 Dongshan An
Dongshan An Xiaoli Dong
Xiaoli Dong Annie An
Annie An Hyung S. Park
Hyung S. Park Marc Strous
Marc Strous Gerrit Voordouw
Gerrit Voordouw- 1Petroleum Microbiology Research Group, Department of Biological Sciences, University of Calgary, Calgary, AB, Canada
- 2Department of Geosciences, University of Calgary, Calgary, AB, Canada
Sodium bisulfite (SBS) is used as an oxygen scavenger to decrease corrosion in pipelines transporting brackish subsurface water used in the production of bitumen by steam-assisted gravity drainage. Sequencing 16S rRNA gene amplicons has indicated that SBS addition increased the fraction of the sulfate-reducing bacteria (SRB) Desulfomicrobium, as well as of Desulfocapsa, which can also grow by disproportionating sulfite into sulfide, sulfur, and sulfate. SRB use cathodic H2, formed by reduction of aqueous protons at the iron surface, or use low potential electrons from iron and aqueous protons directly for sulfate reduction. In order to reveal the effects of SBS treatment in more detail, metagenomic analysis was performed with pipe-associated solids (PAS) scraped from a pipe section upstream (PAS-616P) and downstream (PAS-821TP) of the SBS injection point. A major SBS-induced change in microbial community composition and in affiliated hynL genes for the large subunit of [NiFe] hydrogenase was the appearance of sulfur-metabolizing Epsilonproteobacteria of the genera Sulfuricurvum and Sulfurovum. These are chemolithotrophs, which oxidize sulfide or sulfur with O2 or reduce sulfur with H2. Because O2 was absent, this class likely catalyzed reduction of sulfur (S0) originating from the metabolism of bisulfite with cathodic H2 (or low potential electrons and aqueous protons) originating from the corrosion of steel (Fe0). Overall this accelerates reaction of of S0 and Fe0 to form FeS, making this class a potentially powerful contributor to microbial corrosion. The PAS-821TP metagenome also had increased fractions of Deltaproteobacteria including the SRB Desulfomicrobium and Desulfocapsa. Altogether, SBS increased the fraction of hydrogen-utilizing Delta- and Epsilonproteobacteria in brackish-water-transporting pipelines, potentially stimulating anaerobic pipeline corrosion if dosed in excess of the intended oxygen scavenger function.
Introduction
Pipeline failure caused by corrosion can have serious consequences for the oil and gas industry (Ossai et al., 2015) and understanding the causes of corrosion is therefore important. While chemical reaction between oxygen and iron is the main cause of external pipeline corrosion, microbiologically influenced corrosion (MIC) under mostly anoxic conditions can account for up to 40% of internal pipeline corrosion in the oil and gas industry (Zhu et al., 2003). The application of high-throughput sequencing technologies has indicated that diverse microbes are involved in internal pipeline corrosion, such as sulfate reducing bacteria (SRB), acid-producing fermentative bacteria, including acetogens, as well as methanogens (Dinh et al., 2004; Park et al., 2011; Mand et al., 2014; Okoro et al., 2014; Yang et al., 2014). Among these SRB are often considered to be the major MIC causative agents.
Fundamentally all SRB can corrode iron indirectly by producing the corrosive chemical agent hydrogen sulfide (H2S). This has been referred to as “chemical microbially-influenced corrosion” (CMIC) by Enning and Garrelfs (2014). H2S is produced during sulfate reduction by SRB with electrons usually derived from organic acids, alcohols, or hydrogen (H2), which is formed by fermentation of organic compounds in anoxic settings (Muyzer and Stams, 2008). In the absence of organic electron donors and in the presence of metallic iron, SRB may obtain energy from oxidation of cathodic H2 formed by chemical reaction between protons from water and electrons from anodic dissolution of iron, accelerating corrosion (Mand et al., 2014). Whether SRB are capable of accelerating corrosion by scavenging cathodic H2, which was proposed long ago (Von Wolzogen Kühr and Van der Vlugt, 1934), is still controversial (Enning and Garrelfs, 2014). Instead, some SRB are thought to corrode iron through direct uptake of the anodic electrons with protons from water for sulfate reduction (“electrical microbially-influenced corrosion”; EMIC; Enning and Garrelfs, 2014).
The microbial consumption or production of H2 is catalyzed by hydrogenases that can be divided into two main phylogenetically unrelated groups, the [NiFe]- and [FeFe]-hydrogenases (Vignais and Billoud, 2007). [NiFe]-hydrogenases are common in Archaea and Bacteria. Many are external to the cytoplasm and are primarily associated with H2 oxidation in oxic and anoxic metabolism. [FeFe]-hydrogenases are found in Bacteria and Eukarya. These enzymes are especially prevalent in the cytoplasm of anaerobic fermentative organisms (e.g., the Firmicutes), where they form hydrogen in metabolic reactions coupling the oxidation of reduced electron carriers (NADH, NADPH or reduced ferredoxin, Fdred) to the reduction of protons. SRB of the genus Desulfovibrio are exceptional in having a periplasmic [FeFe]-hydrogenase, which functions in hydrogen oxidation at high hydrogen concentration (Caffrey et al., 2007). Hence, assuming that consumption of cathodic hydrogen is important in MIC, both types of hydrogenases can contribute. However, [NiFe]-hydrogenases may be more important than [FeFe] hydrogenases because these enzymes are more widespread and act at lower hydrogen concentrations. Likewise, if metabolism of anodic electrons and aqueous protons in EMIC involves formation of H2 both types of enzymes may contribute. In addition to SRB, hydrogenotrophic methanogens and acetogens have been found to contribute to MIC by using cathodic H2 or anodic electrons for reduction of CO2 to methane and acetate, respectively (Dinh et al., 2004; Mand et al., 2014).
As indicated previously (Park et al., 2011), the presence of MIC-causing SRB can be promoted by injection of sodium bisulfite (SBS), which is used as an oxygen scavenger to decrease oxygen-mediated corrosion in pipelines and other steel infrastructure. Injection of SBS into pipelines transporting brackish subsurface water to a plant generating steam for production of bitumen by steam-assisted gravity drainage caused a drastic change in microbial community composition of pipe-associated solids (PAS). Relative to solids from a pipe section upstream of the SBS injection point (PAS-616P), solids from a downstream pipe section (PAS-821TP) had a smaller fraction of methanogens of the family Methanobacteriaceae and larger fractions of SRB of the genera Desulfomicrobium and Desulfocapsa (Park et al., 2011). Desulfocapsa can also grow by disproportionating bisulfite into sulfide and sulfate (Finster, 2008). Here we evaluate the genetic potential of the microbial communities in these two PAS samples in more detail by an in depth metagenomic analysis with a focus on hydrogenase genes.
