower rates, such as centromeres and common fragile sited (CFS), are formed during the S phase and can be visualized after chromosome condensation in mitotic cells. They form bridges between sister chromatids that must be removed prior to chromosome segregation. Absence of MUS81-EME1 results in non-processing of UFBs and thus leads to CFS-associated chromosomal instability and mitotic catastrophe (Chan et al., 2009; Chan and Hickson, 2011; Wechsler et al., 2011; Naim et al., 2013; Ying et al., 2013). Accumulation of UFBs therefore causes cell cycle arrest at the G1/S transition. Intriguingly, Vpr targets MUS81 for ubiquitination by VPRBP, leading to decreased levels of MUS81 prior to G2/M arrest (Laguette et al., 2014). Since a stark increase of FANCD2 twin foci that mark the edges of UFBs (Chan et al., 2009) occurs in the presence of Vpr, this indicates that, although not complete, decreased MUS81 levels in Vpr-expressing cells may be sufficient to impair UFBs untangling prior to mitosis (Chan et al., 2009; Wechsler et al., 2011; Naim et al., 2013; Ying et al., 2013). However, Vpr-associated replication stress prevents completion of G2. This likely prevents the occurrence of mitotic catastrophe. While it is possible that additional Vpr-associated functions may prevent cells from exiting mitosis, one may also speculate that steric hindrance imposed by UFBs tying together sister chromatids may also contribute to the extent of G2/M arrest witnessed in Vpr-expressing cells (Figure 1). Thus, the complete sequence of events leading from SLX4 complex premature activation to cell cycle arrest by Vpr requires further investigations.
Viral Protein Regulatory and Innate Immunity
One interesting feature of Vpr is that disruption of the corresponding open reading frame (ORF) results in inefficient viral spread ex vivo particularly in primary macrophages (Connor et al., 1995) while its most studied molecular function is to halt cell cycle progression. This conundrum has puzzled the HIV field for several years but little was understood about how these two observations can be reconciled until recent work (Laguette et al., 2014).
Infection with an HIV-1 molecular clone harboring a deletion of the Vpr ORF causes an increase of IFN production as compared to infection with wild type HIV-1 (Okumura et al., 2008; Doehle et al., 2009; Laguette et al., 2014). This HIV-induced IFN production is augmented following SLX4 complex subunits (SLX4, VPRBP, and MUS81) knock-down, suggesting that the presence of the SLX4 complex is required for inhibition of HIV-dependent type 1 IFN production. Furthermore, the SLX4 complex binds HIV-1-derived reverse transcribed DNA in presence of Vpr, suggesting that Vpr is required for this interaction. In addition, in the absence of SLX4, there is an increase of HIV DNA in infected cells. This further suggests that the SLX4 complex is required to degrade excess HIV-derived nucleic acids susceptible of triggering innate immune responses. While the MUS81-EME1 endonuclease module appears to be required for this process one cannot exclude contribution of additional SLX4-bound endonucleases. Indeed, SLX4–SLX1 interaction is required for SLX4 complex associated resolvase activity and SLX1 expression is required for Vpr-mediated cell cycle arrest (Laguette et al., 2014). Overall, similar to TREX1 and RNaseH2, the SLX4 complex would prevent sensing of excess nucleic acids derived from HIV reverse transcription (Figure 2). Importantly, RNaseH2 and TREX1 preferentially degrade DNA within DNA:RNA hybrids (Haruki et al., 2002) and ssDNA substrates, respectively (Mazur and Perrino, 2001) while SLX4-bound MUS81-EME1 presumably target dsDNA structures (Fadden et al., 2013). While all these nucleic acid species arise in the course of HIV reverse transcription, the relative contribution of these nucleases remains to be evaluated. Furthermore, SLX4-mediated nucleic acids processing would lead to the generation of DNA fragments that likely require further processing to avoid recognition. This leaves several questions open amongst which are: what is the sensor triggered in the absence of SLX4 and what are the nucleases mobilized to clear SLX4-processed DNA fragments? These processes may rely on previously identified key players in viral life cycles. However, one must bear in mind that the SLX4 scaffold can bind to additional proteins involved in DNA metabolism and is as such involved in several additional pathways, including Telomere maintenance and DNA mismatch repair (Svendsen et al., 2009). Whether these may intervene in the degradation of virus derived nucleic acids is also to be explored.

