 Katarzyna M. Bocian-Ostrzycka
Katarzyna M. Bocian-Ostrzycka Magdalena J. Grzeszczuk
Magdalena J. Grzeszczuk Lukasz Dziewit
Lukasz Dziewit Elżbieta K. Jagusztyn-Krynicka
Elżbieta K. Jagusztyn-Krynicka- Department of Bacterial Genetics, Faculty of Biology, Institute of Microbiology, University of Warsaw, Warsaw, Poland
The bacterial proteins of the Dsb family—important components of the post-translational protein modification system—catalyze the formation of disulfide bridges, a process that is crucial for protein structure stabilization and activity. Dsb systems play an essential role in the assembly of many virulence factors. Recent rapid advances in global analysis of bacteria have thrown light on the enormous diversity among bacterial Dsb systems. While the Escherichia coli disulfide bond-forming system is quite well understood, the mechanisms of action of Dsb systems in other bacteria, including members of class Epsilonproteobacteria that contain pathogenic and non-pathogenic bacteria colonizing extremely diverse ecological niches, are poorly characterized. Here we present a review of current knowledge on Epsilonproteobacteria Dsb systems. We have focused on the Dsb systems of Campylobacter spp. and Helicobacter spp. because our knowledge about Dsb proteins of Wolinella and Arcobacter spp. is still scarce and comes mainly from bioinformatic studies. Helicobacter pylori is a common human pathogen that colonizes the gastric epithelium of humans with severe consequences. Campylobacter spp. is a leading cause of zoonotic enteric bacterial infections in most developed and developing nations. We focus on various aspects of the diversity of the Dsb systems and their influence on pathogenicity, particularly because Dsb proteins are considered as potential targets for a new class of anti-virulence drugs to treat human infections by Campylobacter or Helicobacter spp.
Introduction
The oxidation reaction between two cysteine thiol groups, combined with the release of two electrons, results in the formation of a disulfide bond. This bond formation is a rate-limiting step in the protein folding process, and it is catalyzed by proteins of the Dsb (disulfide bond) system. This post-translational protein modification takes place in oxidative environments: either in the periplasm for Gram-negative bacteria or the space between the cytoplasmic membrane and the cell wall for Gram-positive bacteria (Hatahet and Ruddock, 2013). The formation of disulfide bonds (Dsb) plays a key role in bacterial virulence, which often depends on cysteine-rich, extracytoplasmic proteins (Heras et al., 2009, 2015; Denoncin and Collet, 2013). A combination of microbiological, biochemical, biophysical and proteomic approaches has yielded a detailed characterization of the Dsb protein network for the model microorganism, Escherichia coli (EcDsb proteins). In general, as shown on Figure 1, there are two, mostly antagonistic, metabolic pathways acting in the E. coli periplasm: an oxidation pathway and an isomerization/reduction pathway (Messens and Collet, 2006; Gleiter and Bardwell, 2008; Ito and Inaba, 2008; Depuydt et al., 2011).
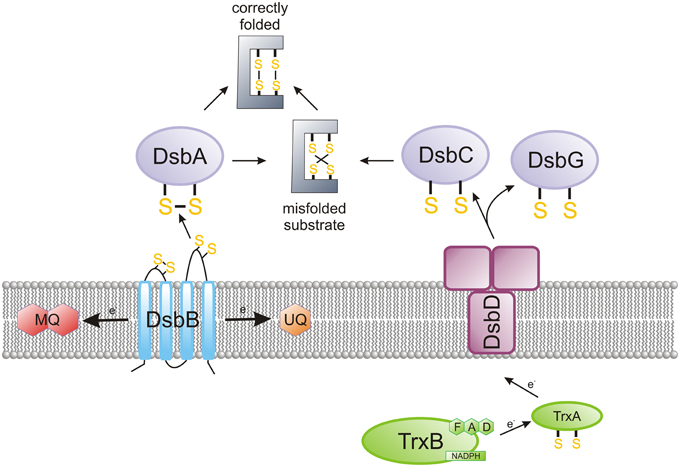
Figure 1. The main features of the oxidized protein folding in Escherichia coli. The bacterial proteins of the Dsb family catalyze the formation of disulfide bridges, a post-translational modification in many extracytoplasmic proteins. EcDsbA is a TRX family member, which forms disulfides in a consecutive manner on polypeptide chains that are entering the periplasm. EcDsbA is reoxidized by integral membrane protein EcDsbB, which catalyzes the electron transfer to the respiratory chain. EcDsbC ensures the rearrangement of improperly introduced disulfides. In E. coli, DsbC is kept in the reduced form by an integral membrane protein, DsbD, which catalyzes the transfer of electrons from the cytoplasm to the periplasm. The Campylobacter Dsb system is more complex than the E. coli system in terms of the number of elements included. Generally, Campylobacter species contain two DsbA and two DsbB proteins. However, several differences are observed not only among various species of the Campylobacter genus but also between strains of the same species (see Figures 2, 4). Helicobacter pylori, the best characterized member of the Helicobacter genus, does not encode the classical DsbA/DsbB oxidoreductases. Instead, it possesses an unusual pair of proteins involved in disulfide bond formation—periplasmic HP0231 (DsbK), with a dimeric structure that resembles EcDsbG/C, and its membrane partner HpDsbI (HP0595), a protein with a β-propeller structure. The mechanism for the rearrangement of incorrectly introduced disulfides was not examined in Campylobacter and Helicobacter cells.
The first reaction (catalyzed by EcDsbA and EcDsbB) is responsible for the formation of disulfide bonds in the newly synthesized proteins, just after they cross the cytoplasmic membrane (Kadokura and Beckwith, 2009). As this process occurs in a non-selective way, a second reaction (driven by EcDsbC and EcDsbD) rearranges improperly introduced disulfide bonds (Gruber et al., 2006). A large-scale bioinformatic analysis of bacterial genomes to search for Dsbs homologs, in conjunction with detailed functional and structural studies of DsbAs, has revealed that the pathways for disulfide bond formation within the bacterial kingdom are extremely diverse. The sets of Dsb proteins involved in the oxidative pathway varies, depending on the microorganism. The differences are observed not only between various bacterial genera and species, but also between bacterial strains of the same species. For instance, certain bacterial genomes encode multiple DsbAs with different substrate specificities, or multiple DsbBs; some other bacterial genomes possess only DsbA homologs; and others encode neither DsbA nor DsbB (Heras et al., 2009). In some microorganisms, the bacterial homolog of the vitamin K epoxide reductase (VKOR) plays a DsbB role (Li et al., 2010; Landeta et al., 2015). There are also some bacteria that possess Dsb proteins that fold into a V-shaped, homodimeric molecule similar to EcDsbC and EcDsbG, but are involved in disulfide bond formation like monomeric DsbAs (Daniels et al., 2010; Roszczenko et al., 2012; Kpadeh et al., 2013). Additionally, there are some microorganisms that do not have any components of the Dsb system (Dutton et al., 2008).
The three-dimensional structures of many DsbA-homologous proteins have recently been determined. There are about 25 DsbA structures deposited in the Protein Data Bank (PDB) (April 2015). These crystallographic studies revealed that even though all DsbAs possess a common thioredoxin fold, containing the active site with a CXXC motif, they display numerous structural differences that influence their biochemical properties, including redox potential and substrate specificity (McMahon et al., 2014). The redox potential of the Dsb proteins is mainly dependent on both the XX dipeptide within the active site CXXC motif and a residue located upstream of the cis-proline loop (which is distant in the linear sequence but close to the CXXC in the three-dimensional structure) (Charbonnier et al., 1999; Lafaye et al., 2009; Ren et al., 2009). Most of the EcDsbA homologs (more than 70%) possess a CXXC motif identical to EcDsbA (CPHC). However, various combinations of amino-acids in the XX dipeptide of the CXXC motif have been observed among members of the DsbA family (Chivers et al., 1996; Quan et al., 2007; Heras et al., 2009). The diverse redox properties of the DsbAs, as well as other TRX-fold proteins, are assumed to be also determined by indirect interactions of polar residues with the side chain of the N-terminal catalytic cysteine residue (Rinaldi et al., 2009). Members of DsbA-family fall into two main DsbA classes, based on comparison of their structural and biochemical features (McMahon et al., 2014).
The epsilon subdivision of the Gram-negative Proteobacteria has two major orders: Campylobacterales and Nautiliales. This taxon has multiple genera and includes both pathogenic and non-pathogenic microorganisms. The pathogenic group mainly colonizes the gastrointestinal tracts of many animal species (including humans); the non-pathogenic group are mainly microorganisms that live freely in water. Some of these occupy exceptional ecological niches, such as deep-sea hydrothermal vents (Miroshnichenko and Bonch-Osmolovskaya, 2006; Nakagawa et al., 2007; Giovannelli et al., 2011). The most prominent representatives of human pathogens are members of the Campylobacterales order belonging to the genus Campylobacter and Helicobacter, and Arcobacter and Wolinella strains are now also being analyzed.
Human infection by Campylobacter constitutes an important public-health problem worldwide. The greatest threat comes from two species, Campylobacter jejuni and Campylobacter coli. Both are inhabitants of the chicken intestinal tract, and each is a major etiological agent of human gastroenteritis (Kirkpatrick and Tribble, 2011; EFSA, 2014). In some immunocompromised humans, infection by these species can result in severe autoimmune disorders such as Guillain-Barré or Miller-Fisher syndromes (Zilbauer et al., 2008). New members of Campylobacter species, such as C. curvus or C. concisus, have recently been identified as emerging human and animal pathogens (Silva et al., 2011).
The genus Arcobacter, closely related phylogentically to Campylobacter, contains pathogenic and non-pathogenic species isolated from various ecological niches. These microorganisms are now recognized as emerging zoonotic enteropathogens, and contaminated food or contaminated water are the major sources of human infections (Collado and Figueras, 2011).
Although numerous identified species of the Helicobacter genus colonize various vertebrates, most of global research has focused on two species: Helicobacter pylori and Helicobacter hepaticus, as they are important predisposing factors in gastric cancers in humans (Yamaoka and Graham, 2014; Segura-Lopez et al., 2015). Infection with H. pylori affects about half of the world's population, yet, its prevalence varies geographically. Currently, infections are highly prevalent in developing countries, but are disappearing in well developed countries. H. pylori infections induce both acute and chronic gastritis and peptic ulcers. H. pylori is also considered to be a high risk factor for the development of mucosa-associated lymphoid tissue lymphoma and adenocarcinoma of the stomach (De Falco et al., 2015). Based on results of clinical studies, the World Health Organization (WHO) has designated H. pylori infections as class I carcinogens (WHO, 1994). However, it should be pointed out that an H. pylori infection has two faces (Bocian and Jagusztyn-Krynicka, 2012). This bacterium has accompanied humans for at least 60,000 years (Linz et al., 2007). The prevalence of H. pylori infection has been decreasing over the past 50 years. At the same time an increased incidence of gastroesophageal reflux disease and esophageal adenocarcinoma, has been noted. Some studies also suggest an inverse correlation of H. pylori infection with childhood asthma or obesity. Thus, it is debatable whether we should consider this bacterium as a colonizer or as a pathogen (Cover and Blaser, 2009; Cid et al., 2013; Otero et al., 2014).
While Wolinella succinogenes is classified as a member of the Helicobacteriaceae, phylogenetic studies show that it is an “intermediate” between the Campylobacteriaceae and Helicobacteriaceae families (Baar et al., 2003).
In this review we present progress that has recently been made to unravel the intricate details of how the Dsb systems of pathogenic Epsilonproteobacteria function. We focus on the roles the Dsb systems play in oxidative protein folding and cytochrome c biogenesis, and we also present potential benefits in therapy that may arise from accumulated knowledge about Dsb systems. The review also encompasses data on the in silico analysis of Dsb proteins of 107 representatives Epsilonproteobacteria, including: (i) Arcobacter spp. (4 strains), (ii) Campylobacter spp. (31 strains), (iii) Helicobacter spp. (71 strains), and W. succinogenes. The results of our in silico analysis (Figure 2, Table S1) summarize the current knowledge and add some novel observations concerning diversity of the Epsilonproteobacteria Dsb systems.
