 Jacqueline Hollensteiner1
Jacqueline Hollensteiner1 Franziska Wemheuer1
Franziska Wemheuer1 Rebekka Harting2
Rebekka Harting2 Anna M. Kolarzyk2†
Anna M. Kolarzyk2† Stefani M. Diaz Valerio1
Stefani M. Diaz Valerio1 Anja Poehlein1
Anja Poehlein1 Elzbieta B. Brzuszkiewicz1
Elzbieta B. Brzuszkiewicz1 Kai Nesemann2
Kai Nesemann2 Susanna A. Braus-Stromeyer2
Susanna A. Braus-Stromeyer2 Gerhard H. Braus2
Gerhard H. Braus2 Rolf Daniel1
Rolf Daniel1 Heiko Liesegang1*
Heiko Liesegang1*- 1Göttingen Genomics Laboratory, Department of Genomic and Applied Microbiology, Institute of Microbiology and Genetics, Georg-August-University, Gottingen, Germany
- 2Department of Molecular Microbiology and Genetics, Institute of Microbiology and Genetics and Göttingen Center for Molecular Biosciences, Georg-August-University, Gottingen, Germany
Verticillium wilt causes severe yield losses in a broad range of economically important crops worldwide. As many soil fumigants have a severe environmental impact, new biocontrol strategies are needed. Members of the genus Bacillus are known as plant growth-promoting bacteria (PGPB) as well as biocontrol agents of pests and diseases. In this study, we isolated 267 Bacillus strains from root-associated soil of field-grown tomato plants. We evaluated the antifungal potential of 20 phenotypically diverse strains according to their antagonistic activity against the two phytopathogenic fungi Verticillium dahliae and Verticillium longisporum. In addition, the 20 strains were sequenced and phylogenetically characterized by multi-locus sequence typing (MLST) resulting in 7 different Bacillus thuringiensis and 13 Bacillus weihenstephanensis strains. All B. thuringiensis isolates inhibited in vitro the tomato pathogen V. dahliae JR2, but had only low efficacy against the tomato-foreign pathogen V. longisporum 43. All B. weihenstephanensis isolates exhibited no fungicidal activity whereas three B. weihenstephanensis isolates showed antagonistic effects on both phytopathogens. These strains had a rhizoid colony morphology, which has not been described for B. weihenstephanensis strains previously. Genome analysis of all isolates revealed putative genes encoding fungicidal substances and resulted in identification of 304 secondary metabolite gene clusters including 101 non-ribosomal polypeptide synthetases and 203 ribosomal-synthesized and post-translationally modified peptides. All genomes encoded genes for the synthesis of the antifungal siderophore bacillibactin. In the genome of one B. thuringiensis strain, a gene cluster for zwittermicin A was detected. Isolates which either exhibited an inhibitory or an interfering effect on the growth of the phytopathogens carried one or two genes encoding putative mycolitic chitinases, which might contribute to antifungal activities. This indicates that chitinases contribute to antifungal activities. The present study identified B. thuringiensis isolates from tomato roots which exhibited in vitro antifungal activity against Verticillium species.
Introduction
Verticillium wilt occurs in a wide range of plant species including economical important crops. Symptoms include wilting, stunting, vascular discoloration, and early senescence, which cause an annual yield loss of billion dollars worldwide (Pegg and Brady, 2002; Fradin and Thomma, 2006). Causative agents are different soil-borne Verticillium species such as Verticillium dahliae and Verticillium longisporum. Verticillium longisporum has a narrow host range mainly infecting Brassicaceae (Zeise and von Tiedemann, 2002). Wilting symptoms or crop losses are only observed in the greenhouse (Zeise and von Tiedemann, 2002). In contrast, V. dahliae is known as a vascular pathogen with a broad host range including trees, legumes, ornamental crops, and vegetables such as tomato (Pegg and Brady, 2002; Luo et al., 2014). This fungus forms extremely outlasting melanized resting structures (microsclerotia) that are able to survive in soil for many years (Schnathorst, 1981). As consequence, the control of this phytopathogenic fungus is difficult. As many soil fumigants have a severe environmental impact, no pesticide which meets the standards of a sustainable agriculture is currently available to control the expansion of V. dahliae and V. longisporum (Frank, 2003; Depotter et al., 2016). Other control strategies such as crop rotation, the usage of resistant plant species, and soil solarization have only limited efficiency (Haas and Défago, 2005; Depotter et al., 2016). They are either ineffective, time-consuming, costly, may have a severe environmental impact, or even could affect human health (Angelopoulou et al., 2014).
Many plant-associated bacteria have beneficial effects on their host plant by increasing nutrient availability (Scherling et al., 2009) or by modulating the plant immunity (Jones and Dangl, 2006). Moreover, some of these plant growth-promoting bacteria (PGPB) have been used as biological control agents against plant diseases and pests (Ahemad and Kibret, 2014). However, common fungal antagonistic bacteria such as Pseudomonas species have only limited inhibitory impact on Verticillium due to the long-term persistence of Verticillium microsclerotia in soil (Angelopoulou et al., 2014). Members of the endospore-forming genus Bacillus possess a high potential as new fungal antagonists. They provide several advantages compared to other biocontrol agents such as (i) a better life-shell, (ii) a dry-product formulation which contains a lower contamination rate, (iii) established large-scale treatment, and finally, (iv) a cheap and easy usage (Katan, 1981; Fravel, 2005; Haas and Défago, 2005). Some Bacilli synthesize antifungal compounds such as cyclic lipopeptides produced by non-ribosomal peptide synthetases (NRPS), polyketide synthases (PKS) or mycolytic enzymes like chitinases (Swiontek Brzezinska et al., 2014; Aleti et al., 2015). Three families of Bacillus lipopeptides are known to confer an antifungal effect: surfactins, iturins, and fengycins (Ongena and Jacques, 2008). Strains of the species Bacillus thuringiensis (Bt) have been used as biological control agent against insecticidal crop pests for decades (Schnepf et al., 1998).
Bacillus thuringiensis (Bt) is a member of the Bacillus cereus sensu lato (Bcsl) complex, which comprises seven species (Priest et al., 2004) including the well-investigated name-giving species B. cereus (Bc), an opportunistic human pathogen. Other Bcsl members are B. anthracis (Ba), the etiological agent of anthrax, and B. weihenstephanensis (Bw). The latter is the only described species, which is able to grow under psychrophilic conditions (Lechner et al., 1998; Helgason et al., 2000). All Bcsl members share a highly conserved chromosomal backbone and have diverse extra-chromosomal elements (Priest et al., 2004). Especially Ba, Bc, and Bt strains showed high identity in a previous study (Priest et al., 1994). Despite their obvious similarity, specific phenotypic characteristics including the presence or absence of virulence genes have been used for differentiation (Helgason et al., 2000). However, the grouping of those species is in a still ongoing debate and it was suggested to reclassify this group to a single species (Okinaka and Keim, 2016). Bt is able to produce parasporal protein crystals consisting of δ-endotoxins (Schnepf et al., 1998). The insecticidal effect of entomopathogenic Bt is attributed to the production of these crystal toxins. In addition to its insecticidal effects, Rocha et al. (2014) reported that Bt serovar kurstaki can prevent the growth of the maize pathogen Fusarium verticilloides. However, a systematic investigation on different Bt strains isolated from root-associated soil as antagonists of Verticillium species has not been performed to date.
