 Feng Xu1,2,3
Feng Xu1,2,3 Saba Ilyas1
Saba Ilyas1 Jeffrey A. Hall1,3
Jeffrey A. Hall1,3 Stephen H. Jones3,4
Stephen H. Jones3,4 Vaughn S. Cooper1,3
Vaughn S. Cooper1,3 Cheryl A. Whistler1,3*
Cheryl A. Whistler1,3*- 1Department of Molecular, Cellular and Biomedical Sciences, University of New Hampshire, Durham, NH, USA
- 2Graduate Program in Genetics, University of New Hampshire, Durham, NH, USA
- 3Northeast Center for Vibrio Disease and Ecology, University of New Hampshire, Durham, NH, USA
- 4Department of Natural Resources and the Environment, University of New Hampshire, Durham, NH, USA
Gastric infections caused by the environmentally transmitted pathogen, Vibrio parahaemolyticus, have increased over the last two decades, including in many parts of the United States (US). However, until recently, infections linked to shellfish from the cool northeastern US waters were rare. Cases have risen in the Northeast, consistent with changes in local V. parahaemolyticus populations toward greater abundance or a shift in constituent pathogens. We examined 94 clinical isolates from a period of increasing disease in the region and compared them to 200 environmental counterparts to identify resident and non-indigenous lineages and to gain insight into the emergence of pathogenic types. Genotyping and multi-locus sequence analysis (MLSA) of clinical isolates collected from 2010 to 2013 in Massachusetts, New Hampshire, and Maine revealed their polyphyletic nature. Although 80% of the clinical isolates harbored the trh hemolysin either alone or with tdh, and were urease positive, 14% harbored neither hemolysin exposing a limitation for these traits in pathogen detection. Resident sequence type (ST) 631 strains caused seven infections, and show a relatively recent history of recombination with other clinical and environmental lineages present in the region. ST34 and ST674 strains were each linked to a single infection and these strain types were also identified from the environment as isolates harboring hemolysin genes. Forty-two ST36 isolates were identified from the clinical collection, consistent with reports that this strain type caused a rise in regional infections starting in 2012. Whole-genome phylogenies that included three ST36 outbreak isolates traced to at least two local sources demonstrated that the US Atlantic coastal population of this strain type was indeed derived from the Pacific population. This study lays the foundation for understanding dynamics within natural populations associated with emergence and invasion of pathogenic strain types in the region.
Introduction
Rare pathogenic variants of Vibrio parahaemolyticus, a ubiquitous yet typically harmless estuarine bacterium, can cause human gastric infections most often from the consumption of raw or improperly handled seafood, and wound infections from recreational aquatic activities (Daniels et al., 2000; Scallan et al., 2011). Infections typically occur seasonally during warmer months when total populations of these bacteria rise, and with them, the risk of exposure to an infectious dose of pathogens increases (Parveen et al., 2008). Even so, temperature and total abundance do not fully explain infection trends as some infections occur when water temperatures and abundance of total V. parahaemolyticus are low (Zimmerman et al., 2007; Johnson et al., 2012; Jones et al., 2012). Furthermore, recurrent infections and outbreaks have also occurred in cooler waters of the Pacific Northwest (PNW) for decades where pathogens re-emerge each year from among diverse residential populations (Altekruse et al., 2000; Johnson et al., 2010; Paranjpye et al., 2012; Turner et al., 2013; Banerjee et al., 2014). A better understanding of conditions that promote emergence and relative abundance of pathogens is necessary to develop appropriate strategies for disease prevention.
Comparatively fewer and sporadic infections have been associated with shellfish harvested from the cooler waters of the Northeastern US. One notable exception was a large multi-state outbreak in 1998, which included oysters harvested from Long Island Sound bordered by New York (NY) and Connecticut (CT) (Figure 1) (CDC, 1999) That outbreak was attributed to a non-indigenous strain from the pandemic clonal complex which are typically serotype O3:K6 and sequence type (ST) 3 and that spread globally from Southeast Asia (CDC, 1999; Depaola et al., 2000; Nair et al., 2007; García et al., 2009; Harth et al., 2009; Martinez-Urtaza et al., 2010; Alikhan et al., 2011). The ensuing low disease incidence in the Northeastern US implied that the regional environmental conditions did not sustain either invasive or endemic pathogenic populations (Mahoney et al., 2010; Ellis et al., 2012). In the last several years, however, reported cases have been increasing in the Northeast, with outbreaks in NY in 2012, and in NY, CT, and Massachusetts (MA) in 2013 (Newton et al., 2014) (Figure 1). This abrupt increase in cases coincided with both warmer than usual ocean temperatures in the region (Figure 1) and the probable Atlantic ecological invasion of a lineage of ST36 strains, which are indigenous to the cooler waters of the PNW (Martinez-Urtaza et al., 2013; Newton et al., 2014). Thus, the emergent disease in the Northeast appears to be unlike other situations under study in the US. Characterizing clinical strains from the region and relating them to native non-pathogens during this period of increased disease incidence could provide insight into how changes in the bacterial population have led to increased disease.
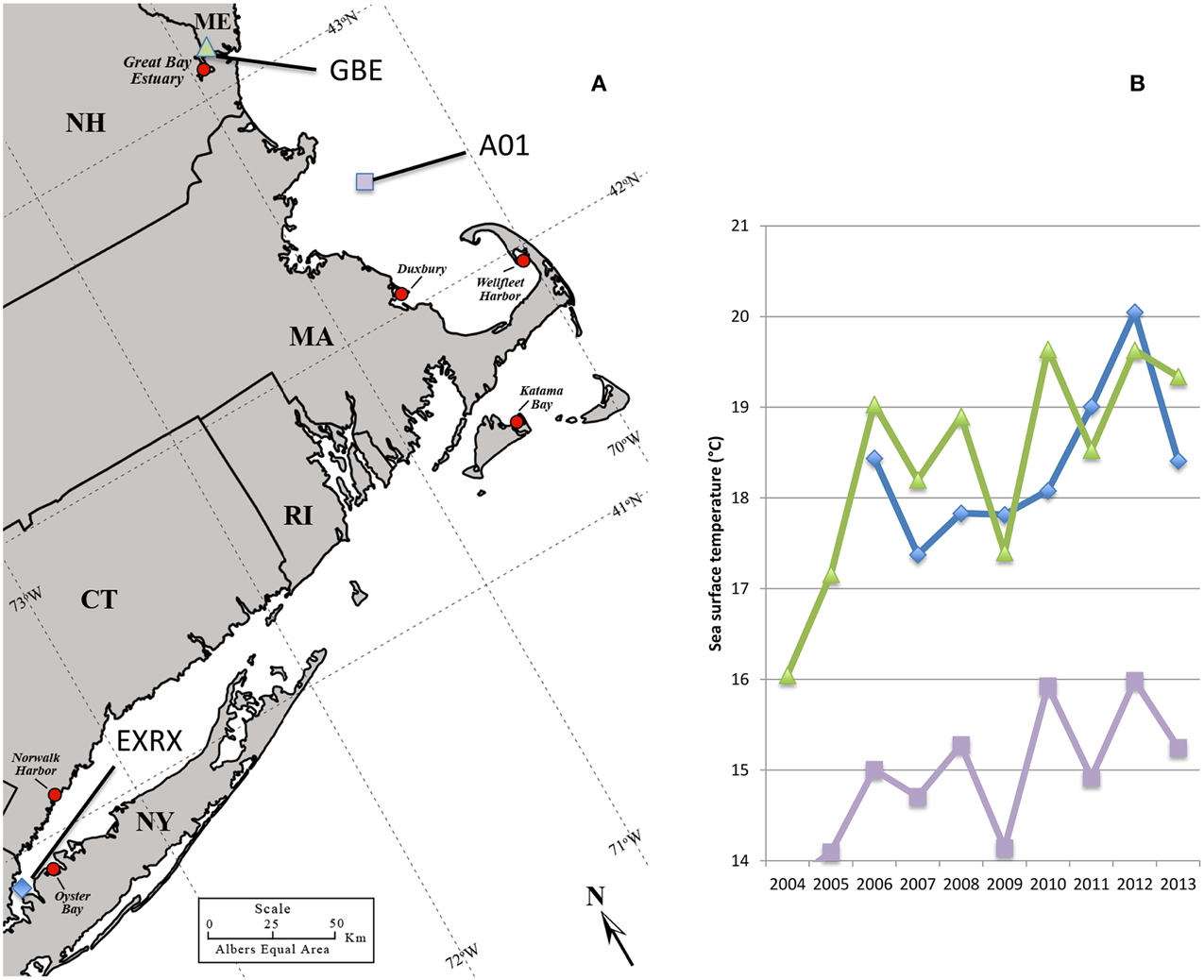
Figure 1. Regional oyster production areas and ocean temperatures. Regional oyster harvest and production areas all identified with red circle, some of which have been linked to cases or outbreaks of V. parahaemolyticus including: Oyster Bay NY in 1998, 2012, and 2013; Norwalk Harbor CT in 1998, and 2013; Katama Bay MA in 2013; and Duxbury MA in 2013. No closures had occurred at the Great Bay Estuary site in NH or Wellfleet Harbor in MA as of 2013 (A). Ocean buoys used for temperature measurements identified by blue diamond, purple square, and green triangle symbols (A,B). Increasing average monthly sea surface water temperatures during June 2004–2013 at offshore buoys in Long Island Sound (EXRX), the Gulf of Maine (A01), and the Great Bay Estuary (GBE) (B). Map was adapted from the US Department of Agriculture Natural Resource Conservation Service.
A significant obstacle for the study of emergent pathogenic strains of V. parahaemolyticus is our lack of understanding of factors that define virulence and that could be used to detect pathogens within mostly non-pathogenic populations. Few of the diagnostic markers most commonly used to define pathogens are actually implicated in disease, including two hemolysin genes (tdh and trh) and a horizontally acquired type-three secretion system (T3SS2) (Honda and Iida, 1993; Hiyoshi et al., 2010). Although hemolysins are sufficient for inducing some disease symptoms, they are not necessary for disease in either mice or humans, indicating they are not the only virulence determinant (Nishibuchi et al., 1992; Xu et al., 1994; García et al., 2009; Thongjun et al., 2013; Banerjee et al., 2014). Perhaps more concerning, the abundance of hemolysin-containing strains in the environment often does not correlate with increased incidence of disease, calling into question the ability of these markers to sufficiently predict risk evaluation (Johnson et al., 2012; Jones et al., 2012).
