 Olivia U. Mason
Olivia U. Mason James Han
James Han Tanja Woyke
Tanja Woyke Janet K. Jansson
Janet K. Jansson- 1Department of Earth, Ocean and Atmospheric Science, Florida State University, Tallahassee, FL, USA
- 2Department of Energy Joint Genome Institute, Walnut Creek, CA, USA
- 3Lawrence Berkeley National Laboratory, Earth Sciences Division, Ecology Department, Berkeley, CA, USA
- 4Department of Plant and Microbial Biology, University of California, Berkeley, CA, USA
During the Deepwater Horizon (DWH) oil spill in the Gulf of Mexico a deep-sea hydrocarbon plume developed resulting in a rapid succession of bacteria. Colwellia eventually supplanted Oceanospirillales, which dominated the plume early in the spill. These successional changes may have resulted, in part, from the changing composition and abundance of hydrocarbons over time. Colwellia abundance peaked when gaseous and simple aromatic hydrocarbons increased, yet the metabolic pathway used by Colwellia in hydrocarbon disposition is unknown. Here we used single-cell genomics to gain insights into the genome properties of a Colwellia enriched during the DWH deep-sea plume. A single amplified genome (SAG) of a Colwellia cell isolated from a DWH plume, closely related (avg. 98% 16S rRNA gene similarity) to other plume Colwellia, was sequenced and annotated. The SAG was similar to the sequenced isolate Colwellia psychrerythraea 34H (84% avg. nucleotide identity). Both had genes for denitrification, chemotaxis, and motility, adaptations to cold environments and a suite of nutrient acquisition genes. The Colwellia SAG may be capable of gaseous and aromatic hydrocarbon degradation, which contrasts with a DWH plume Oceanospirillales SAG which encoded non-gaseous n-alkane and cycloalkane degradation pathways. The disparate hydrocarbon degradation pathways are consistent with hydrocarbons that were abundant at different times in the deep-sea plume; first, non-gaseous n-alkanes and cycloalkanes that could be degraded by Oceanospirillales, followed by gaseous, and simple aromatic hydrocarbons that may have been degraded by Colwellia. These insights into the genomic properties of a Colwellia species, which were supported by existing metagenomic sequence data from the plume and DWH contaminated sediments, help further our understanding of the successional changes in the dominant microbial players in the plume over the course of the DWH spill.
Introduction
The Deepwater Horizon (DWH) oil spill from April to July 2010 was unprecedented due to the extreme depth (1500 m below sea-level; mbsl) and low temperature (4°C), at which it took place. Colwellia species bloomed in the deep-sea hydrocarbon plume, that formed at 1100 mbsl, in early June, 2010 (Valentine et al., 2010; Redmond and Valentine, 2012) after partial capture of the oil began (Dubinsky et al., 2013). At this time the unmitigated flow of oil ceased, and cycloalkanes and non-gaseous n-alkanes, which were dominant until that point, decreased in concentration (Dubinsky et al., 2013). Concomitantly, the concentration of natural gases and simple aromatics increased (Dubinsky et al., 2013). The change in hydrocarbon composition and abundance resulting from partial capture was mirrored by differences in microbial community composition, with a shift in dominance of Oceanospirillales to Colwellia (Dubinsky et al., 2013). Mason et al. (2012) analyzed Oceanospirillales single amplified genomes (SAG) from the deep-sea plume and reported the genome encoded pathways for cyclohexane and non-gaseous n-alkane degradation. These data helped to explain the dominance of Oceanospirillales when cycloalkanes and non-gaseous n-alkanes were abundant, and its subsequent decline when these hydrocarbon constituents decreased. The later appearance of Colwellia in the spill history could be due to its ability to degrade other hydrocarbon constituents in the oil. Microcosm experiments established that Colwellia species were capable of degrading hydrocarbons originating from the oil spill at 4°C (Baelum et al., 2012; Redmond and Valentine, 2012). Specifically, Redmond and Valentine (2012) reported that Colwellia species from the deep-sea plume incorporated labeled ethane, propane, and benzene. The ability to degrade ethane and propane, for example, provides clues as to why Colwellia appears to have increased in abundance when the concentration of these and other gases increased in June 2010.
Hydrocarbon degradation by cultured Colwellia has not previously been reported (e.g., Bowman et al., 1997; Methé et al., 2005). For example, Colwellia psychrerythraea 34H, cultivated from Arctic marine sediments, is psychrophilic, with optimal growth at −1°C to 10°C (Huston, 2003) was not reported to degrade hydrocarbons. The genome of C. psychrerythraea provides insights into the adaptations that enable it to be active at such low temperatures, such as changes to cell membrane fluidity and the use of compounds that provide cryotolerance (Methé et al., 2005). Further, this genome provides a platform for comparison to Colwellia that were enriched during the DWH oil spill. Although the possible role of C. psychrerythraea in bioremediation by aromatic, or C1 contaminant degradation was inferred, the specific contaminants and the pathways catalyzing these reactions have not yet been elucidated (Methé et al., 2005). Thus it remains unresolved as to how Colwellia species that were identified during the DWH spill are capable of growth with ethane, propane, and benzene (Redmond and Valentine, 2012), polycyclic aromatic hydrocarbons (PAH) (Gutierrez et al., 2013), or MC252 crude oil constituents (Baelum et al., 2012). Here our aim was to use single-cell genomics to gain a better understanding of the genomic properties of a deep-sea Colwellia species that enabled it to bloom during the oil spill. Specifically, we present a genome analysis of a Colwellia single-cell isolated directly from the deep-sea plume described in Hazen et al. (2010) and Mason et al. (2012).