The objective of this study was to isolate members of the genus Bacillus to investigate their antifungal potential against two important phytopathogenic fungi differing in their host ranges. For this purpose, we generated a Bacillus strain collection of 267 isolates from tomato root-associated soil by using an enrichment method for Bt. Twenty Bacillus strains from this collection were selected based on unique morphological traits and further investigated for their antifungal activity against V. dahliae and V. longisporum. The phenotypic and taxonomic classification of the bacterial isolates was determined within the Bcsl species complex. The genomes of the 20 selected strains were sequenced and mined for genes related to crystal toxin as well as antifungal compound production. Moreover, the genome sequences were screened for cyclic lipopeptides produced by NRPS/PKS clusters and mycolytic enzymes such as chitinases. The abundance of the genera within the root-associated bacterial community composition of tomato plants was determined based on the bacterial 16S rRNA amplicon sequencing.
Materials and Methods
Soil Sampling and Extraction of Environmental DNA
Soil samples of Solanum lycopersicum were collected in a field plot near Göttingen (Germany, 51° 32′N, 9° 56′O) in June 2014. Samples of topsoil with 10 g each were taken next to roots (<3–5 mm) of three tomato plants and subsequently frozen at −80°C. Prior to DNA extraction, visible roots were removed. Afterwards, samples were treated with mechanical disruption using a microdismembrator (B. Braun Biotech International, Melsungen, Germany) for 3 min with 2000 rpm. Disrupted samples were dissolved in 600 μl sterile water. The DNA was directly extracted using the PowerSoil® DNA isolation kit (MO BIO Laboratories, Inc., Carlsbad, USA) as described by the manufacturer. The quality and purity of extracted DNA was tested with gel electrophoresis and NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, USA), respectively.
Amplification of 16S rRNA Gene
The V6–V8 region of the bacterial 16S rRNA gene was amplified with primers containing the Roche 454 pyrosequencing adaptors, keys as well as one unique MID per sample (underlined): F968 5′-CCATCTCATCCCTGCGTGTCTCCGAC-TCAG-(dN)10-AACGCGAAGAACCTTAC-3′ and R1401 5′-CCATCTCATCCCTGCGTGTCTCCGAC-TCAG-CGGTGTGTACAAGACCC-3′ (Nübel et al., 1996). The PCR (25 μl) contained following final concentrations: one-fold Phusion GC buffer (Thermo Scientific): 0.2 mM of each of the four deoxynucleoside triphosphates (Thermo Scientific), 0.2 μM of each primer, 0.04 U of Phusion high fidelity hot start DNA polymerase (Thermo Scientific), 5% DMSO and ~4 ng of the isolated soil DNA as template. The following thermal cycling scheme was used: initial denaturation at 98°C for 30 s, 30 cycles of denaturation at 98°C for 15 s, annealing at 53°C for 30 s, extension at 72°C for 30 s, followed by an additional extension step at 72°C for 2 min. Negative controls were performed using the reaction mixture without template. Genomic DNA of Bacillus was used as positive control. Three independent PCRs were performed per sample. Obtained PCR products were controlled for appropriate size and subsequently purified using the peqGOLD Gel Extraction Kit (Peqlab, Erlangen, Germany, now VWR) as recommended by the manufacturer. PCR products were quantified using the Quant-iT dsDNA HS assay kit and a Qubit fluorometer as recommended by the manufacturer (Thermo Scientific). Purified PCR products from the three independent PCRs were subsequently pooled in equal amounts. The Göttingen Genomics Laboratory determined the 16S rRNA gene sequences employing the Roche GS-FLX+ pyrosequencer with Titanium chemistry as recommended by the manufacturer (Roche, Mannheim, Germany).
Processing and Analysis of 454 Pyrosequencing Derived Data
Pyrosequencing derived 16S rRNA gene data were preprocessed with QIIME version 1.8 (Caporaso et al., 2010). Preprocessing included the removal of short reads (<300 bp) as well as reads containing long homopolymer stretches (>8 bp) and too many primer mismatches (>3 bp) in the forward primer. Filtered data was subsequently denoised employing Acacia version 1.53b (Bragg et al., 2012). Remaining primer sequences were truncated employing cutadapt version 1.0 (Martin, 2011). Chimeric sequences were removed using USEARCH version 7.0.1090 (Edgar, 2010). For this purpose, sequences were first dereplicated in full-length mode and putative chimeras were initially removed using the UCHIME algorithm in de novo mode and subsequently in reference mode using the most recent SILVA database (SSURef 119 NR) as reference dataset (Edgar et al., 2011; Klindworth et al., 2013). Afterwards, processed sequences of all samples were joined and clustered in operational taxonomic units (OTUs) at 3% genetic dissimilarity according to Wemheuer et al. (2012) employing the UCLUST algorithm (Edgar, 2010). To determine taxonomy, a consensus sequence for each OTU was classified by BLAST alignment against the Silva SSURef 119 NR database (Camacho et al., 2009). Alpha diversity indices were calculated with QIIME as described by Wemheuer et al. (2014). Rarefaction curves were calculated in QIIME and subsequently interpolated in R (R Core Team, 2014) using the “drc” package [https://cran.r-project.org/web/packages/drc/]. The statistical analysis was performed in R.
Isolation and Enrichment of Bacteria
Bacterial strains were obtained from root-associated soil samples. Soil samples were enriched for Bacillus strains, especially for Bt subspecies as described by Patel et al. (2011) using a modified glucose yeast extract salt (GYS) sporulation medium, which contained the following ingredients per liter: 1 g glucose, 2 g yeast extract, 2 g NH4(SO4)2, 0.06 g MnSO4, 0.4 g MgSO4·7H2O, 0.08 g CaCl2 and 5 g KH2PO4. Resulting colonies were picked with sterile toothpicks and inoculated in 96-deep well plates containing 2 ml two-fold concentrated lysogeny broth (TLB; Bone and Ellar, 1989). After overnight shaking at 30°C with 50 rpm (Orbitron S-000119510, Infors HT, Bottmingen, CHE), all colonies were stamped on lysogeny broth (LB; Sambrook and Russell, 2001) agar plates using a sterile 96 deep well plate replicator. The plates were incubated at 30 and 21°C for 24 h.
Phenotypic Analysis of Isolates
Specific growth of cells was monitored after incubation at 30°C for 24 h using Bino Olympus SZX12, Olympus SC30 camera and the Olympus Cell Sense software. Different media were tested, nutrient rich liquid LB as well as modified solid nutrient limiting Simulated Xylem Medium (SXM). SXM was prepared according to (Dixon and Pegg, 1971) which contained following ingredients per liter: 2 g pectin, 4 g casein, 20 ml AspA (50x), 2 ml MgSO4 (1M), 1 ml trace elements (1000x; de Serres and Hollaender, 1982), and 20 g agar. AspA (50x) contained the following ingredients per liter: 300 g NaNO3; 26 g KCl and 76 g KH2PO4. Strains were tested for hemolytic activity on Columbia blood agar with Oxoid sheep-blood “plus” (Thermo Scientific). Hemolytic activity was monitored and measured after 48 h.
