 Jeroen Frank1
Jeroen Frank1 Celia Dingemanse2Arnoud M. Schmitz1Rolf H. A. M. Vossen1Gert-Jan B. van Ommen2Johan T. den Dunnen1,2,3
Celia Dingemanse2Arnoud M. Schmitz1Rolf H. A. M. Vossen1Gert-Jan B. van Ommen2Johan T. den Dunnen1,2,3 Els C. Robanus-Maandag2
Els C. Robanus-Maandag2 Seyed Yahya Anvar1,2*
Seyed Yahya Anvar1,2*- 1Leiden Genome Technology Center, Leiden University Medical Center, Leiden, Netherlands
- 2Department of Human Genetics, Leiden University Medical Center, Leiden, Netherlands
- 3Department of Clinical Genetics, Leiden University Medical Center, Leiden, Netherlands
Background: Immuno-compromised mice infected with Helicobacter typhlonius are used to model microbially inducted inflammatory bowel disease (IBD). The specific mechanism through which H. typhlonius induces and promotes IBD is not fully understood. Access to the genome sequence is essential to examine emergent properties of this organism, such as its pathogenicity. To this end, we present the complete genome sequence of H. typhlonius MIT 97-6810, obtained through single-molecule real-time sequencing.
Results: The genome was assembled into a single circularized contig measuring 1.92 Mbp with an average GC content of 38.8%. In total 2,117 protein-encoding genes and 43 RNA genes were identified. Numerous pathogenic features were found, including a putative pathogenicity island (PAIs) containing components of type IV secretion system, virulence-associated proteins and cag PAI protein. We compared the genome of H. typhlonius to those of the murine pathobiont H. hepaticus and human pathobiont H. pylori. H. typhlonius resembles H. hepaticus most with 1,594 (75.3%) of its genes being orthologous to genes in H. hepaticus. Determination of the global methylation state revealed eight distinct recognition motifs for adenine and cytosine methylation. H. typhlonius shares four of its recognition motifs with H. pylori.
Conclusion: The complete genome sequence of H. typhlonius MIT 97-6810 enabled us to identify many pathogenic features suggesting that H. typhlonius can act as a pathogen. Follow-up studies are necessary to evaluate the true nature of its pathogenic capabilities. We found many methylated sites and a plethora of restriction-modification systems. The genome, together with the methylome, will provide an essential resource for future studies investigating gene regulation, host interaction and pathogenicity of H. typhlonius. In turn, this work can contribute to unraveling the role of Helicobacter in enteric disease.
Introduction
The genus Helicobacter has rapidly expanded since it was first proposed in Goodwin et al. (1989). Today, the genus includes 35 Helicobacter species (Euzeby, 1997), with several (putative) novel species having been discovered recently (Menard et al., 2014). Members of this genus are Gram-negative and are characterized by having highly motile, multiple sheathed flagella and a helical, curved or straight unbranched morphology (Goodwin et al., 1989). All known Helicobacter strains live in human and animal hosts, where they primarily colonize the gastrointestinal tract (Franklin et al., 2001). Infection with Helicobacter sp. has been shown to be endemic in many animal facilities worldwide (Fox et al., 1994; Zenner, 1999; Whary and Fox, 2004; Chichlowski et al., 2008). Although as pathobionts they are benign commensals in immune-competent animals, they can act as opportunistic pathogens in immune-compromised mice.
The Helicobacter genus is well known for its association with enteric-, gastric-, and hepatic disease. The extensively studied human pathobiont, Helicobacter pylori, has been proven capable of causing a persistent inflammatory response in the stomach resulting in a 10–20% lifetime risk of developing peptic ulcers and a 1–2% risk of developing gastric cancer (Graham, 1989; Marshall, 1993; Parsonnet, 1995; Fox et al., 1999). Pathology caused by rodent Helicobacter sp. is often similar to those seen in human enteric diseases, especially inflammatory bowel diseases (IBDs) (Franklin et al., 2001). Consequently, rodent Helicobacter sp. are frequently used to infect immune-compromised mice to study these conditions in more detail.
One species used for IBD modeling is H. typhlonius. This murine Helicobacter, characterized by its lack of urease activity, is a prevalent intestinal colonizer of laboratory and feral mice (Franklin et al., 2001; Parker et al., 2009; Lofgren et al., 2012; Wasimuddin et al., 2012). Infection with H. typhlonius can induce and promote the development of severe IBD and IBD-associated neoplasia in immune-compromised Il10−/− mice (Chichlowski et al., 2008). These characteristics make infection with this species very useful to study IBD pathogenesis and treatment (Franklin et al., 2001; Chichlowski et al., 2008). Recently, we have shown that H. typhlonius infection can also modulate non-colitis-associated intestinal tumor formation as tested in conditional Apc mutant mice (Dingemanse et al., 2015).
To further elucidate the role of Helicobacter in enteric-, gastric-, and hepatic disease, it is increasingly important to determine the genomic sequence of the strain under study. Extensive sequencing efforts have resulted in the complete genomic sequences for at least 9 Helicobacter species, including many different strains (EMBL European Bioinformatics Institute [EMBL-EBI], 2014), while 17 species have been partly sequenced (National Center for Biotechnology Information [NCBI], 2014). Access to the complete genome contributes to the identification of potential virulence factors, permits the investigation of tissue tropism and may help unveil the mechanisms of pathogenesis. In this study, we reveal the complete sequence of the H. typhlonius genome along with its global methylation state at single-nucleotide resolution.
The H. typhlonius MIT 97-6810 genome was sequenced using Pacific Biosciences single-molecule real-time (SMRT) sequencing technology. The resulting long, highly accurate reads were virtually free of context-specific biases (Eid et al., 2009), ensured uniform genome coverage and were capable of resolving large repeats and structural variations. Ensuing de novo assembly and annotation of the genome, we performed base modification and motif identification analysis. It has been shown that methylation is involved in maintaining genome integrity, gene regulation, host interaction, cellular defense and limiting transformation by destroying foreign DNA (Jeltsch, 2003; Wion and Casadesus, 2006; Gonzalez et al., 2014; Krebes et al., 2014; Roberts et al., 2015). Finally, we present our comparative genomic results on closely related murine pathobiont H. hepaticus (Franklin et al., 2001; Fox et al., 2011; Krebes et al., 2014) and human pathobiont H. pylori. In addition, the global methylation state of H. typhlonius is compared to those of H. pylori strains 26695 and J99-R3 (Krebes et al., 2014).
Results and Discussion
Genome Assembly and Annotation
We performed SMRT sequencing to determine the complete genome sequence of H. typhlonius MIT 97-6810. In total 164,030 long (500- 29,940 bp), high-quality single-molecule sequencing reads were obtained (∼338 × coverage) (Table 1). Due to the nature of SMRT sequencing technology, long reads exhibit a relatively high randomly distributed error rate (Eid et al., 2009). Since most assemblers do not tolerate error rates greater than 5–10%, we used the hierarchical genome assembly process (HGAP) to correct sequencing errors. The resulting 4,157 corrected reads (Table 1) were assembled into a single 1,920,832 bp long contig with an average GC content of 38.8% (Table 2; Figure 1). To assess the accuracy and validity of the assembly, all sequencing reads were aligned to the assembled genome. The concordance between reads and reference sequence was found to be over 99.99% and no indication of sequence disagreement or coverage fluctuation could be found.
