 Maozhang He
Maozhang He Shaoming Fang
Shaoming Fang Xiaochang HuangYuanzhang ZhaoShanlin Ke
Xiaochang HuangYuanzhang ZhaoShanlin Ke Hui YangZhuojun LiJun Gao
Hui YangZhuojun LiJun Gao Congying Chen*Lusheng Huang
Congying Chen*Lusheng Huang- State Key Laboratory of Pig Genetic Improvement and Production Technology, Jiangxi Agricultural University, Nanchang, China
Microbial community in gastrointestinal tract participates in the development of the obesity as well as quite a few metabolic diseases in human. However, there are few studies about the relationship between gut microbiota and porcine fatness. Here, we used high-throughput sequencing to perform 16S rRNA gene analysis in 256 cecum luminal samples from Erhualian pigs and 244 stools from Bamaxiang pigs, and adopted a two-part model statistical method to evaluate the association of gut microbes with porcine fatness. As the results, we identified a total of 6 and 108 operational taxonomic units (OTUs), and 9 and 10 bacterial taxa which showed significant associations with fatness traits in the stool and cecum samples, respectively. Cross-validation analysis indicated that gut microbiome showed the largest effect on abdominal adipose by explaining 2.73% phenotypic variance of abdominal fat weight. Significantly more fatness-associated OTUs were identified in the cecum samples than that in the stools, suggesting that cecum luminal samples were better used for identification of fatness-associated microbes than stools. The fatness-associated OTUs were mainly annotated to Lachnospiraceae, Ruminococcaceae, Prevotella, Treponema, and Bacteroides. These microbes have been reported to produce short-chain fatty acids by fermenting dietary indigested polysaccharide and pectin. The short-chain fatty acids can regulate host body energy homeostasis, protect host from inflammation and inhibit fat mass development. Our findings suggested that the gut microbiome may be an important factor modulating fatness in pigs.
Introduction
Obesity has been becoming one of the major health problems for humans, which is characterized by excessive fat accumulation, and imbalanced energy intake and expenditure, and accompanies with low grade of systemic and chronic inflammation. It is associated with a wide range of pathological disturbances in metabolic organs and then predisposes toward type 2 diabetes mellitus (T2DM) (Hanning and Diaz-Sanchez, 2015) and cardiovascular disease (Kahn et al., 2006; Canfora et al., 2015). Further, obesity is also related to certain types of cancer, osteoarthritis, and asthma. In pigs, fatness has been regarded as a typically complex and economic trait in pig production. It brings low feed conversion rate and unfavorable fat mass. Dissection of the mechanism for the fatness in pigs not only benefits the pig industry but also provides important information for understanding human obesity, because pigs possess greater similarity with humans in nutritional and metabolic physiology compared to other animal models (Swindle et al., 2012). Fatness is affected by many factors, such as genetics, nutrition, and lifestyle as well as gut microbiome. More and more studies in humans have shown that obesity is related to the gut microbiota (Ley et al., 2006; Turnbaugh et al., 2009; Munoz-Garach et al., 2016).
Mammalian gastrointestinal tract is resided by a complex, diverse, and dynamic community of symbiotic microbes that continuously interact with the host (Hooper et al., 2002; Backhed et al., 2005). Gut microbiota has demonstrated the great significance to animals by providing a large amount of functions that host lacks, such as fermenting undigested energy substrates, stimulating the host immune system development, participating in the metabolic processes, preventing growth of harmful and pathogenic bacteria and so on (Guarner and Malagelada, 2003). The relationship between human obesity and gut microbiota composition has been established for several decades with the hallmark study by Backhed et al. (2004) which demonstrated that the gut microbiota as an environmental factor modulates fat storage. Since then, the role of the gut microbiota in the pathogenesis of obesity has become a vigorous research area. The further studies showed that obesity is associated with the changes of two dominant phylum-level bacteria of Bacteroidetes and Firmicutes in the gut (Mariat et al., 2009; Gerritsen et al., 2011). A reduction of bacterial diversity and the altered metabolic pathways were demonstrated to associate obesity from a comparison study in obese and lean twins (Turnbaugh et al., 2009). An endotoxin-producing “obese microbe” was isolated from an obese human and was confirmed to cause the obesity in the germfree mice (Fei and Zhao, 2013). In pigs, the studies reported by Guo et al. (2008a,b), Wall et al. (2009), and Luo et al. (2012) inferred that the gut microbiota might participate in the process of fat storage and should be correlated with the porcine adiposity formation. In addition, Yang et al. (2016) identified tens of fatness-associated bacteria including Escherichia spp. which showed a higher relative abundance in high fatness pigs. Overall, accumulating evidences suggest that the endotoxin-induced inflammation, dysbiosis of gut microbiota composition and Firmicutes/Bacteroidetes ratio are involved in the development of obesity. But the mechanism remains controversial (Turnbaugh et al., 2006, 2009; Cani et al., 2007; Wen et al., 2008).
Bamaxiang is a Chinese indigenous mini pig breed, and Erhualian is another Chinese indigenous pig breed which is famous for its high prolificacy. Both breeds show a higher fatness than Western pig breeds (Ai et al., 2013). In this study, we evaluated the association of the gut microbiome with porcine fatness in both faces and cecum luminal samples.
Materials and Methods
Animals and Phenotype Measurement
A total of 244 Bamaxiang and 256 Erhualian pigs were used in this study. The two pig populations were raised in the same farm house. All experimental pigs were fed two times a day using the corn-soybean feed containing 14∼16% of crude protein, 8% of coarse fiber, 3,100 kJ of digestible energy and 0.85% of lysine, and given an ad libitum water. All animals were healthy and did not receive any antibiotic treatment within 2 months before slaughter. The experimental pigs were slaughtered at 300 ± 3 days. The fatness traits including backfat thickness (measured at shoulder, chest and waist, and defined as ShoulderBF, ChestBF, WaistBF, and AverageBF) and fat mass (including LeafFatWt and AbdomenFatWt) (Table 1) were separately measured by the vernier caliper and electronic platform balance. All animal works were conducted according to the guidelines for the care and use of experimental animals established by the Ministry of Agriculture of China. The project was specially approved by Animal Care and Use Committee (ACUC) in Jiangxi Agricultural University.
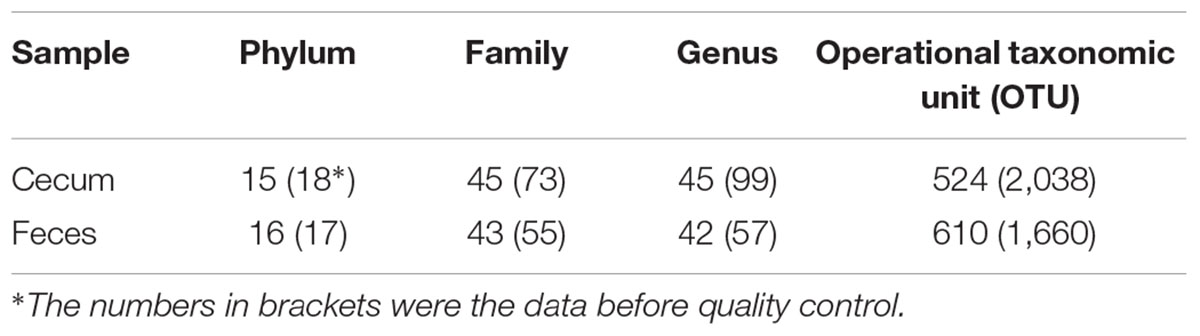
TABLE 1. Summary of gut microbial structure identified in the cecum and feces.
Fecal and Cecum Luminal Sample Collection and DNA Extraction
The luminal contents of cecum were collected from the Erhualian population when pigs were killed in the slaughter house. The fecal samples of the Bamaxiang population were harvested from the rectum before the pigs were transported to the slaughter house. All samples were collected in the 7-ml sterilized plastic centrifuge tubes and dipped in liquid nitrogen immediately. After transported to the laboratory, the samples were stored at -80°C freezer until used. DNA was extracted from fecal and luminal samples with QIAamp DNA Stool Mini Kit (Qiagen, Germany) according to the manufacturer’s protocol (McOrist et al., 2002). The concentration and integrity of DNA were measured by the Nanodrop-1000 and the 0.8% agarose gel electrophoresis.
