 Corey M. Hudson
Corey M. Hudson Britney Y. Lau
Britney Y. Lau Kelly P. Williams
Kelly P. Williams- Sandia National Laboratories, Department of Systems Biology, Livermore, CA, USA
Genes for the RNA tmRNA and protein SmpB, partners in the trans-translation process that rescues stalled ribosomes, have previously been found in all bacteria and some organelles. During a major update of The tmRNA Website (relocated to http://bioinformatics.sandia.gov/tmrna), including addition of an SmpB sequence database, we found some bacteria that lack functionally significant regions of SmpB. Three groups with reduced genomes have lost the central loop of SmpB, which is thought to improve alanylation and EF-Tu activation: Carsonella, Hodgkinia, and the hemoplasmas (hemotropic Mycoplasma). Carsonella has also lost the SmpB C-terminal tail, thought to stimulate the decoding center of the ribosome. We validate recent identification of tmRNA homologs in oomycete mitochondria by finding partner genes from oomycete nuclei that target SmpB to the mitochondrion. We have moreover identified through exhaustive search a small number of complete, but often highly derived, bacterial genomes that appear to lack a functional copy of either the tmRNA or SmpB gene (but not both). One Carsonella isolate exhibits complete degradation of the tmRNA gene sequence yet its smpB shows no evidence for relaxed selective constraint, relative to other genes in the genome. After loss of the SmpB central loop in the hemoplasmas, one subclade apparently lost tmRNA. Carsonella also exhibits gene overlap such that tmRNA maturation should produce a non-stop smpB mRNA. At least some of the tmRNA/SmpB-deficient strains appear to further lack the ArfA and ArfB backup systems for ribosome rescue. The most frequent neighbors of smpB are the tmRNA gene, a ratA/rnfH unit, and the gene for RNaseR, a known physical and functional partner of tmRNA-SmpB.
Introduction
The trans-translation process resolves issues arising when the translating bacterial ribosome reaches the end of an mRNA with no stop codon, chiefly releasing the stalled ribosome but also eliminating both the non-stop mRNA and the encoded incomplete protein. The main agents of trans-translation are the RNA tmRNA (whose gene is named ssrA) and its protein ligand SmpB. tmRNA has a tRNA-like domain (TLD) that lacks an anticodon stem-loop; a bound SmpB occupies this corresponding space, and the complex fills the A site in the stalled ribosome, mimicking tRNA (Bessho et al., 2007; Neubauer et al., 2012). After peptidyl transfer to the alanyl moiety of charged tmRNA, the ribosome switches from the non-stop mRNA to the resume codon on tmRNA and translation continues, adding a short hydrophobic tag peptide to the nascent protein that is the signal for proteolysis (Karzai and Sauer, 2001). Canonical release at the tag reading frame stop codon frees the ribosome. Two back-up systems for trans-translation, ArfA/RF-2 and ArfB, have been described that can allow ribosome release from non-stop mRNA even when ssrA or smpB is inactive; both require the peptidyl-tRNA hydrolase activity of a release factor family member, but not the stop codon recognition usually associated with release factors (Chadani et al., 2010, 2011, 2012; Handa et al., 2011).
The tmRNA-SmpB system is found in bacteria and some organelles and has not yet been identified in archaea or in eukaryotes targeted to the cytoplasm. Aside from one report of a bacterium with a frameshift mutation in smpB, it has generally been considered that all bacteria have the system. Here we present 22 examples of complete bacterial genomes where either ssrA cannot be found, or smpB has an apparently inactivating mutation. A particularly strong case for loss of the system in a bacterial genome comes from a strain of the insect endosymbiont Carsonella ruddii, which, as best as current knowledge can be applied, further appears to lack trans-translation back-up systems. In the course of the exposition we survey bioinformatics tools for tmRNA and SmpB gene searches, and describe a major update of The tmRNA Website (http://bioinformatics.sandia.gov/tmrna).
Materials and Methods
Search Databases
Genomic data were downloaded from four directories (archaea, bacteria, plasmid, and viruses) of RefSeq on November 2012. This dataset consisted of 2031 bacterial and 137 archaeal complete genomes, and 1711 additional bacterial plasmids and 543 bacterial viruses (and 44 additional archaeal plasmids and 38 archaeal viruses) that were not part of chromosomal genome projects. BLAST databases were downloaded on 5 August 2013.
tmRNA Sequence Search
Three primary tmRNA sequence identification tools have been described: the sister programs BRUCE (Laslett et al., 2002) and ARAGORN (Laslett and Canback, 2004) and the Rfam/Infernal system (Griffiths-Jones et al., 2005) that parallels Pfam/HMMER. Rfam has four covariance models for different tmRNA forms. We applied these tools in a combined search for tmRNA and tRNA genes, because the most common false positive tmRNA hits are to legitimate tRNA genes. Our first-pass wrapper tFind.pl (available at bioinformatics.sandia.gov/software) combines tmRNA and tRNA search by running the programs tRNAscan-SE (Lowe and Eddy, 1997), ARAGORN (which also searches for tRNA genes) and BRUCE. It then resolves overlapping calls, divides the tRNAs into the two categories “valid” (those with tRNAscan-SE Cove score above 50 not labeled Pseudo or Undetermined, and also called by ARAGORN) and “questionable” (the remaining tRNA calls), and aims for accurate terminus determinatio Secn (except with two-piece tmRNAs). tmRNA calls in archaea or in bacteria with more than one call were scrutinized manually, rejecting some due to overlaps with better-called tRNAs, poor conservation of alanyl-tRNA synthetase discrimination features or other problems with the TLD. Other rejected bacterial tmRNA duplicate calls were tmRNA pseudogenes (missing one gene end) or tmRNA gene fragments formed by genomic island integration. Rfam/Infernal was not applied in this first pass because of a high false-positive rate (Table 1), but was instead applied when detection failed in a bacterial genome, along with a fourth tmRNA detection system, rFind.pl. This latter script uses our tmRNA full- and terminus-sequence databases with BLASTN to find additional tmRNAs and more accurately determine the termini of two-piece tmRNAs. Attention to the RNA gene termini is important for one method of identifying genomic islands, which favor ssrA and tRNA genes as integration sites (Mantri and Williams, 2004). When the above approaches failed to locate ssrA in a bacterial genome, we searched manually in the vicinity of smpB.