FIGURE 2. Vpr-induced SLX4 complex activation promotes escape from innate immune sensing. (1) Premature delivery of viral genomes in the cytoplasm of host cells may lead to recognition by nucleic acid sensors. (2) Editing by APOBEC3G and dNTP hydrolysis by SAMHD1 induce viral DNA instability and impair reverse transcription. Of note, a nuclease activity has been described for SAMHD1 which may target viral nucleic acids. (3) APOBEC3G and SAMHD1 contribute to generate abortive RT by-products which are taken care of by cellular exonucleases: RNaseH2 degrades DNA:RNA hybrids while TREX1 degrades ssDNA preferentially. (3′) Abortive nucleic acid intermediates may also be directly degraded by RNaseH2 and TREX1. (4) The SLX4 structure specific endonuclease complex is activated by Vpr and likely cleaves dsDNA. Whether these dsDNA fragments are further degraded by cellular exonucleases or may be recognized by nucleic acid sensors remains questioned. (1′) When uncoating is correctly orchestrated, viral nucleic acids are protected from mechanisms described in 1–4, and ensures delivery of viral genomes in the nucleus.
As previously mentioned, FA is associated with high production of pro-inflammatory cytokine in patients. This can be recapitulated in vitro in SLX4-deficient patient cells (Kim et al., 2011) and also in mouse embryonic fibroblasts knocked-out of MUS81 (McPherson et al., 2004) through the activation of NF-κB pathway (Laguette et al., 2014). This leads to the establishment of an antiviral state that likely accounts for the inability of these cells to support efficient HIV replication. While this bears similarities with what is observed in TREX1 deficiency, the endogenous trigger for SLX4 complex activation remains unknown. Those may include, nucleic acids derived from processing of aberrant replication intermediates or endogenous retroviruses. Elucidating the trigger for spontaneous upregulation of pro-inflammatory cytokines in FA is likely to be the next horizon in the field. This would possibly provide insight into the molecular basis of FA-associated chronic inflammation. In addition, Vpr has been shown to modulate immune responses at additional levels. This includes impairment of DC/macrophage maturation, disruption of natural killer T cells effector functions, increased apoptosis of cytotoxic T cells and disruption of T cell activation pathways (reviewed in Ayinde et al., 2010). Thus, Vpr compromises the establishment of adaptive immune responses. How inhibition of pro-inflammatory cytokines by Vpr through activation of SLX4 complex contributes to this process remains to be explored.
DNA Damage Response and Innate Immunity
Initiation of the DNA damage response usually requires recognition of abnormal nucleic acid species in the nucleus and the triggering of a signaling cascade that orchestrates repair. This process bears similarities with what is witnessed when virus-derived nucleic acids are delivered into host cells. Those are recognized by sensors that trigger a signaling cascade aiming at clearing the infection. It has been widely speculated that the nucleic acid-based repository of the information required for de novo virus production is difficultly modified by viruses to promote escape; it constitutes a prime target for cellular sensors. Recent work has placed key players of the DNA damage response on the front line of pathogen recognition. For example DNA-PK has been shown to act as a PRR for DNA and RNA viruses (Zhang et al., 2011; Ferguson et al., 2012). Indeed, DNA-PK is involved in DNA damage response, more particularly in the repair of double-strand breaks and these functions are related to its nuclear localization (for review Davis and Chen, 2013). However, this complex is also found in the cytoplasm where it can bind nucleic acids and activates the production of IFN. This highlights how overlapping mechanisms have evolved for the recognition of pathological nucleic acid species. Furthermore, inflammation impacts every step of tumorigenesis, from initiation to metastatic progression. Tumor-promoting inflammation may either result from environmental factors, as clearly identified in the case of exposure to asbestos for example, or from chronic viral infections and attempts of the immune system to eliminate those. This results in a feed-forward regulatory loop that favors chronic production of pro-inflammatory cytokines, supporting tumorigenesis. Indeed, it is recognized that subclinical, often undetectable, inflammation increases cancer risk (reviewed in Grivennikov et al., 2010). Thus, persistent DNA damage or inability to repair broken DNA may lead to tumor-promoting chronic inflammation (Zheng et al., 2007; Rodier et al., 2009).
Cellular mechanisms exist that serve to avoid the accumulation of pathological nucleic acid species susceptible of triggering innate immune responses. These include the previously mentioned TREX1 exonuclease and the SLX4 complex. Intriguingly, TREX1 that was initially described to be involved in DNA base excision repair (Mazur and Perrino, 2001), also degrades ssDNA derived from aberrant replication intermediates and thus similar to the SLX4 complex is involved in DNA damage response (Yang et al., 2007; Gehrke et al., 2013). Thus, like in TREX1 deficiency, absence of the SLX4 complex may lead to accumulation of pathological nucleic acids in the cytoplasm. Recognition of those by a yet to be identified sensor, activates the immune system.
Importantly, HIV is not the sole virus affected by cellular enzymes involved in DNA metabolism. Indeed, several DNA viruses can be targeted by cellular factors involved in DNA damage response. For example, the genome of the Adenovirus or Herpes Simplex Virus type 1 (HSV-1) can be targeted by protein complexes that control the non-homologous end-joining DNA repair pathway [reviewed in Weitzman et al. (2010)]. These viruses have evolved potent ways of counteracting these proteins that operate as potential restriction factors. This can be compared to what is witnessed during HIV infection in the presence of SAMHD1. Indeed, this HIV restriction factor has been shown, in addition to depleting the dNTP pool (Lahouassa et al., 2012), to have in vitro nuclease activity (Beloglazova et al., 2013).