Classification of Isolated Bacterial Strains
Genomic DNA of each strain was extracted from overnight cultures grown in LB medium at 30°C with 150 rpm (New Brunswick Incubator Shaker Innova 2300, Neu-Isenburg, Germany) by using the MasterPure™ Complete DNA Purification Kit (Epicentre, Madison, USA) according to the manufacturer's instructions. The purified DNA served as template for multi-locus sequence typing (MLST) amplification by PCR according to Priest et al. (2004). Primers for seven house-keeping genes (glpF, gmk, ilvD, pta, purH, pycA, and tpi) are listed in Supplementary Table 1. The PCR mixture (50 μl) contained following final concentrations: one-fold OptiBuffer, 3 mM MgCl2, 0.04 U BIO-X-ACT™ short DNA polymerase (Bioline, London, UK), 1 mM deoxynucleoside triphosphates (Thermo Scientific), 0.4 μM of each primer, sterile water, and ~2 ng of bacterial DNA as template. Thirty cycles were conducted in a Mastercycler (Eppendorf, Hamburg, Germany). An initial denaturation at 98°C for 5 min was followed by 45 s denaturation at 98°C, 45 s annealing at 65°C, and 30 s primer extension at 72°C with a final step of 72°C for 5 min. PCR products were controlled for appropriate size and purified employing the QIAquick PCR purification kit (QIAGEN, Hilden, Germany) and eluted in 35 μl sterile water. Quantification of PCR products was performed via Nanodrop 1000 Spectrophometer (Thermo Scientific). The Göttingen Genomics Laboratory determined the sequences of the PCR products. For classification of the new isolates, all MLST genes from Bcsl representative genomes at time of analysis were considered. The corresponding sequences were obtained from GenBank hosted at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/genome/genomes/486?). To exclude putative Ba isolates, all strains were tested for specific Ba virulence factors. Each MLST gene was aligned to determine the corresponding regions of each housekeeping gene and was trimmed to shortest shared region, resulting in orthologous sequences from 298 to 394 bp, as previously described (Jolley et al., 2004). Analysis was performed with concatenated DNA sequences of all seven gene loci. For construction of a phylogenetic tree, MEGA software 7.0.14 was used (Kumar et al., 2016). Sequence alignment was performed using the ClustalW algorithm and a phylogenetic tree was constructed by using the neighbor-joining method (Saitou and Nei, 1987). The robustness of the tree was evaluated by bootstrap analysis with 1000 resamplings.
Whole Genome Sequencing and Assembly
DNA extracted from isolated bacteria was subjected to whole-genome sequencing (Table 1) using the MiSeq sequencer (Illumina, San Diego, USA). For this purpose, Nextera_XT (Illumina, San Diego, USA) paired-end libraries (2 × 301 bp) were prepared according to the manufacturer's protocols. Resulting reads were quality-filtered with Trimmomatic 0.32 (Bolger et al., 2014) and evaluated using Fastqc (Bahabram Informatics, Babraham Institute; UK). Spades 3.5.0 (Bankevich et al., 2012) was used to assemble processed reads. For scaffolding, the resulting contigs were aligned to reference genomes Bt 407 (CP003889) and Bw KBAB4 (CP000902) using the Mauve Genome Alignment software (Darling et al., 2004). Sequencing results and genome characteristics are summarized in Table 1. Automatic annotation was carried out with the IMG-ER (Integrated Microbial Genomes-Expert Review) system (Markowitz et al., 2009) and with Prokka v1.9 (Seemann, 2014) using Bt 407 (Sheppard et al., 2013) as species reference and a comprehensive toxin protein database (including Cry, Cyt, Vip, Sip proteins). The Prokka pipeline was applied using gene calling by prodigal (Hyatt et al., 2010), rRNA genes and tRNA genes identification with RNAmmer 1.2 (Lagesen et al., 2007) and Aragorn (Laslett and Canback, 2004), respectively. Additionally, signal peptides were identified with SignalP 4.0 (Petersen et al., 2011) and non-coding RNA species with an Infernal 1.1 search against the Rfam database (Eddy, 2011).
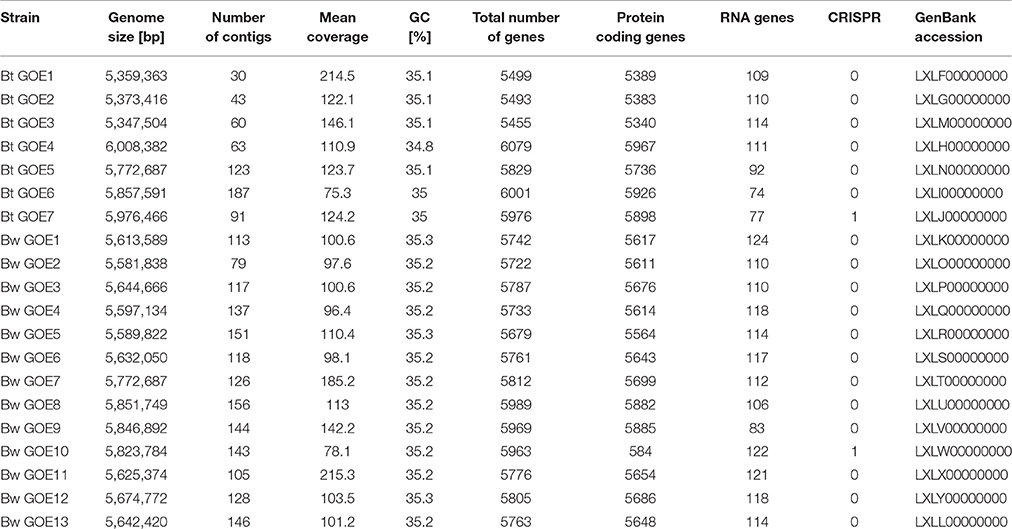
Table 1. Genome statistics of the 20 selected isolates.
Genome Analysis of Bacterial Isolates
The genomes of all strains were screened for genes encoding Bt-specific toxins, such as crystal toxins, cytolytic toxins, vegetative insecticidal protein-toxins, and secreted insecticidal protein toxins (Cry, Cyt, Vip, and Sip toxins), and for candidate-genes for the production of antifungal secondary metabolites and chitinases. For identification of Cry-toxins, all protein sequences derived from the genome sequences were scanned using HMMSCAN implemented in HMMER v.3.1b2 (hmmer.org; Eddy, 1998) against a HMM-profile database. The profile database was generated from holotype Cry toxin sequences retrieved from the official BT Toxin Nomenclature website (http://www.btnomenclature.info/) and Uniprot (Bateman et al., 2015), respectively. To generate toxin-specific HMMs, all toxin sequences were grouped by Markov Cluster Algorithm v.12068 (Enright et al., 2002; van Dongen, 2007) according to the default options with an inflation value (−I) of 10.0. Representative members of each cluster were aligned by ClustalW v.1.83 (Thompson et al., 1994) and used as input to build the model database. After optimization of the models for sensitivity and specificity, the resulting HMMs were used for detection. Resulting group hits were further classified accordingly to the Cry Toxin Nomenclature. For the detection of Sip, Vip, and Cyt toxins, all protein sequences annotated accordingly from Uniprot (Bateman et al., 2015), National Centre for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/), and the holotype-toxin list (http://www.btnomenclature.info/) were controlled for duplicates by 100% sequence identity. Only one representative was kept to create a BLASTp database, which was performed for each genome with an e-value of 1e−50. The analysis for secondary metabolites, i.e., polyketides, alkaloids, terpenes, phenanzines, microlides, and non-ribosomal antibiotic peptides was performed with antiSMASH 3.0 available at (https://antismash.secondarymetabolites.org/) (Weber et al., 2015). Chitinases were identified and compared to chitinase sequences of other Bcsl members. They were aligned using the ClustalW algorithm (Thompson et al., 1994) in MEGA7 (Kumar et al., 2016). To construct a phylogenetic tree, the Neighbor-Joining method (Saitou and Nei, 1987) and evolutionary distances were computed by the Maximum Composite Likelihood method (Tamura et al., 2004). InterPro (Mitchell et al., 2015) was used to group detected chitinases into chitinase families and for the prediction of domains, signatures, and active sites.