Materials and Methods
Single-Cell Sorting, Whole-Genome Amplification, and Screening
Cells were collected following the clean sorting procedures detailed by Rodrigue et al. (2009). Briefly, single cells from the proximal plume water sample, collected on May 29, 2010 from 1207 mbsl (described in Mason et al., 2012), were sorted by the Cytopeia Influx Cell Sorter (BD Biosciences, Franklin Lakes, NJ) into three 96-well plates containing 3 μ l of ultraviolet-treated TE. The cells were stained with SYBR Green I (Invitrogen, Carlsbad, CA) and illuminated by a 488-nm laser (Coherent Inc., Santa Clara, CA). As described by Woyke et al. (2011) the sorting window was based on the size determined by side scatter and green fluorescence (531/40 bp filter). Single cells were lysed for 20 min at room temperature using alkaline solution from the Repli-G UltraFast Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. After neutralization, the samples were amplified using the RepliPHI Phi29 reagents (Epicenter Biotechnologies, Madison, WI). Each 50-μ l reaction contained Phi29 Reaction Buffer (1 × final concentration), 50 μ M random hexamers with the phosphorothioate bonds between the last two nucleotides at the 3′ end (IDT, Coralville, IA), 0.4 mM dNTP, 5% DMSO (Sigma, St Louis, MO), 10 mM DTT (Sigma), 100 U Phi29 and 0.5 mM Syto 13 (Invitrogen). A mastermix of multiple displacement amplification (MDA) reagents minus the Syto 13 sufficient for a 96-well plate was ultraviolet-treated for 60 min for decontamination. Syto 13 was then added to the mastermix, which was added to the single cells for real-time MDA on the Roche LightCycler 480 for 17 h at 30°C. All steps of single-cell handling and amplification were performed under most stringent conditions to reduce the introduction of contamination. Single-cell MDA products were screened using Sanger sequencing of 16S rRNA gene amplicons derived from each MDA product. Two single-cell MDA products were identified as Colwellia, one of which was selected for shotgun sequencing and analysis based on the clean appearance of the 16S rRNA gene Sanger sequencing electropherogram.
Single-Cell Illumina Sequencing, Quality Control, and Assembly
Single-cell amplified DNA of Colwellia was used to generate normalized, indexed Illumina libraries. Briefly, 3 μ g of MDA product was sheared using the Covaris E210 (Covaris, Woburn, MA) with the following protocol: 10% duty cycle, intensity 5 and 200 cycles per burst for 6 min per sample. The fragmented DNA was purified using QIAquick columns (Qiagen) according to the manufacturer's instructions. The sheared DNA was end-repaired, A-tailed and ligated to the Illumina adaptors according to the Illumina standard paired-end protocol. The ligation product was purified using AMPure SPRI beads, then underwent normalization using the Duplex-Specific Nuclease Kit (Axxora, San Diego, CA). The normalized libraries were then amplified by PCR for 12 cycles using a set of two indexed primers and the library pool was sequenced on one lane of an Illumina HiSeq 2000 sequencer according to the manufacturer's protocols (run mode 2 × 76 bp). To ensure that the single-amplified genome was free of contamination, the Illumina sequence data was quality controlled using GC content and blast analysis (for details, see supplemental material).
Bioinformatics
The 16S rRNA gene sequence of the Colwellia SAG was compared to 16S rRNA gene datasets in plume samples and stable isotope probing (SIP) experiments (Valentine et al., 2010; Mason et al., 2012; Redmond and Valentine, 2012). SAG reads were assembled using SPAdes 2.4.0 (Bankevich et al., 2012) with the following parameters: –sc –careful –m 40 –12. The single copy gene analysis was carried out according to Rinke et al. (2013). Unassembled metagenome and metatranscriptome reads from deep-sea plume samples presented by Mason et al. (2012) (available at http://mason.eoas.fsu.edu) and metagenome reads from surface sediments (http://mason.eoas.fsu.edu and MG-RAST IDs 4510162.3-4510175.3), several of which were contaminated during the DWH spill (Mason et al., 2014), were mapped against the assembled Colwellia SAG using Bowtie2 (Langmead and Salzberg, 2012) with default parameters. The SAG assembly was annotated using CAMERA 2.0 (Sun et al., 2011). The reads that went into the SAG assembly were compared to C. psychrerythraea (Methé et al., 2005) using PAUDA (Huson and Xie, 2014), with a bit score of ≥40 serving as the cutoff. JSpecies (Richter and Rosselló-Móra, 2009) was used to compute the average nucleotide identity between the Colwellia SAG assembly and C. psychrerythraea. The average was computed by using values greater than 70%. The lowest observed value that was used in the calculation was 70.34% and the highest was 100%. Additionally, the reads that went into the Colwellia SAG assembly were also compared to a database of genes involved in hydrocarbon degradation (bit score of ≥40 cutoff), similar to the methodology described by Mason et al. (2012) with PAUDA (Huson and Xie, 2014). Last, the Colwellia SAG assembly was compared (blastx) to gene products of the BMO operon of Thauera butanivorans (NCBI locus AY093933). Contigs with bmoR were also compared to the non-redundant protein sequences (nr) in GenBank using blastx. The Colwellia SAG raw reads and assembly are available at: http://mason.eoas.fsu.edu, as are the Oceanospirillales SAG reads and assemblies from Mason et al. (2012).
Results and Discussion
Genomic Features
More than 38 million reads, representing 587X coverage were obtained (assuming a genome size of 5.0 Mb). The Colwellia SAG assembly resulted in 141 contigs with a total assembly size of 1.3 Mb. The estimated genome recovery was 30%, based on single-copy gene analysis (Supplementary Table 1). Average G + C content was 37.5% (Supplementary Results), compared to 37.9% for C. psychrerythraea (Methé et al., 2005). A total of 1163 open reading frame(s) were identified. In the assembled Colwellia SAG 11 rRNA operons and 8 tRNAs were present. All annotated COGs (CAMERA) of the Colwellia SAG assembly are shown in Supplementary Table 2. The average nucleotide identity when comparing the Colwellia SAG to C. psychrerythraea was 84%.
