 Heather N. Buelow1
Heather N. Buelow1 Ara S. Winter1
Ara S. Winter1 David J. Van Horn1
David J. Van Horn1 John E. Barrett2
John E. Barrett2 Michael N. Gooseff3
Michael N. Gooseff3 Egbert Schwartz4
Egbert Schwartz4 Cristina D. Takacs-Vesbach1*
Cristina D. Takacs-Vesbach1*- 1Department of Biology, University of New Mexico, Albuquerque, NM, USA
- 2Department of Biological Sciences, Virginia Tech, Blacksburg, VA, USA
- 3Department of Civil, Architectural, and Environmental Engineering, Institute of Arctic and Alpine Research, University of Colorado Boulder, Boulder, CO, USA
- 4Department of Biological Sciences, Northern Arizona University, Flagstaff, AZ, USA
The soils of the McMurdo Dry Valleys, Antarctica are an extreme polar desert, inhabited exclusively by microscopic taxa. This region is on the threshold of anticipated climate change, with glacial melt, permafrost thaw, and the melting of massive buried ice increasing liquid water availability and mobilizing soil nutrients. Experimental water and organic matter (OM) amendments were applied to investigate how these climate change effects may impact the soil communities. To identify active taxa and their functions, total community RNA transcripts were sequenced and annotated, and amended soils were compared with unamended control soils using differential abundance and expression analyses. Overall, taxonomic diversity declined with amendments of water and OM. The domain Bacteria increased with both amendments while Eukaryota declined from 38% of all taxa in control soils to 8 and 11% in water and OM amended soils, respectively. Among bacterial phyla, Actinobacteria (59%) dominated water-amended soils and Firmicutes (45%) dominated OM amended soils. Three bacterial phyla (Actinobacteria, Proteobacteria, and Firmicutes) were primarily responsible for the observed positive functional responses, while eukaryotic taxa experienced the majority (27 of 34) of significant transcript losses. These results indicated that as climate changes in this region, a replacement of endemic taxa adapted to dry, oligotrophic conditions by generalist, copiotrophic taxa is likely.
Introduction
Microbial species in extreme environments are highly adapted to persist in their niches (Takacs-Vesbach et al., 2013). The specialization of taxa, combined with the relatively low taxonomic richness typically present in extreme environments (Neilson et al., 2012), make these microbial communities susceptible to environmental change (Tilman, 1996; Van Horn et al., 2014). The McMurdo Dry Valleys (MDV) of Antarctica are an extreme polar desert environment, characterized by freezing temperatures (Doran et al., 2002), low soil moisture, low soil organic matter (OM; Burkins et al., 2001), and the absence of higher plants and animals. While the climate of this region has been relatively stable for millennia (Denton et al., 1993), the MDV are currently on the threshold of climate induced change (Fountain et al., 2014), and impacts of these changes are already affecting dry-adapted soil species (Barrett et al., 2008a,b; Knox et al., 2016). In the past decade, increased solar radiation and decreased albedo has accelerated glacial melt, permafrost thaw, and the melting of massive buried ice, thereby increasing soil moisture and nutrient mobilization (Fountain et al., 2014). The study presented here uses experimental water and OM amendments to examine how soil communities in this extreme ecosystem will respond to the expected effects of climate change.
Soils are the largest habitat within the MDV and taxonomic studies have characterized the associated microbial communities revealing extensive bacterial richness (Lee et al., 2012; Van Horn et al., 2013, 2014) but limited eukaryotic diversity, which include nematodes, fungi, rotifers, and tardigrades (Freckman and Virginia, 1997, 1998; Adams et al., 2006). Detecting and distinguishing active taxa in these soils presents challenges. Soil respiration rates, a measure of biological activity in soils, have been shown to be sensitive to temperature (Parsons et al., 2004; Ball et al., 2009; but see Shanhun et al., 2012). Taxonomic shifts in response to water, resource amendments, and changing conditions have been identified using 16S rRNA gene sequencing (Tiao et al., 2012; Schwartz et al., 2014; Van Horn et al., 2014), however, these DNA-based studies cannot distinguish active taxa from dormant or non-viable cells. Non-viable cells are of particular concern in MDV soils, where freezing temperatures can delay degradation. Recently, stable isotope probing with H218O water provided a first look at the taxonomy of definitively active bacteria in MDV soils and revealed a somewhat different community profile than prior 16S rRNA gene studies (Schwartz et al., 2014). The present study uses RNA sequencing as another means of distinguishing the active members of the soil community.
Beyond taxonomy, the next step in understanding this soil ecosystem is to examine the functions of active taxa. Components of MDV soil microbial activities have been investigated using various measures, including CO2 flux and other gas emissions (Burkins et al., 2001; Gregorich et al., 2006; Ball et al., 2009; Van Horn et al., 2014), extracellular enzyme assays (Zeglin et al., 2009; Van Horn et al., 2014), mineralization rate calculations (Barrett et al., 2005; Hopkins et al., 2006b), natural isotopic signature analysis of soil nutrients (Bao et al., 2000; Burkins et al., 2000; Barrett et al., 2006), and stable-isotope tracer techniques (Barrett et al., 2008b). Together these investigations have revealed an active and responsive community, but the functional profile of MDV soils has not been characterized. The goal of this study was to use total community RNA transcripts to characterize active soil taxa and their functions, and further, to determine their response to experimental soil amendments designed to simulate the expected effects of climate change in this region.
Materials and Methods
Site Description
Field experiments took place in dry soils of the Taylor Valley, near the southern shore of Lake Fryxell (77°37′S, 163°12–13′E; Figure 1). Soil surface temperatures average -18.4°C annually (range -52.0 to 22.7°C), and the Fryxell lake basin experiences an average of 25.5 days above freezing per year (Doran et al., 2002). Annual precipitation in the basin is 20–37 mm (Fountain et al., 2010), but sublimation rates are high relative to precipitation inputs (Clow et al., 1988) and the proportion of snowfall that contributes to soil water content remains unknown (Eveland et al., 2013). Liquid water inputs are limited in time and space, occurring during the brief austral summer and concentrated in stream channels and wetted margins of glaciers, streams, and lakes. OM inputs are also limited, with soils averaging 0.03% organic carbon (Burkins et al., 2001). Although relic OM from Glacial Lake Washburn is present in these low elevation soils, most contemporary organic inputs are produced during the austral summer when algal mats proliferate in intermittently wet soils and aquatic habitats and are wind dispersed (Hopkins et al., 2006a; Moorhead, 2007) and in situ production by soil, endolithic, and hypolithic taxa occurs (Friedmann et al., 1993; Moorhead et al., 1999; Cary et al., 2010; Geyer et al., 2013).
FIGURE 1. Landsat image of the sampling location and Lake Fryxell within the McMurdo Dry Valleys.
Sampling and Treatments
The sampling for this experiment was conducted as described previously by Van Horn et al. (2014) for the low salinity treatments of that study. On November 23, 2010, soils were collected to a depth of ∼12 cm using sterile scoops. Soils from this plot have electrical conductivity (a proxy for salinity) of 105 ± 4 μS, a pH of 9.0 ± 0.1, and contain 0.006 ± 0.002 percent nitrogen, and 0.027 ± 0.006 percent organic carbon (Van Horn et al., 2014). Soils were homogenized and placed into sterile cylindrical mesocosms (10 cm diameter × 17 cm deep), which were positioned in the excavated holes such that soil levels within the mesocosms matched the surrounding undisturbed soil level. Although the sampling, homogenization, and mesocosm filling represent disturbance events to the natural soil community, MDV regulations require that all treatments be contained. Therefore, control samples underwent the same sampling and containment processes despite no treatment addition to better assess what community responses resulting from treatments rather than disturbance.
One of three treatments, control, water addition, and OM amendment, was randomly assigned to each of the six mesocosms used for this study, yielding two samples per treatment. Treatments were applied following the methods of Van Horn et al. (2014): prior to treatment additions, soil moisture was measured using a HydroSense soil moisture probe calibrated for the soil in the experimental plots (Campbell Scientific). Sterile deionized water was added to water treatment mesocosms to achieve 10% soil water content by weight. OM was also added to achieve 10% soil water content by weight, which increased organic carbon by 0.005%, roughly one-tenth of the total soil organic carbon. The OM addition was a leachate prepared from native cyanobacterial mat collected from Lake Fryxell. The mat was steeped in sterile DI water and the resulting leachate (2,800 mg/liter dissolved organic carbon) was filter sterilized using 0.2 μm filters. For both treatment additions sterile syringes were used to penetrate the soil in five places within the mesocosm to a depth of 10 cm, distributing the water and/or OM throughout the depth. Soils were treated five times throughout the 30-day incubation period to maintain ∼10% soil moisture by weight. At the conclusion of the incubation period, soils were collected into sterile conical tubes and preserved with an equal volume of sucrose lysis buffer (Giovannoni et al., 1990) and immediately stored at -20°C until processed further.
Nucleic Acid Extraction and Sequencing
A minimum of 15 g soil was extracted from each sample (range = 15–30 g) to yield a minimum of 8 μg RNA per sample for sequencing. Total nucleic acid was extracted from the samples following the cetyltrimethylammonium bromide method of Mitchell and Takacs-Vesbach (2008). To retain only RNA, DNase I and accompanying buffer (Invitrogen) was added to the samples following the manufacturer’s protocol. Sequencing libraries were prepared using Illumina’s mRNA-seq kit following the manufacturer’s protocols, and sequencing was conducted on an Illumina HiSeq 2 × 100.
Data Analysis
Fastq sequence files were uploaded to MG-RAST (Meyer et al., 2008) using the pipeline options: unassembled, allowing for replicates, human sequences screened out, and filtered with dynamic trimming settings (minimum phred score = 5). These fastq sequence files are publicly available on MG-RAST under project 123301 with MG-RAST ID numbers 4614919.3, 4617349.3, 4617350.3, 4617351.3, 4620847.3, and 4620848.3.
Sequences passing MG-RAST pipeline quality controls were then annotated using the M5NR (Wilke et al., 2012) and SEED Subsystems (Overbeek et al., 2005) databases for taxonomic and functional analyses, respectively. Notably, the M5NR database annotates protein-coding reads, while previous taxonomic studies of these soil communities used 16S rRNA gene sequences and rRNA annotation databases (Schwartz et al., 2014; Van Horn et al., 2014). All annotations were made following the MG-RAST default settings of e-value ≤1e - 5, identity ≥60%, and alignment length ≥15 base pairs.
After annotation, biological replicates were pooled by treatment, and analyzed in R (R Development Core Team, 2011), using the phyloseq (McMurdie and Holmes, 2013) and DESeq2 (Love et al., 2014) packages. Differences in the abundance of taxa and the variance in expression of transcripts were characterized using the DESeq2 parameters fitType = “local” and an adjusted p-value threshold of 0.05 to calculate log2 fold changes between treatments. Taxonomy was assigned to transcripts of significantly over-expressed functions. For functions that were under-expressed in water or OM amended samples, taxonomy was assigned to the transcripts of those functions in the control samples because in some cases the transcripts were not present in amendment samples. Similarity percentage analysis (SIMPER), performed in Community Analysis Package 5, was used to determine the OTUs that contributed most to the observed dissimilarity between treatments.
