 Jin-Feng Liu
Jin-Feng Liu Xiao-Bo Sun
Xiao-Bo Sun Guang-Chao Yang
Guang-Chao Yang Serge M. Mbadinga
Serge M. Mbadinga Ji-Dong Gu
Ji-Dong Gu Bo-Zhong Mu
Bo-Zhong Mu- 1State Key Laboratory of Bioreactor Engineering and Institute of Applied Chemistry, East China University of Science and Technology, Shanghai, China
- 2School of Biological Sciences, University of Hong Kong, Hong Kong, China
Sequestration of CO2 in oil reservoirs is considered to be one of the feasible options for mitigating atmospheric CO2 building up and also for the in situ potential bioconversion of stored CO2 to methane. However, the information on these functional microbial communities and the impact of CO2 storage on them is hardly available. In this paper a comprehensive molecular survey was performed on microbial communities in production water samples from oil reservoirs experienced CO2-flooding by analysis of functional genes involved in the process, including cbbM, cbbL, fthfs, [FeFe]-hydrogenase, and mcrA. As a comparison, these functional genes in the production water samples from oil reservoir only experienced water-flooding in areas of the same oil bearing bed were also analyzed. It showed that these functional genes were all of rich diversity in these samples, and the functional microbial communities and their diversity were strongly affected by a long-term exposure to injected CO2. More interestingly, microorganisms affiliated with members of the genera Methanothemobacter, Acetobacterium, and Halothiobacillus as well as hydrogen producers in CO2 injected area either increased or remained unchanged in relative abundance compared to that in water-flooded area, which implied that these microorganisms could adapt to CO2 injection and, if so, demonstrated the potential for microbial fixation and conversion of CO2 into methane in subsurface oil reservoirs.
Introduction
Storage of CO2 in deep geological formations, such as oil reservoirs, is one of the feasible measures to reducing CO2 emissions into the atmosphere. Understanding the fate of CO2 in the subsurface environment is of great scientific interest and significance, and has received increasing attention for more information to assess the feasibility. Due to the fact that abundant microorganisms inhabit in these formations, microbial fixation and conversion of the sequestered CO2 into CH4 are becoming an area of active research and development.
After CO2 injection, characteristics of the formation water may be changed by CO2 dissolution, including pH, the availability of inorganic and organic components in the brine, microbial attachment and biofilm formation as well as the microbial activities at in situ oil reservoirs. Generally, as CO2 is also a potential source of carbon of chemolithoautotrophic microorganisms such as methanogens, the injected CO2 may activate these microorganisms and notably influence the microbial structure and their activity in situ. Studies have been performed on the physical and chemical changes in the CO2 storage sites. The first on-shore CO2 storage site in Europe was established, and the effects and feasibility of CO2 injection and storage in a 650m deep saline aquifer was examined (Wandrey et al., 2011). The potential of microbial conversion of CO2 into CH4 by hydrogenotrophic methanogens isolated from oil reservoirs has been evaluated based on laboratory experiments by Sugai et al. (2012). As to the microbial involvement, six autotrophic CO2 fixation pathways were documented, of which the Calvin–Benson–Bassham (CBB) cycle plays an important role in autotrophic CO2 fixation (Berg, 2011). The CBB biochemical process was reported to occur in Proteobacteria, including some members of Firmicutes, Actinobacteria, and Chloroflexi as well as in plants, algae, and cyanobacteria (Ivanovsky et al., 1999; Zakharchuk et al., 2003; Berg et al., 2005; Caldwell et al., 2007; Lee et al., 2009). Another important pathway of CO2 fixation is the reductive acetyl-CoA pathway that has documented to occur in acetogenic prokaryotes, ammonium-oxidizing Planctomycetes (Strous et al., 2006), sulfidogenic bacteria (Schauder et al., 1988), and autotrophic archaea affiliated with the order Archaeoglobales (Vorholt et al., 1995, 1997). This pathway is also utilized by acetogenic prokaryotes for energy conservation (Ragsdale and Pierce, 2008; Thauer et al., 2008; Biegel and Muller, 2010).
Petroleum reservoirs are known to harbor diverse microorganisms including bacteria such as Proteobacteria, Firmicutes, Actinobacteria, and Chloroflexi and archaea such as methanogens and Archaeoglobales mentioned above (Magot et al., 2000; Li et al., 2010, 2011; Wang et al., 2011; Mbadinga et al., 2012) and they are expected to fix and/or convert CO2 into CH4 more effectively. To investigate whether oil reservoirs have the potential of CO2 biofixation and bioconversion of CO2 into CH4, and to have a better knowledge on microorganisms involved in this process and the impact of long-term CO2 exposure on them, studies from a viewpoint of functional genes are necessary. Functional genes involved in CO2 fixation and conversion into CH4 have been shown to be valuable functional biomarkers for detecting the microbial communities both in environments and enrichment cultures. The genes cbbL and cbbM respectively encoding the key enzymes ribulose 1,5-bisphosphate carboxylase/oxygenase (RubisCO) form I and II of the CBB cycle for CO2 fixation have been used to study microbial communities from hydrothermal vents of the Logatchev field (Hugler et al., 2010). The gene fthfs encoding formyltetrahydrofolate synthetase, a key enzyme in the reductive acetyl-CoA pathway, has been used to investigate the diversity of homoacetogenic bacteria in thermophilic and mesophilic anaerobic sludge (Ryan et al., 2008). Methyl-Coenzyme M reductase (mcr) is vital for CH4 formation, and the α-subunit of MCR (mcrA gene) is commonly used in the detection of specific groups of methanogenic communities (Juottonen et al., 2006). In addition, H2 should be supplied in the process of CO2 conversion into CH4. H2 can be produced by H2-producing prokaryotes which are polyphyletic. [Fe-Fe]-hydrogenases are known to catalyze H2 production in fermentative microorganisms, and thus gene encoding for [Fe-Fe]-hydrogenases represent a good marker gene for the detection of H2-producing anaerobes (Schmidt et al., 2010). These valuable functional biomarkers involved in CO2 fixation and conversion into CH4 are shown in Figure 1.
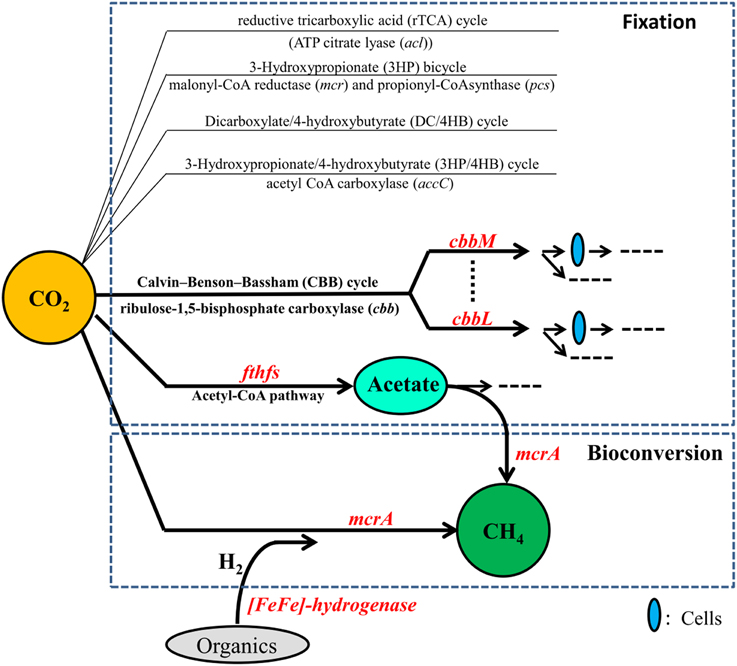
Figure 1. Genes and pathways for CO2 fixation and bioconversion into CH4.
The objectives of this study were to evaluate the potential of in situ microbial fixation and conversion of CO2 into CH4 in subsurface oil reservoir through analysis of functional genes (cbbM, cbbL, fthfs, gene encoding by [FeFe]-hydrogenase, and mcrA) by: (1) characterization of the functional microbial communities involved in this process in the production waters from CO2-flooded and water-flooded areas, respectively, of the same high-temperature oil-bearing bed in Daqing Oilfield; and (2) Analysis of the impact of long-term exposure of CO2 on these functional microbial communities.
Materials and Methods
Sampling Site and Production Water Samples
Production water samples were collected from production wells (designated as C and W) in YSL block of Daqing oilfield, China. At that time, the water cut of fluid from C and W production wells were 15 and 11%, respectively. The CO2 injected had been produced from the sampling well about 1 year before, and the ratio of gas (CO2) to oil was between 22.8 and 145 m3/m3 in production wells. The distance between injection well and the sampling production well is about 250 m. These wells produced oil from the same oil-bearing bed but C is located in the area subjected to CO2 flooding since 2007, whereas W by water-flooding only. To date, about 100,000m3 of liquid CO2 have been injected into the oil-bearing bed with an average injection rate of 10 m3/d per injection well in a manner of CO2-H2O alternate injection for Enhancement of Oil Recovery (EOR). These samples were taken through sampling valves located at the wellhead (average temperature 45°C) and put into 5L sterile bottles, respectively, to fullness, and then capped and sealed to maintain anoxic conditions. The bottles were kept at 4°C before further treatment. The in situ temperature and pressure of the target oil-bearing bed with a depth of about 2000m were about 90°C and 19 MPa, respectively. The average density of oil in this oil-bearing bed is 0.8581 g/cm3 and the information of the production water is listed in Table 1.
