 Saheed Imam
Saheed Imam Sascha Schäuble
Sascha Schäuble Aaron N. Brooks
Aaron N. Brooks Nitin S. Baliga1,3,4,5
Nitin S. Baliga1,3,4,5 Nathan D. Price
Nathan D. Price- 1Institute for Systems Biology, Seattle, WA, USA
- 2Jena University Language and Information Engineering Lab, Friedrich-Schiller-University Jena, Jena, Germany
- 3Departments of Biology and Microbiology, University of Washington, Seattle, WA, USA
- 4Molecular and Cellular Biology Program, University of Washington, Seattle, WA, USA
- 5Lawrence Berkeley National Lab, Berkeley, CA, USA
Microbes are diverse and extremely versatile organisms that play vital roles in all ecological niches. Understanding and harnessing microbial systems will be key to the sustainability of our planet. One approach to improving our knowledge of microbial processes is through data-driven and mechanism-informed computational modeling. Individual models of biological networks (such as metabolism, transcription, and signaling) have played pivotal roles in driving microbial research through the years. These networks, however, are highly interconnected and function in concert—a fact that has led to the development of a variety of approaches aimed at simulating the integrated functions of two or more network types. Though the task of integrating these different models is fraught with new challenges, the large amounts of high-throughput data sets being generated, and algorithms being developed, means that the time is at hand for concerted efforts to build integrated regulatory-metabolic networks in a data-driven fashion. In this perspective, we review current approaches for constructing integrated regulatory-metabolic models and outline new strategies for future development of these network models for any microbial system.
Introduction
Microbial genomes encode a vast repertoire of metabolic pathways that enable physiological adjustment to changing energy sources and nutrient availabilities. The efficient utilization of environmental resources requires selective and timely expression of the metabolic machinery to meet cellular demands. As a consequence, highly interconnected macromolecular networks of metabolic and regulatory components have evolved to control expression of the genome in response to internal and external cues (Figure 1) (Gerosa and Sauer, 2011; Metallo and Vander Heiden, 2013; Chubukov et al., 2014). A primary goal of modern systems biology is to build increasingly accurate representations of these networks that can be used to predict how the macromolecular composition of an organism may change in response to genetic or environmental perturbations. Such models serve as platforms for hypothesis generation that ultimately enable many perturbations to be screened in silico before being tested in vivo, dramatically accelerating the pace and efficiency of scientific discovery (Tomita, 2001; Bonneau et al., 2007; Oberhardt et al., 2009).
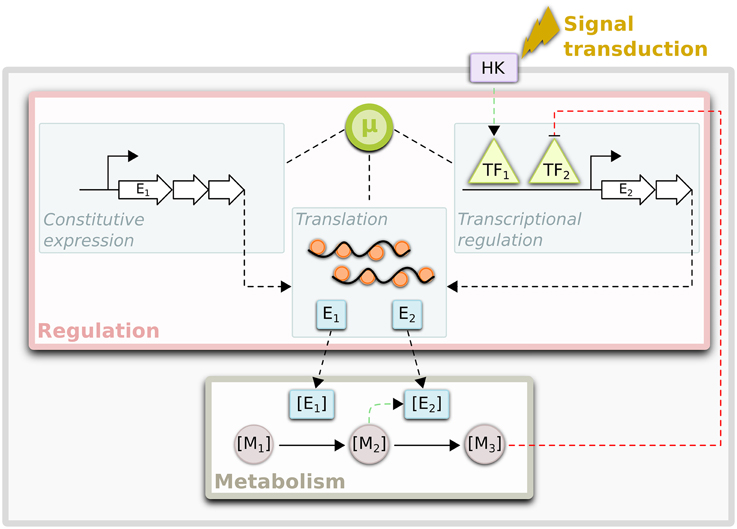
Figure 1. Interconnections between regulation and metabolism. Regulation of flux through metabolic networks is achieved by the control of enzyme levels ([E]) and/or activities. Enzyme levels can be controlled transcriptionally via specific regulation of transcription factors (TFs) or via global mechanisms, which depend on factors such as growth rate (μ). The expression levels of constitutively expressed genes may be solely under control of these global mechanisms. In addition, growth rate also has a significant impact on translation rates. The activities of TFs can be modulated by specific metabolites ([M]) or via post-translational modifications by histidine kinases (HK) that sense environmental cues, among other mechanisms. Enzyme activities can also be modulated via post-translational (allosteric) interactions with metabolites. All these networks are dynamic and in constant communication with one another to determine metabolic state of a cell.
A number of algorithms to construct metabolic, signaling and gene regulatory network models have been developed recently (Thiele et al., 2009; Hyduke and Palsson, 2010; Marbach et al., 2010; Novichkov et al., 2010; Thiele and Palsson, 2010; Yachie et al., 2011). Many were spurred by specific technological advances that enabled comprehensive measurement of microbial cellular components (Thiele et al., 2009; Henry et al., 2010; Thiele and Palsson, 2010; Marbach et al., 2012). These methods have not only been instrumental for contextualizing genome-wide measurements, but have also provided a systems-level perspective of biological organization and function (Oberhardt et al., 2009; Bordbar et al., 2014). Integrated network models that are able to capture these different layers of biological function, on a genome-scale, represent major accomplishments with the potential to revolutionize scientific research (Tomita, 2001).
However, integrating these network models brings about new challenges, both computational and experimental. For instance, algorithms need to be developed to handle the diversity of data types and the various formalisms used to model different biological processes (Machado et al., 2011). Additional challenges also arise from the fact that these processes may occur across vastly different timescales, ranging from milliseconds to weeks. From an experimental standpoint, further technological advancements will be needed to obtain the fine-grained measurements that will be required to build, validate, and refine these models. In this perspective, we briefly review state-of-the-art methods for constructing integrated regulatory-metabolic models, then outline new strategies for constructing data-driven integrated models and suggest how these integrated models could be used to advance basic research, as well as biotechnology.
Advances in the Integration of Metabolic and Regulatory Network Models
Kinetic and constraint-based modeling approaches have enabled quantitative modeling of metabolic processes and played key roles in guiding scientific research (Varma and Palsson, 1994; Palsson, 2000; Steuer et al., 2006; Tran et al., 2008; Bakker et al., 2010; Tan and Liao, 2012; Zielinski and Palsson, 2012). Constraint-based metabolic models (CBMs) have proven to be particularly useful as they enable genome-scale modeling of metabolism. However, these purely metabolic models are limited in their ability to capture condition-dependent changes in metabolic activity (Reed, 2012; Machado and Herrgard, 2014). Thus, to incorporate aspects of the regulatory mechanisms that control metabolism, models that integrate CBMs with known or inferred transcriptional regulatory networks (TRNs) have been developed.