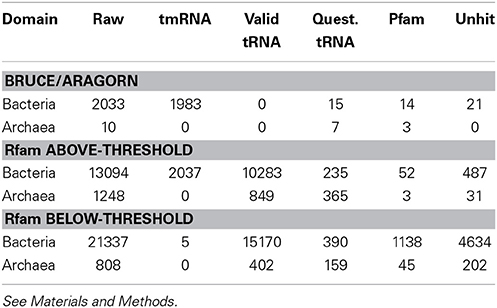
Table 1. Evaluation of primary tmRNA sequence-finding programs.
We evaluated raw output of primary tmRNA-finding software by whether hits overlapped our final sets of tmRNA and other gene types (Table 1). The BRUCE and ARAGORN results were assessed together merging overlapping calls using BEDTools (Quinlan and Hall, 2010), likewise for the results of the four covariance models of Rfam; above-threshold Rfam hits were evaluated separately from intervals unique to the below-threshold hits. These three raw hits datasets were tested for overlap with various gene sets sequentially: our final tmRNAs, the valid tRNAs, the questionable tRNAs, and a set of conserved protein-coding regions. The latter came from six-frame translation of DNAs followed by Pfam-A/HMMER (with cut-TC thresholds) treatment, reporting only the genome segments coding for Pfam-positive portions of proteins. True positive rates for tmRNA discovery were 97.5% for BRUCE/ARAGORN and 15.6% for above-threshold Rfam/Infernal.
smpB Search
The SmpB HMM of Pfam was used with HMMER and its default threshold, and five SmpB profiles (TIGR00086, cd09294, PRK0544, COG0691 and pfam01668) from Conserved Domain Database were used with RPS-TBLASTN and lower thresholds than the default that were nonetheless conservative, set at 1.4-fold above the highest score for a non-SmpB. Sub-threshold hits were examined in cases where a bacterial genome yielded no above-threshold hit. When this approach failed to locate smpB in a bacterial genome, we applied TBLASTN searches, and manual search in the vicinity of ssrA. In the final case of failure (Hodgkinia) we examined newer genomes of the same genus and were able to comparatively identify the gene.
tmRNA/SmpB Sequence Identifiers
For some sequences mentioned here we give the “tmID,” the identifier at The tmRNA Website (http://bioinformatics.sandia.gov/tmrna). Also, the webpage http://bioinformatics.sandia.gov/tmrna/ends.html is devoted to links to all sequences mentioned in this article, comparable to Tables 2, 3.

Table 2. Genomes with unusual ssrA content.
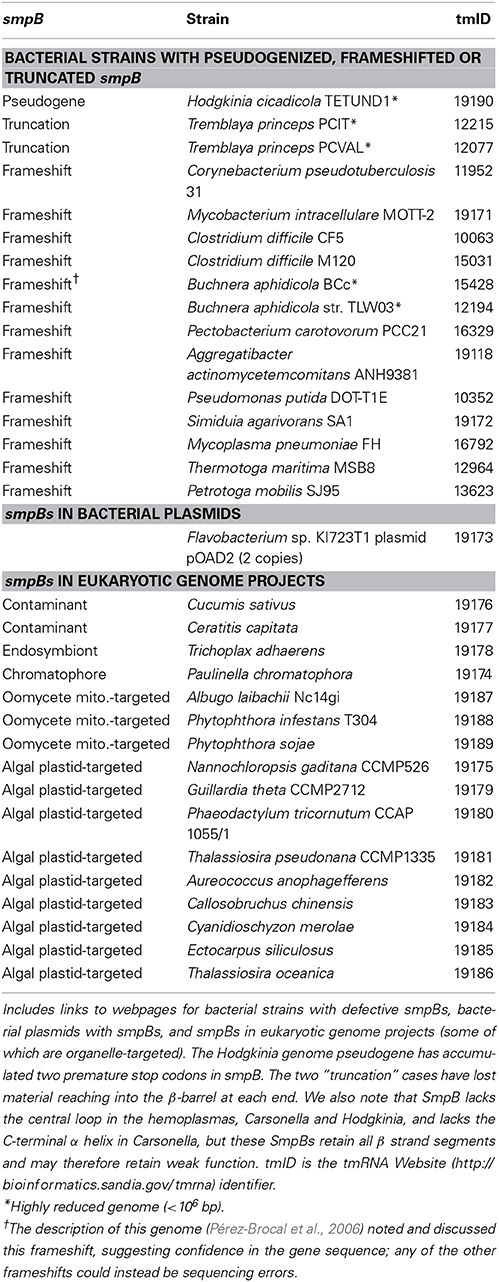
Table 3. Genomes with unusual smpB content.