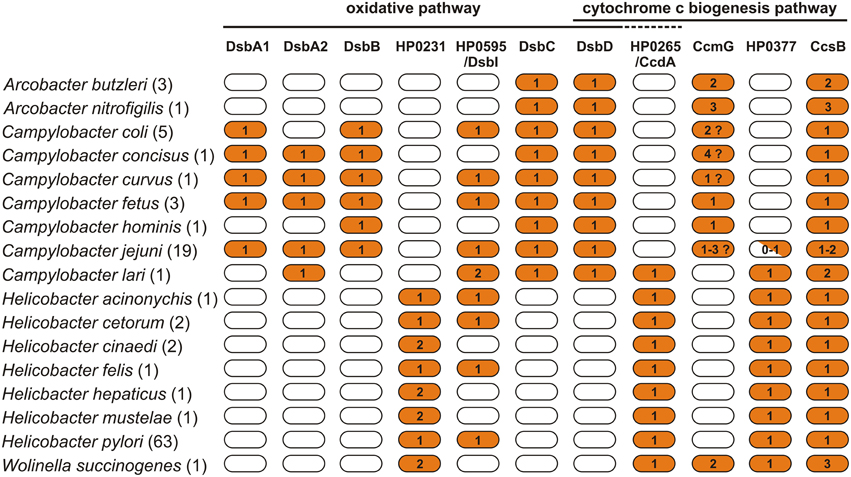
Figure 2. Distribution of proteins of the Dsb family in Arcobacter spp., Campylobacter spp., Helicobacter spp., and Wolinella succinogenes DSM 1740 proteomes. For the analysis, 107 bacterial proteomes were screened for the presence of 11 proteins involved in the oxidative pathway and cytochrome c biogenesis pathway. The exact number of proteomes used for each species is presented in parenthesis next to specie's name. The BLASTp search vs. a manual curated database, including 34 reference proteins, was applied. For the analysis, the following cutoff values were applied (e-value < 10−10 and amino acid sequence identity of at least 25%). Numbers indicate the copy number of proteins in the particular proteomes.
Brief Characteristics of the E. coli Oxidative and Isomerization Dsb System
In the E. coli proteome, the periplasmic DsbA and its partner, an inner-membrane protein DsbB, are encoded by two monocistronic units located in distinct chromosomal loci. DsbA is a 21-kDa monomeric protein in the TRX family, and it directly donates its disulfide bond that is present in the active site to reduce protein substrates. This reaction is conditioned by the presence of two highly conserved motifs: the CXXC (CPHC) active site within the TRX fold and the cis-proline loop. Both motifs are responsible for numerous physico-chemical properties of the DsbA enzyme, and they play a significant role in its folding and stability, as well as in DsbA's interaction with its substrate(s) and its redox partner, DsbB (Kadokura et al., 2004, 2005). In E. coli, DsbA is converted back to the oxidized form by the inner membrane protein DsbB, which has four transmembrane segments and two periplasmic loops, each containing one pair of conserved, essential catalytic cysteine residues. DsbB donates electrons to either ubiquinone during aerobic growth or to menaquinone during anaerobic growth. In contrast to the non-specific oxidoreductase DsbA, DsbB appears to be a very specific enzyme. Under physiological conditions, it is able to oxidize only the reduced form of DsbA, by rapid disulfide exchange (Tapley et al., 2007; Pan et al., 2008).
In E. coli, the periplasmic protein disulfide isomerase, DsbC is involved in rearrangements of incorrectly paired cysteines, so it is required in vivo for full activity of a handful of proteins that contain at least one non-consecutive disulfide bond (Hiniker and Bardwell, 2004). Its X-ray structure has been solved and shows that EcDsbC is a dimeric protein with two 23.3 kDa monomers arranged into a V-shaped homodimeric molecule (McCarthy et al., 2000). Each monomer forms an arm of the V and consists of two domains: a C-terminal catalytic domain with a TRX fold, and an N-terminal dimerization domain. The dimerization of DsbC is crucial for its activity, as it leads to the formation of the substrate binding domain (Segatori et al., 2004; Arredondo et al., 2009). The C- and N-terminal domains are connected via a long α-linker. There are four conserved cysteine residues in each monomer. Two of these residues are arranged in a CXXC motif and are essential for the oxidoreductase activity of the protein (Hiniker et al., 2007).
The E. coli periplasmic oxidoreductase, DsbG, was for a long time considered to be a member of the isomerization/reduction pathway. Although the structure of DsbG resembles that of DsbC, significant structural differences between EcDsbC and EcDsbG were noticed (Heras et al., 2004). Additionally, EcDsbG it is not active in the insulin reduction assay and does not catalyze oxidative refolding of incorrect disulfides present in model protein substrates such as scrambled hirudine or scrambled RNase. Examined in vivo, its isomerizing activity is poor as compared with EcDsbC (Bessette et al., 1999; Hiniker et al., 2007). Depuydt et al. showed that EcDsbG interacts with periplasmic proteins containing a sole cysteine (Depuydt et al., 2009). Thus, it is generally accepted that DsbG is mainly involved in the control of the cysteine sulfenylation level, which protects single cysteine residues from oxidation to sulfenic acid.
In E. coli, DsbC and DsbG are kept in the reduced form by an integral membrane protein, DsbD, that catalyzes the transfer of electrons from the cytoplasm to the periplasm. The E. coli DsbD consists of eight transmembrane segments (β domain), an N-terminal domain (α domain) and a C-terminal domain (γ domain). The N- and C-terminal domains both face the periplasm. It has been established that the electrons flow from NADPH in the cytosol, via the TRX-1, β, γ, and α domains of DsbD, and then to various extracytoplasmic proteins (Stirnimann et al., 2006). Figure 1 presents key elements of the E. coli oxidative protein folding. Several excellent reviews about functioning of the E. coli Dsb system have recently been published (Shouldice et al., 2011; Berkmen, 2012; Denoncin and Collet, 2013; Hatahet et al., 2014).
Oxidative and Isomerization Dsb Pathways of Campylobacter spp. and Arcobacter spp.
Phylogenetic comparisons based on 16sRNA have divided members of the Campylobacter genus into two major groups: the thermotolerant species (group I) and the non-thermotolerant species (group II) (Nothaft et al., 2012; Porcelli et al., 2013). Members of both groups were included in our analysis. Group I is represented by C. jejuni, C. coli, and C. lari; whereas group II is represented by C. fetus, C. curvus, C. concisus, and C. hominis.
We performed in silico analysis of the proteomes of 31 members of the Campylobacter genus, with respect to the presence of Dsb oxidoreductases (Figure 2). A BLASTp search, vs. a manually curated database including 34 reference proteins, was applied. Among the Dsb-like proteins identified, the oxidative folding activity was experimentally investigated for only two model C. jejuni strains: 81-176 and 81116 (Raczko et al., 2005; Grabowska et al., 2011, 2014). Both strains were isolated from human having campylobacteriosis and are commonly used for basic as well applied research by many laboratories (Palmer et al., 1983; Korlath et al., 1985). Genome sequences of both strains were determined (Hofreuter et al., 2006; Pearson et al., 2007). The C. jejuni 81116 genome is slightly smaller than that of C. jejuni 81-176 (1,628,115 bp and 1,641,481 bp, respectively). Sequencing of the C. jejuni 81-176 genome revealed some striking genetic attributes conditioning its high virulence (Hofreuter et al., 2006). It also contains two resident plasmids, pVir and pTet whose products affect virulence (Bacon et al., 2000, 2002; Batchelor et al., 2004). C. jejuni 81116 has small numbers of hypervariable G tracts so it is genetically stable and considered to be appropriate for the genetic research (Manning et al., 2001; Pearson et al., 2007).
The knowledge concerning the Campylobacter Dsb isomerization pathway is extremely limited and comes only from in silico analysis. The analyses performed in this work indicated that all Campylobacter strains contain homologs of EcDsbC and EcDsbD, two proteins that are potentially involved in the isomerization process (Figure 2, Table S1). The next part of this section presents data concerning the Campylobacter Dsb oxidative pathway.
The Dsb oxidative pathway of C. jejuni (CjDsb) differs from the well-characterized oxidative pathway of E. coli. The E. coli pathway has two proteins, DsbA and DsbB, that are encoded by two monocistronic operons located far-apart on the chromosome. In C. jejuni 81116 and C. jejuni 81-176, two model organisms of C. jejuni, the Dsb oxidative pathway consists of four extracytoplasmic proteins. Two are soluble periplasmic proteins (CjDsbA1 and CjDsbA2), and the other two (CjDsbB and CjDsbI) are anchored in the inner membrane (Grabowska et al., 2011).
CjDsbA1 and CjDsbA2 share a high degree of sequence identity (47%); CjDsbA1 and CjDsbA2, respectively, have sequence identities of 24 and 28% with EcDsbA and 28.5 and 39% with EcDsbL (a homolog of EcDsbA present in some pathogenic Enterobacteriaceae (Grabowska et al., 2014). Both proteins have a strong, positively charged electrostatic patch above the active site, similar to that of EcDsbL but not EcDsbA (Grimshaw et al., 2008). However, CjDsbA1 and CjDsbA2 vary considerably with regard to their active sites, and in the charge distribution on the protein surface that is opposite from the active site. These differences are reflected in their phenotypic characteristics. CjDsbA1 is involved in cell motility and the autoagglutation process, and it is responsible for the formation of the disulfide bond of alkaline phosphatase, CjPhoX. The arylsulfotransferase CjAstA is the only substrate of CjDsbA2 identified so far, and it is not a substrate of CjDsbA1 (Grabowska et al., 2014). Similar relationships for multiple DsbAs in one bacterial proteome have also been described in other bacteria. The two redox protein pairs present in the proteome of E. coli UPEC CFT073 (EcDsbA/EcDsbB and the EcDsbL-EcDsbI) also differ in their substrate specificity and only the EcDsbL-EcDsbI redox pair plays a role in introducing disulfide bonds into AstA (Totsika et al., 2009). Another oxidative Dsb pathway with more than one DsbA is found in S. enterica sv. Typhimurium, which contains three DsbAs (i.e., SeDsbA, SeDsbL, and SeSrgA). Only the SeDsbL-SeDsbI redox pair restored full activity of AstA in a triple DsbA-homolog mutant (Heras et al., 2010). The hypothesis of a limited spectra of substrate for both of the C. jejuni DsbAs is also supported by the fact that neither CjDsbA1 nor CjDsb2 is active in insulin reduction (Grabowska et al., 2014).
The biochemical properties of the DsbA proteins are influenced by the cysteine-flanked dipeptide sequence of the CXXC motif and by the nature of the cis-Pro motif. The most common motif of bacterial DsbAs is CPHC paired with VcP. So, we compared the active site CXXC and cis-Pro motifs of the Campylobacter DsbAs. We found that all but one DsbAs of the group I members have the CIHC/CTHC motif paired with TcP, which is atypical for DsbA. The DsbA proteins of two species of group I that have only one DsbA also contain a CIHC motif in the active site paired with TcP. Group II members contain DsbA2 with CPFC, VcP motifs (close to that of the classical DsbA–CPHC, VcP), whereas the residues of the dipeptide in the CXXC motifs of their DsbA1 proteins vary considerably and are paired with either TcP or VcP (CIHC, TcP for C. fetus; CQHC, VcP for C. curvus; and CPHC, VcP for C. concisus) (Table S1).