Co-cultivation Assay
To evaluate the antifungal potential of 20 phenotypically diverse Bacillus strains, co-cultivation assays with the two phytopathogens V. dahliae JR2 (Fradin et al., 2009) and V. longisporum 43 (Zeise and von Tiedemann, 2002) were conducted. All used strains are listed in Table 2. The insecticidal Bt subsp. kurstaki 2 and the Bt subsp. israelensis ONR60A strain were obtained from the Bacillus Genetic Stock Centre (BGSC; Columbus; USA) and served as reference strains. Three nematocidal strains (Bt MYBT18246, Bt Bt18247, and Bt Bt18679) and one insecticidal Bt strain (Btt) were used (Milutinovic et al., 2013; Masri et al., 2015). Escherichia coli DH5α served as negative control. Co-cultivation assays with two phytopathogenic fungi and selected Bacillus strains were performed using three different media. For standard bacterial cultivation, LB was used. The complex pectin containing SXM with limited nutrition tries to simulate the nutrient conditions which are available for a fungus in a plant. Due to the restriction of a synthetic medium it is limited in simulating the natural plant environment, where water, minerals, organic acids and ~500 μm amino acids are available in highly specific concentrations (Singh et al., 2010). The potato dextrose medium (PDM; Carl Roth GmbH, Karlsruhe, Germany with 5 g/L agar), a full medium containing potato starch and glucose, were used for optimal cultivation of Verticillium (Gams et al., 1998). Solid media were prepared containing 2% agar. Approximately 1 × 105 harvested Verticillium conidiospores (Beckman Coulter Counter Size Analyzer, Krefeld, Germany) were homogeneously inoculated per plate using sterile glass beads (2.85–3.45 mm in diameter). A hole (diameter = 0.9 cm) was excised in the middle of the plate. This in vitro setting was designed to study Bacillus/Verticillium interactions under controlled conditions simulating the substrate supply of a plant and animal-associated habitat.

Table 2. Organisms used in this study.
Bacteria were inoculated in 5 ml LB rotating with 120 rpm (New Brunswick Incubator Shaker Innova 2300, Neu-Isenburg, Germany) overnight at 37°C (Bt strains) or 25°C (Bw strains). Isolates were centrifuged for 2 min with 6000 rpm and washed with 2 ml sterile water. Afterwards, 5 ml of LB, PDM, or SXM were inoculated and adjusted to an OD595 of 0.1. Main PDM Bw cultures were inoculated to an OD595 of 0.01 and grown overnight at 25°C to an OD595 of 1. All other main cultures were grown at 30 or 25°C rotating with 120 rpm (New Brunswick Incubator Shaker Innova 2300, Neu-Isenburg, Germany) to an OD595 of 1. When the optical density was reached, 60 μl of the respective bacterial cultures were added into the hole in the plates. Plates were incubated at 25°C for 7 days. Zones of fungal growth inhibition were measured and quantified. Media without bacterial cells were used as positive controls for fungal growth. Each treatment was performed in technical triplicates and biological duplicates. To analyze possible differences between the isolates, a repeated measures ANOVA (Crawley, 2007) was conducted in R due to spatial pseudoreplication.
Deposition of Isolated Strains and Genome Sequences
Sequence data were deposited in the Sequence Read Archive (SRA) of the NCBI under the accession number SRA401353. Genome sequence data were deposited in JGI and GenBank (Table 1). Strains are available on request (Table 2).
Results and Discussion
Soil Bacterial Communities of Tomato Plants
Root-associated bacterial community composition and diversity were assessed by amplicon-based analyses of the V6-V8 region of the bacterial 16S rRNA gene. After quality filtering, denoising, and removal of potential chimeras and non-bacterial sequences, a total of 13,596 high-quality sequences were retrieved and used for further analyses. These sequences were grouped into 3994 OTUs. Calculated rarefaction curves (Supplementary Figure 1) as well as the mean coverage at species level (Table 3) revealed that the majority of bacterial community was recovered by the surveying effort. Richness (number of observed OTUs) and diversity (Shannon indices) for bacterial communities ranged from to 673 to 740.6 and 5.72 to 5.82, respectively (Table 3). All sequences could be classified below phylum level.
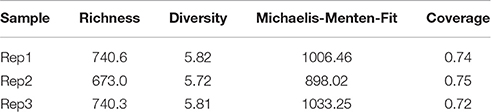
Table 3. Diversity (represented by the Shannon index H') and richness (number of observed OTUs) of tomato-associated bacterial communities.
Eleven abundant phyla (>1% of all sequences across all samples) were present in each soil sample and accounted for more than 96% of all bacterial sequences analyzed (Figure 1). Proteobacteria (30.0%), Acidobacteria (22.6%) and Actinobacteria (15.2%) were the most abundant (>1% abundance of all sequences) bacterial phyla. The Proteobacteria were mainly represented by Beta- and Gammaproteobacteria. Other prominent phyla were Chloroflexi (8.8%), Verrucomicrobia (3.3%) and Firmicutes (1.3%). Although their relative abundance is variable, these phyla have been previously found in different soils (Shange et al., 2012) and in the rhizosphere of different plant species (Romero et al., 2014; Pii et al., 2016). Li J. G. et al. (2014) investigated bacterial communities in roots and rhizosphere soils of healthy and diseased tomato plants and found that Proteobacteria was the most abundant phylum across all samples, followed by Actinobacteria and Bacteroidetes. This is in line with a study of Bulgarelli et al. (2015) on bacterial communities in bulk soil, rhizosphere, and roots of barley. In contrast to the above-mentioned findings, Verrucomicrobia (24%), Acidobacteria (23%) and Proteobacteria (17%) were the major taxonomic groups in rhizosphere bacterial communities of tomato (Romero et al., 2014).
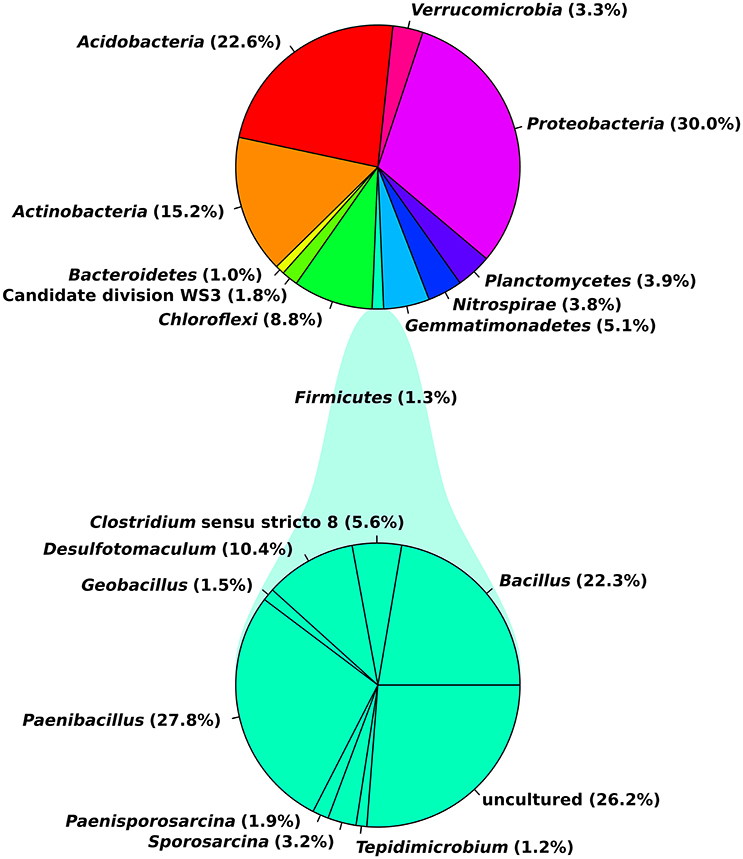
Figure 1. General soil bacterial community composition at phylum level and for the Firmicutes at genus level. Plots were generated in R.