16S rRNA Gene Sequencing and Quality Control of Data
The V4 hypervariable region of 16S rRNA gene was selected and amplified by the fusion primers 515F [GTGCCAGCMGCCG CGGTAA] and 806R [GGACTACHVGGGTWTCTAAT] under the melting temperature of 56°C with 30 cycles. The DNA sequencing procedure was performed on the MiSeq platform (Illumina, USA) according to the manufacturer’s manuals. All 16S rRNA gene sequencing data were submitted to the SRA database in NCBI with the accession numbers SRR4422912, SRR4422947, SRR4422914, SRR4422951, SRR4431318, SRR4431319, SRR4431321, SRR4454082, SRR4454119, and SRR4431322. Data processing and quality control were processed by the standard protocols of bioinformatics analysis. In brief, to obtain the clean sequence reads, we used custom scripts to remove the primer, low-quality, and barcode sequences. According to the report by Fu et al. (2015), we rarefied the library size to 20,000 clean reads depth. And then, FLASH (v.1.2.11) was used to assemble the paired-end clean reads into tags (Magoc and Salzberg, 2011). Unique bacterial sequences with 97% sequence similarity were clustered as operational taxonomic unit (OTU) using the QIIME software (the toolbox for Quantitative Insights Into Microbial Ecology), which uses UCLUST (an algorithm to cluster sequence reads based on similarity) to perform the clustering (Edgar, 2010). Those OTUs which had relative abundance <0.1% and were present in less than 1% of the experimental pigs were removed from further analysis. OTUs were matched to bacteria by using a primer-specific version of the GreenGenes (v13.5) reference database (DeSantis et al., 2006). A total of 234 and 243 pigs which had both phenotypes and 16S rRNA gene sequencing data were remained for the further association study between phenotypic value of fatness and relative abundance of gut microbiota in the two populations.
Statistical Analysis
Microbial Diversity Analysis
The α-diversity indexes of chao1, ACE, observed species, Simpson, and Shannon index were calculated by Mothur software (Schloss et al., 2009). The comparison of α-diversity indexes between Bamaxiang and Erhualian pigs was performed by Wilcoxon t-test. The possible correlations between the relative abundance of bacteria and the variables of environmental and host factors including pen, batch, kinship, and sex were examined by canonical correspondence analysis (CCA) using the R software with vegan package (Dixon, 2003).
Two-Part Model for Association Analysis
To identify the gut microbes which were associated with porcine fatness, we performed the association studies with two-part model (Fu et al., 2015). The two-part model includes a binary model and a quantitative model. The binary model accounted for the effect of the presence/absence of the gut flora on porcine fatness, and the quantitative model analyzed the effect of the abundance of the microbes on porcine fatness.
To further evaluate whether the effect came from the presence/absence or the abundance of the gut microbiota or both, a combination of the binary and quantitative analysis was characterized by a meta-analysis in which the P-value was derived using an unweighted Z method. The formulas of the three models were described as below:
Where y refers to the trait value (backfat thickness, leaf fat weight, and abdominal fat weight) per individual after adjusting for sex and body weight, b is a binary feature, q is a quantitative feature, β1 and β2 are the estimated effects for the binary and abundance effect, and e represents the residuals. Zi is the Z-transform test converting the one tailed P-values, Pi was from each of k independent tests into standard normal deviates. Z is the sum of these Zi divided by the square root of the number of tests, k has a standard normal distribution (Whitlock, 2005). The minimum of the P-values from the binary analysis, quantitative analysis, and meta-analysis was set as the final association P-value. We performed 1,000× permutation tests to control the false discovery rate (FDR). The FDR ≤ 0.1 was set as the significant threshold.
Where N0 is the average number of the detected significance at a certain P cutoff in 1,000 permutations, N1 is the number of the detected positive in the real analysis.
Estimating the Phenotypic Variance Explained by the Gut Microbiome
To test the phenotypic variation of porcine fatness explained by gut microbiome, we conducted a 100 times cross validation. Thus, we split the data randomly into a 70% discovery set and a 30% validation set. In the discovery set, a total of n number of significantly associated OTUs was identified at a certain P-value, and the effect sizes of binary and quantitative features of each OTU (β1 and β2) were estimated before. Then, the risk of the gut microbiome on fatness traits (rm) for each animal in the validation set was calculated using an additive model:
The phenotypic variance explained by the gut microbiome was represented as the squared correlation coefficient (R2) between the trait values corrected for sex and body weight and rm. To ensure the stability of the estimation, we repeated the cross-validation by 100 times and calculated the average value of the explained variations. Considering many microbes that may contribute a small effect but may not be confidently detected at FDR ≤ 0.1, we performed this analysis at a series of different significant P levels ranging from 1.0E – 05 to 0.1.
Results
Microbial Diversity in Porcine Cecum and Feces
The total numbers of sequence reads for fecal and cecum luminal samples were 8,029,976 (an average of 30,766 reads per sample) and 7,320,888 (an average of 28,597 reads per sample), respectively. We rarefied the library size to 20,000 reads per sample to reduce the effect of sequencing depth. The two pig cohorts obtained 1,660 and 2,038 OTUs. After quality control, we focused on the 610 and 524 OTUs in the Bamaxiang and Erhualian population, respectively. These OTUs occupied 99% of the total clean reads in each sample. The tags were annotated to microbial taxa. In the Bamaxiang population, a total of 17 phyla and 57 genera were identified. In the Erhualian population, the numbers of microbial phylum and genus were 18 and 99, respectively (Table 1). We compared the α-diversity of microbiota between cecum and feces samples using the chao1, ACE, observed species, Shannon and Simpson index, and found that all five indexes showed significant difference (P = 6.64E - 52, 1.96E - 50, 2.20E - 55, 5.76E - 05, and 3.90E - 03, respectively). The fecal samples had a significantly higher α-diversity (Figure 1). We further compared the phylogenetic composition of the microbial community at the phylum and genus level. The cecum luminal samples had significantly higher abundances of the phyla Bacteroidetes and Proteobacteria, and the genera Akkermansia, Bacteroides, CF231, Escherichia, and Prevotella. However, the fecal samples showed the higher abundances of the phyla Firmicutes and Spirochaetes, and the genera Lactobacillus, Streptococcus, and Treponema (P < 0.05) (Figure 2). We performed CCA analysis in the tested samples and identified the significant effects of host gender and batch on the microbial composition of both cecum and feces (P < 0.05).
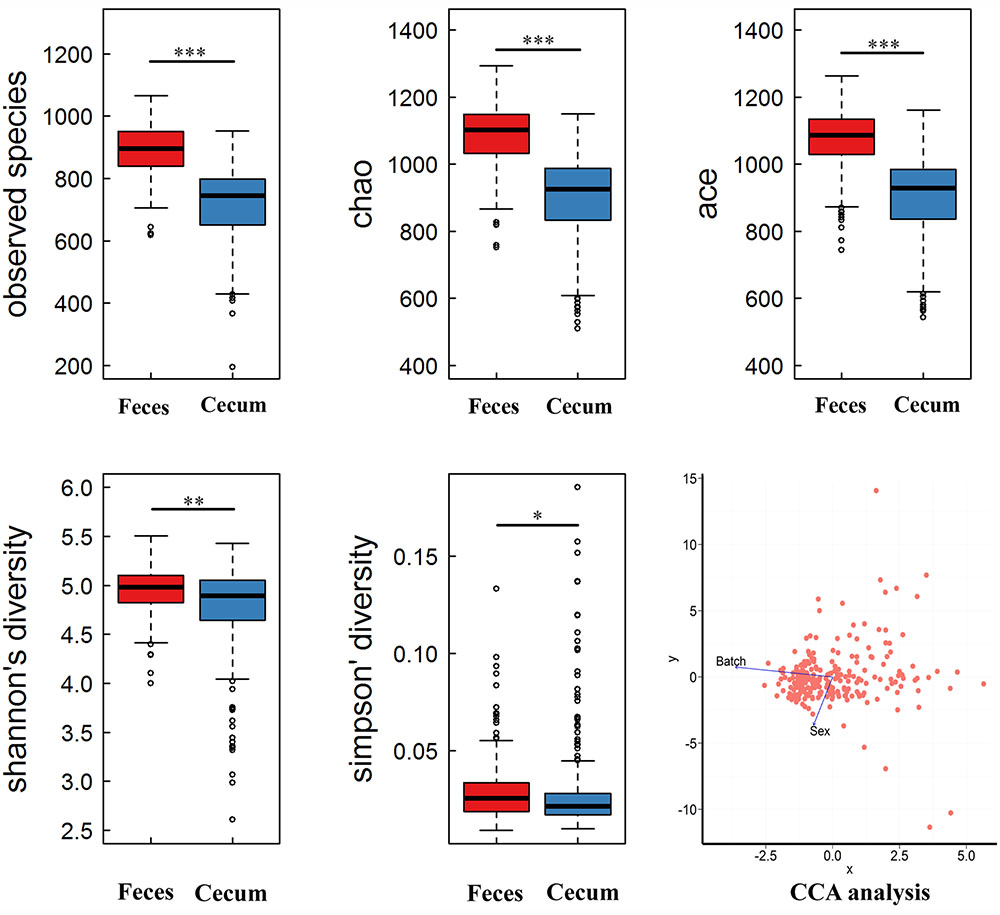
FIGURE 1. Comparison of the α-diversity of gut microbiome between cecum and fecal samples. The gut microbial richness was estimated by observed species, chao and ace index, the diversity was evaluated by Shannon and Simpson index. The microbial richness and diversity were showed significant difference between cecum and feces (∗P < 0.005; ∗∗P < 0.0001; ∗∗∗P < 0.00001; Wilcoxon t-test). Canonical correspondence analysis (CCA) showed that host gender and batch had significant effects on microbial composition (P < 0.05).