Materials and Methods
Sample Collection
Two cutouts from a brackish water-transporting pipeline system were collected upstream (616P) and downstream (821TP) from the SBS injection point. These were the same as described elsewhere (Park et al., 2011). The pipeline cutouts were immersed in pipe-associated water (PAW) from the site, were shipped in sealed, airtight buckets and received in the lab within 24 h. The cutouts and the associated waters were then immediately transferred to a Coy anaerobic hood with an atmosphere of 90% (v/v) N2 and 10% CO2. PAS-616P and PAS-821TP were obtained by scraping the drained surface of the cutouts with a sterile spatula. These were then re-suspended in 260 mL of PAW-616P and PAW-821TP, respectively, filtered using an 0.2 μm Millipore filter (Nylon membrane, USA) prior to use.
Chemical analyses conducted on the samples included the measurement of pH, sulfide (Trüper and Schlegel, 1964), sulfate (ion chromatography with conductivity detector/anion column), ammonium, nitrite (ion chromatography with UV detector/anion column), and organic acids (ion chromatography with UV detector/organic acids column), as detailed else where (Park et al., 2011).
DNA Isolation
DNA was extracted from the PAS samples using a bead-beating procedure outlined by the manufacturer of the FastDNA® Spin Kit for Soil (MP Biomedicals). The extracted DNA was further purified by cesium chloride density gradient centrifugation. The concentration of DNA was quantified using the Qubit Fluorometer, and Quant-iT™ dsDNA HS Assay Kit (Invitrogen). A total of 20.5 and 25.8 μg of CsCl-purified DNAs were obtained from PAS-821TP and PAS-616P, respectively. The purified DNAs were then used for pyrosequencing of 16S rRNA gene (16S) amplicons and for metagenome sequencing.
Pyrosequencing of 16S Amplicons
Amplification of 16S genes was with non-barcoded 16S primers 926Fw (AAACTYAAAKGAATTGRCGG) and 1392R (ACGGGCGGTGTGTRC) in the first PCR and with FLX titanium amplicon primers 454_RL_X and 454T_FwB in the second PCR. The latter primers have the sequences for 926Fw and 1392R as their 3′ ends. Primer 454T_RL_X has a 25 nucleotide A-adaptor (CCATCTCATCCCTGCGTGTCTCCGAC) and a 10 nucleotide multiplex identifier barcode sequence X. Primer 454T_FwB has a 25 nucleotide B-adaptor sequence (CTATGCGCCTTGCCAGCCCGCTCAG). The first PCR was run for 5 min at 95°C, followed by 25 cycles of 30 s at 95°C, 45 s at 55°C, and 90 s at 72°C and finally 10 min at 72°C. The PCR products were used as templates for a second PCR of 10 cycles under the same conditions. PCR products were checked on an agarose gel and purified with a QIAquick PCR Purification kit (Qiagen). The amounts of purified 16S amplicons were then normalized to 20 μl of 20 ng/μl and sent for pyrosequencing to the Genome Quebec and McGill University Innovation Centre (Montreal, QC). Pyrosequencing was performed in a Genome Sequencer FLX Instrument, using a GS FLX Titanium Series Kit XLR70 (Roche Diagnostics Corporation). The 16S sequence reads were analyzed with Phoenix 2 (Soh et al., 2013).
Metagenome Sequencing
Metagenome sequencing was performed with both 454- and Illumina-platforms at the Genome Quebec and McGill University Innovation Centre. For the 454 sequencing, single-end shotgun DNA libraries were prepared from 1.0 μg of purified DNA and sequenced with the 454 Life Sciences GS-FLX genome sequencer using titanium chemistry and standard library construction procedures (Roche Applied Science, Laval, Quebec, Canada).
For the Illumina sequencing, DNA libraries were prepared using the TruSeq DNA Sample Prep Kit v1 (Illumina, San Diego, CA) as per the manufacturer's instructions starting with 2 μg of purified DNA. The libraries were loaded onto the flow cell, one per lane using a cBot (Illumina). Sequencing to obtain paired end 150 bp reads was performed on a HiSeq 2000 instrument (Illumina) as per the manufacturer's instructions.
Metagenomic Analysis and Assembly
All 454 and Illumina metagenomic raw reads were subjected to quality control (QC) and an assembly process using the in-house developed software described by Saidi-Mehrabad et al. (2013) and Tan et al. (2013). Ribosomal RNA genes were identified with Meta-RNA (Huang et al., 2009) from 454 QC reads. All raw 454 and Illumina sequence data were submitted to the Short Read Archive under accession numbers SRX559897, SRX559898, SRX559901, and SRX559902.
Gene Abundance Analysis
Genes for large hydrogenase subunits (referred to as hydL for [FeFe]-hydrogenase and hynL for [NiFe] hydrogenase) and the house keeping gene rpoB, the gene for the RNA polymerase β subunit, were sought in the six-frame translated metagenomic contigs using hmmsearch with a cutoff e-value of e-5. The hydL (PF02906) and hynL (PF00374) hidden Markov models (HMMs) were downloaded from the Pfam database (Finn et al., 2014) and the rpoB (TIGR02013, TIGR03670) HMMs were downloaded from TIGRFAMs (Haft et al., 2013). In the aligned region, the translated amino-acid sequences and the corresponding nucleotide sequences were extracted and included in the further gene abundance and phylogenetic analyses.
The QC metagenomic reads were mapped against the hydL, hynL, and rpoB nucleotide gene sequences using bowtie2 (Langmead and Salzberg, 2012) and the mapping files were processed using Picard with “MarkDuplicates” options (http://broadinstitute.github.io/picard/) to remove PCR duplicates. The BEDTools (Quinlan and Hall, 2010) and in-house perl scripts were then used to calculate coverage information for the extracted genes. The coverages were subsequently normalized to the length of the respective genes to make sure that longer genes did not have higher coverage values just because of their length. The length-normalized coverages were then further normalized to the total number of rpoB genes in the metagenomic samples, yielding an approximation of the number of genes per genome for each extracted hydrogenase genes.
Phylogenetic Analysis
Phylogenetic trees were constructed for hynL genes obtained from the PAS-616P and PAS-821TP metagenomes with a multi-step approach using reference alignments and trees in order to minimize errors and biases introduced by the fragmentary and non-overlapping nature of the metagenomic sequences (Brazelton et al., 2012). The reference multiple sequence alignment was constructed from multiple sequence alignments generated in other work (Vignais et al., 2001) by using mafft version 7.245 with “–merge” option (Katoh and Standley, 2013). Unaligned metagenomic fragments were added to the reference alignment profile using mafft with “–add” option in order to avoid alteration of the relative positions of residues in the reference alignment. Next, a bootstrapped maximum-likelihood tree was constructed from the reference-only alignment using the “-f a” algorithm in RAxML version 8.2.3 (Stamatakis, 2014). The reference-only tree with the highest-likelihood was used as a constraint tree (“-r” flag in RAxML) for 100 inferences from the full alignment (including metagenomic fragments) by RAxML, and bootstrap support values were drawn on the highest-likelihood tree (Brazelton et al., 2012).