Results
Exhaustive Search for ssrA
We applied our tFind.pl search method for ssrA to 2031 bacterial and 137 archaeal complete genomes, and additional RefSeq bacterial and archaeal plasmids and viruses not part of chromosomal genome projects. All ten raw tmRNA hits in Archaea were rejected by criteria noted above, while most bacterial genomes had a single ssrA located on the largest chromosome. Some genomes had a second or third ssrA allele, sometimes on a plasmid. Among plasmid and viral non-chromosomal projects, ssrA was only identified in eight mycobacteriophages Bxz1, Cali, Catera, ET08, Rizal, ScottMcG, Spud and Wildcat, however we can name additional phage tmRNA sequences in genomes that were not in our RefSeq dataset: Bacillus phage G (tmID: 14561) and mycobacteriophage DS6A (tmID: 11587). The DS6A sequence consists of little more than the tmRNA TLD; a similar molecule, whether or not chargeable with alanine, has been shown to strongly inhibit tmRNA, perhaps acting by titrating SmpB (Mao et al., 2009). For six genomes no tmRNA sequence could be identified: Carsonella ruddii PC, the four hemoplasmas of the Mycoplasma suis clade, and the secondary endosymbiont of Ctenarytaina eucalypti (Table 2). For C. ruddii PC, we further examine ssrA pseudogenization below.
Exhaustive Search for smpB
Upon characterization of SmpB as a 7-stranded β barrel, an oligonucleotide-binding (OB) fold was recognized for the region from β3-β7, hinting at possible ancient evolutionary relationships (Dong et al., 2002). However, based on comparisons of backbone coordinates, no other structures at PDB were found to be structurally similar (Dong et al., 2002). Likewise sequence based profiles, specifically the SmpB HMM from Pfam (a standalone family not part of a clan) and a set of 5 SmpB profiles available at the Conserved Domain Database (NCBI) show no interference with other family profiles; the SmpB family is bioinformatically well-behaved. It is a single-domain protein, except that four multi-domain architectures for five (of 4542) SmpBs are reported at Pfam. However, two of these can be explained as an artifactual double-SmpB call due to a 14-aa insert and an artifactual fusion arising from splicing a bacterial gene present in a eukaryotic genome project, while the other three may be explained by sequencing errors not found in related strains, that shifted the smpB frame to that of its upstream neighbor or fused it to the downstream CDS by converting the smpB stop codon to a sense codon.
The above genomes were searched using the SmpB profiles, and for the small number (n = 14) of bacterial genomes for which the profiles failed even below threshold, BLASTX was applied with our SmpB database; for Hodgkinia, comparative analysis with two newer genomes (below) was required to identify smpB (also identifying two new tmRNA sequences). All instances of smpB were on bacterial chromosomes, except for two copies found in Flavobacterium sp. KI723T1 plasmid pOAD2. Some genomes are deficient for smpB (Table 3). Tremblaya has truncations at both ends of smpB, so severe that they may inactivate the protein. Study of newer Hodgkinia genomes as described below identified an isolate that has accumulated two TAA stop codons in smpB. In 13 other strains single frameshifts would inactivate the genes, unless these may be sequencing errors; however in one case the authors discuss the pseudogene, suggesting confidence in its sequencing (Pérez-Brocal et al., 2006).
Some SmpBs show loss of important features, yet may retain some function, given that the β-barrel framework appears intact. The central loop region, which contacts the tmRNA tRNA-like domain and is thought to play roles in alanylation (Dong et al., 2002) and in activating EF-Tu (Miller and Buskirk, 2014), is missing in Carsonella and the hemoplasmas (hemotropic Mycoplasma). The C-terminal tail, of demonstrated importance for SmpB function (Mantri and Williams, 2004; Jacob et al., 2005; Garza-Sánchez et al., 2011), is lost or truncated in Carsonella. In the model Thermus SmpB, this tail is unstructured in solution, but helical when in place in the ribosomal A site with alanine-charged tmRNA (Neubauer et al., 2012). In this location it contacts the 16S rRNA decoding center and continues to follow the path normally occupied by downstream mRNA, yet must undergo major conformational change to make way for the resume codon in later trans-translation steps. Many SmpBs extend variably beyond the helical tail segment of Thermus, raising the question of accommodating this extension in the ribosome. Tropheryma (tmID: 14758) has the longest C-terminal extension, 44 extra residues; when we constrained Tropheryma SmpB to the corresponding Thermus portion (Kelley and Sternberg, 2009), its extension showed continued helical structure with some breaks.
We found 16 smpB instances in eukaryotic genome projects. Four of these can be described as bacterial: two appear to be from enterobacterial microbiome contamininants of the medfly and cucumber genomes, another is from the endosymbiont associated with the placozoan Trichoplax genome (Driscoll et al., 2013), and the fourth is from the quasi-organellar chromatophore of Paulinella that is a recently-captured cyanobacterium. The remaining eukaryotic SmpBs appear to be nuclear-encoded and organelle-targeted. Three are from oomycete genomes and score for the mitochondrial signal peptide, supporting the recent discovery of tmRNA genes in oomycete mitochondria (Hafez et al., 2013). Nine are from algal genomes whose plastids are known to encode tmRNA; for some of these the N-terminal plastid transit peptide sequences have been noted (Jacob et al., 2005), while in others transit peptide identification may require further search for 5′ exons.
smpB Gene Neighborhood
We examined the neighborhood of smpB, and found 11 frequent neighbor gene families (Figure 1A). ssrA is the most frequent neighbor of smpB, yet accounts for fewer than half the cases. The clustering of these neighbors was also examined (Figure 1B). The association with the ubiquitin homolog RnfH and RatA toxin unit genes has been previously noted (Iyer et al., 2006). Several of these common neighbors also interact with the ribosome (RF-2, SecG, and RatA). Furthermore, RNase R is known to be a physical and functional partner with tmRNA-SmpB (Karzai et al., 2000; Liang and Deutscher, 2010; Venkataraman et al., 2014). Transcript analysis has confirmed operon structure for some of these clusters (Mantri and Williams, 2004; Garza-Sánchez et al., 2011).
