 Haifeng Geng
Haifeng Geng Mary B. Tran-Gyamfi2
Mary B. Tran-Gyamfi2 Todd W. Lane
Todd W. Lane Kenneth L. Sale
Kenneth L. Sale- 1Department of Systems Biology, Sandia National Laboratories, Livermore, CA, USA
- 2Department of Biomass Science and Conversion Technology, Sandia National Laboratories, Livermore, CA, USA
- 3Institute of Chemistry, University of the Philippines Diliman, Quezon City, Philippines
Open microalgae cultures host a myriad of bacteria, creating a complex system of interacting species that influence algal growth and health. Many algal microbiota studies have been conducted to determine the relative importance of bacterial taxa to algal culture health and physiological states, but these studies have not characterized the interspecies relationships in the microbial communities. We subjected Nanochroloropsis salina cultures to multiple chemical treatments (antibiotics and quorum sensing compounds) and obtained dense time-series data on changes to the microbial community using 16S gene amplicon metagenomic sequencing (21,029,577 reads for 23 samples) to measure microbial taxa-taxa abundance correlations. Short-term treatment with antibiotics resulted in substantially larger shifts in the microbiota structure compared to changes observed following treatment with signaling compounds and glucose. We also calculated operational taxonomic unit (OTU) associations and generated OTU correlation networks to provide an overview of possible bacterial OTU interactions. This analysis identified five major cohesive modules of microbiota with similar co-abundance profiles across different chemical treatments. The Eigengenes of OTU modules were examined for correlation with different external treatment factors. This correlation-based analysis revealed that culture age (time) and treatment types have primary effects on forming network modules and shaping the community structure. Additional network analysis detected Alteromonadeles and Alphaproteobacteria as having the highest centrality, suggesting these species are “keystone” OTUs in the microbial community. Furthermore, we illustrated that the chemical tropodithietic acid, which is secreted by several species in the Alphaproteobacteria taxon, is able to drastically change the structure of the microbiota within 3 h. Taken together, these results provide valuable insights into the structure of the microbiota associated with N. salina cultures and how these structures change in response to chemical perturbations.
Introduction
Open algal cultures are complex dynamic ecosystems inhabited by diverse microbial communities (Carney et al., 2014). In many cases, microbes in microalgae ecosystems have effects on algal physiology and nutrition as well as play critical roles on stability and homeostasis of algal ecosystems in both lab cultures and field studies (Kayser, 1979; Lee et al., 2000; Geng and Belas, 2010b). These types of algal-microbial interactions have been shown to either promote algal growth (Hold et al., 2001) and protect algae from invading pathogens (Geng and Belas, 2010b), or to inhibit algal growth and destabilize the algal ecosystem (Cole, 1982; Carney et al., 2014). Thus, knowledge of the composition and structure of algal-associated microbiota is important for understanding homeostasis in robust algal cultures, as aberrant microbiota have been reportedly linked with precipitous crashes of algal cultures (Cole, 1982; Carney et al., 2014). These observations argue for finding important relevant factors that contribute to the appropriate composition and proper structuring of this complex biological community (Carney et al., 2014; Sison-Mangus et al., 2014).
Given that microbial communities are enormously diverse, we are only able to characterize their diversity with the recent use of next-generation sequencing technologies (Gonzalez et al., 2012). In the past, algal microbiota researchers have focused on understanding the relationship between collective population diversity and environmental conditions in algal cultures (Alavi et al., 2001; Carney et al., 2014) and on linking individual membership composition to ecosystem descriptors (Costello et al., 2009). However, the overarching property of a microbial consortium may stem from the requisite biological functions of collective groups consisting of multiple interacting bacterial species (known as modules) found in the community. For example, biofilm formation and metabolic complementation are modules in which the collective bacteria species deliver the required functions (Raes and Bork, 2008; Geng and Belas, 2010b). However, limited information is available on the substructure of the microbial communities (e.g., formation of bacterial groups or modularities recapitulated from interacting bacterial species) associated with microalgae. This not only hinders the interpretation of topological structures that oversee the proper function of the microbial community but also reduces the reliability of prediction of the community structure and its effects on the long-term stability of microalgae cultures.
One way of exploring substructures in biological systems is to look for pairs of entity relationships and subsequently using this information to build a correlation network of potentially interacting entities. Correlation networks have been successfully used in studies of cancer (Choi et al., 2005), yeast genetics (Ge et al., 2001), and microbial ecology (Lovejoy et al., 1998; Duran-Pinedo et al., 2011; Gilbert et al., 2012), which focused on searching for groups of lineages that occur together more often than expected by chance. Once networks have been built, several measures and metrics such as node centrality and betweenness can be evaluated to assess and identify the most important and influential nodes in the network (Jeong et al., 2001). Centrality measures the importance of a node based on the number of connections it makes with other nodes (degree centrality) in the network and may include measures of the importance of the neighbors to which it is connected (eigenvalue centrality, Katz centrality, page rank). Nodes with high number of connections to other nodes are perceived as having greater influence over the entire network (Jeong et al., 2001). Betweenness centrality measures the extent to which a node lies on paths between other nodes and is an indicator of the influence a node exerts over other nodes in the network. Nodes with higher betweenness are perceived to have greater influence within a network by virtue of their control over information passing among the other nodes in the network (Yoon et al., 2006). In other microbial ecological systems, network analyses based on Weighted Correlation Network Analysis (WGCNA) were used to identify hub OTUs with influential roles in maintaining a mature biofilm (Duran-Pinedo et al., 2011).
Recently, we found a microbial community associated with Nannochloropsis salina (CCMP, 1776) that displayed community stability and resilience to environmental perturbations at a global level (Geng et al., 2016). To obtain clues about the substructuring of the microbial community associated with N. salina, we subjected N. salina containing communities to various chemicals (e.g., antibiotics, metabolites) and examined changes in the connectivities among taxa in the microbial community. We used longitudinal 16S gene amplicon sequencing of 23 samples descended from a single ancestral microbiota and built correlation networks to reflect the dynamics of the inter-taxa associations and to investigate variations in taxa organization in response to different chemical treatments. As a result, we identified five cohesive modules representing various chemical treatment responses. Subsequently, through network centrality analysis, our data showed that key nodes in the modeled network were primarily from the bacterial species belonging to Alteromonadeles and Alphaproteobacteria, suggesting species from these groups are of particular significance and serve as “keystone” OTUs in the microbial community associate with N. salina.
Species of the Roseobacter clade of Alphaproteobacteria are important symbionts of microalgae (Gonzalez and Moran, 1997; Treangen et al., 2013). One of the key aspects of the Roseobacter–microalgae symbiosis is founded on Roseobacter's ability to produce a distinct set of infochemicals or signaling compounds with specialized functions (Mayali and Azam, 2004; Geng and Belas, 2010b; Seyedsayamdost et al., 2011; Treangen et al., 2013). For example, the antibiotic tropodithietic acid (TDA) produced by Roseobacter played a pivotal role in the bacterial-algal symbiosis by regulating TDA gene expression across various species (Geng and Belas, 2010a; Porsby et al., 2011) and preventing bacterial infection during prosperous algal blooms (Brinkhoff et al., 2004; Bruhn et al., 2007). Inspired by insights from network analysis, we treated the microbiota with various concentrations of TDA, simulating TDA secretion from some Alphaproteobacteria. The introduction of TDA drastically impacted community structure at a global level. Taken together, this experimental metagenomics study, while not fully characterizing OTU interactions in the microbial community associated with N. salina, provides a valuable framework to aid modeling of interactions among the algal microbiota and understanding the important microbiota—microalgae interactions in the context of the complexity of the studied ecosystem.