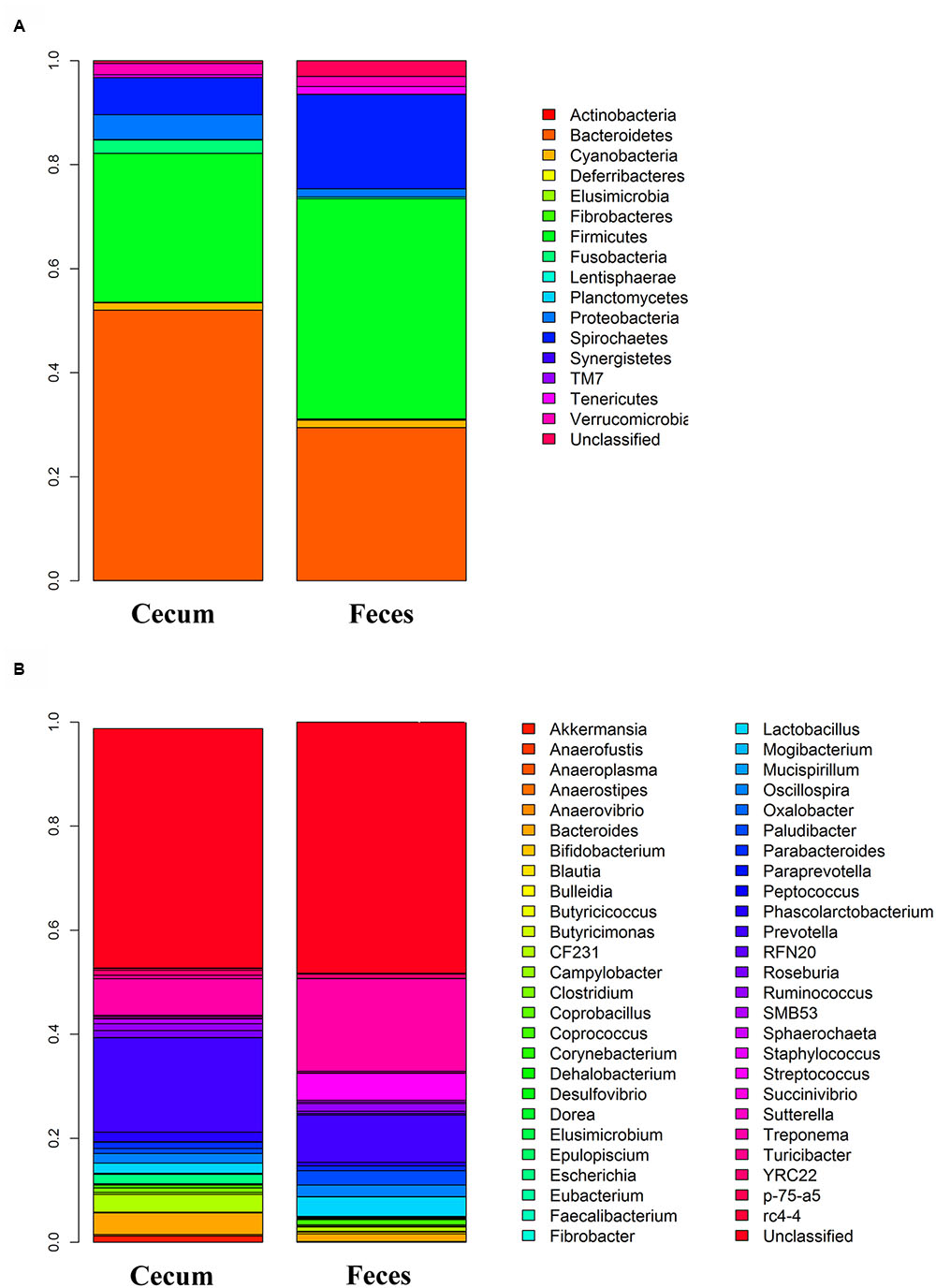
FIGURE 2. Comparison of the relative abundance of gut microbiota between cecum and fecal samples. (A) At phylum level. (B) At genus level.
Identification of Gut Microbes Associated with Porcine Fatness in the Cecum and Fecal Samples
The summary description of phenotypic values of backfat thickness (subcutaneous fat), leaf fat weight and abdominal fat weight is shown in Table 2. The phenotypic values were firstly adjusted for the effects of sex and body weight, and then the residuals were used for association analyses. In the fecal samples, we identified six OTUs (Otu363, Otu393, Otu206, Otu95, Otu1330, and Otu500) that were significantly associated with leaf fat weight at FDR ≤ 0.1. These six OTUs were annotated to unclassified Ruminococcaceae, Ruminococcus gnavus, Lachnospiraceae, Firmicutes, Prevotella, and Clostridiales, respectively. We did not identify any significant associations at OTU level for other fatness traits (Supplementary Table 1). At the taxonomic level, we identified 11 significant associations related to nine unique taxonomies for fatness traits at the significant threshold of FDR ≤ 0.1. Of the 11 associations, two were identified for AverageBF. While only one significant association was found for each of LeafFatWt, ChestBF, and WaistBF (Figure 3A; Supplementary Table 2). The other six associations were detected for abdomimal fat weight (AbdomenFatWt). For more details, Actinobacteria showed a negative association with both WaistBF and AverageBF (P = 6.59E-04 and 7.73E - 04, respectively); Coprobacillus was positively associated with both ChestBF and AverageBF (P = 1.91E - 04 and 2.68E - 04, respectively); and species R. gnavus was positively associated with LeafFatWt (P = 1.00E - 04). The species Mucispirillum schaedleri was the only member of the phylum Deferribacteres identified in this study. The strongly negative associations with AbdomenFatWt were identified on this microbe from phylum to species level (P = 1.10E - 04); In addition, of these 11 significant associations, eight were detected by the binary analysis (presence/absence), two were identified by the quantitative analysis (the abundance of bacteria) and one by meta-analysis.
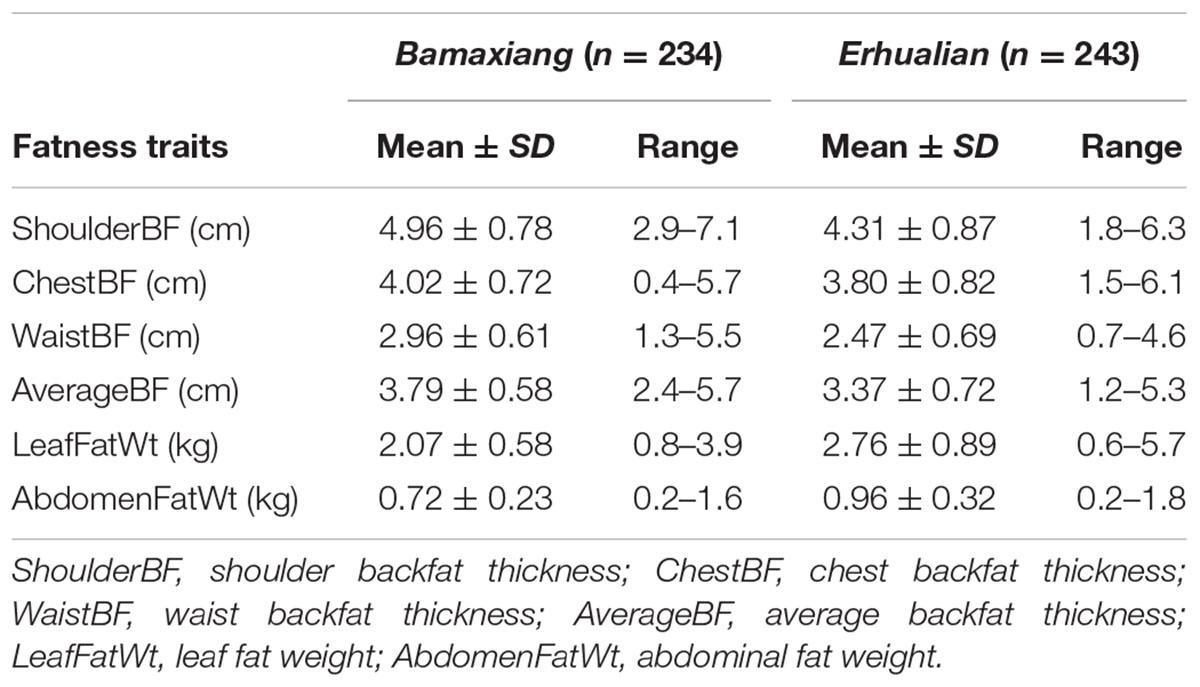
TABLE 2. Summary description of phenotypic values of porcine fatness traits in the Bamaxiang and Erhualian population.