While there is accumulating evidence that proteins involved in DNA repair are involved in viral life cycles, recent work has also shown that proteins previously identified as counteractors of HIV infection are in fact involved in the DNA damage response. This is the case for APOBEC proteins where mutation patterns were found in human cancers (Leonard et al., 2013; Roberts et al., 2013) and SAMHD1. In the case of SAMHD1, recent work has highlighted that SAMHD1 may qualify as a tumor suppressor gene, and thus play roles in DNA damage response, through its ability to regulate the dNTP pool (Clifford et al., 2014; Kretschmer et al., 2014), the levels of these being important for genome stability (Mathews, 2006). Although this SAMHD1 function has been essentially described to be involved in HIV restriction, it may also be related to increased IFN production in AGS. Moreover, RNaseH2, as previously mentioned, is also involved in AGS and degrades RNA in DNA:RNA hybrids and thus may also prevent chronic inflammation (Rice et al., 2007). Importantly, both SAMHD1 and APOBEC3G have been shown to control endogenous retrotransposition (Esnault et al., 2006; Zhao et al., 2013). Since absence of SAMHD1 also correlates with increased pro-inflammatory cytokine production, one may also speculate about the trigger for this response and whether there is a correlation between inefficient DNA repair or endogenous retroelement retrotransposition in the absence of SAMHD1 and chronic inflammation. Similarly, the origin of chronic inflammation in SLX4-deficiency remains to be identified and may include residual nucleic acids resulting from processing of aberrant replication intermediates or endogenous retroelements.
Conclusion
Although host cells oppose numerous blocks to HIV replication, several mechanisms have evolved to counteract those. While defective viruses may elicit pro-inflammatory responses, viruses that establish productive infections remain mostly protected from cellular defenses. Viral accessory proteins are specialized in mediating this escape, in part through counteraction of cellular restriction factors. Interestingly, restriction factors have also been recently reported to play a role in detection of viral infections (PRR). Thus, viral accessory proteins, though their degradation simultaneously achieve escape from recognition and overthrowing of mediators of the antiviral responses. This complex array of interactions between innate immune responses and viral replication is still poorly understood. While new insight into the role of the DNA damage response machinery in this process may add a further layer of complexity, this may also provide with an additional way to identify sensors that detect incoming viruses and escape mechanisms.
In addition to the described role of Vpr in arresting the cell cycle and promoting escape from innate immune sensing, this HIV accessory protein has been shown to play several additional roles in HIV life cycle. Indeed, Vpr contributes to fidelity of reverse transcription (Stark and Hay, 1998), nuclear transport of the pre-integration complex (Heinzinger et al., 1994). Vpr also promotes the transactivation of LTR promoter (Felzien et al., 1998), and induction of apoptosis (Stewart et al., 1997). Do these also result from Vpr-induced activation of the SLX4 complex or do they rely on additional interactions established by Vpr? Importantly, in addition to interacting with structure-specific endonucleases, the SLX4 molecular toolkit also recruits MSH2–MSH3 and TRF2–RAP1 (Svendsen et al., 2009) and is involved in additional cellular functions, including Telomere maintenance, which may be altered upon binding to Vpr. For example, since SLX4 inhibits over-lengthening of telomeric ends, Vpr-induced activation of the SLX4 complex may lead to Telomere shortening and cell death. Whether this is related to increased apoptosis witnessed in Vpr-expressing cells is yet to be explored.