Materials and Methods
Algal Cultures and Experimental Design
Artificial microcosms of N. salina (CCMP, 1776) and the microbiota were generated by acclimating N. salina culture together with seawater microbiota from the coast of Santa Cruz, CA (Geng et al., 2016). After 1:10 culture dilution with fresh sterile artificial seawater media (ESAW) (Berges et al., 2001), algal cultures were grown to exponential phase. For the community restructuring experiments, triplicates were generated by splitting the algal cultures into 8 ml/well in 6-well Corning Costar® cell culture plates. The aliquoted exponential phase N. salina cultures (day 4 post-inoculation) were immediately spiked with two separate doses of sterile-filtered organic compounds (dosage were chosen based on the typical working concentrations for each chemical and a reference concentration (500 nM) for all chemicals; Table 1): (i) mixtures of common forms of bacterial quorum-sensing signaling molecules (Atkinson and Williams, 2009), including acyl-homoserine lactones (AHLs) [Sigma Aldrich (St. Louis, MO)] composed of N-butyryl-DL-homoserine lactone, N-hexanoyl-DL-homoserine lactone, N-octanoyl-DL-homoserine lactone, N-(β-Ketocaproyl)-L-homoserine lactone [31 nM (Wagner-Dobler et al., 2005) and 500 nM]; (ii) tetracycline (500 nM and 104 μM; Sambrook et al., 1989); (iii) ampicillin (500 nM and 134 μM; Sambrook et al., 1989); (iv) tropodithietic acid (TDA), an antibacterial and chemical signaling compound (31 nM and 500 nM; D'Alvise et al., 2012; Enzo Scientific, Farmingdale, NY); (v) D-glucose (500 nM and 300 μM) [Sigma Aldrich (St. Louis, MO)]. Low doses of D-glucose (500 nM) were treated as negative controls, as the effect of glucose on the bacterial community has been found to be minimal compared to antibiotic treatment (Dandekar et al., 2012). The incubation continued at 21°C under constant light conditions (100 μmol photons m−2s−1). An initial 4 ml was taken from each sample after 3 h and centrifuged 10,000 g for 5 min to pellet the bacterial community. The remaining 4 ml in each well were harvested after 24 h. Bacterial community pellets were stored at −80°C prior to DNA extraction.
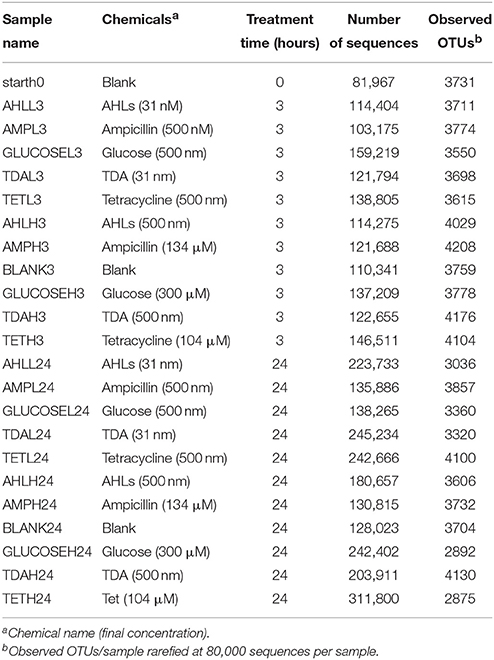
Table 1. Chemical treatments in algal microcosms.
Samples and 16S rRNA Gene Sequencing
Genomic DNA was extracted from algal culture samples with associated microbiota using a ZR Fungal/Bacterial DNA MiniPrep (ZYMO Research, Irvine, CA) following the manufacturer's protocol. 16S gene PCR preparation with standard procedures with barcoded primer set 341F forward and 518R reverse primer as previously described (Bartram et al., 2011). The triplicate PCR products from each sample were pooled and purified using QIAquick PCR purification kit (Qiagen, Valencia, CA) followed by quantification on a Nanodrop ND spectrophotometer (Thermo Science, Wilmington, DE). Twenty-three samples in equal amount with unique index sequence were mixed and further subjected to 2% gel purification using QIAquick gel extraction kit (Qiagen) following by quantification with a Bioanalyser DNA 7500 chip (Agilent Technologies, Santa Clara, CA). The prepared 16S rRNA gene library with addition of 30% PhiX was sequenced for 151-nucleotide paired-end multiplex sequence on MiSeq (Illumina, Hayward, CA) with a loading concentration of 8 pM following manufacturer's protocol.
Sequence Processing
Sequence reads were filtered to remove sequences of poor quality (e.g., two or more continuous base calls below 30 and length <75 bases). Forward and reverse sequences of paired-end sequences were assembled by aligning 3′ ends using SHE-RA software (Rodrigue et al., 2010). Stitched sequences were then clustered using the UCLUST algorithm with 97% similarity and assigned to operational taxonomic units (OTUs) above a 0.80 confidence threshold, which is a homology cutoff value above which typically denotes bacterial species, and taxonomic classifications were assigned in reference to Greengenes taxonomy (RDP-Classifier) in QIIME (DeSantis et al., 2006; Caporaso et al., 2010; Sul et al., 2011). OTUs ascribed to chloroplasts were then excluded from the pre-trimmed OTU table. Differences between samples (beta diversities) were performed using QIIME rarefied at depth 80,000 sequences/sample in post-trimmed OTUs table (DeSantis et al., 2006; Caporaso et al., 2010). To obtain relative abundances of OTUs per sample, the post-trimmed OTU reads were divided by the sum of usable reads. Principal coordinate analysis (PCoA) was applied based on their between-samples weighted UniFrac distances metrics. Jackknifing was performed by resampling 1000 times with replacement of 50,000 sequences per sample in post-trimmed OTUs table and was used to build a rooted pairwise similarity tree using the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) in QIIME (Caporaso et al., 2010)
Correlation Network Analysis
To remove poorly represented OTUs and reduce network complexity, we filtered and retained OTUs that were observed in a minimum of 13 out of total 23 samples, resulting in 1766 OTUs out of 27,298 OTUs being selected. Based on this, we calculated all pairwise Pearson correlations between OTUs using the WGCNA module in R (Team, 2015). Rather than focusing on the significance of the correlation, soft thresholding power was applied in WGCNA to dynamically prune branches off the dendrogram depending on clusters shape (DiLeo et al., 2011). To minimize spurious associations during module identification, the adjacency matrix was transformed to a Topological Overlap Matrix, corresponding dissimilarity was then calculated as a robust measure of interconnectedness in a hierarchical cluster analysis (DiLeo et al., 2011). Average linkage hierarchical cluster analysis was used to construct the corresponding dendrogram (DiLeo et al., 2011).
Network Centralities
Networks were explored, analyzed and visualized with Cytoscape based on the pairwise correlations (Smoot et al., 2011). Global measurements (e.g., betweenness, closeness, neighborhood connectivity, and topological coefficient) assessing the topology and centrality of the resulting network were calculated using Cytoscape with the NetworkAnalyzer plugin (Doncheva et al., 2012). To relate modules to chemical treatments, we correlated the eigengene for each module with the chemicals and looked for significant associations based on p-values.