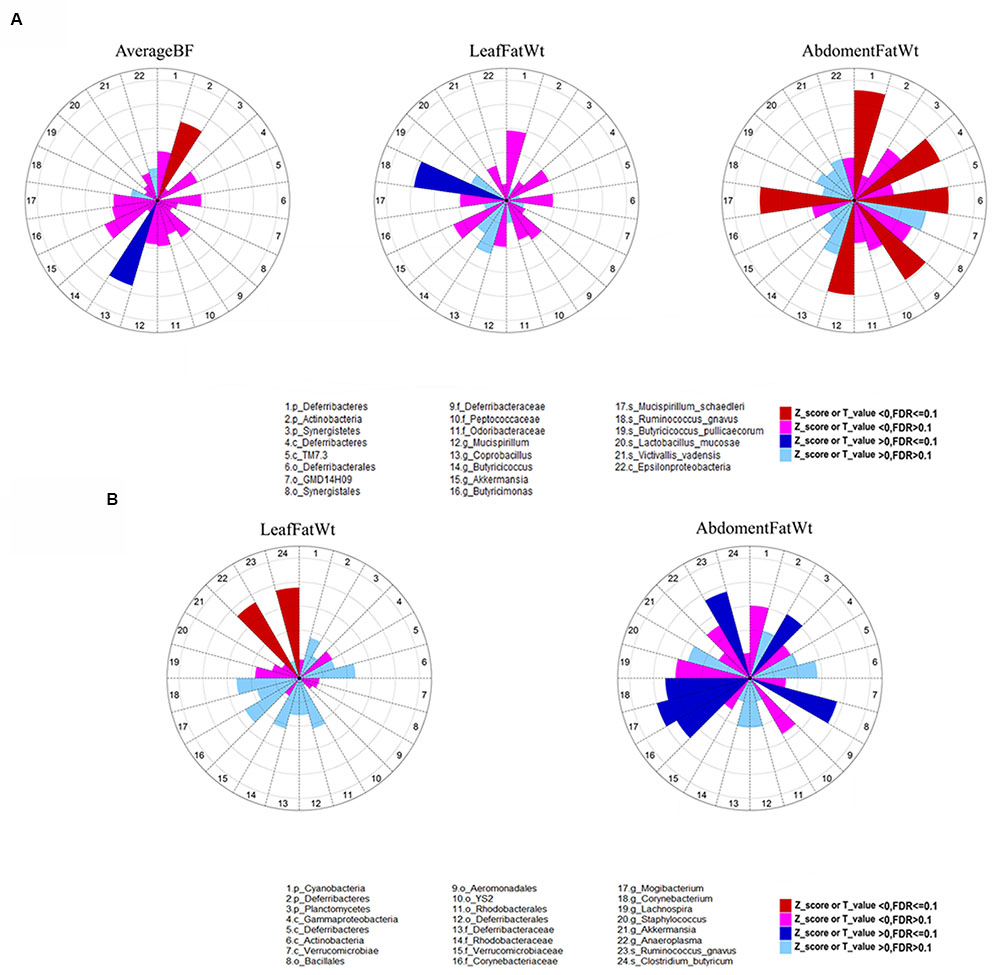
FIGURE 3. The effect of bacterial taxonomies on abdominal fat weight, leaf fat weight, and average backfat thickness. The effects of bacterial taxonomies on abdominal fat weight, leaf fat weight, and average backfat thickness are shown as Z or T scores in the feces (A) and cecum (B), respectively. The different color sectors indicate positive or negative associations and their significance level. Dashed circles indicate the scale of Z or T-values from 1 to 5.
With respect to the cecum luminal samples, a total of 108 significant associations for 80 unique OTUs were found at FDR ≤ 0.1, including four associations with ShoulderBF, 30 with WaistBF, 19 with the AverageBF and 55 with AbdomenFatWt. However, we did not detect any OTUs significantly associated with ChestBF and LeafFatWt. Of the 80 fatness-associated OTUs, two (Otu148 and Otu162) were shared by four traits (AbdomentFat, WaistBF, ShoulderBF, and AverageBF), three OTUs were shared by AbdomentFat, WaistBF and AverageBF, nine by WaistBF and AverageBF, and three by AbdomentFat and WaistBF. Each of the other fatness-associated OTUs was specifically associated with only one phenotype. The detailed annotation results for the fatness-associated OTUs are shown in Supplementary Table 3 and Figure 4. We observed that these OTUs were mainly annotated to the YS2 (Cyanobacteria), Lachnospiraceae, Ruminococcaceae, Prevotella, Treponema, and Bacteroides. Further, the OTUs annotated to the Ruminococcaceae were showed positive associations with fatness traits. Those annotated to the Prevotella, Treponema, and Bacteroides were negatively associated with fatness traits. And both positive and negative associations were observed for the OTUs annotated to the Lachnospiraceae. At the taxonomic level, we identified 10 taxa that were significantly associated with fatness traits at FDR ≤ 0.1, including one for WaistBF, one for ChestBF, six for AbdomenFatWt, and two for LeafFatWt (Figure 3B; Supplementary Table 2). We did not identify any significant associations with ShoulderBF and AverageBF. Of the 10 fatness-associated taxa, five were detected by the binary analysis, one by the quantitative analysis and four by the meta-analysis.
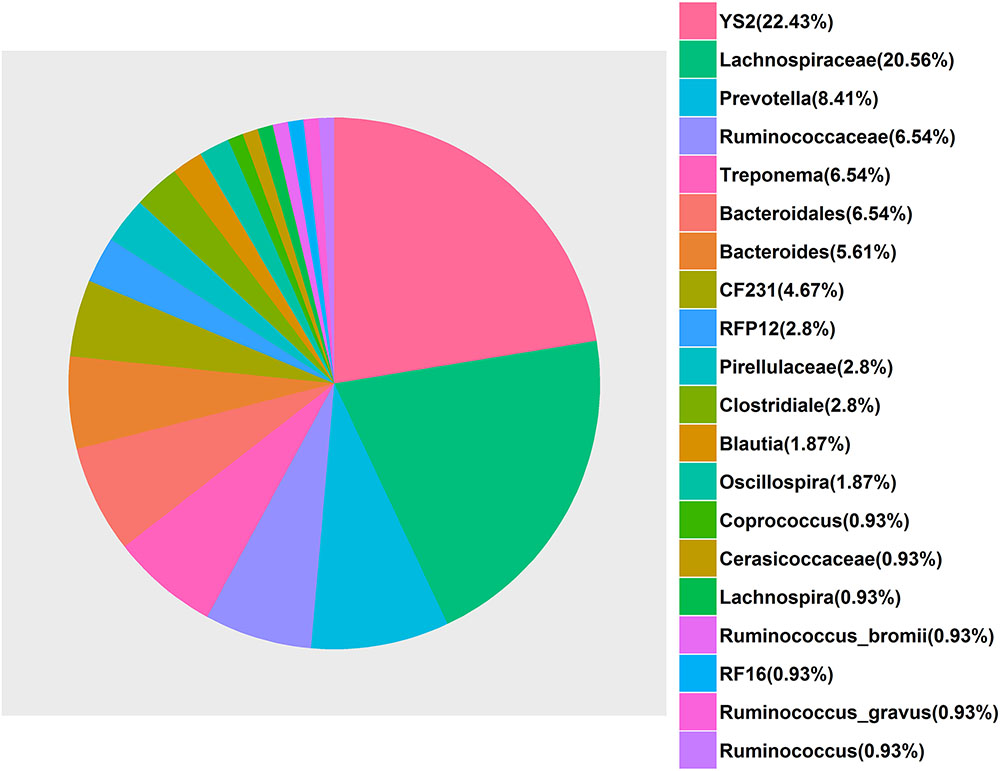
FIGURE 4. The bacterial annotation of the 80 fatness-associated operational taxonomic units (OTUs) in the Erhualian population based on the Greengenes database. The percentages were calculated with (the number of the fatness-associated OTUs annotated to a given bacterial taxonomy/80 × 100%).