In the absence of a definitive diagnostic marker of virulence, identification of related strains or lineages of pathogens that are known to cause infections would aid in the study of seasonal population dynamics associated with emergence of these pathogenic types. The many commonly applied strain typing tools, including serotyping, rep-PCR, pulse field electrophoresis (PFGE), intergenic spacer region (ISR-1), and restriction fragment length polymorphisms can group similar strains but have some limitations for resolving evolutionary relationships, especially when used individually (Chowdhury et al., 2000, 2004; Nair et al., 2007; Jones et al., 2012; Banerjee et al., 2014; Lüdeke et al., 2014). Multi-locus sequence analysis (MLSA) of conserved housekeeping genes can better define relatedness and recombination among strains (Depaola et al., 2003; Jolley et al., 2004; Thompson et al., 2005; Sawabe et al., 2007; González-Escalona et al., 2008; Ellis et al., 2012; Paranjpye et al., 2012; Martinez-Urtaza et al., 2013; Turner et al., 2013; Banerjee et al., 2014). But the analysis of pathogens alone, in the absence of related non-pathogenic relatives, will not provide a complete picture of evolution of virulence.
Here we combine MLSA, genotyping, and phylogenetic analysis to relate clinical strains with environmental isolates from northern New England. The strains analyzed include 94 clinical isolates from reported cases in three northern New England US states and more than 200 environmental isolates identified from the region since 2007 (Mahoney et al., 2010; Ellis et al., 2012). The study demonstrated that disease-causing strains are genetically diverse and polyphyletic. Some infections were caused by pathogens that are resident, but the strains that caused a steep rise in infections in 2013 are derived from the PNW ST36 population as previously suggested (Martinez-Urtaza et al., 2013; Newton et al., 2014). This study lays the foundation for detailed analysis of conditions that have promoted emergence and invasion of pathogenic types in the region.
Materials and Methods
Bacterial Strains, Strain Designations, and Culture Conditions
Ninety-four V. parahaemolyticus clinical isolates, defined as isolated from a clinical (patient) source (e.g., wounds or stool), from 2010 to 2013 were provided by cooperating public health laboratories in MA, New Hampshire (NH), and Maine (ME) (Table 1). For the purposes of this study and in the absence of contradictory data, all these clinical isolates are considered pathogenic, regardless of genotype. Additional environmental strains from the Great Bay Estuary bordered by NH and ME prior to 2011 were also included for comparisons (Supplementary Table 1) (Mahoney et al., 2010; Ellis et al., 2012). A few previously unreported strains collected from the Great Bay Estuary in 2010–2013, and two environmental isolates from oysters harvested in CT that were recalled due to an outbreak were also used in this study (Supplementary Table 1). For the purpose of this study, we define environmental strains that lack hemolysin genes, and that are not phylogenetically related to strains from clinical sources as non-pathogenic. Environmental isolates that harbor either or both hemolysin genes, or are related to isolates from clinical sources (e.g., shared phylogeny or identical ST) are defined as potentially pathogenic. Finally, although not yet identified locally, isolates from the environment that are genetically indistinguishable from a clinical isolate by genotype and ST, and deemed clonal based on whole genome phylogeny are considered pathogenic. Several pathogenic V. parahaemolyticus isolates acquired previously included ST36 strain F11-3A (González-Escalona et al., 2008), pandemic strain MDOH-04-5M732 (Davis et al., 2007) and pre-pandemic strain BB22OP LM5132 (Mccarter, 1998), which were used as controls. Strains were grown in heart infusion (HI) medium with 3% NaCl (Fluka, Buchs, Switzerland) at 28°C (for environmental strains) or 37°C (for clinical strains) for routine culturing. Strains from environmental sources are enriched in APW, isolated on CHROMAgar Vibrio (CHROMagar, Paris, France) as purple colonies and cultured on T-soy agar as previously described (Schuster et al., 2011).
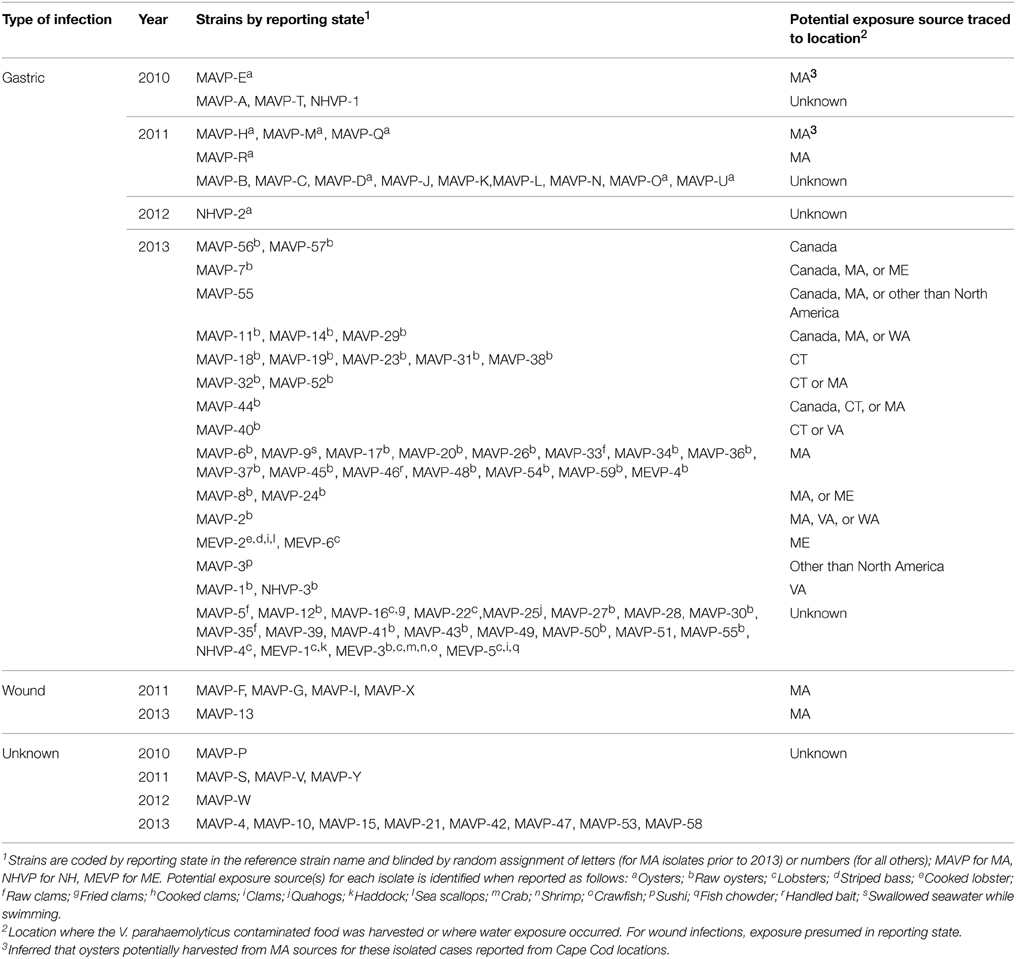
Table 1. Sources and reporting states for clinical isolates available for this study.
Genotypic Analysis
Genotyping (Table 2, Supplementary Table 1) was performed by PCR amplification of template genomic DNA isolated from cultures grown in HI medium for 6 h using either Master Taq (5 PRIME, MD, US) or AccuStart PCR Supermix (Quanta, MD, US). DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega, WI, USA), using columns and manufacturer-provided recipes (Epochlifescience Inc., TX, US), or by cetyltrimethylammonium bromide protein precipitation and organic extraction (Ausubel et al., 1990). The presence of a species-specific gene (tlh), both hemolysin genes (tdh and trh) and the pandemic marker ORF8 was determined using published primers and cycling parameters (Panicker et al., 2004) (Table 2, Supplementary Table 1). The presence of additional virulence-associated genes including a homolog of Escherichia coli cytotoxic necrotizing factor vopC as well as several additional genes located within pathogenicity islands of strain MDOH-04-5M732 including T3SS2 genes vscC2 and effector vopP, was assessed using published primers and cycling parameters (Caburlotto et al., 2009). For each reaction, the presence of an amplicon of the correct size was determined following electrophoresis in 0.7% (for large amplicons) or 1.2% (for small amplicons) Sea Kem LE agarose (Lonza Group Ltd., NH, US) in 1× TAE buffer with 1× GelRed (Phenix Research Products, NC, US) for amplicon visualization. The size of each amplicon was determined by comparison to 1 Kb-plus ladder (Invitrogen Inc., NY, US) and also compared to amplicons from control strains including F11-3A and MDOH-O4-5M732. Presence of the correct size amplicon or gene (for sequenced strains) is denoted by (+), whereas absence of amplicon is denoted by (−).
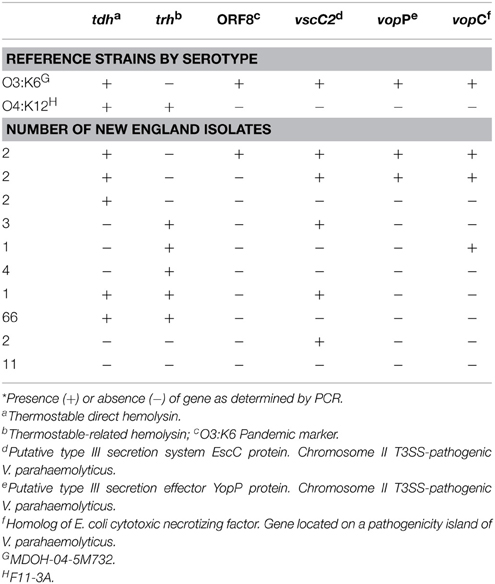
Table 2. Distribution of genotypes* among Northeastern US clinical isolates.
Urease Activity
Urease activity (Table 3, Supplementary Table 1) was determined on strains first grown in HI medium for 6 h at 28°C or 37°C, and then inoculated in triplicate as 10 μl samples onto 200 μl modified Christensen's urea agar, containing 2% NaCl, 0.1% peptone, 0.1% dextrose, 0.2% KH2PO4, 2% urea, 0.12% phenol red, and 2% agar in the wells of a 96-well plate. The plates were sealed with Breathe-Easy membrane (USA scientific Inc., FL, US) and incubated with ventilation at 37°C overnight. A positive reaction is observed as a change in color from yellow to pink. Wells without bacterial inoculum remained yellow. The association of the presence of a hemolysin gene (tdh or trh) and urease activity in isolates was determined using a two-tailed Fisher's exact test (Preacher and Briggs, 2001).