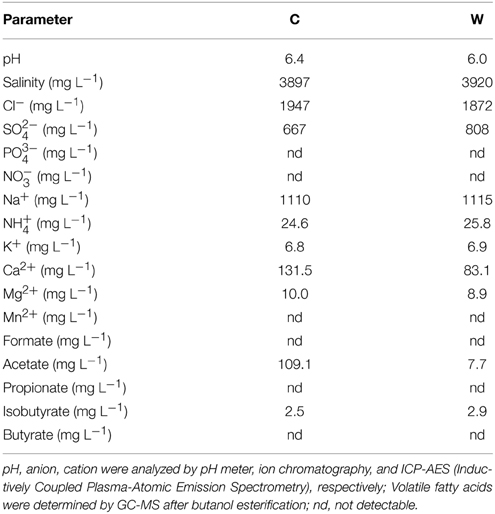
Table 1. Characteristics of the production water samples.
DNA Extraction
DNA was extracted from the oil/water sample according to the method previously described by Wang et al. (2012). Briefly, the water phase was separated from the oil/water mixture by heating the samples to 50°C and by phase separation in sterilized separatory funnels. The microbial biomass in the water fraction was concentrated onto membrane filter (0.22-μ m-pore-size). Total genomic DNA of samples was extracted from 2.0 L of production water samples using AxyPrep™ Bacterial Genomic DNA Miniprep Kit (Axygen Biosciences, Inc., CA, USA) according to the manufacturer's DNA Miniprep spin protocol after concentration onto membrane filters. The genomic DNAs obtained were purified with a DNA purification kit (U-gene, China) according to the manufacturer's instructions. The extracted DNAs were stored at −20°C until PCR amplification of different functional genes (Wang et al., 2012).
PCR Amplifications
Amplifications of the cbbL gene fragment (771 bp) and the cbbM gene fragment (328 bp) were carried out under the conditions described by Campbell and Cary (2004). For amplification of a portion (1102 bp) of the fthfs gene, the PCR conditions used were those described previously by Leaphart and Lovell (2001). For amplification of a fragment (620 bp) of [Fe-Fe]-hydrogenase-encoding gene, the PCR primer set HydH1f/HydH3r was applied using the conditions described by Schmidt et al. (2010). A fragment (470 bp) of the mcrA genes was amplified using the primer set MLf/MLr (Luton et al., 2002) with the conditions as reported previously (Galand et al., 2005). Functional genes fragments were all amplified in five parallel PCR reactions in a Peltier thermal cycler (Bio-Rad, USA), which were subsequently pooled for cloning and construction of genes libraries.
Construction of Functional Genes Clone Libraries
The amplified and pooled PCR products were gel-purified using the Gel Extraction Kit (U-gene, China) and then cloned into Escherichia coli using a pMD19®-T simple vector kit (Takara, Japan) following the instructions of the manufacturer. For each gene clone library, the white colonies obtained were randomly picked and cultured overnight at 37°C in 0.8 ml Luria broth (LB) medium supplemented with ampicillin (50 μg ml−1). The inserted DNAs were amplified by using M13-47 (5′-CGCCAGGGTTTTCCCAGTCACGAC-3′) and RV-M (5′-GAGCGGATAACAATTTCACACAGG-3′) primers targeting the flanking vector sequence, followed by agarose gel electrophoresis with ethidium bromide staining (Guan et al., 2013).
Sequencing and Phylogenetic Analyses
Sequencing was carried out with an ABI 377 automated sequencer. After sequencing, reads were first trimmed for vector before subsequent analyses. Bellerophon was used to check for putative chimeric sequences (Huber et al., 2004). DNA sequences with more than 97% similarity were assembled into the same operational taxonomic units (OTUs) using FastGroup II (Yu et al., 2006), and one representative sequence was chosen from each OTU to compare with sequences in the GenBank Database using the BLASTX algorithm to identify nearest related ones (Altschul et al., 1997). Representative OTUs from clone libraries as well as reference sequences from GenBank were translated into corresponding amino acid sequences using EMBOSS Transeq tool (http://www.ebi.ac.uk/Tools/st/emboss_transeq/) with default parameter (Standard Genetic Code) and then aligned using Clustal Omega (Sievers et al., 2011). Phylogenetic trees were generated using MEGA5 software (Tamura et al., 2011). The topology of the trees was obtained by the Neighbor-Joining method (Saitou and Nei, 1987) with the Poisson correction method and 1000 bootstrap replicates were applied to estimate the support for the nodes in the tree.
Nucleotide Sequence Accession Numbers
Gene sequences data reported here are available in GenBank sequence database under the accession numbers KF111435–KF111455, KF111525–KF111548, KF111456–KF111492, KF111493–KF111501, and KF111502–KF111524 for cbbM gene, cbbL gene, mcrA gene, fthfs gene, and gene encoded by [Fe-Fe]-hydrogenase.
Results
Characterization of Clone Libraries
cbbL and cbbM genes
The cbbL gene types were positively detected in all two kinds of samples (Figure 2). The cbbL gene clone libraries from sample C and W resulted in 11 and 13 OTUs, respectively, and the PCR amplified sequences are spread over the entire tree. Phylogenetic analysis indicates that the cbbL gene sequences obtained are related to those of Alpha-, Beta-, and Gamma-Proteobacteria. One OTU (cbbL-C2-18) is closely related to Hydrogenophaga sp. CL3 affiliated to the family Comamonadaceae within Beta-Proteobacteria (Garcia-Dominguez et al., 2008). The sequences of cbbL-C1-13, cbbL-C1-17, and cbbL-W4-9 all share high similarity with Cupriavidus metallidurans CH34 belonging to the family Burkholderiaceae within Beta-Proteobacteria. The sequence of cbbL-W3-24 shares high identity with endosymbiont of Bathymodiolus azoricus (Spiridonova et al., 2006), a member of Gamma-Proteobacteria. One OTU represented by cbbL-W4-12 shows highest identity with an uncultured bacterium from iron-rich environment (Kellermann et al., 2012).
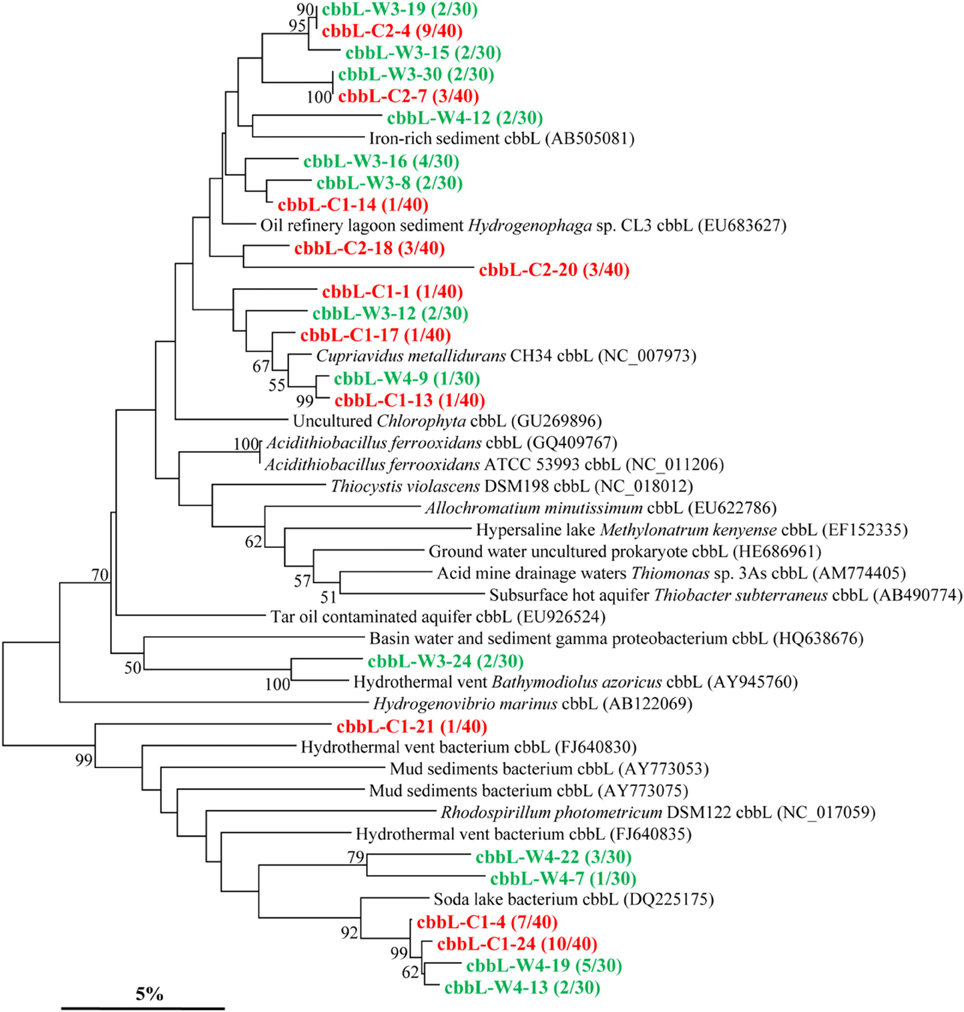
Figure 2. Phylogenetic tree of the cbbL gene retrieved from the water samples (shown in colored) and closely related sequences from GenBank database. Alignments to related sequences (shown with accession number) were performed with MEGA 5 software. The topology of the tree was obtained with the neighbor-joining method. Bootstrap values (n = 1000 replicates) greater than 50% are reported. Scale bar represents 5% amino acid substitution.