To date, only a handful of methods for the genome-scale integration of transcription and metabolism have been described, including regulatory flux balance analysis (rFBA) (Covert and Palsson, 2002), steady-state rFBA (SR-FBA) (Shlomi et al., 2007) and probabilistic regulation of metabolism (PROM) (Chandrasekaran and Price, 2010). The earlier approaches (rFBA and SR-FBA) used Boolean rules to approximate transcriptional control of the metabolic network, permitting only two activity states (on/off) for network components (Covert and Palsson, 2003; Shlomi et al., 2007). With PROM, the Boolean logic is relaxed by introducing probabilistic weights on regulatory influences using gene expression data to estimate the probability that particular TF-gene interactions are functional, allowing for a full range of potential responses from the strength of either activating or repressing regulation (Chandrasekaran and Price, 2010).
These integrated models, however, only consider a static, composite view of a TRN that has dynamic and condition-specific states, thus limiting their utility. To overcome some of these shortcomings, approaches have been developed to identify relevant TRN constraints that allow accurate growth phenotype predictions of a CBM under a given condition, in essence generating condition-specific TRNs (Barua et al., 2010; Chandrasekaran and Price, 2013). Another limitation of these integrated models is that the regulation of metabolic processes occurs at several levels (i.e., transcriptional, post-transcriptional, translational and post-translational), which are not explicitly accounted for in any of these formalisms. As a result, recent efforts have been geared toward integrating some or all of these components into unified models for well-studied microbes (Karr et al., 2012; Lerman et al., 2012; Carrera et al., 2014).
Metabolism and macromolecule expression (ME) models, which integrate stoichiometric representations of gene expression (transcriptional and translational) networks with CBMs, capture important aspects of the mechanisms of macromolecule synthesis (Lerman et al., 2012; O'brien et al., 2013). These models, which impose global growth-related regulatory constraints on metabolism, have been shown to be better predictors of cell phenotypes such as growth, metabolic fluxes and to some extent gene expression levels, than standalone CBMs (O'brien et al., 2013). ME models thus represent a significant advance over CBMs for the holistic modeling of microbial growth. However, ME models currently do not explicitly account for the specific regulatory mechanisms of the TRN or environmental cues, representing an important frontier for enhancing their scope. Recently, Carrera et al. constructed an integrated model for Escherichia coli that combines information from its known transcriptional regulatory, signal transduction and metabolic networks, with high-throughput transcriptomics and phenomics data (Carrera et al., 2014). This integrated network was shown to have greater capabilities than CBMs or ME models for prediction of condition-dependent phenotypes, and provides a useful framework for data-driven integration of genome-scale networks.
A major goal of systems biology is the construction of predictive models of the entire cell or organism (Tomita, 2001). One of the first efforts directed toward achieving this was the E-cell platform for simulation of biological processes based on predefined lists of biomolecules, reaction rules and cell environments (Tomita et al., 1999). A significant advance on this front was the construction of the whole cell model of Mycoplasma genitalium (Karr et al., 2012). While this model also relies on a very large number of detailed molecular measurements, which are unavailable for most organisms, it provides the first glimpse into the future of full-featured, large-scale integrated models that enable dynamic simulation of cellular processes.
Toward Full-Featured Integrated Models
Here we outline the main components that are needed to construct integrated models that capture the key aspects of regulation and metabolism in microbes (Figure 2), with a focus on data-driven approaches that are extensible to any sequenced microbe.
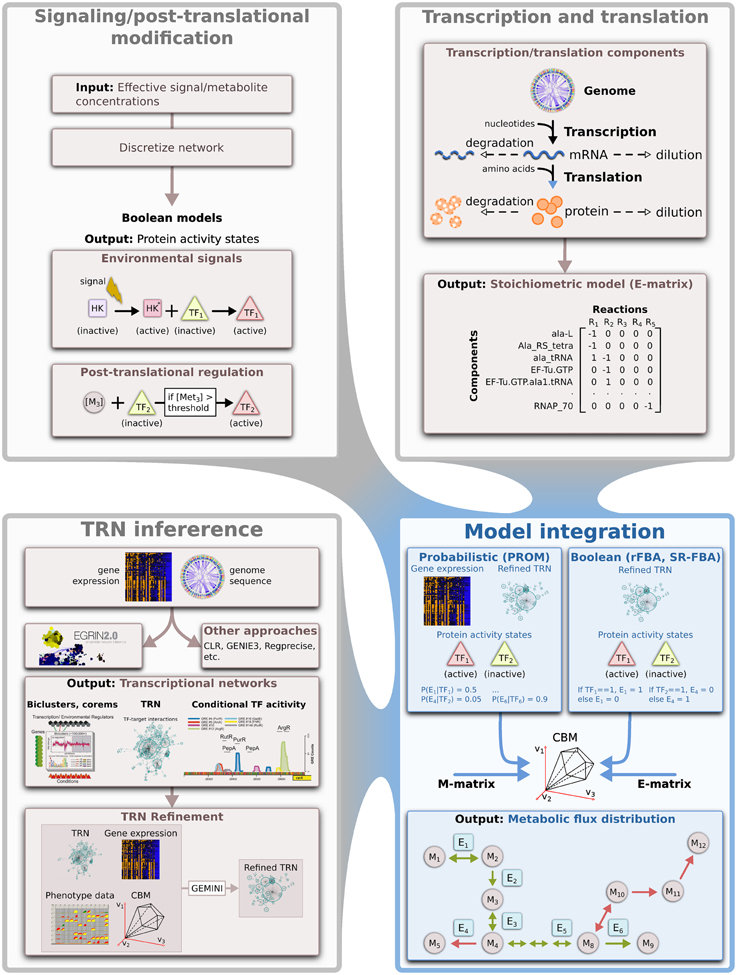
Figure 2. Modeling and integrating of different biological networks. An overview of the approaches used to model disparate biological processes and the computational techniques that could be used for integrating some of these network models. HK, histidine kinase; M, metabolite; E, enzyme; TF, transcription factor; TRN, transcriptional regulatory network.
Genome-scale Metabolic Models
CBMs enable genome-scale modeling of metabolic networks in the absence of kinetic parameters, and provide a platform for integrating multi-omic datasets. While calculations from CBMs often result in a large solution space, which can include many biologically implausible solutions (Reed and Palsson, 2004; Schellenberger et al., 2011), the addition of biologically relevant constraints can significantly improve their predictive accuracy (Reed, 2012). Manually curated genome-scale CBMs have been constructed for many organisms (Oberhardt et al., 2009; Kim et al., 2012) (Table S1). As many metabolic pathways are broadly conserved, these curated CBMs can serve as scaffolds for automated reconstruction of high-quality organism-specific CBMs for closely related organism using genomic information, conceivably making high-quality CBMs available for any sequenced organism, while minimizing the need for manual curation (Henry et al., 2010; Karp et al., 2010; Swainston et al., 2011; Agren et al., 2013; Benedict et al., 2014). Furthermore, computational tools and databases that facilitate automated identification of biomass components (Tervo and Reed, 2013) and minimal media composition (Richards et al., 2014) should further streamline this process.