Results
Response of the Microbial Community Associated with N. salina to Chemical Perturbations
To generate a collection of datasets representing species relative abundance in response to known perturbations, we added a set of chemicals expected to have pervasive effects on N. salina microbial communities to the culture during exponential growth phase. The spiked chemicals include AHLs mixtures, tetracycline, ampicillin, TDA, and D-glucose in low and high concentrations chosen to maximize the likelihood of microbiota perturbations (Section Materials and Methods). The microbial community associated with N. salina from one untreated and four treated samples were collected at 3 and 24 h post inoculation, along with one starting microbiota (0 h), collectively generating 23 samples (Table 1). A total combined 21,029,577 reads from rRNA 16S gene V3 region of 23 microbiota samples were successfully assigned to individual samples. After filtering out low quality reads and OTU assignments (Section Materials and Methods), we obtained an OTU table of the 23 samples with a mean of 158,931 Seqs/sample (minimum: 81,976; maximum: 311,800; median 137,209 Seqs/sample) (Table 1). We assessed inter-sample variability using PCoA based on unweighted Unifrac metric measures in which the distance represents dissimilarity among community structures (Figure 1) (Lozupone and Knight, 2005). Among all tested microbiota, the initial microbiota (start0), blank control (3 h) and glucose treated samples (3 h) were found to be most similar in composition, thus clustering to what we call the “early” microbiota group (Figure 1, labeled with the red bar). The similarities among these groups reflect glucose and short temporal lags have only marginal effects on the structure of the microbiota. These patterns are expected, as glucose is widely used by many bacteria through the hexose monophosphate pathway in glucose metabolism, thereby having minimal effects in restructuring the microbial assemblage as compared to antibiotics (Dandekar et al., 2012). In contrast, short-term 3-h low concentration antibiotics treated-samples (TETL3 and AMPL3) migrate further away from the starting sample (start0) (Figure 1). This shift is further pronounced when comparing the starting microbiota and the high-dose antibiotics-treated samples (TETH3 and AMPH3) at the same 3-h timepoint (Figure 1). In addition, the short-term low TDA treatment for 3 h (TDAL3) resulted in a microbiota that coincided with “early” group microbiotas, which are most similar to the starting microbiota, whereas the high TDA imposed pronounced effects on the microbiota.
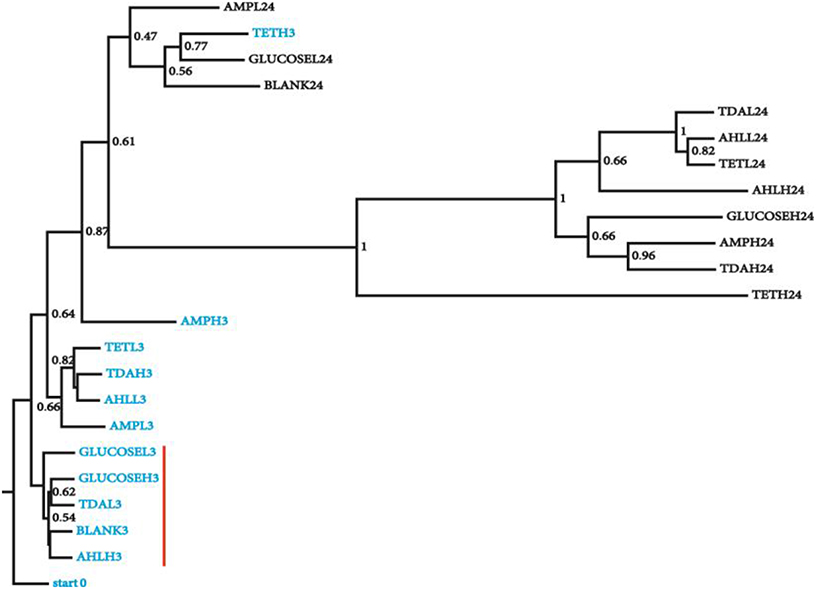
Figure 1. Clustering of microbial diversity (β-diversity) of the starting microbiota with the samples from different chemical treatments at 3 and 24 h. Jackknifing of the UPGMA tree displays the robustness of clustering of the microbiota from the 3 h from 24 h samples. Bootstrap values are shown at the nodes of the tree, indicating percentage of jackknifed data supporting the node. Samples are named as following: chemical name (e.g., GLUCOSE), H/L (high/low concentration, e.g., L) and treatment time (e.g., 3), sample (GLUCOSEL3, treated with low amount of glucose for 3 h). Samples from 3 h (blue) treatment that were most similar to the starting microbiota are highlighted with a red vertical red bar.
The microbiotas from the 3 h time points clustered together, while the 24 h treatment microbiotas formed a separate cluster (Figure 1), revealing groupings in the bacterial community as a function of treatment time. Collectively, 16S gene profiles displayed both culture age-dependent and chemical-responsive structural rearrangement of algal microbiota.
Structural Modulation of Algal Microbiota upon Chemical Treatments
To assess pairwise Pearson correlations among OTU relative-abundance data, we randomly selected 80,000 sequences per sample, built OTU tables, and filtered OTUs that contributed to at least half of 23 microbiota samples. The 1766 qualified OTUs produced from this process were subsequently fitted to a correlation matrix with scale free topology (power of 6, R2 = 0.81) using WGCNA, which has been shown to be robust for analyzing relative abundance data (DiLeo et al., 2011). WGCNA revealed five major co-abundance modules that were arbitrarily given colors yellow, brown, blue, turquoise, and gray (Figure S1). Upon identification of modules in the microbial community associated with N. salina, we associated them with external treatment factors. Figure 2A shows that OTU membership in the yellow module was positively correlated with culture-age associated OTUs (coefficient = 0.54, P < 0.05). Figure 2B illustrates that treatment time was especially important, as three modules were found to be significantly associated with treatment time, whereas the blue (coefficient = −0.80, P < 0.05) and turquoise (coefficient = −0.57, P < 0.05) modules were negatively correlated with treatment time. These data indicate treatment time was a primary factor in contributing to variations in species abundance in these particular studies, which is consistent with beta-diversity analysis using unweighted Unifrac metric measure (Figure 1). Neither the glucose treatment nor the untreated microbiota was significantly associated with any of the five identified modules (P > 0.05). There were also no modules significantly associated with tetracycline under the studied concentrations. Figure 2B shows the brown colored module was selectively correlated with ampicillin treatment (P < 10−7). Taxonomy analysis showed the brown chemotype-specific module was enriched by the Rhodobacteraceae family (13 out of 14, with only 1 coming from the Erythrobacteraceae family, P < 0.05, enrichment analysis), whereas the yellow module was enriched by the Alteromonadaceae family (8 out of 11, P < 0.05, enrichment analysis) (Figure 3). Therefore, the apparent module substructures were associated with the taxonomical coherence of dominant OTUs.
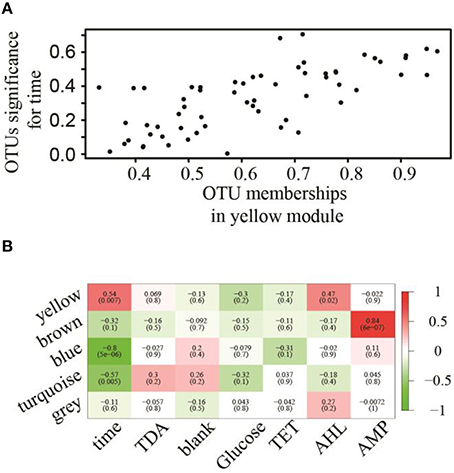
Figure 2. Association of modules with external treatment factors. (A) OTUs associated with treatment–time with respect to their membership significance belonging to the yellow module. (B) Eigengene adjacencies heatmap identifying modules (row) that significantly associated with chemical treatments (column). Negative correlations (green) and positive correlations (red) indicate high adjacency (DiLeo et al., 2011), while white (zero) indicates low adjacency.