SAG Taxonomy
The SAG selected for Illumina sequencing, Colwellia SAG HUGN, was closely related to C. psychrerythraea based on its 16S rRNA gene sequence (99% similar). This clade also includes highly similar Colwellia sequences retrieved from the DWH plume (average 98%; Valentine et al., 2010; Mason et al., 2012) and by stable isotope probing analysis (97–99%; Redmond and Valentine, 2012) (Figure 1). Although the SAG was very similar to the Colwellia sequences identified in situ in the plume sample, Colwellia was not abundant early in the spill history at the time the sample was obtained (Mason et al., 2012). The SAG was also highly similar to Colwellia species found in the June 2010 plume samples (Figure 1), in which Colwellia had a relative abundance of >60% of the community (Valentine et al., 2010).
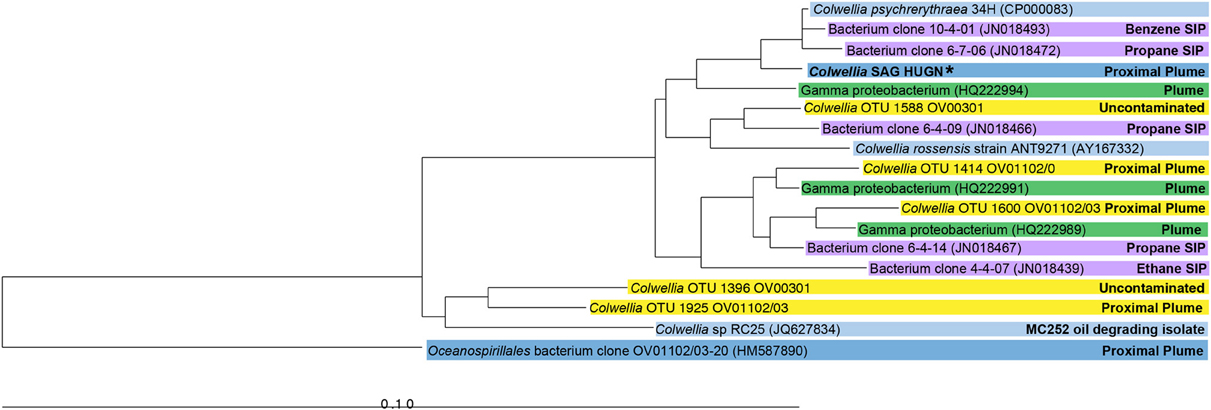
Figure 1. Neighbor-joining phylogenetic tree of 16S rRNA genes of the Colwellia SAG and close relatives. Sequences from a particular study are highlighted with the same color. Information about the environment from which a particular microorganism was found is indicated in the figure. The Colwellia SAG is in bold text and indicated by an asterisk.
Hydrocarbon Degradation
The most frequently observed genes in the reads that went into the Colwellia SAG assembly were aldehyde dehydrogenase, alcohol dehydrogenase, enoyl-CoA hydratase, bmoR, 2-hydroxymuconic semialdehyde dehydrogenase and 4-cresol dehydrogenase. Generally, these genes could be part of several different metabolic pathways, but the observed bmoR, which was highly similar (avg. similarity was 74%) to that of Pseudomonas butanovora [subsequently renamed Thauera butanivorans (Dubbels et al., 2009)] suggested that it may play a similar functional role in the Colwellia species described here. T. butanivorans (Dubbels et al., 2009), isolated from activated sludge from an oil refining plant (Takahashi, 1980), is able to grow on C2–C9 alkanes (Dubbels et al., 2009) using the following pathway: Butane (BMO)→ 1-Butanol (alcohol dehydrogenase)→ Butyraldehyde (aldehyde dehydrogenase)→ Butyric Acid (further metabolized as fatty acid) (Arp, 1999). Although a butane monooxygenase was not identified in the Colwellia SAG presented here, T. butanivorans' bmoR, which encodes a transcriptional regulator (BmoR) for BMO, and is located upstream of the BMO operon (Kurth et al., 2008), was identified (Figure 2). A total of 758 raw reads were annotated as bmoR in the SAG using blastx. The average similarity was 74% to T. butanivorans bmoR (bit score ≥ 40). In a separate analysis this bmoR was also compared to the assembled SAG. When carrying out blastx analysis of the contigs compared to bmoR the percent similarity ranged from 33–42%, with a high bit score average of 172. We acknowledge that similarities in this range are low in terms of a reliable annotation as bmoR. The discrepancy in the % similarity is likely due to methodological differences (short, unassembled 76 bp reads compared to contigs). Further, the GC content of the contigs upon which bmoR was identified was 38%, which is identical to the GC content for the entire SAG assembly (38%). The presence of bmoR, similar to T. butanivorans, in the Colwellia SAG, is not however, diagnostic for presence of the BMO operon. The remaining metabolic pathway used by T. butanivorans in butane oxidation was encoded in the SAG (Figure 2) and was highly similar (≥40 bit score) to that of C. psychrerythraea. C. psychrerythraea does not have a butane monooxygenase thus has a paraphyletic lineage with the Colwellia SAG.