Transcription and Translation Network Models
Growth rate-dependent global regulation functions in concert with TF-controlled specific-regulation to determine the genome-wide expression profiles under a given condition (Berthoumieux et al., 2013) (Figure 1). Global regulation has even been proposed to be the dominant form of regulation under laboratory conditions in some organisms (Price et al., 2013). Because the components of the transcription and translation machinery are relatively well-conserved across bacteria and can be discerned from genomic information, the E. coli gene expression network model (Thiele et al., 2009) provides a template for the construction of similar network models for other sequenced bacteria. Thus, we expect that approaches used for accelerating the generation of genome-scale models will eventually be extended to the more complex task of constructing gene-expression networks for other microbes.
Transcriptional Regulatory Network (TRN) Models
Microbes control the activities and abundance of molecular components to respond quickly to environmental change. A primary mechanism through which microbes exert control over specific cellular processes is through the coordinated transcriptional regulation of gene expression (Gerosa and Sauer, 2011) (Figure 1). Unfortunately, unlike metabolic networks, TRNs are not highly conserved across lineages. Thus, transcriptional regulatory interactions learned in one species may not necessarily exist in others, unless they are related over short phylogenetic distances or share similar lifestyles (Lozada-Chavez et al., 2006; Madan Babu et al., 2006). However, data from high-throughput measurement of global gene expression levels, along with information encoded in the genome of a target organism, can be used for data-driven reconstruction of TRNs (Figure 2) and this has spurred the development of a wide variety of approaches.
The approaches for reconstructing TRN topology (i.e., the set of interactions between TFs and their target genes) vary, ranging from aggregation of experimentally verified interactions (Gama-Castro et al., 2011) to detection of evolutionary conservation among gene targets of related TFs (Novichkov et al., 2010) to data-driven approaches that reverse-engineer TRN topology from relative changes in gene expression (Bonneau et al., 2007; Faith et al., 2007; Huynh-Thu et al., 2010) (Table S1). The advantages and limitations of some of these data-driven approaches have previously been reviewed (De Smet and Marchal, 2010; Marbach et al., 2010). Many of these approaches have also been subjected to unbiased assessments (Stolovitzky et al., 2007; Marbach et al., 2012), systematically identifying their strengths and weaknesses.
To understand TRN function, however, it is also important to know when specific TF-target gene interactions occur. In other words, condition- and/or context-specific interactions determine the consequences of regulation. Such knowledge is particularly important for integrating TRN models with other genome-scale models. Few approaches currently model the condition-specific activities of TFs and their effect on TRNs. A recently published second generation Environmental and Gene Regulatory Influence Network model (EGRIN 2.0) was developed to address this limitation by quantifying the condition-specific regulatory influence of TFs on their target genes and their role in re-organizing the modularity of TRNs for two microbes (Brooks et al., 2014). Since these models specify environmental dependence in addition to topology, TRN models like EGRIN 2.0 are promising candidates for integration with metabolic and other network models.
Signaling Network Models
Microbes respond to constantly changing environments by altering their gene expression patterns. Bacteria achieve this coordination through the use of one-component, two-component and extra-cytoplasmic function sigma factor signal transduction systems, which sense stimuli and orchestrate appropriate cellular responses (Ulrich and Zhulin, 2010). While environmental signals that elicit certain transcriptional responses (e.g., catabolite repression, oxidative stress response etc.) have been well-studied (Farr and Kogoma, 1991; Gorke and Stulke, 2008; Chiang and Schellhorn, 2012), many other signaling systems remain uncharacterized.
Even though signaling networks in bacteria are generally simpler than those employed in eukaryotes, reconstruction of intracellular signaling networks still poses a major challenge. As a result, large-scale signaling networks exist for only a few organisms (Covert et al., 2008; Carrera et al., 2014). Since independent discovery and characterization of these signaling systems would be costly and time consuming, it is desirable to predict the effects of environmental changes based on high-throughput datasets. EGRIN provides one approach to link signaling to internal cellular processes (Bonneau et al., 2007). It achieves this by abstract representation of the biological effect of signaling networks as “environmental factors.” These environmental factors can be associated statistically to internal molecular processes, such as transcription. This feature, however, requires meticulous experimental documentation, including direct measurement of the relevant environmental factors (or their proxies). Unfortunately, publicly available datasets are generally poorly annotated and typically not quantitative, limiting the current utility of this approach. Consequently, a greater emphasis should be placed on thorough experimental annotation to facilitate these data-driven approaches.
Post-translational Regulation
Post-translational mechanisms also play a critical role in regulating metabolic flux. For instance, internal ligand concentrations can alter the activities of TFs that regulate associated pathways (Lim et al., 1987; Ramseier et al., 1995; Leyn et al., 2011). Furthermore, the activities of numerous enzymes are controlled via allosteric interactions (Figure 2). Thus, knowledge of these regulatory metabolites, their effective concentrations and their target proteins will be crucial for achieving predictive control.
For model organisms like E. coli, a number of these regulatory metabolites and their targets are known and approaches exist for incorporating these into integrated models using Boolean rules and/or ordinary differential equations (Covert and Palsson, 2002; Covert et al., 2008). While some allosteric interactions are widely conserved such as fructose-1,6-bisphosphate (FBP) activation of pyruvate kinase, which is conserved from E. coli to humans (Waygood et al., 1976; Jurica et al., 1998; Chubukov et al., 2014), different groups of organisms likely use different strategies and regulatory metabolites. For example, the regulators of glycolysis in γ-Proteobacteria (Cra), α-Proteobacteria (CceR), and β-Proteobacteria (HexR) are post-translationally regulated by FBP, 6-phosphogluconate and 2-keto-3-deoxy-6-phosphogluconate, respectively (Ramseier et al., 1993; Leyn et al., 2011; Imam et al., 2015). Thus, approaches for high-throughput screening of allosteric effectors (Tagore et al., 2008; Li et al., 2010) need to be leveraged to complement standard in vitro approaches to identify post-translational interactions. This process could be facilitated by the development of algorithms that borrow from the field of molecular modeling (Lengauer and Rarey, 1996) to assess the potential of protein-ligand interactions across the network.
Integrating Disparate Network Models
While individual network models have played important roles in improving our understanding of biological systems, recent attention has turned toward integrating them. Such integrated models would encapsulate how regulatory mechanisms control metabolism and how metabolism, in turn, provides feedback regulation on a genome-scale (Figure 1). The motivation for network model integration reflects an acknowledgement that individual models are insufficient to comprehensively describe their respective cellular processes.
One approach to constraining the solution space of CBM predictions is the integration of growth-related constraints on gene expression (i.e., the rates of gene transcription and mRNA translation). Translation and transcriptional network models, which have been constructed for E. coli (Thiele et al., 2009; O'brien et al., 2013) and Thermotoga maritima (Lerman et al., 2012), including their mathematical formulation and integration with CBMs to generate ME-models, provide a basis for construction of similar ME-models for other microbes based mostly on genomic information. As very few parameters need to be specified for integration of these stoichiometric models, construction of ME-models for any sequenced bacterium should become a relatively straightforward task.