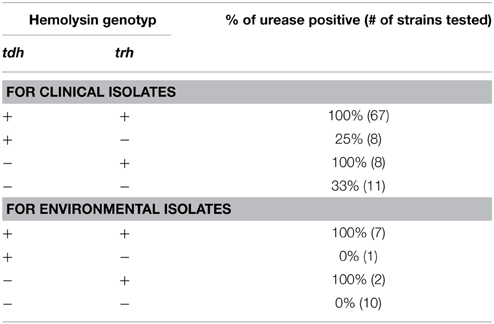
Table 3. Correlation of urease activity with the presence of trh.
Genome Sequencing and Assembly
The V. parahaemolyticus ST631 strain MAVP-E, and four representative ST36 isolates including MAVP-26, MAVP-36, MAVP-45, all traced to shellfish harvest areas in MA, and MAVP-V, from an unknown source, were sequenced using an Illumina-HiSeq2500 device at the Hubbard Center for Genome Studies at the University of New Hampshire. Genomic DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega, WI, USA) as recommended by some sequencing centers or by a cetyltrimethylammonium bromide and organic extraction method (Ausubel et al., 1990) that provides both higher quality and quantity of DNA but requires more technical skill. The DNA quality was assessed visually by electrophoresis. Sequencing libraries were generated from 1 μg of genomic DNA as determined using the Qubit 2.0 fluorimeter (LifeTech, CA, US). DNA was sheared on the Covaris M220 Ultasonicator to a mean size of 500 bp. Libraries were generated using the TruSeq Kit and targeted size selection of 500 bp was completed using the optional gel-extraction method in the TruSeq protocol (Illumina). MAVP-E was sequenced by a high output mode run, 101 bp paired-end whereas MAVP-26 and MAVP-36 were sequenced using a rapid output mode run, 150 bp paired-end with 152-fold coverage for MAVP-E, 249-fold coverage for MAVP-26, 238-fold coverage for MAVP-36, 355-fold coverage for MAVP-45, 847-fold coverage for MAVP-V, 84-fold coverage for MAVP-M, and 167-fold coverage for CT4287. Raw sequences were processed and de novo assemblies performed using the A5 pipeline (Tritt et al., 2012).
Multi-Locus Sequence Analysis and Phylogenies, and Analysis of Population Structure
Phylogenetic analysis was performed from concatenated sequences derived by PCR amplification of multiple house-keeping loci. The amplicons were generated using Master Taq (5 PRIME, MD US), and sequenced by the Sanger method at the UNH Hubbard Center for Genome Studies or by Functional Biosciences (WI, US). For inferring multi-locus phylogeny, we used either seven loci (See Supplementary Figure 1) from two schemes as previously described (Ellis et al., 2012) including three loci adopted to determine the relationships broadly among Vibrio spp. (Sawabe et al., 2007) (gyrB, pryH, and recA) and four loci adopted to closely examine within species relationships (González-Escalona et al., 2008) (dnaE, dtdS, pntA, and tnaA); because these four are the only sequenced loci that overlap with those from strains in the public database (www.pubmlst.org), the phylogenetic relationships of a larger collection of isolates in this study with those of a global distribution were inferred using only four loci (dnaE, dtdS, pntA, and tnaA) (Figure 2). The primer sequences (Supplementary Table 2) and corresponding cycling parameters were used exactly as in published protocols (Sawabe et al., 2007; González-Escalona et al., 2008; Jolley, 2010). For phylogenies inferred from all seven loci, each forward and reverse raw sequence for 25 clinical isolates from 2010 to 2012 was assembled, and the contiguous sequences were then aligned and trimmed to match the length of corresponding sequence of 192 Great Bay Estuary environmental isolates (Ellis et al., 2012), only two of which harbor hemolysin genes. An additional eight isolates collected during 2013 were also included in some analysis. The sequences for individual isolates were then concatenated in alphabetic order. For phylogenies inferred from four loci (dnaE, dtdS, pntA, and tnaA), each raw sequence was assembled, aligned, and trimmed to match the exact corresponding amplicon sequence from the public database. Neighbor-joining trees for concatenated sequence of either four loci (1868 bp) or seven loci (2988 bp) were constructed by Mega 5.0 software (Tamura et al., 2013) using Jukes-Cantor model. The statistical support was assessed by 1000 bootstrap re-assemblies.
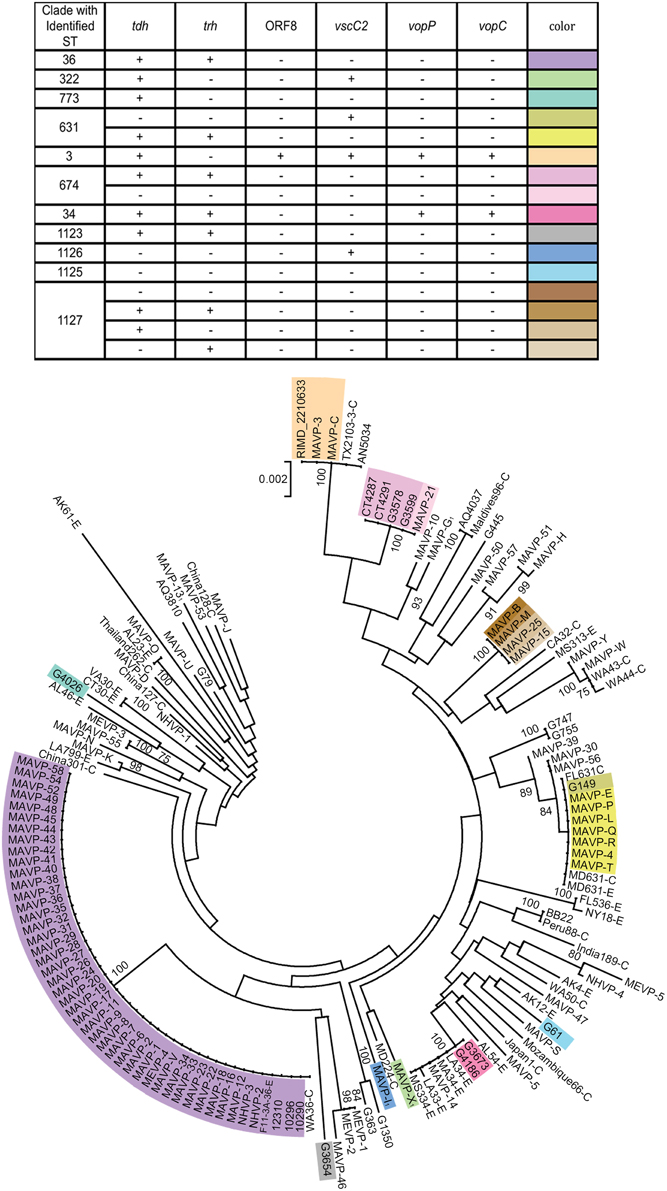
Figure 2. Population structure of Northeast US clinical, related environmental, and some unique worldwide isolates of V. parahaemolyticus. A consensus neighbor-joining tree was constructed from four concatenated housekeeping gene loci including dnaE, dtdS, pntA, and tnaA sequences by using a Jukes-Cantor model. For ease in distinguishing pertinent information associated with worldwide strains from the MLST database, a single representative strain from several sequence types was identified from among the available strains, and the representative strain identified by geographic location (USA by state, international by country name), sequence type number, and as clinical (C) or environmental (E). Environmental strains from the region included isolates from the Great Bay Estuary (all with prefix G) or Connecticut (CT). For seven strains whose draft or complete genomes are publicly available, the loci were recovered from the available assemblies. Among related strains where the probable sequence type of the strains was determined, unique genotypes are indicated by color provided in the key and overlaid upon the tree. The bar indicates 0.2% divergences, and branches with less than 70% bootstrap support are unlabeled. Several clinical strains, for which one or more housekeeping loci were not successfully amplified and sequenced were excluded from the analysis (MAVP-A, MAVP-F, MAVP-59, MEVP-6). 1Isolates were from wound infections.
Comparisons with the published MLST database (http://pubmlst.org) to identify STs were performed on 12 clinical and 16 environmental isolates for which the sequencing of three additional loci (gyrB, pyrC, and recA) were completed as described (González-Escalona et al., 2008). Raw sequences were assembled, aligned, and trimmed as described above. Allele numbers and ST numbers were determined by matching the public database. The STs of sequenced strains were determined from raw short read sequences using the short read sequence typing (SRST2) pipeline (Inouye et al., 2012).
The extent of recombination and mutation within the population was visualized and analyzed by several approaches. The contribution of recombination to phylogeny was evaluated visually using SplitsTree v4 neighbor net analysis of four loci, and the Phi test module was applied for determining statistical support (Huson and Bryant, 2006). The standardized index of association (IAS) was determined from a non-redundant allele database for the collection of 90 clinical and 16 environmental strains using the LIAN 3.5 linkage analysis program (Haubold and Hudson, 2000). This statistic describes the linkage disequilibrium in a multilocus data set where a low rate of recombination relative to mutation is indicative of linkage disequilibrium (IA > 1). The null hypothesis that variation of the observed data (VD) does not differ from that predicted for a population in equilibrium (i.e., experiencing a high rate of recombination relative to mutation) (Ve) was tested by a non-parametric Monte Carlo simulation, with the 5% critical value to determine significant linkage (L). ClonalFrame 1.1 was used to determine the relative influence of recombination compared to mutation (r/m) to nucleotide variation (Didelot and Falush, 2007).
