 Trinity L. Hamilton1*
Trinity L. Hamilton1* Roderick J. Bovee2
Roderick J. Bovee2 Sarah R. Sattin2Wiebke Mohr2†
Sarah R. Sattin2Wiebke Mohr2† William P. Gilhooly III3Timothy W. Lyons4
William P. Gilhooly III3Timothy W. Lyons4 Ann Pearson2*
Ann Pearson2* Jennifer L. Macalady5*
Jennifer L. Macalady5*- 1Department of Biological Sciences, University of Cincinnati, Cincinnati, OH, USA
- 2Department of Earth and Planetary Sciences, Harvard University, Cambridge, MA, USA
- 3Department of Earth Sciences, Indiana University-Purdue University Indianapolis, Indianapolis, IN, USA
- 4Department of Earth Sciences, University of California, Riverside, CA, USA
- 5Penn State Astrobiology Research Center, Department of Geosciences, Pennsylvania State University, University Park, TX, USA
Mahoney Lake in British Columbia is an extreme meromictic system with unusually high levels of sulfate and sulfide present in the water column. As is common in strongly stratified lakes, Mahoney Lake hosts a dense, sulfide-oxidizing phototrophic microbial community where light reaches the chemocline. Below this “plate,” the euxinic hypolimnion is anoxic, eutrophic, saline, and rich in sulfide, polysulfides, elemental sulfur, and other sulfur intermediates. While much is known regarding microbial communities in sunlit portions of euxinic systems, the composition and genetic potential of organisms living at aphotic depths have rarely been studied. Metagenomic sequencing of samples from the hypolimnion and the underlying sediments of Mahoney Lake indicate that multiple taxa contribute to sulfate reduction below the chemocline and that the hypolimnion and sediments each support distinct populations of sulfate reducing bacteria (SRB) that differ from the SRB populations observed in the chemocline. After assembling and binning the metagenomic datasets, we recovered near-complete genomes of dominant populations including two Deltaproteobacteria. One of the deltaproteobacterial genomes encoded a 16S rRNA sequence that was most closely related to the sulfur-disproportionating genus Dissulfuribacter and the other encoded a 16S rRNA sequence that was most closely related to the fatty acid- and aromatic acid-degrading genus Syntrophus. We also recovered two near-complete genomes of Firmicutes species. Analysis of concatenated ribosomal protein trees suggests these genomes are most closely related to extremely alkaliphilic genera Alkaliphilus and Dethiobacter. Our metagenomic data indicate that these Firmicutes contribute to carbon cycling below the chemocline. Lastly, we recovered a nearly complete genome from the sediment metagenome which represents a new genus within the FCB (Fibrobacteres, Chlorobi, Bacteroidetes) superphylum. Consistent with the geochemical data, we found little or no evidence for organisms capable of sulfide oxidation in the aphotic zone below the chemocline. Instead, comparison of functional genes below the chemocline are consistent with recovery of multiple populations capable of reducing oxidized sulfur. Our data support previous observations that at least some of the sulfide necessary to support the dense population of phototrophs in the chemocline is supplied from sulfate reduction in the hypolimnion and sediments. These studies provide key insights regarding the taxonomic and functional diversity within a euxinic environment and highlight the complexity of biogeochemical carbon and sulfur cycling necessary to maintain euxinia.
Introduction
Opposing gradients of light and sulfide support phototrophic sulfur bacteria where those gradients meet. These bacteria contribute to oxidative sulfur cycling and primary productivity through carbon fixation. Mahoney Lake (ML), a small meromictic lake in British Columbia, Canada, supports a remarkably dense 10–20-cm floating “plate” of anoxygenic phototrophs in the chemocline at a depth of ~7 m, below which no light penetrates. The lake characteristics represent an extreme endmember of euxinia: a high concentration of sulfate (400–500 mM) is supplied by local surface waters draining alkaline lavas enriched in Mg2+, Ca2+, Na+, and , and , and the hypolimnion of the lake contains one of the highest levels of sulfide (30–35 mM) observed in a natural system (Northcote and Hall, 1983). The epilimnion is oxic and oligotrophic whereas the hypolimnion is eutrophic (Hall and Northcote, 1986).
At times throughout Earth's history the deep oceans have been oxygen depleted. During several of these events, regional accumulation of sulfide created areas of the ocean that were euxinic. These episodes accompanied significant events such as Phanerozoic biotic crises (e.g., Pancost et al., 2004; Grice et al., 2005) and perhaps were common in the Mesoproterozoic (Canfield, 1998; Brocks et al., 2005; Reinhard et al., 2013). Today, examples of euxinia are rare but permanently sulfidic water is observed in silled basins, meromictic lakes, coastal upwelling zones, and fjords. Of these systems, meromictic lakes are of particular interest due to their shallow phototrophic chemocline communities, extreme geochemical gradients, and laminated sedimentary records under conditions of rapid sedimentation that can preserve biosignatures (Meyer and Kump, 2008). ML is a model system as an extreme endmember of high sulfide concentration and permanent redox stability compared to other meromictic systems. Lake Cadagno, a small meromictic lake in the southern Swiss Alps, for instance is a more dilute system and experiences greater seasonal variation (Bosshard et al., 2000; Del Don et al., 2001; Decristophiris et al., 2009; Gregersen et al., 2009). The high concentration of sulfide in the water column of ML requires overloading of organic matter to support high rates of sulfate reduction. In ML, this process is driven by input of allochthonous carbon from the surrounding catchment as well as autochthonous production. The meromictic redox stability of the ML water column (permanently, rather than seasonally, stratified) results from the high salinity and high sulfur concentrations, which are promoted by the arid climate and surrounding geology (rainfall 40 cm y−1). The maintenance and stability of the euxinic system depends on the combination of these extrinsic factors which may contribute to their apparent rarity in other marine and nonmarine settings (e.g., Meyer and Kump, 2008; Johnston et al., 2009; Canfield, 2013; Leavitt et al., 2013).
Microbial assemblages appear to be unique within each horizon of meromictic lakes, consistent with permanent geochemical and physical partitioning, and these populations function to maintain biogeochemical cycling. In ML, the dense phototrophic plate limits the mixing of nutrients to the epilimnion and absorbs all visible light (Overmann et al., 1991, 1996a). The chemocline maintains a stable ecosystem in which complex cycling of carbon and sulfur occurs (Hamilton et al., 2014). The oxic surface waters support the growth of phytoplankton and aerobic heterotrophic bacteria (Overmann et al., 1996b); however, little is known regarding the microbial community below the chemocline where the sulfide concentration is the highest. Rates of sulfate reduction in the chemocline capable of supplying most of the sulfide necessary for anoxygenic photosynthesis are only observed in late summer (July–September; Overmann et al., 1996a). The rest of the year, molecular diffusion must supply a significant fraction of sulfide to support the dense phototrophic layer, assuming the layer is very active throughout the year. Sulfate concentrations in the monimolimnion were similar throughout the year while the sulfide concentrations ranged from 27 to 60 mM during the study (Overmann et al., 1996a). Collectively, these data provide few clues regarding biological activity in the monimolimnion and sediments of the lake. 16S rRNA analyses of ML indicate that each stratified layer is taxonomically distinct (Klepac-Ceraj et al., 2012). These data also suggest that microbial communities in the deep waters and sediments of Mahoney Lake make important contributions to biogeochemical cycling in the lake through sulfate reduction and fermentation.
Here we report the results of metagenomic sequencing of the ML community from 8 m, just under the phototrophic plate, and from the lake bottom sediment, (15 m) as obtained by grab core. Our data reveal the presence of anaerobic bacteria involved in remineralization of organic matter below the chemocline. Consistent with the high levels of sulfide in ML, we observed populations of sulfate reducing bacteria at 8 m and in the sediments. Genomic reconstruction resulted in two genomes of Deltaproteobacteria species, two genomes of Firmicutes species, and a genome of a species from the FCB (Fibrobacteres, Chlorobi, Bacteroidetes) superphylum which represents a new genus within this superphylum. Collectively, our data provide key insight into biogeochemical cycling in a modern, extreme analog of early Earth euxinia.
Materials and Methods
Environmental Sample Collection and DNA Extraction
Water from 8 m was collected using a Niskin bottle and a grab core was collected of the underlying sediments (15 m) from ML, British Columbia (49° 17′N, 119° 35′W) in July 2008. 50 mL samples of water and sediments were immediately frozen on dry ice for transport to Harvard University, where they were stored at −80°C until further processed. For extraction of genomic DNA, samples were thawed and centrifuged, and DNA was extracted from the resulting pellets using an e.Z.N.A SP Plant Maxi Kit (Omega Bio-tek, Norcross, Georgia) according to the manufacturer's instructions. The yield and quality of the extracted DNA were assessed using gel electrophoresis visualized by ethidium bromide staining and spectrophotometry using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, Delaware).
Metagenomic Sequencing, Assembly, and Binning
Fragmentation and library preparation were performed by the North Carolina State University Genomic Sciences Laboratory. Paired-end 150 bp Illumina (HiSeq 2500) sequencing was performed at the Harvard Center for Systems Biology. Reads were trimmed with Trimmomatic 0.20 (Lohse et al., 2012), and only sequences with at least 50 base pairs in both the forward and reverse direction were retained. Trimmed, screened, paired-end Illumina reads were assembled into contigs with IDBA-UD (ver. 1.1.1) using eight threads with default parameters (Table S1; Peng et al., 2012). Coverage was determined by aligning raw reads to contigs using BWA 0.5.9 (Li and Durbin, 2009).
The assembled contigs were annotated with an in-house annotation pipeline as described previously (Hamilton et al., 2014). Briefly, rRNAs were identified using Meta_RNA (Huang et al., 2009) and Phyloshop (Shah et al., 2010), and tRNAs were identified with tRNAScan (Lowe and Eddy, 1997). Protein-coding genes were identified using the ab initio gene calling tools GeneMark (v.2.6r) (Lukashin and Borodovsky, 1998), MetaGene (v. Aug08) (Noguchi et al., 2006), Prodigal (Hyatt et al., 2010), and FragGeneScan (Rho et al., 2010). Genes were associated with COGs (Clusters of Orthologous Groups of proteins) using rpsblast (Tatusov et al., 2001) and Pfam with hmmsearch (Durbin et al., 1998). Amino acid similarity searches were used for assignment of KO terms (KEGG) (Ogato et al., 2000) and EC numbers to open reading frames.
A custom Python script (available at https://github.com/bovee/Ochre) was used to calculate tetranucleotide frequency of all contigs ≥2500 bp. Corresponding reverse-complement tetranucleotides were combined as described (Dick et al., 2009). Contigs were then binned using emergent self-organizing maps (ESOM) based on tetranucleotide frequency, which resulted in clusters corresponding to taxonomically sorted tetranucleotide usage patterns (Dick et al., 2009). For binning, contigs were split into 5000-bp segments, clustered into taxonomic groups (or “genomic bins”; Voorhies et al., 2012) by tetranucleotide frequency and visualized with Databionic-ESOM (http://databionic-esom.sourceforge.net) using parameters from Dick et al. (2009). Following manual inspection for homogeneous read coverage and further curation by BLASTX/N, phylum-level taxonomic assignment was performed using Phyloshop (Shah et al., 2010) and Megan (Huson et al., 2011).
Well-defined, high coverage bins were selected for in-depth characterization and taxonomic assignment of their predicted genes. Paired reads mapping to scaffolds from each bin were reassembled using Velvet (Zerbino and Birney, 2008) or IDBA-UD (ver. 1.1.1) as previously described (Hug et al., 2013). Scaffolds of each re-assembly were annotated as described above. To estimate genome completeness, the presence of a suite of 76 genes selected from a set of single-copy marker genes that show no evidence for lateral gene transfer (Sorek et al., 2007; Wu and Eisen, 2008) was evaluated (Table S2). Genome coverage was estimated by assuming that the genome size of each phylotype was approximately the same as its closest relative (Whitaker and Banfield, 2006; Jones et al., 2012). Average nucleotide identity (ANI) of protein-coding genes between genomes was calculated using the ANIb BLAST+-based analyses within the JSpeciesWS (Richter et al., 2015).
16S rRNA Gene Reconstruction
Near full-length 16S rRNA sequences were reconstructed from Illumina sequencing reads using EMIRGE (Miller et al., 2011). EMIRGE was run for 100 iterations with default parameters designed to merge reconstructed 16S rRNA genes if candidate consensus sequences share ≥97% sequence identity in any iteration. The non-redundant SILVA SSU reference database version 111 (http://www.arb-silva.de/) was used as the starting database of curated SSU sequences. The relative abundance of each OTU was calculated statistically via the EMIRGE algorithm based on “prior probabilities” of read coverage depth (Miller et al., 2011). Sequences with an estimated abundance of < 0.01% were removed from further analyses. Potential chimeras were identified with UCHIME (Edgar et al., 2011) using Mothur (ver 1.32.1; Schloss et al., 2009) and removed from further analyses. Taxonomic assignment of the EMIRGE-reconstructed 16S rRNA sequences was performed using BLAST and ARB (Ludwig et al., 2004).