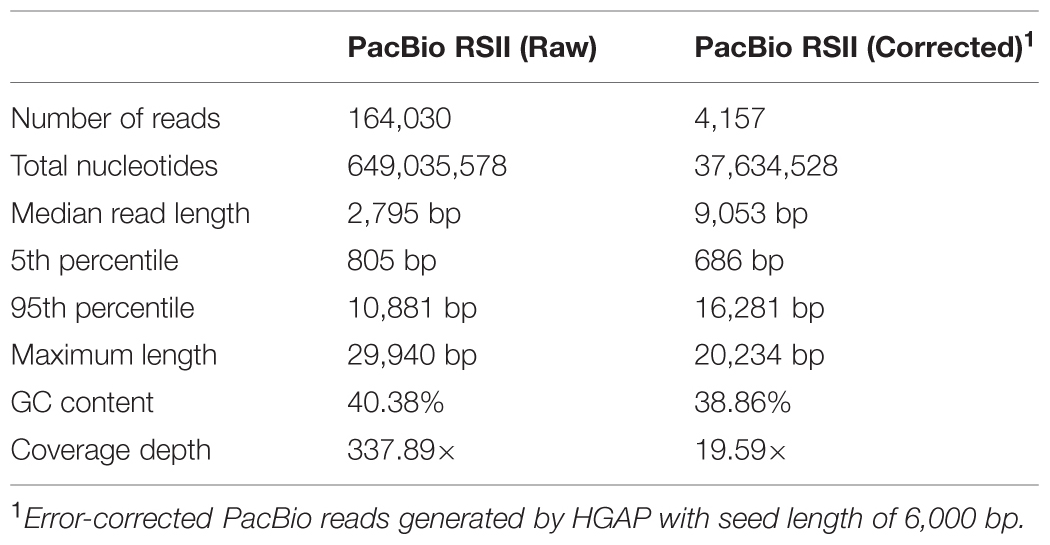
TABLE 1. Read statistics of 3 SMRT sequencing runs pre- and post-correction.

TABLE 2. Single-molecule real-time (SMRT) de novo genome assembly statistics.
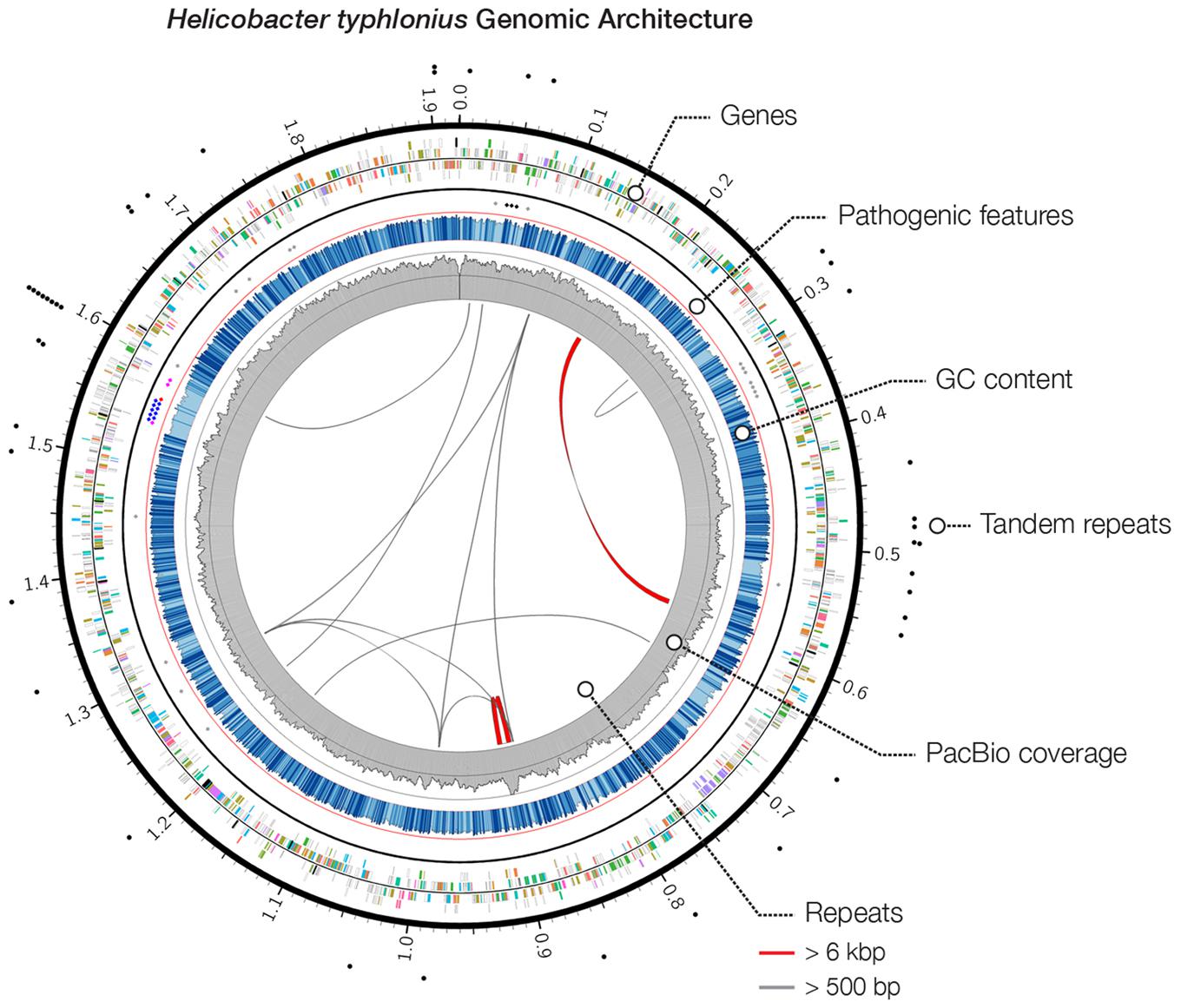
FIGURE 1. Heliobacter typhlonius genomic architecture. Circos plot outlines the genomic features of H. typhlonius. On the outside, sites with short tandem repeats are indicated by black dots. The following ring (outside to inside) illustrates protein-encoding genes for the forward and reverse strand, colored by associated subsystem. Delineated separately (inner spots) are multiple pathogenic features, including cytolethal distending toxins (black), components of the T4SS (blue), virulence-associated protein 2 s (purple), a Cag pathogenicity island protein (red) and sequences that match pathogenic protein families in H. hepaticus (gray). The overall GC content is shown in blue, where light blue regions indicate <5% and dark blue >5% deviation from the average GC content (bin size: 1 Kb). The coverage profile of the SMRT sequencing reads is shown in gray (bin size: 1 Kb). On the inside, repetitive sequences and structural variations (>95% similarity at the nucleotide level) are shown throughout the genome with repeats >6 Kb colored red.
The genome was examined for repeats and structural rearrangements. We found 13 long repeats and 42 short tandem repeats (STRs). There is one distinct region (genomic coordinates ∼885.2–907.9 Kb) that shows a complex repeat structure having relatively high coverage. This particular structure can also be seen in the assembly graph (Supplementary Figure S1). The repeat structure (size 22,672 bp) is slightly larger than the insert size of our sequence library (∼20 Kb), making it a challenging region to assemble. Therefore, although the sequencing reads seem to confirm the final genome sequence, we cannot exclude that the assembler could not fully resolve this region.