Secondary metabolite analysis of the control soils was performed using HMMER v3.1b2 (Finn et al., 2011) with a cutoff bit score of 20. Python scripts and workflow provided by the FOAM database (Prestat et al., 2014) were used to count the best hits from the HMMER run. The following Pfam database (Finn et al., 2013) HMMER profiles were used: ABM (PF03992), Actino_peptide (PF14408), Acyl_transf_1 (PF00698), Acyltransferase (PF01553), Antimicrobial18 (PF08130), Bacteroid_pep (PF14406), Carbam_trans_C (PF16861), Condensation (PF00668), FAE1_CUT1_RppA (PF08392), Herpeto_peptide (PF14409), L_biotic_typeA (PF04604), LANC_like (PF05147), Lantibiotic_a (PF14867), MbtH (PF03621), NRPS (PF08415), Penicil_amidase (PF01804), Chal_sti_synt_C (PF02797), Chal_sti_synt_N (PF00195), Chalcone_3 (PF16036), PP-binding (PF00550), SchA_CurD (PF04486), Strep_pep (PF14404), TfuA (PF07812), Thioesterase (PF00975). The ketosynthase-alpha (KSa) domain from the polyketide synthase type II gene cluster HMMER profiles was from the RDP funcgene repository (Wang Q. et al., 2015). Resistance genes were also identified using HMMER and the Resfam database (Gibson et al., 2014). Full-length HMMER hits from the secondary metabolites were retrieved using esl-fetch. Taxonomy was assigned to the phosphopantetheine- (PP) binding domain of non-ribosomal polyketide synthase (NRPS) gene cluster and the KSa domain hits using GhostKOALA (Kanehisa et al., 2015).
Results and Discussion
Nucleic acid extraction from soil samples resulted in an average of 17.9 ± 8.6 μg RNA per sample (range = 8.5–30.6 μg). Illumina sequencing yielded an average of 11,581,624 ± 4,804,223 reads per sample (range = 6,597,242–19,854,970). After MG-RAST quality control filters, an average of 83 ± 15% of reads were retained for further analysis (range = 59–97%). Of these sequences, an average of 25 ± 12% (range = 12–38%) were rRNA for each sample. Sequences of mRNA able to be annotated as protein-coding averaged 67 ± 14% (range = 46–83%) of reads per sample.
Taxonomic Shifts
Domain-Level Changes
Based on taxonomic assignment of total RNA sequences, the domain Bacteria was dominant in all treatments (53, 82, and 82% of control, water, and OM samples, respectively), followed by Eukaryota, Virus, and Archaea (Table 1). The relative abundance of bacterial sequences increased with both water and OM treatments relative to controls, while eukaryotic abundances declined. Although present, archaeal sequences were rare in all treatments, ranging from 0.03 to 0.07% of the total sequences. Viral sequences were also detected in all treatments, ranging from <1 to 3% of total sequences.
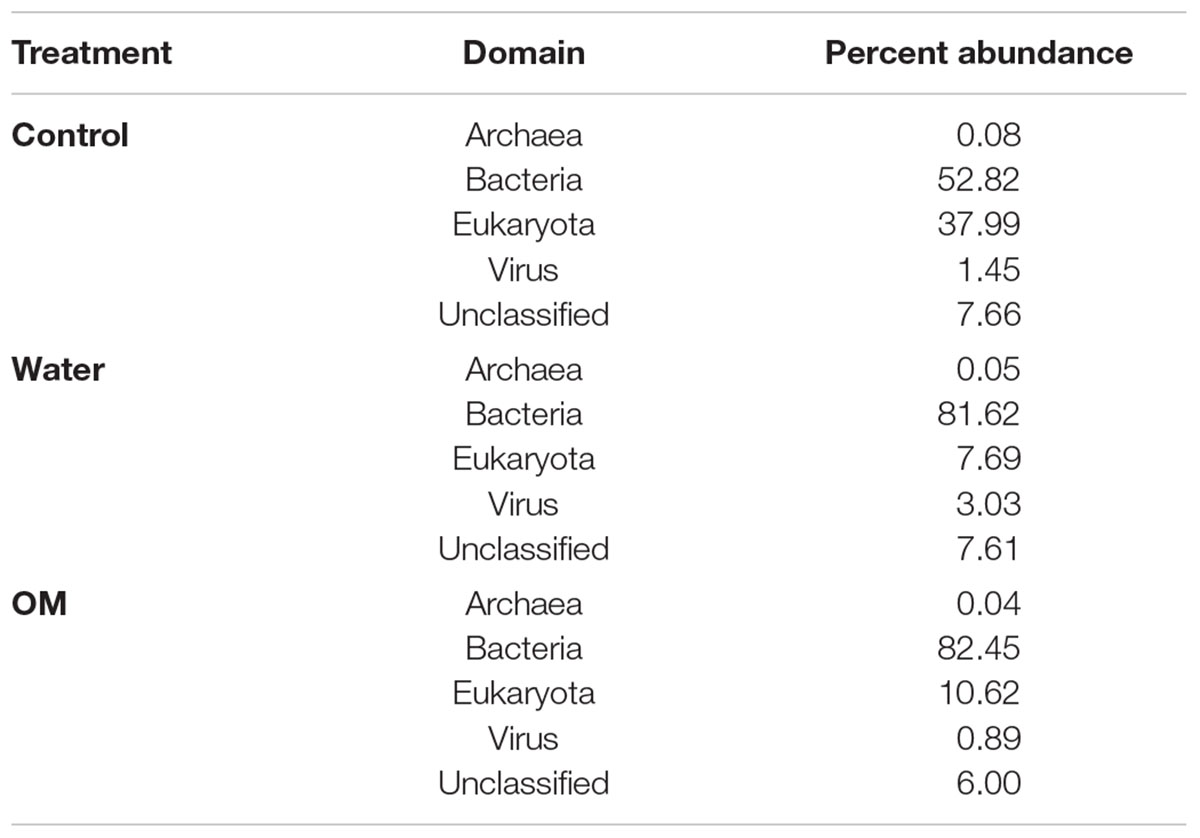
TABLE 1. Percent abundances of sequences from taxonomic domains within treatments.
Bacterial Phyla Responses
Bacterial sequences in all treatments were dominated by three phyla: Actinobacteria, Firmicutes, and Proteobacteria (Figure 2A). The phyla Bacteroidetes, Cyanobacteria, and Tenericutes were also present at >1% abundance, although Cyanobacteria and Tenericutes fell below that threshold in water treatments (Figure 2A). The mean dissimilarity of the bacterial communities across all treatments was 51% (SIMPER analysis, Table 2). Pairwise comparisons between treatments had a mean dissimilarity range of 72–78%. Five OTUs accounted for most of the community compositional differences between treatments, with all other OTUs contributing <5%. The top three OTUs that explained the most variance between each treatment pair were identified as Actinobacteria and Firmicutes in all comparisons. These OTUs were all more abundant in the water and OM treatments than in the control.
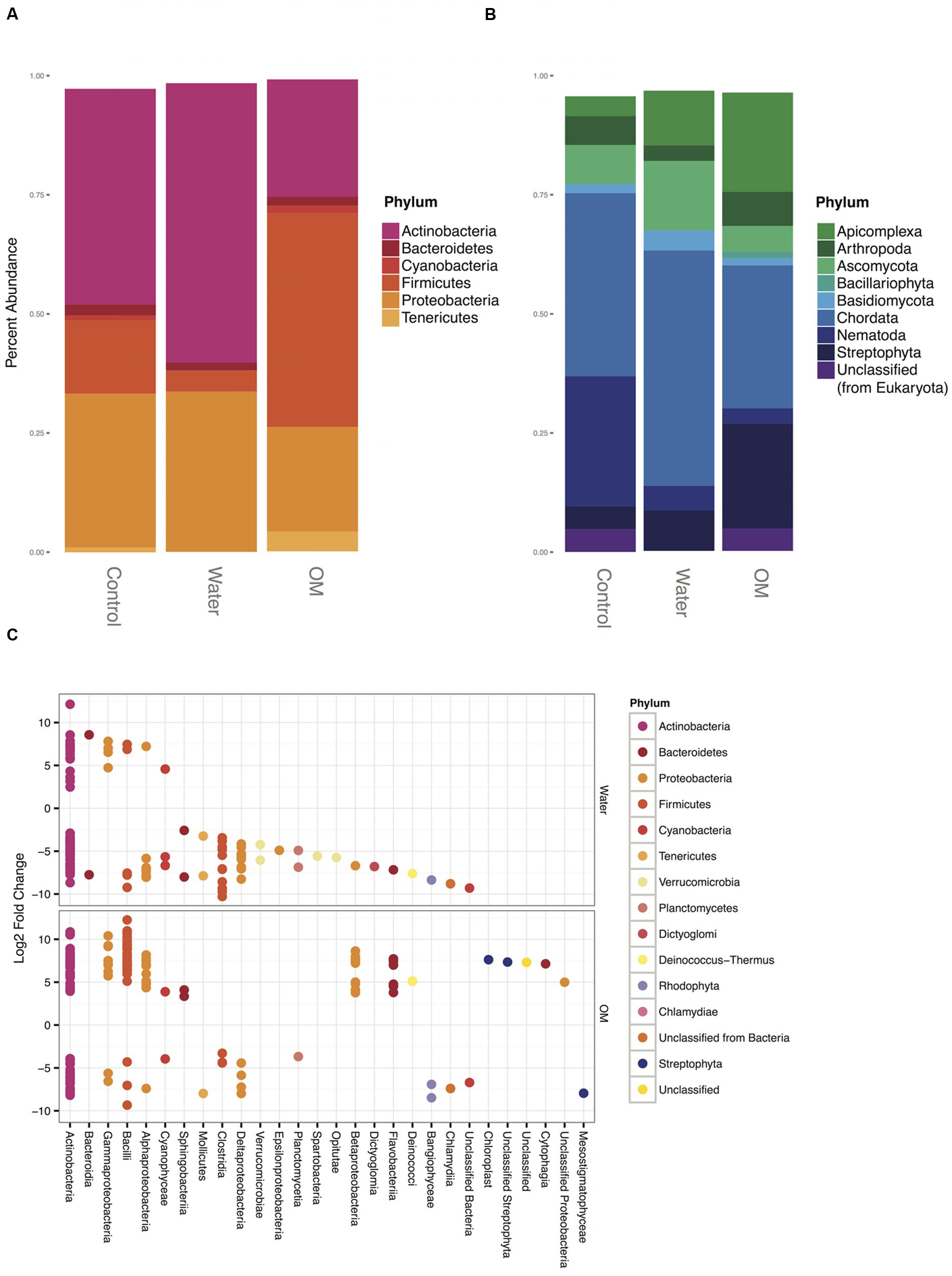
FIGURE 2. Taxonomy of bacterial phyla (A) and eukaryotic phyla (B) representing >1% of sequences in at least two treatments, expressed as the percentage of the number of total sequences. Differential abundance responses from the water and OM amendments versus the control (C), expressed as the significant (p-adjusted value ≥0.05) log2 fold change of sequences in each treatment relative to the control, colored by phylum and grouped by class.