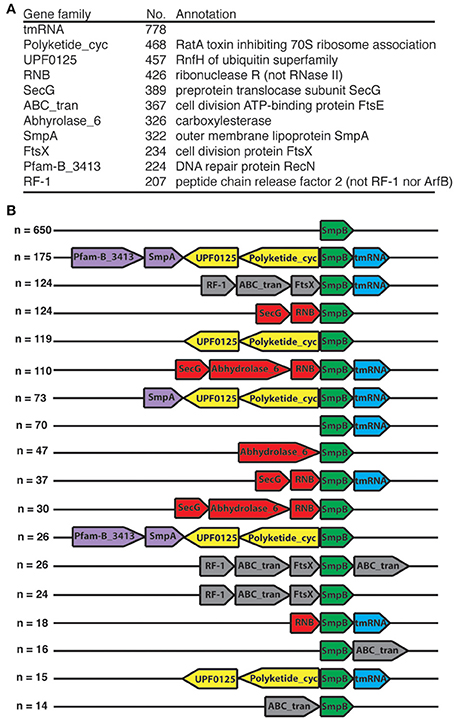
Figure 1. smpB gene neighborhoods. Each neighborhood (n = 2012) in our bacterial complete genome set was taken as the 11-gene window centered at smpB. (A) Frequent neighbors. The tmRNA gene (the only RNA gene encountered) and Pfam families present in more than 200 smpB neighborhoods are listed with a representative annotation for the instances of each family. (B) Clusters. Each neighborhood was summarized as a cluster, considering only the families of (A) (note the more specific gene annotations there). The top clusters are shown with color coding of common subclusters.
The tmRNA Website
The tmRNA Website (De Novoa and Williams, 2004) (http://bioinformatics.sandia.gov/tmrna) provides several research tools. Foremost is the sequence database. The previous instance of the database was updated with the above search results, and with the recently-described oomycete sequences, yielding 1631 unique sequences (1384 encoding one-piece tmRNA and 247 two-piece tmRNA); most are bacterial except for 41 mitochondrial and 22 plastid unique tmRNA sequences. These tmRNAs encode 710 unique proteolysis tag sequences. Each sequence was then used as BLAST query against NCBI est, gss, htgs, nt, other_genomic, patnt, refseq_genomic, tsa_nt and wgs databases, yielding 9167 instances of perfect though occasionally incomplete matches, counting each RefSeq/GenBank cross-reference pair as a single instance. The tmRNA Website provides all these sequences for download or for query by BLAST. These were also provided to RNAcentral (Bateman et al., 2011) and as third-party annotation to the International Nucleotide Sequence Database Archives (GenBank/ENA/DDBJ). Related resources that should be consulted are tmRDB (Andersen et al., 2006), Rfam (Burge et al., 2013), and RNAcentral (Bateman et al., 2011).
The tmRNA Website includes a new SmpB database with 2258 distinct amino acid sequences. These are available for BLAST search and download, as an alignment, as raw sequence and as a database. SmpB sequence is presented together with tmRNA sequences found in the same genome.
Anomalies in Carsonella
Carsonella ruddii is an insect endosymbiont, with extremely small (157–174 kbp) and AT-rich (14–18% GC) genomes, yet virtually no rearrangement of gene order (Sloan and Moran, 2012). The loss of the central loop and C-terminal tail of C. ruddii SmpB were noted above. When only one Carsonella tmRNA sequence was available, it was difficult to identify its tag reading frame. With several new sequences from additional strains, the tag reading has now been identified, standing out as the most conserved reading frame among the strains (Figure 2). C. ruddii is the only species encoding a tag ending in a charged residue (lysine), which hindered previous tag identification, however some strains do have as usual a hydrophobic terminal tag residue.