Next, the genome of H. typhlonius was automatically annotated using the RAST annotation service (Aziz et al., 2008; Overbeek et al., 2014). In total 2,117 protein-encoding genes (PEGs) and 43 RNA genes were identified, from which 890 PEGs (43%) were allocated to 278 annotated subsystems, biological processes or structural complexes realized by a set of functional roles (Overbeek et al., 2005) (Table 3). Subsequently, we estimated the location of the origin of replication (oriC) using Ori-Finder in conjunction with the DoriC database (Gao and Zhang, 2008; Gao et al., 2013). The genome was circularized accordingly with location of the predicted oriC at the start of the genome sequence (Supplementary Figure S2). The dnaA gene was found 11,655 bp upstream of the oriC region.
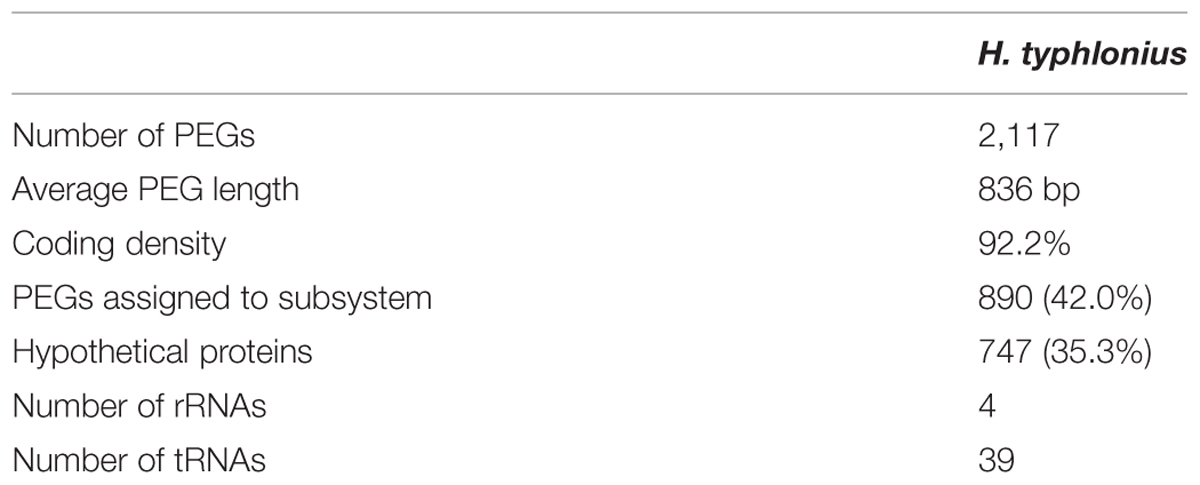
TABLE 3. Annotation statistics.
Recently, Sheh et al. (2014) deposited a draft-genome assembly of H. typhlonius MIT 98-6810 (also known as MIT 97-6810) in GenBank (ASM76576v1). They used the Illumina MiSeq platform to generate short reads that were assembled into 127 contigs which were subsequently scaffolded into 25 scaffolds. Compared to our assembly this assembly is fragmented and contains many scaffolding errors (Supplementary Figure S3). This fragmentation is likely caused by the nature of the Illumina data itself. Based on our assembly of the genome we could identify at least 13 repeated regions longer than 500 bp. Short Illumina reads (up to 300 bp) are unable to span such large repeats and structural variations, making it extremely difficult for the assembler to fully resolve these regions. Furthermore, DNA sequences having high or low GC content are notoriously difficult to PCR and therefore to sequence using second generation sequencing platforms. The PacBio RSII sequencer is not hampered by such characteristics; reads are long and there are virtually no context-specific biases. This enabled us to assemble the entire genome into a single continuous contig. Our assembly provides a comprehensive view of the genetic makeup and architecture of H. typhlonius.
Pathogenicity
Pathogenicity islands (PAIs) are distinct genetic elements that encode virulence-associated factors (Fox et al., 2011). They can often be detected by having a GC content, codon usage and k-mer frequencies, which are distinguishable from the rest of the genome owing to their origin through horizontal gene transfer (Che et al., 2014). The H. typhlonius genome contains one region with markedly lower GC content (∼34.2%) that is flanked by repeats at the 3′ end (∼1.53–1.60 Mb) (Figure 1). The size of this genomic island is estimated to be around 65.5 Kb and is located at 1,532,276–1,597,776 bp. This region contains 75 PEGs that constitute mostly hypothetical proteins (36 PEGs) but also includes many components of type IV secretion system (T4SS). The ability to secrete compounds including toxins is essential for virulence and survival (Fronzes et al., 2009). T4SS families can be divided into three classes based on functionality. First, T4SSs are involved in conjugation, a mechanism that enables the transfer of genetic material such as antibiotic resistance genes among bacteria (Dreiseikelmann, 1994). Second, T4SSs mediate DNA uptake from and release into their surroundings, further enabling genetic exchange (Hamilton and Dillard, 2006). Finally, T4SSs are directly involved in the transfer of protein effectors, including toxins, into eukaryote cells during infection (Fronzes et al., 2009; Terradot and Waksman, 2011). Each of these T4SS classes have been identified in H. pylori (Terradot and Waksman, 2011). T4SS typically consists of a collection of twelve proteins: VirB1–11 and VirD4. The presence of VirB2, VirB4-VirB6, VirB8-VirB11, and VirD4 in H. typhlonius was confirmed via RAST annotation (Supplementary Table S1). Additionally, using BLASTX, we observed strong evidence for the presence of VirB3 and VirB7 in H. typhlonius, whereas VirB1 was absent. Furthermore, cytotoxin-associated gene (Cag) PAI protein and 3 virulence-associated proteins were also present in the genome of H. typhlonius (Figure 2, Supplementary Table S1). The presence of a partial T4SS, a Cag protein and several virulence factors suggests that this region is a putative PAI.
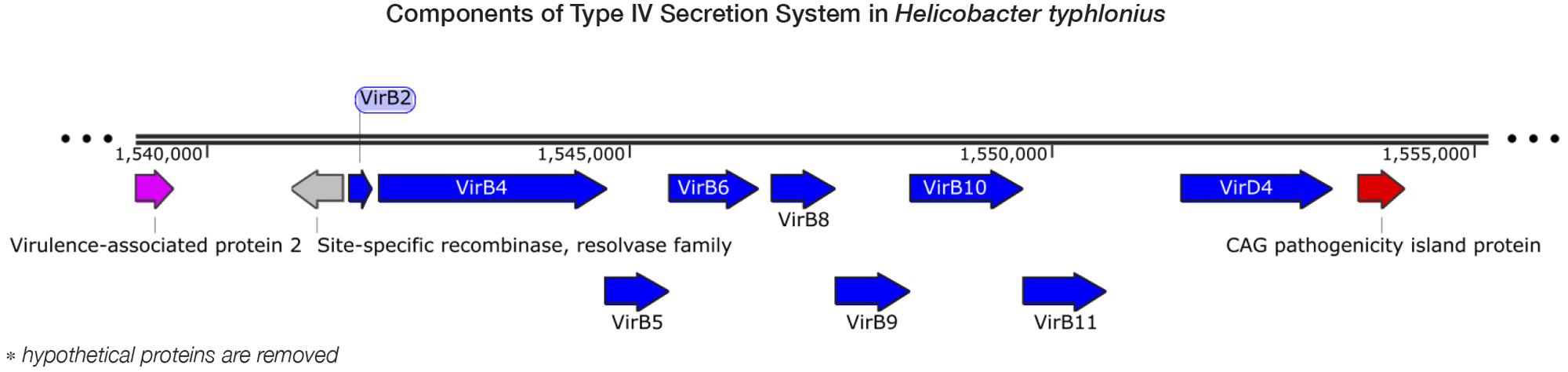
FIGURE 2. Components of type IV secretion system in H. typhlonius. Shown is a part of a putative pathogenicity island on the H. typhlonius genome (∼1.55–1.58 Mb). Components of the T4SS are colored blue. A virulence-associated protein and a Cag pathogenicity island protein are shown in purple and red, respectively. Hypothetical proteins have been omitted from this illustration.