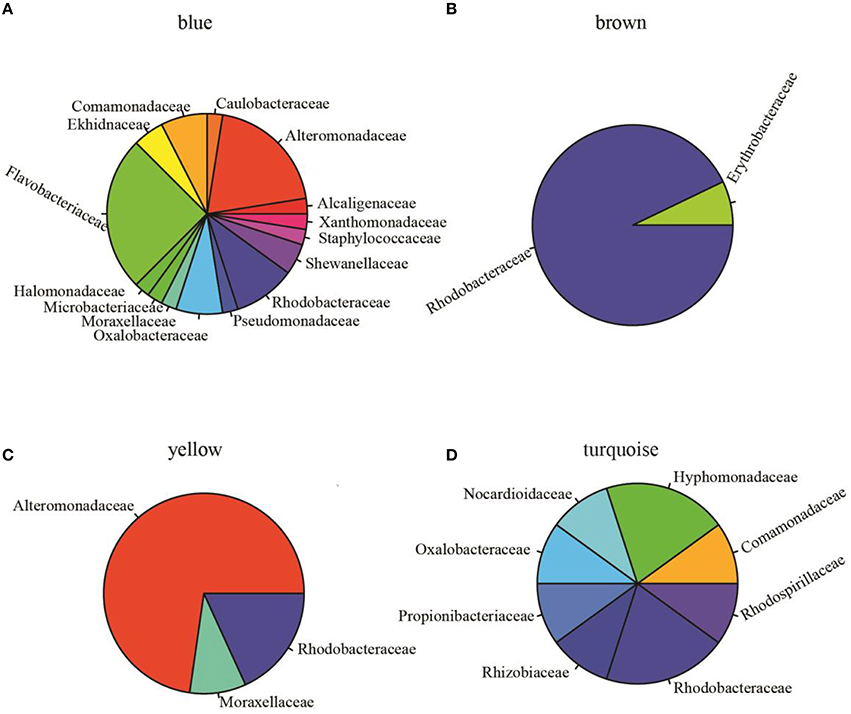
Figure 3. Relative abundance of OTUs at the family level (indicated by different colors) in identified modules. (A) Blue module (total OTUs 40 annotated at family level, 127 OTUs unassigned at family level are not shown) was dominated by Flavobacteriaceae (25%, n = 10) and Alteromonadaceae (20%, n = 8). (B) Brown module (14 OTUs annotated, 63 unassigned) was dominated by Rhodobacteraceae (92%, n = 13). (C) Yellow module (total OTUs 11 annotated at family level) is comprised of Alteromonadaceae (72%, n = 8), Rhodobacteraceae (9.1%, n = 1), and Moraxellaceae (9.1%, n = 1). (D) Turquoise module (10 OTUs assigned to 8 families, 876 unassigned OTUs) is comprised of 8 families. These data showed that Rhodobacteraceae was significantly enriched in the brown module (one-sided Fisher's exact test, P < 0.0001) and Alteromonadaceae was significantly enriched in the yellow module (one-sided Fisher's exact test, P < 0.05).
Network Centralities and Identification of Hubs
To display an empirical base for discovery and to explore specific details of the substructures of the community, associations from the top 1000 OTUs ranked by coefficient were plotted. Figure 4 shows the resulting microbial network consisting of 1000 nodes and 68,675 edges with average clustering network coefficient of 0.856, suggesting the down-selected OTUs constituted a highly connected network. The average number of network neighbors between all pairs of nodes was 137, and the network centralization was 0.768 as determined using NetworkAnalyzer (Doncheva et al., 2012). Hub nodes within respective modules are often proposed to be critical components for the network (Mayali and Azam, 2004). To identify species that may be acting as hubs, and thus be critically important components of the community network structure, we ranked nodes using various network centrality measures, including betweenness, closeness, neighborhood connectivity and topological coefficient (Table 2). Based on these measures, species from the Alteromonas and Rhodobacteraceae families were hypothesized to be key species influencing the inter-OTU relationships.
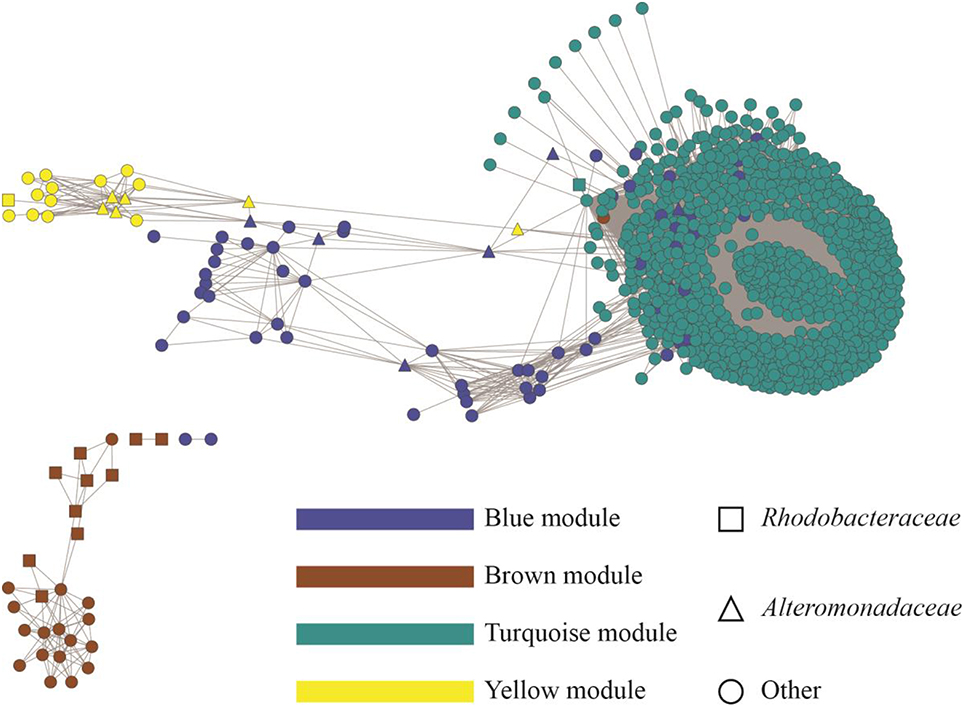
Figure 4. Topology of the microbial community associated with N. salina. The identified modules were colored according to module names (Blue, Brown, Turquoise, and Yellow). The whole network contains 1000 nodes representing bacterial OTUs and 68,675 edges, showing correlations between the OTUs with an edge weight cutoff of 0.20 in the WGCNA network (DiLeo et al., 2011). OTUs were annotated at the family level. Rhodobacteraceae and Alteromonadaceae are shown as squares and triangles, respectively. OTUs from families other than Rhodobacteraceae and Alteromonadaceae are displayed as circle.
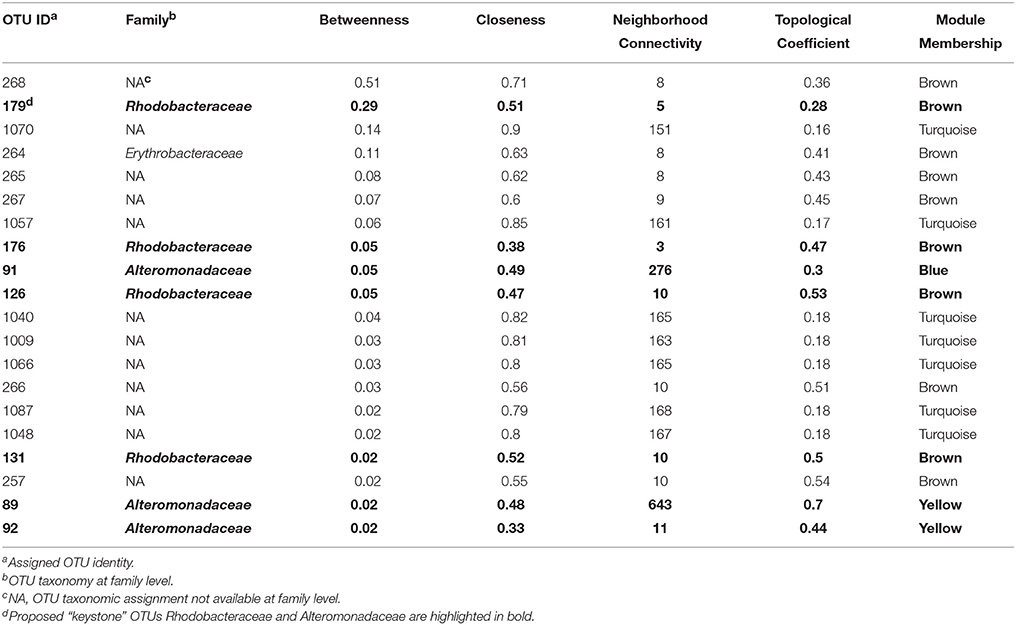
Table 2. Identified “keystone” OTUs in co-abundance networks.