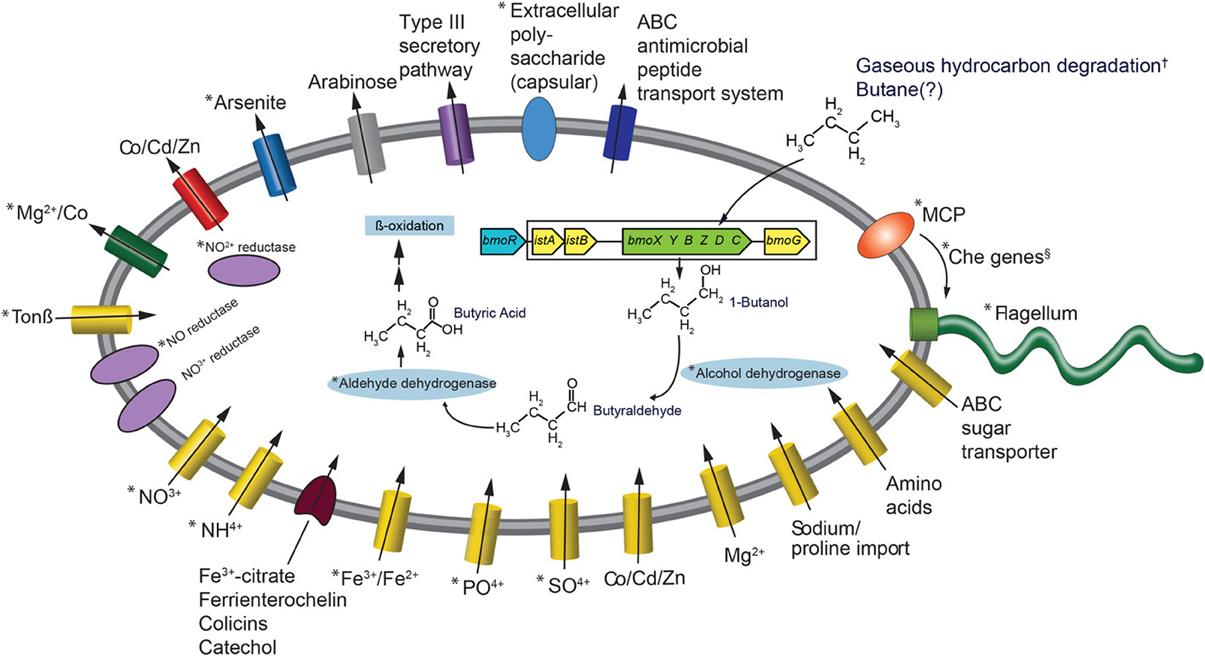
Figure 2. Metabolic reconstruction of the Colwellia SAG. An asterisk indicates Colwellia SAG reads with ≥40 bit score to C. psychrerythraea. The BMO operon and adjacent genes (from Kurth et al., 2008, see Figure 1A) are color coded: blue indicates Cowellia SAG. Additionaly, those genes from the DWH plume and sediment metagenomes (Mason et al., 2012, 2014, respectively) are enclosed by rectangle. Genes shown in yellow indicate DWH plume, those in green indicate sediment metagenomes. These genes were not directly ascribed to the Colwellia SAG described herein. An † indicates that the direct evidence for gaseous hydrocarbon degradation by plume Colwellia was provided by Redmond and Valentine (2012), not from the Colwellia SAG annotation. The Colwellia SAG had the full suite of genes coding for chemotaxis including Che genes, indicated by an §. Che genes include CheA, CheC, CheY, and CheZ.
The lack of a butane monooxygenase in the SAG may indicate that it, does not, in fact have that metabolic capability. Alternatively, it may be due to insufficient coverage of the genome (30% complete) of the Colwellia SAG. To test this hypothesis, we turned to metagenomes from the plume (~0.03 Tb of sequence data) and from sediments (~1 Tb of sequence data), several of which were highly contaminated by the DWH spill (Mason et al., 2012 and 2014, respectively). Although the relative abundance of Colwellia in the DWH plume, from which the single cell was retrieved, was low (1–2%) (Mason et al., 2012), the same bmoR gene was identified in metagenomes from the plume, as were several additional T. butanivorans genes involved in butane oxidation (bit score of ≥40) (Figure 2). However, no butane monooxygenase was recovered in the DWH plume metagenomes. In sediment samples, Colwellia had a high relative abundance of 6% in the more contaminated samples (Mason et al., 2014). The higher abundance of Colwellia in contaminated sediments (Mason et al., 2014) compared to the deep-sea plume may have resulted in better coverage of Colwellia. In the resulting sediment metagenomes, bmoR, bmoG, istA, and istB were identified, all of which had a bit score of ≥40, as was the alpha subunit of butane monooxygenase (bmoX) (Figure 2). The metagenomic data, particularly in surface sediments, suggested that gaseous hydrocarbon degradation via butane monooxygenase was a plausible pathway for degrading DWH hydrocarbon constituents, however this pathway was not directly linked to Colwellia.
Direct evidence for gaseous hydrocarbon degradation by DWH plume Colwellia, which were closely related to the Colwellia SAG described herein, was provided by Redmond and Valentine (2012) who used SIP to examine the uptake of methane, ethane, and propane. They reported that Colwellia enriched from the plume was responsible for the bulk of 13C labeled propane and ethane uptake, but also showed, to a far lesser extent uptake of methane by Colwellia, consistent with the properties of T. butanivorans BMO (Cooley et al., 2009). The putative hydrocarbon degradation pathways discussed above help to resolve several aspects of the microbial response to the oil spill that hitherto were unclear. Specifically, the BMO of T. butanivorans is highly efficient at discriminating against methane when grown on ≥C2 alkanes (Cooley et al., 2009). At the time Colwellia was abundant in mid-June 2010 (Valentine et al., 2010; Redmond and Valentine, 2012), Valentine et al. (2010) reported that, despite the abundance of methane input to the deep-sea during the spill (Joye et al., 2011), propane, ethane (and perhaps butane) fueled microbial respiration. These findings suggested that Colwellia was likely involved in degrading gaseous hydrocarbons, but not methane (Valentine et al., 2010), which was substantiated by the aforementioned SIP experiments.
In addition to gaseous hydrocarbon degradation Redmond and Valentine (2012) also reported that Colwellia enriched in the DWH plume incorporated benzene in SIP experiments. In the Colwellia SAG 4-cresol dehydrogenase (EC 1.17.99.1) and 2-hydroxymuconic semialdehyde dehydrogenase (EC 1.2.1.85) both of which are involved in degradation of several aromatics (e.g., benzene by Pseudomonas putida according to Reardon et al., 2000), were identified. These were also identified in the water column and sediment metagenomes. Together, these data support the uptake of aromatics by plume Colwellia as reported by Redmond and Valentine (2012).