In our study, observed genera included Blastocatella (2.6%), Massilia (1.64%) Gaiella (2.0%), Nitrospira (1.76%), Paenibacillus (0.34%), Bacillus (0.27%), Bradyrhizobium (0.17%), Burkholderia (0.04%), and Pseudomonas (0.006%; Supplementary Table 2). Most sequences of the Firmicutes were assigned to the two genera Paenibacillus (27.8%) and Bacillus (22.3%). Several of the observed genera including Bacillus, Burkholderia, and Pseudomonas are reported as the most significant phosphate-solubilizing bacteria (Bhattacharyya and Jha, 2012). Moreover, members of the genera Bacillus, Paenibacillus, Bradyrhizobium, Pseudomonas, and Burkholderia are known for their plant growth-promoting functions and/or their use as biocontrol agents against different phytopathogens and pests (Bhattacharyya and Jha, 2012; Glick, 2012). In a previous investigation on cultivable bacteria associated with tomato leaves, Bacillus (Firmicutes) showed strong in vitro antifungal activity against three important pathogens of tomato (Botrytis cinerea, Fulvia fulva, and Alternaria solani; Enya et al., 2007).
Classification of Bacterial Isolates
The whole Bcsl taxonomy is recently discussed based on the different methods that are used for strain classification (Okinaka and Keim, 2016). Depending on the specific focus of the study, pathogenic properties or taxonomic features are used for classification resulting in an inconclusive taxonomy. We tried to address this question by performing MLST analysis as described by Priest et al. (2004) in combination with the analysis of phenotypic, biochemical and pathogenic characteristics. Our strain collection comprised 267 Bacillus strains. Twenty isolates with diverse colony morphologies were selected for further analysis.
For the genome analysis of strains, the DNA was isolated and sequenced. Accession numbers and sequencing metadata are summarized in Table 1. The genome sizes ranged from 5.3 to 6.0 MB with a mean GC-content of 35%, which is typical for members of the Bcsl group (http://www.ncbi.nlm.nih.gov/genome/genomes/486?). The total number of genes varied from 5456 to 6080 and the number of identified RNA genes from 157 to 194. As the taxonomy of the Bcsl group is complex, the classical 16S rRNA phylogeny used for sequence based species assignments was ineffective for differentiation within this group. Based on MLST, 7 B. thuringiensis (Bt GOE1-7) and 13 B. weihenstephanensis (Bw GOE1-13) strains were identified (Table 1, Figure 2). None of the isolates clustered in close proximity to the human pathogen Ba or to any of the other known human pathogenic strains from the Bcsl species group. The analysis of similarity using the known Ba toxin genes or capsule genes such as lef, cya, pagA, and capA-capC showed that none of these genes are present in the new isolates. Additionally, there were no similarities to the four species-specific prophages (lambda01-lambda04), which are used for the PCR-based identification of Ba strains (Kolstø et al., 2009). All seven Bt isolates encode genes for the PlcR regulator with 100% sequence identity to Bt-reference sequences whereas no Bw isolate contained a plcR gene (Supplementary Table 3).

Figure 2. Phylogenetic analysis of 20 Bcsl group isolates. The 20 newly isolated strains are depicted in bold. (Blue triangle) Representatives of the Bcsl group species members. Strains which have been tested and showed an effect on V. dahliae JR2 or both V. dahliae JR2 and V. longisporum 43 are indicated by a diamond or square, respectively. Effects on V. dahliae JR2 (red diamond): inhibitory isolates, (beige diamond) inhibitory reference strains. Effect on V. dahliae JR2 and V. longisporum 43 (red square): inhibitory isolate, (beige square) inhibitory reference strain, (white square) no inhibitory effect. The phylogenetic analysis was performed using MEGA7 (Kumar et al., 2016), details are described in the Materials and Methods Section.
The morphology of Bcsl members was previously described in general as similar: the colony shape is irregular with undulate or curled margins (De Vos et al., 2009). Moreover, the colonies are flat to raised and opaque while Bm and Bp exhibit a mycoid to rhizoid growth with hairy-looking adherent colonies (De Vos et al., 2009). The morphological analysis of 20 isolates revealed two different growth-types in liquid as well as on solid media (Figure 3). The first growth type is represented by 7 Bt and 10 Bw isolates which formed circular to weakly irregular colonies with entire or undulate edges. The surface texture was ground glass to granular. The Bt isolates exhibited larger single colony sizes compared to Bw strains. These 17 strains were hemolytic positive on blood agar (Table 4, Figure 3). In liquid LB medium, the bacterial solutions were white yellowish and grew equally cloudy. In contrast, three Bw isolates (Bw GOE8-10) exhibited a Bm-like colony shape, with rhizoid growth on solid medium. The cells grew adherent and covered rapidly the whole agar plate as it is known for Bm which is in contrast to our MLST analysis (Figure 3; Di Franco et al., 2002). Moreover, the isolates displayed an invasive growth into the media plates (Figure 3). To the best of our knowledge, this has not been described previously for Bw strains. In addition, hemolytic activity could not be determined. In liquid medium, the strains produced an aggregation of clumps comparable to Bm (Di Franco et al., 2002). All Bw isolates showed mesophilic growth at 30°C instead of the described psychrophilic growth (Di Franco et al., 2002).

Figure 3. Representative colony morphology phenotypes of tomato isolates. The growth of three different strains, Bw GOE1, Bw GOE8, and Bt GOE7, was tested on several media. (Top row) Strains in liquid LB media, (2nd, 3rd, and 4th row) close up of single colonies on solid SXM media, pre/after scraping and washing with spatula and sterile water, and cross section of the agar. (5th and bottom row) Growth on solid blood agar and close up of single colonies. Strain abbreviations are explained in Table 2.

Table 4. Hemolytic activity assay.
To determine the pathogenic properties of the isolates, we screened for virulence factors specific for the Bt species group (Cry, Cyt Sip, and Vip toxins). Within the MLST tree (Figure 2), the Bt isolates clustered with B. thuringiensis reference strains, with present or absent Cry toxins. In contrast, all Bw isolates clustered with B. weihenstephanensis references (Figure 2). The strains Bt Bc601, Bt BMB171, Bt CTC, Bt HD1011, Bt HD682, Bt str. Al Hakam, and Bt HD571 contained no homologs to any known Cry, Cyt, or Vip toxins (Figure 2). Notably, even in the type strain Bt serovar konkukian str. 97-27, which was isolated from a necrotic human wound, no full-length hits to known Cry, Cyt, or Vip toxins were identified (Han et al., 2006). Only Bt GOE6 contained a gene with similarity to Cry46. In all other isolates, no homologs to any known Cry, Cyt, Sip, or Vip toxins were detected using BLASTp and HMM models. Moreover, proteins sharing a domain with Cry6 and Cry22 were detected in all Bt and Bw isolates. Modeling and detection of known as well as for new Cry toxins is not trivial based on their variable structures. Cry toxins can be sub-grouped into three classes: Three-domain toxins, Bin-toxins and Mtx-toxins (de Maagd et al., 2003). Additionally, some Cry toxins are phylogenetically unrelated and unique such as Cry6, Cry22, Cry34, Cry37, Cry55, and Cry46 (Palma et al., 2014). It is possible that our strains encode novel Cry toxins. The proteins sharing a domain with Cry6 and Cry22 and might represent new toxin candidates. However, Cry toxins are often encoded on mobile elements located on plasmids (González et al., 1982). Cry toxin-encoding Bt plasmids may be instable and thus can get lost within cultivation (Masri et al., 2015; Sheppard et al., 2016).