TDA Alter the Structure of the Microbial Community Associated with N. salina
Correlation network analysis led us to hypothesize that bacterial species from Rhodobacteraceae are key species in structuring the microbial community associated with N. salina. To gather evidence for this hypothesis, we treated N. salina containing microbial communities with varying concentrations of TDA, a hallmark antibiotic in many species of the Roseobacteria clade in Rhodobacteraceae, and sampled the microbiota at 3 and 24 h time periods. Taxonomic distributions at the family-level across TDA-perturbed samples, grouped by exposure time, are illustrated in Figure 5. The largest taxa in all microbiota were populations of Flavobacteriia, Alphaproteobacteria, and Gammaproteobacteria bacterial classes. Figure 5 also shows that the 24 h microbiota decreased in relative abundance of Flavobacteriia (23 ± 4%) and Alphaproteobacteria (14 ± 4%), comparted to 3 h (excluding TDA, 500 nM, 3 h) Flavobacteriia (39 ± 2%) and Alphaproteobacteria (32 ± 2%). Meanwhile, Gammaproteobacteria relative abundance at 24 h increased to 60 ± 9% compared to 3 h (excluding TDA, 500 nM, 3 h) Gammaproteobacteria (25 ± 9%). Of particular note, the microbiota (TDA, 500 nM, 3 h) was composed of Flavobacteriia (31 ± 1%), Alphaproteobacteria (21 ± 0%), and Gammaproteobacteria (45 ± 2%), suggesting it exhibits some compositional similarities to 24 h microbiota (Figure 5). PCoA of the data revealed patterns of beta-diversity (unweighted UniFrac distance metric) primarily forming two clusters differentiated by the time point at which the microbiota was sampled (Figure 6). Specifically, without TDA treatment, the microbiota at the 3-h time point were similar in structure to the starting microbiota (Unifrac distance, 0.03 ± 0.01). The addition of low doses of TDA (31.25 nM) slightly shifted the microbial composition over the 3-h time period (Unifrac distance, 0.05 ± 0.02). In contrast, treatment with higher TDA doses (500 nM) markedly shifted the composition away from the initial microbiota (Unifrac, 0.14 ± 0.02), and at the 3-h time point clustered with the samples obtained after 24 h. Hence, the data imply that microbiota exposed to 500 nM TDA will not only drastically restructure (as compared to the unperturbed community), but also hasten the progression of microbial community structure in a dose-dependent manner toward the more mature microbiota.
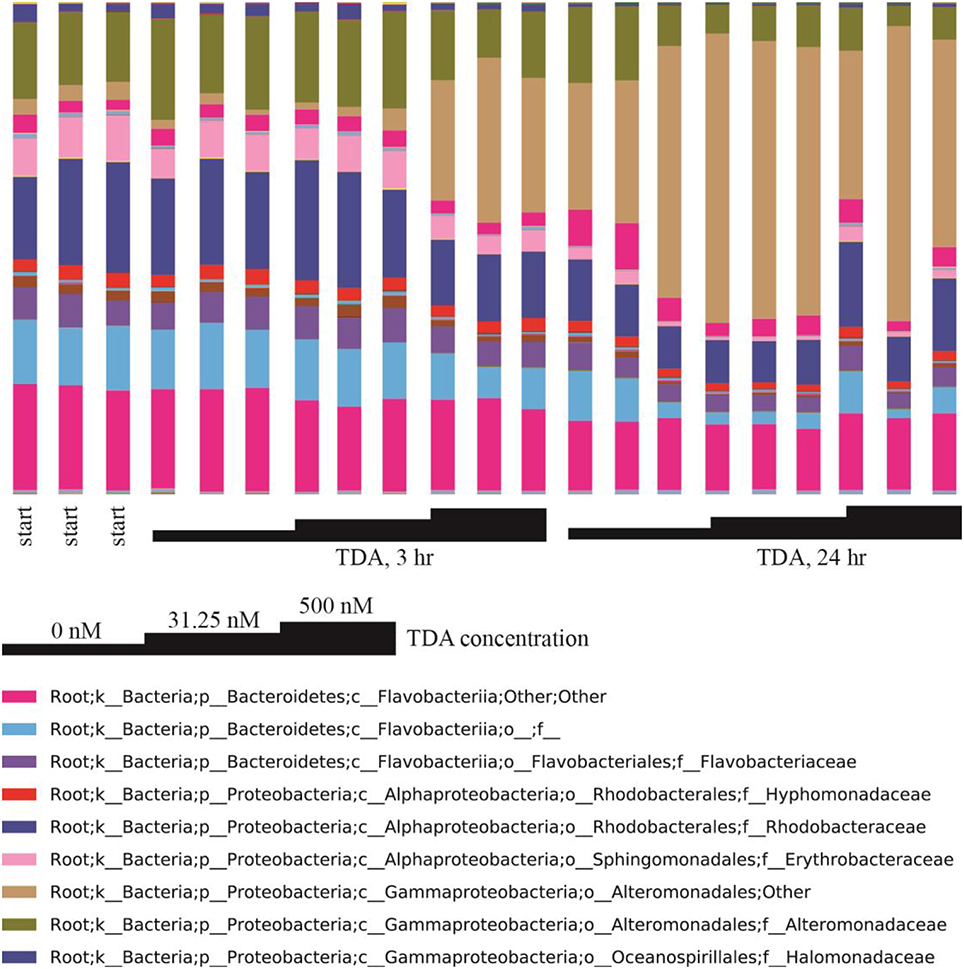
Figure 5. Family-level taxonomic composition of TDA-perturbed microbiota. Triplicate samples are grouped by TDA concentration and exposure time. Taxonomic affiliations of OTUs are shown at the family level (see legend colors) and TDA treatment concentrations are indicated.
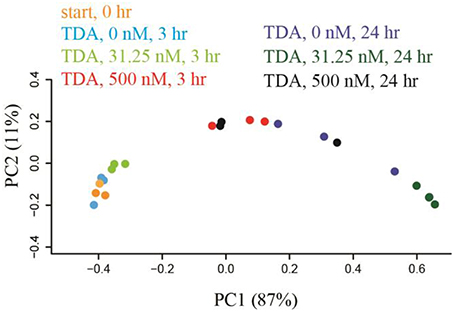
Figure 6. Microbiota were shifted by TDA addition in a dose-dependent manner within 3 h treatment based on weight-Unifrac PCoA with respect to treatment time and TDA concentration. TDA (500 nM)-treated microbiota shifted the microbiota structure away from 3 h-microbiota cluster toward the 24 h-group.
Discussion
Among the efforts to understand the complexity of microalgae microbiota, this current study is unique in that it aimed to chart multispecies relationships underpinning the substructure of microalgae microbiota. To achieve this aim, we experimentally perturbed a multispecies algal microbiota with various chemical treatments, quantified the changes to taxa profiles, and applied network analysis for modeling of interspecies relationship which provided a cohesive overview of the microbial community associated with N. salina structure.
It is well established that antibiotics can cause major disturbances within the ecological balance of microbiomes (Mayali and Azam, 2004). Thus, we postulated that antibiotic treatment provides an inferable means to introduce variations among bacterial species due to differential antibiotic susceptibility among them. Indeed, chemical treatments introduced major variations among the microbial communities within the same treatment time. High doses of antibiotic treatment noticeably shifted the overall microbiota structure. Thus, the generated 16S gene metagenomics profiles generally reflected microbiota changes in response to chemical perturbations.