Results
Chemical Characteristics of Pipe Samples
The pipe associated water (PAW) of the 616P and 821TP cutouts had a neutral pH and a sulfate concentration of 0.01 mM (Table S1). No sulfide was observed in either water sample. Higher concentrations of sulfate were observed in suspended PAS samples PAS-616P (0.33 mM) and PAS-821TP (0.09 mM). Passage through the SBS injection point increased the sulfide concentration from 0 mM in PAS-616P to 90 mM in PAS-821TP and the ferrous iron concentration from 1.0 mM in PAS-616P to 1080 mM in PAS-821TP (Table S1). Acetate (0.85 mM), butyrate (0.20 mM), and propionate (0.17 mM) were detected in PAW-616P, but not in PAS-616P. Acetate (0.80 mM) and propionate (0.10 mM) were also detected in PAS-821TP, but not in PAW-821TP (Table S1). These data indicate that the chemical compositions of PAS-616P and PAS-821TP were different (reflecting SBS injection) and that these differed in turn from those of the corresponding planktonic samples.
Community Compositions from 16S Amplicon Sequencing
The 454 pyrosequencing platform produced 20,451 good 16S reads for the two PAS samples (Table 1). These were grouped into 141 operational taxonomic units (OTUs) at a sequence dissimilarity cut-off of 5%. The average Good's coverage of 99.7% suggested that the majority of the phylotypes present in the samples had been identified. A total of 76 taxa was found in the two samples. The microbial community diversity of PAS-616P was slightly lower than that of PAS-821TP, as reflected by the estimated OTUs (Chao) and Shannon index (Table 1). PAS-616P had 75% Archaea and 20% Deltaproteobacteria, whereas PAS-821TP had 50% Archaea and 46% Deltaproteobacteria (Table 1). The increase in Deltaproteobacteria, ferrous iron, and sulfide from PAS-616P to PAS-821TP, likely resulted from SBS injection.
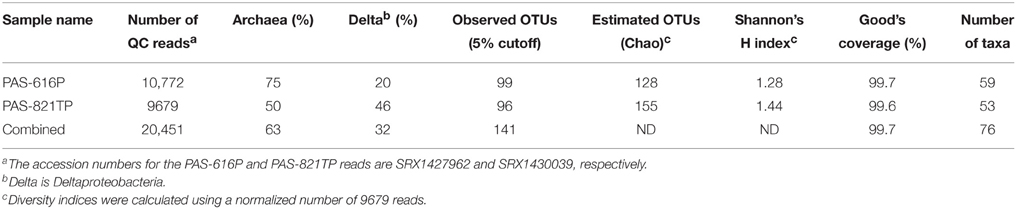
Table 1. Summary of 16S amplicon sequence data and derived diversity parameters for CsCl-purified DNAs from PAS samples also used for metagenomic analysis.
Methanogenic archaea can contribute to MIC in anoxic pipeline systems together with SRB and acetogens. Methanogenic orders in the two PAS samples included the hydrogenotrophic Methanobacteriales and Methanomicrobiales, as well as the Methanosarcinales. This included the Methanobacteriaceae (68.5 and 48.2%), Methanocalculus (0.05 and 0.01%), and the methylotrophic genus Methanolobus (6.4 and 1.6%) in PAS-616P to PAS-821TP, respectively (Table S2).
Five Deltaproteobacterial orders, Desulfovibrionales, Desulfobacterales, Desulfuromonadales, Myxococcales, and Syntrophobacterales, were detected in the 16S amplicons (Figure 1A). Of these the first two represent potential SRB. Desulfomicrobium was the dominant genus within the Desulfovibrionales with proportions of 14.1 and 28.9% in PAS-616P and PAS-821TP amplicon libraries, respectively (Table S2). Desulfomicrobium species use hydrogen or simple organic compounds, including formate, ethanol, lactate, pyruvate, malate, or fumarate, as electron donors for sulfate respiration. However, in the absence of sulfate they grow fermentatively on some of these same organic substrates (Rabus et al., 2015). Within the Desulfobacterales, Desulfocapsa was the dominant taxon with proportions of 5.3 and 16.3% in PAS-616P and PAS-821TP, respectively (Table S2). A unique feature of this genus is its ability to grow by disproportionation of thiosulfate, sulfite, or sulfur (in the presence of ferric iron) in the absence of molecular hydrogen. In the presence of molecular hydrogen, reduction of sulfur and of sulfur oxyanions dominates over disproportionation (Finster, 2008). Desulfuromonadales of the genus Desulfuromonas were detected at 0.14 and 0.01% in the PAS-616P and PAS-821TP amplicons, respectively (Table S1).
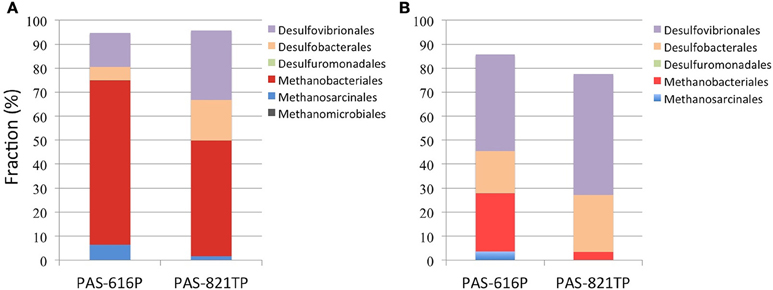
Figure 1. Order-level composition of Archaea and Deltaproteobacteria derived from (A) 16S amplicons and (B) 16S sequences of 454 metagenomes. The deltaproteobacterial orders Myxococcales and Syntrophobacterales are not obvious due to their low abundance in both libraries. The increased fractions of archaeal orders in (A) are likely caused by PCR bias.
Hence a survey of the 16S amplicons of the CsCl-purified DNAs confirmed earlier findings (Park et al., 2011), which indicated a decrease in methanogenic taxa and an increase in sulfate- and sulfite-reducing and sulfite-disproportionating taxa when passing from upstream to downstream of the SBS injection site.