Similarly, the cbbM gene types were also detected in these two samples and yielded 10 and 11 OTUs in C and W, respectively (Figure 3). The cbbM sequences detected are all very similar to those from organisms affiliated with members of Alpha-, Beta-, and Gamma-Proteobacteria. The sequence of cbbM-C1-3 is related to an uncultured bacterium from cave water of Romania (Chen et al., 2009). The OTUs represented by cbbM-W4-22, cbbM-W4-32, cbbM-W3-12, and cbbM-C2-9 are closely related to uncultured bacterium from tar contaminant aquifer and MTBE and ammonium polluted groundwater (Alfreider et al., 2012). Sequences represented by cbbM-C2-21, cbbM-C1-13, and cbbM-C1-7 all share similarities with those recovered from the East China Sea and basin water and sediment. Interestingly, these sequences are also closely related to Halothiobacillus spp., members of sulfur-oxidizing symbionts belonging to Gamma-Proteobacteria. Three OTUs (cbbM-C2-28, cbbM-W3-9, cbbM-W4-38, and cbbM-C1-16) are similar to an uncultured organism from iron-rich environmental samples (Kojima et al., 2009). Sequences represented by both cbbM-W3-14 and cbbM-C1-21 are closely related to Rhodopseudomonas palustris, a member of the order Rhizobiales within the Alpha-Proteobacteria. OTUs cbbM-W4-14 and cbbM-W4-6 representing 29 clones show highest similarities with Phaeospirillum molischianum, affiliated with the family Rhodospirillaceae within Alpha-Proteobacteria and with sequences from methane seep sediment. And cbbM-W3-7, which appeared to forms its own cluster, is related to Thauera spp. within the Beta-Proteobacteria and also to an uncultured bacterium from an environmental sample of paddy soil in China (Yuan et al., 2012).
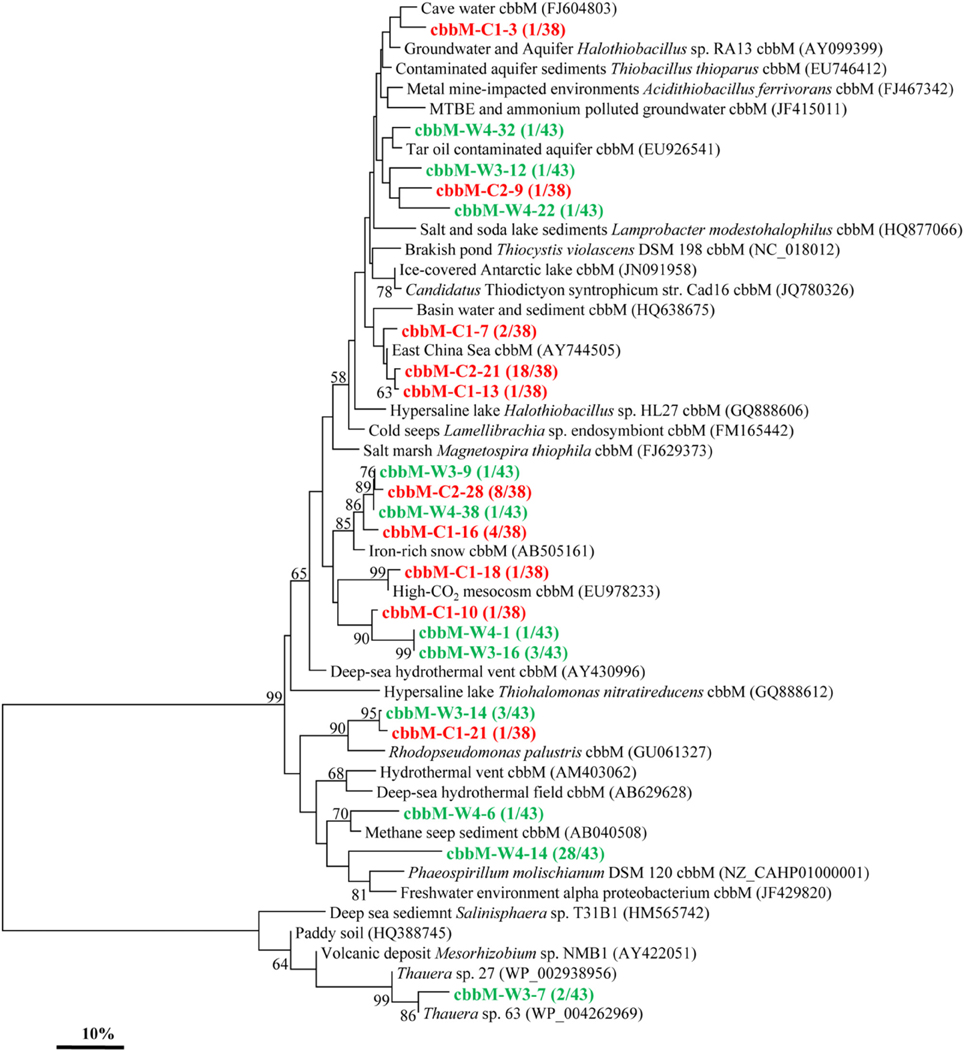
Figure 3. Phylogenetic tree of the cbbM gene retrieved from the water samples (colored) and closely related sequences from GenBank database. Alignments to related sequences (shown with accession number) were performed with MEGA 5 software. The topology of the tree was obtained with the neighbor-joining method. Bootstrap values (n = 1000 replicates) greater than 50% are reported. Scale bar represents 10% amino acid substitution.
fthfs Genes
The fthfs gene sequences were also detected in both samples. However, it showed a less abundant diversity as depicted in the phylogenetic tree (Figure 4) with the screened clones divided into 5 and 4 OTUs in sample C and W, respectively. Phylogenetic analysis shows that most of the fthfs gene sequences are related to members of the Firmicutes. Three OTUs (FTHFS-C2-9, FTHFS-C2-12, and FTHFS-C2-19) of sample C are all most similar to Acetobacterium psammolithicum, a member of the order Clostridiales within Firmicutes while 2 OTUs (FTHFS-C1-7 and FTHFS-C1-5) are obtained in sample C and sharing high similarities with Firmicutes members of the genus Acetobacterium (Xu et al., 2009). OTUs FTHFS-W3-24 and FTHFS-W3-12 are related to sequences from genera Moorella, Desulfitobacterium, and Desulfosporosinus, also members of the Firmicutes. FTHFS-W3-4 is similar to uncultured Alkaliphilus sp. from anaerobic wastewater of Mesa Northwest Wastewater Reclamation Plant (Parameswaran et al., 2010).
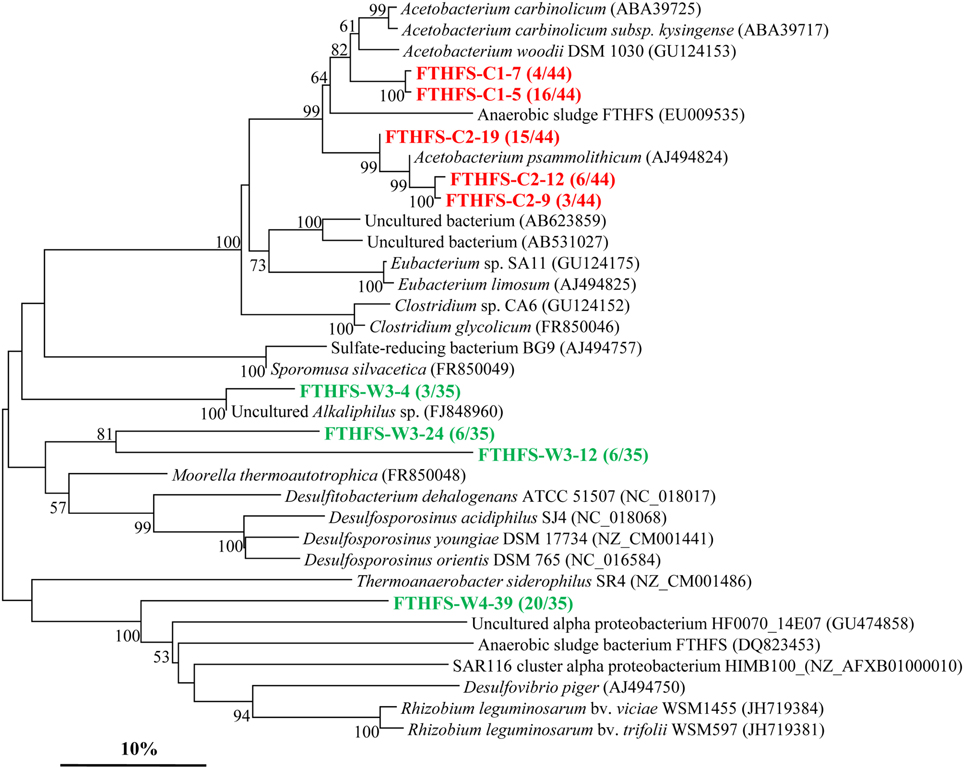
Figure 4. Phylogenetic tree of the fthfs gene retrieved from the water samples (colored) and closely related sequences from GenBank database. Alignments to related sequences (shown with accession number) were performed with MEGA 5 software. The topology of the tree was obtained with the neighbor-joining method. Bootstrap values (n = 1000 replicates) greater than 50% are reported. Scale bar represents 10% amino acid substitution.
[FeFe]-Hydrogenase-Encoding Gene
The [FeFe]-hydrogenase-encoding gene was detected in both C and W samples, and phylogenetic analysis of the sequenced clones were assembled into 9 and 14 OTUs, respectively (Figure 5). The majority of the gene sequences obtained from the two samples cluster with sequences related to Firmicutes. One OTU represented by FeFe-Hyd_W3-6 shares similarity with Syntrophothermus lipocalidus of the Firmicutes. FeFe-Hyd_W4-38 is either related to Shewanella halifaxensis HAW-EB4 within the Gamma-Proteobacteria or to Thermodesulfovibrio yellowstonii within the Nitrospira (Figure 5). FeFe-Hyd_W4-36 is related to Thermodesulfobium narugense belonging to the family Thermodesulfobiaceae within the Firmicutes. FeFe-Hyd_W4-22 shares high identity with Moorella/ thermoacetica affiliated to the family Thermoanaerobacteraceae of Firmicutes. FeFe-Hyd_W4-4, FeFe-Hyd_C2-26, and FeFe-Hyd_W4-32 are all related to Desulfotomaculum kuznetsovii, a member of the order Clostridiales within Firmicutes. FeFe-Hyd_C2-10 and FeFe-Hyd_W4-35 are both similar to Thermotoga lettingae TMO affiliated with the family Thermotogaceae.