These observations highlight two strengths of correlation network approach. First, rather than listing pair-wise correlations, all correlations were integrated into a unified network of interacting species, making it possible to identify small substructures within the microbiota. While this model is generated only by first-order correlations among species, it presents rich, albeit indirect, information on microbiota community structure, allowing us to explore and test species interactions and prioritize hypotheses. Specifically, the bacterial OTU variations resulting from a set of time-series chemical treatments allowed us to identify five modules emerging out of the complex N. salina microbial community. We demonstrated that members of Rhodobacteraceae dominated the brown OTUs module, which was significantly associated with ampicillin treatment (Figure 3). Reports have shown that species of the Roseobacter clade encode intrinsic β-lactamases and are resistant to β-lactam antibiotics such as ampicillin, with tolerance up to 500 μg/ml (Peng et al., 2011; Treangen et al., 2013). This is 10-fold greater than our high dose ampicillin treatment of 50 μg/ml. Meanwhile, bacteriostatic tetracycline, which inhibits bacteria from reproducing (Peng et al., 2011), surprisingly did not result in the formation of modules. Lack of module formation from tetracycline treatment in our experiments can be explained by the possibility of tetracycline activities being compromised by the formation of complexes with divalent cations (such as Mg2+) prevalent in F/2 medium (Lambs et al., 1988; Treangen et al., 2013). In comparison, general metabolites such as glucose did not perturb the network significantly as evidenced by the absence of modules emerging from glucose-supplemented cultures. Overall, these results reflect that an OTU co-abundance network generated correlated associations from empirical data that recapitulates biological information, and therefore has value for further inference analysis.
While correlation analyses do not offer direct mechanistic interpretations, the formation of modules might stem from, for example, between-bacteria metabolic cross-feedings, biofilm formation of aggregated sub-communities, species couplings via chemical signaling or toxic compounds, which could result in bacterial species-species abundance correlations in 16S gene profile data sets. With our data sets, it appears the correlation network has applicability in mining subsets of microbial lineages that share common responsiveness to acute perturbation such as antibiotic treatment and is perhaps less applicable to those stemming from treatments (such as with ubiquitous metabolites) that do not significantly perturb bacterial abundance. Nevertheless, the integration of correlations into interconnected networks may be applied in bridging from bacterial species' activity to microbial community functionality.
Network centrality analysis has been successfully applied in multiple fields (Jeong et al., 2001; Rho et al., 2010; Duran-Pinedo et al., 2011), and we used it to mine influential bacterial species underpinning the interaction network of the microbial community associated with N. salina. The top Rhodobacteraceae OTU has low connectivity, suggesting it has few interaction with other OTUs in the network. Meanwhile, it has higher betweenness values compared to other OTUs. This pattern suggests modular organization of the network via a minimal number of connections required for information flow. These high-betweenness, low-connectivity OTUs may be conceptually thought of as “bridges” in the non-redundant shortest paths, the observation of which resembles previously reported properties of other biological networks (Joy et al., 2005). In comparison, the high-connectivity, low-betweenness case of Alteromonadacea suggests it lies on a large number of redundant shortest paths between other vertices. The difference between the network inferences of Rhodobacteraceae and Alteromonadacea is likely linked to distinct biological and ecological roles of these two groups. Rhodobacteraceae, which contains the Roseobacter clade, are one of the most ubiquitous and abundant bacterial lineages associated with phytoplankton both in environmental and laboratory cultures (Geng and Belas, 2010b; Amin et al., 2012) and has been shown to form close relationships with microalgae (Treangen et al., 2013; Kelder et al., 2014). The interactions among members of the Roseobacter clade (Alphaproteobacteria) and microalgae may occur through physical attachment (Miller and Belas, 2006; Mayali et al., 2008; Krohn-Molt et al., 2013), exchange of nutrients and metabolites (Keshtacher-Liebso et al., 1995; Howard et al., 2006), or antibiotics and signaling molecules (Brinkhoff et al., 2004; Seyedsayamdost et al., 2011; Amin et al., 2015). On the other hand, the marine bacteria from Alteromonadaceae are dominant phylotypes among microbial communities in response to the flux of organic matter in phytoplankton blooms, demonstrating how species of Alteromonadaceae could participate in structuring microalgae bacterial communities (McCarren et al., 2010; Tada et al., 2011).
To explore the possibility of the key species having influential effects on the overall configuration of the microbiota, we looked for evidence of microbiota responsiveness to the proposed key species. We subjected the N. salina microbial communities to varying doses of TDA, which is secreted as a hallmark bioactive compound in many species of the Roseobacter clade from Rhodobacteraceae family (Berger et al., 2011). TDA has both antibiotic and transcription induction activity in a subgroup of algae-associated bacterial genera that include Phaeobacter, Silicibacter, Ruegeria, and Pseudovibrio (Berger et al., 2011). Interestingly, we found that in general the relative abundance of Rhodobacteraceae decreased, while bacteria in Alteromonadales order of unclassified family increased in composition (Figure 5). These observed patterns between TDA and taxa composition are less clear since TDA activity to Alteromonadales has not been documented. Meanwhile the large total number of bacterial populations and differential bacterial growth rates in microbiota might have compounded the observed relative abundance. In terms of community structure, we observed shifting of microalgae microbiota induced by TDA dosage, and a 3 h 500 nM TDA treatment sufficiently skewed the original microbiota structure toward the 24 h unperturbed microbiota. This suggests that, given the myriad complexity in microbiota, microbial community associated with N. salina potentially harbors a dynamic property that responds to the presence of infochemicals exemplified by TDA. While the concentration of TDA produced naturally in either sea water or within the phycosphere is not documented, the involvement of bioactive compounds such as TDA in modulating microbiota structure has profound implications for bacterial community assemblages. Indeed, multiple bioactive compounds with various activities and specificities, such as indole-3-acetic acid (Amin et al., 2015) and indigoidine (Cude et al., 2012), have been characterized in many Roseobacter strains from microalgae microbiota (Buchan et al., 2005; Cude et al., 2012; Leiman et al., 2013; Treangen et al., 2013). In this context, the chemical composition and quantities of bioactive compounds from key species in microbiota may play a role in fine tuning of microbiota to different microbiota compositions and structures.
Our finding from network analysis therefore offers a tangible experimental metagenomics framework to tackle the following question: what is the structure of microbial community associated with N. salina if modeled from the species-species correlation network? Module formation in the microbiota network helps bridge the gap between deep knowledge of individual keystone OTUs and a systems-level view of the microbial community. But such application will have to tackle several issues. Given the connectivity of the correlation network, the correlation network model does not provide detailed mechanisms nor offer regulatory ramification of such variations. While accurate inter-OTU correlation analysis remains an area of active research, methods developed recently such as CoNet (Faust et al., 2012), SparCC (Friedman and Alm, 2012), SPIEC-EASI (Kurtz et al., 2015), and MENA (Deng et al., 2012) could potentially be used to analyze inter-OTUs relationships in microalgae microbiota to improve association accuracy. Meanwhile, many of the remaining OTUs in modules have limited functional annotations due to the nature of 16S amplicon method. Such limitation, however, could be solved using metagenomics combined with metatranscriptomics to better understand microbiota roles in the ecosystem. In the long term, delineation of the substructure of the microbial community associated with N. salina may help us detect, investigate, and assess practical responsiveness of “key” species or probiotic species introduced into the microbiome to withstand environmental perturbations and improve microalgae production.
Author Contributions
KS, TL, EY designed experiments. HG, EY, MT performed experiments. HG, KS analyzed and modeled data. HG, TD, EY, KS wrote manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Laboratory Directed Research and Development Program at Sandia National Laboratories, which is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the US Department of Energy's National Nuclear Security Administration under Contract DE-AC04-94AL85000. Additional funding was provided by the U.S. Department of Energy (DOE) Genomic Science Program under contract SCW1039.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01155
Figure S1. Modules of OTUs in chemical treated microbiotas network. Hierarchical clustering dendrogram of OTUs was built based on dissimilarity based on nodes topological overlap. Modules have been colored by module membership.