The discovery of the SLX4 complex as being involved in inhibition of pro-inflammatory responses opens new avenues in the understanding of the interplay between innate immune responses and HIV infection. This work also opens new perspectives in the understanding of the molecular mechanism underlying cancer related chronic inflammation. Based on the fact that pro-inflammatory cytokine production is witnessed in all cancers, one may anticipate that additional DNA damage repair mechanisms may be involved in pathogen recognition and inhibition of spontaneous pro-inflammatory cytokine production.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ayinde, D., Maudet, C., Transy, C., and Margottin-Goguet, F. (2010). Limelight on two HIV/SIV accessory proteins in macrophage infection: is Vpx overshadowing Vpr? Retrovirology 7, 35. doi: 10.1186/1742-4690-7-35
Beignon, A. S., McKenna, K., Skoberne, M., Manches, O., Dasilva, I., Kavanagh, D. G., et al. (2005). Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Invest. 115, 3265–3275. doi: 10.1172/JCI26032
Beloglazova, N., Flick, R., Tchigvintsev, A., Brown, G., Popovic, A., Nocek, B., et al. (2013). Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J. Biol. Chem. 288, 8101–8110. doi: 10.1074/jbc.M112.431148
Belzile, J. P., Duisit, G., Rougeau, N., Mercier, J., Finzi, A., and Cohen, E. A. (2007). HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 3:e85. doi: 10.1371/journal.ppat.0030085
Blais, V., Gao, H., Elwell, C. A., Boddy, M. N., Gaillard, P. H., Russell, P., et al. (2004). RNA interference inhibition of Mus81 reduces mitotic recombination in human cells. Mol. Biol. Cell 15, 552–562. doi: 10.1091/mbc.E03-08-0580
Boddy, M. N., Gaillard, P. H., McDonald, W. H., Shanahan, P., Yates, J. R. III, and Russell, P. (2001). Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107, 537–548. doi: 10.1016/S0092-8674(01)00536-0
Briot, D., Mace-Aime, G., Subra, F., and Rosselli, F. (2008). Aberrant activation of stress-response pathways leads to TNF-alpha oversecretion in Fanconi anemia. Blood 111, 1913–1923. doi: 10.1182/blood-2007-07-099218
Broz, P., and Monack, D. M. (2013). Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 13, 551–565. doi: 10.1038/nri3479
Castor, D., Nair, N., Declais, A. C., Lachaud, C., Toth, R., Macartney, T. J., et al. (2013). Cooperative control of holliday junction resolution and DNA repair by the SLX1 and MUS81-EME1 nucleases. Mol. Cell 52, 221–233. doi: 10.1016/j.molcel.2013.08.036
Chan, K. L., and Hickson, I. D. (2011). New insights into the formation and resolution of ultra-fine anaphase bridges. Semin. Cell Dev. Biol. 22, 906–912. doi: 10.1016/j.semcdb.2011.07.001
Chan, K. L., Palmai-Pallag, T., Ying, S., and Hickson, I. D. (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 11, 753–760. doi: 10.1038/ncb1882
Ciccia, A., Constantinou, A., and West, S. C. (2003). Identification and characterization of the human mus81-eme1 endonuclease. J. Biol. Chem. 278, 25172–25178. doi: 10.1074/jbc.M302882200
Clifford, R., Louis, T., Robbe, P., Ackroyd, S., Burns, A., Timbs, A. T., et al. (2014). SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood 123, 1021–1031. doi: 10.1182/blood-2013-04-490847
Connor, R. I., Chen, B. K., Choe, S., and Landau, N. R. (1995). Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206, 935–944. doi: 10.1006/viro.1995.1016
Constantinou, A. (2012). Rescue of replication failure by Fanconi anaemia proteins. Chromosoma 121, 21–36. doi: 10.1007/s00412-011-0349-2
Davis, A. J., and Chen, D. J. (2013). DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2, 130–143. doi: 10.3978/j.issn.2218-676X.2013.04.02
Davy, C., and Doorbar, J. (2007). G2/M cell cycle arrest in the life cycle of viruses. Virology 368, 219–226. doi: 10.1016/j.virol.2007.05.043
DeHart, J. L., Zimmerman, E. S., Ardon, O., Monteiro-Filho, C. M., Arganaraz, E. R., and Planelles, V. (2007). HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol. J. 4, 57. doi: 10.1186/1743-422X-4-57
Dehe, P. M., Coulon, S., Scaglione, S., Shanahan, P., Takedachi, A., Wohlschlegel, J. A., et al. (2013). Regulation of Mus81-Eme1 Holliday junction resolvase in response to DNA damage. Nat. Struct. Mol. Biol. 20, 598–603. doi: 10.1038/nsmb.2550nsmb.2550
Di Marzio, P., Choe, S., Ebright, M., Knoblauch, R., and Landau, N. R. (1995). Mutational analysis of cell cycle arrest, nuclear localization and virion packaging of human immunodeficiency virus type 1 Vpr. J. Virol. 69, 7909–7916.