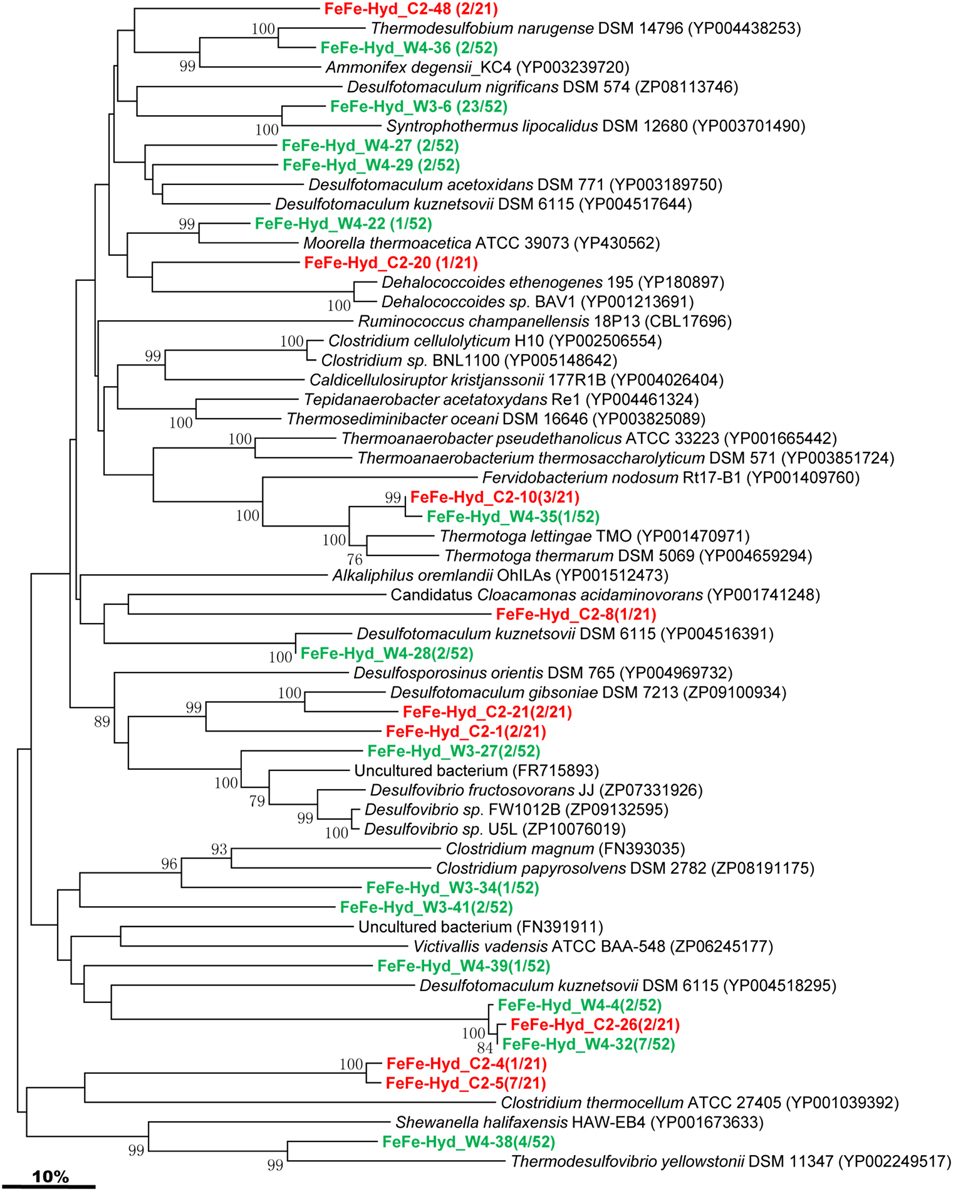
Figure 5. Phylogenetic tree of the [FeFe]-Hydrogenase gene retrieved from the water samples (colored) and closely related sequences from GenBank database. Alignments to related sequences (shown with accession number) were performed with MEGA 5 software. The topology of the tree was obtained with the neighbor-joining method. Bootstrap values (n = 1000 replicates) greater than 50% are reported. Scale bar represents 10% amino acid substitution.
mcrA Genes
By using mcrA-targeted specific PCR primers set, 21 and 16 OTUs (37 overall) were obtained in samples C and W, respectively (Figure 6). Phylogenetic analysis shows that 21 OTUs (13 in C and 8 in W) are all closely related to sequences from members affiliated to the Methanobacteriales, an order known to harbor mostly CO2-reducing methanogens. A total of 7 OTUs (3 in C and 4 in W) shared high identities with mcrA sequences from the Methanomicrobiales. And 9 OTUs (5 in C and 4 in W) are closely related to sequences affiliated to methylotrophic and acetoclastic methanogens within the order Methanosarcinales.
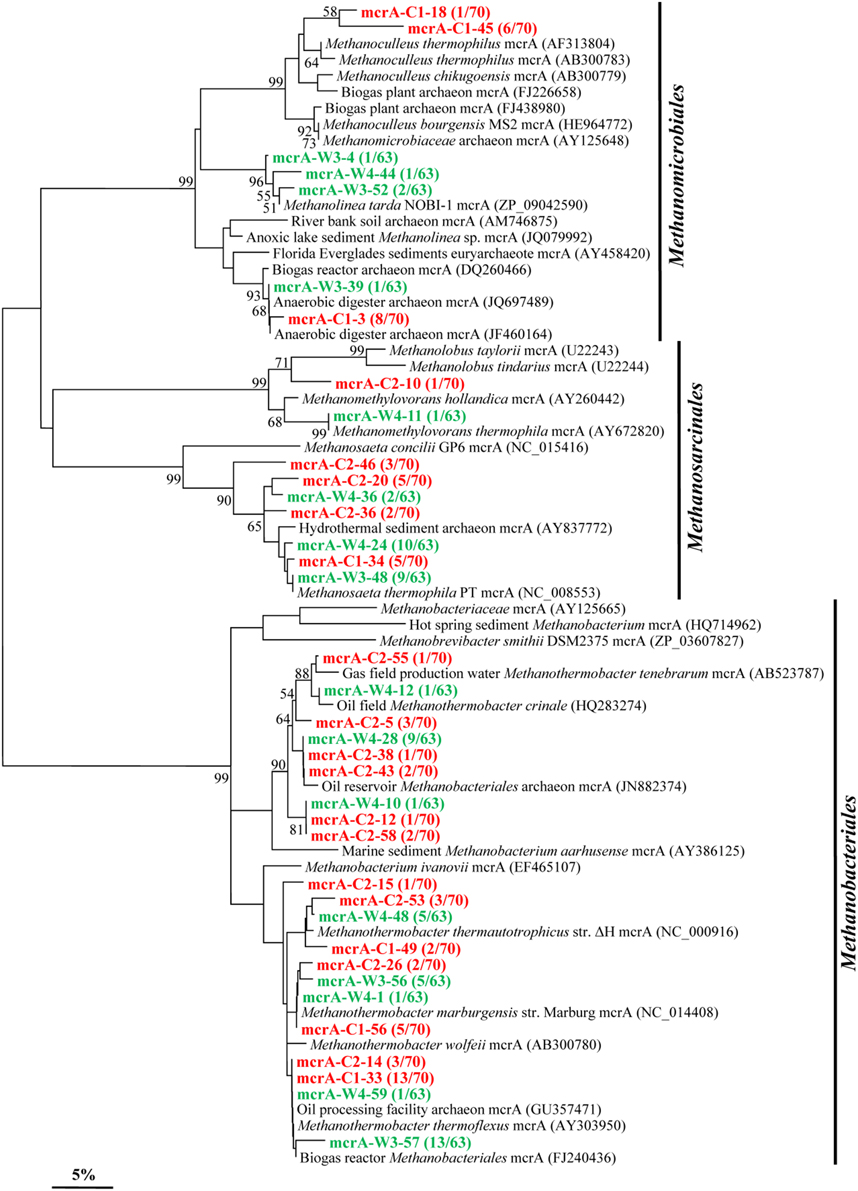
Figure 6. Phylogenetic tree of the mcrA gene retrieved from the water samples (colored) and closely related sequences from GenBank database. Alignments to related sequences (shown with accession number) were performed with MEGA 5 software. The topology of the tree was obtained with the neighbor-joining method. Bootstrap values (n = 1000 replicates) greater than 50% are reported. Scale bar represents 5% amino acid substitution.
