 Bernd Wemheuer1†
Bernd Wemheuer1† Franziska Wemheuer2†
Franziska Wemheuer2† Jacqueline Hollensteiner1Frauke-Dorothee Meyer1
Jacqueline Hollensteiner1Frauke-Dorothee Meyer1 Sonja Voget1
Sonja Voget1 Rolf Daniel1*
Rolf Daniel1*- 1Genomic and Applied Microbiology and Göttingen Genomics Laboratory, Institute of Microbiology and Genetics, Georg-August-University Göttingen, Göttingen, Germany
- 2Department for Crop Sciences, Georg-August-University Göttingen, Göttingen, Germany
Phytoplankton blooms exhibit a severe impact on bacterioplankton communities as they change nutrient availabilities and other environmental factors. In the current study, the response of a bacterioplankton community to a Phaeocystis globosa spring bloom was investigated in the southern North Sea. For this purpose, water samples were taken inside and reference samples outside of an algal spring bloom. Structural changes of the bacterioplankton community were assessed by amplicon-based analysis of 16S rRNA genes and transcripts generated from environmental DNA and RNA, respectively. Several marine groups responded to bloom presence. The abundance of the Roseobacter RCA cluster and the SAR92 clade significantly increased in bloom presence in the total and active fraction of the bacterial community. Functional changes were investigated by direct sequencing of environmental DNA and mRNA. The corresponding datasets comprised more than 500 million sequences across all samples. Metatranscriptomic data sets were mapped on representative genomes of abundant marine groups present in the samples and on assembled metagenomic and metatranscriptomic datasets. Differences in gene expression profiles between non-bloom and bloom samples were recorded. The genome-wide gene expression level of Planktomarina temperata, an abundant member of the Roseobacter RCA cluster, was higher inside the bloom. Genes that were differently expressed included transposases, which showed increased expression levels inside the bloom. This might contribute to the adaptation of this organism toward environmental stresses through genome reorganization. In addition, several genes affiliated to the SAR92 clade were significantly upregulated inside the bloom including genes encoding for proteins involved in isoleucine and leucine incorporation. Obtained results provide novel insights into compositional and functional variations of marine bacterioplankton communities as response to a phytoplankton bloom.
Introduction
Bacteria are major drivers in cycling of nitrogen, carbon, and other elements in marine ecosystems (Azam et al., 1983; Arrigo, 2005; DeLong and Karl, 2005). More than 50% of organic matter produced by phytoplankton is remineralized by marine bacteria (Cole et al., 1988; Karner and Herndl, 1992; Ducklow et al., 1993). Therefore, bacteria play an important role during and after bloom events as large amounts of organic matter are generated by primary production (Azam, 1998).
Recent studies investigating bacterioplankton communities during phytoplankton blooms revealed that community structures and diversity were highly affected (Teeling et al., 2012; Liu et al., 2013; Wemheuer et al., 2014). Observed patterns were correlated to changes of nutrient concentrations and other environmental factors such as water depth or algal species (Fandino et al., 2001; Pinhassi et al., 2004; Grossart et al., 2005; Teeling et al., 2012; Liu et al., 2013; Wemheuer et al., 2014; Gomes et al., 2015). Consequently, understanding the dynamics and interactions between bacterial communities and phytoplankton blooms is crucial to validate the ecological impact of bloom events.
One region with annually recurring spring phytoplankton blooms is the North Sea, a typical coastal shelf sea of the temperate zone. Shelf seas are highly productive due to the continuous nutrient supply by rivers. During the last 40 years, the North Sea and in particular its southern region, the German Bight, underwent high nutrient loading and warming (McQuatters-Gollop et al., 2007; Wiltshire et al., 2008, 2010). Recent studies aimed at understanding bacterial responses to phytoplankton blooms in the North Sea (Alderkamp et al., 2006; Teeling et al., 2012; Wemheuer et al., 2014). A dynamic succession of distinct bacterial clades before, during, and after bloom events in the North Sea was observed in several investigations (Alderkamp et al., 2006; Alonso and Pernthaler, 2006a,b; Teeling et al., 2012). The results indicate that specialized populations occupy ecological niches provided by phytoplankton-derived substrates (Teeling et al., 2012). Klindworth et al. (2014) investigated the diversity and activity of marine bacterioplankton during the same bloom event applying metatranscriptomic techniques. They showed that members of the Rhodobacteraceae and SAR92 clade exhibited high metabolic activity levels. However, recent research focused mainly on changes of community structure as response to phytoplankton blooms, but functional changes and their resulting ecological impacts have been rarely studied. In addition, larger comparative metagenomic and metatranscriptomic studies investigating structural and functional changes of the bacterioplankton during the bloom event are lacking.
In a previous study, we investigated structural differences of the active bacterioplankton community as response toward a Phaeocystis globosa bloom in the southern North Sea in spring 2010 mainly by 16S rRNA pyrotag sequencing (Wemheuer et al., 2014). This microalgae has a cosmopolitan distribution (Schoemann et al., 2005) and is considered to be responsible for harmful algal blooms (Veldhuis and Wassmann, 2005). These blooms have been observed in many marine environments, including the coast of the eastern English Channel, the southern North Sea and the south coast of China (Schoemann et al., 2005). We found that the phytoplankton spring bloom impacted bacterioplankton community structures and the abundance of certain bacterial groups significantly. For example, the Roseobacter RCA cluster and the SAR92 clade were significantly more abundant in the bloom at active community level. For the current study, more than 500 million sequences derived from direct sequencing of environmental DNA and rRNA depleted RNA were added to obtain functional insights into the bloom event. Metatranscriptomic data was mapped on the assembled metagenomic and metatranscriptomic data sets and on the genomes of abundant marine bacteria, e.g., P. temperata RCA23, a member of the Roseobacter RCA cluster. In addition, 16S rRNA genes and transcripts were studied by pyrotag sequencing to obtain insights into structural dynamics of the total and active bacterioplankton community, respectively. The comprehensive experimental design and method combination of this study sheds new light on ecological roles and functions of single members of the bacterioplankton community and the entire community.
Materials and Methods
Sampling and Sample Preparation
Ten water samples for bacterioplankton analyses were collected in the southern North Sea at nine stations in and outside of a P. globosa bloom in May 2010 (Figure 1; Table 1). Six samples were taken in the presence of a phytoplankton bloom (3a, 3b, and 4) and three in bloom absence (5–15). One sample was taken near to a river outfall (1). Stations inside the bloom were located by satellite images and are characterized by their increased chlorophyll content. Note that sample 9 was taken in the bloom area and is considered as a bloom sample despite its relative low chlorophyll content. Sampling and filtration were performed as described previously (Wemheuer et al., 2014). In brief, obtained water samples were initially filtered using a 10 μm nylon net filter and 2.7 μm glass fiber filter. Bacterioplankton was subsequently harvested from a prefiltered 1 l sample on a filter sandwich consisting of a glass fiber and 0.2 μm polycarbonate filter (47 mm diameter). Samples for community analysis were stored at −80°C until further analysis. Several environmental parameters such as chlorophyll a (Chl a), particulate organic nitrogen (PON), salinity, temperature, and nitrate content were determined as described previously (Wemheuer et al., 2014) (Table 2).
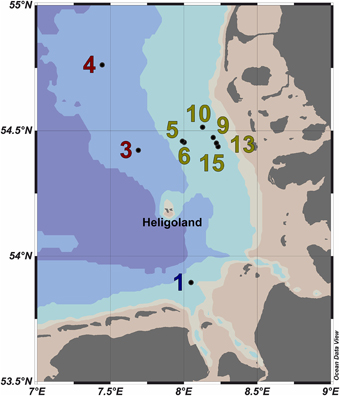
Figure 1. Map of the German Bight showing the locations of the nine sampling stations visited in May 2010. Stations inside the examined phytoplankton bloom are depicted in green; those outside the bloom in red. Station 1 is depicted in blue as it was located in a bloom outside of the area of the examined bloom. Shading of the water masses refers to bottom depth. The map was generated using the Ocean Data View software package [version 4.7.2; Schlitzer, 2015 (http://odv.awi.de/)].