Suerbaum et al. (2003) identified and characterized a PAI (HHGI1) in H. hepaticus ATCC 51449. This PAI spans 71 Kb and has a GC content of 33.2%. HHGI1 contains 70 open reading frames (ORFs) including pathogenic and virulent homologs, but they predominantly encode hypothetical proteins (Suerbaum et al., 2003). We could not confirm the presence of HHGl1 in H. typhlonius. Moreover, BLAST results of all 70 ORFs against the H. typhlonius genome retrieved very limited hits, except for one H. hepaticus gene, HH0237, that was partly found in H. typhlonius. HH0237 is a homolog of a structural component of known bacterial type VI secretion systems (T6SS) (Fox et al., 2011).
We searched for genes encoding subunits of cytolethal distending toxins (CDTs), which are present in several Gram-negative pathogens, including Helicobacters. CdtA and CdtC subunits bind together to subsequently deliver an active subunit of the CdtB toxin (Fox et al., 2011). Each of these three subunits was found in the H. typhlonius genome (Supplementary Table S1). The active CdtB unit has been associated with a variety of biological functions including DNase I-like function, cell-cycle arrest, phosphatase activity, and apoptotic cell death (Ge et al., 2008). Loss of CDT functionality in CDT-deficient isogenic H. hepaticus mutants affects the capability to colonize the large intestine, resulting in milder symptoms of typhlocolitis upon infection in mice (Young et al., 2004; Ge et al., 2005; Pratt et al., 2006).
We used multiple tools to predict and identify additional putative virulence factors, antimicrobial resistance genes or pathogenic features. VirulenceFinder and ResFinder (Zankari et al., 2012; Joensen et al., 2014) did not detect any additional virulence- or antimicrobial resistance genes. PHAST (PHAge Search Tool) (Zhou et al., 2011) was used to detect and annotate prophage sequences, but none were found. PathogenFinder (Cosentino et al., 2013) reported 19 proteins that are linked to pathogenic protein families in H. hepaticus, comprising mostly hypothetical proteins (Supplementary Table S2).
Comparative Genome Analysis
Phylogenetic analysis by Franklin et al. (2001) has demonstrated that H. typhlonius is closely related to H. hepaticus. This latter Helicobacter is a genuine murine pathobiont, capable of causing IBD, chronic hepatitis and liver cancer in numerous mouse models (Suerbaum et al., 2003; Fox et al., 2011). In turn, H. hepaticus is closely related to the human pathobiont and type species H. pylori (Franklin et al., 2001). The genome of H. typhlonius (1,92 Mbp) is somewhat larger than the genome of H. hepaticus ATCC 51449 (1.80 Mbp, accession NC_004917.1) and H. pylori 26695 (1.67 Mbp, accession NC_000915). The GC content is very similar for H. typhlonius (38.8% GC) and H. hepaticus (38.9% GC), while H. pylori (35.9% GC) deviates from the two having a considerably lower GC content.
Although less PEGs are predicted for H. hepaticus ATCC 51449 and H. pylori 26695 (1,879 and 1,620 PEGs respectively), H. typhlonius sequence mostly resembles H. hepaticus as 1,594 (75.3%) of its genes were found as orthologous to genes in H. hepaticus. This number is significantly lower for H. pylori having only 1,170 (55.3%) orthologous genes. This is also evident from the amino acid identity of orthologs in H. hepaticus (76.4% AAI) compared to that of H. pylori (50.6% AAI) (Figure 3). The conservation of major subsystems in H. hepaticus and H. pylori varies, with specific subsystems being conserved higher in one over the other and vice versa (Supplementary Figure S4).
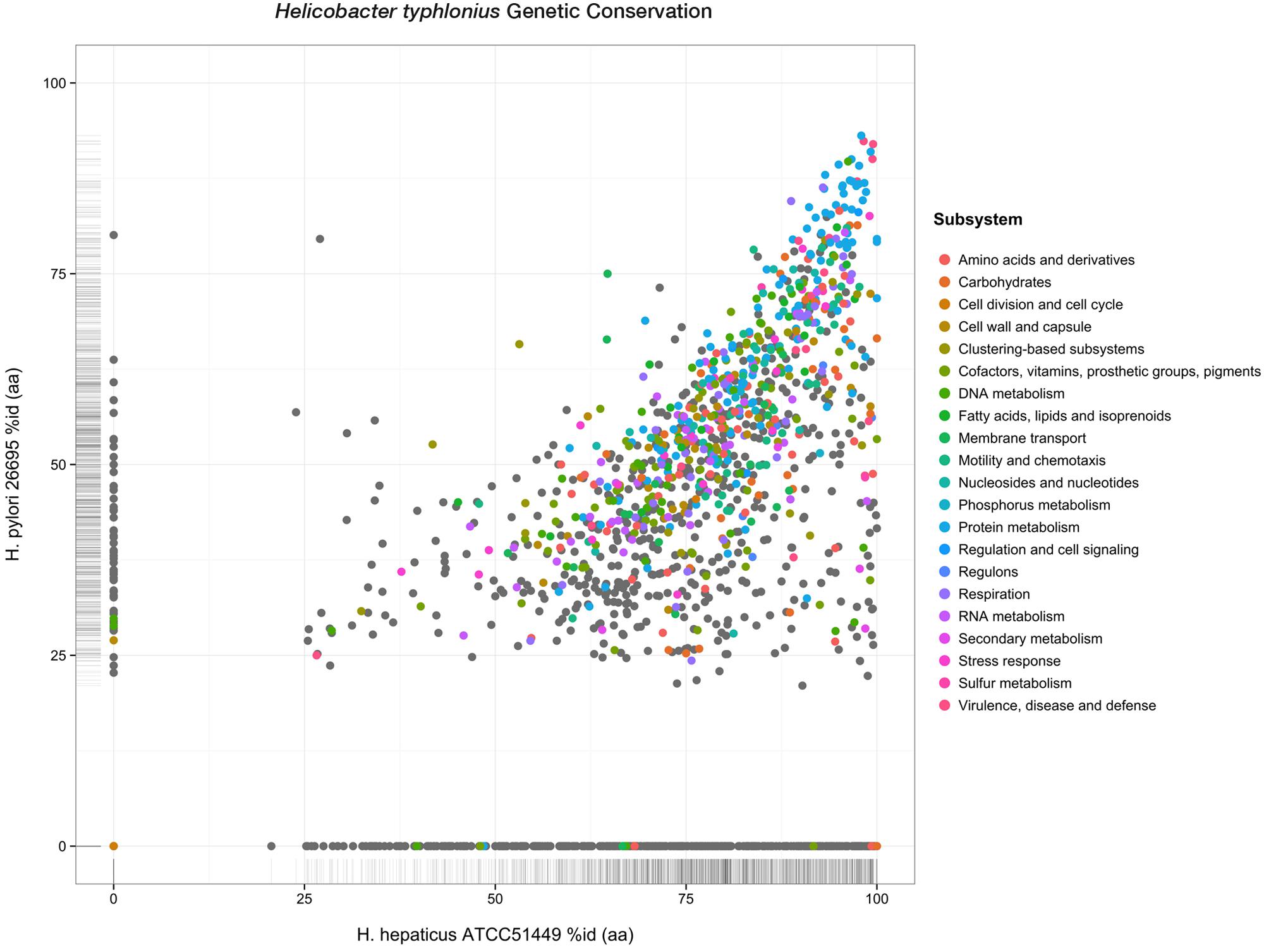
FIGURE 3. Genetic conservation. Scatterplot showing all protein-encoding genes of H. typhlonius and their respective orthologs in H. hepaticus and H. pylori. Similarity is expressed as the percentage of amino acid identity. Protein-coding genes are colored according to subsystem adopting the same color scheme as in Figure 1. PEGs colored gray do not have any association to a subsystem.