In E. coli, the activity of DsbA is restored by the action of EcDsbB, which provides its reoxidation. In EcDsbB, four cysteine residues, located in the periplasmic loops, play a crucial role in EcDsbA re-oxidation (Figure 3) (Kadokura and Beckwith, 2002). In C. jejuni, there are two homologs of EcDsbB: CjDsbB and CjDsbI. As was recently shown in C. jejuni 81-176 and 81116, only the CjDsbA2 activity is fully CjDsbB-dependent, and the possible cooperation between CjDsbA1 and CjDsbB is unclear. CjDsbI is not involved in either CjDsbA1 or CjDsbA2 activities (Grabowska et al., 2014). The “classical” EcDsbB consist of one domain, formed by four trans-membrane segments (TM1-TM4) (Inaba, 2008; Inaba and Ito, 2008). We have attempted to predict the toplogy of the DsbBs of representatives from group I and group II Campylobacter. We found, as previously published, that C. jejuni DsbB and also C. coli DsbB, both contain five membrane-spanning domains compared with four for EcDsbB; whereas the DsbBs of C. fetus, C. concisus, C. curvus, and C. hominis revealed the same topology as EcDsbB (four TMs). Regardless of the number of TMs present, all Campylobacter DsbBs contain the two pairs of conserved Cys residues, which are located within the periplasmic loops connecting TM segments 1–2 and 3–4, as is also seen in EcDsbB. DsbI—atypical DsbB-like protein is present in all Campylobacter proteomes, so far, analyzed. The role of DsbI in the oxidative Dsb pathway is still not completely understood. CjDsbI comprises two domains. The N-terminal domain consists of five transmembrane helices with a conserved oxidoreductase CXXC motif, and it exhibits significant sequence similarity to proteins from the DsbB family. It also contains a conserved Arg residue implicated in the process of transferring electrons (Kadokura et al., 2000). Both CXXC motif and Arg are located in the 1-2 periplasmic loop. A key difference between DsbI and “classical” DsbB proteins is the lack of the second pair of Cys residues. Instead, proteins of the DsbI family possess a C-terminal, periplasm-located domain with a beta-propeller structure, as shown in Figure 3 (Raczko et al., 2005). The role of the C-terminal domain of CjDsbI is still unknown. Proteins with a beta propeller fold, are characterized by extreme sequence diversity, despite similarities in their three-dimensional structures. In eukaryotic cells, they have different functions, whereas in prokaryotic cells they are often involved in redox reactions (Jawad and Paoli, 2002). C. lari does not contain DsbB, but instead it encodes two genes for DsbI, we also checked the topology of these two DsbI proteins. We found no differences between them by the method we used (Figures 2, 3, Table S1).
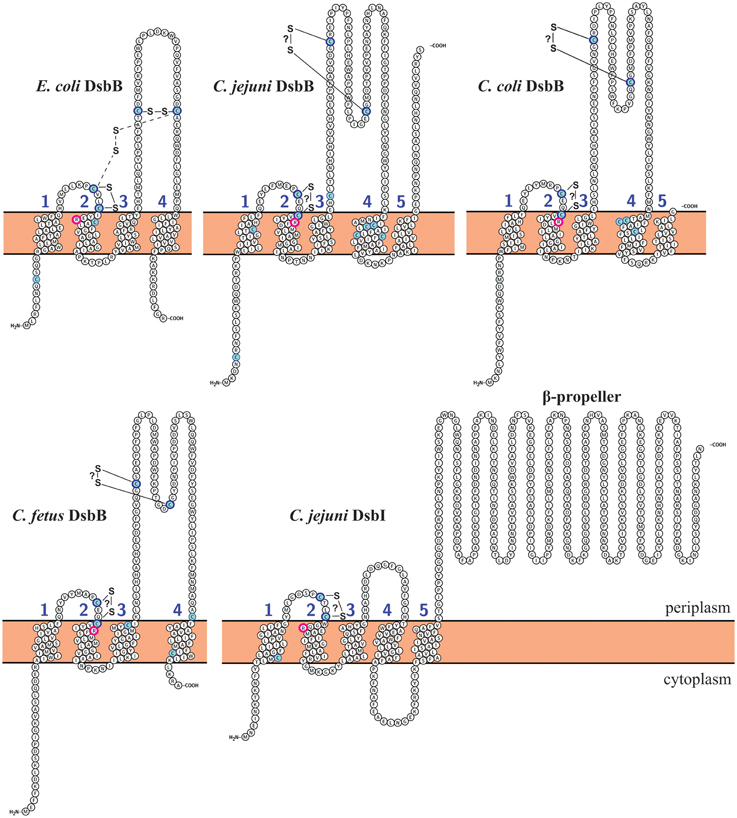
Figure 3. DsbB and DsbI topological model predictions for Campylobacter species and comparison with EcDsbB (E. coli). C. jejuni DsbI model is identical as C. lari DsbI1, DsbI2, and H. pylori DsbI. Transmembrane topology predictions of DsbB and DsbI proteins were determined using Protter (http://wlab.ethz.ch/protter/#) (Omasits et al., 2014). Predictions were confirmed with TOPCONS (http://topcons.cbr.su.se/) (Bernsel et al., 2008, 2009; Viklund et al., 2008), PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) (Buchan et al., 2013) and PredictProtein (http://predictprotein.org) (Rost et al., 2004). Disulfide bonding patterns were predicted using DiANNA software (http://clavius.bc.edu/~clotelab/cgi-bin/DiANNA/DiANNA.py) (Ferre and Clote, 2005). Cysteine residues are marked with blue. Conserved arginine residues are circled with pink. Intermediate disulfide bond between Cys41 and Cys140 in EcDsbB is mark with dashed lines. Disulfide bonds that are not confirmed experimentally are marked with question mark (?). All predictions were compared and manually corrected according to published data.
Our in silico analysis performed on all C. jejuni strains revealed that in respect to their genetic organization they can be classified into three types (Figure 4). The significant differences among the Campylobacter genomes with respect to dsbA2 gene presence were observed. Type A1, represented by C. jejuni 81116 or 81-176, includes strains possessing two functional DsbA proteins (CjDsbA1 and CjDsbA2, respectively). In C. jejuni 81-176 and 81116 the dsb genes are organized into three operons. The monocistronic dsbA1 operon is preceded by the cjdsbA2-cjdsbB-cjastA operon, in which cjastA codes for the Dsb substrate, arylsulfotransferase. The third operon, cjdba-cjdsbI, encodes for the DsbI and its accessory protein Dba, is distant from the other two on the chromosome (Grabowska et al., 2011; Dugar et al., 2013). Type A2 strains contain a truncated variant of cjdsbA2 (encoding a putative TRX-like protein lacking the active CXXC motif), and they additionally lack a functional AstA. The strains classified as type A3 have already lost the functional astA gene, but they still encode a potentially active DsbA2. Regardless of the functionality of dsbA2/astA, the analyzed dsb genes are arranged into two operons, i.e., cjdsbA2-cjdsbB-cjastA, directly followed by a monocistronic unit cjdsbA1 (Figure 4). Additionally two species that are also members of the thermotolerant group I, C. coli and C. lari, have lost one of the dsbA2 genes (Figure 4—types B and C).
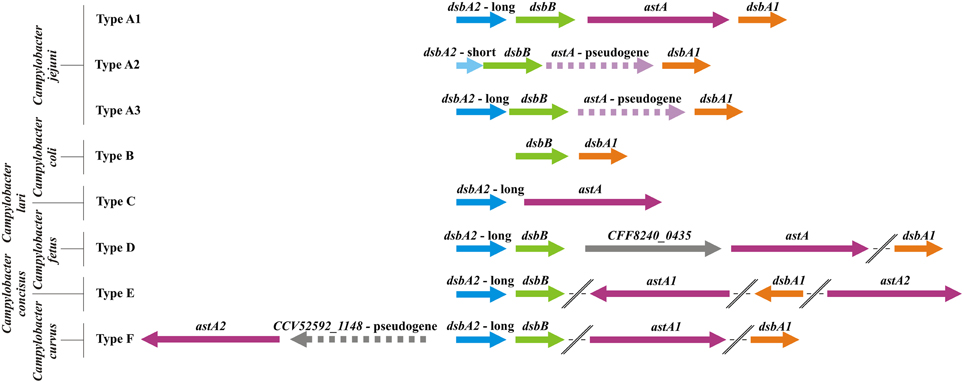
Figure 4. Physical map presenting the genomic location of the Campylobacter Dsb oxidative pathway genes [dsbA1 (orange), dsbA2 (blue), dsbB (green), and astA genes (purple)]. The astA gene was also included as it is a part of the same transcriptional unit with dsbA2 and dsbB, and its product is a substrate for DsbA2. Type A includes C. jejuni strains (A1: C. jejuni subsp. jejuni 81-176, 81116, ICDCCJ07001, MTVDSCj20, and C. jejuni 4031; A2: C. jejuni subsp. jejuni NCTC 11168 = ATCC 700819, 00-2425, 00-2426, 00-2538, 00-2544, IA3902, NCTC 11168-BN148, PT14, R14, and C. jejuni 32488; A3: C. jejuni subsp. jejuni S3 and M1, C. jejuni RM1221, C. jejuni subsp. doylei 269.97). Type B includes strains of C. coli species (C. coli RM5611, 15-537360, CVM N29710, RM1875 and RM4661). Type C is represented by one C. lari strain (C. lari RM2100). Type D includes three members of C. fetus species (C. fetus subsp. fetus 82-40, 04/554 and C. fetus subsp. testudinum 03-427). Type E is represented by one C. concisus strain (C. concisus 13826) and type F by one strain of C. curvus (C. curvus 525.92). The dsbA1 and astA genes of C. concisus, C. curvus, and C. fetus, strains are located on the chromosome apart from dsbA2dsbB genes. Genes and intergenic regions are drawn to scale (based on underlined strains). Pseudogenes or fragmented genes are marked with dashed lines.
In contrast, in the genomes of Campylobacter spp. that belong to the non-thermotolerant group II (C. fetus, C. concisus, and C. curvus), only the dsbA2 and dsbB genes constitute a putative operon. The astA gene, encoding a substrate of DsbA2, never forms an operon with dsbA2/dsbB and is located either in close proximity (C. fetus and C. curvus) to them or is present in a different chromosomal location (there are two copies of the astA gene in C. concisus). Additionally, the dsbA1 genes of the group II Campylobacter spp., encoding oxidoreductases of potentially broad specificity, are located apart from the dsbA2/dsbB operon (Figure 4—types D, E, F). Our previous phylogenetical analysis showed that DsbA1-2, DsbB, and AstA all have very similar evolutionary histories, suggesting horizontal transfer of the entire DsbA1-2/DsbB2/AstA system from the Campylobacter genus to a common ancestor of the Gammaproteobacteria species, followed by its subsequent loss in most organisms of this clade. Moreover, phylogenetic analysis indicates that the divergence of CjDsbA1 and CjDsbA2 occurred after the horizontal transfer event (Grabowska et al., 2014).
All Arcobacter spp. genomes analyzed lack both a classical DsbA and DsbB members of Dsb oxidative pathway. However, they encode classical DsbC and DsbD, which are members of the Dsb isomerization pathway. The mechanism responsible for oxidative folding of proteins in this bacterium requires further investigation (Figure 2, Table S1).
Introduction of the Disulfide Bond into Extracytoplasmic Proteins of Helicobacter spp. and Wolinella succinogenes
H. pylori Dsb oxidizing system is different from that operating in E. coli and in Campylobacter spp. that are phylogenetically related to H. pylori. The genome of H. pylori 26695 has 149 proteins containing the CXXC motif that is characteristic for the thiol:disulfide oxidoreductases identified to date. Only four of these proteins (HP0231, HP0377, HP0824, and HP1458) have a TRX fold (Kaakoush et al., 2007). Additionally, the H. pylori genome lacks classical DsbA and DsbB proteins, members of Dsb oxidative pathway, as well as classical DsbC and DsbD proteins, members of the Dsb isomerization pathway.