ME-models, however, do not currently account for specific regulatory interactions at gene promoters, which are also known to be important drivers of cellular phenotypes. To build comprehensive models, ME-models need to include regulatory constraints from condition-specific TF-gene interactions (O'brien et al., 2013). However, unlike global transcriptional processes, these may not have straightforward stoichiometric representations. Hence, alternative formulations need to be considered. One possibility could involve leveraging a probabilistic formalism such as PROM for integrating inferred TRN models with ME-models. If such TRN models were developed using EGRIN or related approaches, environmental variables could also be integrated using PROM. Extension of ME-models with TRN information represents an exciting frontier that would provide a platform for simulating metabolism with unprecedented detail.
Integration of signaling information poses some unique challenges. For instance, signaling mechanisms are typically dependent on specific (and often unknown) concentrations of relevant molecules, while constraint-based approaches such as FBA do not deal directly with metabolite concentrations. Furthermore, to generate dynamic quantitative signaling network models, kinetic parameters are required, but these are rarely available. This limits the approaches via which these models can be integrated within the paradigm described above. Thus, qualitative representations of signaling networks using Boolean (Klamt et al., 2006) or stoichiometric (Papin and Palsson, 2004) formalisms need to be adopted for integration of these networks with large-scale regulatory-metabolic models (Figure 2, Table S1). These approaches have the advantage of not requiring specification of kinetic parameters or exact molecule concentrations (which can discretized, Klamt et al., 2006), while still being able to capture fundamental properties of signaling networks.
Other challenges to building integrated models are outlined in Box 1, while approaches that may be useful for validation of such models are discussed in Box 2.
Box 1. Challenges to constructing integrated regulatory-metabolic models.
Here we identify some major challenges to building data-driven integrated models of metabolism and regulation. Some of these challenges also represent significant opportunities for algorithmic or experimental breakthroughs.
Comprehensive discovery and characterization of biological components. Reconstruction of biological networks requires an exhaustive list of the components and their functions. However, a large fraction (up to 50%) of the predicted proteins across microbial genomes still have unknown functions (Hanson et al., 2009). This missing information can significantly impact the predictive accuracy of systems biology models. While this process of parts identification is significantly facilitated by comparative genomics and related approaches, this can still be a mitigating factor for groups of bacteria that are not yet well studied.
Greater accuracy of data-driven TRN inference. While TRN inference has played a crucial role in identification of new TFs and novel regulatory interactions, the predictive accuracy and coverage of TRNs constructed from gene expression data is still relatively low. Even for a well-studied bacterium like E. coli for which large compendia of gene expression data exist, state of the art inference approaches only identify a small fraction of the verified interactions in regulonDB with relatively low precision (Marbach et al., 2010, 2012; Gama-Castro et al., 2011). While we anticipate that integration of comparative genomics, constraint-based modeling and other complementary approaches will improve the accuracy and coverage of inferred networks, large gains in predictive accuracy will likely require alternative complementary high-throughput datasets such as ChIP-seq data with tagged TFs (Aldridge et al., 2013; Gasper et al., 2014), DNase I hypersensitivity or genome-wide promoter activity assays.
High-throughput approaches for identifying signaling events. As mentioned above, there is a dearth of both experimental and computational approaches for quick screening and identification of potential signaling interactions. Development of approaches in this area would significantly facilitate reconstruction and integration of signaling network models.
Functional characterization of post-translational modifications. A vast array of metabolic and regulatory proteins are regulated via post-translational modifications. While post-translational modifications are more prevalent in eukaryotes than bacteria, a large and growing number of these modifications are being identified in bacteria, including phosphorylation (Pietack et al., 2010; Schmidl et al., 2010), succinylation (Zhang et al., 2010) and acetylation (Wang et al., 2010)—and each of these can have major impacts on metabolism. While these modifications can easily be identified by mass spectrometry techniques, determination of their functions, if any, is more challenging. However, by combining metabolic flux analysis with mass spectrometry data collected across varying conditions, insights into the function of some these modification can be determined (Wang et al., 2010), though the cost of such analysis may be prohibitive. Integrating such information using stoichiometric representations would relatively straightforward.
Other challenges such as limitations in availability of quantitative data across conditions, tools for visualization of integrated networks and difficulties in integrating different network formalisms at genome-scale are also important considerations.
Box 2. Model validation
Model validation is important both to assess model accuracy and identify shortcomings that can be improved in subsequent versions. However, it is not obvious what validation approaches would be optimal for large-scale integrated models. Traditionally, predictions from CBMs have been validated using substrate utilization and/or gene essentiality data, which has served as a successful approach both for model validation and refinement (Bochner et al., 2001; Feist et al., 2007; Oh et al., 2007; Thiele and Palsson, 2010; Imam et al., 2013). Similarly, initial attempts to validate regulatory-metabolic models have focused on the use of gene essentiality data (Covert et al., 2004; Chandrasekaran and Price, 2010). TRN models, by contrast, have usually been validated by comparison to experimentally derived networks (Stolovitzky et al., 2007; Marbach et al., 2012).
We argue that both of these binary approaches to validation are insufficient to generate key insights that will drive model improvement. Instead, we suggest that quantitative phenotypes may be more appropriate. For instance, deletion of regulatory components such as TFs are typically non-lethal. However, this does not imply that cellular phenotypes are unaffected in these strains. TF deletions may alter growth rates or modify other quantitative cellular phenotypes. In addition, TF deletions may only show their impact across a narrow range of conditions. Thus, simple gene essentiality may be inadequate to assess model performance effectively. More informative would be data from high-throughput growth or fitness assays using deletion mutant libraries (Nichols et al., 2011; Vandersluis et al., 2014) or high-throughput mutagenesis experiments across conditions (Van Opijnen et al., 2009; Khatiwara et al., 2012), which would permit identification and statistical evaluation of genotype-phenotype relationships. Such large-scale datasets should permit robust assessment of the various components of regulatory models and possibly guide the process of model refinement.