We also identified 468 PEGs that are unique to H. typhlonius. The great majority of these PEGs (386) constitute hypothetical proteins. There are nonetheless several annotated PEGs with predicted functions including two virulence-associated proteins, six glycosyl transferases, DNA recombination protein RmuC, DNA sulfur modification protein DndD, several PEGs that are part of restriction-modification (R-M) systems and two CRISPR-associated (Cas) proteins: Cas1 and Cas2. (Supplementary Table S3). Cas1 and Cas2 are part of a complete type II CRISPR-Cas system including Cas9 and a downstream CRISPR array containing 22 spacers that are located at 1,593,570–1,595,058 bp.
Furthermore, we compared the H. typhlonius genome against all other Helicobacter genomes available in The SEED genome database (Overbeek et al., 2005). A collection of 38 annotated PEGs with diverse functions was exclusively found in H. typhlonius (Supplementary Table S4). This set of PEGs determines the uniqueness of the H. typhlonius genome, representing 2.1% of this genome.
Base Modifications and Associated Motifs
We have identified components of R-M systems in the H. typhlonius genome, some of which are present in H. hepaticus ATCC 51449 and H. pylori strain Shi470 as well (Supplementary Table S5). Many putative DNA methyltransferases (MTases) were found, indicating that it should be possible to detect different types of methylation. Of the 18 DNA MTases, 9 orthologs were also present in H. hepaticus ATCC 51449 (average 84.2%AAI) and 11 orthologs were found in H. pylori Shi470 (average 46.6% AAI) (Supplementary Table S6). This suggests that the three organisms may have specific methylation patterns in common and may thus share similar gene regulation, host interaction, pathogenicity or cellular defense systems. Furthermore, we could find 15 putative RNA MTases, all of which were also found in H. hepaticus (average 68.6% AAI), while 13 were seen in H. pylori Shi470 (average 47.5% AAI) (Supplementary Table S7).
Genome-wide analysis of polymerase kinetics during SMRT sequencing enabled the detection of methylated adenine and cytosine bases. The DNA did not receive Tet1 oxidation treatment prior to SMRT sequencing since this requires further fragmentation of the sequencing library, which in turn is not suited for completing the genome of H. typhlonius. Without Tet1 treatment only N6-methyladenine (6mA) and 4-methylcytosine (4mC) signals could be reliably detected (Clark et al., 2013). Adenine bases showed a very distinct modification signal that corresponded well with the overall coverage depth on each strand (Supplementary Figures S5 and S6). We found 28,716 6mAs and 2,049 4mCs base modifications that were distributed across the genome. In total 27,399 methylated adenines (95.4%) and 1,977 methylated cytosines (73.7%) were associated with 8 putative MTase recognition motifs (Figure 4).
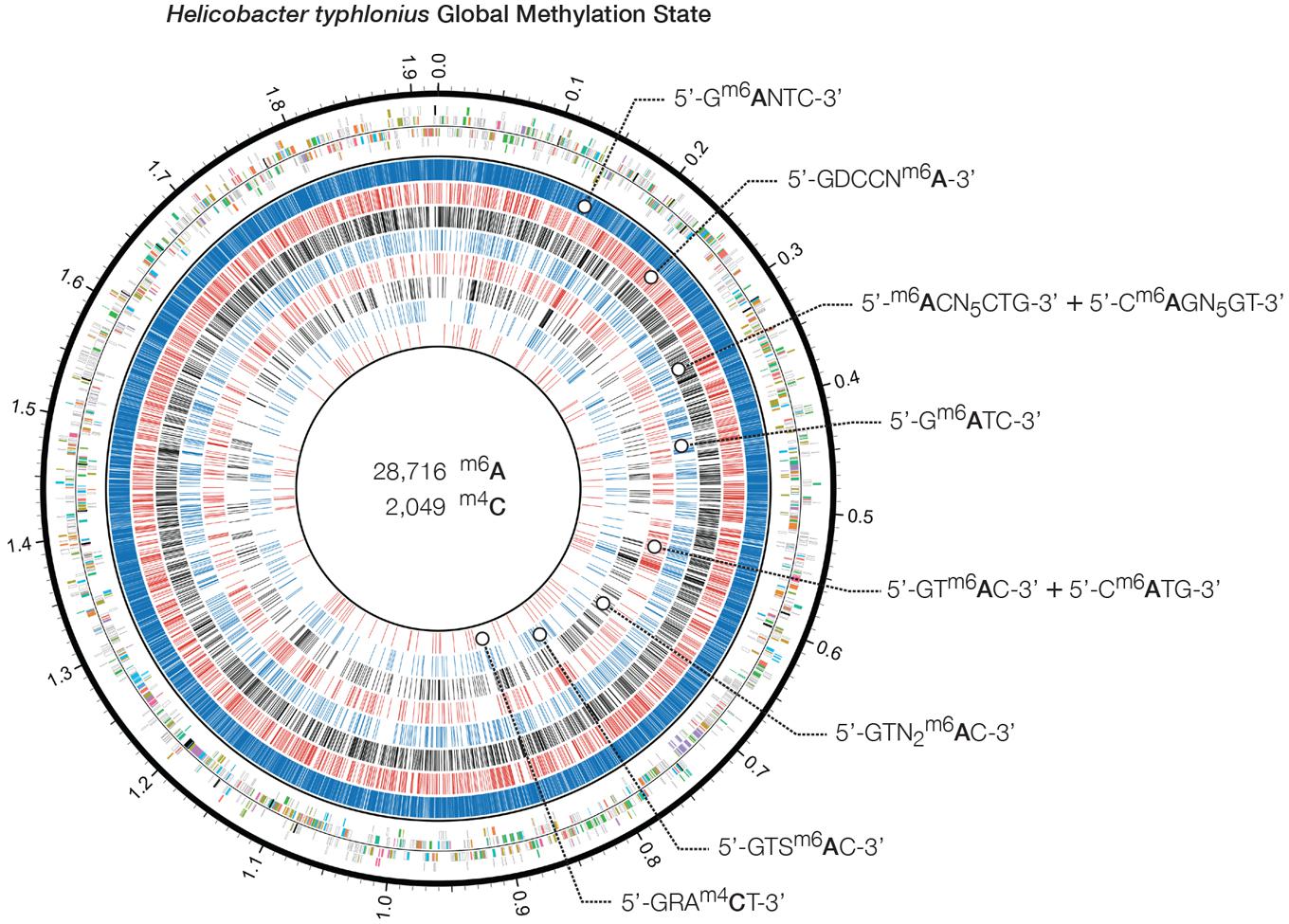
FIGURE 4. Heliobacter typhlonius global methylation state. Circos plot showing in the outermost ring the protein-encoding genes colored by associated subsystem. The following rings depict methylated adenines and cytosines that are associated with specific motifs. From outside to inside: 5′-Gm6ANTC-3′ (blue), 5′-GDCCNm6A-3′ (red), 5′-GRAm4CT-3′ (black), 5′-m6ACN5CTG-3′ and its partner-motif Cm6AGN5GT-3′ (blue), 5′-Gm6ATC-3′ (red), 5′-GTm6AC-3′ and its partner-motif 5′-Cm6ATG-3′ (black), 5′-GTN2m6AC-3′ (blue) and 5′-GTSm6AC-3′ (red).