Taxonomic Assignment of Genome Bins
Several different marker sequences were used to robustly assign taxonomy of the genome bins including 16S rRNA gene sequences (if present in the bin) and ribosomal proteins encoded in a syntenous block (Table S3). When present, the phylogenetic position of 16S rRNA genes was used to make genus-level assignments of genomic bins. The 16S rRNA gene sequences from the genomic bins and closely related sequences identified with BLASTN searches were aligned to the SILVA reference alignment using the SINA Webaligner and merged into the SILVA version 108 database (Pruesse et al., 2007). The alignment was manually refined in ARB (Ludwig et al., 2004), and neighbor-joining analyses were performed in PAUP* v. 4b10 (Swofford, 2001) using Jukes-Cantor-corrected distances and 1000 bootstrap replicates.
Single-copy ribosomal proteins were also analyzed to make genus-level assignments of genomic bins independently from the 16S rRNA gene sequences and in cases where 16S rRNA gene sequences were not present in the bins. Such analyses yield resolution comparable to that for 16S rRNA phylogenetic trees (Hug et al., 2013). A subset of single copy phylogenetic marker proteins (n = 18), identified by Phylo-AMPHORA (Wang and Wu, 2013; Table S3) and verified by annotation and BLAST, were used to assign phylotypes. Reference datasets were further populated with sequences mined from genome sequences in the NCBI databases and JGI IMG-M. Each protein was aligned individually using MEGA (version 6.0, Tamura et al., 2013), and evolutionary models were determined using ProTest (version 3, Darriba et al., 2012). Alignments were then concatenated and neighbor-joining and maximum likelihood phylogeny of each concatenated alignment was calculated using PhyML (Guindon and Gascuel, 2003) and the ProTest-determined evolutionary model with 1000 bootstrap replicates.
Comparative Analysis
Through the annotation pipeline describe above, genes were associated with COGs using rpsblast (Tatusov et al., 2001). The abundance of individual reads matching a particular COG were converted to a fraction representing the relative contribution of each COG count to the total number of sequences assigned to COGs for each dataset (8 m and sediment) to account for different levels of sampling across multiple datasets (Konstantinidis et al., 2009; White et al., 2009; Ferreira et al., 2014). This method has been shown to reduce annotation bias (Delmont et al., 2011). For analysis of carbon, nitrogen, and sulfur cycling pathways, marker genes were defined as previously described (Lauro et al., 2011; Llorens-Marès et al., 2015) and normalized. A full list of the genes is provided in Table S4. Hierarchical cluster analysis and heat map plots were generated with R (R Development Core Team, 2008) using the library “seriation.” The marker genes for dissimilatory sulfate reduction and sulfide oxidation (K00394, K00395, K00396) can operate in both sulfide oxidation and sulfate reduction. Therefore, they were assigned to sulfate reduction or sulfur oxidation based on the best match within KEGG.
Nucleotide Sequence Accession Numbers
The assembled metagenomic sequences can be accessed via IMG/M (http://img.jgi.doe.gov). Raw sequence reads of all samples were deposited at the NCBI Short Read Archive (SRA) and can be accessed under the accession numbers SRR2986055 (8-m sample) and SRR2989655 (sediments). Metagenome bin sequences—ML8_D, MLS_D ML8_F1, ML8_F2, and MLS_C—are deposited at DDBJ/EMBL/GenBank under the accession numbers SAMN04330442, SAMN04330450, SAMN04330440, SAMN04330448, and SAMN04330451, respectively.
Results and Discussion
Community Structure below the Chemocline
Metagenomic data from 8 m water depth and the sediments from Mahoney Lake resulted in 364,727 contigs containing ~496 Mbp and 593,174 contigs containing ~781 Mbp, respectively. Based on BlastX, the majority of contigs (>92%) in each metagenome were assigned to Bacteria while small numbers of sequences affiliated with Archaea, Eukaryota, and viruses were also recovered (Figure S1). Despite changes in the redox chemistry of the water column (Northcote and Hall, 1983; Overmann et al., 1991, 1996a) and the close proximity of the 8-m sample to the dense phototrophic plate at 7 m, the microbial community at 8 m is remarkably similar at the phylum level to the sediments (Figure 1). Within the Bacteria, the largest number of sequences were assigned to the Firmicutes and Alpha-, Delta,- and Gammaproteobacteria at both 8 m and in the sediments (Figure 1). This observation is consistent with the recovery of lipids common to Deltaproteobacteria and heterotrophic, Gram-positive bacteria such as Firmicutes from these samples (Bovee and Pearson, 2014). Sequences affiliated with the Alphaproteobacteria were the most abundant in the 7-m (Hamilton et al., 2014) and sediment samples, accounting for more than 24% and more than 22% of the total, respectively. Sequences affiliated with the Firmicutes were the most abundant group from 8 m, where they accounted for 18% of the total sequences. Sequences affiliated with the Cyanobacteria were present in all samples, while sequences affiliated with Planctomycetes were present only in the sediments. Our previous observations of the chemocline community (Hamilton et al., 2014) and those presented here suggest that phylum-level similarities mask the true level of microbial complexity, which imply variations in the biogeochemical functions being carried out in each environment of the lake.
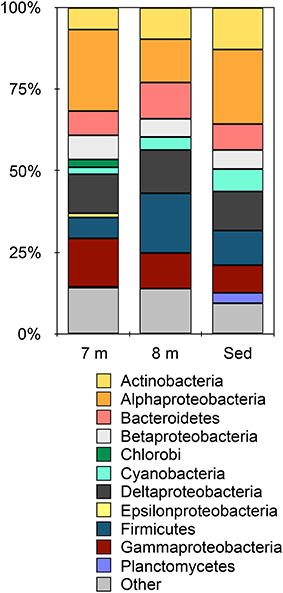
Figure 1. Taxonomic affiliation of sequences in the metagenomes assigned at the phylum-level (except the Proteobacteria which are represented by class). Other indicates all phyla represented by < 5% of the total sequences in any metagenome. Data for 7 m are from Hamilton et al. (2014).
Analysis of full-length 16S rRNA gene sequences reconstructed using EMIRGE resulted in 35 unique operational taxonomic units (OTUs) from both the 8-m and the sediment samples. Rank-abundance curves of the EMIRGE-reconstructed 16S rRNA gene sequences indicate that sequences affiliated with the Deltaproteobacteria and Clostridia are the most abundant in both the 8-m and sediment sample (Figure 2). At 8 m, the relative abundance of 16S rRNA sequences affiliated with Actinobacteria, Cyanobacteria, and Bacilli was higher than in the sediments. In contrast, 16S rRNA sequences affiliated with Planctomycetes and Marine Benthic Group D, an uncultured archaeal clade (Vetriani et al., 1999), were more abundant in the sediments. The recovery of Cyanobacterial 16S rRNA sequences below the sunlit zone suggests sinking biomass from the oxygenated layer of the lake above the chemocline, with preservation of their DNA promoted by the sulfidic, anoxic environment (Coolen and Overmann, 1998). It is worth noting that these 16S rRNA sequences were most closely related to typical phototrophic Cyanobacteria as opposed to the recently discovered non-photosynthetic Melainabacteria which form a novel candidate phylum sibling to Cyanobacteria (Di Rienzi et al., 2013). No observations have been reported of Cyanobacteria thriving in sulfide-rich water below the chemocline in a permanently stratified lake.
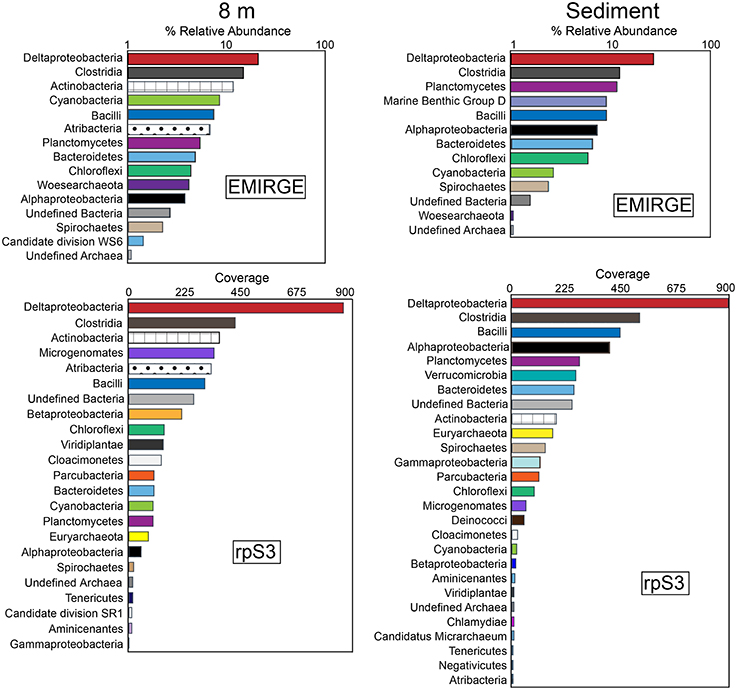
Figure 2. Rank-abundance curves of EMIRGE 16S rRNA sequences and ribosomal protein S3 (rpS3) sequences from the 8 m and sediment metagenomes. The relative abundance of 16S rRNA genes was calculated with EMIRGE (Miller et al., 2011). The rank abundance curves for rpS3 sequences are based on average depth of coverage of the contig where each rpS3 sequence was encoded. Bars represent the sum of the relative abundance of taxonomic groups (at the phylum level except for Proteobacteria and Firmicutes which are summed at the class level).
In addition to the analysis of EMIRGE-reconstructed 16S rRNA gene sequences, we also examined ribosomal protein S3 (rpS3) sequences. rpS3 has a strong phylogenetic signal (Brown et al., 2015) and unlike the EMIRGE-reconstruction that is independent of assembly, provides a direct marker for community composition in the assembled metagenomic data. Using read coverage depth, we also constructed rank-abundance curves of rpS3 sequences from the 8-m and sediment metagenomes. The rank abundance curves for rpS3 sequences are based on average depth of coverage of the contig where each rpS3 sequence was encoded. In general, the rank abundance curves for the rpS3 sequences were in agreement with those observed for the 16S rRNA gene sequences—the highest coverage rpS3 sequences at 8 m and in the sediments were affiliated with Deltaproteobacteria and Clostridia (Figure 2). Actinobacteria and Bacilli rpS3 sequences were abundant in the 8-m sample, and Planctomycetes were abundant in the sediments. In the 8-m sample, rpS3 sequences affiliated with Microgenomates (OP11) and Atribacteria were also abundant.
Some of the highest coverage 16S rRNA and rpS3 sequences were affiliated to lineages with no cultured representatives. For instance, 16S rRNA sequences affiliated with the recently described Woesearchaeota were identified at 8 m. The Woesearchaeota are a new phylum-level lineage within the DPANN superphylum of Archaea with no cultured representatives, despite recovery of multiple genomes of this lineage from groundwater and sediment samples (Castelle et al., 2015) as well as from Sakinaw Lake (Rinke et al., 2013). Sakinaw Lake is a coastal meromictic lake in British Columbia, Canada, with a mixolimnion at 30 m and an anoxic monimolimnion with high concentrations of sodium chloride, sulfate, and sulfide (Perry and Pedersen, 1993; Vagle et al., 2010; Gies et al., 2014). Sequences affiliated with Atribacteria (OP9 and JS1) were also abundant at 8 m. Members of the Atribacteria have been detected in a variety of environments including geothermal systems, petroleum reservoirs, anaerobic digesters, and wastewater treatment facilities. Sequences affiliated to Atribacteria are also found in anaerobic, methane hydrate-bearing sediments (Inagaki et al., 2006; Carr et al., 2015), including those that are low in sulfate. Single-cell and metagenomic sequencing suggests members of the Atribacteria are not involved in sulfate reduction (Dodsworth et al., 2013; Nobu et al., 2015a) but are anaerobic sugar fermenters. Notably, Atribacteria were also recovered from Sakinaw Lake (Rinke et al., 2013; Gies et al., 2014). 16S rRNA sequences affiliated Archaea of Marine Benthic Group D (MBG-D) were abundant in the Mahoney Lake sediments. Members of MBG-D are among the most numerous Archaea in sediments underlying the Earth's oceans (Lloyd et al., 2013). rpS3 sequences affiliated with the Microgenomates (OP11) were present in high coverage at 8 m. Sequences affiliated with Microgenomates have been detected in many environmental samples (Harris et al., 2004) including in anoxic, carbon-rich environments (Briée et al., 2007), but no cultured representatives have been characterized. Metagenomic sequencing suggests a lifestyle based on fermentation (Wrighton et al., 2012).