Ruminococcus gnavus showed a positive association with both leaf fat weight in the fecal samples (P = 1.00E - 04, Z_score = 3.89) and abdominal fat weight in the cecum luminal samples (P = 1.88E - 04, Z_score = 3.73). We did not detect any other taxa that showed the significant association with fatness in both types of samples.
Phenotypic Variance of Porcine Fatness Explained by Gut Microbiome
To investigate how much degree of phenotypic variance of fatness was explained by the gut microbiome, we conducted a 100 times cross-validation analysis by splitting the data set randomly into an 70% discovery set and a 30% validation set at the OTU level. In the Bamaxiang pigs, we found that the OTUs identified at P = 1.0E - 05 level in the discovery set could explain 1.09% phenotypic variation of LeafFatWt in the validation set. In the Erhualian cohort, at P = 1.0E - 05 level, the fatness-associated OTUs can explain 0.73% phenotypic variation of ShoulderBF, 1.53% of WaistBF, 1.11% of AverageBF, and 1.29% of AbdomenFatWt. When the significance threshold of association increased to P = 0.1 and the risk model included more (but less significant) OTUs, the explained variance increased to 1.61% in LeafFatWt for Bamaxiang pigs, and 2.07% in ShoulderBF, 1.75% in WaistBF, 1.55% in AverageBF, and 2.41% in AbdomenFatWt for Erhualian pigs (Figure 5).
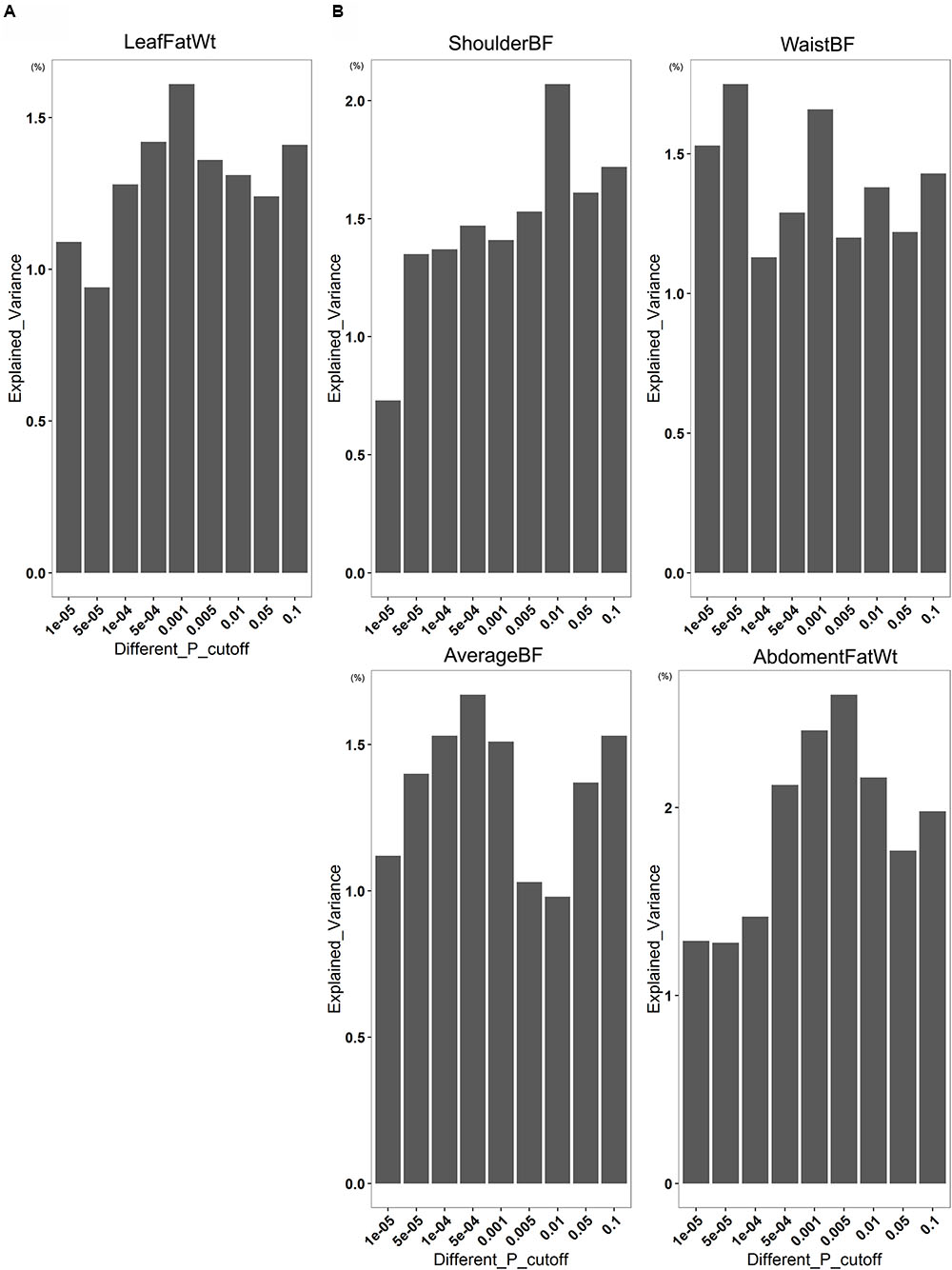
FIGURE 5. The contribution of gut microbiome to pig fatness traits based on the associated OTUs. The figures show the variation of fatness trait values explained by gut microbes at different significance levels in the Bamaxiang (A) and Erhualian (B) pig population.
Discussion
For recent years, more and more researches have concentrated upon the microbiota inhabiting the host gastrointestinal tract since gut microbiome has been reported to associate obesity and metabolic dysfunctional diseases in both humans and mice. Exploring the effect of the gut microbiome on obesity and insulin resistance has gained insight into the role of the microbiota in the development of diabetes mellitus and cardiovascular disease. Fatness is an importantly economic trait in pig production. While no such studies have been reported in pigs. To our knowledge, for the first time, we evaluated the association of the gut microbiome with fatness in swine, especially with the samples from cecum. Moreover, we adopted a novel and powerful two-part model association analysis to interpret effects of both presence/absence and relative abundance of gut microbiota on porcine fatness.
As we expected, although all experimental pigs were raised in the same farm house and fed the similar formula diet, we observed distinct phylogenetic composition of gut microbiome among samples. This should be explained by (1) different sampling sites (cecum vs. feces). Microbiota in stool are a mix mucosally associated microbes, most from the colon and lumenal microbes (Eckburg et al., 2005); (2) different genetic background between two pig cohorts.
We noticed that most of the associations were trait-specific. This heterogeneity may be caused by distinct mechanism of fat deposition for different types of adipose involving different microbes among abdominal adipose, leaf fat and subcutaneous adipose. This condition was similar to that in genetic dissection of porcine fatness traits, in which different genomic loci were identified for each fatness trait (Qiao et al., 2015). To the best of our knowledge, this study first evaluated the fatness-associated microbes in the cecum samples. Compare with the fecal samples, significantly higher number of the fatness-associated OTUs was identified in the cecum samples. As we have well known, cecum has the great diversity and complex of microbiota (Looft et al., 2014). And fermentation of dietary indigestible fiber and polysaccharides occurs at the cecum. We suggested that the samples from the cecum would be better for studying the association between microbiota and fatness than stool samples.