The inconsistent combination of taxonomic relevant features supports the hypothesis of Okinaka and Keim (2016) that the Bcsl species group is complex and that exclusive phenotypic and biochemical characterization may be misleading. The authors suggest that the whole Bcsl complex should be considered as a single species. Investigation of only the phenotypic characteristics of the strains would lead to a mis-classification of Bt isolates, as no known Cry toxins were detected. Moreover, the invasive growing Bw strains would be mis-classified to the species Bm or Bp. However, there are examples for the challenging taxonomic classification of Bcsl members. Bt serovar, navarrensis, bolivia, and vazensis have been classified as Bt based on their ability to produce Cry toxins. They showed the typical psychrotolerant growth and encode the cspA gene signature, which is species specific for Bw (Soufiane and Côté, 2009). We classified our isolates as 7 Bt and 10 Bw based on our MLST study combined with phenotypic features (Figure 2). The three special Bw isolates (Bw GOE8-Bw GOE10) combined several morphological as well as genomic characteristics of the species Bm, Ba, and Bw indicating that they might represent a distinct subspecies within the species Bw (Figure 2). In conclusion, our findings indicate that a combination of MLST analysis and phenotypic, biochemical, and pathogenic classification is the only possibility to distinguish between members of the Bcsl complex.
In vitro Antagonistic Properties of Root-Associated Bacilli toward Verticillium
We further investigated the antagonistic potential of new isolated Bacilli strains against two phytopathogenic Verticillium species with different host ranges. In total, 20 Bacillus isolates and 6 Bt reference strains were tested for their ability to suppress the haploid tomato pathogen V. dahliae JR2 (Fradin et al., 2009) or the diploid rapeseed pathogen V. longisporum 43 (Zeise and von Tiedemann, 2002; Tran et al., 2013) on three different media (Table 2). Not all Bt reference strains were able to suppress V. dahliae JR2 and showed a broad range of different interaction effects against V. longisporum 43 indicating that antifungal activity is a property of a particular Bt strain and not of the whole species (Supplementary Table 4, Supplementary Figures 2, 3). The co-cultivation assays of Bacillus isolates revealed specific phenotypic effects for the antagonistic bacteria as well as for the phytopathogens (Figure 4). The different bacterial isolates varied in their ability to inhibit the mycelia growth of either V. dahliae JR2 or V. longisporum 43. A significant in vitro antagonism was observed for all Bt isolates against V. dahliae JR2 on PDM, excluding Bt GOE6 compared to the tested Bt control strains (Bt Bt18247, Bt MYBT18246, 4Q1) which exhibited no inhibitory effect (Figure 5A, Supplementary Table 5). A clear inhibition zone without Verticillium mycelium, sometimes with a slight formation of microsclerotia, was detected (Supplementary Figure 2). Additionally, V. dahliae JR2 showed an altered phenotype compared to the fungal control by the building of a strong white air mycelium around the bacteria (Figure 4C). The antagonistic effect of Bt varied but was consistent in all biological replicates and had a mean ranging from 1.5 to 8.6 mm (Figure 5). In contrast to all other tested Bt isolates, which exhibited an antagonistic effect on PDM, isolate Bt GOE6 showed a significant antagonism against V. dahliae JR2 only on LB (Figure 4A, Figure 5, Supplementary Table 5). Notably, none of the strains belonging to Bw showed an inhibitory effect on V. dahliae JR2 or V. longisporum 43 (Supplementary Table 4). In comparison, a weaker antagonistic effect of bacteria on V. longisporum 43 was recorded. All strains which had an effect on V. dahliae JR2 lead to an altered phenotype of V. longisporum 43 on PDM. The bacteria induced a stronger melanization in V. longisporum 43, which could be observed on the back of the plate as a black ring around the bacteria (Figure 4D). Only for strain Bt GOE7 an inhibitory effect with high variance in replicates was observed compared to all other tested strains (Figure 5, Supplementary Table 5). On LB, Bt GOE4 and Bt GOE6 exhibited a significant inhibitory effect against V. longisporum 43 compared to all other tested strains (Figure 5, Supplementary Table 5). Additionally, 4D2 showed an inhibitory effect aginst V. longisporum 43 as well but with an increased variance in biological treatments. Moreover, Bt GOE6 had a significant higher inhibitory effect against V. longisporum 43 compared to 4D2 and Bt GOE4. Significant inhibitory effects against both phytopathogens, V. dahliae JR2 and V. longisporum 43, were only observed for Bt GOE4 and Bt GOE6 on LB (Figure 5, Supplementary Table 5). However, two additional phenotypic effects of bacteria were observed: (a) biofilm production of bacteria with varying inhibition effects on fungi and (b) building of hyphae-like growth structures of bacteria leading to a suppression or growth reduction of both phytopathogens. Effect (a) was detected for all Bt isolates, which build moderate biofilms. Bw isolates conferred only a weak biofilm production with no suppression effect, excluding Bw GOE8-GOE10. Effect (b) was observed for isolates Bw GOE8-GOE10 on LB and SXM (Figure 4B, Supplementary Table 4). The hyphae-like structures or rhizoid growth enables the bacterium to invade the same habitat as Verticillium. This might strengthen the competitive access of Bacilli for nutrients, leading to suppression or reduction of fungal growth to different extents. As the isolated Bt strains suppressed V. dahliae JR2 growth, this indicates that tomato-associated Bt strains are able to inhibit phytopathogens. In contrast, the effect on the foreign plant pathogen V. longisporum 43 was in general weaker or even not detectable. We assume that different host-range specificities play an important role in Verticillium species, resulting in two different life strategies. Verticillium dahliae is haploid and comprises strains with a broad host range (Inderbitzin et al., 2011) whereas V. longisporum is amphidiploid (hybrid of V. dahliae and an unknown haploid Verticillium species) with narrow host range infecting mainly Brassicacae (Depotter et al., 2016). This could lead to long-term adaptation of V. longisporum resulting in an increased fitness, as V. longisporum has to persist in soil waiting for its specific host plant. Verticillium dahliae is able to colonize a broad range of host plants and thus has to cope with different environmental conditions. We suggest that this results in a reduced adaptation potential for the different types of host-residing pathogens. An opportunity to enter the next potential host are spores or the formation of resting structures, such as microsclerotia, which are formed under stress conditions and can easily spread by wind or survive in the ground.
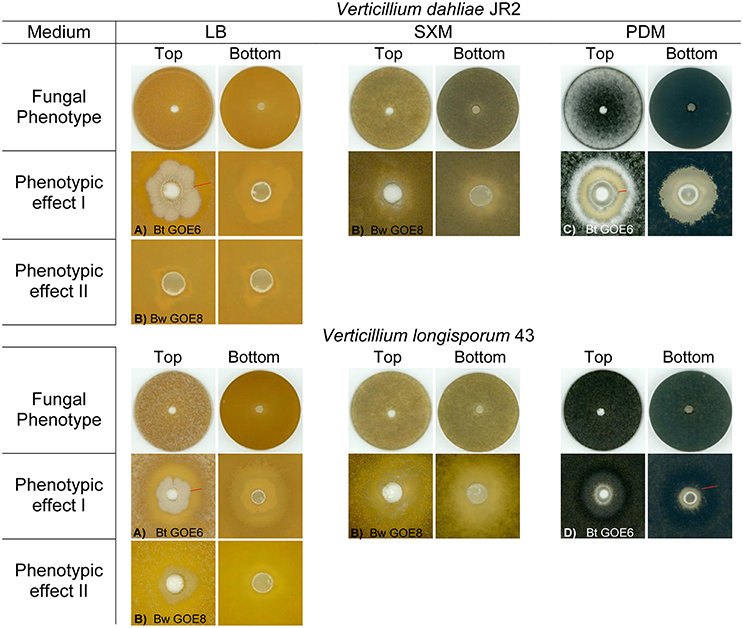
Figure 4. Phenotypic effects of selected bacterial strains against Verticillia. Fungal phenotypes without bacteria are displayed in the 1st and 4th row. Specific interaction effects of V. dahliae JR2 or V. longisporum 43 and antagonistic bacterial strains on different media are depicted: (A) Bacteria produce biofilm with or without inhibition of the fungus, (B) Bacteria produce hyphae-like structures where the growth of the fungus is reduced, (C) White air mycelia is build by the fungus around the bacteria which inhibits V. dahliae JR2, (D) V. longisporum 43 showed a strong melanization surrounding the bacterial application site visible as a dark ring. Inhibition zone and melanization zone of fungi are depicted by a red bar. Strain abbreviations are explained in Table 2.