Reconstruction of Whole Genome Phylogenies
Representative strains within the species V. parahaemolyticus were selected from among the 25 NCBI genome groups (defined as such by ~90% genome identity) from NCBI genomes phylogeny (http://www.ncbi.nlm.nih.gov/genome/691) that had accompanying information on geographic isolation, year, and sample source (environmental or clinical including wound, stool, and ear). The raw sequences from MAVP-E, MAVP-26, MAVP-36, MAVP-45, MAVP-V, MAVP-M, CT4287, (see Table 1 and Supplementary Table 1 for a description of these isolates) were processed and de novo assembled using the A5 pipeline (Tritt et al., 2012). The assembled contigs of all isolates were analyzed using REALPHY v. 1.09 (Bertels et al., 2014). Sequences were analyzed in three separate alignments, each with a unique reference strain including 10290 (GCA_000454205.1), BB22OP(NC_019955.1, NC_019971.1), and RIMD 2210633 (NC_004605.1, NC_004603.1), for phylogenies across a broad distribution of strains, and 10290, 10329 (NZ_AFBW01000001.1 - 33.1), and 10296 (GCA_000500105.1) for analysis of strains within the ST36 clonal complex clade. From these alignments multiple alignment positions were extracted and then merged into a single alignment. Neighbor-joining phylogenies were reconstructed using the maximum likelihood method in PhyML, with a GTR substitution matrix and a gamma-distributed rate heterogeneity model (Guindon et al., 2010). Phylogenies were visualized as trees using FigTree 1.4.2 (Rambaut, 2012). The branch length reflects nucleotide changes per by total number of nucleotides in the sequence.
Environmental Data and Analysis
Sea surface water temperature (SST) data were acquired from US Integrated Ocean Observing System buoys in Long Island Sound and the Gulf of Maine (http://www.neracoos.org/datatools/climatologies) and from the NOAA National Estuarine Research Reserve System Wide Monitoring Program (SWMP) buoys in the Great Bay Estuary (http://cdmo.baruch.sc.edu/get/export.cfm). Monthly average SST data from 2004 to 2013 were compiled from representative buoys for the areas of interest. In Long Island Sound, the EXRX buoy is in close proximity to Norwalk Harbor, CT and Oyster Bay, NY, the AO1 buoy is representative of the southwestern end of the Gulf of Maine, and the Great Bay SWMP buoy is representative of the Great Bay estuary.
Nucleotide Sequences
MLST loci for strains, MAVP-I, MAVP-M, MAVP-E, MAVP-26, MAVP-36, G61, G3654, G4026, G3673, and CT4287 are available at www.pubmlst.org, and Illumina short read genomic sequences for bioproject PRJNA263814 including MAVP-26 (SAMN03107383), MAVP-36 (SAMN03107385), MAVP-E (SAMN03107386), MAVP-V (SAMN03177809), MAVP-M (SAMN03177808), CT4287 (SAMN03177811), and MAVP-45 (SAMN03177810) are available at NCBI.
Results
Genetic Diversity Among Clinical Isolates and Distribution of tdh, trh, and Urease Activity
In light of the recent rise of V. parahaemolyticus infections in the northeastern US, clinical isolates from infections reported in MA, NH, and ME were obtained to identify pathogenic strain types, and determine whether they belong to resident or invasive lineages (See Table 1). A total of 94 clinical V. parahaemolyticus strains from infections reported from 2010 through 2013 were compared to each other and 200 environmental isolates from the region collected since 2007 (Mahoney et al., 2010; Ellis et al., 2012) (Table 1). Even though clinical isolates were not archived from every infection, and information was incomplete, this collection provides an extensive survey of regional infections (Table 1). Prior to 2013, consumption of local shellfish was implicated in only five, isolated infections (Table 1). However, by 2013 many clinical isolates (n = 37) were traced to at least one local source (Table 1 and see Newton et al., 2014). Gastric infections most often were attributed to contaminated oysters (n = 49), some of which were either definitively or potentially locally harvested (n = 27). In 2013, two infections were traced to recreationally harvested seafood in ME, including oysters, cooked fish, and lobster (Table 1). No illnesses were definitively traced to seafood from NH, although it is important to note that at the time of these collections commercial harvesting of oysters from this state was limited. Remarkably, two gastric infections were apparently induced by handling of raw product and casual ingestion of water while swimming, consistent with the presence of highly infective strains with a low infectious dose in near shore waters. Relatively fewer isolates (n = 5) were recovered from wound infections (Table 1).
The distribution of genotypes among clinical isolates indicated that infections were caused by a variety of strains, and although no specific gene or combination of virulence-associated genes defines pathogens reported in the region, certain genotypic profiles were more abundant than others (Table 2, and Supplementary Table 1). Very few isolates contained only the tdh gene (n = 6). Two isogenic, tdh-containing isolates, MAVP-C and MAVP-3, were identified as the pandemic O3:K6-type (Table 2, and Supplementary Table 1). Most clinical isolates from the Northeast US (80%) harbored the trh gene either alone or in combination with tdh (Table 2, Supplementary Table 1). In fact, strains harboring both tdh and trh were highly prevalent among clinical isolates from reported cases in this region even prior to 2013 (Table 1, and Supplementary Table 1). Yet several clinical isolates (n = 13), only three of which were wound isolates, harbored neither hemolysin.
Because urease activity may be useful for easily identifying some pathogenic types due to the proximity of the urease locus only ~7-Kb from the trh gene in the same pathogenicity island of some characterized strains (Park et al., 2000), we investigated whether urease activity correlated with the presence of hemolysin genes and clinical status among northern Northeast isolates (Tables 2, 3). Urease activity significantly correlated with the presence of trh (100% of isolates) either in the absence of tdh in clinical strains (p = 0.012) or in combination with the tdh gene for both clinical (p < 0.0001) and environmental (p < 0.0001) strains. Urease activity did not correlate significantly with tdh alone (p = 1.0). A few environmental isolates of unknown virulence were also urease positive, all of which harbor trh (Table 3).
Phylogenetic Relationships Among Clinical and Environmental Isolates and Identification of Resident Clades
Neighbor-joining phylogenies identified shared lineages of environmental strains and pathogens isolated in the region (Supplementary Figure 1, Figure 2). Related environmental and clinical isolates were first identified by visual inspection of a neighbor-joining phylogeny based on seven genes sequenced from clinical strains isolated prior to 2013 (Supplementary Figure 1). This tree revealed that only eight out of 192 characterized environmental isolates were closely related to clinical isolates as inferred by bootstrap values greater than or equal to 70. The relationships of 90 clinical isolates, and 16 environmental (eight strains isolated prior to 2010, and eight previously unreported hemolysin-encoding strains isolated in 2013) to strains collected worldwide were then examined more broadly by MLSA using four loci shared with the public database (Figure 2). This analysis demonstrated that regional clinical isolates are polyphyletic and include some strains of known STs (Figure 2, Supplementary Table 3). Strains within clades are not always clonal when assessed using virulence-associated genotypes (Table 2, Supplementary Table 1, Figure 2).
Even though definitive traceback data was incomplete prior to 2013, the reporting data (see Table 1) did facilitate identification of the isolates most likely from local sources, and those from outside the region. Both isolates harboring ORF8 grouped with the globally distributed pandemic ST3 strains, but these were not from local sources. The ST631 clade grouped isolates from multiple years that were traced to oysters consumed at various Cape Cod MA locations where oysters are farmed and imported oysters are not typically marketed and thus they were identified as putatively local, and a single isolate was traced to a local source (Table 1, Figure 2). A single environmental ST631 isolate (G149) that based on whole genome phylogeny is a close relative to these strains (Xu, unpublished data) was also isolated in NH in 2007. In agreement with the persistence of this strain type, ST631 are not strict clones (Supplementary Table 1, Figure 2). MAVP-14, and two environmental isolates from NH harboring both tdh and trh grouped with several ST34 environmental strains from the Gulf of Mexico, specifically Louisiana (LA) (González-Escalona et al., 2008) (Supplementary Table 3). MAVP-21 was most closely related to four tdh+/trh+ ST674 environmental isolates, two of which were recovered from oysters harvested from CT and two from the Piscataqua River of the GBE (Figure 1). Finally, even though genetically diverse strains identified as a new ST, ST1127, caused four infections, three were from unknown sources and no environmental isolates of this ST have been identified, thus it is not clear whether they are residents.
We evaluated the MLSA data by several statistical tests to determine the relative contributions of mutation and recombination to population structure. The influence of recombination on clonal structure was supported by the Phi test in SplitsTree v.4 (P < 0.001), and was also indicated by the reticulate structure of the Neighbor Net analysis (Figure 3). However, the LIAN test of recombination indicated the population is in linkage disequilibrium (P < 0.01; L = 0.1009), with a standardized index of association of IAS = 0.2228, which is also visible in the Neighbor Net analysis as long branch lengths emerging from a central network of recombination. Approximately one recombination event for every three mutations is predicted (r/m = 0.337707; 95% credibility region = 0.145408 – 0.571994), which indicates an effect of both recombination and mutation upon population structure.
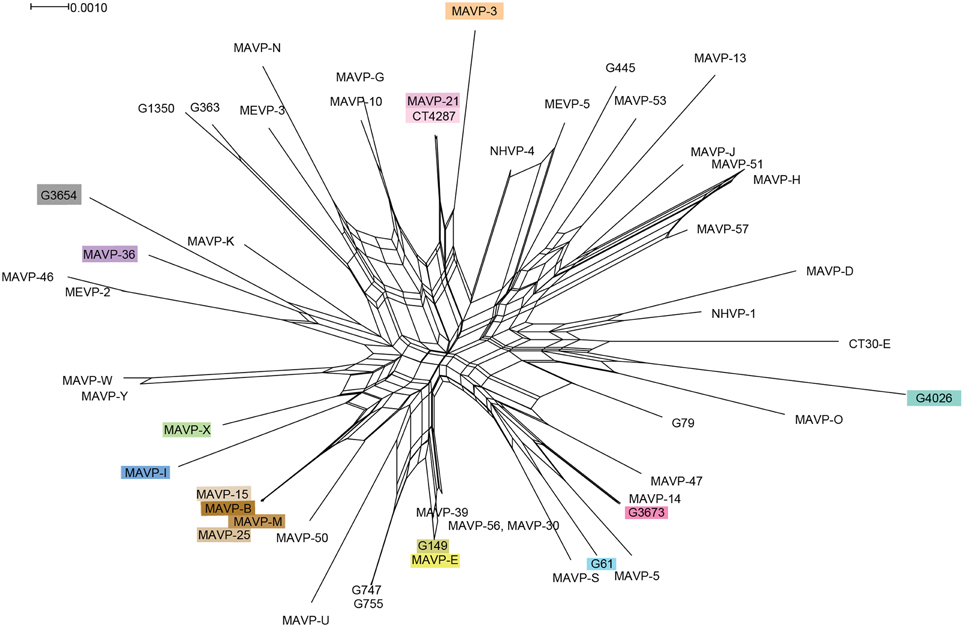
Figure 3. Recombination network revealed by splits decomposition analysis of genotypes of Northeast regional V. parahaemolyticus isolates. The Splits Tree analysis is based on four concatenated gene loci including dnaE, dtdS, pntA, and tnaA (1868 bp) from 54 unique regional clinical and related environmental isolates. Where multiple strains were identified as sharing the same allelic profile or ST, one representative strain per unique genotype was selected for inclusion in analysis. For ease in identifying resident and invasive clades, strains are designated by colors that correspond exactly to color scheme in Figure 2.