Using Integrated Models to Drive Scientific Discovery
Construction and analysis of individual large-scale systems biology models has led to important new biological insights about novel pathways, regulatory interactions and mechanistic details (Bonneau et al., 2007; Oberhardt et al., 2009; Hyduke and Palsson, 2010). Given that these networks are highly interconnected, one might expect that analysis of the properties of integrated models will provide new insights into biological phenomena not achievable with individual network models. Such insights could include how novel inferred transcription-regulatory interactions might redirect flux through apparent suboptimal routes in a metabolic network; identification of synthetic rescues/lethal phenotypes in regulatory components; identification of new knowledge gaps that could guide experimental design; or identification of functional roles for previously redundant network components such as dead-end metabolites (Covert et al., 2008). In addition to this, we expect full-featured regulatory-metabolic models will be crucial in driving scientific research in areas such as:
Metabolic Engineering
CBMs have proved to be very useful tools for guiding the design of genetically modified microbial strains with desired characteristics (Alper et al., 2005; Park et al., 2007; Milne et al., 2009). Many approaches have been developed to identify metabolic or genetic interventions that result in these traits (Segre et al., 2002; Burgard et al., 2003; Pharkya et al., 2004; Shlomi et al., 2005; Kim and Reed, 2010). Currently, these approaches do not consider the contribution of regulation on predicted genetic strategies or the benefits of genetic intervention at the regulatory level. Integrated regulatory-metabolic models will provide these capabilities, permitting: (i) rational strain engineering via modification of regulatory components (e.g., over-expression of TFs); (ii) exclusion of metabolic interventions that are inconsistent with the integrated network structure; or (iii) identification of environmental conditions that might facilitate production of desired products. Thus, integrated regulatory-metabolic models could open up several new avenues for modification of cell phenotypes not currently achievable with CBMs.
Improved Network Inference
While TRNs inferred from high-throughput data have led to the identification of novel interactions and mechanisms, these approaches are error prone (De Smet and Marchal, 2010; Marbach et al., 2010). Recent analysis has shown that known or inferred TRN topology can be refined to achieve consistency with known phenotypes of a target organisms by integration with CBMs (Chandrasekaran and Price, 2013). For instance, the algorithm GEMINI uses PROM formalism to integrate TRN models with CBMs, and then attempts to identify global regulatory interactions that are consistent with condition-specific growth phenotypes, thereby refining the TRN and potentially improving its quality (Chandrasekaran and Price, 2013). While GEMINI was originally used as a post-processing step, there exists the potential of incorporating this or similar approaches into the TRN inference workflow itself to provide inline network refinement (Figure 2).
While a few applications of integrated models have been listed here, this is far from exhaustive and the applications will evolve as new data types and algorithms are developed.
Concluding Remarks
One of the aims of systems biology is to convert system-wide measurements into systems-level biological insight. Computational models that capture the core aspects of biological complexity will be pivotal to achieving this goal. Models of metabolism and regulation can be built from a combination of genomic information, high-throughput measurements, and prior knowledge for any cultured organism. Integrating these models will provide deeper insight into fundamental cellular processes and help contextualize high-throughput experiments.
While full-featured integrated models will be useful to generate biological hypotheses, guide experimental designs and drive biotechnology applications, the level of detail at which these processes are represented within the model will depend on the proposed application. Although additional layers of biological complexity could be included ad infinitum to make a model more closely resemble the reality, greater complexity does not necessarily translate into greater utility.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded in part by DOE-ABY (DEEE0006315) (NSB, NDP), DOE ARPA-E program (DE-AR0000426) (NDP), NIH Center for Systems Biology/2P50 GM076547 (NSB, NDP) and the Camille Dreyfus Teacher-Scholar program (NDP). We also acknowledge financial support from the German Ministry for Research and Education within the framework of the GerontoSys initiative (grant FKZ 0315581D) (SS).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00409/abstract
Abbreviations
FBA, Flux balance analysis; rFBA, Regulatory flux balance analysis; SR-FBA, Steady-state regulatory flux balance analysis; PROM, Probabilistic regulation of metabolism; CBM, Constraint-based metabolic model; TRN, Transcriptional regulatory network; ME, Metabolism and macromolecule expression; EGRIN, Environmental and Gene Regulatory Influence Network model; FBP, Fructose-1,6-bisphosphate; GEMINI, Gene Expression and Metabolism Integrated for Network Inference; ChIP-seq, Chromatin immunoprecipitation followed by high-throughput sequencing.
References
Agren, R., Liu, L., Shoaie, S., Vongsangnak, W., Nookaew, I., and Nielsen, J. (2013). The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput. Biol. 9:e1002980. doi: 10.1371/journal.pcbi.1002980
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aldridge, S., Watt, S., Quail, M. A., Rayner, T., Lukk, M., Bimson, M. F., et al. (2013). AHT-ChIP-seq: a completely automated robotic protocol for high-throughput chromatin immunoprecipitation. Genome Biol. 14:R124. doi: 10.1186/gb-2013-14-11-r124
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alper, H., Jin, Y. S., Moxley, J. F., and Stephanopoulos, G. (2005). Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli. Metab. Eng. 7, 155–164. doi: 10.1016/j.ymben.2004.12.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bakker, B. M., Van Eunen, K., Jeneson, J. A., Van Riel, N. A., Bruggeman, F. J., and Teusink, B. (2010). Systems biology from micro-organisms to human metabolic diseases: the role of detailed kinetic models. Biochem. Soc. Trans. 38, 1294–1301. doi: 10.1042/BST0381294
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barua, D., Kim, J., and Reed, J. L. (2010). An automated phenotype-driven approach (GeneForce) for refining metabolic and regulatory models. PLoS Comput. Biol. 6:e1000970. doi: 10.1371/journal.pcbi.1000970
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benedict, M. N., Mundy, M. B., Henry, C. S., Chia, N., and Price, N. D. (2014). Likelihood-based gene annotations for gap filling and quality assessment in genome-scale metabolic models. PLoS Comput. Biol. 10:e1003882. doi: 10.1371/journal.pcbi.1003882
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Berthoumieux, S., De Jong, H., Baptist, G., Pinel, C., Ranquet, C., Ropers, D., et al. (2013). Shared control of gene expression in bacteria by transcription factors and global physiology of the cell. Mol. Syst. Biol. 9, 634. doi: 10.1038/msb.2012.70
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bochner, B. R., Gadzinski, P., and Panomitros, E. (2001). Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Res. 11, 1246–1255. doi: 10.1101/gr.186501