Community Compositions from Metagenomic 16S Genes
Metagenome sequencing of the PAS samples was performed using both 454 and Illumina platforms. Because of the limitation of fragment sizes in Illumina sequencing, only the 454 16S rRNA gene sequences were used for the analysis of taxonomic profiles of the two metagenomes. A total of 270,379 and 337,937 reads with mean lengths of 659 and 654 bp were obtained for the PAS-616P and PAS-821TP metagenomes, respectively (Table 2). Among these 470 and 703 reads were identified as 16S fragments in the PAS-616P and PAS-821TP metagenomes (Table 2). This corresponds to one 16S read per 575 and 480 total reads, respectively. Taxonomic assignments of these 16S reads revealed that Deltaproteobacteria (58.3%) and Archaea (27.9%) dominated in the PAS-616P metagenome (Table 2). A larger fraction of Deltaproteobacteria (76.1%) and smaller fraction of Archaea (3.4%) was found in the PAS-821TP metagenome (Table 2). Hence these changed similarly as observed for the 16S amplicon libraries (Table 2).
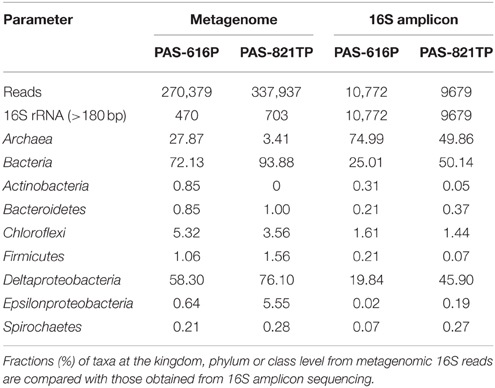
Table 2. Summary of 454 metagenomic data obtained for CsCl purified DNAs from the PAS samples.
Methanobacteriales (24.3%) and Methanosarcinales (3.6%) were the dominant methanogenic orders in the PAS-616P metagenome, while only Methanobacteriales (3.4%) were identified in the PAS-821TP metagenome (Figure 1). These included the Methanobacteriaceae and the genus Methanolobus (Table S2). The SRB were represented by the orders Desulfovibrionales (40.2%), Desulfobacterales (17.4%), and Desulfuromonadales (0.2%) in the PAS-616P metagenome, whereas only Desulfovibrionales (50.4%) and Desulfobacterales (23.8%) were detected in the PAS-821TP metagenome (Figure 1B). Desulfomicrobium and Desulfocapsa were the dominant genera in the orders Desulfovibrionales and Desulfobacterales in these two metagenomes (Table S2).
The frequency of 16S genes representing Epsilonproteobacteria increased from the PAS-616P to the PAS-821TP metagenome from 0.64 to 5.6% (Table 2). This increase was also seen in the 16S amplicon libraries (Table 2: from 0.02 to 0.19%). The smaller values of these fractions reflect the fact that the primers used did not optimally amplify this class (An et al., 2013). The Epsilonproteobacteria consisted mainly of the genera Sulfuricurvum (0.43%) and Arcobacter (0.21%) in the PAS-616P metagenome and of Sulfuricurvum (4.4%) and Sulfurovum (0.85%) in the PAS-821TP metagenome (Table S2). Sulfuricurvum and Sulfurovum are known to oxidize sulfide to sulfate (Kodama and Watanabe, 2004). This group of mainly chemolithotrophic bacteria may also reduce sulfur (S0) to sulfide using H2 as the electron donor (Gevertz et al., 2000).
Abundance of Hydrogenase Genes in PAS Metagenomes
Determining the prevalence of the hynL and hydL genes for the large subunits of [NiFe]- and [FeFe]-hydrogenase indicates the genetic potential of microbial communities for consuming or producing H2. In order to assess the effect of SBS treatment in the brackish water-transporting pipeline system on this potential, we performed hmmsearches for potential homologs of the hynL and hydL genes against contigs of the merged 454 and Illumina assemblies of the PAS-616P and PAS-821TP metagenomes. The aligned region of assembled contigs was extracted and the unassembled metagenomic reads that passed the QC were then mapped. To get the abundance profile of either hydrogenase gene, the total length of the mapped reads was calculated and normalized by the length of the respective genes. For comparative purposes, the length-normalized coverage was further normalized by dividing by the total number of rpoB genes (43,059 in the PAS-616P and 35,808 in the PAS-821TP metagenomes), yielding the fraction of genes per genome (abundance) for each of the extracted hydrogenase gene types (Table 3). The hydrogenase gene sequences, obtained from hmmsearches, were taxonomically identified as the best hit in blastp searches of the NCBI database. The phylogenetic association of these genes is also given as relative abundance (Table 3: %).
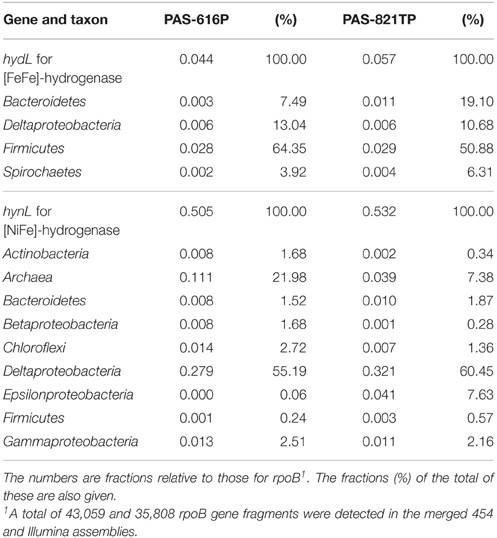
Table 3. Taxonomic origin and abundance of hydL genes for [FeFe]-hydrogenase and of hynL genes for [NiFe]-hydrogenase in the PAS metagenomes.
The PAS-616P and PAS-821TP metagenomes for samples taken upstream and downstream of the SBS injection point in the pipeline system, respectively, had a 10-fold higher abundance of hynL genes for [NiFe]-hydrogenase (Table 3: 0.505 and 0.532) than of hydL genes for [FeFe]-hydrogenase (0.044 and 0.057). The abundance of both types of hydrogenase genes was similar in the PAS-616P and PAS-821TP metagenomes (Table 3). The phylogenetic association of hydL genes was similar with most (Table 3: 64 and 51%) belonging to the Firmicutes, consistent with a function in H2 production in both metagenomes. In contrast, archaeal hynL genes decreased from 22.0 to 7.3%, whereas deltaproteobacterial hynL genes increased from 55.2 to 60.4% in transitioning from the PAS-626P to the PAS-821TP metagenome (Table 3). These changes were similar to those observed for metagenome-derived 16S (Table 2). Bisulfite injection caused a large increase in hynL genes affiliated with Epsilonproteobacteria from 0.06 to 7.63% (Table 3), matching the increase in metagenome-derived 16S (from 0.64 to 5.6%). This indicates this class to have a strong potential for H2 oxidation.