Krebes et al. (2014) performed SMRT sequencing to conduct a comprehensive analysis on two H. pylori strains: 26695 and J99-R3. They demonstrated both pylori genomes are highly methylated, containing a large number of methyltransferases and restriction–modification systems (Krebes et al., 2014). In contrast, no methylation data is available for H. hepaticus, and only two complete R-M systems have been described (Suerbaum et al., 2003). Three out of 8 motifs could be found in both H. typhlonius and H. pylori strains 26695 and J99-R3: 5′-Gm6ANTC-3′, 5′-Gm6ATC-3′ and 5′-GTm6AC-3′ with its partner-motif (reverse-complement) Cm6ATG. One motif was found in H. typhlonius and in pylori strain J99-R3 only: 5′-GTSm6AC-3′. The three remaining motifs were found exclusively in H. typhlonius: 5′-GDCCNm6A-3′, 5′-m6ACN5CTG-3′ and its partner-motif Cm6AGN5GT and GTNNm6AC. Nearly all of the target sequences were completely methylated (>98%) and resided predominantly in the coding regions of the genome (Table 4). Sequence context analysis did not reveal any motifs associated with cytosine methylation.
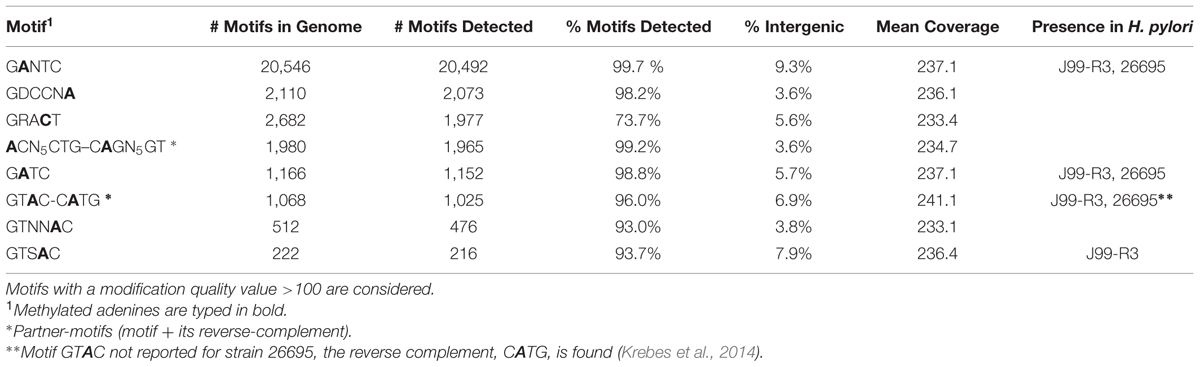
TABLE 4. Base modifications and motifs: adenine and cytosine motif statistics.
Conclusion
In this study, the complete genome sequence of H. typhlonius MIT 97-6810 enabled us to identify many pathogenic features (including a set of 19 possibly pathogenic proteins), the presence of CDTs, a putative PAI (containing components of a T4SS together with a cag protein) and multiple virulence factors. These findings suggest that H. typhlonius has the potential to act as a pathobiont.
Furthermore, we described the global methylation state of the genome. We found many methylated sites and discovered a diverse plethora of R-M systems. Methylation patterns differ among closely related species, nonetheless specific recognition motifs are conserved. Together with the genome, the methylome will provide an essential resource for forthcoming studies investigating gene regulation, host interaction, pathogenicity and cellular defense. Follow-up studies are necessary to investigate the pathophysiologic effects of H. typhlonius and to evaluate the true nature of its pathogenic capabilities. In turn, these findings can contribute to unraveling the role of Helicobacter in enteric disease.
Materials and Methods
Genomic DNA Preparation
The H. typhlonius strain MIT 97-6810 has been isolated from the cecal and fecal content of Il10−/− knockout mice with IBD by Fox et al. (1999). H. typhlonius was obtained from the Culture Collection, University of Gothenburg, Sweden (CCUG 48335T) and was grown micro-aerobically on Biomerieux chocolate agar + PolyViteX (PVX) plates (Mediaproducts, Groningen, The Netherlands) for 2–3 days at 37°C (Franklin et al., 2001). Genomic DNA was extracted using the MOBIO Ultraclean Fecal kit (Sanbio, Uden, The Netherlands) according to the manufacturer’s instructions, combined with phenol–chloroform extraction and RNase A treatment.
Sequencing
Genomic DNA was fragmented with G-tubes (Covaris), end-repaired and SMRTbell DNA template libraries (insert size of ∼20 Kb) were prepared according to the manufacturer’s specification. SMRT sequencing (3 SMRT cells) was performed on the Pacific Biosciences RSII sequencer according to standard protocols (MagBead Standard Seq v2 loading, 1 × 180 min movie) using the P4-C2 chemistry.
De Novo Genome Assembly
Continuous long reads were attained from three SMRT sequencing runs. Reads longer than 500 bp with a quality value over 0.75 were merged together into a single dataset. Next, the hierarchical genome-assembly process (HGAP) pipeline (Chin et al., 2013) was used to correct for random errors in the long seed reads (seed length threshold 6 Kb) by aligning shorter reads from the same library against them. The resulting corrected, preassembled reads were used for de novo assembly using Celera Assembler 8.1 (Myers et al., 2000). Celera Assembler employs an overlap-layout-consensus (OLC) strategy that is well suited for the use of long, corrected PacBio reads. Since SMRT sequencing features very little variations of the quality throughout the reads (Koren et al., 2012), no quality values were used during the assembly. Default parameters were employed while using the BOGART unitigger and setting the merSize to 14 (configuration settings are provided in Supplementary File S1). To validate the quality of the assembly and determine the final genome sequence, the Quiver consensus algorithm (Chin et al., 2013) was used. Quiver takes advantage of all information from the raw pulse and base-calls that are generated during the SMRT sequencing to infer the most accurate consensus sequence (Chin et al., 2013). Finally, the ends of the assembled sequence were trimmed to have the genome circularized.