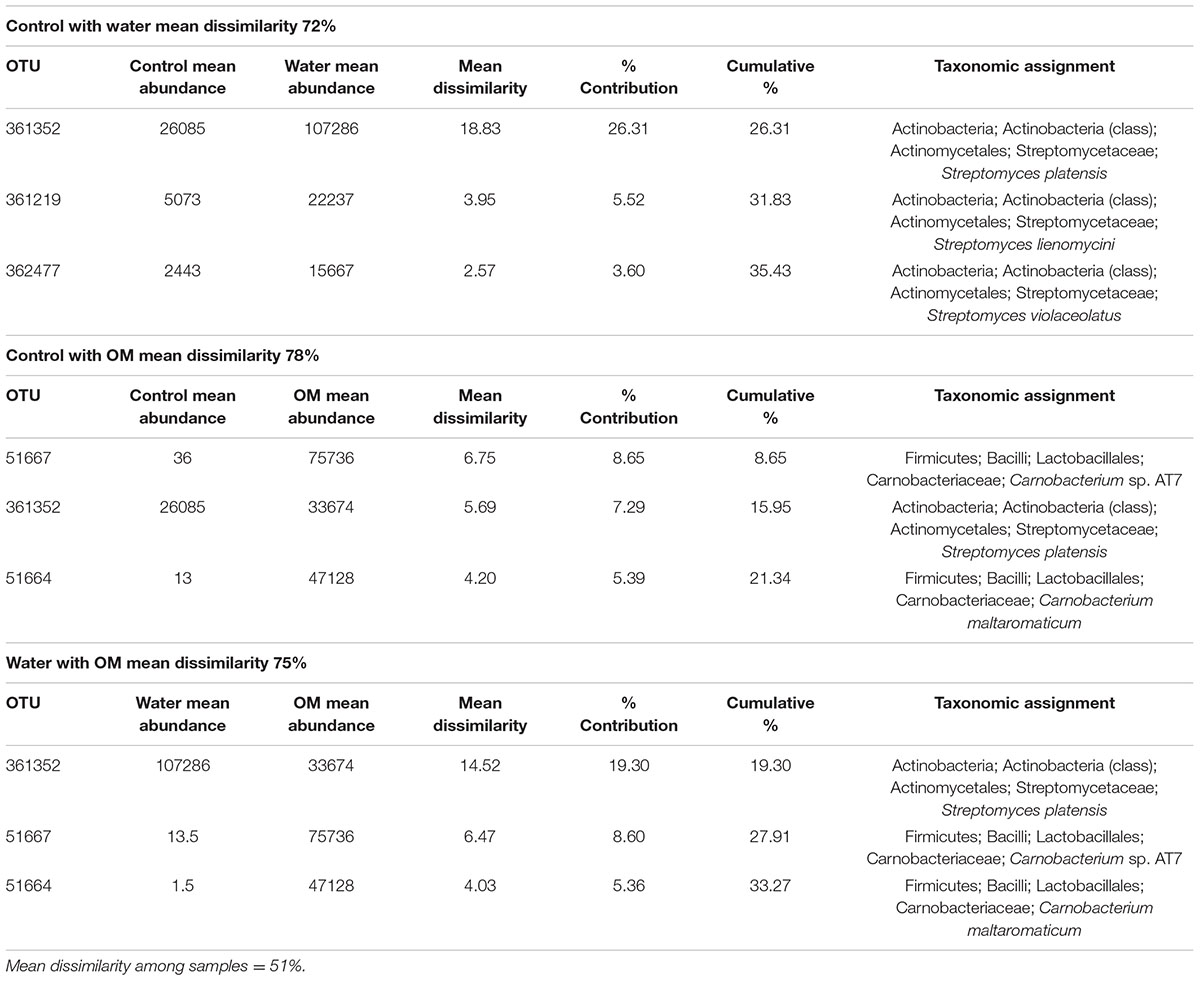
TABLE 2. Similarity percentage analysis (SIMPER) between treatments.
In general, water and OM amendments resulted in decreased abundances of bacterial phyla (Figure 2C). Members of only five phyla (Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria, and Cyanobacteria) had significant differential increases in abundance for water and OM treatments versus controls (Figure 2C). These phyla also had significantly increased functional transcripts (Figure 4). Because these phyla represent the strongest paired taxonomic and functional responses in this study, discussion of each phylum follows.
Actinobacteria
Actinobacteria are globally dominant in arid soils (Costello et al., 2009; Fierer et al., 2009; Pointing et al., 2009; Neilson et al., 2012). In MDV soils, DNA-based studies report declines in Actinobacteria in response to water and OM amendments of Schwartz et al. (2014) and Van Horn et al. (2014). In the present study, Actinobacteria produced the highest relative abundance of bacterial transcripts for the control (45%) and water addition (59%) samples, but declined (25%) in OM addition samples (Figure 2A). However, differential abundance analysis revealed varied responses of individual populations, with significant positive and negative log2 fold changes within the Actinobacteria class for both treatments (Figure 2C). Notably, the Micrococcaceae family had the strongest positive response of any Actinobacteria group in the OM samples and the third highest response (behind Streptomycetaceae and Bifidobacteriaceae) in the water samples of the present study, though the family had significant positive and negative differential abundance responses in both treatments. These strong positive responses are consistent with previous stable isotope work in these soils, that determined members of the Micrococcaceae family had the highest calculated growth rates of any bacterial group in water addition samples (Schwartz et al., 2014), suggesting that members of this family are poised to take advantage of transient water inputs in arid soils.
Bacteroidetes
In MDV soils, Bacteroidetes have been reported as one of the three most dominant phyla (Cary et al., 2010; Lee et al., 2012), using techniques that do not account for active versus inactive cells. In the present study, the relative abundance of Bacteroidetes was 2.3% of the control sample bacterial phyla, and declined in both water (1.6%) and OM (1.9%) samples. These results are consistent with a stable isotope study distinguishing active community members of these soils and amendments, which reported Bacteroidetes as a small portion of the bacterial community across all treatments (Schwartz et al., 2014).
Increased abundance of Bacteroidetes with OM enrichment has been observed in a variety of soils (Fierer et al., 2007, 2011), however, this phylum has shown inconsistent responses to OM amendments in MDV soils: relative abundance increased with algal mat leachate additions to soils (Van Horn et al., 2014), but decreased with mummified seal carcass (Tiao et al., 2012). In the present study, greater taxonomic resolution and differential abundance analysis provided a more detailed account of the taxonomic changes within this phylum. Members of three Bacteroidetes classes (Cytophagia, Flavobacteriia, and Sphingobacteriia) significantly increased in the OM treatments despite the overall decline of the relative abundance of the phylum.
Cyanobacteria
The phylum Cyanobacteria decreased in relative abundance in water treatments and increased in OM treatments (Figure 2A). However, the differential abundance of sequences within the phylum reveals a more complex pattern (Figure 2C). The orders Nostocales and Chroococcales made up the majority (>91%) of cyanobacterial sequences for all treatments and were the only orders contributing to cyanobacterial differential abundance responses across treatments. In both treatments Nostocales increased differential abundance (4.6 and 3.9 log2 fold change in the water and OM treatments, respectively) while Chroococcales decreased (-5.7 and -6.7 log2 fold change in water and -4.0 log2 fold change in OM).
Cyanobacteria are known inhabitants of soils and crusts in both hot and cold deserts, and are well adapted to the stresses of hydration and desiccation cycles, with response to wetting occurring within minutes (Garcia-Pichel and Pringault, 2001; Rajeev et al., 2013). Both Nostocales and Chroococcales are known inhabitants of harsh, cold environments, including MDV (Wood et al., 2008), barren high arctic (Elster et al., 1999; Kastovska et al., 2005), and high altitude Himalayan (Řeháková et al., 2011) soils. While both orders are known to thrive in subnival environments (Řeháková et al., 2011), Nostocales has been detected in greater abundance nearer the soil surface than Chroococcales (Elster et al., 1999). Resistance to disturbance or desiccation tolerance, factors that are correlated with soil depth, may contribute to the differing responses observed in the present study.
Firmicutes
Firmicutes was the third most abundant phylum in the control (15%) and water (4%) samples, but had the greatest increase in abundance in OM samples, contributing 45% of bacterial sequences in OM samples (Figure 2A). This striking increase by Firmicutes has been seen in other studies involving nutrient addition treatments to MDV soils, indicating that this phylum is adept at utilizing organic resources when available (Tiao et al., 2012; Schwartz et al., 2014; Van Horn et al., 2014). A high abundance of Firmicutes has also been reported in a metatranscriptomic study of Alaskan permafrost soils, highlighting the phylum’s ability to function in a frozen environment (Hultman et al., 2015). Firmicutes does not always increase in response to carbon-only amendments, and nitrogen amendments may also be necessary to trigger increases (Ramirez et al., 2010; Cederlund et al., 2014). Although only dissolved organic carbon was measured for the OM leachate added in the present study, the use of native algal mat as a complex nutrient source is presumed to have simultaneously increased nitrogen content in the soils.
Proteobacteria
Proteobacteria contributed 32% of control, 34% of water, and 22% of OM bacterial 16S rRNA gene sequences (Figure 2A). Previous DNA-based OM amendments studies in MDV soils have reported substantial relative abundance increases of only Gammaproteobacteria (Tiao et al., 2012), or of both Beta- and Gammaproteobacteria (Van Horn et al., 2014). The present study found that three classes (Alpha-, Beta-, and Gammaproteobacteria) had significant increases in abundance (Figure 2C), consistent with the findings of the only other study to distinguish active bacterial populations in OM amended MDV soils (Schwartz et al., 2014). Selectively sequencing active community members has increased detection of Proteobacteria in Alaskan soils as well, with a metatranscriptomic approach detecting greater abundances of Proteobacteria than either metagenomic or 16S rRNA gene based sequencing studies (Hultman et al., 2015).
Acidobacteria
Although Acidobacteria are a typical member of soil communities and have been reported in 16S rRNA gene studies of MDV soils (Lee et al., 2012; Van Horn et al., 2014), they represented only 0.04, 0.02, and 0.04% of bacterial sequences for control, water, and OM samples of this study, respectively, and were not significantly different in abundance for any treatment. This phylum was also not a dominant member of the active fraction of a stable isotope probing study of these soils, and it was detected in greater abundance in the inactive fraction of the same study (Schwartz et al., 2014). The Acidobacteria detected in 16S rRNA gene studies of these soils may be preserved non-viable cells, or simply slow or inefficient responders to increasing soil moisture and OM.
Eukaryotic Taxa
The domain Eukaryota contained 38% of sequences in the controls, and declined in relative abundance to 8% in water and 11% in OM amendments (Table 1). Discussion of key taxonomic groups and the challenges of eukaryotic annotation follow.
Chordata
Among eukaryotic phyla, a high proportion (38, 49, and 30% of control, water, and OM, respectively) of sequences were annotated as Chordata (Figure 2B), which was unexpected given the MDV are a microbially dominated ecosystem. The majority of reads annotated as Chordata fall into either the family Muridae (20, 31, and 74% of Chordata sequences in control, water, and OM treatments, respectively) or the family Hominidae (35, 36, 7% of control, water, and OM Chordata, respectively). While the MG-RAST pipeline uses Bowtie (Langmead et al., 2009) to filter out human sequences, reads with >60% identity and an alignment length of <35 bases were not culled from the dataset. As other studies have reported, contamination is typical in next-generation sequencing efforts to date and human reads are discovered in non-primate sequences across many databases (Malmström et al., 2005; Cibulskis et al., 2011; Longo et al., 2011; Kumar et al., 2013; Laurence et al., 2014). Additionally, there is bias toward intensively studied sequences, such as Muridae, in annotation databases (Wang et al., 2010), and unexpected mouse sequences have been detected in human sequencing projects (Robinson et al., 2010; Delviks-Frankenberry et al., 2012) and common DNA extraction and amplification reagents (Erlwein et al., 2011; Tuke et al., 2011). While Chordata-annotated sequences are included in all analyses for transparency, we do not speculate further on their presence or role in situ.