Phylogeny and Functional Diversity of hynL Genes
hynL genes for [NiFe]-hydrogenase detected in the PAS metagenome assemblies were compared with nearest homolog sequences from the literature in phylogenetic trees, in which genes with high similarity were collapsed into the same clade. Four groups of hynL genes, representing [NiFe]-hydrogenases with distinct physiological roles (Vignais et al., 2001), were all observed in the PAS-616P and PAS-821TP trees (Figures 2, 3). Their phylogenetic distribution among Deltaproteobacteria (mostly SRB), Archaea (mostly methanogens), and Epsilonproteobacteria (mostly sulfur-metabolizing bacteria; SMB) is indicated in Table S3.
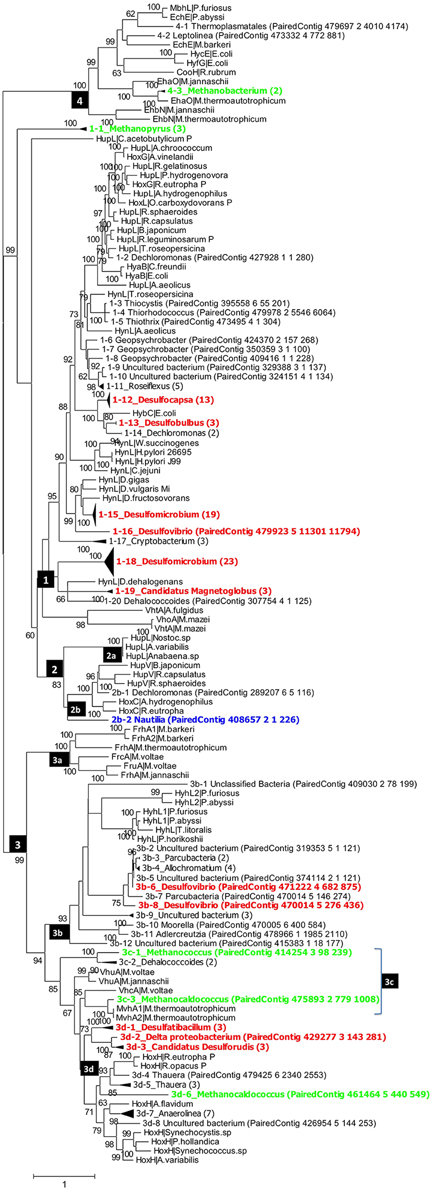
Figure 2. Phylogenetic analysis of putative hynL genes for the large subunit of [NiFe]-hydrogenase detected in the PAS-616P metagenome. Contigs with high similarity and the same taxonomic origin were collapsed into, the same clade and the number of contigs in each clade is indicated in brackets. Clades for putative methanogens, SRB, and chemolithoautotrophic SMB are highlighted in green, red, and blue. Maximum-likelihood bootstrap support values of more than 60 are shown. The numbers in the black boxes are different types of [NiFe] hydrogenases, identified by Vignais et al. (2001) as 1, 2 (2a, 2b), 3 (3a, 3b, 3c, 3d), and 4.
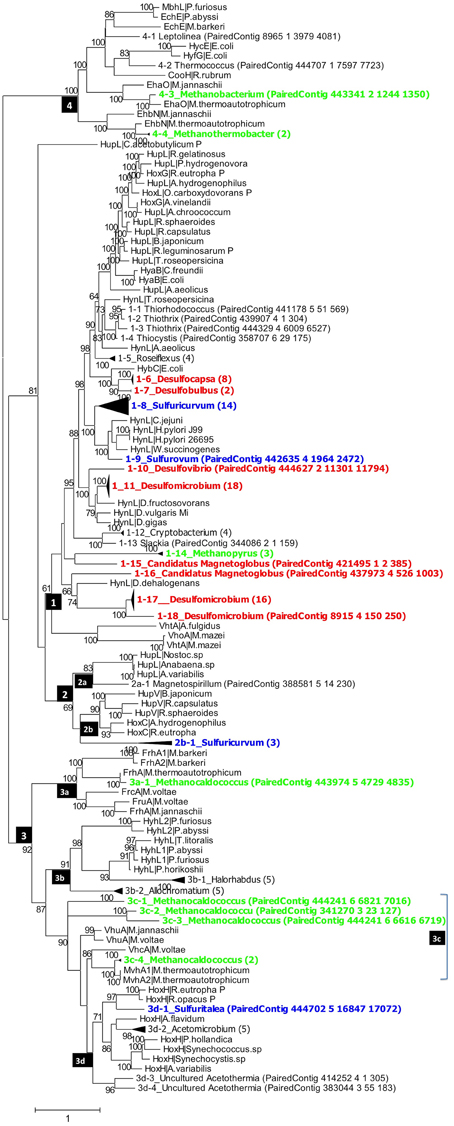
Figure 3. Phylogenetic analysis of putative hynL genes for the large subunit of [NiFe]-hydrogenase detected in the PAS-821TP metagenome. Contigs with high similarity and same taxonomic origin were collapsed into the same clade and the number of contigs in each clade is indicated in brackets. Clades for putative methanogens, SRB, and chemolithoautotrophic SMB are highlighted in green, red, and blue. Maximum-likelihood bootstrap support values of more than 60 are shown. The numbers in the black boxes are different types of [NiFe] hydrogenases, identified by Vignais et al. (2001) as 1, 2 (2a, 2b), 3 (3a, 3b, 3c, 3d), and 4.
Group_1 includes the H2-oxidizing membrane-bound [NiFe] hydrogenases, most of which leave protons on the external side of the membrane while transporting electrons to an internally located b-type cytochrome. Group_1 [NiFe] hydrogenases were represented in the PAS-616PL tree in clades 1-1 to 1-20 (Figure 2). Nine of these harbored multiple contigs (indicated in brackets) with clades 1-12_Desulfocapsa_(13), 1-15_Desulfomicrobium_(19), and 1-18_Desulfomicrobium_(23) being most heavily populated. The abundance (the number of sequences divided by 43,059 observed rpoB sequences) of Group_1 clades was 0.365 with Deltaproteobacteria hynL genes (0.283) outnumbering those for Archaea (0.062). No Group_1 hynL genes for Epsilonproteobacteria were observed (Table S3). Group_1 hynL genes were represented in the PAS-821TP tree in clades 1-1 to 1-18 of which eight harbored multiple contigs (Figure 3), including 1-6_Desulfocapsa_(8), 1-7_Desulfobulbus_(2), 1-11_Desulfomicrobium_(18), and 1-17_Desulfomicrobium_(16) for the Deltaproteobacteria, 1-14_Methanopyrus_(3) for the Archaea and 1-8_Sulfuricurvum_(14) for the Epsilonproteobacteria. Relative to the PAS-616P metagenome, the PAS-821TP metagenome had a decreased abundance of hynL genes from Archaea (Table S3: 0.018) and an increased abundance of hynL genes from Delta- and Epsilonproteobacteria (Table S3: 0.363 and 0.034). The latter included Sulfurovum, in addition to the more dominant Sulfuricurvum. Filamentous Gammaproteobacteria, potentially oxidizing sulfide or sulfur (Chernousova et al., 2009; van der Meer et al., 2010), were observed in both metagenomes, i.e., 1-2_Thiothrix and 1-3_Thiothrix in the PAS-821TP metagenome. Hence, the continuous injection of bisulfite decreased the presence of archaeal hynL genes, while increasing hynL genes for sulfur-metabolizing Epsilonproteobacteria, which may use Group_1 [NiFe]-hydrogenases to oxidize H2 for reduction of sulfur (Campbell et al., 2006; Peters et al., 2015).