Our recent work (using biochemical and genetic approaches) led to the characterization of the first dimeric oxidoreductase (HP0231) that functions in an oxidizing pathway of H. pylori (Roszczenko et al., 2012). The knowledge about the role of HP0231 in cell physiology has been recently expanded by the data published by Lester et al. (2015). H. pylori isogenic hp0231 mutant is non-motile and reveals some changes in the cell morphology. Additionally it is DTT-sensitive and shows reduced growth compared to the wild type parental strain (Roszczenko et al., 2012). Lack of HP0231 also affects H. pylori resistance to oxidative stress (Lester et al., 2015). HP0231 functioning in the oxidative protein folding was shown by proving its ability to complement the lack of DsbA in E. coli when delivered by a low-copy recombinant plasmid (Roszczenko et al., 2012) and by the observation that its over-expression in E. coli results in strong toxic effect (Lester et al., 2015). The structure of HP0231 has recently been solved (Yoon et al., 2011) and it appears to be a V-shaped protein like EcDsbC or EcDsbG. Two domains of HP0231, the catalytic and dimerization domains, resemble the corresponding domains of EcDsbG. However, several substantial structural differences between the two proteins have been shown. HP0231 and EcDsbG vary in the length of the linker helix and in the amino acid residues lining the V-shaped cleft (Yoon et al., 2011). Interestingly, the XX dipeptide from the active CXXC site of dimeric HP0231 is identical to that of monomeric EcDsbA (i.e., CPHC) but different from that of EcDsbC/G (i.e., CGYC/CPYC). Additionally, the cis-Pro loop of HP0231 is VcP, as in EcDsbA, whereas a conserved threonine residue is found in the cis-Pro loops of EcDsbC and EcDsbG. Although the catalytic domain of HP0231 possesses motifs typical for canonical DsbA proteins, evolutionarily it is most closely related to the catalytic domains of DsbG. Similarly, the highly diverged N-terminal dimerization domain is homologous to the dimerization domain of DsbG. The uncommon combination of catalytic and dimerization domains is reflected in the protein's biochemical properties. Some of the properties, such as a redox potential similar to that of EcDsbA, activity in the insulin reduction test (Roszczenko et al., 2012) proved that HP0231 functions in an oxidizing Dsb pathway. On the other hand, HP0231, similarly to EcDsbC or EcDsbG, acts as molecular chaperone, as documented by Lester et al. (2015). HcpE (Helicobacter cysteine-rich protein E) containing nine consecutive disulfide bonds, was identified as a substrate of HP0231. HP0231 affects HcpE production and secretion in the native host and also assists its correct folding in E. coli. So, due to its dual function HP0231 was named DsbK (Lester et al., 2015).
H. pylori does not contain a classical homodimeric DsbC, which in the most Gram-negative bacteria is a periplasmic protein responsible for the rearrangement of incorrectly paired cysteines (Denoncin and Collet, 2013). HP0231 does not complement the deficiency of DsbC in E. coli cells as measured by the copper sensitivity assay (Roszczenko et al., 2012). Lack of isomerization activity in E. coli cells may result from the inability of HP0231 to interact with EcDsbD. Also, H. pylori does not encode a classical DsbD. Instead it has a shortened version of DsbD, CcdA (HP0265) consisting only of the β transmembrane domain of DsbD (Cho et al., 2012). It should be noted at this point that H. pylori encodes other protein of the Dsb family—HP0377 with structure similar to CcmG (cytochrome c maturation), previously annotated as DsbC-like protein (Kaakoush et al., 2007). Its biochemical analysis indicates that it might be also involved in Dsb isomerization/reduction pathway (Yoon et al., 2013) (for details see below). It is unclear how HP0231 is re-oxidized in vivo. H. pylori does not encode a classical DsbB, though it encodes a DsbB-like protein, HpDsbI (HP0595). Mutation of the hpdsbI gene results in the appearance of HP0231 in a reduced form, but the active-oxidized form remains more pronounced (Roszczenko et al., 2012), which may suggest that HP0231-DsbI cooperation is rather collateral.
Our bioinformatic in silico analysis performed on 71 Helicobacter spp. genome revealed a high conservation of the mechanism responsible for protein oxidative folding within two subdivisions of the Helicobacter genus, the first subdivision including gastric species and the second including enterohepatic species (with the exception of H. mustelae) (Gupta, 2006; Porcelli et al., 2013). All members of the former group contain a dimeric oxidoreductase homolog of the HP0231 of H. pylori 26695, with an active site that includes the CPHC motif paired with VcP. Potentially, these oxidoreductases interact with DsbIs, although the mechanism of the process is not completely clear. Interestingly, all enterohepatic species and H. mustelae lack DsbI, and at the same time, they encode two homologs of HP0231 with various CXXC motifs (CPSC and CPYC in the case of H. cinaedi and H. hepaticus, and CPHC and CSFC in the case of H. mustelae). In every case, the CXXC motif is paired with VcP. The observed divergences may reflect differences in the colonized ecological niches that result in various sets of Dsb substrates (Figures 2, 5, Table S1).
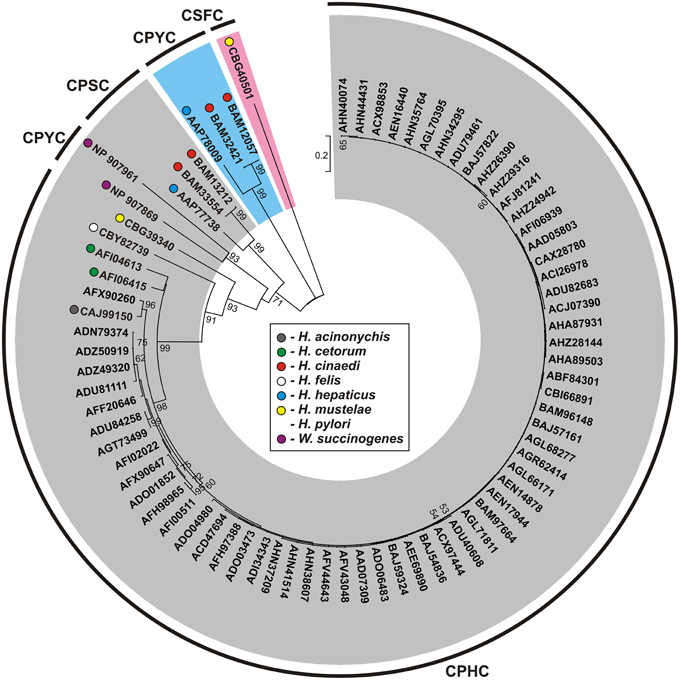
Figure 5. Phylogenetic tree of the HP0231-like proteins of Helicobacter spp. and Wolinella succinogenes DSM 1740. The analysis was based on 77 protein sequences and 265 amino acid positions (highly variable portions of the alignments were eliminated by the use of G-blocks). The unrooted tree was constructed using the neighbor-joining algorithm. The statistical support for the internal nodes was determined by 1000 bootstrap replicates. Values of >50% are shown. Accession numbers of the protein sequences used for the phylogenetic analysis are given. The three main clusters were distinguished with gray, pink, and blue color, respectively. The conserved catalytic motifs (CXXC) are presented.
The Dsb oxidative pathway of W. succinogenes seems to act in a way similar to that of the entrohepatic Helicobacter spp. It also encodes two homologs of HP0231 and lacks DsbI. However, the two W. succinogenes homologs of HP0231 contain active sites with identical motifs: CPYC paired with VcP and were grouped together, within cluster III (Figures 2, 5, Table S1).
Significant Features of Bacterial Cytochrome c Biogenesis
In the highly oxidizing environment of the periplasm, there is a need for selected proteins to be kept in a reduced form, such as c-type cytochromes that are essential for energy metabolism. The cytochrome c maturation process requires the ligation of heme to reduced thiols of the CXXCH motif of the apocytochrome (Thony-Meyer and Kunzler, 1997).
Since DsbA or other periplasmic dithiol-oxidases randomly introduce disulfide bonds into apocytochromes, bacteria have evolved a special redox system to revert these disulfides into reduced cysteine residues while in a highly oxidizing environment. Among the five cytochrome c biogenesis systems identified so far, system I (named the Ccm—cytochrome c maturation process) and system II (designated as the Ccs—cytochrome c synthesis) are the most common pathways in the prokaryotic world (Kranz et al., 2009; Sanders et al., 2010; Simon and Hederstedt, 2011). Figure 6A presents the model of bacterial cytochrome c biogenesis. They comprise two kinds of proteins: those involved in the handling of heme that play a role in its ligation to the apocytochrome, and those contributing to reduction of the disulfide bond of the CXXCH heme-binding motif (Kranz et al., 2009).
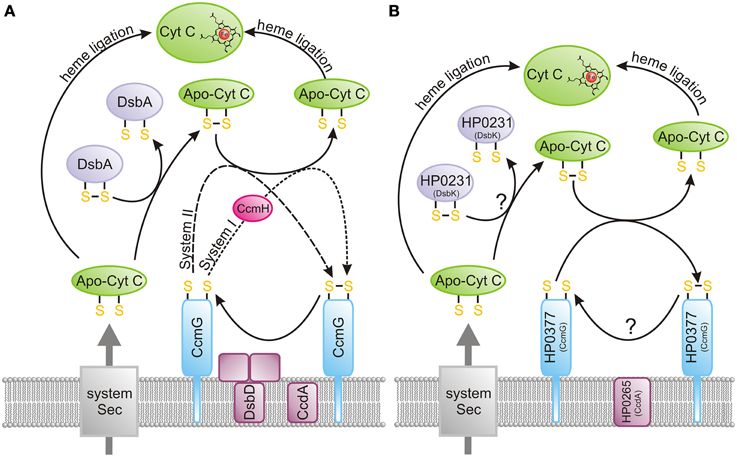
Figure 6. Cytochrome c biogenesis. The heme transport mechanism, which is more complex for system I (CcmA-E) compared to system II (CcsA and CcsB or CcsAB), is not shown on the figure. (A) Model presenting the bacterial cytochrome c biogenesis (systems I and II). Apocytochrome c, just after its transport across the membrane by Sec proteins, is oxidized by the DsbA/DsbA-like oxidoreductases. Subsequently, it is reduced by CcmG and ligated to heme. Potentially, some apocytochrome c molecules escape oxidation, and they attach heme after they enter the periplasm. CcmG is kept in a reduced form by the CcdA/DsbD-like protein, which transfers electrons from the cytoplasm to the periplasm. (B) Model presenting the Helicobacter pylori cytochrome c biogenesis (system II). Apocytochrome c, just after its transport across the membrane by Sec proteins, is oxidized by the dimeric oxidoreductase, HP0231 (DsbK). Subsequently, it is reduced by HP0377 (CcmG) and ligated to heme. Potentially, some apocytochrome c molecules escape oxidation, and they attach heme after they enter the periplasm. HP0377 is kept in a reduced form by the DsbD-like protein HP0265 (CcdA), which transfers electrons from the cytoplasm to the periplasm.