Potential for Sulfate Reduction
Sulfate reducing bacteria and archaea include members of the phyla Proteobacteria (Deltaproteobacteria), Firmicutes, Nitrospirae, Thermodesulfobacteria, Crenarchaeota, and Euryarchaeota. In general, these taxa couple the oxidation of organic matter or H2 to the reduction of sulfate or compounds with intermediate sulfur oxidation states to sulfide. Sulfide concentrations reach 30–35 mM in the hypolimnion of ML (Overmann et al., 1996a), exceeding the concentration thought to inhibit sulfate reduction (~16 mM; (Reis et al., 1992)). At elevated levels, sulfide is reversibly toxic. However, the H2S: ratio in this system still is not high enough to thermodynamically inhibit sulfate reduction because of the very high levels of associated sulfate (Amend and Shock, 2001; Hamilton et al., 2014). Here, we classified rpS3 sequences affiliated with known sulfur reducing bacteria (Deltaproteobacteria and Clostridia were the only ones detected) to better understand the potential for sulfate reduction below the chemocline in ML.
Sequences affiliated with the Desulfuromonadales are present in high coverage at both 8 m and in the sediments based on rank abundance curves of Deltaproteobacterial rpS3 sequences (Figure 3A). The hypolimnion (8 m) and sediments are also rich in both elemental sulfur and polysulfide and most members of the Desulfuromonadales couple the complete oxidation of organic substrates to CO2 with the reduction of S0 or Fe(III). Sequences affiliated with the Desulfobacterales were present in highest coverage at 8 m (Figure 3A). Members of the Desulfobacterales grow best when coupling acetate oxidation to sulfate reduction although some species are capable of fixing CO2. Acetate was the main volatile fatty acid accumulated in ML chemocline incubations when sulfate reduction was inhibited by molybdate, suggesting it is the preferred carbon source of sulfate reducers within the chemocline (Overmann et al., 1996a). Our detection of Desulfobacterales at 8 m suggests that acetate is also important in the hypolimnion. Desulfobacterales species are common in anaerobic marine and brackish sediments (Widdel, 1987).
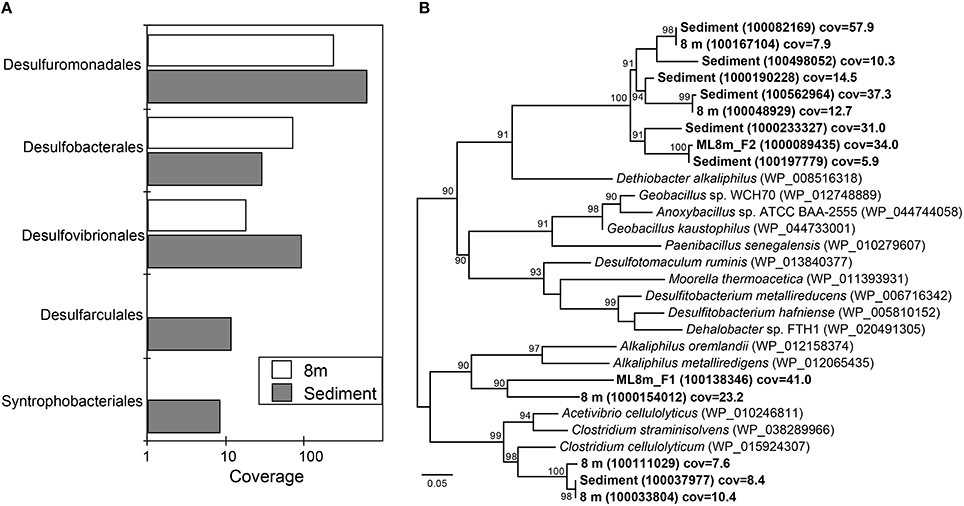
Figure 3. Phylogenetic classification of ribosomal protein S3 (rpS3) sequences affiliated with Deltaproteobacteria (A) and Clostridia (B). (A) Rank-abundance curve of Deltaproteobacterial ribosomal protein S3 (rpS3) sequences from the 8 m and sediment metagenomes classified at the order level. (B) Maximum likelihood phylogenetic tree of rpS3 sequences affiliated with Clostridia recovered from the 8 m and sediment metagenomes and closely related sequences. Sequences recovered in the present study are in bold. NCBI accession numbers or IMG gene ids are shown in parentheses. Bootstrap support values based on 1000 bootstrap samplings >90 are noted. Cov = average depth of coverage of the contig containing the rpS3 sequence.
Desulfovibrionales were also relatively more common in ML sediments than in the water column (Figure 3A). While these taxa also reduce sulfate to sulfide, the Order Desulfovibrionales includes spp. that are capable of switching to a syntrophic lifestyle based on hydrogen consumption (Bryant et al., 1977; McInerney and Bryant, 1981; Stolyar et al., 2007). rpS3 sequences affiliated with the Syntrophobacteriales also were recovered from the sediments (Figure 3A). Members of the Syntrophobacteriales are syntrophic, usually with hydrogen- or formate-utilizing organisms, and are capable of fueling sulfate reduction, and typically degrade fatty or aromatic acids. Together, these results point to the likely importance of H2-based syntrophy in ML sediments. Syntrophic metabolisms are common in many anoxic environments where they are integral to carbon cycling (Morris et al., 2013). In characterized Desulfovibrionales species, the switch to syntrophy occurs in the absence of sulfate. In contrast to this observation, the recovery of sequences affiliated with syntrophic sulfate reducing organisms from the sulfate-rich deep ML environment suggests that syntrophic interactions mediated by Deltaproteobacteria might contribute to the anoxic carbon cycle in Mahoney Lake. Alternatively, members of the Desulfovibrionales and Desulfobacterales are also capable of fueling cell growth via disproportionation of sulfite or thiosulfate to sulfate and sulfide (Bak and Pfennig, 1987). rpS3 sequences affiliated with the Desulfovibrionales and Desulfobacterales were also recovered from the chemocline of Mahoney Lake, where disproportionation rather than (or in addition to) H2-based syntrophy is thought to be important (Hamilton et al., 2014).
We recovered 14 unique rpS3 sequences affiliated with Clostridia below the chemocline. The majority of these sequences were most closely related to the alkaliphilic Dethiobacter alkaliphilus (Figure 3B). Dt. alkaliphilus spp. can grow by sulfur disproportionation, and polysulfides are important intermediates during this reaction (Poser et al., 2013). Geochemical data collected in situ indicates zero valent sulfur, including both intracellular sulfur and polysulfides, are important intermediates in the S cycle in ML (Overmann et al., 1996a). Polysulfide concentrations in the bulk chemocline water of Mahoney Lake are around 50 μM and increase to ~400 μM at a depth of 13–14 m (Overmann et al., 1996a). Other rpS3 sequences were affiliated with Alkaliphilus metalliredigenes, Alkaliphilus oremlandii, and Clostridium celluloyticum (Figure 3B). A. oremlandii is a spore-forming organism that can use arsenate or thiosulfate as an electron acceptor with small organics such as acetate, pyruvate, or lactate as electron donors (Fisher et al., 2008). A. metalliredigenes was isolated from a high-pH borax leachate pond. It reduces metals in the presence of yeast extract at elevated salt concentrations; however use of sulfate or thiosulfate as an electron acceptor in pure culture was not observed (Ye et al., 2004). C. celluloyticum and other closely related Clostridium species degrade cellulose, xylan and polysaccharides.
Community Metabolism
Consistent with the observation of anoxic water below the chemocline, we detected very few genes that encode components of aerobic carbon fixation or aerobic respiration (Figure 4). The genetic potential for fermentation and CO oxidation was moderately more abundant at 8 m than in the sediments. We observed no evidence for methanogenesis or the aerobic oxidation of methane at either depth. Genes for nitrogen mineralization and nitrogen assimilation were abundant at both depths. The 8-m and sediment metagenomes contain a limited diversity of functional genes for assimilatory and dissimilatory sulfate reduction. To further assess the genetic potential for sulfate reduction at 8 m and in the sediments, we identified three genes essential for sulfate reduction: dsrAB, the genes encoding dissimilatory sulfite reductase, and dsrC. The 8-m metagenome contained five copies of dsrAB, and the sediment metagenome contained 12 copies of dsrAB. We also recovered 17 copies of dsrC, that all encode the two strictly conserved cysteines in the C-terminal arm (Santos et al., 2015), from the 8-m metagenome and 17 from the sediments. dsr genes are also encoded in the genomes of non-sulfate-reducing, syntrophic bacteria and thus their presence does not confirm sulfate reduction (Imachi et al., 2006). Regardless, the closest tBlastN hits of all of the dsrAB and dsrC sequences were to dissimilatory sulfate reducing Deltaproteobacteria, suggesting they are not involved in sulfur oxidation. Genes encoding components of the Sox enzyme complex which oxidizes thiosulfate were rare, as were genes coding for sulfide quinone oxidoreductase, a protein that oxidizes sulfide to elemental sulfur (data not shown).
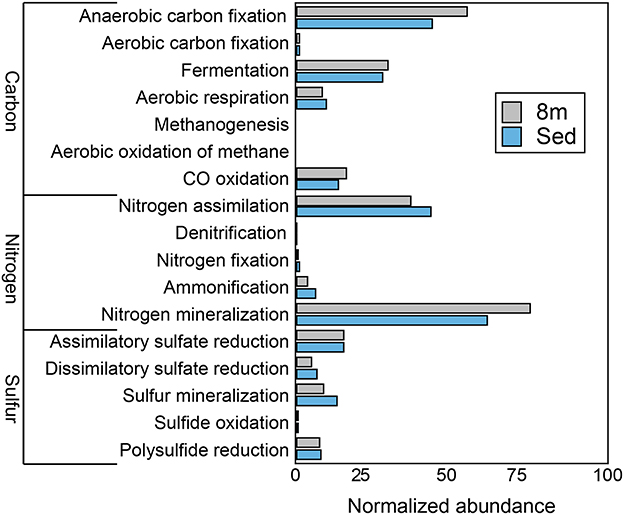
Figure 4. The genetic potential for carbon, nitrogen, and sulfur cycling at 8 m and in the sediments of Mahoney Lake. The genetic potential for each step was estimated using a combination of normalized marker gene ratios as previously described (Lauro et al., 2011). The marker genes for dissimilatory sulfate reduction and sulfide oxidation (K00394, K00395, K00396) can operate in both sulfide oxidation and sulfate reduction. Therefore, they were assigned to sulfate reduction or sulfur oxidation based on the best match within KEGG. Sed, sediments. Marker genes are provided in Table S4.
Organisms such as Clostridia produce H2 to dispose of the excess reductant generated during fermentation, while others may also produce H2S when polysulfide is available (Vignais et al., 2001). In general, the H2 evolution activity of [FeFe]-hydrogenases is 10- to 100-fold greater than [NiFe]-hydrogenases that are typically involved in H2 uptake (Vignais et al., 2001; Frey, 2002). Fermentative organisms can also consume H2 via [NiFe]-hydrogenases to produce the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) for anabolic metabolism. The 8-m metagenome contained 154 copies of genes encoding [NiFe]-hydrogenases and 163 copies of genes encoding [FeFe]-hydrogenases, while the sediment dataset contained 242 genes encoding [NiFe]-hydrogenases and 199 copies of genes encoding [FeFe]-hydrogenases. This distribution is consistent with the high abundance of organisms related to known fermenters and with the presence of other genes involved in fermentation. The 8-m metagenome contained 32 copies of the gene encoding the NifD, a structural component of the Mo-dependent nitrogenase, while 40 copies of nifD were recovered from the sediment metagenome. N2 reduction, catalyzed by the nitrogenase enzyme, results in H2 production concomitant with N2-fixation with a fraction of the electrons used for H2 production (Eady, 1996). In environments where fixed nitrogen is not limiting, an intriguing possibility is that nitrogen fixation is employed for H2 production. The metagenomes also encode numerous proteins that may be involved in the decomposition of recalcitrant plant biomass or cellulose, including glycosyl hydrolyses, glycosyl transferases, endoglucanases, and glyoxal oxidases.
We recovered 12 and 15 full-length, unique sequences of the gene encoding ribulose-1,5 bisphosphate carboxylase-oxygenase (RuBisCO), the key CO2-fixing enzyme of the Calvin-Benson-Bassham (CBB) cycle, from the 8-m metagenome and the sediment metagenome respectively. Of the 27 sequences, eight were identified as type I, five as type II, 10 as type III, and five as type IV (Figure S2). RuBisCO type I is most efficient under low CO2 concentration while Type II is less able to discriminate between O2 and CO2 and thus tends to function better under low oxygen and/or high CO2 concentrations. Type III RuBisCO enzymes are typically associated with Archaea; however, sequences affiliated with archaea were not abundant in either metagenome. In Archaea, the type III enzymes are involved in converting AMP, phosphate, CO2, and H2O to adenine and two molecules of 3-PGA, salvaging the adenine base of AMP (Sato et al., 2007; Wrighton et al., 2012). Type IV RuBisCO or RuBisCO-like protein is thought to be incapable of catalyzing carbon fixation because it lacks several key conserved residues necessary for carboxylase activity; instead, RuBisCO-like protein may play a role in sulfur oxidation or oxidative stress (Tabita et al., 2007).