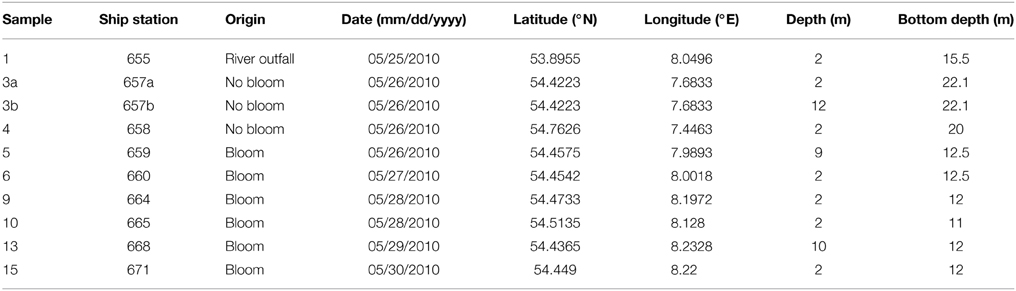
Table 1. Sampling site characteristics.
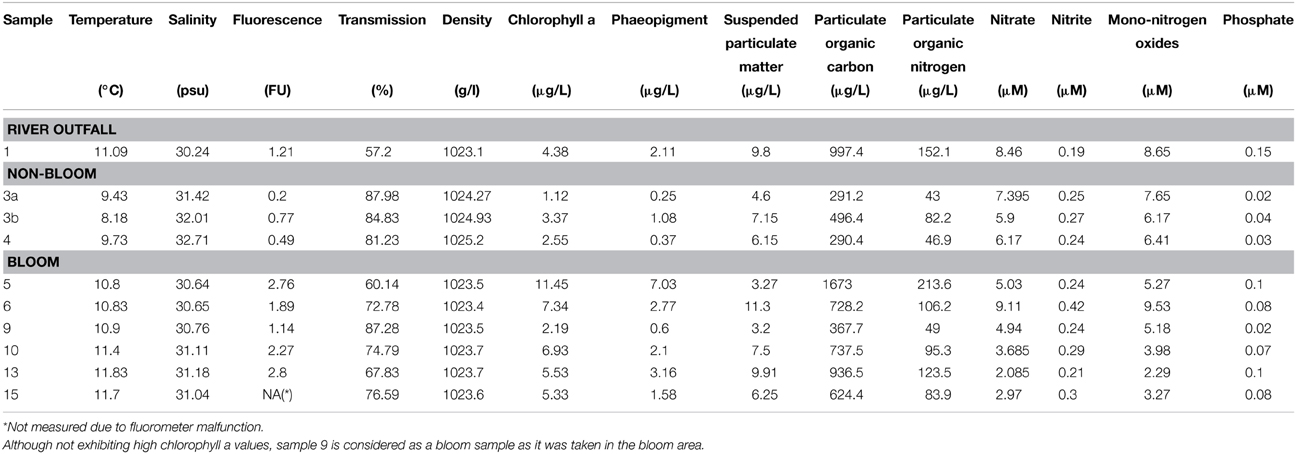
Table 2. Environmental parameters measured for the 10 water samples.
Extraction and Purification of Environmental DNA and RNA
Environmental DNA and RNA were co-extracted from the filter sandwich as described by Weinbauer et al. (2002). DNA and RNA were subsequently purified employing the peqGOLD gel extraction kit (Peqlab, Erlangen, Germany) and the RNeasy Mini Kit (Qiagen, Hilden, Germany), respectively, as recommended by the manufacturers. Residual DNA was removed from RNA samples and its absence was confirmed according to Wemheuer et al. (2012).
To assess bacterioplankton community structures, DNA-free RNA was directly converted to cDNA employing the SuperScript® III reverse transcriptase (Invitrogen™, Carlsbad, USA) using a primer specific for the conserved region downstream to variable region 6 of the 16S rRNA (1063r 5′-CTCACGRCACGAGCTGACG-3′). The reaction mixture (20 μl) contained 4 μl of five-fold reaction buffer, 500 μM of each of the four desoxynucleoside triphosphates, 5 mM DTT, 1 μM of the reverse primer, 1 U RiboLock™ RNase Inhibitor (Thermo Fisher Scientific, Schwerte, Germany), 200 U of the reverse transcriptase and approximately 100 ng DNA-free RNA. The reaction was incubated at 55°C for 1 h and subsequently inactivated by incubation at 70°C for 15 min. To remove the RNA in the RNA/DNA hybrids, 2.5 U RNase H (Thermo Fischer Scientific) were added and the reaction incubated at 37° for 15 min followed by inactivation at 65°C for 10 min. Obtained cDNA was subsequently subjected to 16S rRNA gene PCR (as described below). To assess community functions, environmental mRNA was enriched from total RNA using the RiboMinus™ transcriptome isolation kit for Bacteria (Invitrogen™, Carlsbad, USA) with one modification. The initial denaturation of RNA was performed at 70°C for 10 min. RNA was subsequently converted to cDNA employing the SuperScript™ double-stranded cDNA synthesis kit (Invitrogen™) with slight modifications according to Wemheuer et al. (2014). The Göttingen Genomics Laboratory determined the sequences of the extracted DNA and enriched mRNA-derived cDNA using a Roche 454 GS-FLX+ pyrosequencer with titanium chemistry (Roche, Mannheim, Germany) and an Illumina Genome Analyzer IIx (San Diego, USA), respectively (Table 3).
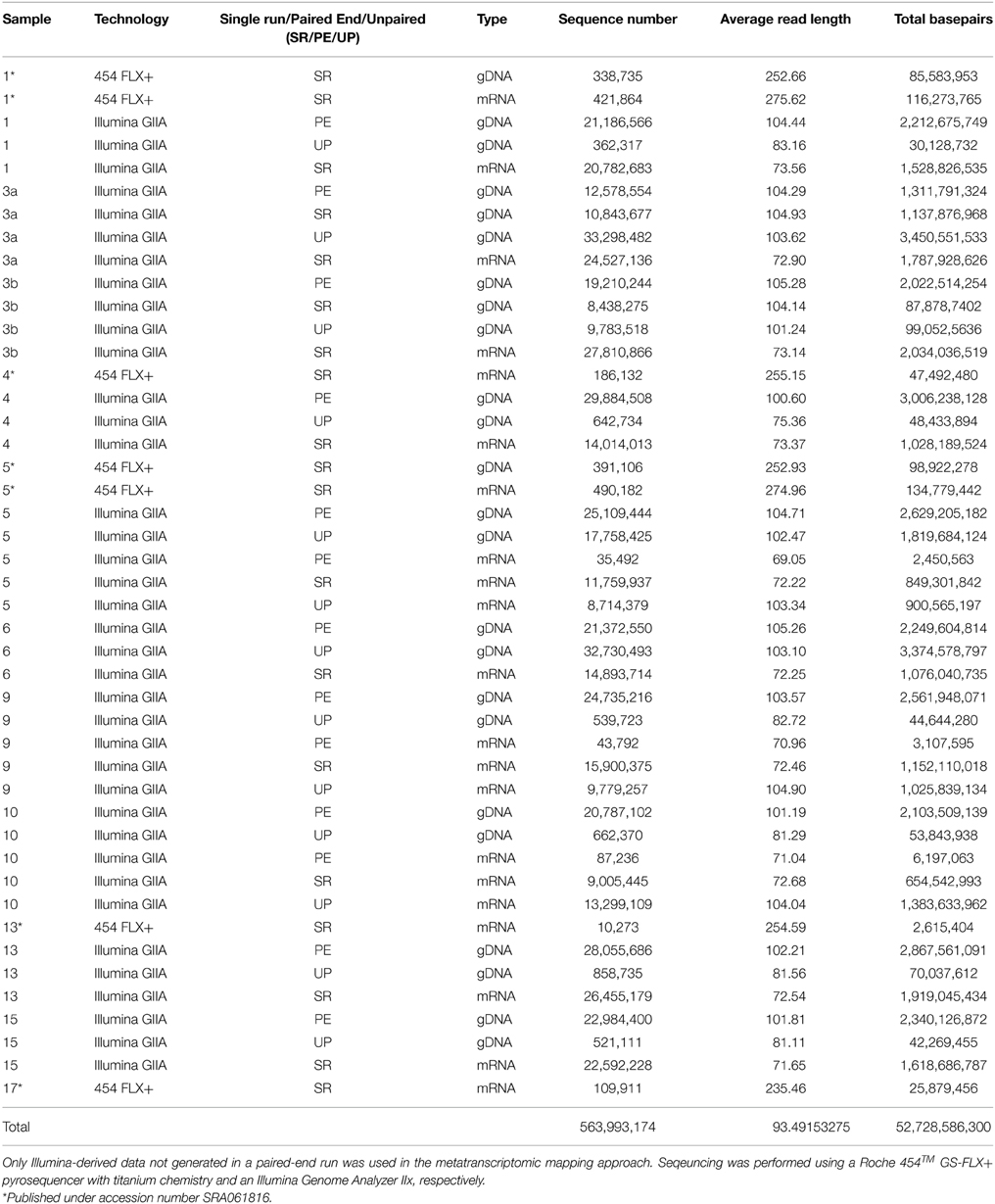
Table 3. Sequence statistics of the quality trimmed metagenome and metatranscriptome data used in this study.