The main difference between system I and II is the process of heme transport and its ligation into reduced apocytochrome, which appears to be more complicated for system I. In E. coli, which uses system I, the CcmABCDEFGH proteins are involved in this process. In contrast, in system II a maximum of four protein components (CcsA, CcsB, CcdA and CcsX) seem to be necessary to achieve cytochrome c biogenesis (Simon and Hederstedt, 2011). The best known system II is the Gram-positive model organism Bacillus subtilis. The B. subtilis system contains four proteins, ResA (CcsX), ResB (CcsB), ResC (CcsA), and CcdA, of which ResB and ResC are two separate polypeptides that form a complex to deliver the heme and function in the periplasmic cytochrome c-heme ligation (Erlendsson et al., 2003; Colbert et al., 2006; Lewin et al., 2006). In some microorganisms, ccsA and ccsB are fused into one large ORF (called ccsBA), whose product contains 10 transmembrane domains (Frawley and Kranz, 2009) (see below). In both systems, there is a need for the reduction of the disulfide bond of the apocytochrome c heme-binding motif (Bonnard et al., 2010). This is accomplished by the action of CcmG (also known as DsbE for system I, ResA for B. subtilis and CcsX for other bacteria). CcmG proteins, both in systems I and II, are kept in the reduced form by an integral membrane protein DsbD, or its shorter analog CcdA. Both proteins catalyze the transfer of electrons from cytoplasmic thioredoxin across the inner membrane to the periplasm. As mentioned above, DsbD consists of eight transmembrane segments (β domain), an N-terminal (α domain) and a C-terminal domain (γ domain). Both, the N- and C-terminal domains face the periplasm (Stirnimann et al., 2006; Rozhkova and Glockshuber, 2008). CcdA is a shorter version of DsbD, consisting only of the β transmembrane domain of DsbD. In contrast to DsbD, which transfers reducing potential to a large numbers of extracytoplasmic proteins, CcdA was thought to only be involved in the cytochrome c maturation process (Katzen et al., 2002). However, some recently published data have shown that in B. subtilis or B. anthracis, CcdA also plays a role in sporulation and virulence (Erlendsson et al., 2004; Han and Wilson, 2013). Recently a new class of DsbD proteins named ScsB, with a domain organization similar to but not identical with that of DsbD, has been described. However, the role of this class of proteins in CcmG reduction was not analyzed (Cho et al., 2012).
Many studies have tried to decipher the cooperation among periplasmic Dsb proteins during c-cytochrome maturation. Initially, it was thought that introduction of the disulfide bonds into the CXXCH motif of the apocytochrome c, just after its transport to periplasm by the Sec system, was an obligatory step of the cytochrome c maturation process because dsbA and dsbB mutants in E. coli were unable to produce cytochrome c (Metheringham et al., 1996; Sambongi and Ferguson, 1996). However, some recent data in the literature contradict this scheme. A lack of the Dsb proteins in the oxidative pathways in B. subtilis or Rhodobacter capsulatus suppresses the cytochrome c deficiency of ccmG or ccdA mutants (Erlendsson and Hederstedt, 2002; Deshmukh et al., 2003; Turkarslan et al., 2008). It was also demonstrated that heterologous expression of the CcsBC of H. pylori or CcsAB from Bordetella pertussis, which both encode cytochrome c synthetase, in E. coli lacking its own cytochrome c machinery results in c-type cytochrome formation. However, the observed effect was significantly enhanced by addition of exogenous reductant (Feissner et al., 2006; Goddard et al., 2010). Detailed analysis of the Paracoccus denitrificans cytochrome maturation led to the conclusion that apocytochrome undergoes a competing process of either heme attachment to, or oxidation of, its cysteine thiols (Mavridou et al., 2012).
Apocytochrome c Reduction in Helicobacter spp. and Wolinella spp.
Our knowledge about the cytochrome c biogenesis processes operating in Epsilonproteobacteria is still limited. There are significant differences among particular members of this class of bacteria, mainly with respect to proteins involved in electron transport from the cytoplasm to the periplasm (DsbD vs. CcdA) (Figure 2, Table S1).
The best described system is that operating in H. pylori cells. This microorganism uses HP0377 (a lipoprotein anchored in the cytoplasmic membrane of the cell) for apocytochrome c reduction. HP0377 is a thioredoxin-fold protein containing the CSYC motif, which indicates that it functions as a disulfide oxidoreductase. Although there is no direct evidence that HP0377 is involved in cytochrome c assembly in vivo, this is likely because its resolved structure is similar, but not identical, to that of other CcmG proteins and because it is able to reduce the oxidized form of apocytochrome c in vitro (Yoon et al., 2013). Additionally, hp0377 is co-transcribed with the ccsBA (hp0378) gene that is involved in heme transport and its ligation to apocytochrome c (Frawley and Kranz, 2009; Sharma et al., 2010). H. pylori or H. hepaticus CcsBA, together with E. coli DsbD/DsbC, can complement an E. coli ccm genes deletion (Goddard et al., 2010). Figure 6B presents the model of H. pylori cytochrome c biogenesis.
HP0377 (CcmG) is potentially re-reduced by HP0265 (CcdA). As H. pylori possess several proteins containing non-consecutive disulfide bonds and lacks the classical DsbC/DsbG, it was postulated that HP0377 is a multifunctional protein involved in the Dsb isomerization process, which contrasts with most CcmGs that are only involved in the cytochrome c maturation process. HP0377 is the first described CcmG protein having an acidic pKa for the N-terminal cysteine of the CXXC motif, similar to the pKa of EcDsbA or EcDsbC; at the same time, HP0377 presents a low redox potential characteristic of a reductant (Yoon et al., 2013). Undoubtedly, further biochemical, genetic and structural experiments are required to clarify the functioning of the atypical Dsb network of H. pylori.
According to our bioinformatics data, an identical cytochrome biogenesis system is potentially active in all of the Helicobacter genus members analyzed so far, where a CcmG protein that is homologous to HP0377 is responsible for apocytochrome c reduction. As the CXXC and cis-Pro motifs significantly influence the redox properties of the Dsb oxidoreductases, we compared the active sites of CXXC motifs present in various members of Helicobacter genus and found that they are strongly conserved (Figure 2, Table S1). All proteins retain Y as the second amino acid of the dipeptide and S as the amino acid most often present in the second position of the motif. Additionally, the H. pylori CcmG proteins are paired with TcP, as is seen for many other oxidoreductases acting as reductants. However, it should be emphasized that dipeptides of the CXXC present in other CcmGs are extremely divergent (Edeling et al., 2002). In all of the analyzed Helicobacter strains, the CcmG proteins are potentially re-reduced by CcdA, and all the strains encode large CcsBA proteins that potentially act as cytochrome c synthetase (Figure 2, Table S1).
There is only one genome of Wolinella spp. that has been sequenced so far. Its analysis indicates that W. succinogenes also uses the system II cytochrome biogenesis; however, it is more expanded than that of H. pylori. It encodes one homolog of HP0377 (CIYC paired with TcP motif) and two homologs of EcCcmG, with CPPC/IcP and CLSC/IcP motifs (Figure 2, Table S1). All of these homologs are potentially re-reduced by the CcdA protein. Wolinella also encodes three CcsBA-type CCHL isoenzymes (Nrfl, CcsA1, and CcsA2), which differ in their specificity for apocytochromes, as they recognize various heme c binding motifs (Kern et al., 2010). It is likely that the three CcmGs interact with specific apocytochrome c substrates. As Wolinella does not encode DsbC, it is likely that at least one of the CcmGs may be also involved in the disulfide bond isomerization process.
Apocytochrome c Reduction in Campylobacter spp. and Arcobacter spp.
Most members of the Campylobacter genus possess system II cytochrome c biogenesis systems. However, in contrast to the H. pylori the cytochrome c biogenesis, those systems resemble the cytochrome c biogenesis system present in B. pertussis as the CcmGs are re-reduced by DsbDs (Kranz et al., 2002; Feissner et al., 2005). In the proteomes of Campylobacter cells, heme is transported through the cytoplasmic membrane and ligated into cytochrome c by a large protein that resulted from the fusion of CcsA and CcsB, as is seen in all genera of Epsilonproteobacteria. In some strains, we found more than one CcsBA, which is also noticeable in the W. succinogenes proteome. The most curious observation derived from in silico analysis is the presence of putative CcmGs of different origins. In some strains, there is more than one gene encoding this protein per genome. The majority of Campylobacter CcmGs are annotated as TlpA. We noticed that they are related to TlpA (thioredoxin-like protein) and to CcmG, and they potentially belong to the TlpA/ResE/CcmG subfamily (Marchler-Bauer et al., 2015). Some members of C. coli species possess two CcmGs: one is related to the H. pylori CcmG (HP0377 in the H. pylori 26695 genome), and the second one is a member of the TlpA/ResE/CcmG subfamily. TlpA was identified for the first time in Bradyrhizobium japonicum, a nitrogen-fixing soil bacterium, as a Dsb protein involved in the biogenesis of apocytochrome aa3 (Loferer et al., 1993). Similar to the classical CcmG, it is anchored to the cytoplasmic membrane and its C-terminal domain contains a thioredoxin domain exposed to the periplasm. Its structure revealed several atypical features that are reflected in the biochemical properties of the protein (Capitani et al., 2001). Analysis of TlpA function in B. japonicum and Neisseria gonorrhoeae revealed that TlpA is a multifunctional oxidoreductase (Achard et al., 2009; Mohorko et al., 2012). In CcmG proteins the critical residues of CXXC motif fluctuate considerably. However, it should be noted that in most cases Campylobacter genus representatives retain the proline as a second amino acid of the XX dipeptide in CXXC motif and IcP motif. When more than one CcmG protein is present in most cases CPSC motif is paired with TcP and CGPC/CSPC motif is paired with IcP. This preliminary analysis suggest differences in the recognized substrates. Evidently, the function of CcmGs in Campylobacter spp. requires experimental investigation, as all bioinformatics predictions should be verified. The cytochrome biogenesis system II of Arcobacter spp. seems to act similarly to bacteria in the Campylobacter genus. However, all the CcmGs of Arcobacter spp. show homology to EcCcmG (Figure 2, Table S1).
Role of Epsilonproteobacteria Dsb Proteins in Pathogenesis, and Potential Therapeutic and Prophylactic Applications
The virulence of many bacterial pathogens depends on extracytoplasmic proteins, many of which contain two or more cysteine residues and achieve their final structure as a result of disulfide bond formation catalyzed by the Dsb proteins. Thus, inactivation of the Dsb system very often results in attenuation of bacterial pathogenicity (Raczko et al., 2005; Heras et al., 2009). Also, although members of Epsilonproteobacteria analyzed so far that are defective in the Dsb oxidative or isomerization pathways do not exhibit growth inhibition, they very often display attenuated virulence in animal models. The hp0595 mutated H. pylori revealed a greatly reduced ability to colonize mice gastric mucosa (Godlewska et al., 2006). Also, it was shown that CjDsbA1 (the main periplasmic oxidoreductase of C. jejuni) plays a crucial role in motility and autoagglutination, mechanisms that are necessary for the bacteria to colonize and spread within a host organism (Grabowska et al., 2014). Moreover, the inactivation of two membrane Campylobacter oxidoreductases (DsbB and DsbI) resulted in significant attenuation of chicken intestine colonization by C. coli 23, and also reduced the invasion/intracellular survival abilities (in the T84 cell line) of the C. jejuni 81-176 strain (Lasica et al., 2010).
The emergence of “new” bacterial pathogens and fast-spreading multidrug resistant strains underscores the urgent need to develop novel therapeutic agents, as well as novel diagnostic and prophylactic tools to fight infectious diseases. Broad knowledge about virulence mechanisms of bacterial pathogens and their interactions with the host, combined with advances in structural biology in the past decade, should help achieve this goal. It is generally accepted that virulence factors are significant targets for therapeutic drugs. Proteins of the Dsb system, which play a key role in the virulence of many pathogenic Gram-negative organisms, represent possible new drug targets (Heras et al., 2015). Their localization in the bacterial periplasm renders them easily accessible to potential small-molecule inhibitors. Furthermore, use of Dsb-protein inhibitors should not create selective pressure for developing resistance. Recently research on the Dsb system of E. coli has resulted in extensive knowledge about protein structure and mechanism of action, and that has enabled the design of specific inhibitors of Dsb protein activity (Duprez et al., 2015; Halili et al., 2015).
Conclusions
Comparison of the Dsb systems of C. jejuni and H. pylori–two well characterized members of Epsilonproteobacteria revealed enormous diversity of the mechanisms involved in oxidative protein folding. The functioning of the Arcobacter and Wolinella genera Dsb systems has so far been poorly understood. Given the importance of disulfide bond formation to achieve native protein structures and the diversity of disulfide bond formation oxidative pathways in bacteria, further research in this field will add to our biochemical and microbiological understanding of these pathways. Additionally, as proteins of the Dsb system play a key role in virulence, they represent possible new drug targets. Inhibition of the Dsb protein interaction with it protein substrates or redox partners could block the formation of virulence factors. An understanding of Epsilonproteobacteria Dsb protein structures and their activities may facilitate discovery of an effective drug against these important human pathogens.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by the National Science Centre (grant no. 2012/05/B/NZ1/00039). We would like to thank Dr. Jeffrey Hansen for critical reading of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00570/abstract
Table S1. Distribution of Dsb-like and associated proteins (with their accession numbers) in Arcobacter spp., Campylobacter spp., Helicobacter spp., and the Wolinella succinogenes DSM 1740 proteomes.