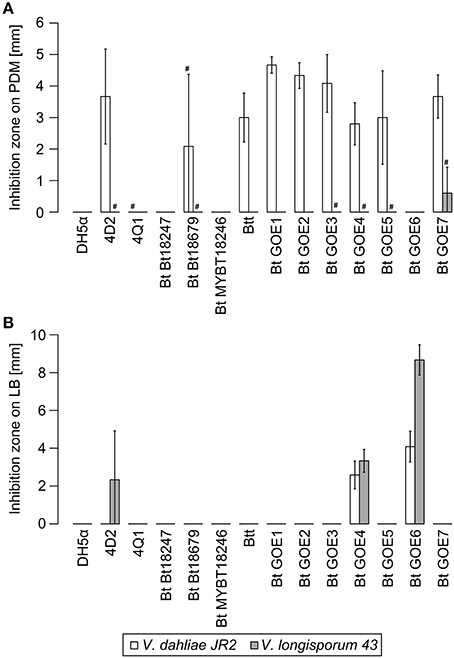
Figure 5. Inhibition of V. dahliae JR2 and V. longisporum 43 by Bacillus isolates. Mean inhibitory effects of all tested strains are displayed in [mm]: (A) Effect on PDM medium, (B) Effect on LB. E. coli DH5α served as negative control. Variation bars on the histogram corresponds to standard deviation. # indicate strains where a phenotypic effect was observed, but no measurable inhibition was quantified due to study limitations. Plots were generated in R. Strain abbreviations are explained in Table 2.
On SXM, only minor and inconsistent effects were observed around the bacterial application site and not further quantified. Instead of an inhibition, a reduced formation of fungal mycelium was observed (Supplementary Figures 2, 3). Nonetheless, the ability of a bacterial strain to inhibit fungal growth differed depending on the medium. Using different phytopathogenic interaction partner had also an impact on phenotypic effects of bacteria (Supplementary Table 4). Generally, the inhibitory effect on V. dahliae JR2 was always more prominent than the effect on V. longisporum 43 (Figure 4, Supplementary Figures 2, 3). In particular, on PDM, the fungus is able to sense the bacteria or antifungal substances. This leads to a strong physiological reaction of V. dahliae JR2 resulting in an increased production of air-mycelium in distance to the bacterial application site. Only in individual cases, a reduced number of microsclerotia was detected (Supplementary Figure 2; Bt GOE5, Bt GOE7). For V. longisporum 43, an increased production of microsclerotia was observed indicating an induced stress response by bacteria. However, SXM is also a rich medium containing the complex heteropolysaccharide pectin, which is more difficult to use for bacteria. We suggest that this reduce or enhance the growth efficiency of bacteria or fungi, respectively. In contrast, LB medium is favored by bacteria, which could explain the biofilm production, whereas both fungi built minor mycelium (Figure 4).
Secondary Metabolites of Root-Associated Bacilli
The secondary metabolite gene prediction tool (AntiSMASH 3.0; Weber et al., 2015) was used to perform a comparative genome analysis and identify candidates contributing to the observed antifungal effects. In total, 304 gene clusters including 101 NRPS gene clusters and 203 RiPP gene clusters were identified (Table 5). In each genome, secondary metabolite gene clusters were detected, including genes assigned to the production of siderophores and terpenes. The gene clusters assigned to siderophore synthesis showed similarity (80–83%, BGC0000942) to a petrobactin biosynthetic gene cluster and a bacillibactin cluster (38–46%, BGC0000309.1). Both are catecholate siderophores, which are produced under iron-limited conditions, and present in Ba and other Bcsl members. Li B. et al. (2014) reported that the transcription of bacillibactin genes was upregulated when strains were confronted with fungal pathogens. Bacillibactin-deficient mutants exhibited no or reduced antagonistic effects. Although the suppression could not be observed in each treatment, the findings indicate that bacillibactin plays a supporting role in the suppression of the two different Verticillium species (Li B. et al., 2014).
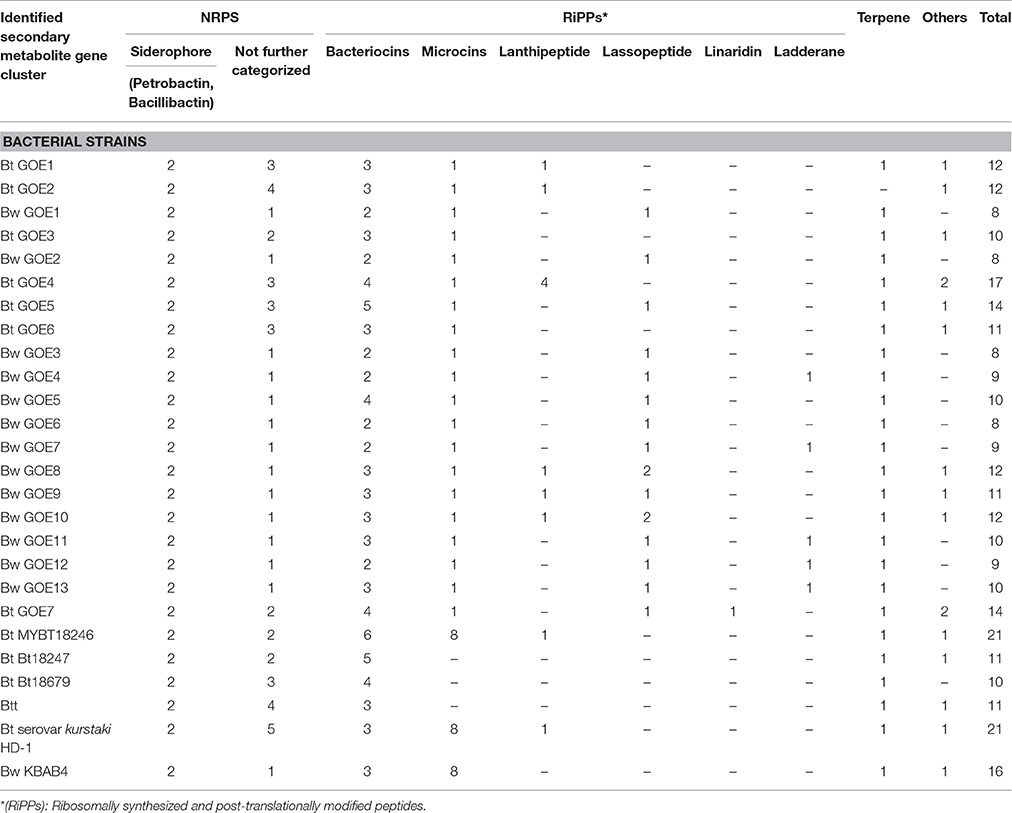
Table 5. Identified secondary metabolite gene clusters of bacterial strains.