Processing and Analysis of Metagenomic and Metatranscriptomic Datasets
Generated metagenomic and metatranscriptomic datasets were initially processed according to Voget et al. (2014). Briefly, fastq files derived from Illumina sequencing were processed employing the Trimmomatic tool version 0.30 (Bolger et al., 2014). Sff files derived from pyrosequencing were converted to fastq files prior to quality filtering. Afterwards, all sequences were combined and assembled at different kmer values (29–109 in 10 bp steps) with Velvet and Metavelvet (Zerbino and Birney, 2008; Namiki et al., 2012). Subsequently, all obtained contigs were joined and resulting sequences were dereplicated employing Usearch version 7.0.190 (Edgar, 2010). Open reading frames (ORFs) were predicted for all remaining contigs using Prodigal version 2.6 (Hyatt et al., 2010). Short contigs (< 150 bp) were removed prior to further analysis.
As the metagenomic and metatranscriptromic datasets are likely to contain algal-derived sequences, we subtracted bacterial genes by blast alignment (Camacho et al., 2009) against 15 reference genomes of abundant marine lineages (Table 4) obtained from the integrated microbial genomes (IMG) platform (Markowitz et al., 2012). Genomes of abundant phylogenetic groups as found in the 16S rRNA analysis were chosen for this additional filtering step. Only sequences with an e-value below 0.001 were used in the subsequent analysis. Remaining ORFs were further classified employing UProC version 1.2 in protein mode (Meinicke, 2015).
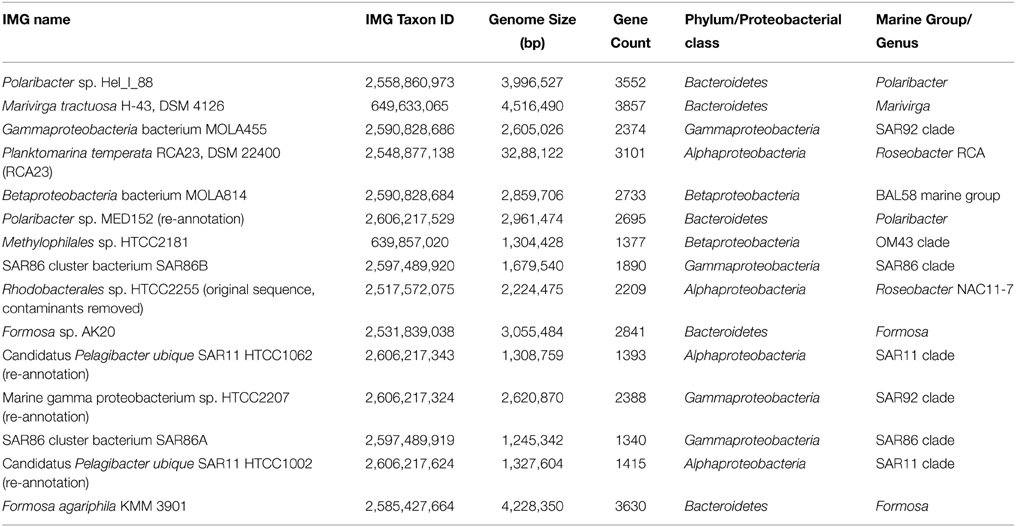
Table 4. Genomes retrieved from the Integrated Microbial Genomes (IMG) database.
Metatranscriptomic datasets were mapped on the 15 genomes and on the assembled contigs using Bowtie 2 version 2.2.4 (Langmead and Salzberg, 2012) with one mismatch in the seed and multiple hits reporting enabled for the metagenomic binning. Ribosomal RNA was removed from metatranscriptomic datasets prior to mapping employing SortMeRNA version 2.0 (Kopylova et al., 2012) (Table 5). The number of unique sequences per gene was calculated, and the overall mapping result was normalized by the total number of unique reads in the sample. The top 23,884 open reading frames based on number of assigned, normalized reads are provided in Supplementary Table S1.
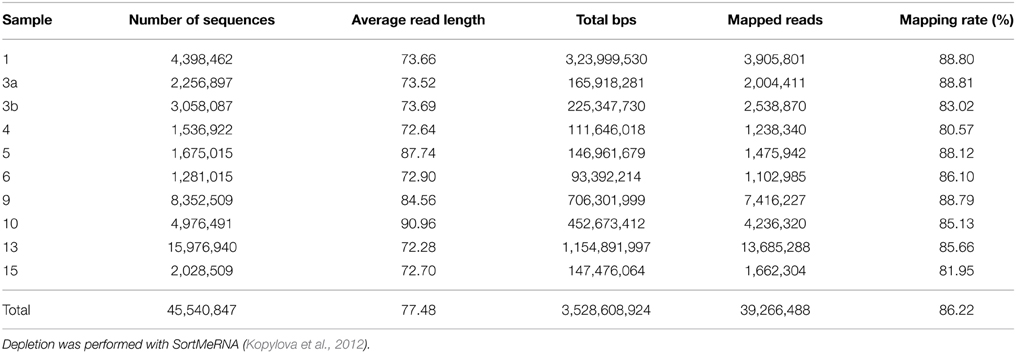
Table 5. Sequence statistics of the quality trimmed and rRNA depleted metatranscriptomic data sets used for mapping.
Results from the genome mapping were additionally normalized by the unique RNA/DNA ratio calculated for each bacterial group in the respective sample (see Campbell and Kirchman, 2013). In detail, the ratio was calculated by dividing the relative abundance of a 16S rRNA transcript by the relative abundance of the corresponding 16S rRNA gene. On the one hand, assuming that the major fraction of the bacterioplankton community is present outside and inside the algal bloom, the abundance at DNA level of single species is mainly linked to cell abundance rather than other factors such as 16S rRNA gene copy number. On the other hand, RNA abundance is correlated with protein synthesis (Blazewicz et al., 2013) and may indirectly serve as approximation for gene expression levels. Therefore, a high RNA/DNA ratio reflects an increased gene expression per cell and vice versa.
Amplification and Sequencing of 16S rRNA
To assess bacterial community structures, the V3–V6 region of the bacterial 16S rRNA was amplified by PCR. The PCR reaction (50 μl) contained 10 μl of five-fold Phusion HF buffer, 200 μM of each of the four desoxynucleoside triphosphates, 1.5 mM MgCl2, 4 μM of each primer, 2.5% DMSO, 2 U of Phusion high fidelity hot start DNA polymerase (Thermo Fisher Scientific), and approximately 50 ng of DNA or 25 ng of cDNA as template. The following thermal cycling scheme was used: initial denaturation at 98°C for 5 min, 25 cycles of denaturation at 98°C for 45 s, annealing at 60°C for 45 s, followed by extension at 72°C for 30 s. The final extension was carried out at 72°C for 5 min. Negative controls were performed by using the reaction mixture without template. The V3–V6 region was amplified with the following set of primers according to Muyzer et al. (1995) containing the Roche 454 pyrosequencing adaptors, keys and one unique MID per sample (underlined): 341f 5′-CCATCTCATCCCTGCGTG TCTCCGAC-TCAG-(dN)10-CCT ACGGRAGGCAGCAG-3′ and 1063r 5′-CCT ATCCCCTGTGTGCCTTGGCAGTC-TCAG-CTCACGRCACGAGCTGAC G-3′. Obtained PCR products were controlled for appropriate size and subsequently purified using the peqGOLD gel extraction kit (Peqlab) as recommended by the manufacturer. Three independent PCR reactions were performed per sample, purified by gel extraction, and pooled in equal amounts. Quantification of the PCR products was performed using the Quant-iT dsDNA HS assay kit and a Qubit fluorometer (Invitrogen™) as recommended by the manufacturer. The Göttingen Genomics Laboratory determined the sequences using a Roche GS-FLX++ 454 pyrosequencer with Titanium chemistry (Roche, Mannheim, Germany).