Doe, C. L., Ahn, J. S., Dixon, J., and Whitby, M. C. (2002). Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J. Biol. Chem. 277, 32753–32759. doi: 10.1074/jbc.M202120200
Doehle, B. P., Hladik, F., McNevin, J. P., McElrath, M. J., and Gale, M. (2009). Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J. Virol. 83, 10395–10405. doi: 10.1128/JVI.00849-09
Dufour, C., Corcione, A., Svahn, J., Haupt, R., Poggi, V., Beka’ssy, A. N., et al. (2003). TNF-alpha and IFN-gamma are overexpressed in the bone marrow of Fanconi anemia patients and TNF-alpha suppresses erythropoiesis in vitro. Blood 102, 2053–2059. doi: 10.1182/blood-2003-01-0114
Esnault, C., Millet, J., Schwartz, O., and Heidmann, T. (2006). Dual inhibitory effects of APOBEC family proteins on retrotransposition of mammalian endogenous retroviruses. Nucleic Acids Res. 34, 1522–1531. doi: 10.1093/nar/gkl054
Fadden, A. J., Schalbetter, S., Bowles, M., Harris, R., Lally, J., Carr, A. M., et al. (2013). A winged helix domain in human MUS81 binds DNA and modulates the endonuclease activity of MUS81 complexes. Nucleic Acids Res. 41, 9741–9752. doi: 10.1093/nar/gkt760
Fekairi, S., Scaglione, S., Chahwan, C., Taylor, E. R., Tissier, A., Coulon, S., et al. (2009). Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138, 78–89. doi: 10.1016/j.cell.2009.06.029
Felzien, L. K., Woffendin, C., Hottiger, M. O., Subbramanian, R. A., Cohen, E. A., and Nabel, G. J. (1998). HIV transcriptional activation by the accessory protein, VPR, is mediated by the p300 co-activator. Proc. Natl. Acad. Sci. U.S.A. 95, 5281–5286. doi: 10.1073/pnas.95.9.5281
Ferguson, B. J., Mansur, D. S., Peters, N. E., Ren, H., and Smith, G. L. (2012). DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 1, e00047. doi: 10.7554/eLife.00047
Gaillard, P. H., Noguchi, E., Shanahan, P., and Russell, P. (2003). The endogenous Mus81-Eme1 complex resolves Holliday junctions by a nick and counternick mechanism. Mol. Cell 12, 747–759. doi: 10.1016/S1097-2765(03)00342-3
Gallo-Fernandez, M., Saugar, I., Ortiz-Bazan, M. A., Vazquez, M. V., and Tercero, J. A. (2012). Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res. 40, 8325–8335. doi: 10.1093/nar/gks599
Gao, D., Wu, J., Wu, Y. T., Du, F., Aroh, C., Yan, N., et al. (2013). Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906. doi: 10.1126/science.1240933
Garcia-Higuera, I., Taniguchi, T., Ganesan, S., Meyn, M. S., Timmers, C., Hejna, J., et al. (2001). Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell 7, 249–262. doi: 10.1016/S1097-2765(01)00173-3
Garner, E., Kim, Y., Lach, F. P., Kottemann, M. C., and Smogorzewska, A. (2013). Human GEN1 and the SLX4-associated nucleases MUS81 and SLX1 are essential for the resolution of replication-induced Holliday junctions. Cell Rep. 5, 207–215. doi: 10.1016/j.celrep.2013.08.041
Garner, E., and Smogorzewska, A. (2011). Ubiquitylation and the Fanconi anemia pathway. FEBS Lett. 585, 2853–2860. doi: 10.1016/j.febslet.2011.04.078
Gatza, M. L., Watt, J. C., and Marriott, S. J. (2003). Cellular transformation by the HTLV-I Tax protein, a jack-of-all-trades. Oncogene 22, 5141–5149. doi: 10.1038/sj.onc.1206549
Gehrke, N., Mertens, C., Zillinger, T., Wenzel, J., Bald, T., Zahn, S., et al. (2013). Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 39, 482–495. doi: 10.1016/j.immuni.2013.08.004
Genovesio, A., Kwon, Y. J., Windisch, M. P., Kim, N. Y., Choi, S. Y., Kim, H. C., et al. (2011). Automated genome-wide visual profiling of cellular proteins involved in HIV infection. J. Biomol. Screen. 16, 945–958. doi: 10.1177/1087057111415521
Grivennikov, S. I., Greten, F. R., and Karin, M. (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. doi: 10.1016/j.cell.2010.01.025
Groschel, B., and Bushman, F. (2005). Cell cycle arrest in G2/M promotes early steps of infection by human immunodeficiency virus. J. Virol. 79, 5695–5704. doi: 10.1128/JVI.79.9.5695-5704.2005
Haruki, M., Tsunaka, Y., Morikawa, M., and Kanaya, S. (2002). Cleavage of a DNA-RNA-DNA/DNA chimeric substrate containing a single ribonucleotide at the DNA-RNA junction with prokaryotic RNases HII. FEBS Lett. 531, 204–208. doi: 10.1016/S0014-5793(02)03503-2
He, J., Choe, S., Walker, R., Di Marzio, P., Morgan, D. O., and Landau, N. R. (1995). Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69, 6705–6711.
Heinzinger, N. K., Bukinsky, M. I., Haggerty, S. A., Ragland, A. M., Kewalramani, V., Lee, M. A., et al. (1994). The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc. Natl. Acad. Sci. U.S.A. 91, 7311–7315. doi: 10.1073/pnas.91.15.7311
Howley, P. M., and Livingston, D. M. (2009). Small DNA tumor viruses: large contributors to biomedical sciences. Virology 384, 256–259. doi: 10.1016/j.virol.2008.12.006
Hrecka, K., Gierszewska, M., Srivastava, S., Kozaczkiewicz, L., Swanson, S. K., Florens, L., et al. (2007). Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. U.S.A. 104, 11778–11783. doi: 10.1073/pnas.0702102104
Jowett, J. B., Planelles, V., Poon, B., Shah, N. P., Chen, M. L., and Chen, I. S. (1995). The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J. Virol. 69, 6304–6313.