Arthropoda
The annotations within the phylum Arthropoda may also be skewed by model organism bias. The order Diptera made up the greatest percentage (39%) of Arthropoda sequences in the control samples. Only two endemic Diptera species have been reported on the Antarctic continent (Convey and Block, 1996), both from the midge family Chironomidae. However, the majority (51%) of control sample Diptera annotations were attributed to the fruit fly family Drosophilidae while Chironomidae annotations represented only 0.1%. Similarly, the common aquatic family Daphniidae comprised the majority of Arthropoda sequences in water (69%) and OM (75%) addition samples.
Nematoda
The eukaryotic phylum Nematoda was the next most abundant in control samples (27%), but declined in both water (5%) and OM (3%) addition samples (Figure 2B). Three nematode species are commonly reported in MDV soils and sediments: Scottnema lindsayae, Eudorylaimus antarcticus, and Plectus antarcticus, though none of these species are currently present in the M5NR database used to annotate this dataset. Rather, all differentially expressed functions linked to nematodes were attributed to the model organism genus Caenorhabditis. Dry MDV soils distant from streams or snowpack are dominated by a single species, S. lindsayae (Freckman and Virginia, 1998; Treonis et al., 1999; Gooseff et al., 2003). Abundance of S. lindsayae generally decreases with greater soil moisture, though this decline is not always statistically significant (Freckman and Virginia, 1997, 1998; Treonis et al., 1999; Gooseff et al., 2003). Additionally, there is no statistically significant relationship between nematode abundance and spatial distribution of soil OM in MDV soils (Freckman and Virginia, 1997; Gooseff et al., 2003). As the soils of this study were not adjacent to streams, the dry-adapted S. lindsayae was expected to be the dominant nematode species as predicted by a logistic regression habitat suitability model (Poage et al., 2008). Despite annotation resolution challenges, the relative abundance of Nematoda declined with both treatments that increased soil moisture in the present study, as would be expected for S. lindsayae.
Fungal phyla
In all samples, the two dominant fungal phyla were Ascomycota (8, 15, and 5% of eukaryotic sequences in control, water, and OM samples, respectively) and Basidiomycota (2, 4, and 2% in control, water, and OM, respectively) consistent with prior DNA-based survey of MDV soils (Fell et al., 2006). Ascomycetes have been reported as the dominant fungal group in a variety of soils across the continent (Lawley et al., 2004; Arenz and Blanchette, 2011), although in MDV soils with <5% soil moisture Basidiomycota were more widely-distributed (Fell et al., 2006). In the present study, relative abundances of both fungal phyla increased in water addition samples, consistent with prior positive correlations between fungal abundance and soil moisture in the MDV (Connell et al., 2006; Fell et al., 2006; Arenz and Blanchette, 2011). However, the relative abundances of both phyla decreased in OM addition samples, which is inconsistent with prior culture-based studies of Antarctic soils. In general, un-amended soil carbon concentrations are positively correlated with total fungal abundance (Arenz and Blanchette, 2011) and filamentous fungal abundance (Connell et al., 2006), and Ascomycota has been found to be an abundant decomposer in Antarctic soils where debris is present (Arenz and Blanchette, 2011). Fungal taxa are known to exhibit varied responses to carbon amendments in temperate soils based on specific substrate composition, and soil bacteria are known to outcompete fungi in some cases (Hanson et al., 2008; Chen et al., 2013). Varied results of fungal richness and diversity may occur due to sample size and number (Ranjard et al., 2003), as fungi are known to be spatially clustered (Foster, 1988; Horton and Bruns, 2001). Future amendment studies in MDV soils will require more robust sampling and additional analyses to better interpret fungal responses.
Streptophyta
The phylum Streptophyta contributed 5% of the control eukaryotic sequences (Figure 2B). The phylum was present in significantly greater relative abundance in the OM (22%) samples than control samples (Figure 2C). However, these sequences were exclusively chloroplast-derived annotations and interpretation of those results should be considered cautiously. Rather than indicative of plant species, the Streptophyta annotations may be considered a proxy for active photosynthetic organisms in these samples.
Investigation of Transcript Functions
Transcript Annotation
The SEED Subsystems functional annotations provide four levels of resolution, given as levels 1–4 in MG-RAST. Level 1, the highest level of functional categorization, referred to here as subsystems, is primarily considered, with further hierarchy described when relevant. A total of 28 subsystems were identified in the dataset. In control samples, representing the baseline functional profile of the soils in this study, the greatest relative abundance of sequences were in the subsystems of RNA metabolism, followed by protein metabolism, carbohydrates, and clustering-based (Figure 3A). RNA and protein metabolism subsystems contain many transcripts related to transcription, translation, and protein management and these subsystems have high representation in metatranscriptome studies (de Menezes et al., 2012), especially relative to metagenome studies (Urich et al., 2008). Annotations within the clustering-based subsystem (CBSS) are putative, based on the proximity of unknown sequences to those in a cluster of genes of known function (Gerdes et al., 2011). CBSS annotations were redundant, categorized into both CBSS and the putative function’s subsystem on the MG-RAST server. Thus, CBSS annotations are only shown in Figure 3, and redundant CBSS annotations are not presented in Figures 4 and 5.
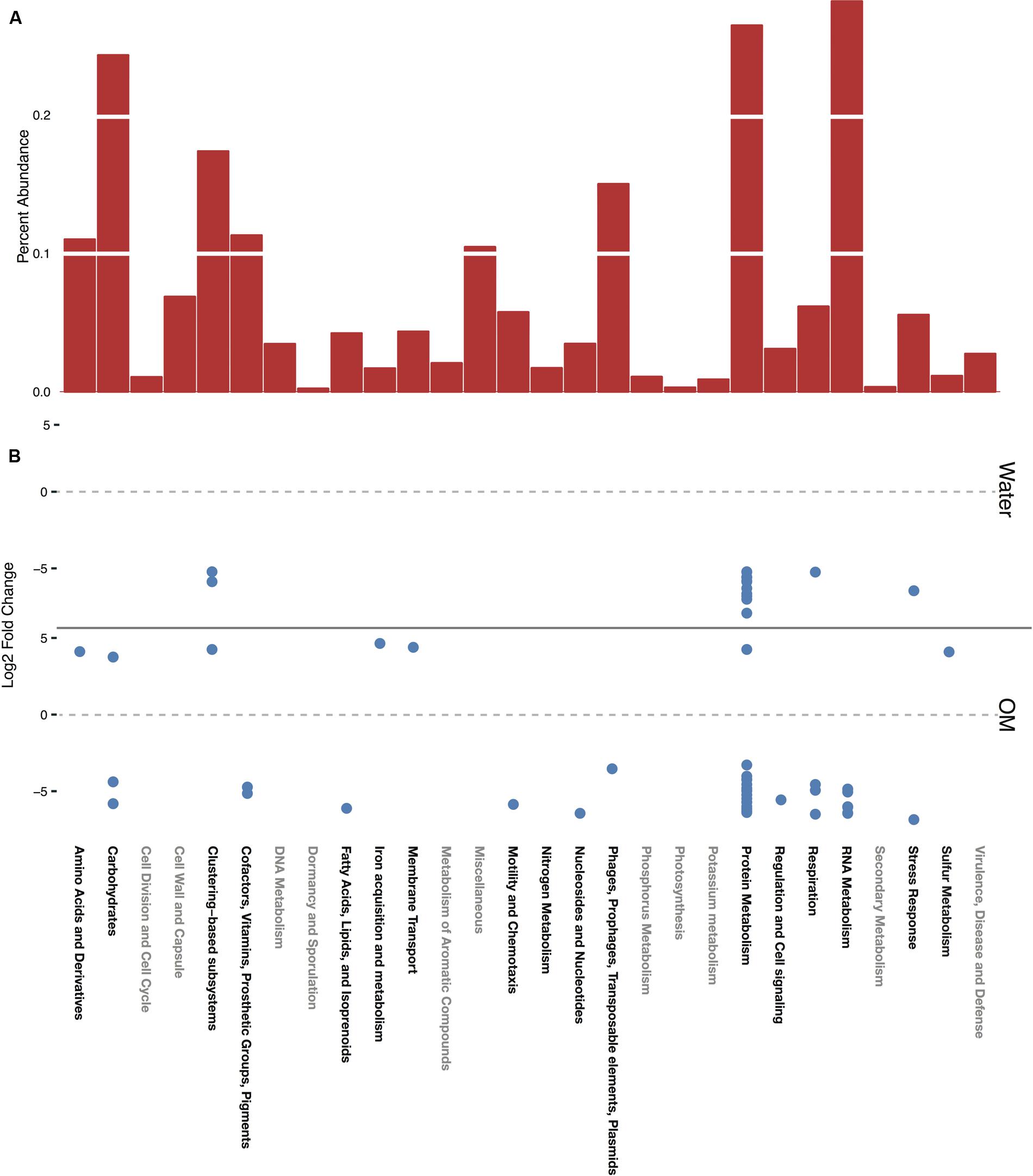
FIGURE 3. Functional profile of the control, expressed as percent abundance of each subsystem (A). All significant (p-adjusted value ≥0.05) differential expression for water and OM treatments, expressed as the log2 fold change of sequences in each treatment relative to the control (B).
Functional Profile of the Control Soils
The functional annotation of mRNA transcripts by the SEED Subsystems database revealed that the active portion of the MDV control soil microbial community is dominated by a chemoorganoheterotrophic bacterial community, but includes photoautotrophic cyanobacteria and algae, and various heterotrophic eukaryotes. Of particular note are the most abundant transcripts in carbohydrate and nitrogen metabolism subsystems, which reflect the community’s adaptation to the low OM content of these soils despite relatively high nitrogen and phosphorous content (Barrett et al., 2006). Complex carbon sources, such as plant cellulose material, are lacking in these soils, and simpler carbon sources are utilized (Barrett et al., 2005, 2006). Transcripts of the serine-glyoxylate cycle, a key component of methylotrophy (Anthony, 2011), comprised 11% of the carbohydrate subsystem, and maltose and maltodextrin utilization transcripts comprised another 11%. Photorespiration transcripts, a component of CO2 fixation, were less abundant (4%). Although organic nitrogen concentrations are low in these soils, inorganic nitrate concentrations are exceptionally high due to years of atmospheric deposition (Wada et al., 1981; Burkins et al., 2000; Barrett et al., 2002, 2007; Witherow et al., 2006). These nitrates appear to be valuable to the community as nitrate and nitrite ammonification transcripts were 31% of the nitrogen metabolism subsystem. Ammonia assimilation transcripts were also abundant (48% of nitrogen metabolism), but denitrification (6%) and nitrogen fixation (<1%) transcripts were rare.