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bonneau, R., Facciotti, M. T., Reiss, D. J., Schmid, A. K., Pan, M., Kaur, A., et al. (2007). A predictive model for transcriptional control of physiology in a free living cell. Cell 131, 1354–1365. doi: 10.1016/j.cell.2007.10.053
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bordbar, A., Monk, J. M., King, Z. A., and Palsson, B. O. (2014). Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 15, 107–120. doi: 10.1038/nrg3643
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brooks, A. N., Reiss, D. J., Allard, A., Wu, W. J., Salvanha, D. M., Plaisier, C. L., et al. (2014). A system-level model for the microbial regulatory genome. Mol. Syst. Biol. 10, 740. doi: 10.15252/msb.20145160
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Burgard, A. P., Pharkya, P., and Maranas, C. D. (2003). Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 84, 647–657. doi: 10.1002/bit.10803
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carrera, J., Estrela, R., Luo, J., Rai, N., Tsoukalas, A., and Tagkopoulos, I. (2014). An integrative, multi-scale, genome-wide model reveals the phenotypic landscape of Escherichia coli. Mol. Syst. Biol. 10, 735. doi: 10.15252/msb.20145108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chandrasekaran, S., and Price, N. D. (2010). Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 107, 17845–17850. doi: 10.1073/pnas.1005139107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chandrasekaran, S., and Price, N. D. (2013). Metabolic constraint-based refinement of transcriptional regulatory networks. PLoS Comput. Biol. 9:e1003370. doi: 10.1371/journal.pcbi.1003370
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chiang, S. M., and Schellhorn, H. E. (2012). Regulators of oxidative stress response genes in Escherichia coli and their functional conservation in bacteria. Arch. Biochem. Biophys. 525, 161–169. doi: 10.1016/j.abb.2012.02.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chubukov, V., Gerosa, L., Kochanowski, K., and Sauer, U. (2014). Coordination of microbial metabolism. Nat. Rev. Microbiol. 12, 327–340. doi: 10.1038/nrmicro3238
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Covert, M. W., Knight, E. M., Reed, J. L., Herrgard, M. J., and Palsson, B. O. (2004). Integrating high-throughput and computational data elucidates bacterial networks. Nature 429, 92–96. doi: 10.1038/nature02456
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Covert, M. W., and Palsson, B. O. (2002). Transcriptional regulation in constraints-based metabolic models of Escherichia coli. J. Biol. Chem. 277, 28058–28064. doi: 10.1074/jbc.M201691200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Covert, M. W., and Palsson, B. O. (2003). Constraints-based models: regulation of gene expression reduces the steady-state solution space. J. Theor. Biol. 221, 309–325. doi: 10.1006/jtbi.2003.3071
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Covert, M. W., Xiao, N., Chen, T. J., and Karr, J. R. (2008). Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 24, 2044–2050. doi: 10.1093/bioinformatics/btn352
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Smet, R., and Marchal, K. (2010). Advantages and limitations of current network inference methods. Nat. Rev. Microbiol. 8, 717–729. doi: 10.1038/nrmicro2419
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Faith, J. J., Hayete, B., Thaden, J. T., Mogno, I., Wierzbowski, J., Cottarel, G., et al. (2007). Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 5:e8. doi: 10.1371/journal.pbio.0050008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farr, S. B., and Kogoma, T. (1991). Oxidative stress responses in Escherichia coli and Salmonella typhimurium. Microbiol. Rev. 55, 561–585.
Feist, A. M., Henry, C. S., Reed, J. L., Krummenacker, M., Joyce, A. R., Karp, P. D., et al. (2007). A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 3, 121. doi: 10.1038/msb4100155
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gama-Castro, S., Salgado, H., Peralta-Gil, M., Santos-Zavaleta, A., Muniz-Rascado, L., Solano-Lira, H., et al. (2011). RegulonDB version 7.0: transcriptional regulation of Escherichia coli K-12 integrated within genetic sensory response units (Gensor Units). Nucleic Acids Res. 39, D98–D105. doi: 10.1093/nar/gkq1110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gasper, W. C., Marinov, G. K., Pauli-Behn, F., Scott, M. T., Newberry, K., Desalvo, G., et al. (2014). Fully automated high-throughput chromatin immunoprecipitation for ChIP-seq: identifying ChIP-quality p300 monoclonal antibodies. Sci. Rep. 4:5152. doi: 10.1038/srep05152
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gerosa, L., and Sauer, U. (2011). Regulation and control of metabolic fluxes in microbes. Curr. Opin. Biotechnol. 22, 566–575. doi: 10.1016/j.copbio.2011.04.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gorke, B., and Stulke, J. (2008). Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6, 613–624. doi: 10.1038/nrmicro1932
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hanson, A. D., Pribat, A., Waller, J. C., and De Crecy-Lagard, V. (2009). ‘Unknown’ proteins and ‘orphan’ enzymes: the missing half of the engineering parts list–and how to find it. Biochem. J. 425, 1–11. doi: 10.1042/BJ20091328
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Henry, C. S., Dejongh, M., Best, A. A., Frybarger, P. M., Linsay, B., and Stevens, R. L. (2010). High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 28, 977–982. doi: 10.1038/nbt.1672
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huynh-Thu, V. A., Irrthum, A., Wehenkel, L., and Geurts, P. (2010). Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 5:e12776. doi: 10.1371/journal.pone.0012776
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hyduke, D. R., and Palsson, B. O. (2010). Towards genome-scale signalling network reconstructions. Nat. Rev. Genet. 11, 297–307. doi: 10.1038/nrg2750
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Imam, S., Noguera, D. R., and Donohue, T. J. (2013). Global insights into energetic and metabolic networks in Rhodobacter sphaeroides. BMC Syst. Biol. 7:89. doi: 10.1186/1752-0509-7-89
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Imam, S., Noguera, D. R., and Donohue, T. J. (2015). CceR and AkgR regulate central carbon and energy metabolism in Alphaproteobacteria. mBio 6:e02461-14. doi: 10.1128/mBio.02461-14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jurica, M. S., Mesecar, A., Heath, P. J., Shi, W., Nowak, T., and Stoddard, B. L. (1998). The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 6, 195–210. doi: 10.1016/S0969-2126(98)00021-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Karp, P. D., Paley, S. M., Krummenacker, M., Latendresse, M., Dale, J. M., Lee, T. J., et al. (2010). Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief. Bioinformatics 11, 40–79. doi: 10.1093/bib/bbp043
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Karr, J. R., Sanghvi, J. C., Macklin, D. N., Gutschow, M. V., Jacobs, J. M., Bolival, B. Jr., et al. (2012). A whole-cell computational model predicts phenotype from genotype. Cell 150, 389–401. doi: 10.1016/j.cell.2012.05.044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khatiwara, A., Jiang, T., Sung, S. S., Dawoud, T., Kim, J. N., Bhattacharya, D., et al. (2012). Genome scanning for conditionally essential genes in Salmonella enterica Serotype Typhimurium. Appl. Environ. Microbiol. 78, 3098–3107. doi: 10.1128/AEM.06865-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, J., and Reed, J. L. (2010). OptORF: optimal metabolic and regulatory perturbations for metabolic engineering of microbial strains. BMC Syst. Biol. 4:53. doi: 10.1186/1752-0509-4-53