Annotations
The location of the origin of replication site (oriC) was predicted using the Ori-Finder web service (Gao and Zhang, 2008). Ori-Finder was configured to search for Helicobacter specific DnaA boxes while allowing for two unmatched sites. In addition, the DoriC database (Gao et al., 2013) holding prokaryote oriC data was used to select the most likely candidate oriC amongst the Ori-Finder results. Annotation of the assembled genome was performed using RAST prokaryotic genome annotation service (Aziz et al., 2008). Additional annotation was carried out using several web services offered by the Center for Genomic Epidemiology. ResFinder 2.1 (Zankari et al., 2012), PathogenFinder 1.1 (Cosentino et al., 2013) and VirulenceFinder 1.2 (Joensen et al., 2014) were used for the prediction of acquired antimicrobial resistance genes, potential pathogenic features and virulence genes respectively. PHAST (PHAge Search Tool) (Zhou et al., 2011) was used to detect and annotate prophage sequences in the assembled genome. CRISPRs were identified using the CRISPRFinder web tool (Grissa et al., 2007). Genomic repeats and other structural variations were identified using NUCmer (Kurtz et al., 2004) and filtered according to length threshold of 500 bp and 95% copy identity. Tandem repeats were identified separately using Tandem Repeat Finder online service (Benson, 1999).
Comparative Genome Analysis
The final genome sequence of H. typhlonius was compared to the genome sequences of two other Helicobacter species: murine H. hepaticus ATCC 51449 (Suerbaum et al., 2003) and human H. pylori 26695 (Krebes et al., 2014). RAST/The SEED was used to infer the conservation of annotated genes and pathways. BLAST (Altschul et al., 1990) searches using default parameters were performed to identify regions of interest.
Base Modification Analysis
The DNA did not receive Tet1 oxidation treatment prior to SMRT sequencing, meaning only N6-methyladenine (6mA) and 4-methylcytosine (4mC) signals could be reliably detected (Clark et al., 2013). All reads were aligned to the assembled genome. Kinetic signals detected during SMRT sequencing were processed for all genomic positions using a previously described protocol (Flusberg et al., 2010; Clark et al., 2012). The Pacific Biosciences SMRT Portal analysis platform 2.3.0 was used to identify modified bases and associated motifs. The DNA base modification analysis uses an in silico kinetic model and a t-test based scoring system to detect modified bases. In order to accurately identify methylated bases, a threshold of 100 for log-transformed P-value was used. The threshold was optimized according to the distribution of P-values for different bases, minimizing the false positive rate. Additional data analysis was performed in R (R Core Team, 2015).
Author Contributions
JF and SA performed the analyses. SA and ER-M designed the study. CD, AS, and RV performed library preparation and SMRT sequencing. SA, ER-M, G-JvO and JdD coordinated the study. JF drafted the manuscript that was subsequently revised by all co-authors.
Data Availability
The whole-genome shotgun SMRT sequencing reads of H. typhlonius MIT 97-6810 are deposited at the European Nucleotide Archive (ENA) under study ID PRJEB10402 (http://www.ebi.ac.uk/ena/data/view/PRJEB10402). The complete genome sequence and annotation can be retrieved using chromosome accession number LN907858 (http://www.ebi.ac.uk/ena/data/view/LN907858). RAST annotation data is accessible through The Seed Viewer, filed under Genome ID 76936.6. RAST guest account can be used to access all the files that were generated during the annotation and comparative genomics (username: guest; password: guest).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work was supported by the Valorisation Fund of the CMSB through grants to ER-M.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.01549
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Aziz, R. K., Bartels, D., Best, A. A., Dejongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Che, D., Hasan, M. S., and Chen, B. (2014). Identifying pathogenicity islands in bacterial pathogenomics using computational approaches. Pathogens 3, 36–56. doi: 10.3390/pathogens3010036
Chichlowski, M., Sharp, J. M., Vanderford, D. A., Myles, M. H., and Hale, L. P. (2008). Helicobacter typhlonius and Helicobacter rodentium differentially affect the severity of colon inflammation and inflammation-associated neoplasia in IL10-deficient mice. Comp. Med. 58, 534–541.
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Clark, T. A., Lu, X., Luong, K., Dai, Q., Boitano, M., Turner, S. W., et al. (2013). Enhanced 5-methylcytosine detection in single-molecule, real-time sequencing via Tet1 oxidation. BMC Biol. 11:4. doi: 10.1186/1741-7007-11-4
Clark, T. A., Murray, I. A., Morgan, R. D., Kislyuk, A. O., Spittle, K. E., Boitano, M., et al. (2012). Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 40, e29. doi: 10.1093/nar/gkr1146
Cosentino, S., Voldby Larsen, M., Moller Aarestrup, F., and Lund, O. (2013). PathogenFinder–distinguishing friend from foe using bacterial whole genome sequence data. PLoS ONE 8:e77302. doi: 10.1371/journal.pone.0077302
Dingemanse, C., Belzer, C., Van Hijum, S. A. F. T., Günthel, M., Salvatori, D., Den Dunnen, J., et al. (2015). Akkermansia muciniphila and Helicobacter typhlonius modulate intestinal tumor development in mice. Carcinogenesis 36, 1388–1396. doi: 10.1093/carcin/bgv120
Dreiseikelmann, B. (1994). Translocation of DNA across bacterial membranes. Microbiol. Rev. 58, 293–316.
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
EMBL European Bioinformatics Institute [EMBL-EBI] (2014). Genomes Pages – Bacteria. Available at: http://www.ebi.ac.uk/genomes/bacteria.html [Accessed November 15, 2015].
Euzeby, J. P. (1997). List of bacterial names with standing in nomenclature: a folder available on the internet. Int. J. Syst. Bacteriol. 47, 590–592. doi: 10.1099/00207713-47-2-590
Flusberg, B. A., Webster, D. R., Lee, J. H., Travers, K. J., Olivares, E. C., Clark, T. A., et al. (2010). Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465. doi: 10.1038/nmeth.1459
Fox, J. G., Dewhirst, F. E., Tully, J. G., Paster, B. J., Yan, L., Taylor, N. S., et al. (1994). Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J. Clin. Microbiol. 32, 1238–1245.
Fox, J. G., Ge, Z., Whary, M. T., Erdman, S. E., and Horwitz, B. H. (2011). Helicobacter hepaticus infection in mice: models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 4, 22–30. doi: 10.1038/mi.2010.61
Fox, J. G., Gorelick, P. L., Kullberg, M. C., Ge, Z., Dewhirst, F. E., and Ward, J. M. (1999). A novel urease-negative Helicobacter species associated with colitis and typhlitis in IL-10-deficient mice. Infect. Immun. 67, 1757–1762.
Franklin, C. L., Gorelick, P. L., Riley, L. K., Dewhirst, F. E., Livingston, R. S., Ward, J. M., et al. (2001). Helicobacter typhlonius sp. nov., a novel murine urease-negative Helicobacter species. J. Clin. Microbiol. 39, 3920–3926. doi: 10.1128/JCM.39.11.3920-3926.2001
Fronzes, R., Christie, P. J., and Waksman, G. (2009). The structural biology of type IV secretion systems. Nat. Rev. Microbiol. 7, 703–714. doi: 10.1038/nrmicro2218
Gao, F., Luo, H., and Zhang, C. T. (2013). DoriC 5.0: an updated database of oriC regions in both bacterial and archaeal genomes. Nucleic Acids Res. 41, D90–D93. doi: 10.1093/nar/gks990
Gao, F., and Zhang, C. T. (2008). Ori-Finder: a web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinformatics 9:79. doi: 10.1186/1471-2105-9-79
Ge, Z., Feng, Y., Whary, M. T., Nambiar, P. R., Xu, S., Ng, V., et al. (2005). Cytolethal distending toxin is essential for Helicobacter hepaticus colonization in outbred Swiss Webster mice. Infect. Immun. 73, 3559–3567. doi: 10.1128/IAI.73.6.3559-3567.2005
Ge, Z., Schauer, D. B., and Fox, J. G. (2008). In vivo virulence properties of bacterial cytolethal-distending toxin. Cell. Microbiol. 10, 1599–1607. doi: 10.1111/j.1462-5822.2008.01173.x
Gonzalez, D., Kozdon, J. B., Mcadams, H. H., Shapiro, L., and Collier, J. (2014). The functions of DNA methylation by CcrM in Caulobacter crescentus: a global approach. Nucleic Acids Res. 42, 3720–3735. doi: 10.1093/nar/gkt1352
Goodwin, C. S., Armstrong, J. A., Chilvers, T., Peters, M., Collins, M. D., Sly, L., et al. (1989). Transfer of Campylobacter pylori and Campylobacter mustelae to Helicobacter gen. nov. as Helicobacter pylori comb. nov. and Helicobacter mustelae comb. nov., Respectively. Int. J. Syst. Bacteriol. 39, 397–405. doi: 10.1099/00207713-39-4-397
Graham, D. Y. (1989). Campylobacter pylori: defining a cause of gastritis and peptic ulcer disease. Proceedings from a symposium of the 13th International Congress of Gastroenterology. Rome, 7 September 1988. Scand. J. Gastroenterol. Suppl. 160, 1–68.