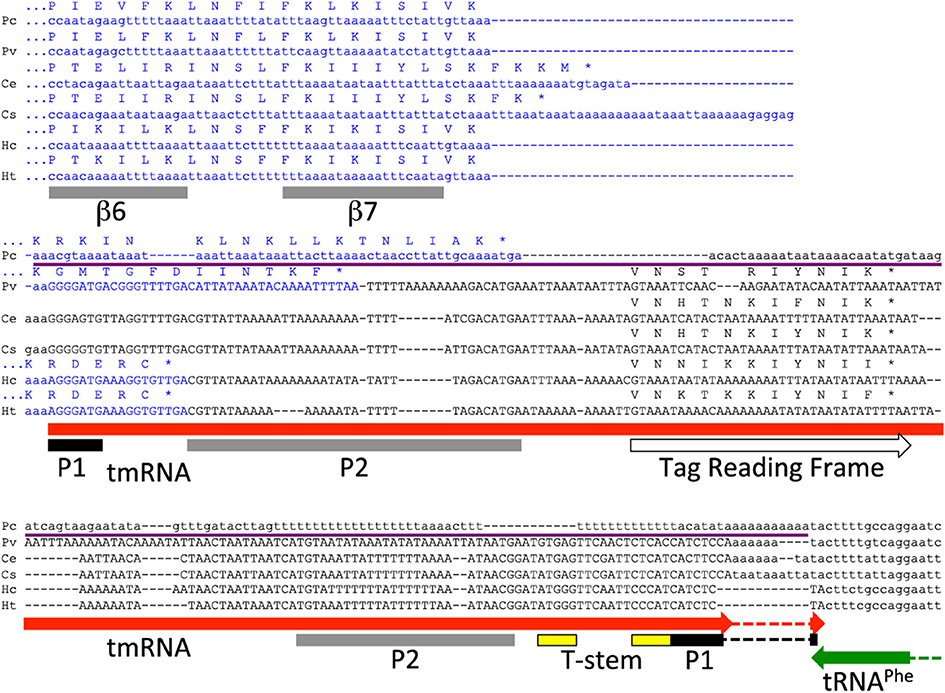
Figure 2. Carsonella smpB-ssrA: pseudogenization, neighbor gene overlap, and comparative detection of the tag reading frame. In strain PC, the three main ssrA conserved regions, at the 5′ and 3′ termini and at the tag reading frame, have all suffered so many nucleotide changes as to be unrecognizable, yet the region is largely still present. The smpB CDS (blue) extends into ssrA (expected to produce non-stop smpB mRNAs) or the ssrA pseudogene in four cases. In the HC/HT lineage, a small deletion has caused ssrA to overlap with its downstream and oppositely-oriented neighboring tRNAPhe gene changing the last tmRNA acceptor stem (P1) nucleotide from C to U, which apparently led to a compensating G to A mutation at the first P1 nucleotide. The tag reading frame has now been determined by comparative analysis as the most conserved reading frame in ssrA, that also shares some amino acid similarity to other tag sequences. Carsonella SmpB lacks the central loop (not shown here) and the C-terminal tail, which in Thermus is a 25-residue segment following β7. The C-terminus of SmpB does extend variably beyond β7 with apparently random amino acid sequence that depends on the extent of intrusion into ssrA, but these extensions are not as long as for normal SmpBs and they do not thread into the α helix model (Kelley and Sternberg, 2009).
It was previously noted that smpB overlaps ssrA in Carsonella (Mao et al., 2009). This sets up an interesting feedback situation where the smpB mRNA would be cleaved by tmRNA maturation, and thereby become a non-stop substrate for the action of its own gene product. However, this situation is not widespread; we found it nowhere else but in Carsonella, and in only half of the Carsonella strains.
All tmRNAs in our database and indeed all bacterial tRNA-Ala at the Genomic tRNA Database (Chan and Lowe, 2009) have a terminal G:C base pair closing the acceptor stem, except for the tmRNAs of the C. ruddii HC/C. ruddii HT lineage. This anomaly is apparently due to a small deletion causing a 2-nt overlap between the 3′ termini of ssrA and the oppositely oriented tRNA-Phe gene, that changed the terminal residue of the tmRNA acceptor stem from the usual C to U (Figure 2). A base substitution mutation reverting this U back to C would have altered the discriminator base of tRNA-Phe; instead the deletion apparently drove the fixation of a compensatory mutation at the far end of ssrA producing the unique A:U closing base pair, which may allow better recognition by alanyl-tRNA synthetase than the post-deletion G:U pair would.
Although there were six complete bacterial genomes in which we failed to find tmRNA sequences, the genome of C. ruddii PC presents an especially clear case of pseudogenization. Because C. ruddii genomes show no rearrangement of gene order (Sloan and Moran, 2012), the site of any ssrA remnant could be predicted. An anchored segment (thin purple line in Figure 2) of the closely related C. ruddii PV genome is 216 bp (within which the tmRNA sequence occupies 202 bp); the corresponding segment in PC is 178 bp. This pseudogenization thus appears to have occurred largely in place and not by major deletion. The thoroughness of obliteration is remarkable; none of the most conserved regions of ssrA have been retained, neither for the 5′ tRNA-like domain, the resume codon region, nor the 3′ tRNA-like domain. Nucleotide bias has increased with this pseudogenization: GC content of the anchored region drops from and 17.6% in PV to 13.5% in PC. We expected that without tmRNA, selective constraint on smpB would relax in PC, but there is no evidence for this. The 181 orthologous protein-coding gene pairs shared between the close relatives C. ruddii PV (which encodes tmRNA) and C. ruddii PC (which does not) have already been evaluated for selective regime, revealing that they are generally under a purifying selection regime with low dN/dS ratios (Sloan and Moran, 2012). For smpB, the dN/dS value is 0.14 (D. Sloan, pers. comm.), in the middle of the peak of the dN/dS distribution for all genes. This indicates that relative to other genes, purifying selection is not relaxed in PC for smpB, even after the loss of its partner ssrA. Perhaps ssrA loss was too recent to detect follow-on relaxation at smpB.
Neither ribosome rescue backup system seems available to compensate for ssrA loss; C. ruddii PC had no detectable ArfA while its two matches to ArfB gave much stronger matches to the better conserved proteins RF-1 and RF-2.
Hodgkinia
Hodgkinia cicadicola is an insect endosymbiont with an extremely reduced (134–144 kbp) genome of balanced nucleotide composition (46–58% GC), and it uses UAG as a Trp codon rather than Stop (McCutcheon and Moran, 2011). Despite applying the profiles and BLAST at highest sensitivity, considering its unusual genetic code, and specifically searching in the ssrA vicinity we could not find smpB when only the H. cicadicola Dsem genome was available. With the recent arrival of two new genomes, one, H. cicadicola TETUND2, gave low but consistent signals with the profiles, identifying smpB and leading to identification in the other two genomes. All three SmpBs lack the central loop. H. cicadicola Dsem may also have lost the C-terminal tail. The H. cicadicola TETUND1 smpB has further accumulated two TAA stop codons and we therefore classify it as a pseudogene.