Genomes
Analysis of tetranucleotide frequency allowed the recovery of several near-complete genomes, including two Firmicutes species from the 8-m sample, a genome of a new member of the FCB superphylum from the sediments and two Deltaproteobacteria species—one from 8 m and one from the sediment sample.
Deltaproteobacteria
Previous work has shown that a significant portion of the sulfide used by the dense layer of photoautotrophs in the ML chemocline at 7 m is generated by sulfate reduction within the chemocline (Overmann et al., 1991, 1996a). However, sulfide flux from below the chemocline has also been observed (Overmann et al., 1991) and sulfide concentrations increase with depth suggesting sulfate reducing bacteria are active below the chemocline (Northcote and Hall, 1983; Overmann et al., 1991). In addition, analysis of 16S rRNA sequences from ML are consistent with sulfate reduction in the hypolimnion and sediments (Klepac-Ceraj et al., 2012). Here, we reconstructed genomes of two Deltaproteobacteria species from the aphotic zone of ML—one from the 8-m sample and one from the sediments. According to phylogenetic analysis of 16S rRNA gene sequences and 18 concatenated ribosomal proteins in the bins (Figure 5A; Figure S3), the ML8_D population is closely related to uncultured deep-sea sediment clones and to Dissulfuribacter thermophilus, a hydrothermal vent chemolithoautotroph that disproportionates elemental sulfur to thiosulfate and sulfite (Figure 5A; Slobodkin et al., 2013). The ML8_D and D. thermophilus 16S rRNA sequences share 90% sequence identity. The closest relatives with fully sequenced genomes are Desulfobulbus spp. (Figure S3). The ML8_D genome is 98% complete and contains ~3.5 Mbp over 93 scaffolds with an average G+C content of 45.2%. The genome encodes a complete dissimilatory sulfate reduction pathway—a sulfate adenylyltransferase (ATP-sulfurylase, Sat), an APS reductase (AprAB), and the dissimilatory sulfite reductase complex (DsrAB, DsrC, and DsrMKJOP). In addition, the genome encodes all of the proteins necessary for dissimilatory nitrate reduction—nitrate reductase (Nar) and nitrite reductase (Nir) as well as genes encoding proteins for gluconeogenesis, ATP synthase, acetyl-CoA synthetase (the key enzyme for the utilization of acetate), some genes encoding homologs of TCA cycle proteins, and genes encoding flagella. The genome also encodes an acetyl-CoA synthase, a carbon monoxide dehydrogenase, a formate dehydrogenase and a Hox [NiFe]-hydrogenase. The presence of hydrogenase and formate dehydrogenase suggest that H2 and formate play important roles in the flow of electrons during sulfate reduction by the ML8_D population. However, the exact function of the NAD+-reducing hydrogenase (Hox) is not known—it has been suggested to play a role in removing excess electrons from fermentation (Horch et al., 2012). Some SRBs use CO dehydrogenase/acetyl-CoA synthase to fix carbon via the reductive acetyl-CoA pathway (Schauder et al., 1989); however, the ML8_D genome does not encode the full pathway. Although the genome is incomplete, the gene content for the ML8_D population suggests that these organisms are heterotrophic, motile, and capable of coupling anaerobic acetate oxidation to the reduction of sulfate.
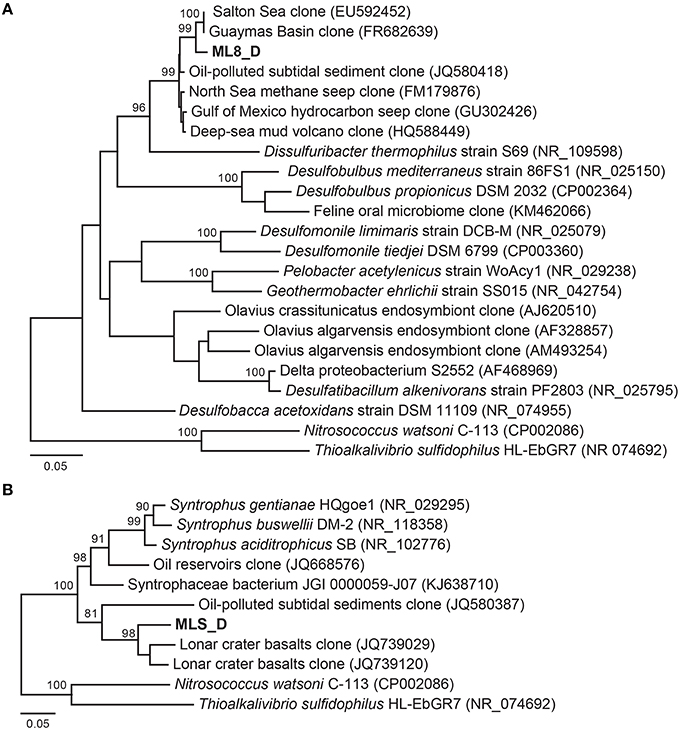
Figure 5. Maximum likelihood based phylogenetic 16S rRNA gene tree of closely related Deltaproteobacteria and the Deltaproteobacteria genomic bins from the 8 m metagenome (A) and the sediment metagenome (B). Bootstrap support values based on 1000 bootstrap samplings >90 are noted.
Analysis of the Deltaproteobacteria bin from the sediments, MLS_D, indicates this population is related to clones from oil-impacted sediments and basalts and to previously characterized members of the genus Syntrophus, including the model organism S. aciditrophicus (Figure 5B; Figure S3). The MLS_D and S. aciditrophicus 16S rRNA sequences share 91% sequence identity. S. aciditrophicus degrades fatty acids and aromatic acids in syntrophic association with hydrogen/formate-using microorganisms (Jackson et al., 1999). MLS_D contained ~2.5 Mbp over 70 scaffolds with an average G+C content of 55.9%. The genome is 98% complete based on the presence of single copy marker genes (Table S2). The genome encodes the genetic machinery necessary for assimilatory sulfate reduction (the Cys system) as well as Nap (a periplasmic nitrate reductase), the genetic machinery necessary for assimilation of ammonia, and biosynthesis of flagella. The genes necessary for an F-type ATPase were also present as were the gluconeogenic and pentose phosphate pathway genes to synthesize hexose- and pentose-phosphates from acetyl-CoA. Genes encoding two of the subunits for the ATP-dependent, benzoyl-CoA reductase were detected as well as genes encoding an Rnf-type, ion-translocating, electron transport complex, and genes for transporters for uptake of branched-chain amino acids. Genes encoding multiple alcohol deyhrogenases were encoded in the MLS_D genome, including one that is closely related to adh1 of Desulfovibrio vulgaris Hildenborough. In D.vulgaris Hildenborough, adh1 is highly expressed in cells grown on lactate, pyruvate, formate, or hydrogen as electron donors for sulfate reduction (Haveman et al., 2003). Genes encoding proton-translocating, NADH:ubiquinone oxidoreductase (complex I) or succinate dehydrogenase (complex II) were not recovered. In addition, MLS_D genome did not contain the genes that would permit the use of external electron acceptors such as oxygen, nitrate, sulfate, iron, or organic molecules. While these genes are also lacking from the genome of the obligate syntroph S. aciditrophicus, the MLS_D genome is incomplete (~98% complete), and thus we cannot conclude that the MLS_D population in Mahoney Lake relies on hydrogen/formate-using microorganisms.
Firmicutes
We reconstructed two Firmicutes genomes from the 8-m sample. Based on phylogenetic analysis of 18 concatenated ribosomal proteins (Figure 6), the ML8_F1 population is most closely related to members of the Alkaliphilus genus, and ML8_F2 is most closely related to D. alkaliphilus. The ML8_F1 genome contained ~2.3 Mbp over 58 scaffolds with an average G+C content of 50.2% (Table 1). The ML8_F2 genome contained ~3.0 Mbp over 143 scaffolds with an average G+C content of 46.6%. Based on the presence of single copy phylogenetic marker genes (Table S2), the ML8_F1 genome is 98% complete while the ML8_F2 genome is 96% complete.
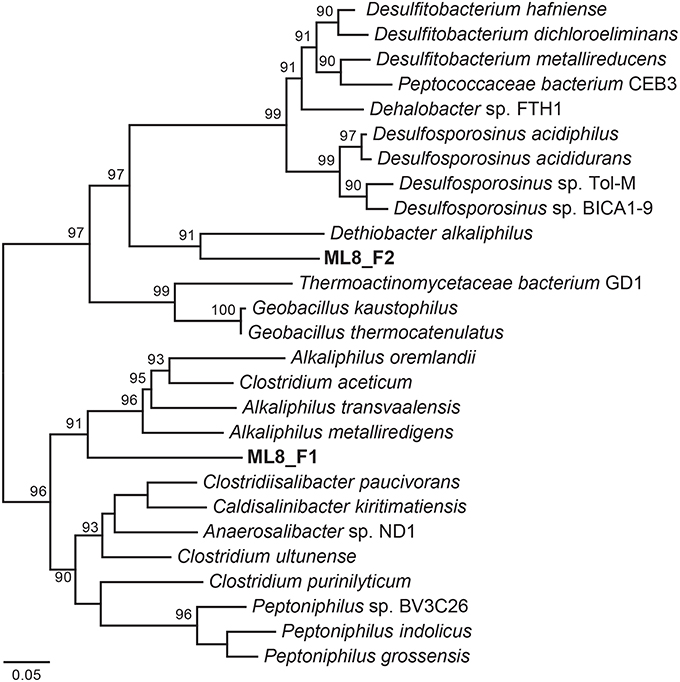
Figure 6. Maximum likelihood phylogenetic tree of 18 concatenated single-copy ribosomal proteins (Table S3) the Clostridia species genomic bins. Bootstrap support values based on 1000 bootstrap samplings >90 are noted.
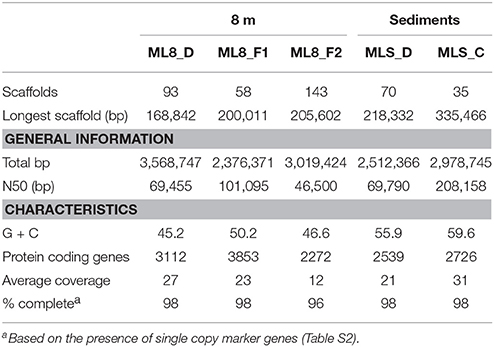
Table 1. Statistics for the genome bins.
The ML8_F1 genome is most closely related to Alkaliphilus spp.—including A. metalliredigens, A. transvaalensis, and A. oremlandii. A. metalliredigens was isolated from a leachate pond and is a metal-reducing alkaliphile (Ye et al., 2004). A. oremlandii can use thiosulfate as an electron acceptor with a variety of organic electron donors, including acetate, pyruvate, and formate (Fisher et al., 2008). The ML8_F2 genome is most closely related to D. alkaliphilus. In addition to sulfur disproportionation, D. alkaliphilus oxidizes H2 and is a facultative autotroph (Sorokin et al., 2008). The ML8_F1 genome encodes a Nuo-type NADH hydrogenase and an F-type ATPase. The ML8_F2 genome encodes components of a Nuo-type NADH dehydrogenase, a V-type ATPase, and an F-type ATPase. The ML8_F2 genome encodes a homolog of acetyl-CoA synthase acetyl-CoA synthase and a carbon monoxide dehydrogenase as well as all of the genes necessary to carry out this activity (Ragsdale and Pierce, 2008). The ML8_F1 genome encodes a carbon monoxide dehydrogenase but not the acetyl-CoA synthase. Both genomes encoded multiple [FeFe]-hydrogenases and one [NiFe]-hydrogenase—plus all the necessary hydrogenase maturation genes. Genes encoding enzymes necessary for assimilatory (Cys) and dissimilatory sulfate reduction (Dsr), as well as nitrogen fixation, denitrification, or nitrate reduction, were absent in both ML8_F genomes which are both >95% complete based on the presence of single copy marker genes. Genes encoding proteins involved in glycolysis and the tricarboxylic acid (TCA) cycle were present in both genomes, along with numerous transporters, particularly metal transporters. While further data are required to test our hypothesis that the ML8_F1 and F2 populations are acetogenic bacteria that use CO or CO2 plus H2 as their sole carbon and energy sources (Drake et al., 1994) our data do suggest a role for Firmicutes populations in hydrogen oxidation and acetate production in Mahoney Lake.