References
Achard, M. E., Hamilton, A. J., Dankowski, T., Heras, B., Schembri, M. S., Edwards, J. L., et al. (2009). A periplasmic thioredoxin-like protein plays a role in defense against oxidative stress in Neisseria gonorrhoeae. Infect. Immun. 77, 4934–4939. doi: 10.1128/IAI.00714-09
Arredondo, S. A., Chen, T. F., Riggs, A. F., Gilbert, H. F., and Georgiou, G. (2009). Role of dimerization in the catalytic properties of the Escherichia coli disulfide isomerase DsbC. J. Biol. Chem. 284, 23972–23979. doi: 10.1074/jbc.M109.010199
Baar, C., Eppinger, M., Raddatz, G., Simon, J., Lanz, C., Klimmek, O., et al. (2003). Complete genome sequence and analysis of Wolinella succinogenes. Proc. Natl. Acad. Sci. U.S.A. 100, 11690–11695. doi: 10.1073/pnas.1932838100
Bacon, D. J., Alm, R. A., Burr, D. H., Hu, L., Kopecko, D. J., Ewing, C. P., et al. (2000). Involvement of a plasmid in virulence of Campylobacter jejuni 81–176. Infect. Immun. 68, 4384–4390. doi: 10.1128/IAI.68.8.4384-4390.2000
Bacon, D. J., Alm, R. A., Hu, L., Hickey, T. E., Ewing, C. P., Batchelor, R. A., et al. (2002). DNA sequence and mutational analyses of the pVir plasmid of Campylobacter jejuni 81–176. Infect. Immun. 70, 6242–6250. doi: 10.1128/IAI.70.11.6242-6250.2002
Batchelor, R. A., Pearson, B. M., Friis, L. M., Guerry, P., and Wells, J. M. (2004). Nucleotide sequences and comparison of two large conjugative plasmids from different Campylobacter species. Microbiology 150, 3507–3517. doi: 10.1099/mic.0.27112-0
Berkmen, M. (2012). Production of disulfide-bonded proteins in Escherichia coli. Protein Expr. Purif. 82, 240–251. doi: 10.1016/j.pep.2011.10.009
Bernsel, A., Viklund, H., and Elofsson, A. (2008). Remote homology detection of integral membrane proteins using conserved sequence features. Proteins 71, 1387–1399. doi: 10.1002/prot.21825
Bernsel, A., Viklund, H., Hennerdal, A., and Elofsson, A. (2009). TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res. 37, W465–W468. doi: 10.1093/nar/gkp363
Bessette, P. H., Cotto, J. J., Gilbert, H. F., and Georgiou, G. (1999). In vivo and in vitro function of the Escherichia coli periplasmic cysteine oxidoreductase DsbG. J. Biol. Chem. 274, 7784–7792. doi: 10.1074/jbc.274.12.7784
Bocian, K. M., and Jagusztyn-Krynicka, E. K. (2012). The controversy over anti-Helicobacter pylori therapy. Pol. J. Microbiol. 61, 239–246.
Bonnard, G., Corvest, V., Meyer, E. H., and Hamel, P. P. (2010). Redox processes controlling the biogenesis of c-type cytochromes. Antioxid. Redox Signal. 13, 1385–1401. doi: 10.1089/ars.2010.3161
Buchan, D. W., Minneci, F., Nugent, T. C., Bryson, K., and Jones, D. T. (2013). Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357. doi: 10.1093/nar/gkt381
Capitani, G., Rossmann, R., Sargent, D. F., Grutter, M. G., Richmond, T. J., and Hennecke, H. (2001). Structure of the soluble domain of a membrane-anchored thioredoxin-like protein from Bradyrhizobium japonicum reveals unusual properties. J. Mol. Biol. 311, 1037–1048. doi: 10.1006/jmbi.2001.4913
Charbonnier, J. B., Belin, P., Moutiez, M., Stura, E. A., and Quemeneur, E. (1999). On the role of the cis-proline residue in the active site of DsbA. Protein Sci. 8, 96–105. doi: 10.1110/ps.8.1.96
Chivers, P. T., Laboissiere, M. C., and Raines, R. T. (1996). The CXXC motif: imperatives for the formation of native disulfide bonds in the cell. EMBO J. 15, 2659–2667.
Cho, S. H., Parsonage, D., Thurston, C., Dutton, R. J., Poole, L. B., Collet, J. F., et al. (2012). A new family of membrane electron transporters and its substrates, including a new cell envelope peroxiredoxin, reveal a broadened reductive capacity of the oxidative bacterial cell envelope. MBio 3:e00291-11 doi: 10.1128/mBio.00291-11
Cid, T. P., Fernandez, M. C., Benito Martinez, S., and Jones, N. L. (2013). Pathogenesis of Helicobacter pylori infection. Helicobacter 18(Suppl. 1), 12–17. doi: 10.1111/hel.12076
Colbert, C. L., Wu, Q., Erbel, P. J., Gardner, K. H., and Deisenhofer, J. (2006). Mechanism of substrate specificity in Bacillus subtilis ResA, a thioredoxin-like protein involved in cytochrome c maturation. Proc. Natl. Acad. Sci. U.S.A. 103, 4410–4415. doi: 10.1073/pnas.0600552103
Collado, L., and Figueras, M. J. (2011). Taxonomy, epidemiology, and clinical relevance of the genus Arcobacter. Clin. Microbiol. Rev. 24, 174–192. doi: 10.1128/CMR.00034-10
Cover, T. L., and Blaser, M. J. (2009). Helicobacter pylori in health and disease. Gastroenterology 136, 1863–1873. doi: 10.1053/j.gastro.2009.01.073
Daniels, R., Mellroth, P., Bernsel, A., Neiers, F., Normark, S., von Heijne, G., et al. (2010). Disulfide bond formation and cysteine exclusion in Gram-positive bacteria. J. Biol. Chem. 285, 3300–3309. doi: 10.1074/jbc.M109.081398
De Falco, M., Lucariello, A., Iaquinto, S., Esposito, V., Guerra, G., and De Luca, A. (2015). Molecular Mechanisms of Helicobacter pylori Pathogenesis. J. Cell. Physiol. 230, 1702–1707. doi: 10.1002/jcp.24933
Denoncin, K., and Collet, J. F. (2013). Disulfide bond formation in the bacterial periplasm: major achievements and challenges ahead. Antioxid. Redox Signal. 19, 63–71. doi: 10.1089/ars.2012.4864
Depuydt, M., Leonard, S. E., Vertommen, D., Denoncin, K., Morsomme, P., Wahni, K., et al. (2009). A periplasmic reducing system protects single cysteine residues from oxidation. Science 326, 1109–1111. doi: 10.1126/science.1179557
Depuydt, M., Messens, J., and Collet, J. F. (2011). How proteins form disulfide bonds. Antioxid. Redox Signal. 15, 49–66. doi: 10.1089/ars.2010.3575
Deshmukh, M., Turkarslan, S., Astor, D., Valkova-Valchanova, M., and Daldal, F. (2003). The dithiol:disulfide oxidoreductases DsbA and DsbB of Rhodobacter capsulatus are not directly involved in cytochrome c biogenesis, but their inactivation restores the cytochrome c biogenesis defect of CcdA-null mutants. J. Bacteriol. 185, 3361–3372. doi: 10.1128/JB.185.11.3361-3372.2003
Dugar, G., Herbig, A., Forstner, K. U., Heidrich, N., Reinhardt, R., Nieselt, K., et al. (2013). High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet. 9:e1003495. doi: 10.1371/journal.pgen.1003495
Duprez, W., Premkumar, L., Halili, M. A., Lindahl, F., Reid, R. C., Fairlie, D. P., et al. (2015). Peptide inhibitors of the Escherichia coli DsbA oxidative machinery essential for bacterial virulence. J. Med. Chem. 58, 577–587. doi: 10.1021/jm500955s
Dutton, R. J., Boyd, D., Berkmen, M., and Beckwith, J. (2008). Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. U.S.A. 105, 11933–11938. doi: 10.1073/pnas.0804621105
Edeling, M. A., Guddat, L. W., Fabianek, R. A., Thony-Meyer, L., and Martin, J. L. (2002). Structure of CcmG/DsbE at 1.14 A resolution: high-fidelity reducing activity in an indiscriminately oxidizing environment. Structure 10, 973–979. doi: 10.1016/S0969-2126(02)00794-3
EFSA. (2014). The European union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2012. EFSA J. 12, 3547–3859. doi: 10.2903/j.efsa.2014.3547
Erlendsson, L. S., Acheson, R. M., Hederstedt, L., and Le Brun, N. E. (2003). Bacillus subtilis ResA is a thiol-disulfide oxidoreductase involved in cytochrome c synthesis. J. Biol. Chem. 278, 17852–17858. doi: 10.1074/jbc.M300103200
Erlendsson, L. S., and Hederstedt, L. (2002). Mutations in the thiol-disulfide oxidoreductases BdbC and BdbD can suppress cytochrome c deficiency of CcdA-defective Bacillus subtilis cells. J. Bacteriol. 184, 1423–1429. doi: 10.1128/JB.184.5.1423-1429.2002
Erlendsson, L. S., Moller, M., and Hederstedt, L. (2004). Bacillus subtilis StoA Is a thiol-disulfide oxidoreductase important for spore cortex synthesis. J. Bacteriol. 186, 6230–6238. doi: 10.1128/JB.186.18.6230-6238.2004
Feissner, R. E., Beckett, C. S., Loughman, J. A., and Kranz, R. G. (2005). Mutations in cytochrome assembly and periplasmic redox pathways in Bordetella pertussis. J. Bacteriol. 187, 3941–3949. doi: 10.1128/JB.187.12.3941-3949.2005
Feissner, R. E., Richard-Fogal, C. L., Frawley, E. R., Loughman, J. A., Earley, K. W., and Kranz, R. G. (2006). Recombinant cytochromes c biogenesis systems I and II and analysis of haem delivery pathways in Escherichia coli. Mol. Microbiol. 60, 563–577. doi: 10.1111/j.1365-2958.2006.05132.x
Ferre, F., and Clote, P. (2005). DiANNA: a web server for disulfide connectivity prediction. Nucleic Acids Res. 33, W230–W232. doi: 10.1093/nar/gki412
Frawley, E. R., and Kranz, R. G. (2009). CcsBA is a cytochrome c synthetase that also functions in heme transport. Proc. Natl. Acad. Sci. U.S.A. 106, 10201–10206. doi: 10.1073/pnas.0903132106
Giovannelli, D., Ferriera, S., Johnson, J., Kravitz, S., Perez-Rodriguez, I., Ricci, J., et al. (2011). Draft genome sequence of Caminibacter mediatlanticus strain TB-2, an Epsilonproteobacterium isolated from a deep-sea hydrothermal vent. Stand. Genomic Sci. 5, 135–143. doi: 10.4056/sigs.2094859
Gleiter, S., and Bardwell, J. C. (2008). Disulfide bond isomerization in prokaryotes. Biochim. Biophys. Acta 1783, 530–534. doi: 10.1016/j.bbamcr.2008.02.009
Goddard, A. D., Stevens, J. M., Rondelet, A., Nomerotskaia, E., Allen, J. W., and Ferguson, S. J. (2010). Comparing the substrate specificities of cytochrome c biogenesis systems I and II: bioenergetics. FEBS J. 277, 726–737. doi: 10.1111/j.1742-4658.2009.07517.x