Because Actinobacteria were a large portion of the control soil bacteria, we specifically searched for secondary metabolite genes (Amoutzias et al., 2008; Wang H. et al., 2014, 2015). The analysis focused on the PKSα domain of the PKSII gene cluster and the PCP domain of the NRPS gene cluster as these domains are highly conserved which make them good targets for analysis (Bachmann and Ravel, 2009). The majority of the transcripts detected were for proteins responsible for the assembly of polyketides (PKSII, 32% identified as ketoacyl synthase (KSα) genes) or non-ribosomal peptide synthetases (NRPS, 29% associated with the peptidyl carrier protein domain or PCP) which are known to produce a wide variety of secondary products. The taxonomic assignments of the KSα domain genes were largely from Actinobacteria (44%), but also Proteobacteria (27%) and Firmicutes (14%). The NRPS transcripts were similarly distributed among phyla Actinobacteria (50%), Proteobacteria (19%), and Firmicutes (16%). Resistance genes (including a range of anti-microbial compounds) were also considered, and the majority (84%) were for TEM beta-lactamase, a commonly occurring class of genes globally.
Differentially Expressed Transcripts
Transcripts of four subsystems were under-expressed in the water treatment relative to the control and transcripts of 14 subsystems were under-expressed in the OM treatment (Figure 3B). Significant over-expression was only detected in OM treatments: transcripts of seven subsystems were over-expressed (Figure 3B), but removal of redundant CBSS transcripts reduced the over-expressed subsystems to six (Figure 4). The over-expressed transcripts were predominately attributed to three bacterial phyla: Actinobacteria, Proteobacteria, and Firmicutes (Figure 4). Conversely, under-expressed transcripts were attributed largely to eukaryotic taxa in the control soils (Figure 5). Patterns within these results and their taxonomic designations are discussed below.
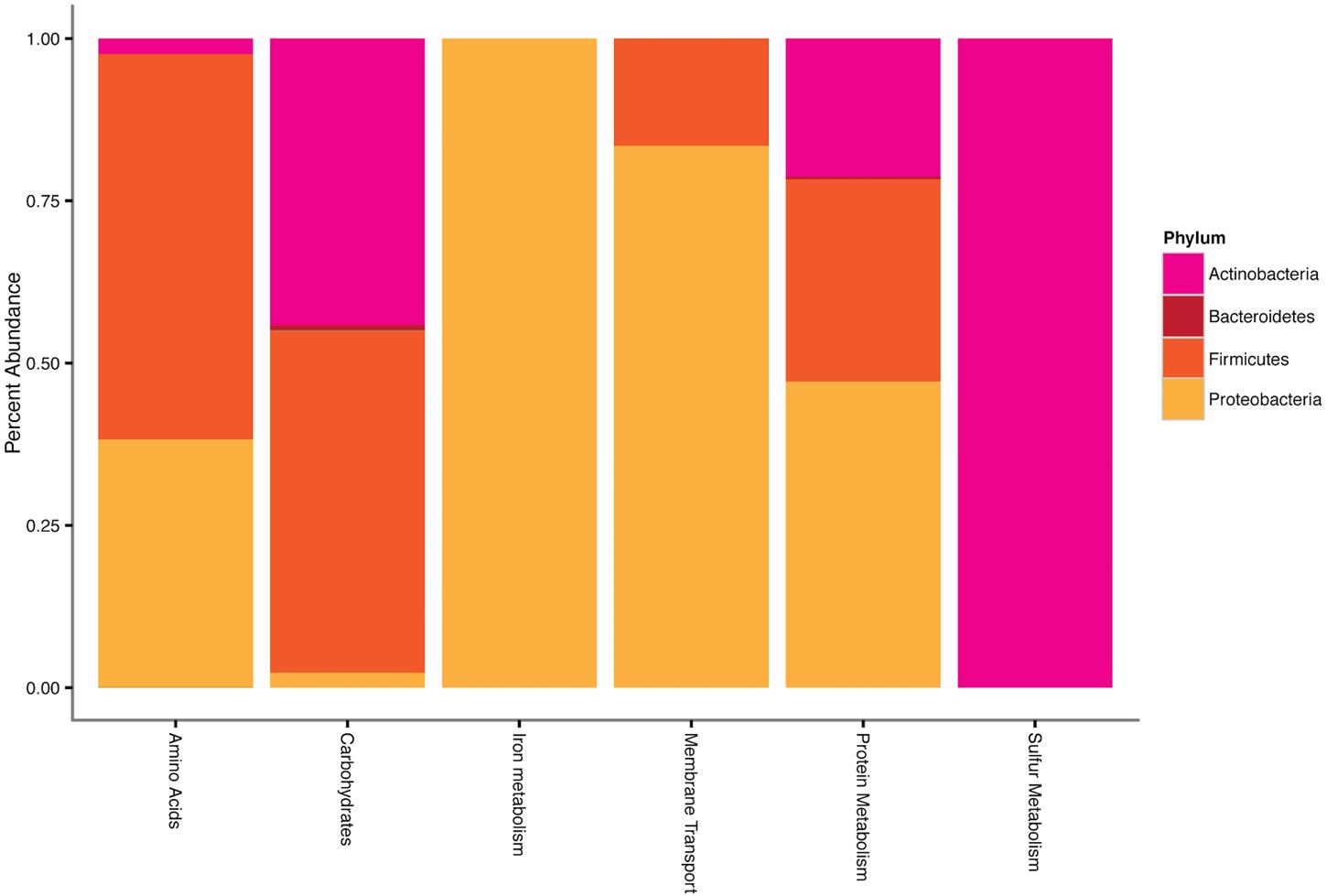
FIGURE 4. Phylum-level taxonomy of sequences that had significant positive differential expression (log2 fold change), expressed as the percentage of total sequences for each differential expression result. Phyla that contributed <1% of total sequences have been excluded.
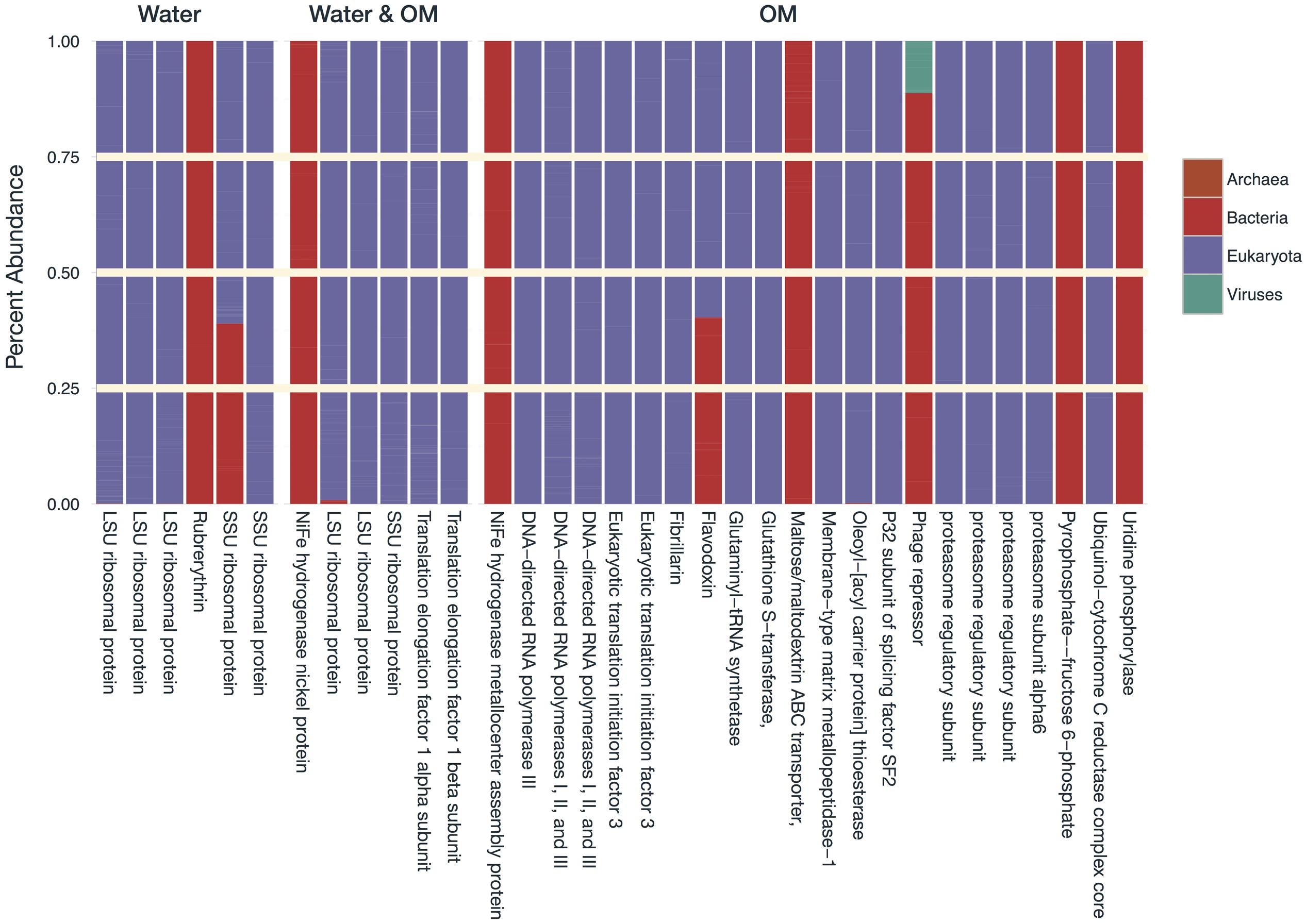
FIGURE 5. Domain-level taxonomy of sequences that had significant negative differential expression (log2 fold change), expressed as the percentage of total sequences for each differential expression result. Taxonomic assignments are based on the sequences in the control samples.
Carbohydrate utilization
All over- and under-expressed transcripts of the carbohydrate subsystem were identified as bacterial (Figures 4 and 5). The over-expressed transcript was that of 6-phosphogluconate dehydrogenase, decarboxylating (EC 1.1.1.44), which is the rate-limiting enzyme of the pentose phosphate pathway (PPP) that generates fructose-6-phosphate and reducing power. In addition, the PPP is responsible for generating metabolic intermediates for biosynthesis, catabolizing sugars that cannot be utilized by any other pathway (Sprenger, 1995), and NADPH, which may be used for mediating oxidative stress (Wang Y. P. et al., 2014). This transcript was over-expressed by several groups of bacteria: Actinobacteria (44%), Bacteroidetes (1%), Cyanobacteria (<1%), Firmicutes (53%), and Proteobacteria (2%; Figure 4). The Proteobacteria response was attributed to Gammaproteobacteria (98%) and Deltaproteobacteria (2%). The two under-expressed carbohydrate subsystem transcripts were of glycolysis and maltose/maltodextrin utilization pathways (Figure 5). Actinobacteria were solely responsible for the under-expressed glycolysis transcripts, while Alpha- (1%) and Gammaproteobacteria (30%), Cyanobacteria (<1%), and Actinobacteria (67%) all contributed to the under-expression of maltose/maltodextrin utilization. These functional shifts in the carbohydrate subsystem suggest that bacteria are responding to the carbon sources provided by the OM amendment.