MLS_C Genome
We reconstructed a genome from the ML sediment metagenome that contains ~2.9 Mbp over 35 scaffolds with an average G+C content of 59.6%. The genome is nearly complete, encoding 98% of single copy phylogenetic marker genes (Table S2). The 16S rRNA gene sequence is most closely related to sequences recovered from Yuncheng Salt Lake of Shanxi Province, China which is rich in sulfate, chloride and magnesium; submarine methane seeps (Beal et al., 2009; Pachiadaki et al., 2010); and Zodletone Spring (anaerobic spring rich in methane, sulfide, and sulfur in Oklahoma, USA; Elshahed et al., 2007) as well as to the isolates Caldithrix abyssi and Caldithrix paleocroryensis, which are thermophilic, mixotrophic anaerobes from hydrothermal deep-sea environments (Miroshnichenko et al., 2003, 2010). The MLS_C 16S rRNA sequence shares only 83 and 84% sequence identity with the C. abyssi and C. paleocroryensis 16S rRNA sequences, respectively. In addition, the MLS_C 16S rRNA sequence shares 79% sequence identity with the 16S rRNA sequence from Fibrobacter succinogenes S85, a cellulolytic luminal mesophilic bacterium (Weimer, 1993). The predicted MLS_C proteins shared the highest percent sequence identity to proteins from many different phyla (Figure 7). The majority were affiliated with Deltaproteobacteria and Candidatus Cloacimonetes (WWE1 clade). Most Caldithrix sequences have been recovered from sulfide-rich marine niches, such as those found near hydrothermal vents, but they have also been recovered from mangrove soil, sulfidic caves, farm soil, methanogenic granular sludges, and in a benzoate-degrading consortium (Huang et al., 2010; Zhou et al., 2011; Alauzet and Jumas-Bilak, 2014). Sequences affiliated with Candidatus Cloacimonetes were first recovered from a wastewater treatment plant (Pelletier et al., 2008). Analysis of the single copy marker genes in these related isolates was similar—the highest percent sequence identity of individual proteins were from many different phyla, even though the majority of these sequences are on the same contig (Table S5).
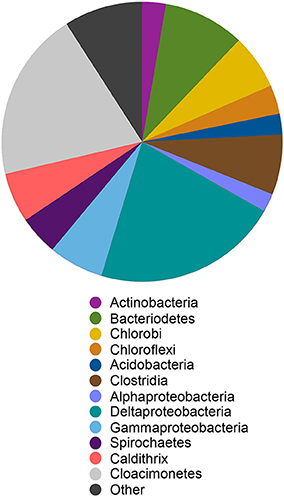
Figure 7. Best BlastP hits for all predicted protein sequences in the Candidatus Aegiribacteria MLS_C genome. Other indicates all phyla represented by < 5% of the total sequences in the genome. The relative abundance of taxonomic groups is summed at the phylum level except for Proteobacteria and Firmicutes which are summed at the class level and Caldithrix which are summed at the genus level.
Phylogenetic analysis of the MLS_C 16S rRNA gene places the lineage into a monophyletic branch within the FCB superphylum MLS_C represents a deep-branching, monophyletic lineage (90% bootstrap support) with F. succinogenes S85 as the next relative (Figure 8). Phylogenetic analysis of the rpS3 sequences places the MLC_S lineage into a monophyletic branch (95% bootstrap support) with Candidatus Cloacimonetes acidaminovorans (Figure S4). The highest ANI observed in this limited data set was between the MLS_C genome and a Marinimicrobia bacterium genome (76.9%) whereas the average nucleotide identity between the MLS_C genome and the genomes of F. succinogenes and Candidatus Cloacimonetes acidaminovorans was 64.3 and 62.4%, respectively (Figure S4). Members of the clade Marinimicrobia (formally SAR406; Rinke et al., 2013) are common in marine environments (Yilmaz et al., 2015). Based on these data, the MLS_C population appears to be only distantly related to the Caldithrix genus, Candidatus Cloacimonetes, and F. succinogenes S85. Because the 16S rRNA gene from MLS_C shares low sequence identity (79–84%) to described bacterial phyla, and the sequence represents a deep-branching, monophyletic lineage, we propose that it represents a new genus within the FCB superphylum. In agreement with recently proposed guidelines for taxonomic classification of environmentally-derived genome sequences with currently accepted nomenclature standards (Hedlund et al., 2015), we propose the name Candidatus Aegiribacteria MLS_C after the Norse God of the Sea, Aegir, a renowned undersea brewer, reflecting its presumed fermentative metabolism. The FCB superphylum now includes the original phyla Fibrobacteres, Chlorobi, Bacteroidetes, and Ignavibacteriae as well as the candidate phyla Cloacimonetes (WWE1), Marinimicrobia (SAR406), Latescibacteria (WS3), Gemmatimonadetes, Hydrogenendentes (NKB19) (Rinke et al., 2013), the Zixibacteria RBG1 (Castelle et al., 2013), the recently discovered “Candidatus Kryptonia” (Eloe-Fadrosh et al., 2016), the Caldithrix genus and the new genus described here, Candidatus Aegiribacteria MLS_C.
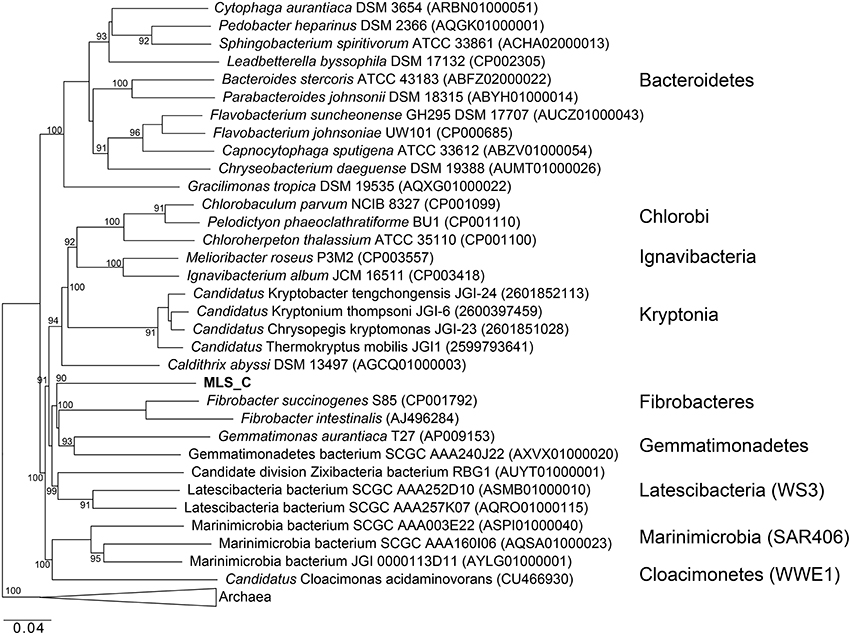
Figure 8. Maximum likelihood based phylogenetic 16S rRNA gene tree of members of the FCB superphylum and MLS_C. Bootstrap support values based on 1000 bootstrap samplings >90 are noted. NCBI accession numbers or IMG gene ids are shown in parentheses.
The G+C content of the MLS_C genome is elevated (59.6%) compared to the C. abyssi genome (43.3%) and the C. palaeochoryensis genome (42.5%), as well as the reconstructed Candidatus Cloacamonas acidaminovorans genome (37.9%; Pelletier et al., 2008) and the F. succinogenes S85 genome (48%; Suen et al., 2011). There are only two characterized representatives of the Caldithrix genus, C. abyssi, and C. palaeochoryensis. C. abyssi oxidizes molecular hydrogen and acetate in the presence of nitrate, while C. palaeochoryensis is an obligately fermentative chemoorganotroph that can grow on di- and polysaccharides, as well as proteinaceous substrates (Alauzet and Jumas-Bilak, 2014). Ca. Cloacimonetes sequences have been recovered from anaerobic digesters (Pelletier et al., 2008; Lykidis et al., 2011; Wu et al., 2013; Limam et al., 2014; Nobu et al., 2015b). Ca. Cloacimonetes have been inferred to perform syntrophic propionate degradation (Nobu et al., 2015b) and the anaerobic digestion of cellulose (Limam et al., 2014), but no isolates have been fully characterized. Selective enrichment and a reconstructed genome sequence of Ca. Cloacamonas acidaminovorans suggest the organism is syntrophic (Pelletier et al., 2008). F. succinogenes S85 produces succinate, acetate, and formate as major fermentative end products and is specialized to derive energy from cellulose (Weimer, 1993; Suen et al., 2011).
Genes encoding pili and flagella were present in the ML_C genome suggesting that MLS_C is motile. Genes encoding a periplasmic [FeFe]-hydrogenase and a [NiFe]-hydrogenase and all necessary hydrogenase maturation genes were present, as well as a heterodisulfide reductase that is widespread in anaerobic bacteria. The genome encodes homologs of D-lactate dehydrogenase (gene ldhA) and pyruvate dehydrogenase (gene pdh) for conversion of lactate to acetyl-CoA, as well as carbon monoxide dehydrogenase/acetyl-CoA synthetase (CODH/ACS). The MLS_C genomes is nearly complete (~98%) and similar to the Ca. Cloacamonas acidaminovorans genome, the genome does not contain genes encoding the enzymes necessary to synthesize certain amino acids. The genome encodes all of the machinery necessary for degradation of organic matter for acquisition of carbon and nitrogen including multiple proteases and peptidases. The genome also encodes the proteins necessary for the oxidation of propionate into acetate and carbon dioxide via methylmalonyl-CoA, succinate, fumarate, malate, oxaloacetate, pyruvate, and acetyl-CoA as intermediates. This pathway is found in obligate syntrophic bacteria (Schink, 1997) and is also encoded in the Ca. Cloacamonas acidaminovorans genome (Pelletier et al., 2008). This pathway is only thermodynamically favorable under low H2 conditions, such as in syntrophic consortia with H2-scavenging bacteria (Megonigal et al., 2004; Ariesyady et al., 2007). Populations capable of scavenging H2, such as sulfate reducing bacteria and acetogenic bacteria, are present in the sediment metagenome and could fulfill this partnership.
Carbon and Sulfur Cycling in Mahoney Lake
16S rRNA sequencing indicated that different layers of ML are host to distinct microbial assemblages (Klepac-Ceraj et al., 2012). Our metagenomic sequencing suggests these microbial assemblages are similar phylogenetically at the phylum or class level (Figure 1) despite marked differences in geochemistry throughout the stratified water column—the epilimnion is oxic and oligotrophic whereas the hypolimnion is anoxic, more saline, eutrophic and rich in sulfate, sulfide, and polysulfide (Hall and Northcote, 1986). To assess potential differences in functional diversity at the chemocline and below, we compared COG functional gene assignments between the 7-m (chemocline), 8-m, and sediment communities. These data indicate similar COG distributions in assemblages from the three horizons (Figure 9A; Figure S5A). COGs represented at high abundance included common functions such as translation, ribosomal structure, and biogenesis; energy production and conversion; and replication, recombination, and repair. The sediment and 8-m communities contain a more similar distribution of COG categories compared to the 7-m community. Because this analysis is based on COG categories, we examined the 50 most abundant COGs to determine what functional differences might be detectable between the horizons. In this analysis, the 7- and 8-m communities were more similar than the sediment community (Figure S5B). Putative proteins with lower relative abundance in the 7- and 8-m assemblages compared to the sediments included COG0840 and COG1372. COG0840 is involved in signal transduction and cell motility, while COG1372 is an endonuclease with a role in DNA replication, recombination, and repair. Sequences with lower relative abundance in the sediments included proteins involved in signal transduction (COG0745) and DNA replication, recombination, and repair (COG4974). COG1595, a DNA-directed RNA polymerase, had a higher relative abundance at 7 and 8 m compared to the sediments. A putative NAD(P)-dependent dehydrogenase (COG1028) was also more abundant in and near the chemocline.
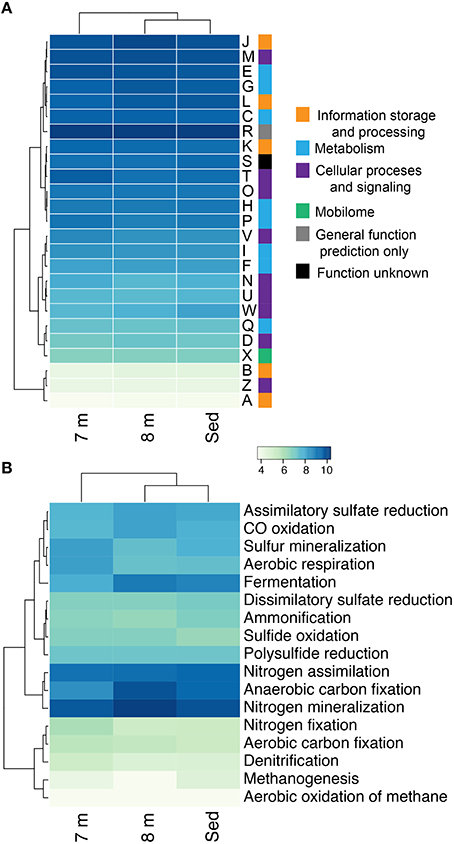
Figure 9. Hierarchical clustering of the relative abundance of COG categories (A) and the genetic potential for carbon, nitrogen, and sulfur cycling in each assemblage at 7, 8 m, and in the sediments of Mahoney Lake (B). Values within each category are normalized across samples (see Section Materials and Methods). Clustering analyses is based on the normalized abundance profiles of COGs. The genetic potential for each step in (B) was estimated using a combination of normalized marker gene ratios as previously described (Lauro et al., 2011). Marker genes are provided in Table S4. The 7 m data set is from Hamilton et al. (2014). Sed, Sediment; A, RNA processing and modification; B, chromatin structure and dynamics; C, energy production and conversion; D, cell cycle control, cell division, chromosome partitioning; E, amino-acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; J, translation, ribosomal structure, and biogenesis; K, transcription; L, replication, recombination, and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, post-translational modification, protein turnover, chaperones; P, inorganic ion transport and metabolism; and Q, secondary metabolites biosynthesis, transport, and catabolism; R, general function prediction only; S, function unknown; T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; W, extracellular structures; X, nuclear structure; V, defense mechanisms; Z, cytoskeleton.