Nitrogen
Previously we reported that nitrate was significantly lower in plume samples from which the Colwellia SAG was isolated compared to non-plume samples (p-value 0.003) (Hazen et al., 2010). One hypothesis is that nitrate was consumed as an electron acceptor during hydrocarbon degradation. As the previously sequenced C. psychrerythraea strain was reported to have genes coding for denitrification (Methé et al., 2005), we sought to determine whether the Cowellia SAG also possessed the genetic capacity for denitrification. First, a direct comparison was made between C. psychrerythraea and unassembled reads from the Colwellia SAG that went into the final assembly. This comparison revealed a high sequence similarity (≥40 bit score) of genes involved in denitrification in both the Colwellia SAG and C. psychrerythraea (Figure 2). For example, C. psychrerythraea's nitrate permease (GI71146856), nitrite reductases (small and large subunits; GI71146751 and GI71143641), nitric-oxide reductase (GI71148260 and GI71144846) and an anaerobic nitric-oxide reductase transcription regulator (GI71282073) were present in the SAG (Figure 2).
Although many genes in the denitrification pathway were common between the Colwellia SAG and C. psychrerythraea, there were differences. For example, nitrate reduction appears to be encoded by different genes. In the Colwellia SAG reduction of nitrate could be carried out via a nitrate/TMAO reductase (COG3005) and a periplasmic nitrate reductase system, NapE component (COG4459). Further an uncharacterized protein involved in response to nitric oxide was present in the SAG (COG3213). The Colwellia SAG lacked a nitrous-oxide reductase gene, which may be an artifact of an incomplete genome for the Colwellia SAG. It is, however, plausible that the SAG does in fact lack the capability to reduce nitrous oxide. Given the importance of nitrous oxide as a greenhouse gas (Wuebbles, 2009), further elucidating the role of Colwellia in denitrifying processes and end-products is necessary, particularly in the context of an unprecedented ecosystem perturbation, such as the DWH oil spill.
Iron
Baelum et al. (2012) tested the hypothesis put forth by Hazen et al. (2010), that iron could have limited microbial growth, in microcosm experiments with MC252 crude oil. The results suggested that iron limitation resulted in floc formation of which Colwellia was the most abundant microorganism. They also reported that in the first 20 days of incubation, lower growth, and hydrocarbon degradation rates were observed, although the total amount of oil degraded after 20 days was the same as in microcosms with sufficient iron. Further, Joung and Shiller (2013) reported that iron concentrations in the plume compared to non-plume samples indicate an enhanced microbial iron demand. This suggests that in the deep-sea plume, at least initially, iron could have affected both the growth and degradation rate of hydrocarbons by Colwellia, although is unlikely that iron concentrations were a growth-limiting factor nor a factor in the total amount of hydrocarbons degraded during the spill (Joung and Shiller, 2013).
To better understand Colwellia's iron acquisition strategy we first compared the Colwellia SAG to C. psychrerythraea, followed by direct annotation of Colwellia SAG contigs using CAMERA. Similar to C. psychrerythraea the Colwellia SAG lacked genes for siderophore production. Yet, highly similar genes when comparing the two (≥40 bit score) were found for iron transport and uptake (Figure 2). Iron(III) ABC transporter proteins (GIs 71277892 and 71143632) and ferrous iron transport proteins (GIs 71144726 and 71143643) were identified, yet the two genomes had different genes coding for interactions with siderophore complexes (Figure 2). Annotation of the Colwellia SAG assembly with CAMERA revealed the genome encoded functions involved in iron acquisition, including an outer membrane receptor for the siderophore-iron complex ferrienterochelin (Lundrigan and Kadner, 1986) (COG4771), and an outer membrane receptor for Fe3+-dicitrate (COG4772) (Figure 2). Other functions involved in iron transport were also annotated (COGs 0370, 1629, and 1918) (Figure 2). Taken together, the data suggested that the Colwellia SAG could have been at a physiological disadvantage if it was, in fact, unable to synthesize siderophores. Conceivably, Colwellia could have relied on scavenging siderophores produced by other microorganisms in the plume for remediating iron limitation. Although we acknowledge that missing genes for siderophore production could be due to the incompleteness of the Colwellia SAG, C. psychrerythraea also lacks the ability to produce siderophores.
Floc Formation
The C. psychrerythraea genome encodes a capsular polysaccharide biosynthesis protein (GI71145258), as does the Colwellia SAG (Figure 2). Polysaccharide capsules are found on the cell surface of many bacteria and are involved in, among other processes, adherence of one bacterium to another (Roberts, 1996). Further, C. psychrerythraea's putative polysaccharide biosynthesis glycosyltransferase, which likely functions in extracellular polysaccharide biosynthesis (Methé et al., 2005), was identified in the Colwellia SAG. As discussed above, Baelum et al. (2012) observed flocs largely comprised of Colwellia that formed under iron limiting conditions in microcosms incubated with MC252 oil. Our findings provide preliminary evidence for a plausible mechanism by which cells could have aggregated together in the face of nutrient, and particularly, iron limitation in the deep-sea plume.
Chemotaxis and Motility
A full suite of genes involved in signal transduction, chemotaxis, and motility were present in the Colwellia SAG (Figure 2), many of which were highly similar to C. psychrerythraea. For example C. psychrerythraea's chemotaxis proteins CheA, CheY, and CheZ, numerous methyl-accepting chemotaxis proteins (MCP) and the full suite of genes coding for flagellum synthesis and operation were observed in the Colwellia SAG. Parales et al. (2000) reported that several strains of bacteria exhibited a chemotactic response to benzene and toluene and suggested MCPs were involved. More direct evidence was provided regarding the role of MCPs in the ability of Pseudomonas putida to detect naphthalene (Grimm and Harwood, 1999). Given the suite of genes, including MCP, encoded in the Colwellia SAG, it is plausible that the Colwellia SAG was able to sense and move toward a chemo-attractant, such as degradable hydrocarbons that accumulated in the deep-sea plume during the DWH spill.