Notably, Actinobacteria was the only phylum linked to each of the differentially expressed carbohydrate transcripts. This phylum has been linked with carbohydrate subsystem transcripts in other soils responding to disturbance events, including deforestation (Mendes et al., 2015) and inoculation after sterilization (Delmont et al., 2014). Additionally, all of the over-expressed sulfur metabolism transcripts were attributed to Actinobacteria (Figure 4). These transcripts were for the enzyme neuraminidase NanP, which is traditionally associated with pathogenicity but has been detected in three non-pathogenic genera of Actinobacteria in soils, and is thought to be used to metabolize sialic acids as a nutrient source in those cases (Gruteser et al., 2012). Sialic acid degrading enzymes have been observed previously in MDV soils, and were speculated to aid in degrading water-binding polymers of soil microorganisms (Gallikowski and Hirsch, 1988).
Transporters
All of the over-expressed transcripts in amino acids and membrane transport subsystems were of ABC-transport systems. Although a small portion (2%) was attributed to Actinobacteria, the phyla Firmicutes (59%) and Proteobacteria (38%) were largely responsible for over-expressed amino acid transcripts and were solely responsible for over-expressed membrane transport transcripts (Figure 4). The Proteobacteria response in amino acid transcripts was attributed to Alpha- (4%), Beta- (4%), and Gammaproteobacteria (91%), while that of membrane transport was attributed exclusively to Gammaproteobacteria. Transcripts in the amino acid subsystem were for polyamine transport, while those in the membrane transport subsystem were for oligopeptides. An increase in transport transcripts suggests increased nutrient availability and uptake potential, which is consistent with the complex OM addition of this study. Similarly, a comparison of frozen and thawed Alaskan soils also found higher abundances of transporter functions in thawed samples, which the authors deemed indicative of increased nutrient mobilization (Hultman et al., 2015).
Cellular Growth
All of the over-expressed protein metabolism transcripts were for protein biosynthesis, specifically bacterial SSU ribosomal protein S4p. Members of Actinobacteria (21%), Bacteroidetes (<1%), Firmicutes (31%), and Proteobacteria (47%) all contributed to the over-expression of these transcripts (Figure 4). As with the over-expressed amino acid transcripts, the Proteobacterial response was attributed to Alpha- (1%), Beta- (8%), and Gammaproteobacteria (91%). The increased expression of biosynthesis transcripts highlights that these bacteria were actively growing. Conversely, many of the under-expressed transcripts attributed to eukaryotic taxa were basic components of cellular growth, including ribosomal protein biosynthesis, polymerases, and translation factors (Figure 5). The over-expression of cellular growth transcripts in Bacteria and under-expression in Eukaryota likely attributed to the relative abundance shifts of these domains with treatments (Table 1).
Iron Acquisition
The over-expressed transcript in the iron metabolism subsystem (Figure 4) was for an enterobactin synthase component. Enterobactins are high-affinity iron-chelating compounds used for iron uptake, characteristically by the Enterobacteriaceae family (Fiedler et al., 2001). Indeed the over-expressed iron acquisition transcripts were attributed exclusively to that family of Proteobacteria.
Conclusion
As part of the MDV coastal thaw zone (Marchant and Head, 2007; Fountain et al., 2014), the soils of this study are part of an “at risk landscape” in the Antarctic due to low elevation, proximity to the Ross Sea coast, permafrost ice abundance, and warm summer temperatures (Fountain et al., 2014). These soils are expected to experience increased water availability and mobilized nutrients due to thaw of permafrost and buried ice deposits (Fountain et al., 2014). The experimental amendments of the present study were designed to simulate expected climate change impacts, and they appear to be stressors in these soils, resulting in losses of taxonomic and functional diversity. The impending increases of soil moisture and OM due to climate change may diminish the taxonomic richness and functional capacity of these soil communities. The most significant positive responses were observed in the Bacteria (Figures 2C and 4), whereas negative responses were largely among the Eukaryota (Figure 5). Many of the transcripts under-expressed by Eukaryota are indicative of basic cellular growth (Figure 5), thus repressed growth accounted for the observed taxonomic decline in abundance of Eukaryota in response to amendments (Table 1). Conversely, the transcripts lost by Bacteria were largely components of specialized pathways, rather than basic cellular functions (Figure 5). However, significant taxonomic losses occurred within the Bacteria as well. Three phyla (Actinobacteria, Proteobacteria, and Firmicutes) dominated the few positive transcript responses (Figure 4), and taxonomic diversity loss within Bacteria was apparent (Figures 2A,C). Dry soil bacterial communities of the MDV have been reported to contain distinct endemic taxa relative to wetted soils (Zeglin et al., 2011), and increased water and OM availability decreased diversity of dry soil bacterial communities (Schwartz et al., 2014; Van Horn et al., 2014). As climate change impacts the region, a loss of dry-adapted endemic oligotrophic taxa and dominance by generalist taxa is likely.
Author Contributions
Experimental design by DV, JB, and CT-V. Project proposal and funding secured by DV, JB, MG, ES, and CT-V. Sample collection by DV. Sample processing by HB. Data analysis by HB and AW. Manuscript by HB, AW, CT-V, and DV.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was funded by NSF grants 0838879 to JB, MG, and CT-V, 1142102 to CT-V, DV, and ES, and 1245991 to DV. Additional support was provided by the McMurdo LTER, NSF grant 1115245. Illumina sequencing was completed at the Next Generation Sequencing Facility at the BioFrontiers Institute, University of Colorado, Boulder.
Footnotes
References
Adams, B. J., Bardgett, R. D., Ayres, E., Wall, D. H., Aislabie, J., Bamforth, S., et al. (2006). Diversity and distribution of Victoria Land biota. Soil Biol. Biochem. 38, 3003–3018. doi: 10.1016/j.soilbio.2006.04.030
Amoutzias, G. D., Van de Peer, Y., and Mossialos, D. (2008). Evolution and taxonomic distribution of nonribosomal peptide and polyketide synthases. Future Microbiol. 3, 361–370. doi: 10.2217/17460913.3.3.361
Anthony, C. (2011). How half a century of research was required to understand bacterial growth on C1 and C2 compounds; the story of the serine cycle and the ethylmalonyl-CoA pathway. Sci. Prog. 94, 109–137. doi: 10.3184/003685011X13044430633960
Arenz, B., and Blanchette, R. (2011). Distribution and abundance of soil fungi in Antarctica at sites on the Peninsula, Ross Sea Region and McMurdo Dry Valleys. Soil Biol. Biochem. 43, 308–315. doi: 10.1016/j.soilbio.2010.10.016
Bachmann, B. O., and Ravel, J. (2009). Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 458, 181–217. doi: 10.1016/S0076-6879(09)04808-3
Ball, B. A., Virginia, R. A., Barrett, J., Parsons, A. N., and Wall, D. H. (2009). Interactions between physical and biotic factors influence CO2 flux in Antarctic dry valley soils. Soil Biol. Biochem. 41, 1510–1517. doi: 10.1016/j.soilbio.2009.04.011
Bao, H., Campbell, D. A., Bockheim, J. G., and Thiemens, M. H. (2000). Origins of sulphate in Antarctic dry-valley soils as deduced from anomalous 17O compositions. Nature 407, 499–502. doi: 10.1038/35035054
Barrett, J., Virginia, R., Wall, D., Doran, P., Fountain, A., Welch, K., et al. (2008a). Persistent effects of a discrete warming event on a polar desert ecosystem. Glob. Chang. Biol. 14, 2249–2261. doi: 10.1111/j.1365-2486.2008.01641.x
Barrett, J., Virginia, R. A., Wall, D. H., and Adams, B. J. (2008b). Decline in a dominant invertebrate species contributes to altered carbon cycling in a low-diversity soil ecosystem. Glob. Chang. Biol. 14, 1734–1744. doi: 10.1111/j.1365-2486.2008.01611.x
Barrett, J. E., Virginia, R. A., Hopkins, D. W., Aislabie, J., Bargagli, R., Bockheim, J. G., et al. (2006). Terrestrial ecosystem processes of Victoria Land. Antarctica 38, 3019–3034.
Barrett, J. E., Virginia, R. A., Lyons, W. B., McKnight, D. M., Priscu, J. C., Doran, P. T., et al. (2007). Biogeochemical stoichiometry of Antarctic Dry Valley ecosystems. J. Geophys. Res. 112, 1–12. doi: 10.1029/2005JG000141
Barrett, J. E., Virginia, R. A., Parsons, A. N., and Wall, D. H. (2005). Potential soil organic matter turnover in Taylor Valley, Antarctica. Arctic Antarctic Alpine Res. 37, 108–117. doi: 10.1657/1523-0430(2005)037[0108:PSOMTI]2.0.CO;2
Barrett, J. E., Virginia, R. A., and Wall, D. H. (2002). Trends in resin and KCl-extractable soil nitrogen across landscape gradients in Taylor Valley, Antarctica. Ecosystems 5, 289–299. doi: 10.1007/s10021-001-0072-6
Burkins, M. B., Virginia, R. A., Chamberlain, C. P., and Wall, D. H. (2000). Origin and distribution of soil organic matter in Taylor Valley, Antarctica. Ecology 81, 2377–2391. doi: 10.1890/0012-9658 (2000)081[2377:OADOSO]2.0.CO;2
Burkins, M. B., Virginia, R. A., and Wall, D. H. (2001). Organic carbon cycling in Taylor Valley, Antarctica: quantifying soil reservoirs and soil respiration. Global Change Biol. 7, 113–125. doi: 10.1046/j.1365-2486.2001.00393.x
Cary, S. C., McDonald, I. R., Barrett, J. E., and Cowan, D. A. (2010). On the rocks: the microbiology of Antarctic Dry Valley soils. Nat. Rev. Microbiol. 8, 129–138. doi: 10.1038/nrmicro2281
Cederlund, H., Wessén, E., Enwall, K., Jones, C. M., Juhanson, J., Pell, M., et al. (2014). Soil carbon quality and nitrogen fertilization structure bacterial communities with predictable responses of major bacterial phyla. Appl. Soil Ecol. 84, 62–68. doi: 10.1016/j.apsoil.2014.06.003
Chen, J., Liu, X., Zheng, J., Zhang, B., Lu, H., Chi, Z., et al. (2013). Biochar soil amendment increased bacterial but decreased fungal gene abundance with shifts in community structure in a slightly acid rice paddy from Southwest China. Appl. Soil Ecol. 71, 33–44. doi: 10.1016/j.apsoil.2013.05.003
Cibulskis, K., McKenna, A., Fennell, T., Banks, E., DePristo, M., and Getz, G. (2011). ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics 27, 2601–2602. doi: 10.1093/bioinformatics/btr446
Clow, G. D., McKay, C. P., Simmons, G. M. Jr., and Wharton, R. A. Jr. (1988). Climatological observations and predicted sublimation rates at lake hoare, Antarctica. J. Clim. 1, 715–728. doi: 10.1175/1520-0442(1988)001<0715:COAPSR>2.0.CO;2
Connell, L., Redman, R., Craig, S., and Rodriguez, R. (2006). Distribution and abundance of fungi in the soils of Taylor Valley, Antarctica. Soil Biol. Biochem. 38, 3083–3094. doi: 10.1016/j.soilbio.2006.02.016
Convey, P., and Block, W. (1996). Antarctic Diptera: ecology, physiology and distribution. Eur. J. Entomol. 93, 1–14.