Our recent analysis of the Mahoney Lake chemocline indicated functional redundancy in both the oxidative and reductive arms of the sulfur cycles within the narrow phototrophic plate at 7 m (Hamilton et al., 2014). Comparison of COGs from 7, 8 m, and the sediments reveals no difference in the abundance of assimilatory or dissimilatory sulfate reduction, sulfur mineralization, or polysulfide reduction. These data suggest that similar processes are also active below the chemocline or that our data include genetic material that settled from the chemocline (Figure 9B). In the COG analysis, the marker genes for dissimilatory sulfate reduction and sulfide oxidation (K00394, K00395, K00396) are equivalent and can operate in either process. In the 8-m and sediment metagenomes, the majority of sequences cluster with those involved in dissimilatory sulfate reduction, consistent with recovery of multiple populations capable of reducing oxidized sulfur below the chemocline (data not shown). In contrast, the majority of the same COGs from the 7-m metagenome clustered with sequences involved in sulfur oxidation (Hamilton et al., 2014). The hypolimnion (8 m) and sediments are also rich in polysulfides, elemental sulfur, and other sulfur intermediates that can be disproportionated (Bak and Cypionka, 1987; Finster, 2008; Poser et al., 2013). We observed multiple populations capable of reducing oxidized sulfur compounds in the highly sulfidic (35 mM) hypolimnion but very little evidence for sulfur oxidation. Autotrophic growth by sulfite/thiosulfate disproportionation appears to be a common trait among haloalkaliphilic SRB (Sorokin et al., 2011). Based on the recovery of taxonomic markers genes (16S rRNA and rpS3 sequences) related to Deltaproteobacteria that disproportionate sulfur compounds and the abundance of sulfur cycle intermediates below the chemoline in Mahoney Lake, it is plausible that disproportionation is an important process in ML.
The redox stability of ML can be attributed to its geochemistry—that is, high salinity and high sulfur concentrations—and a physiographic position that results in minimal wind mixing and the delivery of allochthonous carbon required to supply the extra flux of electrons needed to sustain sulfate reduction (Overmann, 1997). Oxygenic primary production in the epilimnion is not sufficient to supply electrons for sulfate reduction in the form of phytoplanktonic carbon fixed in situ—instead, organic matter is supplied from the surrounding landscape (Overmann et al., 1996a,b; Overmann, 1997). The strong density stratification and the related redox profile help retain organic matter in neutrally buoyant layers, supporting remineralization of organic matter in the water column (Overmann, 1997)—a feature that is also common in some marine systems (MacIntyre et al., 1995; Sorokin, 2002). ML hosts one of the densest chemocline phototroph communities observed to date, yet direct export of this planktonic organic matter from the chemocline to the sediments is low (Overmann, 1997). Based on fatty acid profiles and bulk organic carbon isotopes, organic matter from the water column is distinguishable from sediment organic matter (Bovee and Pearson, 2014). The latter has characteristics dominantly of allochthonous, higher-plant sources and is most likely sourced from the margins of the lake basin. In contrast, most of the planktonic organic matter is lost from the shallow water column through respiration. We found abundant evidence for anaerobic respiration below the chemocline, while proteins necessary for aerobic respiration were elevated at 7 m compared to 8 m and the sediments (Figure 9B).
In this study we recovered abundant evidence for heterotrophic—and especially fermentative—organisms at 8 m and in the sediments. The relative abundance of sequences for proteins putatively involved in anaerobic carbon fixation and fermentation were elevated relative to the chemocline (Figure 9B). Our data also indicate the presence of syntrophic populations and heterotrophs capable of degrading cellulose in the sediments. These observations are consistent with intense organic matter remineralization in the water column and delivery of more complex carbon substrates to the sediments.
Conclusion
Mahoney Lake is an extreme meromictic system with unusually high levels of sulfate and sulfide. Here, we observed diverse microbial assemblages that are taxonomically similar at the phylum level yet functionally distinct in different layers of the lake. Metagenomic sequencing recovered abundant anaerobic bacteria from below the chemocline that are putatively involved in remineralizing organic matter and genes encoding for hydrogenases presumably due to organic matter loading and the need to dispose of excess electrons. Although the precise biochemical pathway of sulfur disproportionation remains uncharacterized, we recovered sequences most closely related to Deltaproteobacteria spp. known to perform this reaction in pure cultures. These data, along with the abundance of zero valent sulfur below the chemocline (Overmann et al., 1996a), suggest a role for sulfur disproportionation in highly euxinic systems despite the observation that elemental sulfur disproportionation becomes thermodynamically unfavorable under high sulfide concentrations (Canfield and Thamdrup, 1994; Rabus et al., 2000). We recovered a breadth of taxonomic and functional diversity from below the chemocline in an extreme euxinic environment including a nearly complete genome of Candidatus Aegiribacteria MLS_C, a new genus within the FCB superphylum. Our data highlight the role of these diverse assemblages in maintaining euxinia and suggest undiscovered taxa and diversity exist in other dark, sulfur-rich environments.
Author Contributions
TH, WG, TL, AP, and JM designed the study. AP, WG, and TL collected the samples. TH, RB, SS, WM, and AP performed the analyses. TH, RB, SS, WM, WG, TL, AP, and JM contributed to writing, editing and finalizing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the contributions of the reviewers whose comments improved the manuscript. This project was funded by the National Science Foundation (NSF EAR-0525503 to AP and JM). TH graciously acknowledges support from the NAI Postdoctoral Program.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00598
References
Alauzet, C., and Jumas-Bilak, E. (2014). “The phylum Deferribacteres and the genus Caldithrix,” in The Prokaryotes – Other Major Lineages of Bacteria and the Archaea, eds E. Rosenbrug, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin; Heidelberg: Springer-Verlag), 595–611.
Amend, J. P., and Shock, E. S. (2001). Energetics of overall metabolic reactions of thermophilic and hyperthermophilic Archaea and Bacteria. FEMS Microbiol. Rev. 25, 175–243. doi: 10.1111/j.1574-6976.2001.tb00576.x
Ariesyady, H. D., Ito, T., Yoshiguchi, K., and Okabe, S. (2007). Phylogenetic and functional diversity of propionate-oxidizing bacteria in an anaerobic digester sludge. Appl. Microbiol. Biotechnol. 75, 673–683. doi: 10.1007/s00253-007-0842-y
Bak, F., and Cypionka, H. (1987). A novel type of energy-metabolism involving fermentation of inorganic sulfur-compounds. Nature 326, 891–892. doi: 10.1038/326891a0
Bak, F., and Pfennig, N. (1987). Chemolithotrophic growth of Desulfovibrio sulfodismutans sp. nov. by disproportionation of inorganic sulfur compounds. Arch. Microbiol. 147, 184–189. doi: 10.1007/BF00415282
Beal, E. J., House, C. H., and Orphan, V. J. (2009). Manganese- and iron-dependent marine methane oxidation. Science 325, 184–187. doi: 10.1126/science.1169984
Bosshard, P. P., Santini, Y., Grüter, D., Stettler, R., and Bachofen, R. (2000). Bacterial diversity and community composition in the chemocline of the meromictic alpine Lake Cadagno as revealed by 16S rDNA analysis. FEMS Microbiol. Ecol. 31, 173–182. doi: 10.1111/j.1574-6941.2000.tb00682.x
Bovee, R., and Pearson, A. (2014). Strong influence of the littoral zone on sedimentary lipid biomarkers in a meromictic lake. Geobiology 12, 529–541. doi: 10.1111/gbi.12099
Briée, C., Moreira, D., and López-García, P. (2007). Archaeal and bacterial community composition of sediment and plankton from a suboxic freshwater pond. Res. Microbiol. 158, 213–227. doi: 10.1016/j.resmic.2006.12.012
Brocks, J. J., Love, G. D., Summons, R. E., Knoll, A. H., Logan, G. A., and Bowden, S. A. (2005). Biomarker evidence for green and purple sulphur bacteria in a stratified Palaeoproterozoic sea. Nature 437, 866–870. doi: 10.1038/nature04068
Brown, C. T., Hug, L. A., Thomas, B. C., Sharon, I., Castelle, C. J., Singh, A., et al. (2015). Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523, 208–2011. doi: 10.1038/nature14486
Bryant, M. P., Campbell, L. L., Reddy, C. A., and Crabill, M. R. (1977). Growth of Desulfovibrio in lactate or ethanol media low in sulfate in association with H2-utilizing methanogenic bacteria. Appl. Environ. Microbiol. 33, 1162–1169.
Canfield, D. E. (1998). A new model for Proterozoic ocean chemistry. Nature 396, 450–453. doi: 10.1038/24839
Canfield, D. E. (2013). Sulfur isotopes in coal constrain the evolution of the Phanerozoic sulfur cycle. Proc. Natl. Acad. Sci. U.S.A. 110, 8443–8446. doi: 10.1073/pnas.1306450110
Canfield, D. E., and Thamdrup, B. (1994). The production of 34S-depleted sulfide during bacterial disproportionation of elemental sulfur. Science 266, 1973–1975. doi: 10.1126/science.11540246
Carr, S. A., Orcutt, B. N., Mandernack, K. W., and Spear, J. R. (2015). Abundant atribacteria in deep marine sediment from the Adélie Basin, Antarctica. Front. Microbiol. 6:872. doi: 10.3389/fmicb.2015.00872
Castelle, C. J., Hug, L. A., Wrighton, K. C., Thomas, B. C., Williams, K. H., Wu, D., et al. (2013). Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat. Commun. 4, 2120. doi: 10.1038/ncomms3120
Castelle, C. J., Wrighton, K. C., Thomas, B. C., Hug, L. A., Brown, C. T., Wilkins, M. J., et al. (2015). Genomic expansion of domain Archaea highlights roles for organisms from new Phyla in anaerobic carbon cycling. Curr. Biol. 25, 690–701. doi: 10.1016/j.cub.2015.01.014
Coolen, M. J. L., and Overmann, J. (1998). Analysis of subfossil molecular remains of purple sulfur bacteria in a Lake Sediment. Appl. Environ. Microbiol. 64, 4513–4521.
Darriba, D., Taboada, G., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. doi: 10.1038/nmeth.2109
Decristophiris, P. M. A., Peduzzi, S., Ruggeri-Bernardi, N., Hahn, D., and Tonolla, M. (2009). Fine scale analysis of shifts in bacterial community structure in the chemocline of meromictic Lake Cadagno, Switzerland. J. Limnol. 68, 16–24. doi: 10.4081/jlimnol.2009.16
Del Don, C., Hanselmann, K. W., Peduzzi, R., and Bachofen, R. (2001). The meromictic alpine Lake Cadagno: orographical and biogeochemical description. Aquat. Sci. 63, 70–90. doi: 10.1007/PL00001345
Delmont, T. O., Malandain, C., Prestat, E., Larose, C., Monier, J. M., Simonet, P., et al. (2011). Metagenomic mining for microbiologists. ISME J. 5, 1837–1843. doi: 10.1038/ismej.2011.61
Dick, G. J., Andersson, A. F., Baker, B. J., Simmons, S. L., Thomas, B. C., Yelton, A. P., et al. (2009). Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10:R85. doi: 10.1186/gb-2009-10-8-r85
Di Rienzi, S. C., Sharon, I., Wrighton, K. C., Koren, O., Hug, L. A., Thomas, B. C., et al. (2013). The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. Elife. 2:e01102. doi: 10.7554/eLife.01102
Dodsworth, J. A., Blainey, P. C., Murugapiran, S. K., Swingley, W. D., Ross, C. A., Tringe, S. G., et al. (2013). Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for members of the OP9 lineage. Nat. Commun. 4, 1854. doi: 10.1038/ncomms2884
Drake, H. L., Daniel, S. L., Matthies, C., and Küsel, K. (1994). “Acetogenesis, acetogenic bacteria, and the acetyl-CoA pathway: past and current perspectives,” in Acetogenesis, ed H. L. Drake (New York, NY: Chapman & Hall), 3–60.