Group_2a including cyanobacterial [NiFe]-hydrogenases (Peters et al., 2015) was not significantly present. Group_2b includes regulatory [NiFe] hydrogenases, which can induce expression of hyn genes, when H2 is present. These were represented in a single clade 2b-1_Sulfuricurvum_(3) in the PAS-821TP metagenome, suggesting that expression of Group_1 hyn genes in Epsilonproteobacteria is regulated. The abundance of regulatory Group_2b hynL genes was lower than of Group_1 hynL genes (Table S3: 0.006 and 0.036, respectively).
Group_3a [NiFe]-hydrogenases are cytoplasmic enzymes involved in reduction of coenzyme F420 in methanogenic Archaea. This group was found in low abundance in the PAS-821TP metagenome (Table S3). Group_3b [NiFe]-hydrogenases had a diverse phylogenetic distribution, which included the SMB Allochromatium (both metagenomes) and the SRB Desulfovibrio (PAS-616P only). Group_3c includes methyl viologen-reducing [NiFe]-hydrogenases (MvhAgD), which function in the reduction of heterodisulfide with H2 in methanogens (Peters et al., 2015). This group included hynL genes from Methanocaldococcus (Figures 2, 3) and was more abundant in the PAS-616P than in the PAS-821TP metagenome (Table S3: 0.026 and 0.012, respectively). Group_3d includes bidirectional heteromultimeric [NiFe]-hydrogenases (HoxHY), which associate with NADH dehydrogenase for energy generation in aerobic bacteria (Peters et al., 2015). The hydL genes from the Betaproteobacteria Sulfuritalea in the PAS-821TP metagenome also belonged to this group (Figure 3: 3d-1_Sulfuritalea). In the PAS-616P metagenome Group_3d included Firmicutes, such as Candidatus Desulforudis.
The [NiFe] hydrogenases included in group 4 are typically responsible for cytoplasmic, flavodoxin-dependent H2 production in Bacteria, and methanogenic Archaea. The latter were represented through clade 4-3_Methanobacterium in both metagenomes. The abundance of Group_4 hynL genes of the genus Methanobacterium decreased in transitioning from the PAS-616P to the PAS-821TP metagenome (Table S3). This genus includes hydrogenotrophic methanogens with potential for iron corrosion (Dinh et al., 2004).
Discussion
The continuous injection of bisulfite into a pipeline system transporting brackish water with little sulfate (Table 1), but high concentrations of bicarbonate (0.5–1.5 g/L; Park et al., 2011) had a significant effect on microbial community composition of the pipe wall. Analysis of metagenomic 16S reads, obtained by 454 sequencing, indicated that SBS decreased the fraction of methanogenic Archaea (Methanobacteriaceae and Methanolobus), but increased the fraction of Deltaproteobacteria (Desulfomicrobium and Desulfocapsa) and Epsilonproteobacteria (Sulfuricurvum and Sulfurovum), as indicated in Table 2 and Table S2.
The more extensive dataset on the distribution of hydL and hynL genes in metagenomic reads, obtained by 454 and Illumina sequencing, confirmed the shift in taxonomic distribution seen in 454 16S reads while allowing more detailed assigment of taxa involved in H2 metabolism. First of all hydL genes for [FeFe]-hydrogenase were approximately ten-fold less frequent than hynL genes for [NiFe] hydrogenases. Most hydL genes were affiliated with Firmicutes (Table 3), which were only a minor fraction (Table 2: 1.1–1.6%) in both metagenomes. We focused, therefore, on analysis of the phylogenetic distribution of hynL genes. This includes Archaea, which have hynL but do not have hydL genes, but may exclude Firmicutes, i.e., only 0.24–0.57% of all hynL genes was affiliated with this phylum.
Injection of bisulfite decreased the abundance of hynL genes from methanogenic Archaea of Group_1 (0.062–0.018, Methanopyrus in both metagenomes), Group_3c (0.026–0.012) and Group_4 (0.023–0.006), while increasing the abundance of hynL genes from Deltaproteobacteria of Group_1 (0.283–0.363), but not of Group_3b (0.006–0) and Group_3d (0.020–0). Group_1 hynL genes were mostly affiliated with the genera Desulfomicrobium and Desulfocapsa in both metagenomes. Other Group_1 hynL genes found in both metagenomes included those from Candidatus Magnetoglobus, which are affiliated with the Deltaproteobacteria class and may also be SRB (Abreu et al., 2007). Group_1 hynL genes from Dechloromonas and Dehalococcoides, which use H2 for inorganic or organic dechlorination, disappeared upon bisulfite injection (Figures 2, 3).
Injection of bisulfite strongly increased the abundance of Group_1 hynL genes from the Epsilonproteobacteria Sulfuricurvum and Sulfurovum (from 0 to 0.034) and of Group_2b hynL genes from Sulfuricurvum. This class also includes the phylogenetically related genera Sulfurimonas, Sulfurospirillum, Campylobacter, and Arcobacter, as well as the more distantly related Nautilia (Han et al., 2012). Most Epsilonproteobacteria are chemolithoautotrophs, which oxidize reduced sulfur species (sulfide, sulfur, and thiosulfate) with O2 (under microaerophilic conditions) and nitrate (Kodama and Watanabe, 2004; Campbell et al., 2006). Many also reduce sulfur with H2 (Campbell et al., 2006), a reaction first documented for Arcobacter strain FWKO_B (Gevertz et al., 2000). This is in contrast to Betaproteobacteria of the genus Sulfuritalea (Figure 3: 3d-1), which oxidize both H2 and reduced sulfur species with O2 and/or nitrate, but do not reduce sulfur with H2 (Kojima and Fukui, 2010). Because O2 is absent following SBS injection, Group_1 [NiFe]-hydrogenase of the Epsilonproteobacteria likely functioned in reduction of sulfur with H2 with the Group_2b regulatory proteins ensuring that the enzyme is expressed under these conditions. The Group_2b HoxBC [NiFe] hydrogenase sensor was first discovered in Alcaligenes eutrophus (now Ralstonia eutrophus), where HoxBC functions in H2 recognition and transmits the signal to HoxJ, which modulates response regulator HoxA to activate transcription of Group_1 hyn genes (Lenz and Friedrich, 1998).