Costello, E. K., Halloy, S. R., Reed, S. C., Sowell, P., and Schmidt, S. K. (2009). Fumarole-supported islands of biodiversity within a hyperarid, high-elevation landscape on Socompa Volcano, Puna de Atacama, Andes. Appl. Environ. Microbiol. 75, 735–747. doi: 10.1128/AEM.01469-08
de Menezes, A., Clipson, N., and Doyle, E. (2012). Comparative metatranscriptomics reveals widespread community responses during phenanthrene degradation in soil. Environ. Microbiol. 14, 2577–2588. doi: 10.1111/j.1462-2920.2012.02781.x
Delmont, T. O., Francioli, D., Jacquesson, S., Laoudi, S., Mathieu, A., Nesme, J., et al. (2014). Microbial community development and unseen diversity recovery in inoculated sterile soil. Biol. Fert. Soils 50, 1069–1076. doi: 10.1007/s00374-014-0925-8
Delviks-Frankenberry, K., Cingöz, O., Coffin, J. M., and Pathak, V. K. (2012). Recombinant origin, contamination, and de-discovery of XMRV. Curr. Opin. Virol. 2, 499–507. doi: 10.1016/j.coviro.2012.06.009
Denton, G. H., Sugden, D. E., Marchant, D. R., Hall, B. L., and Wilch, T. I. (1993). East Antarctic ice sheet sensitivity to Pliocene climatic change from a Dry Valleys perspective. Geografiska Annaler 75, 155–204. doi: 10.2307/521200
Doran, P. T., McKay, C. P., Clow, G. D., Dana, G. L., Fountain, A. G., Nylen, T., et al. (2002). Valley floor climate observations from the McMurdo dry valleys, Antarctica, 1986-2000. J. Geophys. Res. 107, ACL13-1–ACL13-2. doi: 10.1029/2001JD002045
Elster, J., Lukesová, A., Svoboda, J., Kopecky, J., and Kanda, H. (1999). Diversity and abundance of soil algae in the polar desert, Sverdrup Pass, central Ellesmere Island. Polar Record 35, 231–254. doi: 10.1017/S0032247400015515
Erlwein, O., Robinson, M. J., Dustan, S., Weber, J., Kaye, S., and McClure, M. O. (2011). DNA extraction columns contaminated with murine sequences. PLoS ONE 6:e23484. doi: 10.1371/journal.pone.0023484
Eveland, J., Gooseff, M. N., Lampkin, D. J., Barrett, J., and Takacs-Vesbach, C. (2013). Spatial and temporal patterns of snow accumulation and aerial ablation across the McMurdo Dry Valleys, Antarctica. Hydrol. Process. 27, 2864–2875.
Fell, J. W., Scorzetti, G., Connell, L., and Craig, S. (2006). Biodiversity of micro-eukaryotes in Antarctic Dry Valley soils with < 5% soil moisture. Soil Biol. Biochem. 38, 3107–3119. doi: 10.1016/j.soilbio.2006.01.014
Fiedler, H.-P., Krastel, P., Müller, J., Gebhardt, K., and Zeeck, A. (2001). Enterobactin: the characteristic catecholate siderophore of Enterobacteriaceae is produced by Streptomyces species. FEMS Microbiol. Lett. 196, 147–151. doi: 10.1111/j.1574-6968.2001.tb10556.x
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364. doi: 10.1890/05-1839
Fierer, N., Lauber, C. L., Ramirez, K. S., Zaneveld, J., Bradford, M. A., and Knight, R. (2011). Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 6, 1007–1017. doi: 10.1038/ismej.2011.159
Fierer, N., Strickland, M. S., Liptzin, D., Bradford, M. A., and Cleveland, C. C. (2009). Global patterns in belowground communities. Ecol. Lett. 12, 1238–1249. doi: 10.1111/j.1461-0248.2009.01360.x
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2013). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Foster, R. (1988). Microenvironments of soil microorganisms. Biol. Fertil. Soils 6, 189–203. doi: 10.1007/BF00260816
Fountain, A. G., Levy, J. S., Gooseff, M. N., and Van Horn, D. (2014). The McMurdo Dry Valleys: a landscape on the threshold of change. Geomorphology 225, 25–35. doi: 10.1016/j.geomorph.2014.03.044
Fountain, A. G., Nylen, T. H., Monaghan, A., Basagic, H. J., and Bromwich, D. (2010). Snow in the McMurdo Dry Valleys, Antarctica. Int. J. Climatol. 30, 633–642. doi: 10.1002/joc.1933
Freckman, D. W., and Virginia, R. A. (1997). Low-diversity Antarctic soil nematode communities: distribution and response to disturbance. Ecology 78, 363–369. doi: 10.1890/0012-9658(1997)078[0363:LDASNC]2.0.CO;2
Freckman, D. W., and Virginia, R. A. (1998). Soil biodiversity and community structure in the McMurdo Dry Valleys, Antarctica. Antarctic Res. Ser. 72, 323–335.
Friedmann, E. I., Kappen, L., Meyer, M. A., and Nienow, J. A. (1993). Long-term productivity in the cryptoendolithic microbial community of the Ross Desert, Antarctica. Microb. Ecol. 25, 51–69. doi: 10.1007/BF00182129
Gallikowski, C., and Hirsch, P. (1988). 1.4 Preliminary Characterization and Identification of 1984/85 continental antarctic soil microorganisms of linnaeus terrace (Altitude 1600 m; McMurdo Dry Valleys). Polarforschung 58, 93–101.
Garcia-Pichel, F., and Pringault, O. (2001). Microbiology: cyanobacteria track water in desert soils. Nature 413, 380–381. doi: 10.1038/35096640
Gerdes, S., El Yacoubi, B., Bailly, M., Blaby, I. K., Blaby-Haas, C. E., Jeanguenin, L., et al. (2011). Synergistic use of plant-prokaryote comparative genomics for functional annotations. BMC Genomics 12:S2. doi: 10.1186/1471-2164-12-S1-S2
Geyer, K. M., Altrichter, A. E., Van Horn, D. J., Takacs-Vesbach, C. D., Gooseff, M. N., and Barrett, J. E. (2013). Environmental controls over bacterial communities in polar desert soils. Ecosphere 4, 1–17. doi: 10.1890/ES13-00048.1
Gibson, M. K., Forsberg, K. J., and Dantas, G. (2014). Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 9, 207–216.
Giovannoni, S. J., Delong, E. F., Schmidt, T. M., and Pace, N. R. (1990). Tangential flow filtration and preliminary phylogenetic analysis of marine picoplankton. Appl. Environ. Microbiol. 56, 2572–2575.
Gooseff, M. N., Barrett, J. E., Doran, P. T., Fountain, A. G., Lyons, W. B., Parsons, A. N., et al. (2003). Snow-patch influence on soil biogeochemical processes and invertebrate distribution in the McMurdo Dry Valleys, Antarctica. Arctic Antarctic Alpine Res. 35, 91–99. doi: 10.1657/1523-0430(2003)035[0091:SPIOSB]2.0.CO;2
Gregorich, E. G., Hopkins, D. W., Elberling, B., Sparrow, A. D., Novis, P., Greenfield, L. G., et al. (2006). Emission of CO2, CH4 and N2O from lakeshore soils in an Antarctic dry valley. Soil Biol. Biochem. 38, 3120–3129.
Gruteser, N., Marin, K., Krämer, R., and Thomas, G. H. (2012). Sialic acid utilization by the soil bacterium Corynebacterium glutamicum. FEMS Microbiol. Lett. 336, 131–138. doi: 10.1111/j.1574-6968.2012.02663.x
Hanson, C. A., Allison, S. D., Bradford, M. A., Wallenstein, M. D., and Treseder, K. K. (2008). Fungal taxa target different carbon sources in forest soil. Ecosystems 11, 1157–1167. doi: 10.1007/s10021-008-9186-4
Hopkins, D. W., Sparr, A. D., Novis, P. M., Gregorich, E. G., Elberling, B., and Greenfield, L. G. (2006a). Controls on the distribution of productivity and organic resources in Antarctic Dry Valley soils. Proc. Biol. Sci. 273, 2687–2695.
Hopkins, D. W., Sparrow, A. D., Elberling, B., Gregorich, E. G., Novis, P. M., Greenfield, L. G., et al. (2006b). Carbon, nitrogen and temperature controls on microbial activity in soils from an Antarctic dry valley. Soil Biol. Biochem. 38, 3130–3140. doi: 10.1016/j.soilbio.2006.01.012
Horton, T. R., and Bruns, T. D. (2001). The molecular revolution in ectomycorrhizal ecology: peeking into the black-box. Mol. Ecol. 10, 1855–1871. doi: 10.1046/j.0962-1083.2001.01333.x
Hultman, J., Waldrop, M. P., Mackelprang, R., David, M. M., McFarland, J., Blazewicz, S. J., et al. (2015). Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 521, 208–212. doi: 10.1038/nature14238
Kanehisa, M., Sato, Y., and Morishima, K. (2015). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kastovska, K., Elster, J., Marek, S., and Santruckova, H. (2005). Microbial assemblages in soil microbial succession after glacial retreat in svalbard (High Arctic). Microb. Ecol. 50, 396–407. doi: 10.1007/s00248-005-0246-4
Knox, M. A., Wall, D. H., Virginia, R. A., Vandegehuchte, M. L., San Gil, I., and Adams, B. J. (2016). Impact of diurnal freeze–thaw cycles on the soil nematode Scottnema lindsayae in Taylor Valley, Antarctica. Polar Biol. 39, 583–592. doi: 10.1007/s00300-015-1809-6
Kumar, S., Jones, M., Koutsovoulos, G., Clarke, M., and Blaxter, M. (2013). Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front. Genet. 4:237. doi: 10.3389/fgene.2013.00237
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Laurence, M., Hatzis, C., and Brash, D. E. (2014). Common Contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLoS ONE 9:e97876. doi: 10.1371/journal.pone.0097876
Lawley, B., Ripley, S., Bridge, P., and Convey, P. (2004). Molecular analysis of geographic patterns of eukaryotic diversity in Antarctic soils. Appl. Environ. Microbiol. 70, 5963–5972. doi: 10.1128/AEM.70.10.5963-5972.2004
Lee, C. K., Barbier, B. A., Bottos, E. M., McDonald, I. R., and Cary, S. C. (2012). The inter-valley soil comparative survey: the ecology of Dry Valley edaphic microbial communities. ISME J. 6, 1046–1057. doi: 10.1038/ismej.2011.170
Longo, M. S., O’Neill, M. J., and O’Neill, R. J. (2011). Abundant human DNA contamination identified in non-primate genome databases. PLoS ONE 6:e16410. doi: 10.1371/journal.pone.0016410
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 15:550.