Adaptation to Cold Temperatures
Direct comparison of the SAG to the C. psychrerythraea genome revealed shared genomic features (≥40 bit score similarity) thought to be involved in adaptation to life at cold temperatures; for example a C. psychrerythraea fatty acid cis/trans isomerase, which would allow the cell to alter the ratio of cis- to trans-esterified fatty acids in phospholipids (Methé et al., 2005). The ability to increase cis-isomerization, could enhance membrane fluidity at low temperatures (Methé et al., 2005), which would be an important adaption to life at low temperatures. Further, C. psychrerythraea's putative 3-oxoacyl-(acyl-carrier-protein) reductase and putative 3-oxoacyl-(acyl-carrier-protein) synthase III, which serve key roles in fatty acid and phospholipid biosynthesis (Methé et al., 2005), were identified in the SAG, and could also play a role regulating membrane fluidity at cold temperatures (Russell, 1997).
Biosynthesis of Compatible Solutes
The Colwellia SAG had genes coding for choline dehydrogenase and betaine aldehyde dehydrogenase, which were highly similar (≥40 bit score) to those of C. psychrerythraea. In the genome sequence of the psychrophilic hydrocarbon degrader, Oleispira antarctica these were the two mechanisms for biosynthesis of compatible solutes (Kube et al., 2013). Choline dehydrogenase and betaine aldehyde dehydrogenase are involved in catalyzing the reaction that leads to synthesis of the osmolyte betaine (Kube et al., 2013). Genes coding for choline dehydrogenase, and others, were suggested to be involved in synthesis of betaine, which was cited as an osmoprotectant, and also a cryoprotectant in C. psychrerythraea (Methé et al., 2005). The dual functionality of an osmoprotectant and cryoprotectant in Colwellia would be an important adaptation for the Colwellia SAG to life in the low temperature, deep-sea marine environment.
Comparison of DWH Deep-Sea Plume Colwellia SAG to Oceanospirillales SAG
Mason et al. (2012) presented an annotated Oceanospirillales SAG that was isolated from the proximal plume sample (OV01102/03 in Hazen et al., 2010) from which the Colwellia SAG presented herein was also isolated. Oceanospirillales had a higher relative abundance in the proximal plume sample compared to Colwellia (16S rRNA gene sequence relative abundance was 81% vs. 1% percent, respectively) (Mason et al., 2012). Thus most of the genes and transcripts from the proximal plume sample were thought to derive from Oceanospirillales and the role that Colwellia played in oil disposition has been largely unexplored. Comparison of the SAGs revealed several differences; namely the dominant hydrocarbon pathways, iron acquisition strategies, and respiration capabilities. First, the Oceanospirillales genome encoded cyclohexane degradation by an alkane monooxygenase (Mason et al., 2012), also recently identified in the genome sequence of the closely related Oleispira antarctica (Kube et al., 2013). As discussed above, the Colwellia SAG encoded bmoR, 4-cresol dehydrogenase and 2-hydroxymuconic semialdehyde dehydrogenase, the latter of which are involved in degradation of several aromatics. Thus, the hydrocarbon constituents potentially degraded by these two gammaproteobacteria, that increased in abundance in the plume, are disparate. Second, the ability to synthesize siderophores to acquire iron, which may have affected hydrocarbon respiration rates early on (but was not likely limiting) by the microbial community in the deep-sea plume during the oil spill, would have given the dominant Oceanospirillales a competitive advantage over Colwellia. As discussed above, the Colwellia described here may have been able to scavenge siderophores produced by other microorganisms, perhaps those of Oceanospirillales, to acquire iron. Finally, the genome of Oceanospirillales did not encode a pathway for denitrification, in contrast to the Colwellia presented here. Therefore, when faced with reduced oxygen concentrations Colwellia could have continued to carry out respiration, while Oceanospirillales may not have been able to do so.
Comparison of DWH Deep-Sea Plume Metagenomes and Metatranscriptomes and Sediment Metagenomes to the Colwellia SAG
Mason et al. (2012) reported relative abundances of 1–2% of Colwellia in metagenomes from the proximal plume closest to the DWH wellhead (1.8 km away) and at a more distant location, referred to as the distal plume (10.8 km away), collected May 26-June 2, 2010. Of these metagenome sequence reads 0.30% mapped to the Colwellia SAG. To determine if the Colwellia SAG was active in the deep-sea plume, 5S, 16S, and 23S rRNA sequences were subtracted from the plume metatranscriptomes. These reads were then mapped against the assembled Colwellia SAG. Comparison of the Colwellia SAG to proximal plume station metatranscriptome sequences revealed that 0.20% of the metatranscriptome reads mapped to the single-cell assembly. Fewer metatranscriptome reads from the distal station mapped to the Colwellia SAG (0.01%). Thus a representative of plume Colwellia may have been actively degrading gaseous and simple aromatic hydrocarbons, albeit at low levels, in late May 2010, when these hydrocarbons were less abundant. In contrast Rivers et al. (2013) reported that 16% of their metatranscriptome reads from two DWH plume stations (6–8 km from the wellhead, collected May 26-June 3, 2010; at the same time we sampled) mapped to C. psychrerythraea, although these authors used blastx while we used Bowtie2. Further, the abundance of Colwellia was significantly higher in Rivers et al. (2013; see Figure 1B for abundance) 16S rRNA gene survey as compared to the samples presented in Mason et al. (2012).
Previously, we characterized the microbial community in 64 surface sediment samples, which had varying degrees of hydrocarbon contamination from the DWH spill (Mason et al., 2014). As discussed above, Colwellia was up to 6% (relative abundance) of the microbial community in these sediments, particularly those with the highest hydrocarbon concentrations (Mason et al., 2014). From these samples 14 were selected for metagenomic sequencing. Of these 14 samples, 7 were determined a priori to exceed the Environmental Protection Agency (EPA)'s PAH benchmarks for aquatic life. The Colwellia SAG assembly was mapped to the sediment metagenomes with 0.25–0.04% of reads mapping. The most SAG reads mapped to 6 of the 7 samples that exceeded the EPA's PAH benchmarks. These results are similar to the plume metagenome and indicate that the Colwellia SAG sequenced was represented in both the water column and sediments that were contaminated during the DWH spill.