References
Alavi, M., Miller, T., Erlandson, K., Schneider, R., and Belas, R. (2001). Bacterial community associated with Pfiesteria-like dinoflagellate cultures. Environ. Microbiol. 3, 380–396. doi: 10.1046/j.1462-2920.2001.00207.x
Amin, S. A., Hmelo, L. R., Van Tol, H. M., Durham, B. P., Carlson, L. T., Heal, K. R., et al. (2015). Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 522, 98–101. doi: 10.1038/nature14488
Amin, S. A., Parker, M. S., and Armbrust, E. V. (2012). Interactions between diatoms and bacteria. Microbiol. Mol. Biol. Rev. 76, 667–684. doi: 10.1128/MMBR.00007-12
Atkinson, S., and Williams, P. (2009). Quorum sensing and social networking in the microbial world. J. R. Soc. Interface 6, 959–978. doi: 10.1098/rsif.2009.0203
Bartram, A. K., Lynch, M. D. J., Stearns, J. C., Moreno-Hagelsieb, G., and Neufeld, J. D. (2011). Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl. Environ. Microbiol. 77, 3846–3852. doi: 10.1128/AEM.02772-10
Berger, M., Neumann, A., Schulz, S., Simon, M., and Brinkhoff, T. (2011). Tropodithietic acid production in Phaeobacter gallaeciensis is regulated by N-acyl homoserine lactone-mediated quorum sensing. J. Bacteriol. 193, 6576–6585. doi: 10.1128/JB.05818-11
Berges, J. A., Franklin, D. J., and Harrison, P. J. (2001). Evolution of an artificial seawater medium: improvements in enriched seawater, artificial water over the last two decades. J. Phycol. 37, 1138–1145. doi: 10.1046/j.1529-8817.2001.01052.x
Brinkhoff, T., Bach, G., Heidorn, T., Liang, L., Schlingloff, A., and Simon, M. (2004). Antibiotic production by a Roseobacter clade-affiliated species from the German Wadden Sea and its antagonistic effects on indigenous isolates. Appl. Environ. Microbiol. 70, 2560–2565. doi: 10.1128/AEM.70.4.2560-2565.2003
Bruhn, J. B., Gram, L., and Belas, R. (2007). Production of antibacterial compounds and biofilm formation by Roseobacter species are influenced by culture conditions. Appl. Environ. Microbiol. 73, 442–450. doi: 10.1128/AEM.02238-06
Buchan, A., Gonzalez, J. M., and Moran, M. A. (2005). Overview of the marine Roseobacter lineage. Appl. Environ. Microbiol. 71, 5665–5677. doi: 10.1128/AEM.71.10.5665-5677.2005
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Carney, L. T., Reinsch, S. S., Lane, P. D., Solberg, O. D., Jansen, L. S., Williams, K. P., et al. (2014). Microbiome analysis of a microalgal mass culture growing in municipal wastewater in a prototype OMEGA photobioreactor. Algal Res. 4, 52–61. doi: 10.1016/j.algal.2013.11.006
Choi, J. K., Yu, U., Yoo, O. J., and Kim, S. (2005). Differential coexpression analysis using microarray data and its application to human cancer. Bioinformatics 21, 4348–4355. doi: 10.1093/bioinformatics/bti722
Cole, J. J. (1982). Interactions between bacteria and algae in aquatic ecosystems. Annu. Rev. Ecol. Syst. 13, 291–314. doi: 10.1146/annurev.es.13.110182.001451
Costello, E. K., Lauber, C. L., Hamady, M., Fierer, N., Gordon, J. I., and Knight, R. (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. doi: 10.1126/science.1177486
Cude, W. N., Mooney, J., Tavanaei, A. A., Hadden, M. K., Frank, A. M., Gulvik, C. A., et al. (2012). Production of the antimicrobial secondary metabolite indigoidine contributes to competitive surface colonization by the marine roseobacter Phaeobacter sp. Strain Y4I. Appl. Environ. Microbiol. 78, 4771–4780. doi: 10.1128/AEM.00297-12
D'Alvise, P. W., Lillebo, S., Prol-Garcia, M. J., Wergeland, H. I., Nielsen, K. F., Bergh, O., et al. (2012). Phaeobacter gallaeciensis reduces Vibrio anguillarum in cultures of microalgae and rotifers, and prevents vibriosis in cod larvae. PLoS ONE 7:e43996. doi: 10.1371/journal.pone.0043996
Dandekar, A. A., Chugani, S., and Greenberg, E. P. (2012). Bacterial quorum sensing and metabolic incentives to cooperate. Science 338, 264–266. doi: 10.1126/science.1227289
Deng, Y., Jiang, Y. H., Yang, Y., He, Z., Luo, F., and Zhou, J. (2012). Molecular ecological network analyses. BMC Bioinformatics 13:113. doi: 10.1186/1471-2105-13-113
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
DiLeo, M. V., Strahan, G. D., Den Bakker, M., and Hoekenga, O. A. (2011). Weighted correlation network analysis (WGCNA) applied to the tomato fruit metabolome. PLoS ONE 6:e26683. doi: 10.1371/journal.pone.0026683
Doncheva, N. T., Assenov, Y., Domingues, F. S., and Albrecht, M. (2012). Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc. 7, 670–685. doi: 10.1038/nprot.2012.004
Duran-Pinedo, A. E., Paster, B., Teles, R., and Frias-Lopez, J. (2011). Correlation network analysis applied to complex biofilm communities. PLoS ONE 6:e28438. doi: 10.1371/journal.pone.0028438
Faust, K., Sathirapongsasuti, J. F., Izard, J., Segata, N., Gevers, D., Raes, J., et al. (2012). Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 8:e1002606. doi: 10.1371/journal.pcbi.1002606
Friedman, J., and Alm, E. J. (2012). Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8:e1002687. doi: 10.1371/journal.pcbi.1002687
Ge, H., Liu, Z., Church, G. M., and Vidal, M. (2001). Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat. Genet. 29, 482–486. doi: 10.1038/ng776
Geng, H., and Belas, R. (2010a). Expression of tropodithietic acid biosynthesis is controlled by a novel autoinducer. J. Bacteriol. 192, 4377–4387. doi: 10.1128/JB.00410-10
Geng, H., and Belas, R. (2010b). Molecular mechanisms underlying roseobacter-phytoplankton symbioses. Curr. Opin. Biotechnol. 21, 332–338. doi: 10.1016/j.copbio.2010.03.013
Geng, H., Sale, K. L., Tran-Gyamfi, M. B., Lane, T. W., and Yu, E. T. (2016). Longitudinal analysis of microbiota in microalga Nannochloropsis salina cultures. Microb. Ecol. 72, 14–24. doi: 10.1007/s00248-016-0746-4
Gilbert, J. A., Steele, J. A., Caporaso, J. G., Steinbruck, L., Reeder, J., Temperton, B., et al. (2012). Defining seasonal marine microbial community dynamics. ISME J. 6, 298–308. doi: 10.1038/ismej.2011.107
Gonzalez, A., King, A., Robeson Ii, M. S., Song, S., Shade, A., Metcalf, J. L., et al. (2012). Characterizing microbial communities through space and time. Curr. Opin. Biotechnol. 23, 431–436. doi: 10.1016/j.copbio.2011.11.017
Gonzalez, J. M., and Moran, M. A. (1997). Numerical dominance of a group of marine bacteria in the alpha-subclass of the class Proteobacteria in coastal seawater. Appl. Environ. Microbiol. 63, 4237–4242.
Hold, G. L., Smith, E. A., Birkbeck, T. H., and Gallacher, S. (2001). Comparison of paralytic shellfish toxin (PST) production by the dinoflagellates Alexandrium lusitanicum NEPCC 253 and Alexandrium tamarense NEPCC 407 in the presence and absence of bacteria. FEMS Microbiol. Ecol. 36, 223–234. doi: 10.1111/j.1574-6941.2001.tb00843.x
Howard, E. C., Henriksen, J. R., Buchan, A., Reisch, C. R., Bürgmann, H., Welsh, R., et al. (2006). Bacterial taxa that limit sulfur flux from the Ocean. Science 314, 649–652. doi: 10.1126/science.1130657
Jeong, H., Mason, S. P., Barabasi, A. L., and Oltvai, Z. N. (2001). Lethality and centrality in protein networks. Nature 411, 41–42. doi: 10.1038/35075138
Joy, M. P., Brock, A., Ingber, D. E., and Huang, S. (2005). High-betweenness proteins in the yeast protein interaction network. J. Biomed. Biotechnol. 2005, 96–103. doi: 10.1155/JBB.2005.96
Kayser, H. (1979). Growth interactions between marine dinoflagellates in multispecies culture experiments. Mar. Biol. 52, 357–369. doi: 10.1007/BF00389077
Kelder, T., Stroeve, J. H. M., Bijlsma, S., Radonjic, M., and Roeselers, G. (2014). Correlation network analysis reveals relationships between diet-induced changes in human gut microbiota and metabolic health. Nutr. Diabetes 4, e122. doi: 10.1038/nutd.2014.18
Keshtacher-Liebso, E., Hadar, Y., and Chen, Y. (1995). Oligotrophic bacteria enhance algal growth under iron-deficient conditions. Appl. Environ. Microbiol. 61, 2439–2441.