Processing and Analysis of 16S rRNA Datasets
Generated 16S rRNA gene and rRNA datasets were processed as described by Wietz et al. (2015). In brief, sequences were preprocessed with QIIME and subsequently denoised employing Acacia (Bragg et al., 2012). Remaining primer sequences were truncated employing cutadapt (Martin, 2011). To remove chimeras, sequences were first dereplicated and putative chimeras were removed using UCHIME in de novo mode and subsequently in reference mode using the most recent SILVA database (SSURef 119 NR) as reference dataset (Edgar et al., 2011; Quast et al., 2013). Processed sequences of all samples were joined and clustered in operational taxonomic units (OTUs) at 3 and 20% genetic dissimilarity according to Wemheuer et al. (2013) employing the UCLUST algorithm with optimal flag (Edgar, 2010). To determine taxonomy, a consensus sequence for each OTU at 97% genetic similarity was classified by BLAST alignment against the Silva SSURef 119 NR database (Camacho et al., 2009). All non-bacterial OTUs were removed. Sequences statistics are shown in Table 6. The curated OTU table is provided as Supplemental Table S2. The final Alpha diversity indices were calculated with QIIME as described by Wemheuer et al. (2013) (see Table 7).
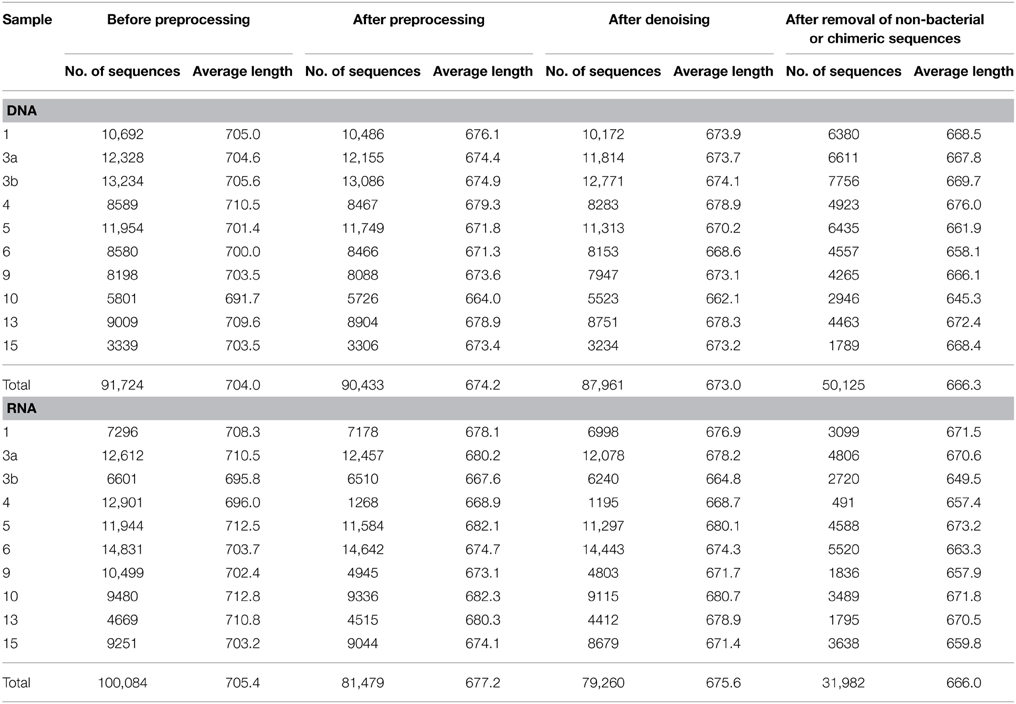
Table 6. Statistics of the 16S rRNA analysis.
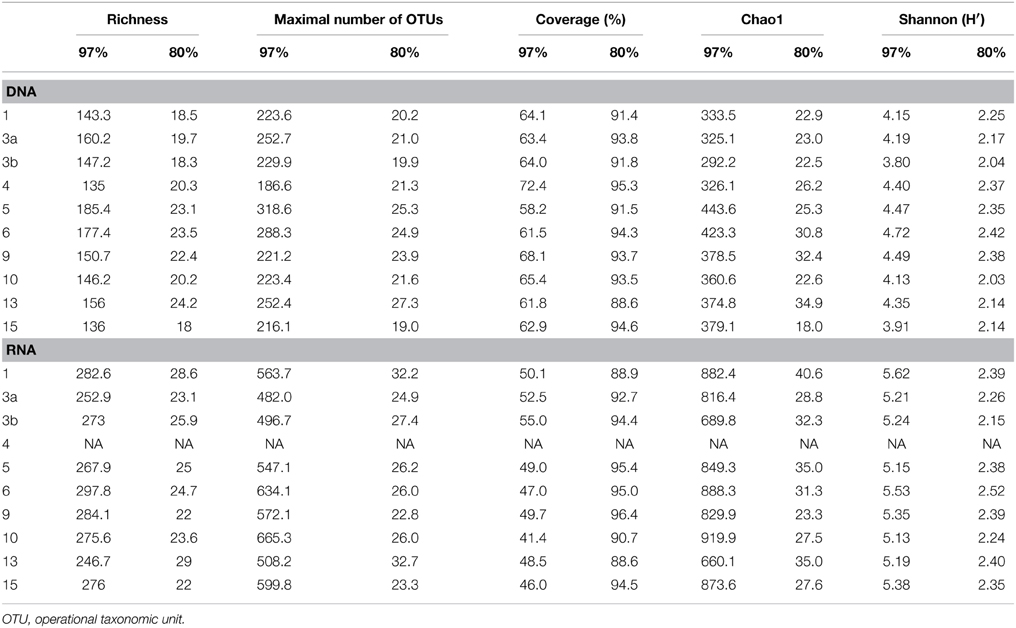
Table 7. Alpha diversity indices at 97 and 80% genetic similarity derived from the 16S rRNA analysis.
Statistical Analysis
All statistical analyses were conducted employing R [version 3.1.2; R Core Team, 2014 (http://www.R-project.org/]. Possible correlations between phytoplankton bloom presence and richness (number of OTUs) as well Shannon indices, abundance, and gene expression were determined employing the non-parametric Wilcox rank-sum test (Gifford et al., 2013). Correlations were considered as significant with P ≤ 0.05. Sample 1 was excluded from the statistical analysis because it was taken in another bloom event.
Sequence Data Deposition
Sequence data were deposited in the sequence read archive of the National Center for Biotechnology Information under accession numbers SRA061816 and SRA060677, respectively (for details see Table 3).
Results and Discussion
Characteristics of the Samples
In the current survey, we examined structural and functional responses of the bacterioplankton community toward a phytoplankton bloom. Samples for community analysis were taken randomly at different locations and different depths within a P. globosa bloom in the German Bight (Figure 1, Table 1). Six samples were taken in presence of the phytoplankton bloom (samples 5, 6, 9, 10, 13, and 15) and three in bloom absence (samples 3a, 3b, and 4). One sample was taken near the Weser river outfall (sample 1). Salinity ranged from 30.7 to 32.7 psu. Fluorescence was approximately 0.45 and 2.2 mg/m3 outside and inside the algal bloom, respectively. Temperatures ranged from 8.2 to 11.8°C. All environmental parameters are listed in Table 2. Based on our previous analysis, most measured parameters were significantly linked to algal bloom presence (see Wemheuer et al., 2014). Only the suspended particulate matter content (SPM) and the nitrite concentration exhibited no direct correlation to bloom presence.