Characterization of Functional Microbial Communities
Changes in microbial structure were analyzed by their relative abundance calculated from the number of clones and the results were showed in Figure 7. The community structure of microorganisms with most similarity to the retrieved amino acid sequences of cbbM gene was distinct in W and C samples (Figure 7A). The genera Phaeospirillum (67.4%), Leptothrix (14.0%), Rhodopseudomonas (7.0%), and Thiobacillus (7.0%) were dominant in W sample, whereas, Halothiobacillus (55.3%) and Leptothrix (36.8%) were dominant in C sample. In the cbbL clones libraries (Figure 7B), the genera Rhodospirillum 36.7% and 42.5%, Hydrogenophaga 46.7% and 47.5%, Cupriavidus 10.0% and 7.5% were dominant in W and C sample, respectively. As for the composition of fthfs communities (Figure 7C), in W sample, the community was mainly composed by microorganisms related to genera Nisaea (57.1%), Moorella (34.3%), and Alkaliphilus (8.6%), however, only by microorganisms related to genus Acetobacterium (100%) in C sample. It can be seen from Figure 7D that C sample was dominantly composed by microbes related to members of genera Clostridium (38.1%), Desulfotomaculum (28.6%), and Thermotoga (14.3%), while the W sample by Syntrophothermus (44.2%) and Desulfotomaculum (30.8%). Meanwhile, those related to Ammonifex, Dehalococcoides, and Cloacamonas all rose in relative abundance from undetectable in W sample to 4.8% in C sample. The methanogen community was demonstrated in Figure 7E. As shown in Figure 7E, thermophilic Methanothermobacter (55.7% and 57.1% in C and W sample, respectively), Methanolinea (11.4% and 7.9% in C and W sample, respectively), and Methanosaeta (21.4% and 33.3% in C and W sample, respectively) were the predominant methanogens. Methanoculleus (10.0%) were only detected in the C sample.
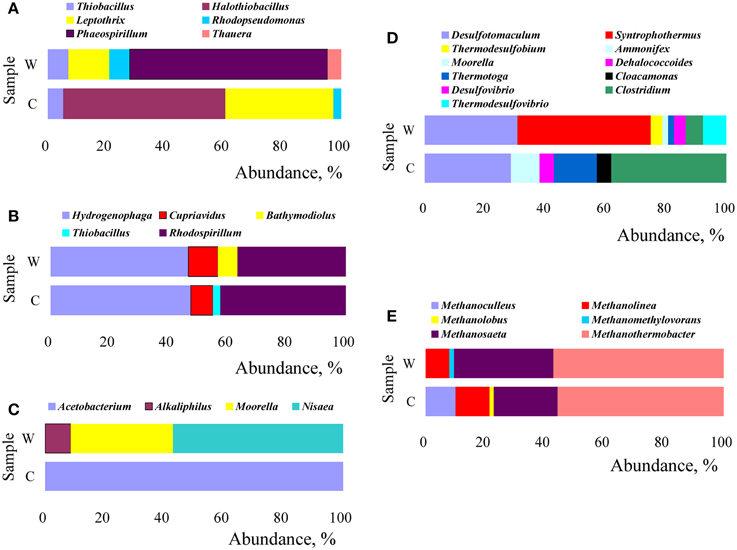
Figure 7. Relative abundance of functional microbes (at the genus level) with respect to the sequences retrieved by functional marker genes of cbbM (A), cbbL (B), fthfs (C), FeFe-hydrogenase(D), and mcrA (E).
Discussion
Occurrence of Microorganisms Associated with CO2 Sequestration in Oil Reservoirs
The microbial community structure in production water samples in Daqing oilfield of China was analyzed by means of a suite of functional genes as biomarkers. Our results indicate that members of the Proteobacteria (Halothiobacillus, Leptothrix, Hydrogenophaga, and Rhodospirillum) were the predominant ones with the ability of fixation of CO2 in in situ oil reservoirs. It has been reported that the CBB cycle for CO2 fixation operates in Proteobacteria belonging to the alpha-, beta-, and gamma-subgroups, and some members of the Firmicutes (Zakharchuk et al., 2003; Caldwell et al., 2007). In addition, the acetogens belonging to Clostridiaceae within Firmicutes can use the reductive acetyl-CoA pathway not only for CO2 fixation but also for the production of acetic acid, which is substrate for methanogenesis. Other major bacterial sequences in the clone libraries of sample W are related to those of Hydrogenophilaceae, and similar microorganisms were reported to use the rTCA cycle for autotrophic CO2 fixation (Schauder et al., 1987; Thauer et al., 1989). For the archaeal mcrA gene clone libraries, the predominance of the genus Methanothermobacter belonging to hydrogenotrophic methanogens is notable.
The majority of cbbL gene types obtained were very similar to the microorganisms belonging to Alpha-, Beta-, and Gamma-Proteobacteria. And some members of these phyla have been reported in previous studies, but of which Hydrogenophaga sp. and Cupriavidus sp. were rarely documented (Alfreider et al., 2003). The cbbM gene types detected are also related to those of Alpha-, Beta-, and Gamma-Proteobacteria, and this is consistent with the research results of Hugler et al. (2010). All above data suggest that microorganisms within Proteobacteria mainly use the CBB cycle for CO2 fixation in the oil reservoirs studied.
Acetogenic bacteria are among the most phylogenetically diverse bacterial functional groups. To date, approximately hundreds of homoacetogenic species have been identified and phylogenetically classified into 21 different genera. The fthfs gene sequences obtained from CO2-flooded fraction of the reservoir shared high similarities with those from members of the Firmicutes with most of the sequences related to the order Clostridiales, deducing that microorganisms affiliated with Firmicutes inhabiting the herein investigated oil reservoirs have the ability to fix CO2 as well as convert CO2 into acetic acid via the acetyl-CoA pathway.
H2 is necessary to in situ CH4 production by hydrogenotrophic methanogens in oil reservoirs. In the present study, we found that sequences from microorganisms similar with those from the Firmicutes, Gamma-Proteobacteria, and Thermotogae were the most encountered in clone libraries established for [FeFe]-hydrogenase-encoding gene, and these results are consistent with those of Schmidt et al. (2010), who found that members of the order Clostridiales and Thermoanaerobacter sp. were likewise all capable of fermentative production of H2 (Schmidt et al., 2010).
Methanogenesis is the terminal step of organic compound degradation and plays a major role in the global carbon cycle (Garrity and Holt, 2001; Liu and Whitman, 2008). The most important precursors for methane production during anaerobic digestion of organic matter are H2-CO2 and acetate, which are converted into methane by hydrogenotrophic and aceticlastic methanogens (Mayumi et al., 2011), respectively. Interestingly, it is proposed that syntrophic acetate oxidation coupled to hydrogenotrophic methanogenesis is an alternative methanogenic pathway in petroleum reservoirs (Mayumi et al., 2011). Analysis based on the mcrA gene types indicates 12 OTUs detected share high identity with those of the genus Methanothermobacter.
To the best of our knowledge, the collection of functional genes described in the present work has not yet been investigated in oil reservoir systems, although some of them have been reported in geothermal environments. The detection of CO2 fixation genes as well as hydrogenases-encoding and fthfs genes in production fluids of high temperature oil reservoirs provides new insights on the diversity and composition of microorganisms involved in the microbial fixation of CO2 and its subsequent conversion to methane.
Impact of CO2 Injection on Specific Microbial Communities with Respect to Microbial Fixation and Bioconversion of CO2
Microbial fixation and conversion of CO2 into methane in oil reservoir by indigenous microorganisms is one of the most promising solutions to the mitigation of CO2 emission. We explored the potential for autotrophic CO2 fixation and bioconversion with microbial communities in oil reservoir by detection of relative functional biomarker genes such as CO2 fixation (cbbM, cbbL), acetogenesis (fthfs), hydrogen formation ([FeFe]-hydrogenase-encoding gene), and methanogenesis (mcrA). Microbial fixation and conversion of CO2 are usually implemented by chemolithoautotrophic microorganisms, which usually obtain their energy through the oxidation of inorganic compounds and utilization of CO2 as their sole source of carbon. Thus, the CO2 injected as well as the subsequent changes in pH and other geochemical parameters induced by CO2 have an influence on the metabolism of the both heterotrophic and lithoautotrophic microorganisms (Ramos, 2003). Therefore, injection of CO2 may cause some changes in microbial populations as well as their activities, and it is important to characterize these changes with respect to CO2 fixation and bioconversion to methane.
Methanogens use molecular hydrogen (H2) anaerobically by transferring electrons from H2 to CO2 to form methane. As demonstrated in Figure 7E, Thermophilic Methanothermobacter, Methanolinea, and Methanosaeta were predominant methanogens both in W and C samples. With comparison to W sample, the promotion in relative abundance of Methanolinea (from 7.9 to 11.4%) and Methanoculleus (from undetectable to 10.0%) as well as the reduction in relative abundance of Methanosaeta (from 33.3 to 21.4%) were observed, which implied that the injected CO2 influenced negatively on Methanosaeta but positively on Methanoculleus and Methanolinea. Considering that Methanothermobacter, Methanolinea, and Methanoculleus are known to be hydrogenotrophic methanogens, Methanosaeta to aceticlastic methanogens, and Methanomethylovorans to methylotrophic methanogens, it is reasonable to conclude that injection of CO2 either increase or maintain the relative abundance of hydrogentropic methanogens, but it decreases that of aceticlastic methanogens and methylotrophic methanogens.
More interestingly, Methanoculleus was detected only in C sample. The genus has been found in different habitats including oil reservoir (Berdugo-Clavijo and Gieg, 2014), deep marine sediments (Mikucki et al., 2003), and swine manure storage tank (Barret et al., 2012, 2013). The occurrence of this genus in C sample implies that it may be related to CO2 injection driven high acetate concentration. This assumption is consistent with the fact that Methanoculleus spp. consume acetate while carrying out hydrogenotrophic methanogenesis and the growth of some Methanoculleus members requires acetate even though they do not convert it to methane (Mikucki et al., 2003; Barret et al., 2013, 2015). Also, Berdugo-Clavijo and Gieg found that the relative abundance of Methanoculleus decreased substantially with acetate (Berdugo-Clavijo and Gieg, 2014). In this study, the C-water is highly enriched in acetate relative to W, which one might normally assume favors aceticlastic methanogens. Based on the known properties of Methanoculleus spp., it seems that the acetate is favoring acetate assimilating methanogens.