Interestingly, many fatness-associated microbiota identified in this study have potential functions related to metabolisms. At the taxonomic level, some of the fatness-associated bacterial taxa have been reported to participate in the process of the utilization of undigested carbohydrates from the diets or the host polysaccharide. For examples, R. gnavus was positively associated with fatness traits in both fecal and cecum samples. R. gnavus plays a pivotal role in UDCA formation in the colon, which regarded as a supplement of the bile acid (Lee et al., 2013). A recent study found that the α-galactosidase 1 (Aga1) and α-galactosidase 2 (Aga2) which are two kinds of the glycoside hydrolase (GH) family from R. gnavus played an indispensable role in the degradation of dietary oligosaccharides and exerted a tremendous fascination on designing of galacto-oligosaccharide (GOS) prebiotics (Cervera-Tison et al., 2012). The studies in human and rat found that R. gnavus was enriched in the obese rats and humans (Petriz et al., 2014; Andoh et al., 2016). Furthermore, both R. gnavus and Coprobacillus identified in this study could ferment the indigested polysaccharide into the SCFAs from the food in gastrointestinal tract, and then the SCFAs were absorbed by the host and could regulate host body energy homeostasis (Layden et al., 2013). Both Anaerovibrio and Clostridium butyricum were negatively associated with LeafFatWt. A previous study indicated that the Anaerovibrio lipolytica from Anaerovibrio can produce lipase in hydrolysis of triglyceride (Henderson, 1971). Previous studies have summarized that, as a kind of probiotics, C. butyricum can produce butyrate that provides the majority energy to the gut epithelial and repairs the intestinal mucosa damaged by virus (Araki et al., 2002, 2004; Zhang et al., 2011). The M. schaedleri was negatively associated with AbdomenFatWt in fecal samples. Interestingly, in the diet induced obesity (DIO) mice, the abundance of Mucispirillum was decreased (Clarke et al., 2013). Actinobacteria which was negatively associated with both average and waist backfat, has been recognized as the producer of many bioactive metabolites including antibacterials and antivirals for humans (Mahajan and Balachandran, 2011), and growth promoting substances for plants and animals (Atta and Ahmad, 2009).
At the OTU level, the fatness-associated OTUs were mainly annotated to YS2 (Cyanobacteria), Lachnospiraceae, Ruminococcaceae, Prevotella, Treponema, and Bacteroidales (Figure 3). Lachnospiraceae is abundant in the digestive tract of many mammals and relatively rare elsewhere. Members of this family have been linked to obesity in humans (Cho et al., 2012), mainly due to the association of many species within the group with the production of butyric acid (Duncan et al., 2002). Ruminococcaceae has been reported to play the role of biohydrogenation, and is capable of producing butyrate via fermenting various substrates, which may exert potential physiological functions in host health (Huws et al., 2011; Onrust et al., 2015). Kim et al. (2012) observed that Ruminococcaceae was enriched in mice fed high fat diet. Geurts et al. (2011) also observed a higher abundance of Ruminococcaceae in the db/db mice compared to lean mice. Prevotella and Treponema belong to xylan-degrading bacteria, which contain a set of bacterial genes for cellulose and xylan hydrolysis, and act as a host mutualistic component to help to degradate dietary fiber which could produce significantly more short-fatty acids (Warnecke et al., 2007; Flint et al., 2008). De Filippo et al. (2010) proposed that the degrading polysaccharide-rich diet allowed gut microbiota to maximize energy intake from fibers diets, but also protect them from inflammation and noninfectious colonic diseases. Besides Prevotella and Treponema, Bacteroides was also annotated to the fatness-associated OTUs. These microbes ferment polysaccharides to short-chain fatty acids, such as propionate by Bacteroides spp. from succinate pathway and acetate by Prevotella spp. from pyruvate via acetyl-CoA. Maslowski et al. (2009) and Smith et al. (2013) showed that the interactions of acetate and propionate with GPR43 have an important role in anti-inflammatory effects via the modulation of cTReg cell. In addition, the SCFAs can influence host homeostasis, inhibit the accumulation of fat mass development in adipose tissue and promote leptin level. In diet-induced obese mice, Bacteroides-prevotella also showed a negative correction with fat mass development and inflammation (Neyrinck et al., 2011; Ridaura et al., 2013; Ivarsson et al., 2014). In the cecum samples, Prevotella, Bacteroides, and Treponema were negatively associated with fatness traits.
In addition, Blautia, Coprococcus, Ruminococcus, and Clostridiales were also annotated to the fatness-associated OTUs in the cecum samples. These bacteria were identified in a phylo-functional core of gut microbiota in healthy young Chinese cohorts (Zhang J. et al., 2015). The members of Blautia and Ruminococcus have been reported to produce acetate via acetyl-CoA from pyruvate and Wood–Ljungdahl pathway by fermenting glucose and indigestible diet fiber (Miller and Wolin, 1996; Barcenilla et al., 2000; Pryde et al., 2002; Liu et al., 2008; Kovatcheva-Datchary et al., 2009; Crost et al., 2013). Perry et al. (2016) reported that the increased acetate could promote hyperphagia and increase energy storage as fat. In mice, ingestion of high fat diet was associated with the higher abundance of Clostridiales compared to the low fat diet regardless of propensity for obesity (de La Serre et al., 2010; Delzenne and Cani, 2011).
We estimated that gut microbiome could explain 1.55–2.73% of the variation in the six fatness-associated traits. This effect size was similar to that of most quantitative trait loci (QTL) previously reported for porcine fatness traits (Ai et al., 2012). However, in humans, gut microbiota has been implicated as a pivotal contributing factor in diet-related obesity (Zhang C. et al., 2015). This different contribution size should be due to the reason that all experimental pigs were raised in the same farm house and provided the same diet. The environmental factors, such as diets displayed less effects on gut microbiome and the subsequent fat deposition. Although the effect size of gut microbiome on fatness is small, we have established that gut microbiome should be a risk factor for porcine fatness.
A recent study reported by Ross et al. (2013) described an efficient methodology for predicting complex traits from quantitative microbiome profiles, and demonstrated that microbiome profiles can be used to predict human inflammatory bowel disease (IBD) status and BMI, and methane production in cattle with high accuracy. And based on the phylotypes of gut microbiota, Zhang et al. (2010) used partial least square discriminate analysis (PLS-DA) to predict host genotypes, diets or obesity phenotypes. In the futural study, the construction of prediction methodology based on the gut microbiome profiles for porcine fatness traits would greatly promote the pig production by reducing fat mass and improving the feed efficiency. Furthermore, the results from this study gave important cues for isolation of the causative microbes for porcine fatness that would provide basic information for regulating the gut microbiome to reduce fat deposition in pigs.
Conclusion
We identified a number of taxa and OTUs that showed significant associations with porcine fatness traits in the cecum luminal samples and feces. The fatness-associated microbiota were mainly involved in fermenting dietary indigestible fiber and polysaccharides to produce short-fatty acids. The short-fatty acids have been reported to inhibit fat mass development and inflammation. Significantly higher number of fatness-associated OTUs were identified in the cecum suggesting that cecum luminal samples would be better used for investigation of fatness-associated microbes than stool samples. Although the effect size of gut microbiome on porcine fatness is not very large in this study, we established that gut microbiome should be a risk factor for porcine fatness. These results help us to better understand the structure and functional potential of swine gastrointestinal microbiota, and provide a new insight into the role of gut microbes in affecting the porcine fatness traits.
Ethics Statement
All samples were collected according to the guidelines for the care and use of experimental animals established by the Ministry of Agriculture of China. Animal Care and Use Committee (IACUC) in Jiangxi Agricultural University specifically approved this study. All the experimental pigs used in this study were raised in the farm house affiliated to State Key Laboratory for Pig Genetic Improvement and Production Technology, Jiangxi Agricultural University. The pig owners consented to this study. No vulnerable populations were involved in this study.
Author Contributions
CC conceived and designed the experiments, analyzed the data, wrote, and revised the manuscript; MH performed the experiments, analyzed the data and wrote the manuscript; SF, XH, HY, ZL, and JG performed the experiments; YZ and SK collected the samples; LH conceived and designed the experiments, and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Natural Science Foundation of China (31472071) and Natural Science Foundation of Jiangxi province (2013BAB20006).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.02108/full#supplementary-material
References
Ai, H., Huang, L., and Ren, J. (2013). Genetic diversity, linkage disequilibrium and selection signatures in chinese and Western pigs revealed by genome-wide SNP markers. PLoS ONE 8:e56001. doi: 10.1371/journal.pone.0056001
Ai, H., Ren, J., Zhang, Z., Ma, J., Guo, Y., Yang, B., et al. (2012). Detection of quantitative trait loci for growth-and fatness-related traits in a large-scale White Duroc × Erhualian intercross pig population. Anim. Genet. 43, 383–391. doi: 10.1111/j.1365-2052.2011.02282.x
Andoh, A., Nishida, A., Takahashi, K., Inatomi, O., Imaeda, H., Bamba, S., et al. (2016). Comparison of the gut microbial community between obese and lean peoples using 16S gene sequencing in a Japanese population. J. Clin. Biochem. Nutr. 59, 65–70. doi: 10.3164/jcbn.15-152
Araki, Y., Andoh, A., Fujiyama, Y., Takizawa, J., Takizawa, W., and Bamba, T. (2002). Oral administration of a product derived from Clostridium butyricum in rats. Int. J. Mol. Med. 9, 53–57.