Bloom Presence Affects Bacterial Community Structures
Total and active bacterioplankton community structures were assessed by pyrosequencing-based analysis of the V3–V6 region of the 16S rRNA amplified from environmental DNA and RNA, respectively. A total of 50,125 and 31,982 high-quality bacterial 16S rRNA sequences were obtained across all 10 samples at DNA and RNA level, respectively (Table 6). Calculated rarefaction curves (data not shown) as well as diversity indices revealed that the majority of the bacterial community was recovered by the surveying effort (Table 7).
Classification of the obtained 16S rRNA sequences revealed that Proteobacteria and Bacteroidetes were the most abundant bacterial phyla across all samples (approximately 78% and 20%, respectively). At higher taxonomic resolution, the majority of the obtained sequences was affiliated to 17 bacterial groups, clades, and genera (Figure 2). These groups represented different lineages within the Alpha-, Beta-, and Gammaproteobacteria and the Bacteroidetes. These results are in accordance with our previous study (Wemheuer et al., 2014) and recent investigations of bacterial communities in the North Sea (Alderkamp et al., 2006; Sapp et al., 2007; Teeling et al., 2012). However, in our previous study, the number of Bacteroidetes was rather low which can be attributed to the differences in primer pairs and variable regions of the 16S rRNA gene used in our previous study.
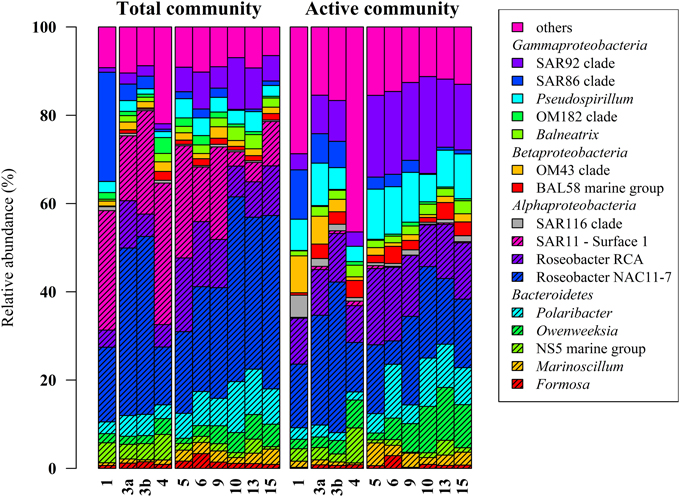
Figure 2. Relative distribution of abundant bacterial lineages in the total (DNA-based) and active (RNA-based) bacterioplankton community at stations outside (1–4) and inside (5–15) the examined phytoplankton bloom. Only groups with an average abundance of more than 1% either at DNA or RNA level are shown. Station 1 is separated from the oher samples because it was located in a bloom outside of the area of the examined bloom.
Alphaproteobacteria accounted for 50% of all sequences with a higher abundance at DNA and RNA level (59 and 41%, respectively). The opposite was recorded for the Gammaproteobacteria, which accounted for 16% (DNA) and 31% (RNA), respectively. The increased abundance of the Gammaproteobacteria was mainly attributed to the higher abundances of Pseudospirrilii and the SAR92 clade. Changes in the abundances of the different alphapoteobacterial taxa were mainly attributed to the overall low abundance of the SAR11 clade at RNA level. A low activity of SAR11 is supported by other studies (West et al., 2008; Lamy et al., 2010; Klindworth et al., 2014). For example, Lamy et al. found an overall low abundance and activity of the SAR11 clade in a P. globosa bloom in the eastern English Channel. In another study, Alonso and Pernthaler (2006b) showed that SAR11 is highly abundant but not very active in costal North Sea waters. In addition, West et al. (2008) demonstrated that SAR11 was more abundant at DNA level than at RNA level in the Southern Ocean.
Several groups responded significantly toward algal bloom presence at DNA and/or RNA level, e.g., the abundance of the SAR92 clade was three times higher at RNA level and in bloom presence. This is in accordance with previous studies (Pinhassi et al., 2005; West et al., 2008; Klindworth et al., 2014; Wemheuer et al., 2014). For example, a phytoplankton bloom induced by inorganic nutrient enrichment influenced SAR92 in a mesocosm experiment (Pinhassi et al., 2005). Klindworth et al. (2014) found that members of the Rhodobacteraceae and SAR92 clade exhibited high metabolic activity levels during a bloom succession, which indicates their important role during bloom events. In addition, the 16S cDNA estimates for SAR11 were notably lower in the earlier bloom sample. The authors suggest that members of this clade could not profit from the increasing availability of nutrients in the decaying bloom and thus were outcompeted by other clades. This is in line with our study in which we found significant lower abundances of SAR11 in the bloom presence and at RNA level.
Members of the two genera Marinoscillum and Polaribacter were significantly more abundant in bloom samples both at DNA and RNA level. Bacteroidetes are widespread in marine systems and play an important role in organic matter degradation (Gómez-Pereira et al., 2010). The higher abundance of this phylum during the phytoplankton bloom was verified by recent findings (Alderkamp et al., 2006; Lamy et al., 2010; Tada et al., 2012; Teeling et al., 2012). The strongest increase in activity during the senescent stage of a P. globosa bloom in the North Sea was observed for Bacteroidetes (Alderkamp et al., 2006) A mesocosm experiment targeting bacterial succession patterns during a diatom bloom revealed that Bacteroidetes had a relatively high growth potential as the bloom peaked (Tada et al., 2012). The authors suggested that the early development contributed to the initial stage of bloom decomposition. Therefore, this phylum seems to benefit from the conditions provided by the algal bloom and might play an important role in the degradation of phytoplankton-derived organic matter. Klindworth et al. (2014) mapped metatranscriptomic data on assembled and taxonomically classified metagenomic data and found that Formosa and Polaribacter acted as major algal polymer degraders. A similar conclusion was drawn in a study of a P. globosa bloom in the eastern English Channel. Here, members of Bacteroidetes group dominated the activities and the abundances during the growth phase of the algae (Lamy et al., 2010).
Bloom Presence Affects Bacterioplankton Gene Expression
After removal of ribosomal RNA, nearly 45 million Illumina reads remained and were used for environmental gene expression analysis. Generated mRNA datasets were initially mapped on the 15 reference genomes belonging to abundant marine genera and lineages. However, only 10% of the sequences mapped to these reference genomes. Most of these sequences were affiliated to the genome of P. temperata RCA23 (see Supplementary Table S3). This strain was isolated in the German Wadden Sea (Giebel et al., 2011, 2013), and its genome was recently described (Voget et al., 2014).
Mapping mRNA datasets on genomes is a common approach when analyzing metatranscriptomic data (e.g., Gifford et al., 2013). The advantage of this approach is that community functions can be linked to a certain organism. However, most reads are not included in the analysis because reference genomes for many marine lineages are still missing. Another problem is the data normalization when mapping metatranscriptomic data on genomes. In a transcriptomic approach, all sequences derive from a single organism and data can be normalized by the number of reads mapped. However, in a metatranscriptomic approach, the amount of sequences affiliated to an organism is not only linked to its gene expression but also to the gene expression of all other community members. Thus, a decrease in abundance can be caused either by a lower gene expression of the organism or by an increased expression of other community members.
Here, we mapped the mRNA data on reference genomes representing marine lineages, which were abundant in the 16S rRNA gene analysis. These genomes included data from P. temperata RCA23 and HTCC2255, belonging to two Roseobacter lineages, the RCA and NAC11-7 clusters. Other reference genomes used were the genomes of HTCC2207 und MOLA455, both members of the SAR92 clade (Stingl et al., 2007; Courties et al., 2014). Mapping the datasets on assembled metagenomic and metatranscriptomic data resulted in an overall alignment rate of more than 86.22% (see Table 5). This overall high coverage is higher than in other studies. For example, Kopf et al. (2015) mapped up to 80% of their metatranscriptomic data on the corresponding metagenome. We were able to map our data on approximately 600,000 genes with almost 800,000 different functions.