Kawai, T., and Akira, S. (2006). Innate immune recognition of viral infection. Nat. Immunol. 7, 131–137. doi: 10.1038/ni1303
Kim, C. M., Koike, K., Saito, I., Miyamura, T., and Jay, G. (1991). HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 351, 317–320. doi: 10.1038/351317a0
Kim, J. M., Kee, Y., Gurtan, A., and D’Andrea, A. D. (2008). Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood 111, 5215–5222. doi: 10.1182/blood-2007-09-113092
Kim, Y., Lach, F. P., Desetty, R., Hanenberg, H., Auerbach, A. D., and Smogorzewska, A. (2011). Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 43, 142–146. doi: 10.1038/ng.750
Kretschmer, S., Wolf, C., Konig, N., Staroske, W., Guck, J., Hausler, M., et al. (2014). SAMHD1 prevents autoimmunity by maintaining genome stability. Ann. Rheum. Dis. doi: 10.1136/annrheumdis-2013-204845
Kutok, J. L., and Wang, F. (2006). Spectrum of Epstein-Barr virus-associated diseases. Annu. Rev. Pathol. 1, 375–404. doi: 10.1146/annurev.pathol.1.110304.100209
Laguette, N., Bregnard, C., Hue, P., Basbous, J., Yatim, A., Larroque, M., et al. (2014). Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 156, 134–145. doi: 10.1016/j.cell.2013.12.011
Lahouassa, H., Daddacha, W., Hofmann, H., Ayinde, D., Logue, E. C., Dragin, L., et al. (2012). SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 13, 223–228. doi: 10.1038/ni.2236
Le Rouzic, E., Belaidouni, N., Estrabaud, E., Morel, M., Rain, J. C., Transy, C., et al. (2007). HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle 6, 182–188. doi: 10.4161/cc.6.2.3732
Lebon, P., Badoual, J., Ponsot, G., Goutieres, F., Hemeury-Cukier, F., and Aicardi, J. (1988). Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J. Neurol. Sci. 84, 201–208. doi: 10.1016/0022-510X(88)90125-6
Lee, S. H., and Rozee, K. R. (1970). Variation of interferon production during the cell cycle. Appl. Microbiol. 20, 11–15.
Leonard, B., Hart, S. N., Burns, M. B., Carpenter, M. A., Temiz, N. A., Rathore, A., et al. (2013). APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 73, 7222–7231. doi: 10.1158/0008-5472.CAN-13-1753
Li, G., Park, H. U., Liang, D., and Zhao, R. Y. (2010). Cell cycle G2/M arrest through an S phase-dependent mechanism by HIV-1 viral protein R. Retrovirology 7, 59. doi: 10.1186/1742-4690-7-59
Lobrich, M., and Jeggo, P. A. (2007). The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat. Rev. Cancer 7, 861–869. doi: 10.1038/nrc2248
Lopez-Girona, A., Furnari, B., Mondesert, O., and Russell, P. (1999). Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature 397, 172–175. doi: 10.1038/16488
Malim, M. H., and Bieniasz, P. D. (2012). HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2, a006940. doi: 10.1101/cshperspect.a006940
Mathews, C. K. (2006). DNA precursor metabolism and genomic stability. FASEB J. 20, 1300–1314. doi: 10.1096/fj.06-5730rev
Matos, J., Blanco, M. G., Maslen, S., Skehel, J. M., and West, S. C. (2011). Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147, 158–172. doi: 10.1016/j.cell.2011.08.032
Matos, J., Blanco, M. G., and West, S. C. (2013). Cell-cycle kinases coordinate the resolution of recombination intermediates with chromosome segregation. Cell Rep. 4, 76–86. doi: 10.1016/j.celrep.2013.05.039
Matsuoka, S., Ballif, B. A., Smogorzewska, A., Mcdonald, E. R. III, Hurov, K. E., Luo, J., et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166. doi: 10.1126/science.1140321
Mazur, D. J., and Perrino, F. W. (2001). Structure and expression of the TREX1 and TREX2 3’ –> 5’ exonuclease genes. J. Biol. Chem. 276, 14718–14727. doi: 10.1074/jbc.M010051200
McPherson, J. P., Lemmers, B., Chahwan, R., Pamidi, A., Migon, E., Matysiak-Zablocki, E., et al. (2004). Involvement of mammalian Mus81 in genome integrity and tumor suppression. Science 304, 1822–1826. doi: 10.1126/science.1094557304/5678/1822
Monroe, K. M., Yang, Z., Johnson, J. R., Geng, X., Doitsh, G., Krogan, N. J., et al. (2013). IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343, 428–432. doi: 10.1126/science.1243640
Mu, J. J., Wang, Y., Luo, H., Leng, M., Zhang, J., Yang, T., et al. (2007). A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J. Biol. Chem. 282, 17330–17334. doi: 10.1074/jbc.C700079200
Munoz, I. M., Hain, K., Declais, A. C., Gardiner, M., Toh, G. W., Sanchez-Pulido, L., et al. (2009). Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 35, 116–127. doi: 10.1016/j.molcel.2009.06.020
Naim, V., and Rosselli, F. (2009). The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 11, 761–768. doi: 10.1038/ncb1883ncb1883
Naim, V., Wilhelm, T., Debatisse, M., and Rosselli, F. (2013). ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 15, 1008–1015. doi: 10.1038/ncb2793ncb2793
Okumura, A., Alce, T., Lubyova, B., Ezelle, H., Strebel, K., and Pitha, P. M. (2008). HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology 373, 85–97. doi: 10.1016/j.virol.2007.10.042
Poon, B., Jowett, J. B., Stewart, S. A., Armstrong, R. W., Rishton, G. M., and Chen, I. S. (1997). Human immunodeficiency virus type 1 vpr gene induces phenotypic effects similar to those of the DNA alkylating agent, nitrogen mustard. J. Virol. 71, 3961–3971.