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, T. Y., Sohn, S. B., Kim, Y. B., Kim, W. J., and Lee, S. Y. (2012). Recent advances in reconstruction and applications of genome-scale metabolic models. Curr. Opin. Biotechnol. 23, 617–623. doi: 10.1016/j.copbio.2011.10.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Klamt, S., Saez-Rodriguez, J., Lindquist, J. A., Simeoni, L., and Gilles, E. D. (2006). A methodology for the structural and functional analysis of signaling and regulatory networks. BMC Bioinformatics 7:56. doi: 10.1186/1471-2105-7-56
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lengauer, T., and Rarey, M. (1996). Computational methods for biomolecular docking. Curr. Opin. Struct. Biol. 6, 402–406. doi: 10.1016/S0959-440X(96)80061-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lerman, J. A., Hyduke, D. R., Latif, H., Portnoy, V. A., Lewis, N. E., Orth, J. D., et al. (2012). In silico method for modelling metabolism and gene product expression at genome scale. Nat. Commun. 3, 929. doi: 10.1038/ncomms1928
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leyn, S. A., Li, X., Zheng, Q., Novichkov, P. S., Reed, S., Romine, M. F., et al. (2011). Control of proteobacterial central carbon metabolism by the HexR transcriptional regulator: a case study in Shewanella oneidensis. J. Biol. Chem. 286, 35782–35794. doi: 10.1074/jbc.M111.267963
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, X., Gianoulis, T. A., Yip, K. Y., Gerstein, M., and Snyder, M. (2010). Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell 143, 639–650. doi: 10.1016/j.cell.2010.09.048
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lim, D. B., Oppenheim, J. D., Eckhardt, T., and Maas, W. K. (1987). Nucleotide sequence of the argR gene of Escherichia coli K-12 and isolation of its product, the arginine repressor. Proc. Natl. Acad. Sci. U.S.A. 84, 6697–6701. doi: 10.1073/pnas.84.19.6697
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lozada-Chavez, I., Janga, S. C., and Collado-Vides, J. (2006). Bacterial regulatory networks are extremely flexible in evolution. Nucleic Acids Res. 34, 3434–3445. doi: 10.1093/nar/gkl423
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Machado, D., Costa, R. S., Rocha, M., Ferreira, E. C., Tidor, B., and Rocha, I. (2011). Modeling formalisms in Systems Biology. AMB Express 1:45. doi: 10.1186/2191-0855-1-45
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Machado, D., and Herrgard, M. (2014). Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism. PLoS Comput. Biol. 10:e1003580. doi: 10.1371/journal.pcbi.1003580
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Madan Babu, M., Teichmann, S. A., and Aravind, L. (2006). Evolutionary dynamics of prokaryotic transcriptional regulatory networks. J. Mol. Biol. 358, 614–633. doi: 10.1016/j.jmb.2006.02.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marbach, D., Costello, J. C., Kuffner, R., Vega, N. M., Prill, R. J., Camacho, D. M., et al. (2012). Wisdom of crowds for robust gene network inference. Nat. Methods 9, 796–804. doi: 10.1038/nmeth.2016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marbach, D., Prill, R. J., Schaffter, T., Mattiussi, C., Floreano, D., and Stolovitzky, G. (2010). Revealing strengths and weaknesses of methods for gene network inference. Proc. Natl. Acad. Sci. U.S.A. 107, 6286–6291. doi: 10.1073/pnas.0913357107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Metallo, C. M., and Vander Heiden, M. G. (2013). Understanding metabolic regulation and its influence on cell physiology. Mol. Cell 49, 388–398. doi: 10.1016/j.molcel.2013.01.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Milne, C. B., Kim, P. J., Eddy, J. A., and Price, N. D. (2009). Accomplishments in genome-scale in silico modeling for industrial and medical biotechnology. Biotechnol. J. 4, 1653–1670. doi: 10.1002/biot.200900234
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nichols, R. J., Sen, S., Choo, Y. J., Beltrao, P., Zietek, M., Chaba, R., et al. (2011). Phenotypic landscape of a bacterial cell. Cell 144, 143–156. doi: 10.1016/j.cell.2010.11.052
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Novichkov, P. S., Rodionov, D. A., Stavrovskaya, E. D., Novichkova, E. S., Kazakov, A. E., Gelfand, M. S., et al. (2010). RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 38, W299–W307. doi: 10.1093/nar/gkq531
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oberhardt, M. A., Palsson, B. O., and Papin, J. A. (2009). Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol. 5, 320. doi: 10.1038/msb.2009.77
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
O'brien, E. J., Lerman, J. A., Chang, R. L., Hyduke, D. R., and Palsson, B. O. (2013). Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 9, 693. doi: 10.1038/msb.2013.52
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oh, Y. K., Palsson, B. O., Park, S. M., Schilling, C. H., and Mahadevan, R. (2007). Genome-scale reconstruction of metabolic network in Bacillus subtilis based on high-throughput phenotyping and gene essentiality data. J. Biol. Chem. 282, 28791–28799. doi: 10.1074/jbc.M703759200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Palsson, B. (2000). The challenges of in silico biology. Nat. Biotechnol. 18, 1147–1150. doi: 10.1038/81125
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Papin, J. A., and Palsson, B. O. (2004). Topological analysis of mass-balanced signaling networks: a framework to obtain network properties including crosstalk. J. Theor. Biol. 227, 283–297. doi: 10.1016/j.jtbi.2003.11.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, J. H., Lee, K. H., Kim, T. Y., and Lee, S. Y. (2007). Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc. Natl. Acad. Sci. U.S.A. 104, 7797–7802. doi: 10.1073/pnas.0702609104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pharkya, P., Burgard, A. P., and Maranas, C. D. (2004). OptStrain: a computational framework for redesign of microbial production systems. Genome Res. 14, 2367–2376. doi: 10.1101/gr.2872004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pietack, N., Becher, D., Schmidl, S. R., Saier, M. H., Hecker, M., Commichau, F. M., et al. (2010). In vitro phosphorylation of key metabolic enzymes from Bacillus subtilis: PrkC phosphorylates enzymes from different branches of basic metabolism. J. Mol. Microbiol. Biotechnol. 18, 129–140. doi: 10.1159/000308512
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Price, M. N., Deutschbauer, A. M., Skerker, J. M., Wetmore, K. M., Ruths, T., Mar, J. S., et al. (2013). Indirect and suboptimal control of gene expression is widespread in bacteria. Mol. Syst. Biol. 9, 660. doi: 10.1038/msb.2013.16
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramseier, T. M., Bledig, S., Michotey, V., Feghali, R., and Saier, M. H. Jr. (1995). The global regulatory protein FruR modulates the direction of carbon flow in Escherichia coli. Mol. Microbiol. 16, 1157–1169. doi: 10.1111/j.1365-2958.1995.tb02339.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramseier, T. M., Negre, D., Cortay, J. C., Scarabel, M., Cozzone, A. J., and Saier, M. H. Jr (1993). In vitro binding of the pleiotropic transcriptional regulatory protein, FruR, to the fru, pps, ace, pts and icd operons of Escherichia coli and Salmonella typhimurium. J. Mol. Biol. 234, 28–44. doi: 10.1006/jmbi.1993.1561