Durbin, R., Eddy, S., Krogh, A., and Mitchison, G. (1998). Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, UK: Cambridge University Press.
Eady, R. R. (1996). Structure-function relationships of alternative nitrogenase. Chem. Rev. 96, 3013–3030. doi: 10.1021/cr950057h
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Eloe-Fadrosh, E. A., Paez-Espino, D., Jarett, J., Dunfield, P. F., Hedlund, B. P., Dekas, A. E., et al. (2016). Global metagenomic survey reveals a new bacterial candidate phylum in geothermal springs. Nat. Commun. 7:10476. doi: 10.1038/ncomms10476
Elshahed, M. S., Youssef, N. H., Luo, Q., Najar, F. Z., Roe, B. A., Sisk, T. M., et al. (2007). Phylogenetic and metabolic diversity of Planctomycetes from anaerobic, sulfide- and sulfur-rich Zodletone Spring, Oklahoma. Appl. Environ. Microbiol. 73, 4707–4716. doi: 10.1128/AEM.00591-07
Ferreira, A. J. S., Siam, R., Setubal, J. C., Moustafa, A., Sayed, A., Chambergo, F. S., et al. (2014). Core microbial functional activities in ocean environments revealed by global metagenomic profiling analyses. PLoS ONE 9:e97338. doi: 10.1371/journal.pone.0097338
Finster, K. (2008). Microbial disproportionation of inorganic sulfur compounds. J. Sulfur Chem. 29, 281–292. doi: 10.1080/17415990802105770
Fisher, E., Dawson, A. M., Polshyna, G., Lisak, J., Crable, B., Perera, E., et al. (2008). Transformation of inorganic and organic arsenic by Alkaliphilus oremlandii sp. nov. Strain OhILAs. Ann. N.Y. Acad. Sci. 1125, 230–241. doi: 10.1196/annals.1419.006
Frey, M. (2002). Hydrogenases: hydrogen-activating enzymes. ChemBioChem 3, 153–160. doi: 10.1002/1439-7633(20020301)3:2/3<153::AID-CBIC153>3.0.CO;2-B
Gies, E. A., Konwar, K. M., Beatty, J. T., and Hallam, S. J. (2014). Illuminating microbial dark matter in meromictic sakinaw lake. Appl. Environ. Microbiol. 80, 6807–6818. doi: 10.1128/AEM.01774-14
Gregersen, L. H., Habicht, K. S., Peduzzi, S., Tonolla, M., Canfield, D. E., Miller, M., et al. (2009). Dominance of a clonal green sulfur bacterial population in a stratified lake. FEMS Microbiol. Ecol. 70, 30–41. doi: 10.1111/j.1574-6941.2009.00737.x
Grice, K., Cao, C. Q., Love, G. D., Bottcher, M. E., Twitchett, R. J., Grosjean, E., et al. (2005). Photic zone euxinia during the Permian-Triassic superanoxic event. Science 307, 706–709. doi: 10.1126/science.1104323
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Hall, K. J., and Northcote, T. G. (1986). Conductivity-temperature standardization and dissolved solids estimation in a meromictic saline lake. Can. J. Fish. Aquat. Sci. 43, 2450–2454. doi: 10.1139/f86-304
Hamilton, T. L., Bovee, R. J., Thiel, V., Sattin, S. R., Mohr, W., Schaperdoth, I., et al. (2014). Coupled reductive and oxidative sulfur cycling in the phototrophic plate of a meromictic lake. Geobiology 12, 451–468. doi: 10.1111/gbi.12092
Harris, J. K., Kelley, S. T., and Pace, N. R. (2004). New perspectives on uncultured bacterial phylogenetic division OP11. Appl. Environ. Microbiol. 70, 845–849. doi: 10.1128/AEM.70.2.845-849.2004
Haveman, S. A., Brunelle, V., Voordouw, J. K., Voordouw, G., Heidelberg, J. F., and Rabus, R. (2003). Gene expression analysis of energy metabolism mutants of Desulfovibrio vulgaris Hildenborough indicates an important role for alcohol dehydrogenase. J. Bacteriol. 185, 4345–4353. doi: 10.1128/JB.185.15.4345-4353.2003
Hedlund, B. P., Dodsworth, J. A., and Staley, J. T. (2015). The changing landscape of microbial biodiversity exploration and its implications for systematics. Syst. Appl. Microbiol. 38, 231–236. doi: 10.1016/j.syapm.2015.03.003
Horch, M., Lauterbach, L., Lenz, O., Hildebrandt, P., and Zebger, I. (2012). NAD(H)-coupled hydrogen cycling – structure–function relationships of bidirectional [NiFe] hydrogenases. FEBS Lett. 586, 545–556. doi: 10.1016/j.febslet.2011.10.010
Huang, Y., Gilna, P., and Li, W. (2009). Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25, 1338–1340. doi: 10.1093/bioinformatics/btp161
Huang, Y. J., Kim, E., Cox, M. J., Brodie, E. L., Brown, R., Wiener-Kronish, J. P., et al. (2010). A persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. OMICS 14, 9–59. doi: 10.1089/omi.2009.0100
Hug, L. A., Castelle, C. J., Wrighton, K. C., Thomas, B. C., Sharon, I., Frischkorn, K. R., et al. (2013). Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1:22. doi: 10.1186/2049-2618-1-22
Huson, D. H., Mitra, S., Weber, N., Ruscheweyh, H., and Schuster, S. C. (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560. doi: 10.1101/gr.120618.111
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Imachi, H., Sekiguchi, Y., Kamagata, Y., Loy, A., Qiu, Y.-L., Hugenholtz, P., et al. (2006). Non-sulfate-reducing, syntrophic bacteria affiliated with Desulfotomaculum Cluster I are widely distributed in methanogenic environments. Appl. Environ. Microbiol. 72, 2080–2091. doi: 10.1128/AEM.72.3.2080-2091.2006
Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M., Lauer, A., et al. (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc. Natl. Acad. Sci. U.S.A. 103, 2815–2820. doi: 10.1073/pnas.0511033103
Jackson, B. E., Bhupathiraju, V. K., Tanner, R. S., Woese, C. R., and McInerney, M. J. (1999). Syntrophus aciditrophicus sp. nov., a new anaerobic bacterium that degrades fatty acids and benzoate in syntrophic association with hydrogen-using microorganisms. Arch. Microbiol. 171, 107–114. doi: 10.1007/s002030050685
Johnston, D. T., Wolfe-Simon, F., Pearson, A., and Knoll, A. H. (2009). Anoxygenic photosynthesis modulated Proterozoic oxygen and sustained Earth's middle age. Proc. Natl. Acad. Sci. U.S.A. 106, 16925–16929. doi: 10.1073/pnas.0909248106
Jones, D. S., Albrecht, H. L., Dawson, K. S., Schaperdoth, I., Freeman, K. H., Pi, Y., et al. (2012). Community genomic analysis of an extremely acidophilic sulfur-oxidizing biofilm. ISME J. 6, 158–170. doi: 10.1038/ismej.2011.75
Klepac-Ceraj, V., Hayes, C. A., Gilhooly, W. P., Lyons, T. W., Kolter, R., and Pearson, A. (2012). Microbial diversity under extreme euxinia: Mahoney Lake, Canada. Geobiology 10, 223–235. doi: 10.1111/j.1472-4669.2012.00317.x
Konstantinidis, K. T., Braff, J., Karl, D. M., and DeLong, E. F. (2009). Comparative metagenomic analysis of a microbial community residing at a depth of 4,000 meters at station ALOHA in the North Pacific subtropical gyre. Appl. Environ. Microbiol. 75, 5345–5355. doi: 10.1128/AEM.00473-09
Lauro, F. M., DeMaere, M. Z., Yau, S., Brown, M. V., Ng, C., Wilkins, D., et al. (2011). An integrative study of a meromictic lake ecosystem in Antarctica. ISME J. 5, 879–895. doi: 10.1038/ismej.2010.185
Leavitt, W. D., Halevy, I., Bradley, A. S., and Johnston, D. T. (2013). Influence of sulfate reduction rates on the Phanerozoic sulfur isotope record. Proc. Natl. Acad. Sci. U.S.A. 110, 11244–11249. doi: 10.1073/pnas.1218874110
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Limam, R. D., Chouari, R., Mazéas, L., Wu, T.-D., Li, T., Grossin-Debattista, J., et al. (2014). Members of the uncultured bacterial candidate division WWE1 are implicated in anaerobic digestion of cellulose. Microbiologyopen 3, 157–167. doi: 10.1002/mbo3.144
Llorens-Marès, T., Yooseph, S., Goll, J., Hoffman, J., Vila-Costa, M., Borrego, C. M., et al. (2015). Connecting biodiversity and potential functional role in modern euxinic environments by microbial metagenomics. ISME J. 9, 1648–1661. doi: 10.1038/ismej.2014.254
Lloyd, K. G., Schreiber, L., Peterson, D. G., Kjeldson, K. U., Lever, M. A., Steen, A. D., et al. (2013). Predominant Archaea in marine sediments degrade detrital proteins. Nature 496, 215–218. doi: 10.1038/nature12033
Lohse, M., Bolger, A. M., Nagel, A., Fernie, A. R., Lunn, J. E., Stitt, M., et al. (2012). RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 40, W622–W627. doi: 10.1093/nar/gks540
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.0955
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Buchner, A., et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Lukashin, A., and Borodovsky, M. (1998). GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26, 1107–1115. doi: 10.1093/nar/26.4.1107
Lykidis, A., Chen, C. L., Tringe, S. G., McHardy, A. C., Copeland, A., Kyrpides, N. C., et al. (2011). Multiple syntrophic interactions in a terephthalate-degrading methanogenic consortium. ISME J. 5, 122–130. doi: 10.1038/ismej.2010.125
MacIntyre, S., Alldredge, A. L., and Gotschalk, C. C. (1995). Accumulation of marine snow at density discontinuities in the water column. Limnol. Oceanogr. 40, 449–468. doi: 10.4319/lo.1995.40.3.0449
McInerney, M. J., and Bryant, M. P. (1981). Anaerobic degradation of lactate by syntrophic associations of Methanosarcina barkeri and Desulfovibrio species and effect of H2 on acetate degradation. Appl. Environ. Microbiol. 41, 346–354.
Megonigal, J. P., Hines, M. E., and Visscher, P. T. (2004). “Anaerobic metabolism: linkages to trace gases and aerobic processes,” in Biochemistry, ed W. H. Schlesinger (Oxford, UK: Elsevier-Pergamon), 317–424.
Meyer, K. M., and Kump, L. R. (2008). Oceanic euxinia in Earth history: causes and consequences. Annu. Rev. Earth Planet. Sci. 36, 251–288. doi: 10.1146/annurev.earth.36.031207.124256
Miller, C. S., Baker, B. J., Thomas, B. C., Singer, S. W., and Banfield, J. F. (2011). EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 12:R44. doi: 10.1186/gb-2011-12-5-r44
Miroshnichenko, M. L., Kolganova, T. V., Spring, S., Chernyh, N., and Bonch-Osmolovskaya, E. A. (2010). Caldithrix palaeochoryensis sp. nov., a thermophilic, anaerobic, chemo-organotrophic bacterium from a geothermally heated sediment, and emended description of the genus Caldithrix. Int. J. Syst. Evol. Microbiol. 60, 2120–2123. doi: 10.1099/ijs.0.016667-0
Miroshnichenko, M. L., Kostrikina, N. A., Chernyh, N. A., Pimenov, N. V., Tourova, T. P., Antipov, A. N., et al. (2003). Caldithrix abyssi gen. nov., sp. nov., a nitrate-reducing, thermophilic, anaerobic bacterium isolated from a Mid-Atlantic Ridge hydrothermal vent, represents a novel bacterial lineage. Int. J. Syst. Evol. Microbiol. 53, 323–329. doi: 10.1099/ijs.0.02390-0
Morris, B. E. L., Henneberger, R., Huber, H., and Moissl-Eichinger, C. (2013). Microbial syntrophy: interaction for the common good. FEMS Microbiol. Rev. 37, 384–406. doi: 10.1111/1574-6976.12019
Nobu, M. K., Dodsworth, J. A., Murugapiran, S. K., Rinke, C., Gies, E. A., Webster, G., et al. (2015a). Phylogeny and physiology of candidate phylum ‘Atribacteria’ (OP9/JS1) inferred from cultivation-independent genomics. ISME J. 5, 273–286. doi: 10.1038/ismej.2015.97
Nobu, M. K., Narihiro, T., Rinke, C., Kamagata, Y., Woyke, T., and Liu, W.-T. (2015b). Microbial dark matter ecogenomics reveals complex synergistic networks in a methanogenic bioreactor. ISME J. 8, 1710–1722. doi: 10.1038/ismej.2014.256
Noguchi, H., Park, J., and Takagi, T. (2006). MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 34, 5623–5630. doi: 10.1093/nar/gkl723
Northcote, T. G., and Hall, K. J. (1983). Limnological contrasts and anomalies in 2 adjacent saline lakes. Hydrobiologia 105, 179–194. doi: 10.1007/BF00025187
Ogato, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., and Kanehisa, M. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34. doi: 10.1093/nar/27.1.29
Overmann, J. (1997). “Mahoney Lake: a case study of the ecological significance of phototrophic purple sulfur bacteria,” in Advances in Microbial Ecology, ed J. G. Jones (New York, NY: Plenum Press), 251–288.