Conclusion
Analyses of the microbial community response to the oil spill revealed a clear story of microbial succession, however, few detailed descriptions of the cellular physiology of the indigenous microbes that responded to the hydrocarbon inputs have been presented. Herein we describe several aspects of a Colwellia single-cell genome that furthers our understanding of the potential role that Colwellia played during microbial succession in the deep-sea hydrocarbon plume that formed during the DWH spill. At the time that Colwellia was most abundant in the plume, methane was highly abundant but largely not degraded in favor of other gaseous hydrocarbons. This could have been due to the genetic capacity of the Colwellia to preferentially degrade gaseous hydrocarbons at the expense of methane, if it does, in fact, have a butane monooxygenase. Additionally, genes involved in BTEX degradation in the Colwellia SAG were identified. These preliminary findings may help to resolve the simultaneous increase in Colwellia abundance when these hydrocarbon compounds accumulated in the plume. The Colwellia SAG was captured in metagenomes from the water column and surface sediments that were contaminated by the DWH spill. In addition, both Oceanospirillales and Colwellia SAGs possessed genes for chemotaxis and motility, suggesting that this is an advantage for rapid response to hydrocarbon inputs in the deep-sea. We also provide preliminary evidence that the Colwellia species described herein may have been able to produce polysaccharides for floc production that corresponds to the high abundance of flocs in microcosms under iron limited conditions as reported by Baelum et al. (2012). This could represent a novel property enabling Colwellia to cope with conditions of low iron concentrations, but further experiments are necessary to confirm this hypothesis.
Author Contributions
Olivia U. Mason participated in the research cruise during which the deep-sea plume sample for single-cell isolation and sequencing was obtained. Olivia U. Mason carried out bioinformatics analyses and wrote the manuscript. James Han carried out bioinformatics analyses and helped write the manuscript. Tanja Woyke carried out single-cell genome isolation and sequencing and helped write the manuscript. Janet K. Jansson conceived the experimental design and helped write the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a subcontract from the University of California at Berkeley, Energy Biosciences Institute (EBI) to Lawrence Berkeley National Laboratory under its U.S. Department of Energy contract DE-AC02-05CH11231. We would like to thank the captain and crew of the R/V Ocean Veritas.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00332/abstract
References
Arp, D. J. (1999). Butane metabolism by butane-grown “Pseudomonas butanovora.” Microbiology 145, 1173–1180. doi: 10.1099/13500872-145-5-1173
Baelum, J., Borglin, S., Chakraborty, R., Fortney, J. L., Lamendella, R., Mason, O. U., et al. (2012). Deep-sea bacteria enriched by oil and dispersant from the Deepwater Horizon spill. Environ. Microbiol. 14, 2405–2416. doi: 10.1111/j.1462-2920.2012.02780.x
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bowman, J. P., Gosinkt, J. J., McCammon, S. A., Skerratt, H., Lewis, T. E., Nichols, D. S., et al. (1997). Colwellia demingiae sp. nov., Colwellia hornerae sp. nov., Colwellia rossensis sp. nov. and Colwellia psychrotropica sp. nov.: psychrophilic Antarctic species with the ability to synthesize docosahexaenoic acid (22:ω 63). Int. J. Syst. Bacteriol. 48, 1171–1180. doi: 10.1099/00207713-48-4-1171
Cooley, R. B., Dubbels, B. L., Sayavedra-Soto, L. A., Bottomley, P. J., and Arp, D. J. (2009). Kinetic characterization of the soluble butane monooxygenase from Thauera butanivorans, formerly “Pseudomonas butanovora.” Microbiology 155, 2086–2096. doi: 10.1099/mic.0.028175-0
Dubbels, B. L., Sayavedra-Soto, L. A., Bottomley, P. J., and Arp, D. J. (2009). Thauera butanivorans sp. nov., a C2-C9 alkane-oxidizing bacterium previously referred to as “Pseudomonas butanovora.” Int. J. Syst. Evol. Microbiol. 59, 1576–8. doi: 10.1099/ijs.0.000638-0
Dubinsky, E. A., Conrad, M. E., Chakraborty, R., Bill, M., Borglin, S. E., Hollibaugh, J. T., et al. (2013). Succession of hydrocarbon-degrading bacteria in the aftermath of the deepwater horizon oil spill in the gulf of Mexico. Environ. Sci. Technol. 47, 10860–10867. doi: 10.1021/es401676y
Grimm, A. C., and Harwood, C. S. (1999). NahY, a catabolic plasmid-encoded receptor required for chemotaxis of pseudomonas putida to the aromatic hydrocarbon naphthalene. J. Bacteriol. 181, 3310.
Gutierrez, T., Singleton, D. R., Berry, D., Yang, T., Aitken, M. D., and Teske, A. (2013). Hydrocarbon-degrading bacteria enriched by the Deepwater Horizon oil spill identified by cultivation and DNA-SIP. ISME J. 7, 2091–2104. doi: 10.1038/ismej.2013.98
Hazen, T. C., Dubinsky, E. A., DeSantis, T. Z., Andersen, G. L., Piceno, Y. M., Singh, N., et al. (2010). Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science 330, 204–208. doi: 10.1126/science.1195979
Huson, D. H., and Xie, C. (2014). A poor man's BLASTX - high-throughput metagenomic protein database search using PAUDA. Bioinformatics 30, 38–39. doi: 10.1093/bioinformatics/btt254
Huston, A. L. (2003). Bacterial Adaptation to the Cold: in situ Activities of Extracellular Enzymes in the North Water Polynya and Characterization of a Cold-Active Aminopeptidase from Colwellia Psychrerythraea Strain 34H. Ph.D. thesis, Seattle, WA: University of Washington.