As indicated previously (Park et al., 2011), injection of bisulfite removes O2 by chemical reaction:
Both bisulfite (when dosed in excess) and sulfate can be reduced by SRB of the genus Desulfomicrobium, using cathodic H2 (or low potential electrons) from steel (Fe0) as the electron donor:
Alternatively, is disproportionated into sulfide or sulfur and sulfate by Deltaproteobacteria of the genus Desulfocapsa:
Assuming little H2 production by fermentation of organics, the balance between reduction (Reactions 2 and 3) and disproportionation (Reactions 5 and 6) will depend on the availability of cathodic hydrogen (or of low potential electrons in an EMIC scenario). The generated sulfur (S0) is then used by Epsilonproteobacteria, which were clearly stimulated by injection of bisulfite and which may also use cathodic H2:
Reactions (2) and (7) can be combined with formation of FeS from Fe2+ and HS− to:
which indicates that Epsilonproteobacteria may gain energy for growth by accelerating the reaction between metallic iron and elemental sulfur, which would take place more slowly in their absence. Both FeS and S0 were significant components of the pipeline scale downstream from the SBS injection point (Park et al., 2011). The transported brackish water had little organic carbon (Table S1; Park et al., 2011), which could have served as an alternative electron donor for sulfur reduction. Indeed, Deltaproteobacteria of the genus Desulfuromonas, which specialize in this activity, were found in low fractions (Table S2: up to 0.14%) and decreased upon SBS injection.
Hence, the metagenomic studies presented here have uncovered a potential role for Epsilonproteobacteria in MIC in pipelines subjected to injection of bisulfite for the scavenging of oxygen, which is commonly used. Clearly injection of excess bisulfite should be avoided and the emergence of Epsilonproteobacteria, which were nearly absent upstream from the SBS injection point, may serve as an indicator of increased MIC threat.
Author Contributions
DA: Contributed to design of the study and isolation and purification of DNA for 16S amplicon and metagenomic analysis, performed data analysis of 16S amplicons and metagenomes, interpretation of data, and drafting the paper. XD: Conducted metagenomic data analysis which included processing of QC and assembly of reads, generating gene abundance, and phylogeny trees. AA: Participated in data analysis of 16S amplicons and drafting the paper. HP: Contributed to sample collections. MS: Contributed to metagenomic data analysis. GV: Contributed to design of the study, interpretation of data, drafting the paper, final approval of the version to be published.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported through a Natural Sciences and Engineering Research Council (NSERC) Industrial Research Chair (IRC) awarded to GV. The NSERC IRC is also supported by Baker Hughes, BP, Computer Modeling Group Ltd., ConocoPhillips, Intertek, Dow Microbial Control, Enbridge, Enerplus Corporation, Oil Search Limited, Shell Global Solutions International, Suncor Energy Inc., Yara Norge, and Alberta Innovates Energy and Environment Solutions (AIEES). We are grateful for the administrative support provided by Rhonda Clark.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00028
References
Abreu, F., Martins, J. L., Silveira, T. S., Keim, C. N., De Barros, H. G. P. L., Filho, F. J. G., et al. (2007). ‘Candidatus Magnetoglobus multicellularis’, a multicellular, magnetotactic prokaryote from a hypersaline environment. Int. J. Syst. Evol. Microbiol. 57, 1318–1322. doi: 10.1099/ijs.0.64857-0
An, D., Caffrey, S. M., Soh, J., Agrawal, A., Brown, D., Budwill, K., et al. (2013). Metagenomics of hydrocarbon resource environments indicates aerobic taxa and genes to be unexpectedly common. Environ. Sci. Techmol. 47, 10708–10717. doi: 10.1021/es4020184
Brazelton, W. J., Nelson, B., and Schrenk, M. O. (2012). Metagenomic evidence for h(2) oxidation and h(2) production by serpentinite-hosted subsurface microbial communities. Front. Microbiol. 2:268. doi: 10.3389/fmicb.2011.00268
Caffrey, S. M., Park, H.-S., Voordouw, J., He, Z., Zhou, J., and Voordouw, G. (2007). Function of periplasmic hydrogenases in the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 189, 6159–6167. doi: 10.1128/JB.00747-07
Campbell, B. J., Engel, A. S., Porter, M. L., and Takai, K. (2006). The versatile epsilon-proteobacteria: key players in sulphidic habitats. Nat. Rev. Microbiol. 4, 458–468. doi: 10.1038/nrmicro1414
Chernousova, E., Gridneva, E., Grabovich, M., Dubinina, G., Akimov, V., Rossetti, S., et al. (2009). Thiothrix caldifontis sp. nov. and Thiothrix lacustris sp. nov., gammaproteobacteria isolated from sulfide springs. Int. J. Syst. Evol. Microbiol. 59, 3135. doi: 10.1099/ijs.0.009456-0
Dinh, H. T., Kuever, J., Mußmann, M., Hassel, A. W., Stratmann, M., and Widdel, F. (2004). Iron corrosion by novel anaerobic microorganisms. Nature 427, 829–832. doi: 10.1038/nature02321
Enning, D., and Garrelfs, J. (2014). Corrosion of iron by sulfate-reducing bacteria: new views of an old problem. Appl. Environ. Microbiol. 80, 1226–1236. doi: 10.1128/AEM.02848-13
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). The Pfam protein families database. Nucleic Acids Res. 42, 222–230. doi: 10.1093/nar/gkt1223
Finster, K. (2008). Microbiological disproportionation of inorganic sulfur compounds. J. Sulfur Chem. 29, 281–292. doi: 10.1080/17415990802105770
Gevertz, D., Telang, A. J., Voordouw, G., and Jenneman, G. E. (2000). Isolation and characterization of strains CVO and FWKO B: two novel nitrate-reducing, sulfide-oxidizing bacteria isolated from oil field brine. Appl. Environ. Microbiol. 66, 2491–2501. doi: 10.1128/AEM.66.6.2491-2501.2000
Haft, D. H., Selengut, J. D., Richter, R. A., Harkins, D., Basu, M. K., and Beck, E. (2013). TIGRFAMs and Genome Properties in 2013. Nucleic Acids Res. 41, 387–395. doi: 10.1093/nar/gks1234