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reed, J. L. (2012). Shrinking the metabolic solution space using experimental datasets. PLoS Comput. Biol. 8:e1002662. doi: 10.1371/journal.pcbi.1002662
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reed, J. L., and Palsson, B. O. (2004). Genome-scale in silico models of E. coli have multiple equivalent phenotypic states: assessment of correlated reaction subsets that comprise network states. Genome Res. 14, 1797–1805. doi: 10.1101/gr.2546004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Richards, M. A., Cassen, V., Heavner, B. D., Ajami, N. E., Herrmann, A., Simeonidis, E., et al. (2014). MediaDB: a database of microbial growth conditions in defined media. PLoS ONE 9:e103548. doi: 10.1371/journal.pone.0103548
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schellenberger, J., Lewis, N. E., and Palsson, B. O. (2011). Elimination of thermodynamically infeasible loops in steady-state metabolic models. Biophys. J. 100, 544–553. doi: 10.1016/j.bpj.2010.12.3707
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schmidl, S. R., Gronau, K., Pietack, N., Hecker, M., Becher, D., and Stulke, J. (2010). The phosphoproteome of the minimal bacterium Mycoplasma pneumoniae: analysis of the complete known Ser/Thr kinome suggests the existence of novel kinases. Mol. Cell. Proteomics 9, 1228–1242. doi: 10.1074/mcp.M900267-MCP200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Segre, D., Vitkup, D., and Church, G. M. (2002). Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. U.S.A. 99, 15112–15117. doi: 10.1073/pnas.232349399
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shlomi, T., Berkman, O., and Ruppin, E. (2005). Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc. Natl. Acad. Sci. U.S.A. 102, 7695–7700. doi: 10.1073/pnas.0406346102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shlomi, T., Eisenberg, Y., Sharan, R., and Ruppin, E. (2007). A genome-scale computational study of the interplay between transcriptional regulation and metabolism. Mol. Syst. Biol. 3, 101. doi: 10.1038/msb4100141
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steuer, R., Gross, T., Selbig, J., and Blasius, B. (2006). Structural kinetic modeling of metabolic networks. Proc. Natl. Acad. Sci. U.S.A. 103, 11868–11873. doi: 10.1073/pnas.0600013103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stolovitzky, G., Monroe, D., and Califano, A. (2007). Dialogue on reverse-engineering assessment and methods: the DREAM of high-throughput pathway inference. Ann. N.Y. Acad. Sci. 1115, 1–22. doi: 10.1196/annals.1407.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Swainston, N., Smallbone, K., Mendes, P., Kell, D., and Paton, N. (2011). The SuBliMinaL Toolbox: automating steps in the reconstruction of metabolic networks. J. Integr. Bioinform. 8:186. doi: 10.2390/biecoll-jib-2011-186
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tagore, R., Thomas, H. R., Homan, E. A., Munawar, A., and Saghatelian, A. (2008). A global metabolite profiling approach to identify protein-metabolite interactions. J. Am. Chem. Soc. 130, 14111–14113. doi: 10.1021/ja806463c
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tan, Y., and Liao, J. C. (2012). Metabolic ensemble modeling for strain engineers. Biotechnol. J. 7, 343–353. doi: 10.1002/biot.201100186
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tervo, C. J., and Reed, J. L. (2013). BioMog: a computational framework for the de novo generation or modification of essential biomass components. PLoS ONE 8:e81322. doi: 10.1371/journal.pone.0081322
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thiele, I., Jamshidi, N., Fleming, R. M., and Palsson, B. O. (2009). Genome-scale reconstruction of Escherichia coli's transcriptional and translational machinery: a knowledge base, its mathematical formulation, and its functional characterization. PLoS Comput. Biol. 5:e1000312. doi: 10.1371/journal.pcbi.1000312
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thiele, I., and Palsson, B. O. (2010). A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121. doi: 10.1038/nprot.2009.203
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tomita, M. (2001). Whole-cell simulation: a grand challenge of the 21st century. Trends Biotechnol. 19, 205–210. doi: 10.1016/S0167-7799(01)01636-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tomita, M., Hashimoto, K., Takahashi, K., Shimizu, T. S., Matsuzaki, Y., Miyoshi, F., et al. (1999). E-CELL: software environment for whole-cell simulation. Bioinformatics 15, 72–84. doi: 10.1093/bioinformatics/15.1.72
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tran, L. M., Rizk, M. L., and Liao, J. C. (2008). Ensemble modeling of metabolic networks. Biophys. J. 95, 5606–5617. doi: 10.1529/biophysj.108.135442
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ulrich, L. E., and Zhulin, I. B. (2010). The MiST2 database: a comprehensive genomics resource on microbial signal transduction. Nucleic Acids Res. 38, D401–D407. doi: 10.1093/nar/gkp940
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vandersluis, B., Hess, D. C., Pesyna, C., Krumholz, E. W., Syed, T., Szappanos, B., et al. (2014). Broad metabolic sensitivity profiling of a prototrophic yeast deletion collection. Genome Biol. 15:R64. doi: 10.1186/gb-2014-15-4-r64
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Opijnen, T., Bodi, K. L., and Camilli, A. (2009). Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6, 767–772. doi: 10.1038/nmeth.1377
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Varma, A., and Palsson, B. O. (1994). Metabolic flux balancing: basic concepts, scientific and practical use. Nat. Biotechnol. 12, 994–998. doi: 10.1038/nbt1094-994
Wang, Q., Zhang, Y., Yang, C., Xiong, H., Lin, Y., Yao, J., et al. (2010). Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007. doi: 10.1126/science.1179687
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Waygood, E. B., Mort, J. S., and Sanwal, B. D. (1976). The control of pyruvate kinase of Escherichia coli. Binding of substrate and allosteric effectors to the enzyme activated by fructose 1,6-bisphosphate. Biochemistry 15, 277–282. doi: 10.1021/bi00647a006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yachie, N., Saito, R., Sugiyama, N., Tomita, M., and Ishihama, Y. (2011). Integrative features of the yeast phosphoproteome and protein-protein interaction map. PLoS Comput. Biol. 7:e1001064. doi: 10.1371/journal.pcbi.1001064
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, Z., Tan, M., Xie, Z., Dai, L., Chen, Y., and Zhao, Y. (2010). Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63. doi: 10.1038/nchembio.495
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: metabolic networks, transcriptional networks, constraint-based modeling, network integration, flux balance analysis, signaling, regulation, metabolism
Citation: Imam S, Schäuble S, Brooks AN, Baliga NS and Price ND (2015) Data-driven integration of genome-scale regulatory and metabolic network models. Front. Microbiol. 6:409. doi: 10.3389/fmicb.2015.00409
Received: 02 January 2015; Paper pending published: 12 February 2015;
Accepted: 20 April 2015; Published: 05 May 2015.
Edited by:
Christopher Scott Henry, Argonne National Laboratory, USAReviewed by:
Shunichi Ishii, J Craig Venter Institute, USARichard Splivallo, University of Frankfurt, Germany
Copyright © 2015 Imam, Schäuble, Brooks, Baliga and Price. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathan D. Price, Institute for Systems Biology, 401 Terry Ave. N., Seattle, WA 98109, USA, nprice@systemsbiology.org