Anomalies in Mycoplasma
The third group we find lacking the SmpB central loop is the hemoplasmas (hemotropic Mycoplasma), which also have reduced genomes. We prepared a genome-based phylogenetic tree for Mycoplasma (Figure 3) that included 7 hemoplasmas, which formed a clade in the tree with two main subclades, in agreement with (Guimaraes et al., 2014) who named the two subclades haemofelis and suis. We were unable to identify the tmRNA gene nor its trace in any of the four genomes of the suis clade. The haemofelis clade did not help locate it because the haemofelis ssrA region (greA/ssrA/Hyp/rplQ/rpoA) is rearranged in the suis clade as greA/X/trmD/rpoA (where X is an 18 kbp insert of 26 hypothetical genes in M. wenyonii).
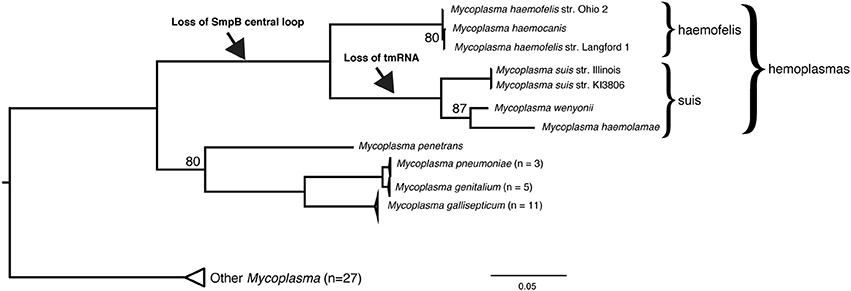
Figure 3. smpB and ssrA in hemoplasmas. The hemoplasmas have lost the SmpB central loop and for the suis subclade we cannot find the tmRNA gene. Genomes of 54 Mycoplasma strains were aligned using Mugsy (Angiuoli and Salzberg, 2011), yielding only the rRNA operon region as alignable for all strains; this was trimmed to 1679 bp using GBlocks requiring at least half the taxa per column (Castresana, 2000), then a maximum likelihood tree was prepared using a GTR+Γ model and autoFC bootstopping in RAxML 7.2.8 (Stamatakis, 2006). The hemoplasma clade and phylogenetic surroundings agree with recent 32-protein and 16S rRNA phylogenies (Guimaraes et al., 2014).
Non-stop mRNAs Due to t(m)RNA Gene Overlap
The observation of smpB overlap with ssrA in Carsonella led us to ask how many mRNAs might become non-stop due to maturation of CDS-overlapping tmRNA or tRNA genes (Table 4). Others have found high-frequency non-stop mRNA caused by an RNase III site in arfA (Garza-Sánchez et al., 2011). We considered only the proteins positive for Pfam-A families, which account for 75.0% of the bacterial proteins studied, and for comparison included “questionable” tRNAs (probably mostly false positives) and oppositely oriented CDS/RNA gene pairs. We consider the 379 same-orientation overlaps of valid t(m)RNA genes as candidates for producing high-frequency non-stop mRNAs, although those with the CDS downstream of the RNA gene are suspicious; they may result from calling the start codon too far upstream. This represents an exceedingly small fraction of mRNAs tested (~1 in 15000). The top Pfam families among these candidates represent few evolutionary events, mostly affecting the same tRNA gene in a closely related group of genomes.
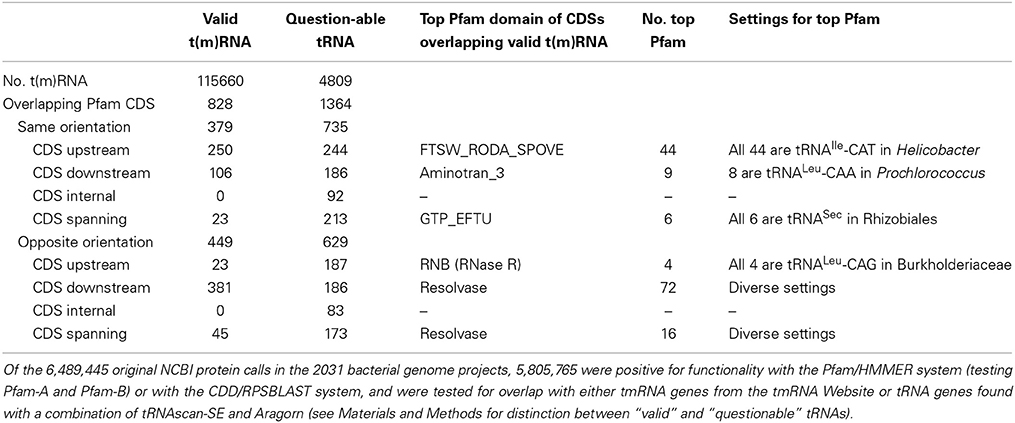
Table 4. Functional protein CDSs that overlap t(m)RNA genes.
Discussion
It is generally thought that neither tmRNA nor SmpB can function without the other (Sundermeier and Karzai, 2007; Felden and Gillet, 2011), although there are some counter-examples; e.g., smpB but not ssrA can be knocked out in Mycobacterium tuberculosis (Personne and Parish, 2014). Among the six bacteria that appear to lack tmRNA and 16 that appear to lack SmpB, none lack both; cofunction would predict eventual concomitant loss. In one case of tmRNA loss that we examined, selective constraint did not appear to relax for the remaining smpB. Both for tmRNA and SmpB, there may be more independent function than has been recognized.