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Hamilton, H. L., and Dillard, J. P. (2006). Natural transformation of Neisseria gonorrhoeae: from DNA donation to homologous recombination. Mol. Microbiol. 59, 376–385. doi: 10.1111/j.1365-2958.2005.04964.x
Jeltsch, A. (2003). Maintenance of species identity and controlling speciation of bacteria: a new function for restriction/modification systems? Gene 317, 13–16. doi: 10.1016/S0378-1119(03)00652-8
Joensen, K. G., Scheutz, F., Lund, O., Hasman, H., Kaas, R. S., Nielsen, E. M., et al. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. doi: 10.1128/JCM.03617-13
Koren, S., Schatz, M. C., Walenz, B. P., Martin, J., Howard, J. T., Ganapathy, G., et al. (2012). Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 30, 693–700. doi: 10.1038/nbt.2280
Krebes, J., Morgan, R. D., Bunk, B., Sproer, C., Luong, K., Parusel, R., et al. (2014). The complex methylome of the human gastric pathogen Helicobacter pylori. Nucleic Acids Res. 42, 2415–2432. doi: 10.1093/nar/gkt1201
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5, R12. doi: 10.1186/gb-2004-5-2-r12
Lofgren, J. L., Esmail, M., Mobley, M., Mccabe, A., Taylor, N. S., Shen, Z., et al. (2012). Prevalence of murine Helicobacter spp. Infection is reduced by restocking research colonies with Helicobacter-free mice. J. Am. Assoc. Lab. Anim. Sci. 51, 436–442.
Marshall, B. J. (1993). Treatment strategies for Helicobacter pylori infection. Gastroenterol. Clin. North Am. 22, 183–198.
Menard, A., Pere-Vedrenne, C., Haesebrouck, F., and Flahou, B. (2014). Gastric and enterohepatic Helicobacters other than Helicobacter pylori. Helicobacter 19(Suppl. 1), 59–67. doi: 10.1111/hel.12162
Myers, E. W., Sutton, G. G., Delcher, A. L., Dew, I. M., Fasulo, D. P., Flanigan, M. J., et al. (2000). A whole-genome assembly of Drosophila. Science 287, 2196–2204. doi: 10.1126/science.287.5461.2196
National Center for Biotechnology Information [NCBI] (2014). Genome Information by Organism. Available at: http://www.ncbi.nlm.nih.gov/genome/browse/ [Accessed November 15 2014].
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H. Y., Cohoon, M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702. doi: 10.1093/nar/gki866
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Parker, S. E., Malone, S., Bunte, R. M., and Smith, A. L. (2009). Infectious diseases in wild mice (Mus musculus) collected on and around the University of Pennsylvania (Philadelphia) Campus. Comp. Med. 59, 424–430.
Parsonnet, J. (1995). Bacterial infection as a cause of cancer. Environ. Health Perspect. 103(Suppl. 8), 263–268. doi: 10.2307/3432323
Pratt, J. S., Sachen, K. L., Wood, H. D., Eaton, K. A., and Young, V. B. (2006). Modulation of host immune responses by the cytolethal distending toxin of Helicobacter hepaticus. Infect. Immun. 74, 4496–4504. doi: 10.1128/IAI.00503-06
R Core Team (2015). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Roberts, R. J., Vincze, T., Posfai, J., and Macelis, D. (2015). REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 43, D298–D299. doi: 10.1093/nar/gku1046
Sheh, A., Shen, Z., and Fox, J. G. (2014). Draft genome sequences of eight enterohepatic Helicobacter species isolated from both laboratory and wild rodents. Genome Announc. 2, e1218–e1214. doi: 10.1128/genomeA.01218-14
Suerbaum, S., Josenhans, C., Sterzenbach, T., Drescher, B., Brandt, P., Bell, M., et al. (2003). The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc. Natl. Acad. Sci. U.S.A. 100, 7901–7906. doi: 10.1073/pnas.1332093100
Terradot, L., and Waksman, G. (2011). Architecture of the Helicobacter pylori Cag-type IV secretion system. FEBS J. 278, 1213–1222. doi: 10.1111/j.1742-4658.2011.08037.x
Wasimuddin, Cizkova, D., Bryja, J., Albrechtova, J., Hauffe, H. C., and Pialek, J. (2012). High prevalence and species diversity of Helicobacter spp. detected in wild house mice. Appl. Environ. Microbiol. 78, 8158–8160. doi: 10.1128/AEM.01989-12
Whary, M. T., and Fox, J. G. (2004). Natural and experimental Helicobacter infections. Comp. Med. 54, 128–158.
Wion, D., and Casadesus, J. (2006). N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 4, 183–192. doi: 10.1038/nrmicro1350
Young, V. B., Knox, K. A., Pratt, J. S., Cortez, J. S., Mansfield, L. S., Rogers, A. B., et al. (2004). In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect. Immun. 72, 2521–2527. doi: 10.1128/IAI.72.5.2521-2527.2004
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zenner, L. (1999). Pathology, diagnosis and epidemiology of the rodent Helicobacter infection. Comp. Immunol. Microbiol. Infect. Dis. 22, 41–61. doi: 10.1016/S0147-9571(98)00018-6
Keywords: Helicobacter typhlonius, genome assembly, single-molecule real-time sequencing, Pacific Biosciences, pathogenicity, methylation
Citation: Frank J, Dingemanse C, Schmitz AM, Vossen RHAM, van Ommen G-JB, den Dunnen JT, Robanus-Maandag EC and Anvar SY (2016) The Complete Genome Sequence of the Murine Pathobiont Helicobacter typhlonius. Front. Microbiol. 6:1549. doi: 10.3389/fmicb.2015.01549
Received: 22 September 2015; Accepted: 21 December 2015;
Published: 08 January 2016.
Edited by:
Jae-Ho Shin, Kyungpook National University, South KoreaReviewed by:
Seong Woon Roh, Korea Basic Science Institute, South KoreaEmiley Eloe-Fadrosh, Joint Genome Institute, USA
Copyright © 2016 Frank, Dingemanse, Schmitz, Vossen, van Ommen, den Dunnen, Robanus-Maandag and Anvar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seyed Yahya Anvar, s.y.anvar@lumc.nl