Occurrence of Non-Indigenous ST36 Clinical Isolates in the Northeast US
Most clinical isolates from 2013 (n = 42) were closely related to each other and to ST36 clonal complex strains from the PNW (Figure 2) (Newton et al., 2014). The ecological invasion of the ST36-clade strain in the Atlantic from the Pacific was suggested when these genotypes were associated with infections reported in NY in 2012 (Martinez-Urtaza et al., 2013). All isolates from the northern New England region in this ST36-clade shared the same virulence associated genotype; however, MAVP-V of unknown source and that was isolated in 2011, is distinct from the others from the Northeast in that it is missing a phage-encoding island that is also missing in closely related strain 12310 from Washington state (Haendiges et al., 2014) (Table 1, Supplementary Table 1, Figure 2, and Supplementary Figure 1). Remarkably, the ST36-clade isolates were not only traced to oysters harvested south of Cape Cod proximal to Long Island Sound and Oyster Bay NY, but also from north of Cape Cod in the Gulf of Maine (See Figure 1). This geographical distance suggests that the New England ST36-clade strains both spread northward and grew sufficiently to increase infections (Newton et al., 2014). MAVP-26, MAVP-36, and MAVP-45 (traced to at least two, and potentially three shellfish harvest areas in MA), and MAVP-V (from an unknown source) were subsequently confirmed to be ST36 by whole genome sequencing.
Whole Genome Phylogeny of V. parahaemolyticus and Relationships Between ST36 Atlantic and Pacific Populations
From the collection of available draft genomes of strains of known identity (i.e., clinical and environmental source) and geographic location of isolation, we examined phylogenetic relationships among isolates of V. parahaemolyticus. Although the selected strains likely underrepresent the diversity and distribution of pathogen types, they nevertheless provide insight into the evolution of different lineages. Distinctive pathogen lineages grouped within three major nodes, one of which included the pandemic RIMD 2210633, BB22OP, and northern New England resident ST631 clades, a second of which grouped the ST36 clade and other clinical and environmental strains from both the Atlantic and Pacific, and a third that grouped fewer and more distantly-related representative strains from the Atlantic and Gulf of Mexico (Figure 4). This broad phylogenetic relationship illustrates that although infectious strains of V. parahaemolyticus are polyphyletic, they may yet belong to genetic clusters that can be diagnostically and epidemiologically informative.
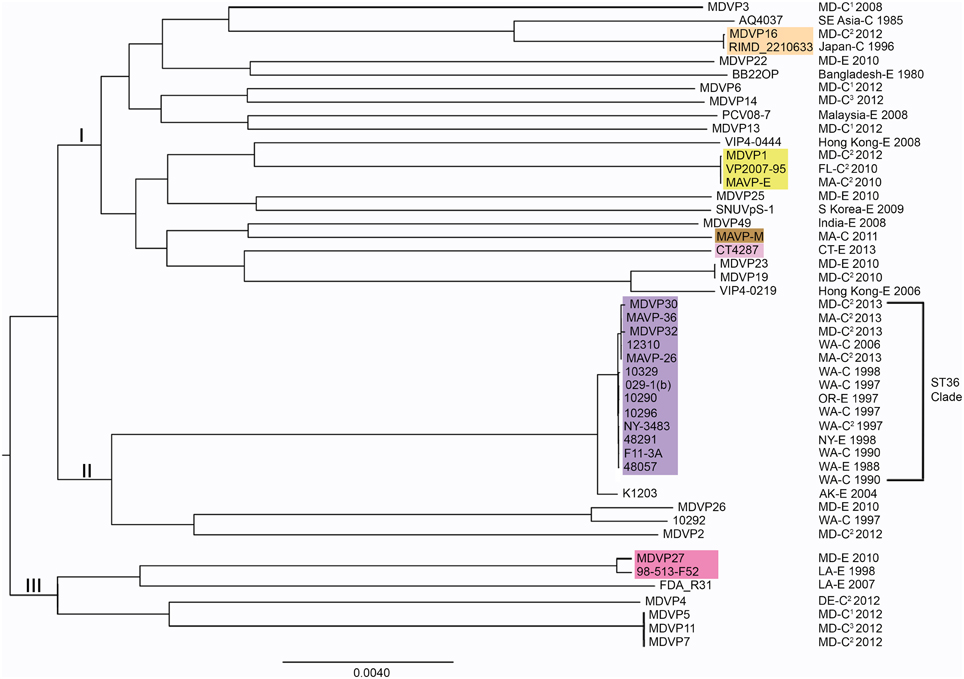
Figure 4. Phylogenies of pathogenic lineages of V. parahaemolyticus. Multiple genome reference-sequence alignment based phylogeny were reconstructed using REALPHY v1.09 to evaluate the evolution and relatedness of pathogenic lineages, where related pathogens grouped within three major nodes (I, II, and III). Maximum likelihood phylogenies of strains of broad phylogenetic distribution were reconstructed based on merged reference strains 10329, BB22OP, and RIMD 2210633, where the merged alignment represents 63% coverage of sites of the largest reference genome (Vp10329). The branch length reflects nucleotide changes per by total number of nucleotides in the sequence. Representative strains are identified by geographic location (USA by state, international by country name), as clinical (C) or environmental (E) and year isolated. For ease in identifying strains or sequenced types identified in the Northeast, select strains are designated by colors that correspond exactly to color scheme in Figure 2. 1Isolates were from wound infections. 2Isolates were from gastric infections. 3Isolates were from ear infections.
To test this hypothesis, the genomes and relationships of related ST36-clade strains were compared to gain insight into the patterns of microevolution within this clade (Figure 5). As expected from a dynamic of recent ecological invasion in the Atlantic, most strains within this clade isolated prior to 2011 were from the PNW, where this strain is indigenous. One exception was a single environmental isolate from NY in 1998 NY-3483. NY-3483 grouped with these historic PNW ST36 strains. More recent clinical isolates from 2013 traced to Atlantic sources were closely related to and share a common ancestor with strain 12310, a Washington State (WA) isolate from 2006. This topology strongly supports that the Atlantic ST36 strains are non-indigenous, and were derived from the PNW ST36 population.
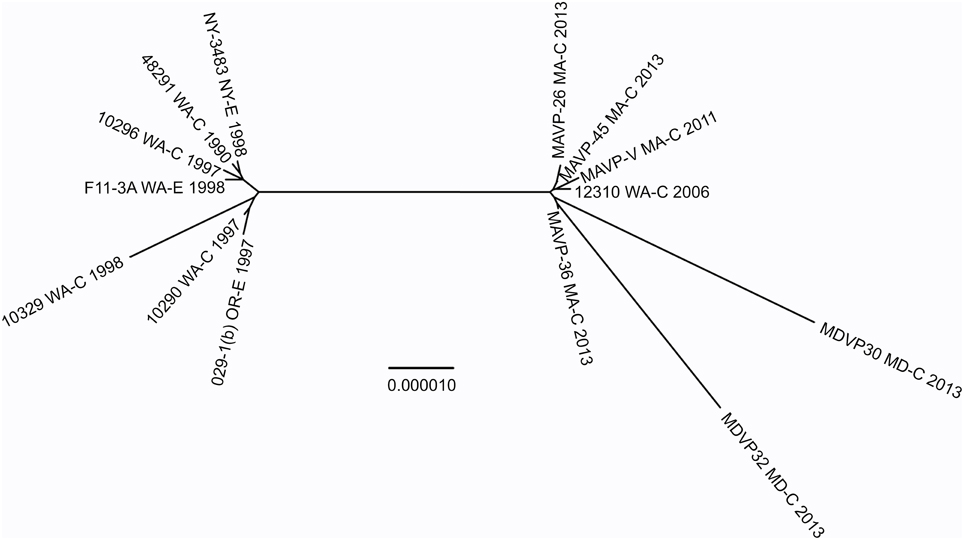
Figure 5. Phylogenetic relationship between Atlantic and Pacific ST36 populations. Multiple genome reference-sequence alignment based phylogeny were reconstructed using REALPHY v1.09 to evaluate the evolution and relatedness of ST36 pathogenic lineages. Maximum likelihood phylogenies within the ST36 clade were reconstructed based on merged reference strains 10329, 10290, and 10296 where the merged alignment represents 92% coverage of sites of the largest reference genome (Vp10329). The branch length reflects nucleotide changes per by total number of nucleotides in the sequence. Representative strains are identified by geographic location [USA by state, as clinical (C) or environmental (E) and year isolated].
Discussion
V. parahaemolyticus has become the most common bacterial infection acquired from seafood in the US, with an estimated 35,000 cases each year (Scallan et al., 2011). Even with protective measures in place in the Gulf States, and the PNW, reported cases continue to increase nationwide (Crim et al., 2014). But disease burden in the northeastern US had until recently remained relatively low, even with these national trends. Over the last several years, assumptions that pathogens could not thrive in the cooler waters of the Northeast have given way as several unprecedented outbreaks occurred across multiple states leading to the implementation of protective measures in both CT and MA. The water temperatures in the Long Island Sound and the Gulf of Maine were at or near 11-year maxima from October 2011 through September 2012, and again in July 2013 (Dawicki, 2013; NERACOOS, 2014) (Figure 1). The first outbreaks coincided with these unusually mild conditions that have also had profound effects on the Gulf of Maine ecosystem (Mills et al., 2013). Likewise, many previous US outbreaks and the spread of the pandemic strain type on the Pacific US coast have also been linked to warmer ocean temperatures (Mclaughlin et al., 2005; Martinez-Urtaza et al., 2008), a predicted effect of global climate change, indicating that these recent outbreaks in the Northeast US may herald a continuing trend of increased disease in this region, and for others in more northern latitudes (Baker-Austin et al., 2013).