The tmRNA literature cautions against reporting failure to find genes, and it is of course possible that our detection methods were inadequate or that genome sequences have errors, but we may be starting to identify bacteria that truly lack tmRNA or SmpB. These bacteria tend to have highly reduced genomes that have lost many genes otherwise widely conserved. It can morever be noted that tmRNA-SmpB is lacking in most mitochondria and plastids, which likewise have highly reduced genomes derived from bacteria. Thus, tmRNA-SmpB is not always required in bacteria or their descendents. Those organelles where we can detect the system fit this pattern: the RNA gene is retained in the organelle and can be traced to the organelle's ancestral bacterial group, while the partner protein gene resides in the nucleus, encoding the appropriate organellar import peptide. Intracellular but non-organellar bacteria do not have this luxury of passing genes to the nucleus for safekeeping. However, nucleus-stored organellar proteins need not always derive from the organelle's ancestor. In our preliminary phylogenetic tree of SmpB (not shown), the plastid SmpBs did cluster with Cyanobacteria, but the mitochondrial SmpBs clustered apart from the Alphaproteobacteria.
The ArfA and ArfB backup systems for ribosome rescue are not of wide enough phylogenetic distribution to explain all the tmRNA or SmpB losses noted here, although a mitochondrial ArfB homolog has been reported (Richter et al., 2010), and additional analogs, homologs or backup systems may yet be discovered. The current data suggest that neither the primary nor the backup ribosome rescue systems are required in all bacteria.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Daniel Sloan (Yale U.) for detailed data on Carsonella dN/dS values. This research was fully supported by the Laboratory Directed Research and Development program at Sandia National Laboratories. Sandia National Laboratories is a multi-program laboratory managed and operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Corporation, for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-AC04-94AL85000.
References
Andersen, E. S., Rosenblad, M. A., Larsen, N., Westergaard, J. C., Burks, J., Wower, I. K., et al. (2006). The tmRDB and SRPDB resources. Nucleic Acids Res. 34, D163–D168. doi: 10.1093/nar/gkj142
Angiuoli, S. V., and Salzberg, S. L. (2011). Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics 27, 334–342. doi: 10.1093/bioinformatics/btq665
Bateman, A., Agrawal, S., Birney, E., Bruford, E. A., Bujnicki, J. M., Cochrane, G., et al. (2011). RNAcentral: a vision for an international database of RNA sequences. RNA 17, 1941–1946. doi: 10.1261/rna.2750811
Bessho, Y., Shibata, R., Sekine, S.-I., Murayama, K., Higashijima, K., Hori-Takemoto, C., et al. (2007). Structural basis for functional mimicry of long-variable-arm tRNA by transfer-messenger RNA. Proc. Natl. Acad. Sci. U.S.A. 104, 8293–8298. doi: 10.1073/pnas.0700402104
Burge, S. W., Daub, J., Eberhardt, R., Tate, J., Barquist, L., Nawrocki, E. P., et al. (2013). Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 41, D226–D232. doi: 10.1093/nar/gks1005
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Bio. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chadani, Y., Ito, K., Kutsukake, K., and Abo, T. (2012). ArfA recruits release factor 2 to rescue stalled ribosomes by peptidyl-tRNA hydrolysis in Escherichia coli. Mol. Microbiol. 86, 37–50. doi: 10.1111/j.1365-2958.2012.08190.x
Chadani, Y., Ono, K., Kutsukake, K., and Abo, T. (2011). Escherichia coli YaeJ protein mediates a novel ribosome−rescue pathway distinct from SsrA-and ArfA-mediated pathways. Mol. Microbiol. 80, 772–785. doi: 10.1111/j.1365-2958.2011.07607.x
Chadani, Y., Ono, K., Ozawa, S. I., Takahashi, Y., Takai, K., Nanamiya, H., et al. (2010). Ribosome rescue by Escherichia coli ArfA (YhdL) in the absence of trans-translation system. Mol. Microbiol. 78, 796–808. doi: 10.1111/j.1365-2958.2010.07375.x
Chan, P. P., and Lowe, T. M. (2009). GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 37, D93–D97. doi: 10.1093/nar/gkn787
De Novoa, P. G., and Williams, K. P. (2004). The tmRNA website: reductive evolution of tmRNA in plastids and other endosymbionts. Nucleic Acids Res. 32, D104–D108. doi: 10.1093/nar/gkh102
Dong, G., Nowakowski, J., and Hoffman, D. W. (2002). Structure of small protein B: the protein component of the tmRNA–SmpB system for ribosome rescue. EMBO J. 21, 1845–1854. doi: 10.1093/emboj/21.7.1845
Driscoll, T., Gillespie, J. J., Nordberg, E. K., Azad, A. F., and Sobral, B. W. (2013). Bacterial DNA sifted from the Trichoplax adhaerens (Animalia: Placozoa) genome project reveals a putative rickettsial endosymbiont. Genome Biol. Evol. 5, 621–645. doi: 10.1093/gbe/evt036
Felden, B., and Gillet, R. (2011). SmpB as the handyman of tmRNA during trans-translation. RNA Biol. 8, 440–449. doi: 10.4161/rna.8.3.15387
Garza-Sánchez, F., Schaub, R. E., Janssen, B. D., and Hayes, C. S. (2011). tmRNA regulates synthesis of the ArfA ribosome rescue factor. Mol. Microbiol. 80, 1204–1219. doi: 10.1111/j.1365-2958.2011.07638.x