Most of the genes affiliated to the two members of the SAR92 clade were upregulated which corresponds to their increasing abundance at 16S rRNA transcipt level. For example, one leucyl-tRNA synthetase and three isoleucyl-tRNA synthetases affiliated to MOLA455 were significantly upregulated in the bloom (Figure 3A). Morover, two leucine-tRNAs were significantly upregulated in the bloom in P. temperata RCA23 (Figure 3B). In addition, an isoleucyl-tRNA synthetase affiliated to P. temperata RCA23 (Figure 3A) and a isoleucyl-tRNA synthetase and a leucyl-tRNA synthetase of P. temperata RCA23 (Figure 3B) were marginal significantly upregulated in the bloom (P < 0.1). This is in line with a study by West et al. (2008). The authors found that the Roseobacter groups NAC11-7 and RCA as well as the SAR92 clade were the most important contributors to leucine incorporation during the peak of a naturally iron-fertilized phytoplankton bloom in the Southern Ocean. This result is confirmed by a study about a P. globosa bloom in the English Channel (Lamy et al., 2010). Here, Bacteroidetes and Gammaproteobacteria were the most abundant and active groups during the growth period of the algae. Gammaproteobacteria and Alphaproteobacteria dominated by the Roseobacter clade accounted for the major part of leucine incorporation after the disappearance of the bloom. In addition, the contributions of different bacterial groups to bulk abundance and leucine incorporation were partly correlated with cell-specific exoproteolytic and exoglucosidic activities and with particulate organic carbon. This indicates some specificity of these bacterial groups with respect to their ecological role in the environment. Interestingly, we identified two beta-glucosidases affiliated to HTCC2207 being expressed only in bloom samples (Figure 3A). In the study of Teeling et al. (2012), metagenomic and metaproteomic data indicated the presence of distinct sets of carbohydrate-active enzymes (CAZymes) and transporters, which suggested a positive selection for bacteria with the capacity to decompose phytoplankton biomass. Four HTCC2207 indicator genes have been described to contain cadherin domains involved in complex carbohydrate degradation via cell aggregation and direct binding to cellulose, xylan, and related compounds (Gifford et al., 2013). This might explain the increase of the SAR92 clade as observed in the present study. In addition, the role of Gammprotaobacteria during polysaccharide degradation has been recently addressed in a study by Wietz et al. (2015). The authors showed that alginate and agarose degradation covaried with the abundance of different lineages within the Gammaproteobacteria.
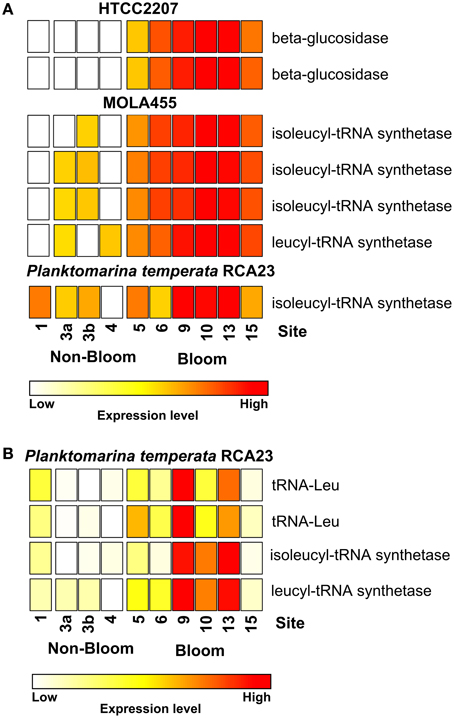
Figure 3. Expression of certain gene involved in leucine/isoleucine incorporation and polymer degradation inside and outside the examined phytoplankton bloom. Data either derives from the mapping on the assembled metagenomic and metatranscriptomic data (A) or from the mapping on reference genomes (B). Gene expressions levels are log2 transformed and standardized. To obtain only positive values, normalized gene expression values were increased by one prior to log transformation.
Interestingly, a major fraction of the expressed genes affiliated to SAR92 clade and other bacterial lineages was linked to several genes such as RNA polymerases, heat shock proteins, chaperons, sigma factors, and ribosomal proteins. This is consistent with a study from Klindworth et al. (2014). In this study, the most abundant mRNA transcripts with known functions coded for housekeeping genes including DNA gyrase, elongation factors, and sigma factors. The authors indicated that this reflects differences in nutritional ecological strategies of the dominant bacterial classes.
However, the high number of heat shock and other stress-related genes overexpressed in bloom samples in members of the SAR92 clade might not necessarily reflect its ecological role as a polymer degrader but might also be caused by a higher stress tolerance toward the rapidly changing conditions during the phytoplankton bloom (Wemheuer et al., 2014). PON, Chl a, and phaeopigments of stations in the bloom area were significantly higher than that outside the bloom area. An increasing pH from 7.9 to 8.7 was observed as a result of CO2 net fixation into the alga during a phytoplankton bloom (Brussaard et al., 1996). Members of SAR92 clade might benefit from bloom conditions due to their high stress tolerance level rather than filling one of the specialized ecological niches formed during a phytoplankton bloom.
Adaption to Environmental Changes by Higher Genome Plasticity
The overall expression level of P. temperata RCA23 was higher inside the bloom (Figure 4). Numerous genes encoding for transposases in its genome were highly overexpressed. Transposases are the most abundant and most ubiquitous genes in nature (Aziz et al., 2010). In addition, investigation of the metatranscriptomic bins revealed the presence of several transposases that were affiliated to P. temperata and overexpressed in the bloom (Figure 4). It has been shown that some bacteria expressed transposases under changing environmental conditions to rearrange genome architecture. For example, up to 81 genes encoded for transposases were upregulated in Microcystis aeruginosa relative to the control when grown on urea (Steffen et al., 2014). Genome rearrangements and the resulting genome mosaics have been also found in other members of the Roseobacter clade. The genomes of Octadecabacter arcticus and O. antarcticus are highly different despite their similarity on 16S rRNA gene sequence level and the presence of some unique gene features (Vollmers et al., 2013). This is attributed to genomic rearrangements caused by an unusually high number of transposases in the genomes of both Octadecabacter strains. We assume that the recorded overexpression could result in a higher genome plasticity/heterogeneity of this population and thus might be a possible adaptation strategy of P. temperata to environmental changes. Moreover, as found in other members of the Roseobacter clade, it might be one of the key features of this group explaining its high abundance in marine ecosystems and its ability to adapt to various marine niches. However, comparative genome studies are missing because only one genome of the genus Planktomarina is currently available. Consequently, this issue cannot been fully answered yet.
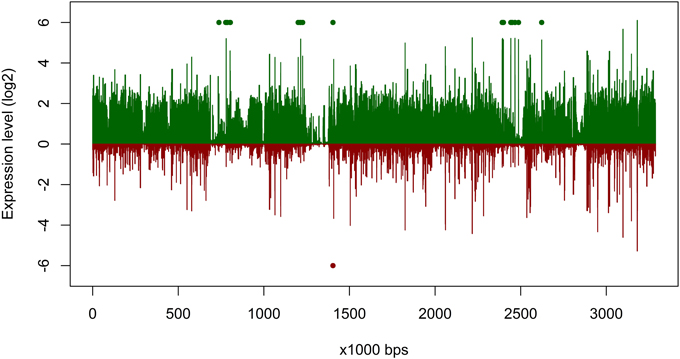
Figure 4. Gene expression of Planktomarina temperata RCA 23 inside and outside the examined phytoplankton bloom. Gene expressions are log2 transformed mean values from the three non-bloom and six bloom stations, respectively. To obtain only positive values, normalized gene expression values were increased by one prior to log transformation. Expression inside the bloom is depicted in green, outside the bloom in red. Green/red dots mark the position of transposases, which were highly upregulated.
Conclusions
Active bacterial communities in the North Sea are dominated by only a few marine groups such as the Roseobacter RCA cluster. Some of these lineages responded significantly toward the P. globosa bloom investigated in this study. For example, the SAR92 clade was three times more abundant at active bacterial community level and in bloom presence. The metatranscriptomic approach revealed that these groups are not dominated by well-studied isolates or type species as only 10% of all metatranscriptomic sequences mapped on reference genomes. Therefore, in situ experiments employing available isolates do not necessarily reflect environmental conditions and, thus, only provide limited information on the ecological role of the studied isolates. However, mapping these reads on assembled metagenomic and metatranscriptomic sequences led to an overall mapping rate of more than 85% demonstrating the power of this combined approach. The functional analysis performed in this study provides insights into gene expression patterns of the abundant community members. The high abundance of the SAR92 clade, which is supposed to be involved in polymer-degradation during and after the bloom, is attributed to a higher stress tolerance indicated by the high number of heat shock expressed in the bloom. Although the number of field studies targeting the active bacterial community either by metatranscriptomic or metaproteomic approaches has been increased over the past years, the complex dynamics of marine environments are still largely unexplored. This study provides a deep insight into structural and functional responses of the bacterioplankton community toward a phytoplankton bloom. Therefore, it paved the way for a better understanding of the complex dynamics of marine bacteria and their interactions with the surrounding environment.