The increase in recent infections in the Northeast was associated with both resident and non-indigenous V. parahaemolyticus lineages. The establishment of the ST36 strain, first reported in the region during an outbreak in Oyster Bay NY in 2012 explains the abrupt increase in cases traced to several different locations of northern New England in 2013, including one location in CT proximal to Oyster Bay NY, and two geographically distant locations in MA, to the south and north of Cape Cod (Martinez-Urtaza et al., 2013; Newton et al., 2014) (See Results Section – Occurrence of Non-Indigenous ST36 Clinical Isolates in the Northeast US, Figure 1, and Table 1). However, even before these unprecedented multi-state outbreaks, cases were on the rise in the region from potentially emergent residential lineages including ST631, ST34, and ST674 (See Table 1, Supplementary Figure 1, and Figure 2). Genetically diverse ST631 strains were responsible for seven isolated infections between 2010 and 2013 (Figures 1, 2, Table 1, Supplementary Figure 1). Previously identified pathogenic ST631 isolates have been traced to Florida (FL) (Noriea Iii et al., 2010), and Maryland (MD) (Haendiges et al., 2014) indicating a fairly wide Atlantic US-coastal distribution of this ST (Supplementary Table 3). The identification of a single environmental ST631 isolate from NH (G149) lacking tdh and trh, that was closely related to the clinical isolates based on whole genome maximum likelihood phylogeny (Xu and Whistler, unpublished) indicates the clade is resident rather than transient in New England (Ellis et al., 2012). A single clinical isolate potentially from a local oyster and two environmental isolates were identified as ST34 (See Table 1, Figure 2). Although environmental ST34 isolates of unknown virulence are broadly distributed from the Pacific and Gulf of Mexico waters, two previously identified ST34 isolates are from clinical sources, indicating the potential for virulence among this clade (Supplementary Table 3) (González-Escalona et al., 2008). Finally, within the ST674 lineage, a single clinical isolate from an unknown source and four environmental isolates from CT and NH oysters were identified (Table 1, Figure 2). These are the first ever reported strains of this ST and their presence in more than one location in New England suggests these related strains are resident and potentially endemic. Further analysis of the clinical and environmental strains from these resident lineages may provide insight into evolution and recent emergence of pathogenic types in the region.
The recent appearance of ST36-clade strains in the Atlantic is apparently not the first invasion: one isolate, NY-3483, was identified in NY in 1998, when one of the largest multi-state outbreaks of V. parahaemolyticus in the US occurred in NY and CT (CDC, 1999). That outbreak was attributed to the invasive pandemic O3:K6 strain type, not the ST36 strain type. This coincidence could indicate that certain conditions promoted the ecological invasion of non-resident pathogenic strain types, but neither strain was apparently able to establish residency. The whole genome phylogeny of these ST36 strains indicates that current ST36 strains causing infections in the Northeast are not derived from NY-3483 (Figure 5). Rather, the current ecologically invasive population shares a common ancestor with a quite distinctive 2006 isolate from the PNW (Figure 5). Curiously, the serologically unique ST36 strains responsible for several outbreaks in the PNW in 2006 (both WA and Canada) did not persist in that region (Banerjee et al., 2014). Furthermore, current trends in the PNW suggest that infections by ST36 strains have declined locally for as yet unknown reasons (http://www.cdc.gov/vibrio/investigations/). In the Atlantic, the non-indigenous ST36 lineage has persisted for several years, and spread northward into the Gulf of Maine (Figure 1). This strain recurrence suggests that yet-undefined environmental factors, perhaps in combination with a particular genetic predisposition, allowed it to compete with resident V. parahaemolyticus strains. Furthermore, two unique MD ST36 isolates chosen for this analysis for comparison (Haendiges et al., 2014) also share a common ancestor with the MA isolates (and more specifically MAVP-36), suggesting the clinical populations in the Northern and Mid-Atlantic could undergo admixture. However, the greater genetic distance of the MD isolates from other strains could reflect subsequent evolution following an earlier introduction or could indicate more rapid reproduction for this subpopulation as would be anticipated in the warmer ocean waters compared to the Northeast (Figure 5).
The analysis of the contributions of mutation and recombination indicates a significant effect of both processes upon population structure (Figure 3) (Vos and Didelot, 2009). The reticulate nature of the SplitsTree phylogeny is consistent with multiple evolving subpopulations of pathogens undergoing recombination, but not frequently enough to break up the distinct population structure of the major lineages (Figure 3) (Huson and Bryant, 2006). This data and its interpretation may appear to contrast with those inferred from other Vibrio sp. for which mutation is lower, not higher than recombination (Schuster et al., 2011; Turner et al., 2013); however, the subpopulations under scrutiny are often more closely related, and sometimes exclude environmental counterparts. When isolates that were not traced specifically to Northeast sources were excluded from this analysis, the population structure of regional isolates agreed with other studies that indicate substantial recombination among V. parahaemolyticus (González-Escalona et al., 2008; Ellis et al., 2012) (Figure 3). As one example, the SplitsTree analysis indicates a striking and more recent recombination history among ST631 and related non-ST631 environmental and clinical strains that could have influenced the evolution and potentially the emergence of this and new pathogenic types in the region (Figure 3). In contrast, the Northeast ST36 population has evidently evolved primarily through mutation, but this recent population expansion likely has provided insufficient time for the effects of recombination to be apparent. Even so, several resident strain lineages from local sources, including G3654, MAVP-46, and MEVP-2, were identified as potentially having a past recombinatorial history with the ST36 lineage.
Pathogenic strains from the Northeast, even those that are resident, are genotypically diverse (Table 2, Supplementary Table 1, Figure 2), making the development of an optimal detection strategy for all pathogens extremely challenging. The majority of infections in the Northeast were caused by trh-containing, urease positive strains (Tables 2, 3, Supplementary Table 1), and both attributes are used for assignment of environmental isolates as pathogens since trh-containing strains have increased among clinical isolates in North America in recent years (Tables 2, 3) (Jones et al., 2012; Martinez-Urtaza et al., 2013; Banerjee et al., 2014). Utility of urease activity as a surrogate for trh detection in Northeast V. parahaemolyticus populations is well supported by our analysis (Table 3). Yet the reliability of trh as a virulence marker for environmental surveillance is not thoroughly validated because the pathogenic potential of environmental isolates with this trait from any coastal population is still untested. Directed experimentation must determine whether environmental isolates that harbor hemolysins are in fact virulent and, conversely, that those lacking these genes are not virulent. However, virulence studies with environmental isolates are uncommon and reveal potential shortcomings in virulence models currently in use (Caburlotto et al., 2009; Mahoney et al., 2010). For instance, Mahoney et al. examined the cytotoxicity, a commonly-used proxy for virulence, of a variety of known pathogenic strains compared to environmental isolates lacking any of the classic virulence determinants used in surveillance, including tdh, trh, and T3SS2 (Mahoney et al., 2010). Surprisingly, many environmental isolates that would otherwise be identified as non-pathogenic are more cytotoxic to human CaCo-2 cells than most clinical strains, indicating relative cytotoxicity as a measurement for virulence may only be useful for studies comparing known pathogens, and not for environmental isolates (Mahoney et al., 2010). Thus, the determination of virulence potential and assignment of an environmental isolate as a pathogen is not an easy task given the limitations of current models for disease and infection. Ultimately, a better understanding of the prevalence of any potential diagnostic trait among non-pathogens in the environment is necessary.
The current state of knowledge supports multiple points of view on the utility of hemolysins as a diagnostic trait, even with their strong correlation with clinical isolation (Nishibuchi et al., 1992; Honda and Iida, 1993; Xu et al., 1994; García et al., 2009; Hiyoshi et al., 2010; Thongjun et al., 2013; Banerjee et al., 2014). It is concerning that 14% of clinical isolates, 11% if counting those from gastric infections only, harbored neither hemolysin gene (Table 2), and therefore, these strains would evade detection as human pathogens in any monitoring program relying upon only these markers. A similar prevalence of clinical strains lacking these virulence markers has been observed in other regions of North America (Jones et al., 2012; Banerjee et al., 2014). One explanation for the lack of tdh or trh in these stool isolates evokes the possibility of misidentification of non-pathogens consumed along with pathogens in an oyster or other food, since standard practices limit analysis to only one isolated colony in confirmed laboratory tests. However, we see this as improbable given that such isolates would need to both colonize and proliferate in order to achieve a high enough population to allow detection at this relatively high frequency. Furthermore, the detection of the same strain repeatedly during an outbreak would be unlikely if other, non-pathogenic V. parahaemolyticus were so abundant during infection. Even though published reports support that hemolysins contribute to virulence and are sufficient for some symptoms (Nishibuchi et al., 1992), other studies demonstrate virulence is unaffected by the deletion of these genes (Xu et al., 1994; Park et al., 2000). Thus this relationship is unresolved and supports the view that non-hemolysin producers isolated from humans may be pathogenic. More thorough analyses of the diversity of isolates from single infections are necessary to address this limitation in our knowledge of the defining characteristics of pathogens. If non-hemolysin producers are pathogenic, analysis of their genomic attributes may provide useful insight into fundamental attributes that promote their virulence. Even in the absence of a definitive (or even a few) virulence marker(s) harbored by all pathogenic V. parahaemolyticus strains, there is promise for monitoring particular lineages of concern (see Figure 2), by the use of markers or pathogenicity islands identified by whole genome comparisons. Assays informed by genomics comparisons could be tailored to different regions, but would need to include new strain diagnostic loci as populations continue to evolve and are influenced by invasive and introduced strains. Ultimately, the above limitations suggest that a combined trait or multi-locus-based assessment that includes hemolysins may be necessary for monitoring how changing population structure correlates with increased disease incidence, and for assessing public health risk.