Ribulose 1, 5-bisphosphate carboxylase (Rubisco, specifically, cbbL, cbbM) are usually used as a biomarker for the CBB CO2 fixation pathway (Campbell and Cary, 2004). Specifically, in subsurface environments, CO2 fixation is usually conducted by chemolithotrophs through the CBB pathway (Kellermann et al., 2012). As Figure 7A showed, the most dominant genus Phaeospirillum(67.4%) in W sample was not detected in C sample and the abundance of Thiobacillus and Rhodopseudomonas in C sample decreased notably while compared to W sample. In addition, the Halothiobacillus (undetected in W sample) appeared to be the most prevalent in C sample. Also, the relative percentage of Leptothrix in C sample increased compared to that in W sample. In the cbbL clones libraries, the abundance of Rhodospirillum increased in abundance from 36.7% in W sample to 42.5% in C sample, members of the genus Hydrogenophaga increased in abundance slightly in C sample compared to that in W sample, while those affiliated to genus Cupriavidus decraed from 10.0% in W sample to 6.5% in C sample (shown in Figure 7B). Alfreider et al. (2003) also detected Hydrogenophaga, Thiobacillus, and others related cbb sequences in a contaminated aquifer. The abundance and diversity of the detected cbb genes hint at a significant potential for CO2 fixation via the Calvin cycle within oil reservoir microbial communities.
Most acetogens are obligate anaerobic bacteria that use the reductive acetyl-CoA pathway as their main mechanism for energy conservation and for synthesis of acetyl-CoA and cell carbon from CO2. Formyltetrahydrofolate synthetase (fthfs) is used to detect acetogenic, fermentative bacteria (Leaphart and Lovell, 2001). In the present work, notable changes were observed in the composition of fthfs communities (Figure 7C). The community dominated by microorganisms related to genera Nisaea, Moorella, and Alkaliphilus in W sample was changed completely to be dominated only by microorganisms related to genus Acetobacterium in C sample. The mechanism for the change of Alkaliphilus from dominance in sample W to undetectable in sample C is not very clear. Generally, this genus is known to be extremely alkaliphilic and thus would not be prone to survive in the acidic conditions caused by the injection of CO2. Although the ability of acetate production on CO2+H2 by Acetobacterium woodii and Moorella were systematically studied (Ragsdale and Pierce, 2008; Demler and Weuster-Botz, 2011), surprisingly, Moorella-like microorganisms were not detected in C sample. This observation implies that Acetobacterium-like microbes are probably more suitable for acetogenesis in CO2-injected oil reservoirs.
Hydrogen is an alternative energy source for autotrophic microbes in a variety of subsurface environments. When hydrogen and carbon dioxide are present, development of autotrophic microorganisms would be possible. For example, methanogens and acetogens may produce organic matter from hydrogen by means of respiring carbon dioxide. As it can be seen from our study (Figure 7D), the composition of [FeFe]-hydrogenase-encoding gene clones libraries at the genus level shows interesting differences in relative abundance between W and C samples. The microbes related to Syntrophothermus predominated in W sample (44.2%) disappeared in C sample, and Desulfotomaculum decreased from 30.8% in W sample to 28.6% in C sample. The relative abundance of members of genera Clostridium, Thermotoga, and Ammonifex increased from 5.8%, 1.9% and undetectable (0.0%) in W sample to 38.1%, 14.3% and 9.5% in C sample, respectively. Interestingly, all the three sulfate reducing bacteria were influenced very markedly as either decreased in relative abundance (Desulfotomaculum) or became undetectable in C sample (Thermodesulfobium and Thermodesulfovibrio). Meanwhile, those related to Dehalococcoides and Cloacamonas all rose in relative abundance from undetectable in W sample to 4.8% in C sample. Morozova et al. (2010) also showed that CO2 injection caused a decrease in the diversity of microorganisms and revealed temporal out-competition of sulfate-reducing bacteria by methanogenic bacteria. Morozova's experiments showed that after CO2 injection the SRB population declined until it was no longer detected while the archaeal population increased, which indicates that archaea may be able to adapt more readily to the more acidic conditions after CO2 injection. Our results reached the same conclusion. But, Morozova found that after a 5 month period of exposure to CO2, the SRB population returned in numbers greater than that prior to CO2 injection. This phenomenon was not observed in our study at present. The reason for this was not quite clear although it was assumed to be resulted partly from the water–gas alternative injection and long-term exposure of CO2 (about 5 years) in our study which were quite different from that in Morozova's experiments.
We found great differences in relative abundance among all the five functional gene clone libraries established from W and C samples, as showed in Figure 7. This phenomenon of previously undetectable and/or rare members of microbial communities becoming dominant after exposure to CO2 has been reported previously (Gulliver and Gregory, 2011). Microorganisms with increasing abundance implies that they may be better withstanding or adapting to exposure to CO2 and subsequent changes in physical and biochemical conditions resulted by CO2 injection.
Analysis of functional genes shows that microbial communities were strongly influenced and the diversity reduced by CO2 injection. For example, there were eight different genera in W sample whereas only six were retrieved from C sample for [FeFe]-hydrogenase-encoding gene library. Also, for fthfs library, three different genera were detected in W sample but only one was found in C sample. Our data agree with Gulliver and Gregory (2011) which showed that different families of bacteria presided with variation in CO2 partial pressure. Knowledge of surviving and thriving microbial populations may help in better understanding of the fate of CO2 following injection and to make better strategy for use of microorganisms in subsurface environments for improving the efficiency of injection and microbial fixation of CO2, and hence ensuring the security for long-term CO2 storage in subsurface petroleum reservoirs.
Primers used for mcrA amplification are divided into different groups: MCR, ME, ML, and these primers are able to amplify most methanogens. It has been reported that the ME-related primers are also able to amplify anaerobic methane-oxidizing archaea (ANME) (Narihiro and Sekiguchi, 2011). The primers used for mcrA amplification to target the methanogenic communities in the samples investigated in the present study were described by Luton et al. (2002) which belonged to the ML group. To the best of our knowledge, the ML group ability to amplify ANME's remains to be demonstrated.
Due to the fact that the CO2 injected had been produced about 1 year before the collection of these samples when the ratio of gas (CO2) to oil was between 22.8 and 145 m3/m3 in production wells, the changes in the relative abundance of five genes relevant to CO2 utilization and methane production by microorganisms can be considered mainly attributed to CO2 injection. The small size of the clone library and the number of clones sequenced would influence, to some extent, on the analysis of microorganisms with low frequency. Nevertheless, the major functional microorganisms and their changes in relative abundance can still be recognized, even with certain biases, as demonstrated in the present study. The analysis of the changes in microbial community may be influenced by the following factors: (1) The samples were all collected from the sampling valve located at the wellhead of production well and hence, these samples may contain microbes from oil reservoir as well as that survived in oil tubes between the well bottoms to the sampling valve; (2) The sampling water may be produced both from oil-bearing layers or sub-layers with CO2 production (CO2-impacted water) and that with no CO2 production even they received CO2(non CO2-impacted water); (3) The retention time of CO2 in oil reservoir is relatively short, i.e., while CO2 was injected through injection wells into target oil reservoir, part of them would be produced afterwards from the production well about 250–300m away from the injector; (4) CO2 was injected into the target oil reservoir with water-CO2 alternative injection manner.
For a more accurate characterization of microbial community and their changes caused by CO2 injection in oil reservoir, the collection of produced water from only the CO2-impacted zones, the qualitative and quantitative analysis of microbial community, the physiochemical changes of subsurface water such as pH, volatile acids over time, as well as the analysis of the origin of volatile acids (by isotopic analysis) and etc. are very important.
Methane Formation Potential from Injected CO2 in Oil Reservoirs
Bioconversion of CO2 into CH4 in situ oil reservoirs by indigenous methanogens is an area of active research and development. Hydrogenotrophic methanogens need not only CO2 but also H2 to produce CH4; therefore, H2 should be supplied to them in reservoirs for this process. It has been reported that there are several kinds of microorganisms capable of producing H2 by degrading crude oil in reservoir environments. The potential of the microbial conversion of CO2 into CH4 by enrichment culture experiments using microorganisms indigenous to oil reservoirs has been studied (Sugai et al., 2012). Different from that mentioned above, we evaluated the potential of this process from the viewpoint of functional genes. In our study, both the functional genes of H2-producing and CH4-producing were detected in the CO2-flooding oil reservoirs, and the water-flooding oil reservoirs as well. Furthermore, some H2-producing microorganisms (e.g., Clostridium and Thermotoga) and hydrogenotrophic methanogens such as Methanothermobacter and Methanolinea as well as Methanoculleus remained or evolving to be predominant after long term exposure to CO2 in CO2-flooding area compared to that in water-flooding area. Meanwhile, these H2-producing bacteria and hydrogenotrophic methanogens were both identified in the 16S rRNA genes cloning libraries (data not shown in this paper). It is assumed that these hydrogenotrophic methanogens live in symbiosis with hydrogen-producing bacteria and convert CO2 into CH4 in oil reservoirs. These results indicate that indigenous microbial conversion process of CO2 into CH4 has high potential.
The detection of CO2 fixation potential is alternative evidence to autotrophic activity in situ oil reservoirs. Therefore, attentions should be further paid on the evaluation of the activities of those microorganisms in subsurface ecosystems with the potential of microbial fixation of CO2 and its subsequent bioconversion into methane. Once those microorganisms are activated by means of nutrient injection and etc., taking into consideration of the tremendous capacity of CO2 sequestration in oil and gas reservoir (totally about 9 × 1011 tons in the world), it seems more reasonable to believe that the in situ fixation and reclamation of CO2 sequestrated in oil reservoir will play an notable role in mitigating atmospheric CO2 building up as well as energy shortage.