Author Contributions
RD and BW conceived and designed the experiments; BW, FW, JH, SV, and FM performed the experiments and analyzed the data; BW, FW, and RD wrote the paper; all authors reviewed, edited, and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the crew of the research vessel Heincke for their valuable support during the sampling campaign. We are grateful to Peter Meinicke and Heiko Liesegang for the help during data analysis. This work was funded by the Deutsche Forschungsgemeinschaft (DFG) as part of the collaborative research center TRR51 and the Alfred Wegener Institute under grant number AWI-HE327_00. Additionally, we acknowledge support by DFG and the Open Access Publication Funds of the Göttingen University.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00805
References
Alderkamp, A. C., Sintes, E., and Herndl, G. J. (2006). Abundance and activity of major groups of prokaryotic plankton in the coastal North Sea during spring and summer. Aquat. Microb. Ecol. 45, 237–246. doi: 10.3354/ame045237
Alonso, C., and Pernthaler, J. (2006a). Concentration-dependent patterns of leucine incorporation by coastal picoplankton. Appl. Environ. Microbiol. 72, 2141–2147. doi: 10.1128/AEM.72.3.2141-2147.2006
Alonso, C., and Pernthaler, J. (2006b). Roseobacter and SAR11 dominate microbial glucose uptake in coastal North Sea waters. Environ. Microbiol. 8, 2022–2030. doi: 10.1111/j.1462-2920.2006.01082.x
Arrigo, K. R. (2005). Marine microorganisms and global nutrient cycles. Nature 437, 349–355. doi: 10.1038/nature04159
Azam, F. (1998). Microbial control of oceanic carbon flux: the plot thickens. Science 280, 694–696. doi: 10.1126/science.280.5364.694
Azam, F., Fenchel, T., Field, J. G., Gray, J. S., Meyer-Reil, L. A., and Thingstad, F. (1983). The ecological role of water-column microbes in the sea. Mar. Ecol. Prog. Ser. 10, 257–263. doi: 10.3354/meps010257
Aziz, R. K., Breitbart, M., and Edwards, R. A. (2010). Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Res. 38, 4207–4217. doi: 10.1093/nar/gkq140
Blazewicz, S. J., Barnard, R. L., Daly, R. A., and Firestone, M. K. (2013). Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J. 7, 2061–2068. doi: 10.1038/ismej.2013.102
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bragg, L., Stone, G., Imelfort, M., Hugenholtz, P., and Tyson, G. W. (2012). Fast, accurate error-correction of amplicon pyrosequences using Acacia. Nat. Methods 9, 425–426. doi: 10.1038/nmeth.1990
Brussaard, C. P. D., Gast, G. J., van Duyl, F. C., and Riegman, R. (1996). Impact of phytoplankton bloom magnitude on a pelagic microbial food web. Mar. Ecol. Prog. Ser. 144, 211–221. doi: 10.3354/meps144211
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Campbell, B. J., and Kirchman, D. L. (2013). Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J. 7, 210–220. doi: 10.1038/ismej.2012.93
Cole, J. J., Findlay, S., and Pace, M. L. (1988). Bacterial production in fresh and saltwater ecosystems: a cross-system overview. Mar. Ecol. Prog. Ser. 43, 1–10. doi: 10.3354/meps043001
Courties, A., Riedel, T., Jarek, M., Papadatou, M., Intertaglia, L., Lebaron, P., et al. (2014). Draft genome sequence of the gammaproteobacterial strain MOLA455, a representative of a ubiquitous proteorhodopsin-producing group in the ocean. Genome Announc. 2, e01203–e01213. doi: 10.1128/genomea.01203-13
DeLong, E. F., and Karl, D. M. (2005). Genomic perspectives in microbial oceanography. Nature 437, 336–342. doi: 10.1038/nature04157
Ducklow, H., Kirchman, D. L., Quinby, H. L., Carlson, C. A., and Dam, H. G. (1993). Stocks and dynamics of bacterioplankton carbon during the spring bloom in the eastern North Atlantic Ocean. Deep Sea Res. II 40, 245–263. doi: 10.1016/0967-0645(93)90016-G
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fandino, L. B., Riemann, L., Steward, G. F., Long, R. A., and Azam, F. (2001). Variations in bacterial community structure during a dinoflagellate bloom analyzed by DGGE and 16S rDNA sequencing. Aquat. Microb. Ecol. 23, 119–130. doi: 10.3354/ame023119
Giebel, H.-A., Kalhoefer, D., Gahl-Janssen, R., Choo, Y.-J., Lee, K., Cho, J.-C., et al. (2013). Planktomarina temperata gen. nov., sp. nov., belonging to the globally distributed RCA cluster of the marine Roseobacter clade, isolated from the German Wadden Sea. Int. J. Syst. Evol. Microbiol. 63, 4207–4217. doi: 10.1099/ijs.0.053249-0
Giebel, H.-A., Kalhoefer, D., Lemke, A., Thole, S., Gahl-Janssen, R., Simon, M., et al. (2011). Distribution of Roseobacter RCA and SAR11 lineages in the North Sea and characteristics of an abundant RCA isolate. ISME J. 5, 8–19. doi: 10.1038/ismej.2010.87
Gifford, S. M., Sharma, S., Booth, M., and Moran, M. A. (2013). Expression patterns reveal niche diversification in a marine microbial assemblage. ISME J. 7, 281–298. doi: 10.1038/ismej.2012.96
Gomes, A., Gasol, J. M., Estrada, M., Franco-Vidal, L., Díaz-Pérez, L., Ferrera, I., et al. (2015). Heterotrophic bacterial responses to the winter–spring phytoplankton bloom in open waters of the NW Mediterranean. Deep Sea Res. I 96, 59–68. doi: 10.1016/j.dsr.2014.11.007
Gómez-Pereira, P. R., Fuchs, B. M., Alonso, C., Oliver, M. J., van Beusekom, J. E., and Amann, R. (2010). Distinct flavobacterial communities in contrasting water masses of the North Atlantic Ocean. ISME J. 4, 472–487. doi: 10.1038/ismej.2009.142
Grossart, H. P., Levold, F., Allgaier, M., Simon, M., and Brinkhoff, T. (2005). Marine diatom species harbour distinct bacterial communities. Environ. Microbiol. 7, 860–873. doi: 10.1111/j.1462-2920.2005.00759.x
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Karner, M., and Herndl, G. (1992). Extracellular enzymatic activity and secondary production in free-living and marine-snow-associated bacteria. Mar. Biol. 113, 341–347.