Krohn-Molt, I., Wemheuer, B., Alawi, M., Poehlein, A., Gullert, S., Schmeisser, C., et al. (2013). Metagenome survey of a multispecies and alga-associated biofilm revealed key elements of bacterial-algal interactions in photobioreactors. Appl. Environ. Microbiol. 79, 6196–6206. doi: 10.1128/AEM.01641-13
Kurtz, Z. D., Muller, C. L., Miraldi, E. R., Littman, D. R., Blaser, M. J., and Bonneau, R. A. (2015). Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput. Biol. 11:e1004226. doi: 10.1371/journal.pcbi.1004226
Lambs, L., Venturini, M., Decock-Le Reverend, B., Kozlowski, H., and Berthon, G. (1988). Metal ion-tetracycline interactions in biological fluids. Part 8. Potentiometric and spectroscopic studies on the formation of Ca(II) and Mg(II) complexes with 4-dedimethylamino-tetracycline and 6-desoxy-6-demethyl-tetracycline. J. Inorg. Biochem. 33, 193–210. doi: 10.1016/0162-0134(88)80049-7
Lee, S.-O., Kato, J., Takiguchi, N., Kuroda, A., Ikeda, T., Mitsutani, A., et al. (2000). Involvement of an extracellular protease in algicidal activity of the marine bacterium Pseudoalteromonas sp. strain A28. Appl. Environ. Microbiol. 66, 4334–4339. doi: 10.1128/AEM.66.10.4334-4339.2000
Leiman, S. A., May, J. M., Lebar, M. D., Kahne, D., Kolter, R., and Losick, R. (2013). D-amino acids indirectly inhibit biofilm formation in Bacillus subtilis by interfering with protein synthesis. J. Bacteriol. 195, 5391–5395. doi: 10.1128/JB.00975-13
Lovejoy, C., Bowman, J. P., and Hallegraeff, G. M. (1998). Algicidal effects of a novel marine pseudoalteromonas isolate (class Proteobacteria, gamma subdivision) on harmful algal bloom species of the genera Chattonella, Gymnodinium, and Heterosigma. Appl. Environ. Microbiol. 64, 2806–2813.
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Mayali, X., and Azam, F. (2004). Algicidal bacteria in the Sea and their impact on algal blooms1. J. Eukaryot. Microbiol. 51, 139–144. doi: 10.1111/j.1550-7408.2004.tb00538.x
Mayali, X., Franks, P. J. S., and Azam, F. (2008). Cultivation and ecosystem role of a marine roseobacter clade-affiliated cluster bacterium. Appl. Environ. Microbiol. 74, 2595–2603. doi: 10.1128/AEM.02191-07
McCarren, J., Becker, J. W., Repeta, D. J., Shi, Y., Young, C. R., Malmstrom, R. R., et al. (2010). Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc. Natl. Acad. Sci. U.S.A. 107, 16420–16427. doi: 10.1073/pnas.1010732107
Miller, T. R., and Belas, R. (2006). Motility is involved in Silicibacter sp. TM1040 interaction with dinoflagellates. Environ. Microbiol. 8, 1648–1659. doi: 10.1111/j.1462-2920.2006.01071.x
Peng, Y., Leung, H. C., Yiu, S. M., and Chin, F. Y. (2011). Meta-IDBA: a de novo assembler for metagenomic data. Bioinformatics 27, i94–i101. doi: 10.1093/bioinformatics/btr216
Porsby, C. H., Webber, M. A., Nielsen, K. F., Piddock, L. J., and Gram, L. (2011). Resistance and tolerance to tropodithietic acid, an antimicrobial in aquaculture, is hard to select. Antimicrob. Agents Chemother. 55, 1332–1337. doi: 10.1128/AAC.01222-10
Raes, J., and Bork, P. (2008). Molecular eco-systems biology: towards an understanding of community function. Nat. Rev. Microbiol. 6, 693–699. doi: 10.1038/nrmicro1935
Rho, M., Tang, H., and Ye, Y. (2010). FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38, e191. doi: 10.1093/nar/gkq747
Rodrigue, S., Materna, A. C., Timberlake, S. C., Blackburn, M. C., Malmstrom, R. R., Alm, E. J., et al. (2010). Unlocking short read sequencing for metagenomics. PLoS ONE 5:e11840. doi: 10.1371/journal.pone.0011840
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Seyedsayamdost, M. R., Case, R. J., Kolter, R., and Clardy, J. (2011). The Jekyll-and-Hyde chemistry of Phaeobacter gallaeciensis. Nat. Chem. 3, 331–335. doi: 10.1038/nchem.1002
Sison-Mangus, M. P., Jiang, S., Tran, K. N., and Kudela, R. M. (2014). Host-specific adaptation governs the interaction of the marine diatom, Pseudo-nitzschia and their microbiota. ISME J. 8, 63–76. doi: 10.1038/ismej.2013.138
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L., and Ideker, T. (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432. doi: 10.1093/bioinformatics/btq675
Sul, W. J., Cole, J. R., Jesus Eda, C., Wang, Q., Farris, R. J., Fish, J. A., et al. (2011). Bacterial community comparisons by taxonomy-supervised analysis independent of sequence alignment and clustering. Proc. Natl. Acad. Sci. U.S.A. 108, 14637–14642. doi: 10.1073/pnas.1111435108
Tada, Y., Taniguchi, A., Nagao, I., Miki, T., Uematsu, M., Tsuda, A., et al. (2011). Differing growth responses of major phylogenetic groups of marine bacteria to natural phytoplankton blooms in the western North Pacific Ocean. Appl. Environ. Microbiol. 77, 4055–4065. doi: 10.1128/AEM.02952-10
Team, R. C. (2015). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Treangen, T. J., Koren, S., Sommer, D. D., Liu, B., Astrovskaya, I., Ondov, B., et al. (2013). MetAMOS: a modular and open source metagenomic assembly and analysis pipeline. Genome Biol. 14:R2. doi: 10.1186/gb-2013-14-1-r2
Wagner-Dobler, I., Thiel, V., Eberl, L., Allgaier, M., Bodor, A., Meyer, S., et al. (2005). Discovery of complex mixtures of novel long-chain quorum sensing signals in free-living and host-associated marine alphaproteobacteria. Chembiochem 6, 2195–2206. doi: 10.1002/cbic.200500189
Keywords: correlation network, microbiota, algae
Citation: Geng H, Tran-Gyamfi MB, Lane TW, Sale KL and Yu ET (2016) Changes in the Structure of the Microbial Community Associated with Nannochloropsis salina following Treatments with Antibiotics and Bioactive Compounds. Front. Microbiol. 7:1155. doi: 10.3389/fmicb.2016.01155
Received: 24 December 2015; Accepted: 11 July 2016;
Published: 26 July 2016.
Edited by:
Xavier Mayali, Lawrence Livermore National Laboratory, USAReviewed by:
Russell T. Hill, University of Maryland Center for Environmental Science, USAChristopher S. Ward, Duke University, USA
Copyright © 2016 Geng, Tran-Gyamfi, Lane, Sale and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenneth L. Sale, klsale@sandia.gov