Araki, Y., Andoh, A., Takizawa, J., Takizawa, W., and Fujiyama, Y. (2004). Clostridium hutyricum, a probiotic derivative, suppresses dextran sulfate sodium-induced experimental colitis in rats. Int. J. Mol. Med. 13, 577–580.
Atta, H. M., and Ahmad, M. S. (2009). Antimycin-A antibiotic biosynthesis produced by Streptomyces sp. AZ-AR-262: taxonomy, fermentation, purification and biological activities. Aust. J. Basic Appl. Sci. 3, 126–135.
Backhed, F., Ding, H., Wang, T., Hooper, L. V., Koh, G. Y., Nagy, A., et al. (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U.S.A. 101, 15718–15723. doi: 10.1073/pnas.0407076101
Backhed, F., Ley, R. E., Sonnenburg, J. L., Peterson, D. A., and Gordon, J. I. (2005). Host-bacterial mutualism in the human intestine. Science 307, 1915–1920. doi: 10.1126/science.1104816
Barcenilla, A., Pryde, S. E., Martin, J. C., Duncan, S. H., Stewart, C. S., Henderson, C., et al. (2000). Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl. Environ. Microbiol. 66, 1654–1661. doi: 10.1128/AEM.66.4.1654-1661.2000
Canfora, E. E., Jocken, J. W., and Blaak, E. E. (2015). Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 11, 577–591. doi: 10.1038/nrendo.2015.128
Cani, P. D., Amar, J., Iglesias, M. A., Poggi, M., Knauf, C., Bastelica, D., et al. (2007). Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes Metab. Res. Rev. 56, 1761–1772.
Cervera-Tison, M., Tailford, L. E., Fuell, C., Bruel, L., Sulzenbacher, G., Henrissat, B., et al. (2012). Functional analysis of family GH36 alpha-galactosidases from Ruminococcus gnavus E1: insights into the metabolism of a plant oligosaccharide by a human gut symbiont. Appl. Environ. Microbiol. 78, 7720–7732. doi: 10.1128/AEM.01350-12
Cho, I., Yamanishi, S., Cox, L., Methé, B. A., Zavadil, J., Li, K., et al. (2012). Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488, 621–626. doi: 10.1038/nature11400
Clarke, S. F., Murphy, E. F., O’sullivan, O., Ross, R. P., O’toole, P. W., Shanahan, F., et al. (2013). Targeting the microbiota to address diet-induced obesity: a time dependent challenge. PLoS ONE 8:e65790. doi: 10.1371/journal.pone.0065790
Crost, E. H., Tailford, L. E., Le Gall, G., Fons, M., Henrissat, B., and Juge, N. (2013). Utilisation of mucin glycans by the human gut symbiont Ruminococcus gnavus is strain-dependent. PLoS ONE 8:e76341. doi: 10.1371/journal.pone.0076341
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696. doi: 10.1073/pnas.1005963107
de La Serre, C. B., Ellis, C. L., Lee, J., Hartman, A. L., Rutledge, J. C., and Raybould, H. E. (2010). Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am. J. Physiol.Gastrointest. Liver Physiol. 299, G440–G448. doi: 10.1152/ajpgi.00098.2010
Delzenne, N. M., and Cani, P. D. (2011). Interaction between obesity and the gut microbiota: relevance in nutrition. Annu. Rev. Nutr. 31, 15–31. doi: 10.1146/annurev-nutr-072610-145146
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Dixon, P. (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x
Duncan, S. H., Barcenilla, A., Stewart, C. S., Pryde, S. E., and Flint, H. J. (2002). Acetate utilization and butyryl coenzyme A (CoA): acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl. Environ. Microbiol. 68, 5186–5190. doi: 10.1128/AEM.68.10.5186-5190.2002
Eckburg, P. B., Bik, E. M., Bernstein, C. N., Purdom, E., Dethlefsen, L., Sargent, M., et al. (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638. doi: 10.1126/science.1110591
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Fei, N., and Zhao, L. (2013). An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 7, 880–884. doi: 10.1038/ismej.2012.153
Flint, H. J., Bayer, E. A., Rincon, M. T., Lamed, R., and White, B. A. (2008). Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat. Rev. Microbiol. 6, 121–131. doi: 10.1038/nrmicro1817
Fu, J., Bonder, M. J., Cenit, M. C., Tigchelaar, E. F., Maatman, A., Dekens, J. A., et al. (2015). The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ. Res. 117, 817–824. doi: 10.1161/CIRCRESAHA.115.306807
Gerritsen, J., Smidt, H., Rijkers, G. T., and De Vos, W. M. (2011). Intestinal microbiota in human health and disease: the impact of probiotics. Genes Nutr. 6, 209–240. doi: 10.1007/s12263-011-0229-7
Geurts, L., Lazarevic, V., Derrien, M., Everard, A., Van Roye, M., Knauf, C., et al. (2011). Altered gut microbiota and endocannabinoid system tone in obese and diabetic leptin-resistant mice: impact on apelin regulation in adipose tissue. Front. Microbiol. 2:149. doi: 10.3389/fmicb.2011.00149
Guarner, F., and Malagelada, J. R. (2003). Gut flora in health and disease. Lancet 361, 512–519. doi: 10.1016/S0140-6736(03)12489-0
Guo, X., Xia, X., Tang, R., and Wang, K. (2008a). Real-time PCR quantification of the predominant bacterial divisions in the distal gut of Meishan and Landrace pigs. Anaerobe 14, 224–228. doi: 10.1016/j.anaerobe.2008.04.001
Guo, X., Xia, X., Tang, R., Zhou, J., Zhao, H., and Wang, K. (2008b). Development of a real-time PCR method for Firmicutes and Bacteroidetes in faeces and its application to quantify intestinal population of obese and lean pigs. Lett. Appl. Microbiol. 47, 367–373. doi: 10.1111/j.1472-765X.2008.02408.x
Hanning, I., and Diaz-Sanchez, S. (2015). The functionality of the gastrointestinal microbiome in non-human animals. Microbiome 3, 51. doi: 10.1186/s40168-015-0113-6
Henderson, C. (1971). A study of the lipase produced by Anaerovibrio lipolytica, a rumen bacterium. J. Gen. Microbiol. 65, 81–89. doi: 10.1099/00221287-65-1-81
Hooper, L. V., Midtvedt, T., and Gordon, J. I. (2002). How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 22, 283–307. doi: 10.1146/annurev.nutr.22.011602.092259
Huws, S. A., Kim, E. J., Lee, M. R., Scott, M. B., Tweed, J. K., Pinloche, E., et al. (2011). As yet uncultured bacteria phylogenetically classified as Prevotella, Lachnospiraceae incertae sedis and unclassified Bacteroidales, Clostridiales and Ruminococcaceae may play a predominant role in ruminal biohydrogenation. Environ. Microbiol. 13, 1500–1512. doi: 10.1111/j.1462-2920.2011.02452.x
Ivarsson, E., Roos, S., Liu, H. Y., and Lindberg, J. E. (2014). Fermentable non-starch polysaccharides increases the abundance of Bacteroides-Prevotella-Porphyromonas in ileal microbial community of growing pigs. Animal 8, 1777–1787. doi: 10.1017/S1751731114001827
Kahn, S. E., Hull, R. L., and Utzschneider, K. M. (2006). Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–846. doi: 10.1038/nature05482
Kim, K.-A., Gu, W., Lee, I.-A., Joh, E.-H., and Kim, D.-H. (2012). High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 7:e47713. doi: 10.1371/journal.pone.0047713
Kovatcheva-Datchary, P., Egert, M., Maathuis, A., Rajilic-Stojanovic, M., De Graaf, A. A., Smidt, H., et al. (2009). Linking phylogenetic identities of bacteria to starch fermentation in an in vitro model of the large intestine by RNA-based stable isotope probing. Environ. Microbiol. 11, 914–926. doi: 10.1111/j.1462-2920.2008.01815.x
Layden, B. T., Angueira, A. R., Brodsky, M., Durai, V., and Lowe, WL Jr (2013). Short chain fatty acids and their receptors: new metabolic targets. Transl. Res. 161, 131–140. doi: 10.1016/j.trsl.2012.10.007
Lee, J. Y., Arai, H., Nakamura, Y., Fukiya, S., Wada, M., and Yokota, A. (2013). Contribution of the 7beta-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J. Lipid Res. 54, 3062–3069. doi: 10.1194/jlr.M039834
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Liu, C., Finegold, S. M., Song, Y., and Lawson, P. A. (2008). Reclassification of Clostridium coccoides, Ruminococcus hansenii, Ruminococcus hydrogenotrophicus, Ruminococcus luti, Ruminococcus productus and Ruminococcus schinkii as Blautia coccoides gen. nov., comb. nov., Blautia hansenii comb. nov., Blautia hydrogenotrophica comb. nov., Blautia luti comb. nov., Blautia producta comb. nov., Blautia schinkii comb. nov. and description of Blautia wexlerae sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 58, 1896–1902. doi: 10.1099/ijs.0.65208-0
Looft, T., Allen, H. K., Cantarel, B. L., Levine, U. Y., Bayles, D. O., Alt, D. P., et al. (2014). Bacteria, phages and pigs: the effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 8, 1566–1576. doi: 10.1038/ismej.2014.12
Luo, Y. H., Su, Y., Wright, A. D., Zhang, L. L., Smidt, H., and Zhu, W. Y. (2012). Lean breed Landrace pigs harbor fecal methanogens at higher diversity and density than obese breed Erhualian pigs. Archaea 2012, 605289. doi: 10.1155/2012/605289
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mahajan, G. B., and Balachandran, L. (2011). Antibacterial agents from actinomycetes-a review. Front. Biosci. (Elite Ed) 4:240–253.