Conclusions
Analysis of a suite of functional genes shows that a diverse microbial community with potential for fixation and conversion of CO2 into methane inhabits oil reservoir. Microorganisms affiliated with members of the genera Methanothemobacter (hydrogenotrophic CO2-reducing methanogens), Acetobacterium and Halothiobacillus as well as hydrogen producers (Firmicutes) seem to be more adaptable to CO2 injection and present the potential for microbial fixation and bioconversion of CO2 into methane in subsurface oil reservoirs. Due to the limitation of clone numbers and the co-production nature of CO2-impacted and non-impacted water in the C sampling well, the impact of CO2 injection on microbial community may be not fully characterized and presented in this study. Even so, the present results showing the response, to some extent, of microbial community on the CO2 injection are of some help in predicting the fate of CO2 following injection and making better strategies for use of microorganisms in subsurface environments for microbial CO2 fixation and bioconversion of CO2 into sustainable energy in subsurface oil reservoirs.
Author Contributions
This study was designed by JL and BM. XS and GY performed all the laboratory experiments. SM analyzed the functional genes data and constructed the phylogenetic trees of these functional genes. JG provided valuable suggestions in the design of the experiments and the preparation of the manuscript. The manuscript was written by JL, assisted by all co-authors. All authors reviewed the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 41273084) and the NSFC/RGC Joint Research Fund (No. 41161160560).
References
Alfreider, A., Schirmer, M., and Vogt, C. (2012). Diversity and expression of different forms of RubisCO genes in polluted groundwater under different redox conditions. FEMS Microbiol. Ecol. 79, 649–660. doi: 10.1111/j.1574-6941.2011.01246.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alfreider, A., Vogt, C., Hoffmann, D., and Babel, W. (2003). Diversity of ribulose-1,5-bisphosphate carboxylase/oxygenase large-subunit genes from groundwater and aquifer microorganisms. Microb. Ecol. 45, 317–328. doi: 10.1007/s00248-003-2004-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Barret, M., Gagnon, N., Kalmokoff, M. L., Topp, E., Verastegui, Y., Brooks, S. P. J., et al. (2013). Identification of Methanoculleus spp. as active methanogens during anoxic incubations of swine manure storage tank samples. Appl. Environ. Microbiol. 79, 424–433. doi: 10.1128/AEM.02268-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barret, M., Gagnon, N., Morissette, B., Kalmokoff, M. L., Topp, E., Brooks, S. P. J., et al. (2015). Phylogenetic identification of methanogens assimilating acetate-derived carbon in dairy and swine manures. Syst. Appl. Microbiol. 38, 56–66. doi: 10.1016/j.syapm.2014.11.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barret, M., Gagnon, N., Morissette, B., Topp, E., Kalmokoff, M., Brooks, S. P. J., et al. (2012). Methanoculleus spp. as a biomarker of methanogenic activity in swine manure storage tanks. FEMS Microbiol. Ecol. 80, 427–440. doi: 10.1111/j.1574-6941.2012.01308.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berdugo-Clavijo, C., and Gieg, L. M. (2014). Conversion of crude oil to methane by a microbial consortium enriched from oil reservoir production waters. Front. Microbiol. 5:197. doi: 10.3389/fmicb.2014.00197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berg, I. A. (2011). Ecological aspects of the distribution of different autotrophic CO2 fixation pathways. Appl. Environ. Microbiol. 77, 1925–1936. doi: 10.1128/AEM.02473-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berg, I. A., Keppen, O. I., Krasil'nikova, E. N., Ugol'kova, N. V., and Ivanovsky, R. N. (2005). Carbon metabolism of filamentous anoxygenic phototrophic bacteria of the family Oscillochloridaceae. Microbiology 74, 258–264. doi: 10.1007/s11021-005-0060-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Biegel, E., and Muller, V. (2010). Bacterial Na+-translocating ferredoxin:NAD+ oxidoreductase. Proc. Natl. Acad. Sci. U.S.A. 107, 18138–18142. doi: 10.1073/pnas.1010318107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caldwell, P. E., Maclean, M. R., and Norris, P. R. (2007). Ribulose bisphosphate carboxylase activity and a Calvin cycle gene cluster in Sulfobacillus species. Microbiology 153, 2231–2240. doi: 10.1099/mic.0.2007/006262-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Campbell, B. J., and Cary, S. C. (2004). Abundance of reverse tricarboxylic acid cycle genes in free-living microorganisms at deep-sea hydrothermal vents. Appl. Environ. Microbiol. 70, 6282–6289. doi: 10.1128/AEM.70.10.6282-6289.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y., Wu, L., Boden, R., Hillebrand, A., Kumaresan, D., Moussard, H., et al. (2009). Life without light: microbial diversity and evidence of sulfur- and ammonium-based chemolithotrophy in Movile Cave. ISME J. 3, 1093–1104. doi: 10.1038/ismej.2009.57
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Demler, M., and Weuster-Botz, D. (2011). Reaction engineering analysis of hydrogenotrophic production of acetic acid by Acetobacterium woodii. Biotechnol. Bioeng. 108, 470–474. doi: 10.1002/bit.22935
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galand, P. E., Juottonen, H., Fritze, H., and Yrjala, K. (2005). Methanogen communities in a drained bog: effect of ash fertilization. Microb. Ecol. 49, 209–217. doi: 10.1007/s00248-003-0229-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garcia-Dominguez, E., Mumford, A., Rhine, E. D., Paschal, A., and Young, L. Y. (2008). Novel autotrophic arsenite-oxidizing bacteria isolated from soil and sediments. FEMS Microbiol. Ecol. 66, 401–410. doi: 10.1111/j.1574-6941.2008.00569.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garrity, G., and Holt, J. (2001). “Phylum AII. Euryarchaeota phy. nov,” in Bergey's Manual® of Systematic Bacteriology, eds. D. Boone and R. Castenholz (New York, NY: Springer), 211–355. doi: 10.1007/978-0-387-21609-6_17
Guan, J., Xia, L.-P., Wang, L.-Y., Liu, J.-F., Gu, J.-D., and Mu, B.-Z. (2013). Diversity and distribution of sulfate-reducing bacteria in four petroleum reservoirs detected by using 16S rRNA and dsrAB genes. Int. Biodeterior. Biodegradation 76, 58–66. doi: 10.1016/j.ibiod.2012.06.021
Gulliver, D., and Gregory, K. (2011). “CO2 gradient affects on deep subsurface microbial ecology during carbon sequestration,” in American Geophysical Union (AGU), Fall Meeting (San Francisco, CA).
Huber, T., Faulkner, G., and Hugenholtz, P. (2004). Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20, 2317–2319. doi: 10.1093/bioinformatics/bth226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hugler, M., Gartner, A., and Imhoff, J. F. (2010). Functional genes as markers for sulfur cycling and CO2 fixation in microbial communities of hydrothermal vents of the Logatchev field. FEMS Microbiol. Ecol. 73, 526–537. doi: 10.1111/j.1574-6941.2010.00919.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ivanovsky, R. N., Fal, Y. I., Berg, I. A., Ugolkova, N. V., Krasilnikova, E. N., Keppen, O. I., et al. (1999). Evidence for the presence of the reductive pentose phosphate cycle in a filamentous anoxygenic photosynthetic bacterium, Oscillochloris trichoides strain DG-6. Microbiology 145, 1743–1748. doi: 10.1099/13500872-145-7-1743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Juottonen, H., Galand, P. E., and Yrjala, K. (2006). Detection of methanogenic Archaea in peat: comparison of PCR primers targeting the mcrA gene. Res. Microbiol. 157, 914–921. doi: 10.1016/j.resmic.2006.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kellermann, C., Selesi, D., Lee, N., Hugler, M., Esperschutz, J., Hartmann, A., et al. (2012). Microbial CO2 fixation potential in a tar-oil-contaminated porous aquifer. FEMS Microbiol. Ecol. 81, 172–187. doi: 10.1111/j.1574-6941.2012.01359.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kojima, H., Fukuhara, H., and Fukui, M. (2009). Community structure of microorganisms associated with reddish-brown iron-rich snow. Syst. Appl. Microbiol. 32, 429–437. doi: 10.1016/j.syapm.2009.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leaphart, A. B., and Lovell, C. R. (2001). Recovery and analysis of formyltetrahydrofolate synthetase gene sequences from natural populations of acetogenic bacteria. Appl. Environ. Microbiol. 67, 1392–1395. doi: 10.1128/AEM.67.3.1392-1395.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, J., Park, D., Park, S., Hwang, E., Oh, J., and Kim, Y. (2009). Expression and regulation of ribulose 1,5-bisphosphate carboxylase/oxygenase genes in Mycobacterium sp. strain JC1 DSM 3803. J. Microbiol. 47, 297-307. doi: 10.1007/s12275-008-0210-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, H., Chen, S., Mu, B.-Z., and Gu, J.-D. (2010). Molecular detection of anaerobic ammonium-oxidizing (Anammox) bacteria in high-temperature petroleum reservoirs. Microb. Ecol. 60, 771–783. doi: 10.1007/s00248-010-9733-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, H., Mu, B.-Z., Jiang, Y., and Gu, J.-D. (2011). Production processes affected prokaryotic amoA gene abundance and distribution in high-temperature petroleum reservoirs. Geomicrobiol. J. 28, 692–704. doi: 10.1080/01490451.2010.514026
Liu, Y., and Whitman, W. B. (2008). Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. N.Y. Acad. Sci. 1125, 171–189. doi: 10.1196/annals.1419.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luton, P. E., Wayne, J. M., Sharp, R. J., and Riley, P. W. (2002). The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology 148, 3521–3530.