Han, C., Kotsyurbenko, O., Chertkov, O., Held, B., Lapidus, A., Nolan, M., et al. (2012). Complete genome sequence of the sulfur compounds oxidizing chemolithoautotroph Sulfuricurvum kujiense type strain (YK-1T). Stand Genomic Sci. 19, 94–103. doi: 10.4056/sigs.2456004
Huang, Y., Gilna, P., and Li, W. (2009). Annotation of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25, 1338–1340. doi: 10.1093/bioinformatics/btp161
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kodama, Y., and Watanabe, K. (2004). Sulfuricurvum kujiense gen. nov., sp. nov., a facultatively anaerobic, chemolithoautotrophic, sulphur-oxidizing bacterium isolated from an underground crude-oil storage cavity. Int. J. Syst. Evol. Microbiol. 54, 2297–2300. doi: 10.1099/ijs.0.63243-0
Kojima, H., and Fukui, M. (2010). Sulfuritalea hydrogenivorans gen. nov., sp. nov., a facultative autotroph isolated from a freshwater lake. Int. J. Syst. Evol. Microbiol. 61, 1651–1655. doi: 10.1099/ijs.0.024968-0
Langmead, B., and Salzberg, S. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lenz, O., and Friedrich, B. (1998). A novel multicomponent regulatory system mediates H2 sensing in Alcaligenes eutrophus. Proc. Natl. Acad. Sci. U.S.A. 95, 12474–12479. doi: 10.1073/pnas.95.21.12474
Mand, J., Park, H. S., Jack, T. R., and Voordouw, G. (2014). The role of acetogens in microbially influenced corrosion of steel. Front. Microbiol. 5:268. doi: 10.3389/fmicb.2014.00268
Muyzer, G., and Stams, A. J. M. (2008). The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. Microbiol. 6, 441–454. doi: 10.1038/nrmicro1892
Okoro, C., Smith, S., Chiejina, L., Lumacud, R., An, D., Park, H. S., et al. (2014). Comparison of microbial communities involved in souring and corrosion in offshore and onshore oil production facilities in Nigeria. J. Ind. Microbiol. Biotechnol. 41, 665–678. doi: 10.1007/s10295-014-1401-z
Ossai, C. I., Boswell, B., and Davies, I. J. (2015). Pipeline failures in corrosive environments - a conceptual analysis of trends and effects. Eng. Fail. Anal. 53, 36–58. doi: 10.1016/j.engfailanal.2015.03.004
Park, H. S., Chatterjee, I., Dong, X., Wang, S. H., Sensen, C. W., Caffrey, S. M., et al. (2011). Effect of sodium bisulfite injection on the microbial community composition in a brackish-water-transporting pipeline. Appl. Environ. Microbiol. 77, 6908–6917. doi: 10.1128/AEM.05891-11
Peters, J., Schut, G., Boyd, E., Mulder, D., Shepard, E., Broderick, J., et al. (2015). [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta. 1853, 1350–1369. doi: 10.1016/j.bbamcr.2014.11.021
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rabus, R., Venceslau, S. S., Wöhlbrand, L., Voordouw, G., Wall, J. D., and Pereira, I. A. (2015). A post-Genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. Adv. Microb. Physiol. 66, 55–321. doi: 10.1016/bs.ampbs.2015.05.002
Saidi-Mehrabad, A., He, Z., Tamas, I., Sharp, C. E., Brady, A. L., Rochman, R. F., et al. (2013). Methanotrophic bacteria in oilsands tailings ponds of northern Alberta. ISME J. 7, 908–921. doi: 10.1038/ismej.2012.163
Soh, J., Dong, X., Caffrey, S. M., Voordouw, G., and Sensen, C. W. (2013). Phoenix 2: a locally installable large-scale 16S rRNA gene sequence analysis pipeline with Web interface. J. Biotechnol. 167, 393–403. doi: 10.1016/j.jbiotec.2013.07.004
Stamatakis, A. (2014). RAxML Version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tan, B., Dong, X., Sensen, C. W., and Foght, J. (2013). Metagenomic analysis of an anaerobic alkane-degrading microbial culture: potential hydrocarbon-activating pathways and inferred roles of community members. Genome 56, 599–611. doi: 10.1139/gen-2013-0069
Trüper, H. G., and Schlegel, H. G. (1964). Sulphur metabolism in Thiohodanceae I. Quantitative measurements in growing cells of Chromatium okenii. Antonie van Leeuwenhoek 30, 225—238. doi: 10.1007/BF02046728
van der Meer, M. T., Klatt, C. G., Wood, J., Bryant, D. A., Bateson, M. M., Lammerts, L., et al. (2010). Cultivation and genomic, nutritional, and lipid biomarker characterization of Roseiflexus strains closely related to predominant in situ populations inhabiting Yellowstone hot spring microbial mats. J. Bacteriol. 192, 3033–3042. doi: 10.1128/JB.01610-09
Vignais, P. M., and Billoud, B. (2007). Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107, 4206–4272. doi: 10.1021/cr050196r
Vignais, P. M., Billoud, B., and Meyer, J. (2001). Classification and phylogeny of hydrogenases. FEMS Microbiol. Rev. 25, 455–501. doi: 10.1111/j.1574-6976.2001.tb00587.x
Von Wolzogen Kühr, C. A. H., and Van der Vlugt, L. S. (1934). De grafiteering van gietijzer als electrobiochemich process inanaerobe Gronden. Water 18, 147–165.
Yang, F., Shi, B., Bai, Y., Sun, H., Lytle, D. A., and Wang, D. (2014). Effect of sulfate on the transformation of corrosion scale composition and bacterial community in cast iron water distribution pipes. Water Res. 59, 46–57. doi: 10.1016/j.watres.2014.04.003
Keywords: corrosion, pipeline, microbiologically influenced corrosion (MIC), microbial community analysis, metagenomics, sulfate reducing bacteria (SRB), Epsilonproteobacteria, hydrogenase
Citation: An D, Dong X, An A, Park HS, Strous M and Voordouw G (2016) Metagenomic Analysis Indicates Epsilonproteobacteria as a Potential Cause of Microbial Corrosion in Pipelines Injected with Bisulfite. Front. Microbiol. 7:28. doi: 10.3389/fmicb.2016.00028
Received: 17 November 2015; Accepted: 11 January 2016;
Published: 28 January 2016.
Edited by:
Wael Ahmed Ismail, Arabian Gulf University, BahrainReviewed by:
Christopher L. Hemme, University of Rhode Island, USAJan Kuever, Bremen Institute for Materials Testing, Germany
Huda Mahmoud Mahmoud, Kuwait University, Kuwait
Copyright © 2016 An, Dong, An, Park, Strous and Voordouw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerrit Voordouw, voordouw@ucalgary.ca