Mariat, D., Firmesse, O., Levenez, F., Guimaraes, V., Sokol, H., Dore, J., et al. (2009). The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 9:123. doi: 10.1186/1471-2180-9-123
Maslowski, K. M., Vieira, A. T., Ng, A., Kranich, J., Sierro, F., Yu, D., et al. (2009). Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286. doi: 10.1038/nature08530
McOrist, A. L., Jackson, M., and Bird, A. R. (2002). A comparison of five methods for extraction of bacterial DNA from human faecal samples. J. Microbiol. Methods 50, 131–139. doi: 10.1016/S0167-7012(02)00018-0
Miller, T. L., and Wolin, M. J. (1996). Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl. Environ. Microbiol. 62, 1589–1592.
Munoz-Garach, A., Diaz-Perdigones, C., and Tinahones, F. J. (2016). Gut microbiota and type 2 diabetes mellitus. Endocrinol. Nutr. 63, 560–568. doi: 10.1016/j.endonu.2016.07.008
Neyrinck, A. M., Possemiers, S., Druart, C., Van De Wiele, T., De Backer, F., Cani, P. D., et al. (2011). Prebiotic effects of wheat arabinoxylan related to the increase in bifidobacteria, Roseburia and Bacteroides/Prevotella in diet-induced obese mice. PLoS ONE 6:e20944. doi: 10.1371/journal.pone.0020944
Onrust, L., Ducatelle, R., Van Driessche, K., De Maesschalck, C., Vermeulen, K., Haesebrouck, F., et al. (2015). Steering endogenous butyrate production in the intestinal tract of broilers as a tool to improve gut health. Front. Vet. Sci. 2:75. doi: 10.3389/fvets.2015.00075
Perry, R. J., Peng, L., Barry, N. A., Cline, G. W., Zhang, D., Cardone, R. L., et al. (2016). Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature 534, 213–217. doi: 10.1038/nature18309
Petriz, B. A., Castro, A. P., Almeida, J. A., Gomes, C. P., Fernandes, G. R., Kruger, R. H., et al. (2014). Exercise induction of gut microbiota modifications in obese, non-obese and hypertensive rats. BMC Genomics 15:511. doi: 10.1186/1471-2164-15-511
Pryde, S. E., Duncan, S. H., Hold, G. L., Stewart, C. S., and Flint, H. J. (2002). The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 217, 133–139. doi: 10.1111/j.1574-6968.2002.tb11467.x
Qiao, R., Gao, J., Zhang, Z., Li, L., Xie, X., Fan, Y., et al. (2015). Genome-wide association analyses reveal significant loci and strong candidate genes for growth and fatness traits in two pig populations. Genet. Sel. Evol. 47:17. doi: 10.1186/s12711-015-0089-5
Ridaura, V. K., Faith, J. J., Rey, F. E., Cheng, J., Duncan, A. E., Kau, A. L., et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214. doi: 10.1126/science.1241214
Ross, E. M., Moate, P. J., Marett, L. C., Cocks, B. G., and Hayes, B. J. (2013). Metagenomic predictions: from microbiome to complex health and environmental phenotypes in humans and cattle. PLoS ONE 8:e73056. doi: 10.1371/journal.pone.0073056
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Smith, P. M., Howitt, M. R., Panikov, N., Michaud, M., Gallini, C. A., Bohlooly, Y. M., et al. (2013). The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573. doi: 10.1126/science.1241165
Swindle, M. M., Makin, A., Herron, A. J., Clubb, F. J. Jr., and Frazier, K. S. (2012). Swine as models in biomedical research and toxicology testing. Vet. Pathol. 49, 344–356. doi: 10.1177/0300985811402846
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi: 10.1038/nature07540
Turnbaugh, P. J., Ley, R. E., Mahowald, M. A., Magrini, V., Mardis, E. R., and Gordon, J. I. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031. doi: 10.1038/nature05414
Wall, R., Ross, R. P., Shanahan, F., O’mahony, L., O’mahony, C., Coakley, M., et al. (2009). Metabolic activity of the enteric microbiota influences the fatty acid composition of murine and porcine liver and adipose tissues. Am. J. Clin. Nutr. 89, 1393–1401. doi: 10.3945/ajcn.2008.27023
Warnecke, F., Luginbuhl, P., Ivanova, N., Ghassemian, M., Richardson, T. H., Stege, J. T., et al. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565. doi: 10.1038/nature06269
Wen, L., Ley, R. E., Volchkov, P. Y., Stranges, P. B., Avanesyan, L., Stonebraker, A. C., et al. (2008). Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455, 1109–1113. doi: 10.1038/nature07336
Whitlock, M. C. (2005). Combining probability from independent tests: the weighted Z-method is superior to Fisher’s approach. J. Evol. Biol. 18, 1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x
Yang, H., Huang, X., Fang, S., Xin, W., Huang, L., and Chen, C. (2016). Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci. Rep. 6, 27427. doi: 10.1038/srep27427
Zhang, B., Xie, X.-H., and Wang, R.-Q. (2011). The effect of Clostridium butyricum powder in treatment of virus enteritis in infants. Chin. J. Microecol. 23, 340–341.
Zhang, C., Yin, A., Li, H., Wang, R., Wu, G., Shen, J., et al. (2015). Dietary modulation of gut microbiota contributes to alleviation of both genetic and simple obesity in children. EBioMedicine 2, 966–982. doi: 10.1016/j.ebiom.2015.07.007
Zhang, C., Zhang, M., Wang, S., Han, R., Cao, Y., Hua, W., et al. (2010). Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 4, 232–241. doi: 10.1038/ismej.2009.112
Keywords: gut microbiome, fatness, swine, two-part model analysis, 16S rRNA gene
Citation: He M, Fang S, Huang X, Zhao Y, Ke S, Yang H, Li Z, Gao J, Chen C and Huang L (2016) Evaluating the Contribution of Gut Microbiota to the Variation of Porcine Fatness with the Cecum and Fecal Samples. Front. Microbiol. 7:2108. doi: 10.3389/fmicb.2016.02108
Received: 31 July 2016; Accepted: 13 December 2016;
Published: 23 December 2016.
Edited by:
George Tsiamis, University of Patras, GreeceReviewed by:
Spyridon Ntougias, Democritus University of Thrace, GreeceZakee L. Sabree, Ohio State University, USA
Copyright © 2016 He, Fang, Huang, Zhao, Ke, Yang, Li, Gao, Chen and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Congying Chen, chcy75@hotmail.com