Conclusions
Environmental conditions that create a warmer and longer season conducive for rapid growth do not entirely explain the current trend of increasing numbers of V. parahaemolyticus infections from local northern New England sources. This study suggests that changes in the bacterial populations underlie enhanced disease risk in the region. Yet, other factors may also contribute to increases in the reported number of cases. First, commercial shellfish harvesting has risen steeply in both MA and CT in recent years, with overall increased summer harvesting and consumption of product from the area (MADMF, 2013). But this increase does not explain the rise in cases in ME from recreationally harvested shellfish. Second, Vibriosis is now a reportable disease in all New England states, and an increased awareness of the pathogen by the public and health practitioners may contribute to the rise in reported cases. Regardless, the status of V. parahaemolyticus as an emergent pathogen of significant concern in the region is justified by projections of changing climate that may be conducive to V. parahaemolyticus growth. This concern is amplified by the finding that a non-indigenous pathogen invaded the Atlantic during a period with warmer than usual ocean temperature and has now likely established populations in the Northeast (Newton et al., 2014). Since Vibrio species are known to undergo recombination readily (Turner et al., 2013), and recombination influenced the population structure of these isolates (Figure 3) it is not yet clear how the now likely-established population of the Atlantic ST36 strain could further shape the Northeast resident pathogen population. Our findings lay a foundation for future research aimed at understanding the interplay between pathogen genotype and environment that leads to disease emergence.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful for the assistance provided by public health and management agency partners for clinical strains and isolate information including: J. C. Mahoney formerly of the New Hampshire Public Health Laboratories; J. K. Kanwit of the Maine Department of Marine Resources and A. Robbins of the Maine Center for Disease Control and Prevention; Associate Commissioner S. Condon and K. Foley of the Massachusetts Department of Public Health, and M. Hickey and C. Schillaci from the Massachusetts Division of Marine Fisheries. Valuable information and guidance was also provided by N. Gonzalez-Escalona, J. Jones, and A. DePaola. Technical assistance, samples, sample processing, sample analysis, and production of visuals was provided by P. Siwokoti, A. Normandin, A. Parker, M. Hartwick, A. Marcinkiewicz, M. Taylor, E. Jones, J. Yu, R. Donner, M. Gerding, E. Urquhart, S. Pankey, T. Howell, K. DeRosia-Banick, J. Brawley, and C. Sherman. Partial funding for this was provided by the USDA National Institute of Food and Agriculture Hatch NH00574, NH00609 (accession 233555), and NH00625 (accession 1004199). Additional funding provided by the National Oceanic and Atmospheric Administration College Sea Grant program and grants R/CE-137, R/SSS-2, R/HCE-3. Support also provided through the National Institutes of Health 1R03AI081102-01, and National Science Foundation DBI 1229361 NSF MRI and EPSCoR IIA-1330641. This is Scientific Contribution Number 2577 for the New Hampshire Agricultural Experiment Station.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2015.00272/abstract
References
Alikhan, N.-F., Petty, N. K., Zakour, N. L. B., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Altekruse, S., Bishop, R., Baldy, L., Thompson, S., Wilson, S., Ray, B., et al. (2000). Vibrio gastroenteritis in the US Gulf of Mexico region: the role of raw oysters. Epidemiol. Infect. 124, 489–495. doi: 10.1017/S0950268899003714
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ausubel, F., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A., et al. (1990). Current Protocols in Molecular Biology. New York, NY: Wiley and Sons, Inc.
Baker-Austin, C., Trinanes, J. A., Taylor, N. G., Hartnell, R., Siitonen, A., and Martinez-Urtaza, J. (2013). Emerging Vibrio risk at high latitudes in response to ocean warming. Nat. Clim. Change 3, 73–77. doi: 10.1038/nclimate1628
Banerjee, S. K., Kearney, A. K., Nadon, C. A., Peterson, C.-L., Tyler, K., Bakouche, L., et al. (2014). Phenotypic and genotypic characterization of Canadian clinical isolates of Vibrio parahaemolyticus collected from 2000 to 2009. J. Clin. Microbiol. 52, 1081–1088. doi: 10.1128/JCM.03047-13
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bertels, F., Silander, O. K., Pachkov, M., Rainey, P. B., and Van Nimwegen, E. (2014). Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol. Biol. Evol. 31, 1077–1088. doi: 10.1093/molbev/msu088
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Caburlotto, G., Gennari, M., Ghidini, V., Tafi, M., and Lleo, M. M. (2009). Presence of T3SS2 and other virulence−related genes in tdh−negative Vibrio parahaemolyticus environmental strains isolated from marine samples in the area of the Venetian Lagoon, Italy. FEMS Microbiol. Ecol. 70, 506–514. doi: 10.1111/j.1574-6941.2009.00764.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Centers for Disease Control and Prevention (CDC). (1999). Outbreak of Vibrio parahaemolyticus infection associated with eating raw oysters and clams harvested from long island sound–connecticut, New Jersey, and New York, 1998. MMWR Morb. Mortal. Wkly. Rep. 48, 48–51.
Chowdhury, N. R., Chakraborty, S., Ramamurthy, T., Nishibuchi, M., Yamasaki, S., Takeda, Y., et al. (2000). Molecular evidence of clonal Vibrio parahaemolyticus pandemic strains. Emerg. Infect. Dis. 6, 631–636. doi: 10.3201/eid0606.000612
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chowdhury, N. R., Stine, O. C., Morris, J. G., and Nair, G. (2004). Assessment of evolution of pandemic Vibrio parahaemolyticus by multilocus sequence typing. J. Clin. Microbiol. 42, 1280–1282. doi: 10.1128/JCM.42.3.1280-1282.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Crim, S. M., Iwamoto, M., Huang, J. Y., Griffin, P. M., Gilliss, D., Cronquist, A. B., et al. (2014). Incidence and trends of infection with pathogens transmitted commonly through food—Foodborne Diseases Active Surveillance network, 10 US sites, 2006–2013. MMWR Morb. Mortal. Wkly. Rep. 63, 328–332.
Daniels, N. A., Mackinnon, L., Bishop, R., Altekruse, S., Ray, B., Hammond, R. M., et al. (2000). Vibrio parahaemolyticus infections in the United States, 1973–1998. J. Infect. Dis. 181, 1661–1666. doi: 10.1086/315459
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Davis, C. R., Wingfield, D. L., Peak, K. K., Veguilla, W., Amuso, P. T., Cannons, A. C., et al. (2007). Molecular characterization of Vibrio parahaemolyticus strains associated with foodborne illness in Florida. J. Food Prot. 70, 2396–2401.
Dawicki, S. (2013). NOAA Science Spotlight. Available online at: http://www.nefsc.noaa.gov/press_release/2013/SciSpot/SS1304/ (Accessed September 1, 2014).
Depaola, A., Kaysner, C. A., Bowers, J., and Cook, D. W. (2000). Environmental investigations of Vibrio parahaemolyticus in oysters after outbreaks in Washington, Texas, and New York (1997 and 1998). Appl. Environ. Microbiol. 66, 4649–4654. doi: 10.1128/AEM.66.11.4649-4654.2000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Depaola, A., Ulaszek, J., Kaysner, C. A., Tenge, B. J., Nordstrom, J. L., Wells, J., et al. (2003). Molecular, serological, and virulence characteristics of Vibrio parahaemolyticus isolated from environmental, food, and clinical sources in North America and Asia. Appl. Environ. Microbiol. 69, 3999–4005. doi: 10.1128/AEM.69.7.3999-4005.2003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Didelot, X., and Falush, D. (2007). Inference of bacterial microevolution using multilocus sequence data. Genetics 175, 1251–1266. doi: 10.1534/genetics.106.063305
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ellis, C. N., Schuster, B. M., Striplin, M. J., Jones, S. H., Whistler, C. A., and Cooper, V. S. (2012). Influence of seasonality on the genetic diversity of Vibrio parahaemolyticus in New Hampshire shellfish waters as determined by multilocus sequence analysis. Appl. Environ. Microbiol. 78, 3778–3782. doi: 10.1128/AEM.07794-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
García, K., Torres, R., Uribe, P., Hernández, C., Rioseco, M. L., Romero, J., et al. (2009). Dynamics of clinical and environmental Vibrio parahaemolyticus strains during seafood-related summer diarrhea outbreaks in southern Chile. Appl. Environ. Microbiol. 75, 7482–7487. doi: 10.1128/AEM.01662-09
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
González-Escalona, N., Martinez-Urtaza, J., Romero, J., Espejo, R. T., Jaykus, L.-A., and Depaola, A. (2008). Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J. Bacteriol. 190, 2831–2840. doi: 10.1128/JB.01808-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haendiges, J., Timme, R., Allard, M., Myers, R. A., Payne, J., Brown, E. W., et al. (2014). Draft genome sequences of clinical Vibrio parahaemolyticus strains isolated in Maryland (2010 to 2013). Genome Announc. 2:e00776–14. doi: 10.1128/genomeA.00776-14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Harth, E., Matsuda, L., Hernández, C., Rioseco, M. L., Romero, J., González-Escalona, N., et al. (2009). Epidemiology of Vibrio parahaemolyticus outbreaks, southern Chile. Emerg. Infect. Dis. 15, 163–168. doi: 10.3201/eid1502.071269
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haubold, B., and Hudson, R. R. (2000). LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics 16, 847–849. doi: 10.1093/bioinformatics/16.9.847
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hiyoshi, H., Kodama, T., Iida, T., and Honda, T. (2010). Contribution of Vibrio parahaemolyticus virulence factors to cytotoxicity, enterotoxicity, and lethality in mice. Infect. Immun. 78, 1772–1780. doi: 10.1128/IAI.01051-09
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Honda, T., and Iida, T. (1993). The pathogenicity of Vibrio parahaemolyticus and the role of the thermostable direct haemolysin and related haemolysins. Rev. Med. Microbiol. 4, 106–113. doi: 10.1097/00013542-199304000-00006
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Inouye, M., Conway, T. C., Zobel, J., and Holt, K. E. (2012). Short read sequence typing (SRST): multi-locus sequence types from short reads. BMC Genomics 13:338. doi: 10.1186/1471-2164-13-338
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johnson, C., Flowers, A., Noriea, N., Zimmerman, A., Bowers, J., Depaola, A., et al. (2010). Relationships between environmental factors and pathogenic vibrios in the northern Gulf of Mexico. Appl. Environ. Microbiol. 76, 7076–7084. doi: 10.1128/AEM.00697-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johnson, C. N., Bowers, J. C., Griffitt, K. J., Molina, V., Clostio, R. W., Pei, S., et al. (2012). Ecology of Vibrio parahaemolyticus and Vibrio vulnificus in the coastal and estuarine waters of Louisiana, Maryland, Mississippi, and Washington (United States). Appl. Environ. Microbiol. 78, 7249–7257. doi: 10.1128/AEM.01296-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jolley, K. (2010). BIGSdb: Scalable Analysis of Bacterial Genome Variation at the Population Level. Available online at: pubmlst.http://pubmlst.org/vparahaemolyticus/info/protocol.shtml (Accessed February 1, 2013).