Magot, M., Ollivier, B., and Patel, B. K. (2000). Microbiology of petroleum reservoirs. Antonie Van Leeuwenhoek 77, 103–116. doi: 10.1023/A:1002434330514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mayumi, D., Mochimaru, H., Yoshioka, H., Sakata, S., Maeda, H., Miyagawa, Y., et al. (2011). Evidence for syntrophic acetate oxidation coupled to hydrogenotrophic methanogenesis in the high-temperature petroleum reservoir of Yabase oil field (Japan). Environ. Microbiol. 13, 1995–2006. doi: 10.1111/j.1462-2920.2010.02338.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mbadinga, S. M., Li, K. P., Zhou, L., Wang, L. Y., Yang, S. Z., Liu, J. F., et al. (2012). Analysis of alkane-dependent methanogenic community derived from production water of a high-temperature petroleum reservoir. Appl. Microbiol. Biotechnol. 96, 531–542. doi: 10.1007/s00253-011-3828-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mikucki, J. A., Liu, Y. T., Delwiche, M., Colwell, F. S., and Boone, D. R. (2003). Isolation of a methanogen from deep marine sediments that contain methane hydrates, and description of Methanoculleus submarinus sp nov. Appl. Environ. Microbiol. 69, 3311–3316. doi: 10.1128/AEM.69.6.3311-3316.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morozova, D., Wandrey, M., Alawi, M., Zimmer, M., Vieth, A., Zettlitzer, M., et al. (2010). Monitoring of the microbial community composition in saline aquifers during CO2 storage by fluorescence in situ hybridisation. Int. J. Greenhouse Gas Control 4, 981–989. doi: 10.1016/j.ijggc.2009.11.014
Narihiro, T., and Sekiguchi, Y. (2011). Oligonucleotide primers, probes and molecular methods for the environmental monitoring of methanogenic archaea. Microb. Biotechnol. 4, 585–602. doi: 10.1111/j.1751-7915.2010.00239.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parameswaran, P., Zhang, H., Torres, C. I., Rittmann, B. E., and Krajmalnik-Brown, R. (2010). Microbial community structure in a biofilm anode fed with a fermentable substrate: the significance of hydrogen scavengers. Biotechnol. Bioeng. 105, 69–78. doi: 10.1002/bit.22508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ragsdale, S. W., and Pierce, E. (2008). Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 1784, 1873–1898. doi: 10.1016/j.bbapap.2008.08.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramos, J.-L. (2003). Lessons from the genome of a lithoautotroph: making biomass from almost nothing. J. Bacteriol. 185, 2690–2691. doi: 10.1128/JB.185.9.2690-2691.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ryan, P., Forbes, C., and Colleran, E. (2008). Investigation of the diversity of homoacetogenic bacteria in mesophilic and thermophilic anaerobic sludges using the formyltetrahydrofolate synthetase gene. Water Sci. Technol. 57, 675–680. doi: 10.2166/wst.2008.059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Schauder, R., Preuß, A., Jetten, M., and Fuchs, G. (1988). Oxidative and reductive acetyl CoA/carbon monoxide dehydrogenase pathway in Desulfobacterium autotrophicum. Arch. Microbiol. 151, 84–89. doi: 10.1007/BF00444674
Schauder, R., Widdel, F., and Fuchs, G. (1987). Carbon assimilation pathways in sulfate-reducing bacteria II. Enzymes of a reductive citric acid cycle in the autotrophic Desulfobacter hydrogenophilus. Arch. Microbiol. 148, 218–225. doi: 10.1007/BF00414815
Schmidt, O., Drake, H. L., and Horn, M. A. (2010). Hitherto unknown [Fe-Fe]-hydrogenase gene diversity in anaerobes and anoxic enrichments from a moderately acidic fen. Appl. Environ. Microbiol. 76, 2027–2031. doi: 10.1128/AEM.02895-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539–544. doi: 10.1038/msb.2011.75
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spiridonova, E. M., Kuznetsov, B. B., Pimenov, N. V., and Tourova, T. P. (2006). Phylogenetic characterization of endosymbionts of the hydrothermal vent mussel Bathymodiolus azoricus by analysis of the 16S rRNA, cbbL, and pmoA genes. Microbiology 75, 694–701. doi: 10.1134/S0026261706060129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strous, M., Pelletier, E., Mangenot, S., Rattei, T., Lehner, A., Taylor, M. W., et al. (2006). Deciphering the evolution and metabolism of an anammox bacterium from a community genome. Nature 440, 790–794. doi: 10.1038/nature04647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sugai, Y., Purwasena, I. A., Sasaki, K., Fujiwara, K., Hattori, Y., and Okatsu, K. (2012). Experimental studies on indigenous hydrocarbon-degrading and hydrogen-producing bacteria in an oilfield for microbial restoration of natural gas deposits with CO2 sequestration. J. Nat. Gas Sci. Eng. 5, 31–41. doi: 10.1016/j.jngse.2012.01.011
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thauer, R. K., Kaster, A.-K., Seedorf, H., Buckel, W., and Hedderich, R. (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591. doi: 10.1038/nrmicro1931
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thauer, R. K., Moller-Zinkhan, D., and Spormann, A. M. (1989). Biochemistry of acetate catabolism in anaerobic chemotrophic bacteria. Annu. Rev. Microbiol. 43, 43–67. doi: 10.1146/annurev.mi.43.100189.000355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vorholt, J., Kunow, J., Stetter, K., and Thauer, R. (1995). Enzymes and coenzymes of the carbon monoxide dehydrogenase pathway for autotrophic CO2 fixation in Archaeoglobus lithotrophicus and the lack of carbon monoxide dehydrogenase in the heterotrophic A. profundus. Arch. Microbiol. 163, 112–118. doi: 10.1007/s002030050179
Vorholt, J. A., Hafenbradl, D., Stetter, K. O., and Thauer, R. K. (1997). Pathways of autotrophic CO2 fixation and of dissimilatory nitrate reduction to N2O in Ferroglobus placidus. Arch. Microbiol. 167, 19–23. doi: 10.1007/s002030050411
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wandrey, M., Pellizari, L., Zettlitzer, M., and Würdemann, H. (2011). Microbial community and inorganic fluid analysis during CO2 storage within the frame of CO2SINK–long-term experiments under in situ conditions. Energy Proc. 4, 3651–3657. doi: 10.1016/j.egypro.2011.02.296
Wang, L. Y., Duan, R. Y., Liu, J. F., Yang, S. Z., Gu, J. D., and Mu, B. Z. (2012). Molecular analysis of the microbial community structures in water-flooding petroleum reservoirs with different temperatures. Biogeosciences 9, 4645–4659. doi: 10.5194/bg-9-4645-2012
Wang, L.-Y., Gao, C.-X., Mbadinga, S. M., Zhou, L., Liu, J.-F., Gu, J.-D., et al. (2011). Characterization of an alkane-degrading methanogenic enrichment culture from production water of an oil reservoir after 274 days of incubation. Int. Biodeterior. Biodegradation 65, 444–450. doi: 10.1016/j.ibiod.2010.12.010
Xu, K., Liu, H., Du, G., and Chen, J. (2009). Real-time PCR assays targeting formyltetrahydrofolate synthetase gene to enumerate acetogens in natural and engineered environments. Anaerobe 15, 204–213. doi: 10.1016/j.anaerobe.2009.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, Y., Breitbart, M., McNairnie, P., and Rohwer, F. (2006). FastGroupII: a web-based bioinformatics platform for analyses of large 16S rDNA libraries. BMC Bioinformatics 7:57. doi: 10.1186/1471-2105-7-57
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yuan, H., Ge, T., Wu, X., Liu, S., Tong, C., Qin, H., et al. (2012). Long-term field fertilization alters the diversity of autotrophic bacteria based on the ribulose-1,5-biphosphate carboxylase/oxygenase (RubisCO) large-subunit genes in paddy soil. Appl. Microbiol. Biotechnol. 95, 1061–1071. doi: 10.1007/s00253-011-3760-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zakharchuk, L. M., Egorova, M. A., Tsaplina, I. A., Bogdanova, T. I., Krasil'nikova, E. N., Melamud, V. S., et al. (2003). Activity of the enzymes of carbon metabolism in Sulfobacillus sibiricus under various conditions of cultivation. Microbiology 72, 553–557. doi: 10.1023/A:1026039132408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: CO2 fixation, bioconversion, methane, functional genes, oil reservoir, microbial communities
Citation: Liu J-F, Sun X-B, Yang G-C, Mbadinga SM, Gu J-D and Mu B-Z (2015) Analysis of microbial communities in the oil reservoir subjected to CO2-flooding by using functional genes as molecular biomarkers for microbial CO2 sequestration. Front. Microbiol. 6:236. doi: 10.3389/fmicb.2015.00236
Received: 25 December 2014; Accepted: 10 March 2015;
Published: 31 March 2015.
Edited by:
John Joseph Kilbane, Illinois Institute of Technology/Intertek, USAReviewed by:
Naresh Singhal, The University of Auckland, New ZealandThomas E. Hanson, University of Delaware, USA
John Joseph Kilbane, Illinois Institute of Technology/Intertek, USA
Copyright © 2015 Liu, Sun, Yang, Mbadinga, Gu and Mu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Dong Gu, School of Biological Sciences, The University of Hong Kong, Pokfulam Road, Hong Kong, China jdgu@hkucc.hku.hk;
Bo-Zhong Mu, State Key Laboratory of Bioreactor Engineering and Institute of Applied Chemistry, East China University of Science and Technology, Meilong Road 130, Shanghai, 200237, China bzmu@ecust.edu.cn