Godlewska, R., Dzwonek, A., Mikula, M., Ostrowski, J., Pawlowski, M., Bujnicki, J. M., et al. (2006). Helicobacter pylori protein oxidation influences the colonization process. Int. J. Med. Microbiol. 296, 321–324. doi: 10.1016/j.ijmm.2005.11.010
Grabowska, A. D., Wandel, M. P., Lasica, A. M., Nesteruk, M., Roszczenko, P., Wyszynska, A., et al. (2011). Campylobacter jejuni dsb gene expression is regulated by iron in a Fur-dependent manner and by a translational coupling mechanism. BMC Microbiol. 11:166. doi: 10.1186/1471-2180-11-166
Grabowska, A. D., Wywial, E., Dunin-Horkawicz, S., Lasica, A. M., Wosten, M. M., Nagy-Staron, A., et al. (2014). Functional and bioinformatics analysis of two Campylobacter jejuni homologs of the thiol-disulfide oxidoreductase, DsbA. PLoS ONE 9:e106247. doi: 10.1371/journal.pone.0106247
Grimshaw, J. P., Stirnimann, C. U., Brozzo, M. S., Malojcic, G., Grutter, M. G., Capitani, G., et al. (2008). DsbL and DsbI form a specific dithiol oxidase system for periplasmic arylsulfate sulfotransferase in uropathogenic Escherichia coli. J. Mol. Biol. 380, 667–680. doi: 10.1016/j.jmb.2008.05.031
Gruber, C. W., Cemazar, M., Heras, B., Martin, J. L., and Craik, D. J. (2006). Protein disulfide isomerase: the structure of oxidative folding. Trends Biochem. Sci. 31, 455–464. doi: 10.1016/j.tibs.2006.06.001
Gupta, R. S. (2006). Molecular signatures (unique proteins and conserved indels) that are specific for the epsilon Proteobacteria (Campylobacterales). BMC Genomics 7:167. doi: 10.1186/1471-2164-7-167
Halili, M. A., Bachu, P., Lindahl, F., Bechara, C., Mohanty, B., Reid, R. C., et al. (2015). Small molecule inhibitors of disulfide bond formation by the bacterial DsbA-DsbB dual enzyme system. ACS Chem. Biol. 10, 957–964. doi: 10.1021/cb500988r
Han, H., and Wilson, A. C. (2013). The two CcdA proteins of Bacillus anthracis differentially affect virulence gene expression and sporulation. J. Bacteriol. 195, 5242–5249. doi: 10.1128/JB.00917-13
Hatahet, F., Boyd, D., and Beckwith, J. (2014). Disulfide bond formation in prokaryotes: history, diversity and design. Biochim. Biophys. Acta 1844, 1402–1414. doi: 10.1016/j.bbapap.2014.02.014
Hatahet, F., and Ruddock, L. W. (2013). Topological plasticity of enzymes involved in disulfide bond formation allows catalysis in either the periplasm or the cytoplasm. J. Mol. Biol. 425, 3268–3276. doi: 10.1016/j.jmb.2013.04.034
Heras, B., Edeling, M. A., Schirra, H. J., Raina, S., and Martin, J. L. (2004). Crystal structures of the DsbG disulfide isomerase reveal an unstable disulfide. Proc. Natl. Acad. Sci. U.S.A. 101, 8876–8881. doi: 10.1073/pnas.0402769101
Heras, B., Scanlon, M. J., and Martin, J. L. (2015). Targeting virulence not viability in the search for future antibacterials. Br. J. Clin. Pharmacol. 79, 208–215. doi: 10.1111/bcp.12356
Heras, B., Shouldice, S. R., Totsika, M., Scanlon, M. J., Schembri, M. A., and Martin, J. L. (2009). DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 7, 215–225. doi: 10.1038/nrmicro2087
Heras, B., Totsika, M., Jarrott, R., Shouldice, S. R., Guncar, G., Achard, M. E., et al. (2010). Structural and functional characterization of three DsbA paralogues from Salmonella enterica serovar typhimurium. J. Biol. Chem. 285, 18423–18432. doi: 10.1074/jbc.M110.101360
Hiniker, A., and Bardwell, J. C. (2004). In vivo substrate specificity of periplasmic disulfide oxidoreductases. J. Biol. Chem. 279, 12967–12973. doi: 10.1074/jbc.M311391200
Hiniker, A., Ren, G., Heras, B., Zheng, Y., Laurinec, S., Jobson, R. W., et al. (2007). Laboratory evolution of one disulfide isomerase to resemble another. Proc. Natl. Acad. Sci. U.S.A. 104, 11670–11675. doi: 10.1073/pnas.0704692104
Hofreuter, D., Tsai, J., Watson, R. O., Novik, V., Altman, B., Benitez, M., et al. (2006). Unique features of a highly pathogenic Campylobacter jejuni strain. Infect. Immun. 74, 4694–4707. doi: 10.1128/IAI.00210-06
Inaba, K. (2008). Protein disulfide bond generation in Escherichia coli DsbB-DsbA. J. Synchrotron Radiat. 15, 199–201. doi: 10.1107/S090904950706061X
Inaba, K., and Ito, K. (2008). Structure and mechanisms of the DsbB-DsbA disulfide bond generation machine. Biochim. Biophys. Acta 1783, 520–529. doi: 10.1016/j.bbamcr.2007.11.006
Ito, K., and Inaba, K. (2008). The disulfide bond formation (Dsb) system. Curr. Opin. Struct. Biol. 18, 450–458. doi: 10.1016/j.sbi.2008.02.002
Jawad, Z., and Paoli, M. (2002). Novel sequences propel familiar folds. Structure 10, 447–454. doi: 10.1016/S0969-2126(02)00750-5
Kaakoush, N. O., Kovach, Z., and Mendz, G. L. (2007). Potential role of thiol:disulfide oxidoreductases in the pathogenesis of Helicobacter pylori. FEMS Immunol. Med. Microbiol. 50, 177–183. doi: 10.1111/j.1574-695X.2007.00259.x
Kadokura, H., Bader, M., Tian, H., Bardwell, J. C., and Beckwith, J. (2000). Roles of a conserved arginine residue of DsbB in linking protein disulfide-bond-formation pathway to the respiratory chain of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 97, 10884–10889. doi: 10.1073/pnas.97.20.10884
Kadokura, H., and Beckwith, J. (2002). Four cysteines of the membrane protein DsbB act in concert to oxidize its substrate DsbA. EMBO J. 21, 2354–2363. doi: 10.1093/emboj/21.10.2354
Kadokura, H., and Beckwith, J. (2009). Detecting folding intermediates of a protein as it passes through the bacterial translocation channel. Cell 138, 1164–1173. doi: 10.1016/j.cell.2009.07.030
Kadokura, H., Nichols, L. N., and Beckwith, J. (2005). Mutational alterations of the key cis proline residue that cause accumulation of enzymatic reaction intermediates of DsbA, a member of the thioredoxin superfamily. J. Bacteriol. 187, 1519–1522. doi: 10.1128/JB.187.4.1519-1522.2005
Kadokura, H., Tian, H., Zander, T., Bardwell, J. C., and Beckwith, J. (2004). Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science 303, 534–537. doi: 10.1126/science.1091724
Katzen, F., Deshmukh, M., Daldal, F., and Beckwith, J. (2002). Evolutionary domain fusion expanded the substrate specificity of the transmembrane electron transporter DsbD. EMBO J. 21, 3960–3969. doi: 10.1093/emboj/cdf405
Kern, M., Scheithauer, J., Kranz, R. G., and Simon, J. (2010). Essential histidine pairs indicate conserved haem binding in epsilonproteobacterial cytochrome c haem lyases. Microbiology 156, 3773–3781. doi: 10.1099/mic.0.042838-0
Kirkpatrick, B. D., and Tribble, D. R. (2011). Update on human Campylobacter jejuni infections. Curr. Opin. Gastroenterol. 27, 1–7. doi: 10.1097/MOG.0b013e3283413763
Korlath, J. A., Osterholm, M. T., Judy, L. A., Forfang, J. C., and Robinson, R. A. (1985). A point-source outbreak of campylobacteriosis associated with consumption of raw milk. J. Infect. Dis. 152, 592–596. doi: 10.1093/infdis/152.3.592
Kpadeh, Z. Z., Jameson-Lee, M., Yeh, A. J., Chertihin, O., Shumilin, I. A., Dey, R., et al. (2013). Disulfide bond oxidoreductase DsbA2 of Legionella pneumophila exhibits protein disulfide isomerase activity. J. Bacteriol. 195, 1825–1833. doi: 10.1128/JB.01949-12
Kranz, R. G., Beckett, C. S., and Goldman, B. S. (2002). Genomic analyses of bacterial respiratory and cytochrome c assembly systems: Bordetella as a model for the system II cytochrome c biogenesis pathway. Res. Microbiol. 153, 1–6. doi: 10.1016/S0923-2508(01)01278-5
Kranz, R. G., Richard-Fogal, C., Taylor, J. S., and Frawley, E. R. (2009). Cytochrome c biogenesis: mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol Mol Biol Rev 73, 510–528. Table of Contents. doi: 10.1128/MMBR.00001-09
Lafaye, C., Iwema, T., Carpentier, P., Jullian-Binard, C., Kroll, J. S., Collet, J. F., et al. (2009). Biochemical and structural study of the homologues of the thiol-disulfide oxidoreductase DsbA in Neisseria meningitidis. J. Mol. Biol. 392, 952–966. doi: 10.1016/j.jmb.2009.07.056
Landeta, C., Blazyk, J. L., Hatahet, F., Meehan, B. M., Eser, M., Myrick, A., et al. (2015). Compounds targeting disulfide bond forming enzyme DsbB of Gram-negative bacteria. Nat. Chem. Biol. 11, 292–298. doi: 10.1038/nchembio.1752
Lasica, A. M., Wyszynska, A., Szymanek, K., Majewski, P., and Jagusztyn-Krynicka, E. K. (2010). Campylobacter protein oxidation influences epithelial cell invasion or intracellular survival as well as intestinal tract colonization in chickens. J. Appl. Genet. 51, 383–393. doi: 10.1007/BF03208868
Lester, J., Kichler, S., Oickle, B., Fairweather, S., Oberc, A., Chahal, J., et al. (2015). Characterization of Helicobacter pylori HP0231 (DsbK): role in disulfide bond formation, redox homeostasis and production of Helicobacter cystein-rich protein HcpE. Mol. Microbiol. 96, 110–133. doi: 10.1111/mmi.12923
Lewin, A., Crow, A., Oubrie, A., and Le Brun, N. E. (2006). Molecular basis for specificity of the extracytoplasmic thioredoxin ResA. J. Biol. Chem. 281, 35467–35477. doi: 10.1074/jbc.M607047200
Li, W., Schulman, S., Dutton, R. J., Boyd, D., Beckwith, J., and Rapoport, T. A. (2010). Structure of a bacterial homologue of vitamin K epoxide reductase. Nature 463, 507–512. doi: 10.1038/nature08720
Linz, B., Balloux, F., Moodley, Y., Manica, A., Liu, H., Roumagnac, P., et al. (2007). An African origin for the intimate association between humans and Helicobacter pylori. Nature 445, 915–918. doi: 10.1038/nature05562
Loferer, H., Bott, M., and Hennecke, H. (1993). Bradyrhizobium japonicum TlpA, a novel membrane-anchored thioredoxin-like protein involved in the biogenesis of cytochrome aa3 and development of symbiosis. EMBO J. 12, 3373–3383.