Klindworth, A., Mann, A. J., Huang, S., Wichels, A., Quast, C., Waldmann, J., et al. (2014). Diversity and activity of marine bacterioplankton during a diatom bloom in the North Sea assessed by total RNA and pyrotag sequencing. Mar. Genomics 18(Pt B), 185–192. doi: 10.1016/j.margen.2014.08.007
Kopf, A., Kostadinov, I., Wichels, A., Quast, C., and Glöckner, F. O. (2015). Metatranscriptome of marine bacterioplankton during winter time in the North Sea assessed by total RNA sequencing. Mar. Genomics 19, 45–46. doi: 10.1016/j.margen.2014.11.001
Kopylova, E., Noé, L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Lamy, D., Obernosterer, I., Laghdass, M., Artigas, L. F., Breton, E., Grattepanche, J. D., et al. (2010). Temporal changes of major bacterial groups and bacterial heterotrophic activity during a Phaeocystis globosa bloom in the eastern English Channel. Aquat. Microb. Ecol. 58, 95–107. doi: 10.3354/ame01359
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Liu, M., Dong, Y., Zhang, W., Sun, J., Zhou, F., Ren, J., et al. (2013). Diversity of bacterial community during spring phytoplankton blooms in the central Yellow Sea. Can. J. Microbiol. 59, 324–332. doi: 10.1139/cjm-2012-0735
Markowitz, V. M., Chen, I.-M. A., Palaniappan, K., Chu, K., Szeto, E., Grechkin, Y., et al. (2012). IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 40, D115–D122. doi: 10.1093/nar/gkr1044
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. doi: 10.14806/ej.17.1.200
McQuatters-Gollop, A., Raitsos, D. E., Edwards, M., Pradhan, Y., Mee, L. D., and Lavender, S. J. (2007). A long-term chlorophyll data set reveals regime shift in North Sea phytoplankton biomass unconnected to nutrient trends. Limnol. Oceanogr. 52, 635–648. doi: 10.4319/lo.2007.52.2.0635
Meinicke, P. (2015). UProC: tools for ultra-fast protein domain classification. Bioinformatics 31, 1382–1388. doi: 10.1093/bioinformatics/btu843
Muyzer, G., Teske, A., Wirsen, C. O., and Jannasch, H. W. (1995). Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch. Microbiol. 164, 165–172. doi: 10.1007/BF02529967
Namiki, T., Hachiya, T., Tanaka, H., and Sakakibara, Y. (2012). MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 40, e155. doi: 10.1093/nar/gks678
Pinhassi, J., Sala, M. M., Havskum, H., Peters, F., Guadayol, O., and Malits, A. (2004). Changes in bacterioplankton composition under different phytoplankton regimens. Appl. Environ. Microbiol. 70, 6753–6766. doi: 10.1128/AEM.70.11.6753-6766.2004
Pinhassi, J., Simó, R., González, J. M., Vila, M., Alonso-Sáez, L., Kiene, R. P., et al. (2005). Dimethylsulfoniopropionate turnover is linked to the composition and dynamics of the bacterioplankton assemblage during a microcosm phytoplankton bloom. Appl. Environ. Microbiol. 71, 7650–7660. doi: 10.1128/AEM.71.12.7650-7660.2005
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2014). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://www.R-project.org/
Sapp, M., Wichels, A., Wiltshire, K. H., and Gerdts, G. (2007). Bacterial community dynamics during the winter–spring transition in the North Sea. FEMS Microbiol. Ecol. 59, 622–637. doi: 10.1111/j.1574-6941.2006.00238.x
Schlitzer, R. (2015). Ocean Data View. Available online at: http://odv.awi.de
Schoemann, V., Becquevort, S., Stefels, J., Rousseau, V., and Lancelot, C. (2005). Phaeocystis blooms in the global ocean and their controlling mechanisms: a review. J. Sea Res. 53, 43–66. doi: 10.1016/j.seares.2004.01.008
Steffen, M. M., Dearth, S. P., Dill, B. D., Li, Z., Larsen, K. M., Campagna, S. R., et al. (2014). Nutrients drive transcriptional changes that maintain metabolic homeostasis but alter genome architecture in Microcystis. ISME J. 8, 2080–2092. doi: 10.1038/ismej.2014.78
Stingl, U., Desiderio, R. A., Cho, J. C., Vergin, K. L., and Giovannoni, S. J. (2007). The SAR92 clade: an abundant coastal clade of culturable marine bacteria possessing proteorhodopsin. Appl. Environ. Microbiol. 73, 2290–2296. doi: 10.1128/AEM.02559-06
Tada, Y., Taniguchi, A., Sato-Takabe, Y., and Hamasaki, K. (2012). Growth and succession patterns of major phylogenetic groups of marine bacteria during a mesocosm diatom bloom. J. Oceanogr. 68, 509–519. doi: 10.1007/s10872-012-0114-z
Teeling, H., Fuchs, B. M., Becher, D., Klockow, C., Gardebrecht, A., Bennke, C. M., et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611. doi: 10.1126/science.1218344
Veldhuis, M. J. W., and Wassmann, P. (2005). Bloom dynamics and biological control of a high biomass HAB species in European coastal waters: a Phaeocystis case study. Harmful Algae 4, 805–809. doi: 10.1016/j.hal.2004.12.004
Voget, S., Wemheuer, B., Brinkhoff, T., Vollmers, J., Dietrich, S., Giebel, H.-A., et al. (2014). Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J. 9, 371–384. doi: 10.1038/ismej.2014
Vollmers, J., Voget, S., Dietrich, S., Gollnow, K., Smits, M., Meyer, K., et al. (2013). Poles apart: arctic and antarctic Octadecabacter strains share high genome plasticity and a new type of Xanthorhodopsin. PLoS ONE 8:e63422. doi: 10.1371/journal.pone.0063422
Weinbauer, M. G., Fritz, I., Wenderoth, D. F., and Höfle, M. G. (2002). Simultaneous extraction from bacterioplankton of total RNA and DNA suitable for quantitative structure and function analyses. Appl. Environ. Microbiol. 68, 1082–1087. doi: 10.1128/AEM.68.3.1082-1087.2002
Wemheuer, B., Güllert, S., Billerbeck, S., Giebel, H.-A., Voget, S., Simon, M., et al. (2014). Impact of a phytoplankton bloom on the diversity of the active bacterial community in the southern North Sea as revealed by metatranscriptomic approaches. FEMS Microbiol. Ecol. 87, 378–389. doi: 10.1111/1574-6941.12230
Wemheuer, B., Wemheuer, F., and Daniel, R. (2012). RNA-based assessment of diversity and composition of active archaeal communities in the German Bight. Archaea 2012:695826. doi: 10.1155/2012/695826
Wemheuer, B., Taube, R., Akyol, P., Wemheuer, F., and Daniel, R. (2013). Microbial diversity and biochemical potential encoded by thermal spring metagenomes derived from the Kamchatka Peninsula. Archaea 2013, 13. doi: 10.1155/2013/136714
West, N. J., Obernosterer, I., Zemb, O., and Lebaron, P. (2008). Major differences of bacterial diversity and activity inside and outside of a natural iron-fertilized phytoplankton bloom in the Southern Ocean. Environ. Microbiol. 10, 738–756. doi: 10.1111/j.1462-2920.2007.01497.x
Wietz, M., Wemheuer, B., Simon, H., Giebel, H.-A., Seibt, M. A., Daniel, R., et al. (2015). Bacterial community dynamics during polysaccharide degradation at contrasting sites in the Southern and Atlantic Oceans. Environ. Microbiol. doi: 10.1111/1462-2920.12842. [Epub ahead of print].
Wiltshire, K. H., Kraberg, A., Bartsch, I., Boersma, M., Franke, H.-D., Freund, J., et al. (2010). Helgoland roads, North Sea: 45 years of change. Estuaries Coasts 33, 295–310. doi: 10.1007/s12237-009-9228-y
Wiltshire, K. H., Malzahn, A. M., Wirtz, K., Janisch, S., Mangelsdorf, P., and Manly, B. F. J. (2008). Resilience of North Sea phytoplankton spring bloom dynamics: an analysis of long-term data at Helgoland Roads. Limnol. Oceanogr. 53, 1294–1302. doi: 10.4319/lo.2008.53.4.1294
Keywords: bacterioplankton, metagenomics, metatranscriptomics, algal bloom, functional changes, Planktomarina temperata, SAR92
Citation: Wemheuer B, Wemheuer F, Hollensteiner J, Meyer F-D, Voget S and Daniel R (2015) The green impact: bacterioplankton response toward a phytoplankton spring bloom in the southern North Sea assessed by comparative metagenomic and metatranscriptomic approaches. Front. Microbiol. 6:805. doi: 10.3389/fmicb.2015.00805
Received: 11 May 2015; Accepted: 22 July 2015;
Published: 11 August 2015.
Edited by:
Eamonn P. Culligan, University College Cork, IrelandCopyright © 2015 Wemheuer, Wemheuer, Hollensteiner, Meyer, Voget and Daniel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rolf Daniel, Department of Genomic and Applied Microbiology and Göttingen Genomics Laboratory, Institute of Microbiology and Genetics, Georg-August University Göttingen, Grisebachstr. 8, D-37077 Göttingen, Germany, rdaniel@gwdg.de
†These authors have contributed equally to this work.