Malmström, H., Storå, J., Dalén, L., Holmlund, G., and Götherström, A. (2005). Extensive human DNA contamination in extracts from ancient dog bones and teeth. Mol. Biol. Evol. 22, 2040–2047. doi: 10.1093/molbev/msi195
Marchant, D. R., and Head, J. W. (2007). Antarctic dry valleys: microclimate zonation, variable geomorphic processes, and implications for assessing climate change on Mars. Icarus 192, 187–222. doi: 10.1016/j.icarus.2007.06.018
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. doi: 10.1371/journal.pone.0061217
Mendes, L. W., Tsai, S. M., Navarrete, A. A., de Hollander, M., van Veen, J. A., and Kuramae, E. E. (2015). Soil-Borne microbiome: linking diversity to function. Microb. Ecol. 70, 255–265. doi: 10.1007/s00248-014-0559-2
Meyer, F., Paarmann, D., D’Souza, M., Olson, R., Glass, E. M., Kubal, M., et al. (2008). The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386
Mitchell, K. R., and Takacs-Vesbach, C. D. (2008). A comparison of methods for total community DNA preservation and extraction from various thermal environments. J. Ind. Microbiol. Biotechnol. 35, 1139–1147. doi: 10.1007/s10295-008-0393-y
Moorhead, D. L. (2007). Mesoscale dynamics of ephemeral wetlands in the antarctic dry valleys: implications to production and distribution of organic matter. Ecosystems 10, 86–94. doi: 10.1007/s10021-006-9005-8
Moorhead, D. L., Doran, P. T., Fountain, A. G., Lyons, W. B., Mcknight, D. M., Priscu, J. C., et al. (1999). Ecological legacies: impacts on ecosystems of the McMurdo Dry Valleys. Bioscience 49, 1009–1019. doi: 10.2307/1313734
Neilson, J. W., Quade, J., Ortiz, M., Nelson, W. M., Legatzki, A., Tian, F., et al. (2012). Life at the hyperarid margin: novel bacterial diversity in arid soils of the Atacama Desert, Chile. Extremophiles 16, 553–566. doi: 10.1007/s00792-012-0454-z
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H.-Y., Cohoon, M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702. doi: 10.1093/nar/gki866
Parsons, A. N., Barrett, J. E., Wall, D. H., and Virginia, R. A. (2004). Soil carbon dioxide flux in Antarctic Dry Valley ecosystems. Ecosystems 7, 286–295. doi: 10.1007/s10021-003-0132-1
Poage, M. A., Barrett, J. E., Virginia, R. A., and Wall, D. H. (2008). The influence of soil geochemistry on nematode distribution, mcmurdo dry Valleys, Antarctica. Arctic Antarctic Alpine Res. 40, 119–128. doi: 10.1657/1523-0430(06-051)[POAGE]2.0.CO;2
Pointing, S. B., Chan, Y. K., Lacap, D. C., Lau, M. C. Y., Jurgens, J. A., and Farrell, R. L. (2009). Highly specialized microbial diversity in hyper-arid polar desert. Proc. Natl. Acad. Sci. U.S.A. 106, 19964–19969. doi: 10.1073/pnas.0908274106
Prestat, E., David, M. M., Hultman, J., Taş, N., Lamendella, R., Dvornik, J., et al. (2014). FOAM (functional ontology assignments for metagenomes): a hidden markov model (HMM) database with environmental focus. Nucleic Acids Res. 42:e145. doi: 10.1093/nar/gku702
R Development Core Team (2011). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rajeev, L., da Rocha, U. N., Klitgord, N., Luning, E. G., Fortney, J., Axen, S. D., et al. (2013). Dynamic cyanobacterial response to hydration and dehydration in a desert biological soil crust. ISME J. 7, 2178–2191. doi: 10.1038/ismej.2013.83
Ramirez, K. S., Lauber, C. L., Knight, R., Bradford, M. A., and Fierer, N. (2010). Consistent effects of nitrogen fertilization on soil bacterial communities in contrasting systems. Ecology 91, 3463–3470. doi: 10.1890/10-0426.1
Ranjard, L., Lejon, D. P., Mougel, C., Schehrer, L., Merdinoglu, D., and Chaussod, R. (2003). Sampling strategy in molecular microbial ecology: influence of soil sample size on DNA fingerprinting analysis of fungal and bacterial communities. Environ. Microbiol. 5, 1111–1120. doi: 10.1046/j.1462-2920.2003.00521.x
Řeháková, K., Chlumská, Z., and Doležal, J. (2011). Soil cyanobacterial and microalgal diversity in dry mountains of Ladakh, NW Himalaya, as related to site, altitude, and vegetation. Microb. Ecol. 62, 337–346. doi: 10.1007/s00248-011-9878-8
Robinson, M. J., Erlwein, O. W., Kaye, S., Weber, J., Cingoz, O., Patel, A., et al. (2010). Mouse DNA contamination in human tissue tested for XMRV. Retrovirology 7:108. doi: 10.1186/1742-4690-7-108
Schwartz, E., Van Horn, D. J., Buelow, H. N., Okie, J. G., Gooseff, M. N., Barrett, J. E., et al. (2014). Characterization of growing bacterial populations in McMurdo Dry Valley soils through stable isotope probing with 18O-water. FEMS Microbiol. Ecol. 89, 415–425. doi: 10.1111/1574-6941.12349
Shanhun, F. L., Almond, P. C., Clough, T. J., and Smith, C. M. S. (2012). Abiotic processes dominate CO2 fluxes in Antarctic soils. Soil Biol. Biochem. 53, 99–111. doi: 10.1016/j.soilbio.2012.04.027
Sprenger, G. A. (1995). Genetics of pentose-phosphate pathway enzymes of Escherichia coli K-12. Arch. Microbiol. 164, 324–330. doi: 10.1007/BF02529978
Takacs-Vesbach, C., Inskeep, W. P., Jay, Z. J., Herrgard, M. J., Rusch, D. B., Tringe, S. G., et al. (2013). Metagenome sequence analysis of filamentous microbial communities obtained from geochemically distinct geothermal channels reveals specialization of three Aquificales lineages. Front. Microbiol. 4:84. doi: 10.3389/fmicb.2013.00084
Tiao, G., Lee, C. K., McDonald, I. R., Cowan, D. A., and Cary, S. C. (2012). Rapid microbial response to the presence of an ancient relic in the Antarctic Dry Valleys. Nat. Commun. 3:660. doi: 10.1038/ncomms1645
Tilman, D. (1996). Biodiversity: population versus ecosystem stability. Ecology 77, 350–363. doi: 10.2307/2265614
Treonis, A. M., Wall, D. H., and Virginia, R. A. (1999). Invertebrate biodiversity in Antarctic dry valley soils and sediments. Nature 2, 482–492.
Tuke, P. W., Tettmar, K. I., Tamuri, A., Stoye, J. P., and Tedder, R. S. (2011). PCR master mixes harbour murine DNA sequences, Caveat emptor! PLoS ONE 6:e19953. doi: 10.1371/journal.pone.0019953
Urich, T., Lanzén, A., Qi, J., Huson, D. H., Schleper, C., and Schuster, S. C. (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE 3:e2527. doi: 10.1371/journal.pone.0002527
Van Horn, D. J., Okie, J. G., Buelow, H. N., Gooseff, M. N., Barrett, J. E., and Takacs-Vesbach, C. D. (2014). Soil microbial responses to increased moisture and organic resources along a salinity gradient in a polar desert. Appl. Environ. Microbiol. 80, 3034–3043. doi: 10.1128/AEM.03414-13
Van Horn, D. J., Van Horn, M. L., Barrett, J. E., Gooseff, M. N., Altrichter, A. E., Geyer, K. M., et al. (2013). Factors controlling soil microbial biomass and bacterial diversity and community composition in a cold desert ecosystem: role of geographic scale. PLoS ONE 8:e66103. doi: 10.1371/journal.pone.0066103
Wada, E., Shibata, R., and Torii, T. (1981). 15N abundance in Antarctica: origin of soil nitrogen and ecological implications. Ecosystems 292, 327–329.
Wang, H., Fewer, D. P., Holm, L., Rouhiainen, L., and Sivonen, K. (2014). Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc. Natl. Acad. Sci. U.S.A. 111, 9259–9264. doi: 10.1073/pnas.1401734111
Wang, H., Sivonen, K., and Fewer, D. P. (2015). Genomic insights into the distribution, genetic diversity and evolution of polyketide synthases and nonribosomal peptide synthetases. Curr. Opin. Genet. Dev. 35, 79–85. doi: 10.1016/j.gde.2015.10.004
Wang, J., Zhou, X., Zhu, J., Zhou, C., and Guo, Z. (2010). Revealing and avoiding bias in semantic similarity scores for protein pairs. BMC Bioinformatics 11:290. doi: 10.1186/1471-2105-11-290
Wang, Q., Fish, J. A., Gilman, M., Sun, Y., Brown, C. T., Tiedje, J. M., et al. (2015). Xander: employing a novel method for efficient gene-targeted metagenomic assembly. Microbiome 3:1. doi: 10.1186/s40168-015-0093-6
Wang, Y. P., Zhou, L. S., Zhao, Y. Z., Wang, S. W., Chen, L. L., Liu, L. X., et al. (2014). Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 33, 1304–1320. doi: 10.1002/embj.201387224
Wilke, A., Harrison, T., Wilkening, J., Field, D., Glass, E. M., Kyrpides, N., et al. (2012). The M5nr: a novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinformatics 13:141. doi: 10.1186/1471-2105-13-141
Witherow, R. A., Lyons, W. B., Bertler, N. A., Welch, K. A., Mayewski, P. A., Sneed, S. B., et al. (2006). The aeolian flux of calcium, chloride and nitrate to the McMurdo Dry Valleys landscape: evidence from snow pit analysis. Antarctic Sci. 18, 497–505. doi: 10.1017/S095410200600054X
Wood, S. A., Rueckert, A., Cowan, D. A., and Cary, S. C. (2008). Sources of edaphic cyanobacterial diversity in the Dry Valleys of Eastern Antarctica. ISME J. 2, 308–320. doi: 10.1038/ismej.2007.104
Zeglin, L. H., Dahm, C. N., Barrett, J. E., Gooseff, M. N., Fitpatrick, S. K., and Takacs-Vesbach, C. D. (2011). Bacterial community structure along moisture gradients in the parafluvial sediments of two ephemeral desert streams. Microb. Ecol. 61, 543–556. doi: 10.1007/s00248-010-9782-7
Zeglin, L. H., Sinsabaugh, R. L., Barrett, J. E., Gooseff, M. N., and Takacs-Vesbach, C. D. (2009). Landscape distribution of microbial activity in the McMurdo Dry Valleys: linked biotic processes, hydrology, and geochemistry in a cold desert ecosystem. Ecosystems 12, 562–573. doi: 10.1007/s10021-009-9242-8
Keywords: metatranscriptomics, microbial ecology, Antarctica, soils in hyper-arid regions, amendments
Citation: Buelow HN, Winter AS, Van Horn DJ, Barrett JE, Gooseff MN, Schwartz E and Takacs-Vesbach CD (2016) Microbial Community Responses to Increased Water and Organic Matter in the Arid Soils of the McMurdo Dry Valleys, Antarctica. Front. Microbiol. 7:1040. doi: 10.3389/fmicb.2016.01040
Received: 03 February 2016; Accepted: 21 June 2016;
Published: 18 July 2016.
Edited by:
Brian D. Lanoil, University of Alberta, CanadaCopyright © 2016 Buelow, Winter, Van Horn, Barrett, Gooseff, Schwartz and Takacs-Vesbach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina D. Takacs-Vesbach, cvesbach@unm.edu