Rathbun, R. K., Faulkner, G. R., Ostroski, M. H., Christianson, T. A., Hughes, G., Jones, G., et al. (1997). Inactivation of the Fanconi anemia group C gene augments interferon-gamma-induced apoptotic responses in hematopoietic cells. Blood 90, 974–985.
Re, F., Braaten, D., Franke, E. K., and Luban, J. (1995). Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J. Virol. 69, 6859–6864.
Rice, G., Patrick, T., Parmar, R., Taylor, C. F., Aeby, A., Aicardi, J., et al. (2007). Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 81, 713–725. doi: 10.1086/521373
Roberts, S. A., Lawrence, M. S., Klimczak, L. J., Grimm, S. A., Fargo, D., Stojanov, P., et al. (2013). An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 45, 970–976. doi: 10.1038/ng.2702ng.2702
Rodier, F., Coppe, J. P., Patil, C. K., Hoeijmakers, W. A., Munoz, D. P., Raza, S. R., et al. (2009). Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979. doi: 10.1038/ncb1909ncb1909
Rogel, M. E., Wu, L. I., and Emerman, M. (1995). The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J. Virol. 69, 882–888.
Roshal, M., Kim, B., Zhu, Y., Nghiem, P., and Planelles, V. (2003). Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J. Biol. Chem. 278, 25879–25886. doi: 10.1074/jbc.M303948200M303948200
Sanchez, Y., Wong, C., Thoma, R. S., Richman, R., Wu, Z., Piwnica-Worms, H., et al. (1997). Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277, 1497–1501. doi: 10.1126/science.277.5331.1497
Sasaki, M. S., and Tonomura, A. (1973). A high susceptibility of Fanconi’s anemia to chromosome breakage by DNA cross-linking agents. Cancer Res. 33, 1829–1836.
Saugar, I., Vazquez, M. V., Gallo-Fernandez, M., Ortiz-Bazan, M. A., Segurado, M., Calzada, A., et al. (2013). Temporal regulation of the Mus81-Mms4 endonuclease ensures cell survival under conditions of DNA damage. Nucleic Acids Res. 41, 8943–8958. doi: 10.1093/nar/gkt645
Schwartz, E. K., and Heyer, W. D. (2011). Processing of joint molecule intermediates by structure-selective endonucleases during homologous recombination in eukaryotes. Chromosoma 120, 109–127. doi: 10.1007/s00412-010-0304-7
Shostak, L. D., Ludlow, J., Fisk, J., Pursell, S., Rimel, B. J., Nguyen, D., et al. (1999). Roles of p53 and caspases in the induction of cell cycle arrest and apoptosis by HIV-1 Vpr. Exp. Cell Res. 251, 156–165. doi: 10.1006/excr.1999.4568
Sirbu, B. M., and Cortez, D. (2013). DNA damage response: three levels of DNA repair regulation. Cold Spring Harb. Perspect. Biol. 5, a012724. doi: 10.1101/cshperspect.a012724a012724
Smith, J., Tho, L. M., Xu, N., and Gillespie, D. A. (2010). The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108, 73–112. doi: 10.1016/B978-0-12-380888-2.00003-0
Stark, G. R., and Taylor, W. R. (2006). Control of the G2/M transition. Mol. Biotechnol. 32, 227–248. doi: 10.1385/MB:32:3:227
Stark, L. A., and Hay, R. T. (1998). Human immunodeficiency virus type 1 (HIV-1) viral protein R (Vpr) interacts with Lys-tRNA synthetase: implications for priming of HIV-1 reverse transcription. J. Virol. 72, 3037–3044.