Griffiths-Jones, S., Moxon, S., Marshall, M., Khanna, A., Eddy, S. R., and Bateman, A. (2005). Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124. doi: 10.1093/nar/gki081
Guimaraes, A. M. S., Santos, A. P., Do Nascimento, N. C., Timenetsky, J., and Messick, J. B. (2014). Comparative genomics and phylogenomics of hemotrophic Mycoplasmas. PLoS ONE 9:e91445. doi: 10.1371/journal.pone.0091445
Hafez, M., Burger, G., Steinberg, S. V., and Lang, F. (2013). A second eukaryotic group with mitochondrion-encoded tmRNA: in silico identification and experimental confirmation. RNA Biol. 10, 1117–1124. doi: 10.4161/rna.25376
Handa, Y., Inaho, N., and Nameki, N. (2011). YaeJ is a novel ribosome-associated protein in Escherichia coli that can hydrolyze peptidyl–tRNA on stalled ribosomes. Nucleic Acids Res. 39, 1739–1748. doi: 10.1093/nar/gkq1097
Iyer, L. M., Burroughs, A. M., and Aravind, L. (2006). The prokaryotic antecedents of the ubiquitin-signaling system and the early evolution of ubiquitin-like β-grasp domains. Genome Biol. 7, R60. doi: 10.1186/gb-2006-7-7-r60
Jacob, Y., Sharkady, S. M., Bhardwaj, K., Sanda, A., and Williams, K. P. (2005). Function of the SmpB tail in transfer-messenger RNA translation revealed by a nucleus-encoded form. J. Biol. Chem. 280, 5503–5509. doi: 10.1074/jbc.M409277200
Karzai, A. W., Roche, E. D., and Sauer, R. T. (2000). The SsrA–SmpB system for protein tagging, directed degradation and ribosome rescue. Nat. Struct. Mol. Biol. 7, 449–455. doi: 10.1038/75843
Karzai, A. W., and Sauer, R. T. (2001). Protein factors associated with the SsrA· SmpB tagging and ribosome rescue complex. Proc. Natl. Acad. Sci. U.S.A. 98, 3040–3044. doi: 10.1073/pnas.051628298
Kelley, L. A., and Sternberg, M. J. E. (2009). Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protocol. 4, 363–371. doi: 10.1038/nprot.2009.2
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Laslett, D., Canback, B., and Andersson, S. (2002). BRUCE: a program for the detection of transfer−messenger RNA genes in nucleotide sequences. Nucleic Acids Res. 30, 3449–3453. doi: 10.1093/nar/gkf459
Liang, W., and Deutscher, M. P. (2010). A novel mechanism for ribonuclease regulation transfer-messenger RNA (tmRNA) and its associated protein SmpB regulate the stability of RNase R. J. Biol. Chem. 285, 29054–29058. doi: 10.1074/jbc.C110.168641
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 0955–0964. doi: 10.1093/nar/25.5.0955
Mantri, Y., and Williams, K. P. (2004). Islander: a database of integrative islands in prokaryotic genomes, the associated integrases and their DNA site specificities. Nucleic Acids Res. 32, D55–D58. doi: 10.1093/nar/gkh059
Mao, C., Bhardwaj, K., Sharkady, S. M., Fish, R. I., Driscoll, T., Wower, J., et al. (2009). Variations on the tmRNA gene. RNA Biol. 6, 355–361. doi: 10.4161/rna.6.4.9172
McCutcheon, J. P., and Moran, N. A. (2011). Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13–26. doi: 10.1038/nrmicro2670
Miller, M. R., and Buskirk, A. R. (2014). An unusual mechanism for EF-Tu activation during tmRNA-mediated ribosome rescue. RNA 20, 228–235. doi: 10.1261/rna.042226.113
Neubauer, C., Gillet, R., Kelley, A. C., and Ramakrishnan, V. (2012). Decoding in the absence of a codon by tmRNA and SmpB in the ribosome. Science 335, 1366–1369. doi: 10.1126/science.1217039
Pérez-Brocal, V., Gil, R., Ramos, S., Lamelas, A., Postigo, M., Michelena, J. M., et al. (2006). A small microbial genome: the end of a long symbiotic relationship? Science 314, 312–313. doi: 10.1126/science.1130441
Personne, Y., and Parish, T. (2014). Mycobacterium tuberculosis possesses an unusual tmRNA rescue system. Tuberculosis 94, 34–42. doi: 10.1016/j.tube.2013.09.007
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Richter, R., Rorbach, J., Pajak, A., Smith, P. M., Wessels, H. J., Huynen, M. A., et al. (2010). A functional peptidyl-tRNA hydrolase, ICT1, has been recruited into the human mitochondrial ribosome. EMBO J. 29, 1116–1125. doi: 10.1038/emboj.2010.14
Sloan, D. B., and Moran, N. A. (2012). Genome reduction and co-evolution between the primary and secondary bacterial symbionts of psyllids. Mol. Bio. Evol. 29, 3781–3792. doi: 10.1093/molbev/mss180
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Sundermeier, T. R., and Karzai, A. W. (2007). Functional SmpB-ribosome interactions require tmRNA. J. Biol. Chem. 282, 34779–34786. doi: 10.1074/jbc.M707256200
Keywords: tmRNA, SmpB, trans-translation, Carsonella, Mycoplasma
Citation: Hudson CM, Lau BY and Williams KP (2014) Ends of the line for tmRNA-SmpB. Front. Microbiol. 5:421. doi: 10.3389/fmicb.2014.00421
Received: 01 June 2014; Accepted: 24 July 2014;
Published online: 13 August 2014.
Edited by:
Kenneth C. Keiler, Pennsylvania State University, USAReviewed by:
Pavel V. Baranov, University College Cork, IrelandTorsten Hain, University of Giessen, Germany
Copyright © 2014 Hudson, Lau and Williams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelly P. Williams, Sandia National Laboratories, Department of Systems Biology, 7011 East Ave., Livermore, CA 94550, USA e-mail: kpwilli@sandia.gov