Overmann, J., Beatty, J. T., and Hall, K. J. (1996b). Purple sulfur bacteria control the growth of aerobic heterotrophic bacterioplankton in a meromictic salt lake. Appl. Environ. Microbiol. 62, 3251–3258.
Overmann, J., Beatty, J. T., Hall, K. J., Pfennig, N., and Northcote, T. G. (1991). Characterization of a dense, purple sulfur bacterial layer in a meromictic salt lake. Limnol. Oceanogr. 36, 846–859. doi: 10.4319/lo.1991.36.5.0846
Overmann, J., Beatty, J. T., Krouse, H. R., and Hall, K. J. (1996a). The sulfur cycle in the chemocline of a meromictic salt lake. Limnol. Oceanogr. 41, 147–156. doi: 10.4319/lo.1996.41.1.0147
Pachiadaki, M. G., Lykousis, V., Stefanou, E. G., and Kormas, K. A. (2010). Prokaryotic community structure and diversity in the sediments of an active submarine mud volcano (Kazan mud volcano, East Mediterranean Sea). FEMS Microbiol. Ecol. 72, 429–444. doi: 10.1111/j.1574-6941.2010.00857.x
Pancost, R. D., Crawford, N., Magness, S., Turner, A., Jenkyns, H. C., and Maxwell, J. R. (2004). Further evidence for the development of photic-zone euxinic conditions during Mesozoic oceanic anoxic events. J. Geol. Soc. 161, 353–364. doi: 10.1144/0016764903-059
Pelletier, E., Kreimeyer, A., Bocs, S., Rouy, Z., Gyapay, G., Chouari, R., et al. (2008). “Candidatus Cloacamonas Acidaminovorans”: genome sequence reconstruction provides a first glimpse of a new bacterial division. Appl. Environ. Microbiol. 190, 2572–2579. doi: 10.1128/jb.01248-07
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, R69. doi: 10.1093/bioinformatics/bts174
Perry, K. A., and Pedersen, T. F. (1993). Sulphur speciation and pyrite formation in meromictic ex-fjords. Geochim. Cosmochim. Acta 57, 4405–4418. doi: 10.1016/0016-7037(93)90491-E
Poser, A., Lohmayer, R., Vogt, C., Knoeller, K., Planer-Friedrich, B., Sorokin, D., et al. (2013). Disproportionation of elemental sulfur by haloalkaliphilic bacteria from soda lakes. Extremophiles 17, 1003–1012. doi: 10.1007/s00792-013-0582-0
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Rabus, R., Hansen, T., and Widdel, F. (2000). “The dissimilatory sulfate and sulfur reducing bacteria,” in The Prokaryotes: An Evolving Electronic Resource for the Microbiological Community, 3rd Edn., ed M. Dworkin (New York, NY: Springer Verlag), 1–87.
Ragsdale, A. W., and Pierce, E. (2008). Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 1784, 1873–1898. doi: 10.1016/j.bbapap.2008.08.012
R Development Core Team (2008). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://www.R-project.org
Reinhard, C. T., Planavsky, N. J., Robbins, L. J., Partin, C. A., Gill, B. C., Lalonde, S. V., et al. (2013). Proterozoic ocean redox and biogeochemical stasis. Proc. Natl. Acad. Sci. U.S.A. 110, 5357–5362. doi: 10.1073/pnas.1208622110
Reis, M. A. M., Almeida, J. S., Lemos, P. C., and Carrondo, M. J. T. (1992). Effect of hydrogen sulfide on growth of sulfate reducing bacteria. Biotechnol. Bioeng. 40, 593–600. doi: 10.1002/bit.260400506
Rho, M., Tang, H., and Ye, Y. (2010). Fraggenescan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38:e191. doi: 10.1093/nar/gkq747
Richter, M., Rosselló-Móra, R., Glöckner, F. O., and Peplies, J. (2015). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N. N., Anderson, I. J., Cheng, J. F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter Nature 499, 431–437. doi: 10.1038/nature12352
Santos, A. A., Venceslau, S. S., Grein, F., Leavitt, W. D., Dahl, C., Johnston, D. T., et al. (2015). A protein trisulfide couples dissimilatory sulfate reduction to energy conservation. Science 350, 1541–1545. doi: 10.1126/science.aad3558
Sato, T., Atomi, H., and Imanaka, T. (2007). Archaeal type III RuBisCOs function in a pathway for AMP metabolism. Science 315, 1003–1006. doi: 10.1126/science.1135999
Schauder, R., Preub, A., Jetten, M., and Fuchs, G. (1989). Oxidative and reductive acetyl CoA/carbon monoxide dehydrogenase pathway in Desulfostrain autotrophicum. Arch. Microbiol. 151, 84–89. doi: 10.1007/BF00444674
Schink, B. (1997). Energetics of syntrophic cooperation in methanogenic degradation. Microbiol. Mol. Biol. Rev. 61, 262–280.
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Shah, N., Tang, H., Doak, T. G., and Ye, Y. (2010). Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. Pac. Symp. Biocomp. 165–176. doi: 10.1142/9789814335058_0018
Slobodkin, A. I., Reysenbach, A.-L., Slobodkin, G. B., Kolagnova, T. V., Kostrikina, N. A., and Bonch-Osmolovskaya, E. A. (2013). Dissulfuribacter thermophilus gen. nov., sp. nov., a thermophilic, autotrophic, sulfur-disproportionating, deeply branching deltaproteobacterium from a deep-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 63, 1967–1971. doi: 10.1099/ijs.0.046938-0
Sorek, R., Zhu, Y., Creevey, C. J., Francino, M. P., Bork, P., and Rubin, E. M. (2007). Genome-wide experimental determination of barriers to horizontal gene transfer. Science 318, 1449–1452. doi: 10.1126/science.1147112
Sorokin, D. Y., Kuenen, J. G., and Muyzer, G. (2011). The microbial sulfur cycle at extremely haloalkaline conditions of Soda Lakes. Front. Microbiol. 2:44. doi: 10.3389/fmicb.2011.00044
Sorokin, D. Y., Tourova, T. P., Mußmann, M., and Muyzer, G. (2008). Dethiobacter alkaliphilus gen. nov. sp. nov., and Desulfurivibrio alkaliphilus gen. nov. sp. nov.: two novel representatives of reductive sulfur cycle from soda lakes. Extremophiles 12, 431–439. doi: 10.1007/s00792-008-0148-8
Stolyar, S., Van Dien, S., Hillesland, K. L., Pinel, N., Lie, T. J., Leigh, J. A., et al. (2007). Metabolic modeling of a mutualistic microbial community. Mol. Syst. Biol. 3, 92. doi: 10.1038/msb4100131
Suen, G., Weimer, P. J, Stevenson, D. M., Aylward, F. O., Boyum, J., Deneke, J., et al. (2011). The complete genome sequence of Fibrobacter succinogenes S85 reveals a cellulolytic and metabolic specialist. PLoS ONE 6:e18814. doi: 10.1371/journal.pone.0018814
Swofford, D. L. (2001). Paup: Phylogenetic Analysis Using Parsimony (and Other Methods) 4.0b10 Edn. Sunderland, MA: Sinauer Associate.
Tabita, F. R., Hanson, T. E., Li, H., Statgopan, S., Singh, J., and Chan, S. (2007). Function, structure, and evolution of the RubisCO-like proteins and their RubisCO homologs. Microbiol. Mol. Biol. Rev. 71, 576–599. doi: 10.1128/MMBR.00015-07
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tatusov, R. L., Natale, D. A., Garkavtsev, I. V., Tatusova, T. A., Shankavaram, U. T., Rao, B. S., et al. (2001). The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29, 22–28. doi: 10.1093/nar/29.1.22
Vagle, S., Hume, J., McLuaghlin, F., MacIsaac, E., and Shortreed, K. (2010). A methane bubble curtain in meromictic Sakinaw Lake, British Columbia. Limnol. Oceanogr. 55, 1313–1326. doi: 10.4319/lo.2010.55.3.1313
Vetriani, C., Jannasch, H. W., MacGregor, B. J., Stahl, D. A., and Reysenbach, A.-L. (1999). Population structure and phylogenetic characterization of marine benthic Archaea in deep-sea sediments. Appl. Environ. Microbiol. 65, 4375–4384.
Vignais, P. M., Billoud, B., and Meyer, J. (2001). Classification and phylogeny of hydrogenases. FEMS Microbiol. Rev. 25, 455–501. doi: 10.1111/j.1574-6976.2001.tb00587.x
Voorhies, A. A., Biddanda, B. A., Kendall, T., Jain, S., Marcus, D. N., Nold, S. C., et al. (2012). Cyanobacterial life at low O2: community genomics and function reveal metabolic versatility and extremely low diversity in a Great Lakes sinkhole mat. Geobiology 10, 250–267. doi: 10.1111/j.1472-4669.2012.00322.x
Wang, Z., and Wu, M. (2013). A phylum-level bacterial phylogenetic marker database. Mol. Biol. Evol. 30, 1258–1262. doi: 10.1093/molbev/mst059
Weimer, P. J. (1993). Effects of dilution rate and pH on the ruminal cellulolytic bacterium Fibrobacter succinogenes S85 in cellulose-fed continuous culture. Arch. Microbiol. 160, 288–294. doi: 10.1007/BF00292079
Whitaker, R., and Banfield, J. (2006). Population genomics in natural microbial communities. Trends Ecol. Evol. 21, 508–516. doi: 10.1016/j.tree.2006.07.001
White, J. R., Nagarajan, N., and Pop, M. (2009). Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5:e1000352. doi: 10.1371/journal.pcbi.1000352
Widdel, F. (1987). New types of acetate-oxidizing, sulfate-reducing Desulfobacter species, D. hydrogenophilus sp. nov., D. latus sp. nov., and D. curvatus sp. nov. Arch. Microbiol. 148, 286–291. doi: 10.1007/BF00456706
Wrighton, K. C., Thomas, B. C., Sharon, I., Miller, C. S., Castelle, C. J., VerBerkmoes, N. C., et al. (2012). Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337, 1661–1665. doi: 10.1126/science.1224041
Wu, J. H., Wu, F. Y., Chuang, H. P., Chen, W. Y., Huang, H. J., Chen, S. H., et al. (2013). Community and proteomic analysis of methanogenic consortia degrading terephthalate. Appl. Environ. Microbiol. 79, 105–112. doi: 10.1128/AEM.02327-12
Wu, M., and Eisen, J. A. (2008). A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 9:R151. doi: 10.1186/gb-2008-9-10-r151
Ye, Q., Roh, Y., Carroll, S. L., Blair, B., Zhou, J., Zhang, C. L., et al. (2004). Alkaline anaerobic respiration: isolation and characterization of a novel alkaliphilic and metal-reducing bacterium. Appl. Environ. Microbiol. 70, 5595–5602. doi: 10.1128/AEM.70.9.5595-5602.2004
Yilmaz, P., Yarza, P., Rapp, J. Z., and Glöckner, F. O. (2015). Expanding the world of marine bacterial and archaeal clades. Front. Microbiol. 6:1524. doi: 10.3389/fmicb.2015.01524
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Keywords: meromictic, euxinia, DSR, Clostridia, sulfate reducing bacteria, Deltaproteobacteria, sulfide, sulfate
Citation: Hamilton TL, Bovee RJ, Sattin SR, Mohr W, Gilhooly III WP, Lyons TW, Pearson A and Macalady JL (2016) Carbon and Sulfur Cycling below the Chemocline in a Meromictic Lake and the Identification of a Novel Taxonomic Lineage in the FCB Superphylum, Candidatus Aegiribacteria. Front. Microbiol. 7:598. doi: 10.3389/fmicb.2016.00598
Received: 22 December 2015; Accepted: 11 April 2016;
Published: 27 April 2016.
Edited by:
Andreas Teske, University of North Carolina at Chapel Hill, USAReviewed by:
John R. Spear, Colorado School of Mines, USAAlexander Loy, University of Vienna, Austria
Copyright © 2016 Hamilton, Bovee, Sattin, Mohr, Gilhooly, Lyons, Pearson and Macalady. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Trinity L. Hamilton, tlh42@psu.edu;
Ann Pearson, pearson@eps.harvard.edu;
Jennifer L. Macalady, jlm80@psu.edu
†Present Address: Wiebke Mohr, Department of Biogeochemistry, Max-Planck-Institute for Marine Microbiology, Bremen, Germany