Joung, D., and Shiller, A. M. (2013). Trace element distributions in the water column near the Deepwater Horizon well blowout. Environ. Sci. Technol. 47, 2161–2168. doi: 10.1021/es303167p
Joye, S. B., MacDonald, I. R., Leifer, I., and Asper, V. (2011). Magnitude and oxidation potential of hydrocarbon gases released from the BP oil well blowout. Nat. Geosci. 4, 160–164. doi: 10.1038/ngeo1067
Kube, M., Chernikova, T. N., Al-Ramahi, Y., Beloqui, A., Lopez-Cortez, N., Guazzaroni, M.-E., et al. (2013). Genome sequence and functional genomic analysis of the oil-degrading bacterium Oleispira antarctica. Nat. Commun. 4, 2156. doi: 10.1038/ncomms3156
Kurth, E. G., Doughty, D. M., Bottomley, P. J., Arp, D. J., and Sayavedra-Soto, L. A. (2008). Involvement of BmoR and BmoG in n-alkane metabolism in “Pseudomonas butanovora.” Microbiology 154, 139–147. doi: 10.1099/mic.0.2007/012724-0
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lundrigan, M. D., and Kadner, R. J. (1986). Nucleotide sequence of the gene for the ferrienterochelin receptor FepA in Escherichia coli. Homology among outer membrane receptors that interact with TonB. J. Biol. Chem. 261, 10797–10801.
Mason, O. U., Hazen, T. C., Borglin, S., Chain, P. S. G., Dubinsky, E. A., Fortney, J. L., et al. (2012). Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J. 6, 1715–1727. doi: 10.1038/ismej.2012.59
Mason, O. U., Scott, N. M., Gonzalez, A., Robbins-Pianka, A., Bælum, J., Kimbrel, J., et al. (2014). Metagenomics reveals sediment microbial community response to Deepwater Horizon oil spill. ISME J. 8, 1464–1475. doi: 10.1038/ismej.2013.254
Methé, B. A., Nelson, K. E., Deming, J. W., Momen, B., Melamud, E., Zhang, X., et al. (2005). The psychrophilic lifestyle as revealed by the genome sequence of Colwellia psychrerythraea 34H through genomic and proteomic analyses. Proc. Natl. Acad. Sci. U.S.A. 102, 10913–10918. doi: 10.1073/pnas.0504766102
Parales, R. E., Ditty, J. L., and Harwood, C. S. (2000). Toluene-Degrading bacteria are chemotactic toward the environmental pollutants benzene, toluene, and trichloroethylene. Appl. Environ. Microbiol. 66, 4098. doi: 10.1128/AEM.66.9.4098-4104.20
Reardon, K. F., Mosteller, D. C., and Bull Rogers, J. D. (2000). Biodegradation kinetics of benzene, toluene, and phenol as single and mixed substrates for Pseudomonas putida F1. Biotechnol. Bioeng. 69, 385–400. doi: 10.1002/1097-0290(20000820)69:4<385::AID-BIT5>3.0.CO;2-Q
Redmond, M. C., and Valentine, D. L. (2012). Natural gas and temperature structured a microbial community response to the Deepwater Horizon oil spill. Proc. Natl. Acad. Sci. U.S.A. 109, 20292–20297. doi: 10.1073/pnas.1108756108
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N. N., Anderson, I. J., Cheng, J.-F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437. doi: 10.1038/nature12352
Rivers, A. R., Sharma, S., Tringe, S. G., Martin, J., Joye, S. B., and Moran, M. A. (2013). Transcriptional response of bathypelagic marine bacterioplankton to the Deepwater Horizon oil spill. ISME J. 7, 2315–2329. doi: 10.1038/ismej.2013.129
Roberts, I. S. (1996). The biochemistry and genetics of capsular polysaccharide production in bacteria. Annu. Rev. Microbiol. 50, 285–315. doi: 10.1146/annurev.micro.50.1.285
Rodrigue, S., Malmstrom, R. R., Berlin, A. M., Birren, B. W., Henn, M. R., and Chisholm, S. W. (2009). Whole genome amplification and de novo assembly of single bacterial cells. PLoS ONE 4:e6864. doi: 10.1371/journal.pone.0006864
Russell, N. J. (1997). Psychrophilic bacteria–molecular adaptations of membrane lipids. Comp. Biochem. Physiol. A Physiol. 118, 489–493. doi: 10.1016/S0300-9629(97)87354-9
Sun, S., Chen, J., Li, W., Altintas, I., Lin, A., Peltier, S., et al. (2011). Community cyberinfrastructure for Advanced Microbial Ecology Research and Analysis: the CAMERA resource. Nucleic Acids Res. 39, D546–D551. doi: 10.1093/nar/gkq1102
Takahashi, J. (1980). Production of intracellular protein from n-butane by Pseudomonas butanovora sp. nov. Adv. Appl. Microbiol. 26, 117–128.
Valentine, D. L., Kessler, J. D., Redmond, M. C., Mendes, S. D., Heintz, M. B., Farwell, C., et al. (2010). Propane respiration jump-starts microbial response to a deep oil spill. Science 330, 208–211. doi: 10.1126/science.1196830
Woyke, T., Sczyrba, A., Lee, J., Rinke, C., Tighe, D., Clingenpeel, S., et al. (2011). Decontamination of MDA reagents for single cell whole genome amplification. PLoS ONE 6:e26161. doi: 10.1371/journal.pone.0026161
Keywords: DWH oil spill, Colwellia, single-cell genomics, deep-sea plume, hydrocarbon degradation, bacteria
Citation: Mason OU, Han J, Woyke T and Jansson JK (2014) Single-cell genomics reveals features of a Colwellia species that was dominant during the Deepwater Horizon oil spill. Front. Microbiol. 5:332. doi: 10.3389/fmicb.2014.00332
Received: 01 November 2013; Accepted: 16 June 2014;
Published online: 08 July 2014.
Edited by:
Ian M. Head, Newcastle University, UKReviewed by:
Vanessa Karel Michelou, University of Hawaii, USAKasper Urup Kjeldsen, Aarhus University, Denmark
Copyright © 2014 Mason, Han, Woyke and Jansson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivia U. Mason, Department of Earth, Ocean and Atmospheric Science, Florida State University, 117 N Woodward Avenue, Rogers Building Rm. 307, Tallahassee, FL, USA e-mail: omason@fsu.edu;
Janet K. Jansson, 1 Cyclotron Road, Berkeley, CA, USA e-mail: jrjansson@lbl.gov