Jolley, K. A., Chan, M.-S., and Maiden, M. C. (2004). mlstdbNet–distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics 5:86. doi: 10.1186/1471-2105-5-86
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jones, J. L., Lüdeke, C. H., Bowers, J. C., Garrett, N., Fischer, M., Parsons, M. B., et al. (2012). Biochemical, serological, and virulence characterization of clinical and oyster Vibrio parahaemolyticus isolates. J. Clin. Microbiol. 50, 2343–2352. doi: 10.1128/JCM.00196-12
Lüdeke, C. H., Fischer, M., Lafon, P., Cooper, K., and Jones, J. L. (2014). Suitability of the molecular subtyping methods intergenic spacer region, direct genome restriction analysis, and Pulsed-Field Gel Electrophoresis for clinical and environmental Vibrio parahaemolyticus Isolates. Foodborne Pathog. Dis. 11, 520–528. doi: 10.1089/fpd.2013.1728
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mahoney, J. C., Gerding, M. J., Jones, S. H., and Whistler, C. A. (2010). Comparison of the pathogenic potentials of environmental and clinical Vibrio parahaemolyticus strains indicates a role for temperature regulation in virulence. Appl. Environ. Microbiol. 76, 7459–7465. doi: 10.1128/AEM.01450-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martinez-Urtaza, J., Baker-Austin, C., Jones, J. L., Newton, A. E., Gonzalez-Aviles, G. D., and Depaola, A. (2013). Spread of Pacific Northwest Vibrio parahaemolyticus strain. N. Engl. J. Med. 369, 1573–1574. doi: 10.1056/NEJMc1305535
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martinez-Urtaza, J., Bowers, J. C., Trinanes, J., and Depaola, A. (2010). Climate anomalies and the increasing risk of Vibrio parahaemolyticus and Vibrio vulnificus illnesses. Food Res. Int. 43, 1780–1790. doi: 10.1016/j.foodres.2010.04.001
Martinez-Urtaza, J., Huapaya, B., Gavilan, R. G., Blanco-Abad, V., Ansede-Bermejo, J., Cadarso-Suarez, C., et al. (2008). Emergence of asiatic Vibrio diseases in South America in phase with El Niño. Epidemiology 19, 829–837. doi: 10.1097/EDE.0b013e3181883d43
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Massachusettes Department of Marine Fischeries (MADMF). (2013). Massachusetts 2013 Vp Control Plan [Online]. Available online at: http://www.mass.gov/eea/docs/dfg/dmf/programsandprojects/2013-vp-plan-041013.pdf (Accessed June 15, 2014).
Mccarter, L. L. (1998). OpaR, a homolog of Vibrio harveyi LuxR, controls opacity of Vibrio parahaemolyticus. J. Bacteriol. 180, 3166–3173.
Mclaughlin, J. B., Depaola, A., Bopp, C. A., Martinek, K. A., Napolilli, N. P., Allison, C. G., et al. (2005). Outbreak of Vibrio parahaemolyticus gastroenteritis associated with Alaskan oysters. N. Engl. J. Med. 353, 1463–1470. doi: 10.1056/NEJMoa051594
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mills, K. E., Pershing, A. J., Brown, C. J., Chen, Y., Chiang, F.-S., Holland, D. S., et al. (2013). Fisheries management in a changing climate lessons from the 2012 ocean heat wave in the northwest Atlantic. Oceanography 26, 191–195. doi: 10.5670/oceanog.2013.27
Nair, G. B., Ramamurthy, T., Bhattacharya, S. K., Dutta, B., Takeda, Y., and Sack, D. A. (2007). Global dissemination of Vibrio parahaemolyticus serotype O3: K6 and its serovariants. Clin. Microbiol. Rev. 20, 39–48. doi: 10.1128/CMR.00025-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
NERACOOS (2014). NERACOOS Ocean and Weather Climate Display. Available online at: http://neracoos.org/datatools/climatologies_display (Accessed August 15, 2014).
Newton, A. E., Garrett, N., Stroika, S. G., Halpin, J. L., Turnsek, M., Mody, R. K., et al. (2014). Notes from the field: increase in Vibrio parahaemolyticus infections associated with consumption of Atlantic coast shellfish—2013. MMWR Morb. Mortal. Wkly. Rep. 63, 335–336.
Nishibuchi, M., Fasano, A., Russell, R., and Kaper, J. (1992). Enterotoxigenicity of Vibrio parahaemolyticus with and without genes encoding thermostable direct hemolysin. Infect. Immun. 60, 3539–3545.
Noriea Iii, N., Johnson, C., Griffitt, K., and Grimes, D. J. (2010). Distribution of type III secretion systems in Vibrio parahaemolyticus from the northern Gulf of Mexico. J. Appl. Microbiol. 109, 953–962. doi: 10.1111/j.1365-2672.2010.04722.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Panicker, G., Call, D. R., Krug, M. J., and Bej, A. K. (2004). Detection of pathogenic Vibrio spp. in shellfish by using multiplex PCR and DNA microarrays. Appl. Environ. Microbiol. 70, 7436–7444. doi: 10.1128/AEM.70.12.7436-7444.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Paranjpye, R., Hamel, O. S., Stojanovski, A., and Liermann, M. (2012). Genetic diversity of clinical and environmental Vibrio parahaemolyticus strains from the Pacific Northwest. Appl. Environ. Microbiol. 78, 8631–8638. doi: 10.1128/AEM.01531-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, K.-S., Iida, T., Yamaichi, Y., Oyagi, T., Yamamoto, K., and Honda, T. (2000). Genetic characterization of DNA region containing the trh and ure genes of Vibrio parahaemolyticus. Infect. Immun. 68, 5742–5748. doi: 10.1128/IAI.68.10.5742-5748.2000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parveen, S., Hettiarachchi, K. A., Bowers, J. C., Jones, J. L., Tamplin, M. L., Mckay, R., et al. (2008). Seasonal distribution of total and pathogenic Vibrio parahaemolyticus in Chesapeake Bay oysters and waters. Int. J. Food Microbiol. 128, 354–361. doi: 10.1016/j.ijfoodmicro.2008.09.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Preacher, K. J., and Briggs, N. E. (2001). Calculation for Fisher's Exact Test: An Interactive Calculation Tool for Fisher's Exact Probability Test for 2 × 2 Tables [Computer Software] [Online].
Rambaut, A. (2012). FigTree v1. 4: Tree figure drawing tool. Available online at: http://tree.bio.ed.ac.uk/software/figtree
Sawabe, T., Kita-Tsukamoto, K., and Thompson, F. L. (2007). Inferring the evolutionary history of vibrios by means of multilocus sequence analysis. J. Bacteriol. 189, 7932–7936. doi: 10.1128/JB.00693-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Scallan, E., Hoekstra, R. M., Angulo, F. J., Tauxe, R. V., Widdowson, M.-A., Roy, S. L., et al. (2011). Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 17, 7–15. doi: 10.3201/eid1701.P11101
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schuster, B. M., Tyzik, A. L., Donner, R. A., Striplin, M. J., Almagro-Moreno, S., Jones, S. H., et al. (2011). Ecology and genetic structure of a northern temperate Vibrio cholerae population related to toxigenic isolates. Appl. Environ. Microbiol. 77, 7568–7575. doi: 10.1128/AEM.00378-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thompson, F., Gevers, D., Thompson, C., Dawyndt, P., Naser, S., Hoste, B., et al. (2005). Phylogeny and molecular identification of vibrios on the basis of multilocus sequence analysis. Appl. Environ. Microbiol. 71, 5107–5115. doi: 10.1128/AEM.71.9.5107-5115.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thongjun, J., Mittraparp-Arthorn, P., Yingkajorn, M., Kongreung, J., Nishibuchi, M., and Vuddhakul, V. (2013). The trend of Vibrio parahaemolyticus infections in southern Thailand from 2006 to 2010. Trop. Med. Health 41, 151. doi: 10.2149/tmh.2013-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tritt, A., Eisen, J. A., Facciotti, M. T., and Darling, A. E. (2012). An integrated pipeline for de novo assembly of microbial genomes. PLoS ONE 7:e42304. doi: 10.1371/journal.pone.0042304
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Turner, J. W., Paranjpye, R. N., Landis, E. D., Biryukov, S. V., González-Escalona, N., Nilsson, W. B., et al. (2013). Population structure of clinical and environmental Vibrio parahaemolyticus from the Pacific Northwest coast of the United States. PLoS ONE 8:e55726. doi: 10.1371/journal.pone.0055726
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vos, M., and Didelot, X. (2009). A comparison of homologous recombination rates in bacteria and archaea. ISME J. 3, 199–208. doi: 10.1038/ismej.2008.93
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, M., Yamamoto, K., and Honda, T. (1994). Construction and characterization of an isogenic mutant of Vibrio parahaemolyticus having a deletion in the thermostable direct hemolysin-related hemolysin gene (trh). J. Bacteriol. 176, 4757–4760.
Zimmerman, A., Depaola, A., Bowers, J., Krantz, J., Nordstrom, J., Johnson, C., et al. (2007). Variability of total and pathogenic Vibrio parahaemolyticus densities in northern Gulf of Mexico water and oysters. Appl. Environ. Microbiol. 73, 7589–7596. doi: 10.1128/AEM.01700-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: disease ecology, emergent pathogen, MLSA, Vibriosis, population structure, pathogen evolution, hemolysin
Citation: Xu F, Ilyas S, Hall JA, Jones SH, Cooper VS and Whistler CA (2015) Genetic characterization of clinical and environmental Vibrio parahaemolyticus from the Northeast USA reveals emerging resident and non-indigenous pathogen lineages. Front. Microbiol. 6:272. doi: 10.3389/fmicb.2015.00272
Received: 07 December 2014; Accepted: 19 March 2015;
Published: 07 April 2015.
Edited by:
Iddya Karunasagar, Nitte University, IndiaReviewed by:
Yan Ling, Beijing Institute of Biotechnology, ChinaJaime Martinez-Urtaza, University of Bath, UK
Copyright © 2015 Xu, Ilyas, Hall, Jones, Cooper and Whistler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheryl A. Whistler, Department of Molecular, Cellular and Biomedical Sciences, University of New Hampshire, 46 College Rd., Durham, NH 03824, USA cheryl.whistler@unh.edu