Stetson, D. B., and Medzhitov, R. (2006). Antiviral defense: interferons and beyond. J. Exp. Med. 203, 1837–1841. doi: 10.1084/jem.20061377
Stewart, S. A., Poon, B., Jowett, J. B., and Chen, I. S. (1997). Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol. 71, 5579–5592.
Stoepker, C., Hain, K., Schuster, B., Hilhorst-Hofstee, Y., Rooimans, M. A., Steltenpool, J., et al. (2011). SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 43, 138–141. doi: 10.1038/ng.751
Su, X., and Huang, J. (2011). The Fanconi anemia pathway and DNA interstrand cross-link repair. Protein Cell 2, 704–711. doi: 10.1007/s13238-011-1098-y
Svendsen, J. M., Smogorzewska, A., Sowa, M. E., O’Connell, B. C., Gygi, S. P., Elledge, S. J., et al. (2009). Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138, 63–77. doi: 10.1016/j.cell.2009.06.030
Szakal, B., and Branzei, D. (2013). Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 32, 1155–1167. doi: 10.1038/emboj.2013.67
Tan, L., Ehrlich, E., and Yu, X. F. (2007). DDB1 and Cul4A are required for human immunodeficiency virus type 1 Vpr-induced G2 arrest. J. Virol. 81, 10822–10830. doi: 10.1128/JVI.01380-07
Wechsler, T., Newman, S., and West, S. C. (2011). Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature 471, 642–646. doi: 10.1038/nature09790nature09790
Weitzman, M. D., Lilley, C. E., and Chaurushiya, M. S. (2010). Genomes in conflict: maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 64, 61–81. doi: 10.1146/annurev.micro.112408.134016
Wen, X., Duus, K. M., Friedrich, T. D., and De Noronha, C. M. (2007). The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VprBP/DCAF1 as an adaptor. J. Biol. Chem. 282, 27046–27057. doi: 10.1074/jbc.M703955200
Whitney, M. A., Royle, G., Low, M. J., Kelly, M. A., Axthelm, M. K., Reifsteck, C., et al. (1996). Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood 88, 49–58.
Wu, L., and Hickson, I. D. (2003). The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 426, 870–874. doi: 10.1038/nature02253nature02253
Wyatt, H. D., Sarbajna, S., Matos, J., and West, S. C. (2013). Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell 52, 234–247. doi: 10.1016/j.molcel.2013.08.035
Yamamoto, K. N., Kobayashi, S., Tsuda, M., Kurumizaka, H., Takata, M., Kono, K., et al. (2011). Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 6492–6496. doi: 10.1073/pnas.1018487108
Yan, N., Regalado-Magdos, A. D., Stiggelbout, B., Lee-Kirsch, M. A., and Lieberman, J. (2010). The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 11, 1005–1013. doi: 10.1038/ni.1941
Yang, Y. G., Lindahl, T., and Barnes, D. E. (2007). Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131, 873–886. doi: 10.1016/j.cell.2007.10.017
Ying, S., Minocherhomji, S., Chan, K. L., Palmai-Pallag, T., Chu, W. K., Wass, T., et al. (2013). MUS81 promotes common fragile site expression. Nat. Cell Biol. 15, 1001–1007. doi: 10.1038/ncb2773
Zhang, X., Brann, T. W., Zhou, M., Yang, J., Oguariri, R. M., Lidie, K. B., et al. (2011). Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 186, 4541–4545. doi: 10.4049/jimmunol.1003389
Zhao, K., Du, J., Han, X., Goodier, J. L., Li, P., Zhou, X., et al. (2013). Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutieres syndrome-related SAMHD1. Cell Rep. 4, 1108-1115. doi: 10.1016/j.celrep.2013.08.019
Keywords: DNA damage response, innate immunity, HIV, Vpr, G2/M arrest, SLX4 complex
Citation: Brégnard C, Benkirane M and Laguette N (2014) DNA damage repair machinery and HIV escape from innate immune sensing. Front. Microbiol. 5:176. doi: 10.3389/fmicb.2014.00176
Received: 22 February 2014; Accepted: 31 March 2014;
Published online: 22 April 2014.
Edited by:
Frank Kirchhoff, University Clinic Ulm, GermanyReviewed by:
Youichi Suzuki, National University of Singapore, SingaporeWiesmüller Lisa, Ulm University, Germany
Copyright © 2014 Brégnard, Benkirane and Laguette. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nadine Laguette and Monsef Benkirane, Laboratoire de Virologie Moléculaire, Institut de Génétique Humaine, CNRS UPR1142, 141, Rue de la Cardonille, Montpellier 34000, France e-mail: nadine.laguette@igh.cnrs.fr; monsef.benkirane@igh.cnrs.fr