Manning, G., Duim, B., Wassenaar, T., Wagenaar, J. A., Ridley, A., and Newell, D. G. (2001). Evidence for a genetically stable strain of Campylobacter jejuni. Appl. Environ. Microbiol. 67, 1185–1189. doi: 10.1128/AEM.67.3.1185-1189.2001
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI's conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Mavridou, D. A., Ferguson, S. J., and Stevens, J. M. (2012). The interplay between the disulfide bond formation pathway and cytochrome c maturation in Escherichia coli. FEBS Lett. 586, 1702–1707. doi: 10.1016/j.febslet.2012.04.055
McCarthy, A. A., Haebel, P. W., Torronen, A., Rybin, V., Baker, E. N., and Metcalf, P. (2000). Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli. Nat. Struct. Biol. 7, 196–199. doi: 10.1038/73295
McMahon, R. M., Premkumar, L., and Martin, J. L. (2014). Four structural subclasses of the antivirulence drug target disulfide oxidoreductase DsbA provide a platform for design of subclass-specific inhibitors. Biochim. Biophys. Acta 1844, 1391–1401. doi: 10.1016/j.bbapap.2014.01.013
Messens, J., and Collet, J. F. (2006). Pathways of disulfide bond formation in Escherichia coli. Int. J. Biochem. Cell Biol. 38, 1050–1062. doi: 10.1016/j.biocel.2005.12.011
Metheringham, R., Tyson, K. L., Crooke, H., Missiakas, D., Raina, S., and Cole, J. A. (1996). Effects of mutations in genes for proteins involved in disulphide bond formation in the periplasm on the activities of anaerobically induced electron transfer chains in Escherichia coli K12. Mol. Gen. Genet. 253, 95–102. doi: 10.1007/pl00013815
Miroshnichenko, M. L., and Bonch-Osmolovskaya, E. A. (2006). Recent developments in the thermophilic microbiology of deep-sea hydrothermal vents. Extremophiles 10, 85–96. doi: 10.1007/s00792-005-0489-5
Mohorko, E., Abicht, H. K., Buhler, D., Glockshuber, R., Hennecke, H., and Fischer, H. M. (2012). Thioredoxin-like protein TlpA from Bradyrhizobium japonicum is a reductant for the copper metallochaperone ScoI. FEBS Lett. 586, 4094–4099. doi: 10.1016/j.febslet.2012.10.026
Nakagawa, S., Takaki, Y., Shimamura, S., Reysenbach, A. L., Takai, K., and Horikoshi, K. (2007). Deep-sea vent epsilon-proteobacterial genomes provide insights into emergence of pathogens. Proc. Natl. Acad. Sci. U.S.A. 104, 12146–12150. doi: 10.1073/pnas.0700687104
Nothaft, H., Scott, N. E., Vinogradov, E., Liu, X., Hu, R., Beadle, B., et al. (2012). Diversity in the protein N-glycosylation pathways within the Campylobacter genus. Mol. Cell. Proteomics 11, 1203–1219. doi: 10.1074/mcp.M112.021519
Omasits, U., Ahrens, C. H., Muller, S., and Wollscheid, B. (2014). Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886. doi: 10.1093/bioinformatics/btt607
Otero, L. L., Ruiz, V. E., and Perez Perez, G. I. (2014). Helicobacter pylori: the balance between a role as colonizer and pathogen. Best Pract. Res. Clin. Gastroenterol. 28, 1017–1029. doi: 10.1016/j.bpg.2014.09.003
Palmer, S. R., Gully, P. R., White, J. M., Pearson, A. D., Suckling, W. G., Jones, D. M., et al. (1983). Water-borne outbreak of Campylobacter gastroenteritis. Lancet 1, 287–290. doi: 10.1016/S0140-6736(83)91698-7
Pan, J. L., Sliskovic, I., and Bardwell, J. C. (2008). Mutants in DsbB that appear to redirect oxidation through the disulfide isomerization pathway. J. Mol. Biol. 377, 1433–1442. doi: 10.1016/j.jmb.2008.01.058
Pearson, B. M., Gaskin, D. J., Segers, R. P., Wells, J. M., Nuijten, P. J., and van Vliet, A. H. (2007). The complete genome sequence of Campylobacter jejuni strain 81116 (NCTC11828). J. Bacteriol. 189, 8402–8403. doi: 10.1128/JB.01404-07
Porcelli, I., Reuter, M., Pearson, B. M., Wilhelm, T., and Van Vliet, A. H. (2013). Parallel evolution of genome structure and transcriptional landscape in the Epsilonproteobacteria. BMC Genomics 14:616. doi: 10.1186/1471-2164-14-616
Quan, S., Schneider, I., Pan, J., Von Hacht, A., and Bardwell, J. C. (2007). The CXXC motif is more than a redox rheostat. J. Biol. Chem. 282, 28823–28833. doi: 10.1074/jbc.M705291200
Raczko, A. M., Bujnicki, J. M., Pawlowski, M., Godlewska, R., Lewandowska, M., and Jagusztyn-Krynicka, E. K. (2005). Characterization of new DsbB-like thiol-oxidoreductases of Campylobacter jejuni and Helicobacter pylori and classification of the DsbB family based on phylogenomic, structural and functional criteria. Microbiology 151, 219–231. doi: 10.1099/mic.0.27483-0
Ren, G., Stephan, D., Xu, Z., Zheng, Y., Tang, D., Harrison, R. S., et al. (2009). Properties of the thioredoxin fold superfamily are modulated by a single amino acid residue. J. Biol. Chem. 284, 10150–10159. doi: 10.1074/jbc.M809509200
Rinaldi, F. C., Meza, A. N., and Guimaraes, B. G. (2009). Structural and biochemical characterization of Xylella fastidiosa DsbA family members: new insights into the enzyme-substrate interaction. Biochemistry 48, 3508–3518. doi: 10.1021/bi801899x
Rost, B., Yachdav, G., and Liu, J. (2004). The PredictProtein server. Nucleic Acids Res. 32, W321–W326. doi: 10.1093/nar/gkh377
Roszczenko, P., Radomska, K. A., Wywial, E., Collet, J. F., and Jagusztyn-Krynicka, E. K. (2012). A novel insight into the oxidoreductase activity of Helicobacter pylori HP0231 protein. PLoS ONE 7:e46563. doi: 10.1371/journal.pone.0046563
Rozhkova, A., and Glockshuber, R. (2008). Thermodynamic aspects of DsbD-mediated electron transport. J. Mol. Biol. 380, 783–788. doi: 10.1016/j.jmb.2008.05.050
Sambongi, Y., and Ferguson, S. J. (1996). Mutants of Escherichia coli lacking disulphide oxidoreductases DsbA and DsbB cannot synthesise an exogenous monohaem c-type cytochrome except in the presence of disulphide compounds. FEBS Lett. 398, 265–268. doi: 10.1016/S0014-5793(96)01256-2
Sanders, C., Turkarslan, S., Lee, D. W., and Daldal, F. (2010). Cytochrome c biogenesis: the Ccm system. Trends Microbiol. 18, 266–274. doi: 10.1016/j.tim.2010.03.006
Segatori, L., Paukstelis, P. J., Gilbert, H. F., and Georgiou, G. (2004). Engineered DsbC chimeras catalyze both protein oxidation and disulfide-bond isomerization in Escherichia coli: reconciling two competing pathways. Proc. Natl. Acad. Sci. U.S.A. 101, 10018–10023. doi: 10.1073/pnas.0403003101
Segura-Lopez, F. K., Guitron-Cantu, A., and Torres, J. (2015). Association between Helicobacter spp. infections and hepatobiliary malignancies: a review. World J. Gastroenterol. 21, 1414–1423. doi: 10.3748/wjg.v21.i5.1414
Sharma, C. M., Hoffmann, S., Darfeuille, F., Reignier, J., Findeiss, S., Sittka, A., et al. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255. doi: 10.1038/nature08756
Shouldice, S. R., Heras, B., Walden, P. M., Totsika, M., Schembri, M. A., and Martin, J. L. (2011). Structure and function of DsbA, a key bacterial oxidative folding catalyst. Antioxid. Redox Signal. 14, 1729–1760. doi: 10.1089/ars.2010.3344
Silva, J., Leite, D., Fernandes, M., Mena, C., Gibbs, P. A., and Teixeira, P. (2011). Campylobacter spp. as a foodborne pathogen: a review. Front. Microbiol. 2:200. doi: 10.3389/fmicb.2011.00200
Simon, J., and Hederstedt, L. (2011). Composition and function of cytochrome c biogenesis system II. FEBS J 278, 4179–4188. doi: 10.1111/j.1742-4658.2011.08374.x
Stirnimann, C. U., Grutter, M. G., Glockshuber, R., and Capitani, G. (2006). nDsbD: a redox interaction hub in the Escherichia coli periplasm. Cell. Mol. Life Sci. 63, 1642–1648. doi: 10.1007/s00018-006-6055-1
Tapley, T. L., Eichner, T., Gleiter, S., Ballou, D. P., and Bardwell, J. C. (2007). Kinetic characterization of the disulfide bond-forming enzyme DsbB. J. Biol. Chem. 282, 10263–10271. doi: 10.1074/jbc.M611541200
Thony-Meyer, L., and Kunzler, P. (1997). Translocation to the periplasm and signal sequence cleavage of preapocytochrome c depend on sec and lep, but not on the ccm gene products. Eur. J. Biochem. 246, 794–799. doi: 10.1111/j.1432-1033.1997.t01-1-00794.x
Totsika, M., Heras, B., Wurpel, D. J., and Schembri, M. A. (2009). Characterization of two homologous disulfide bond systems involved in virulence factor biogenesis in uropathogenic Escherichia coli CFT073. J. Bacteriol. 191, 3901–3908. doi: 10.1128/JB.00143-09
Turkarslan, S., Sanders, C., Ekici, S., and Daldal, F. (2008). Compensatory thio-redox interactions between DsbA, CcdA and CcmG unveil the apocytochrome c holdase role of CcmG during cytochrome c maturation. Mol. Microbiol. 70, 652–666. doi: 10.1111/j.1365-2958.2008.06441.x
Viklund, H., Bernsel, A., Skwark, M., and Elofsson, A. (2008). SPOCTOPUS: a combined predictor of signal peptides and membrane protein topology. Bioinformatics 24, 2928–2929. doi: 10.1093/bioinformatics/btn550
WHO. (1994). Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monogr. Eval. Carcinog. Risks Hum. 61, 1–241.
Yamaoka, Y., and Graham, D. Y. (2014). Helicobacter pylori virulence and cancer pathogenesis. Future Oncol. 10, 1487–1500. doi: 10.2217/fon.14.29
Yoon, J. Y., Kim, J., An, D. R., Lee, S. J., Kim, H. S., Im, H. N., et al. (2013). Structural and functional characterization of HP0377, a thioredoxin-fold protein from Helicobacter pylori. Acta Crystallogr. D Biol. Crystallogr. 69, 735–746. doi: 10.1107/S0907444913001236
Yoon, J. Y., Kim, J., Lee, S. J., Kim, H. S., Im, H. N., Yoon, H. J., et al. (2011). Structural and functional characterization of Helicobacter pylori DsbG. FEBS Lett. 585, 3862–3867. doi: 10.1016/j.febslet.2011.10.042
Keywords: Dsb proteins, disulfide bonds, cytochrome c biogenesis, Epsilonproteobacteria, Campylobacter, Helicobacter, Arcobacter, Wolinella
Citation: Bocian-Ostrzycka KM, Grzeszczuk MJ, Dziewit L and Jagusztyn-Krynicka EK (2015) Diversity of the Epsilonproteobacteria Dsb (disulfide bond) systems. Front. Microbiol. 6:570. doi: 10.3389/fmicb.2015.00570
Received: 18 March 2015; Accepted: 24 May 2015;
Published: 09 June 2015.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
Joachim Reidl, Karl-Franzens-Universität Graz, AustriaCarole Creuzenet, University of Western Ontario, Canada
Copyright © 2015 Bocian-Ostrzycka, Grzeszczuk, Dziewit and Jagusztyn-Krynicka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elżbieta K. Jagusztyn-Krynicka, Department of Bacterial Genetics, Faculty of Biology, Institute of Microbiology, University of Warsaw, Miecznikowa, 1 str. 02-096 Warsaw, Poland, kjkryn@biol.uw.edu.pl
†These authors have contributed equally to this work.