It was suggested that petrobactin could contribute to virulence of Ba (Lee et al., 2011), but antifungal activity was not discussed. A number of plants including tomato are known to produce terpenes for deterring or attracting herbivores, parasites, and predators (Lange et al., 2000; Martin et al., 2003). Terpene assigned gene clusters were identified in all strains and shared only low similarity (11–17%, BGC0000916.1) with molybdenum cofactor biosynthesis genes. Bt GOE4 showed the highest number of detected secondary metabolites and antimicrobial peptides with similarity to known gene clusters with antifungal activity (Figure 6). One gene cluster putatively producing the antifungal zwittermicin A (BGC0001059) was detected. It belongs to the class of antibiotics of type I PKS gene clusters, which is known to suppress plant diseases (Handelsman et al., 1990). Zwittermicin A is a linear aminopolyol, which was first identified in B. cereus UW85 (Silo-Suh et al., 1994). The cluster was only identified in Bt GOE4 and in the reference genome Bt serovar kurstaki HD-1. In Bt GOE4, the cluster differs by lacking the five kab (kabR; kabA-kabD) genes, which are important for the kanosamine production. Kanosamine is not important for the synthesis of zwittermicin A, but it is also known to exhibit fungicidal activities (Janiak and Milewski, 2001). In these strains, a thuricin-like cluster (66% similarity, BGC0000626.1) and a thuricin H biosynthetic gene cluster (90% similarity, BGC0000600.1) were detected. Thuricin H is described as member of a small subclass of bacteriocins, which act on related bacterial strains and not on fungi (Mathur et al., 2015). A cerecidin cluster was detected (47%, BGC0000502) in Bt GOE4. Cerecidin is a novel antibiotic with high activity against a broad range of Gram-positive bacteria (Wang et al., 2014). In addition, a putative paenilamicin (BGC0001033.1) producing type I PKS gene cluster was exclusively identified in Bt18679 and Bw KBAB4 with shared similarity of 35%. Paenilamicins are known to act antibacterial and antifungal (Müller et al., 2014). As this gene cluster was not detected in all suppressing isolates, it is not exclusively the paenilamicins which lead to the inhibitory effect in our experiments.
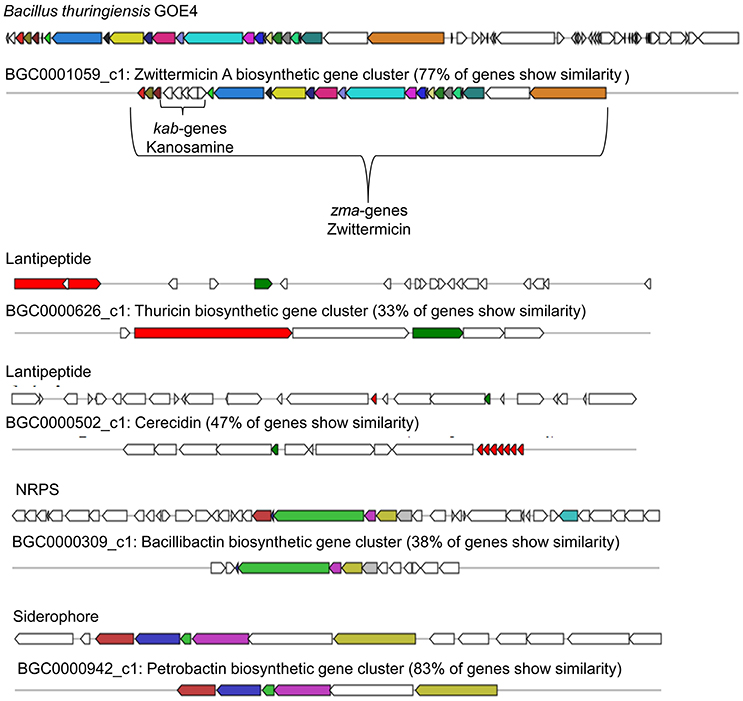
Figure 6. Biosynthetic gene clusters of Bt GOE4. The analysis was performed with AntiSMASH3.0 (Weber et al., 2015). Same colored genes display similarity to published and known gene clusters. A minimum of 30% cutoff was set for visualization of gene clusters.
Gene clusters encoding ribosomally synthesized and post-translationally modified peptides (RiPPs) such as bacteriocins, microcins, lanthipeptides, lassopeptides, ladderane, and linaride were identified as well, but could not be further characterized due to weak similarity to known clusters (Table 5). Only in case of the microcin bacitracin (BGC0000310.1), genes with similarities of 44% have been identified in the strains Bt subsp. kurstaki HD-1 and Bw KBAB4. Bacitracin is not known for a fungicidal effect, but rather for the effect on spore size and crystal production (García-Patrone, 1985). Gene clusters with the potential to encode antifungal compounds, such as zwittermicin A, are present in some strains. Remarkably, the genes are not shared by all isolates acting as fungal antagonists. In contrast, bacillibactin producing gene clusters were identified in each isolate independently of the ability to suppress the growth of V. dahliae JR2 or V. longisporum 43. We think that NRPS/PKS synthesized secondary metabolites or RiPPS could contribute to the observed growth inhibition. We strongly suggest that different complex mechanisms and substances act in combination and are important for an efficient suppression of the fungi investigated.
Chitinases of Root-Associated Bacilli
To elucidate the second class of fungicidal substances, we focused on chitinases as the mycolytic activity of many Bacilli is known (Swiontek Brzezinska et al., 2014). Genome analysis revealed the presence of three different classes of chitinases shared by the genomes of the strains with antifungal activities. Notably, genes encoding a chitinolytic polypeptide with 674 (ChiA) and 360 (ChiB) amino acids were identified in all inhibiting isolates (Table 6, Figure 7) but not in Bw isolates, with exception of the invasive-growing Bw strains (Bw GOE8-10). These isolates contained a gene encoding a chitinase-like enzyme of 314 amino acids. Comparison of the amino acid sequences of all detected chitinases with reference chitinases with known antifungal activity lead to a classification of three chitinase groups: Group I: ChiB; Group II: ChiA and Group III: ChiC (Figure 7B). Identified ChiA chitinases showed sequence similarity to known mycolytic chitinases of Bt subsp. tenebrionis DSM-2803 (de la Fuente-Salcido et al., 2016), B. cereus strain 28-9 (Huang et al., 2005) and Bt subsp. kurstaki (Driss et al., 2005; Figure 7A). They grouped into glycoside hydrolase family 18 with a catalytic domain and an active site. ChiB chitinases clustered to reference chitinases, including ChiB of Bc AH621 and the mycolytic chitinase chiA of Bt serovar colmeri (Liu et al., 2010). They also grouped into glycoside hydrolase family 18, but in addition to the catalytic domain and the active site, the enzymes contained a chitin insertion domain and a carbohydrate-binding type2 domain, which are separated by a fibronectin type III domain.
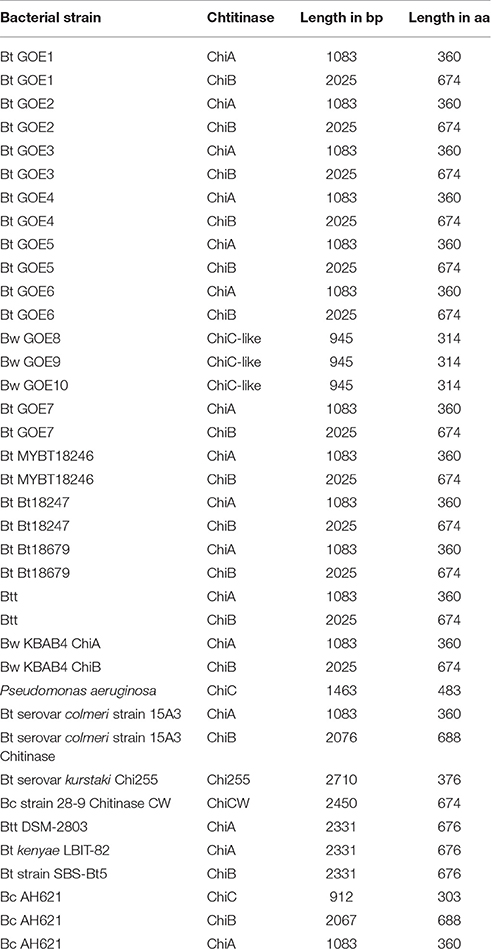
Table 6. Identified chitinases in bacterial strains.
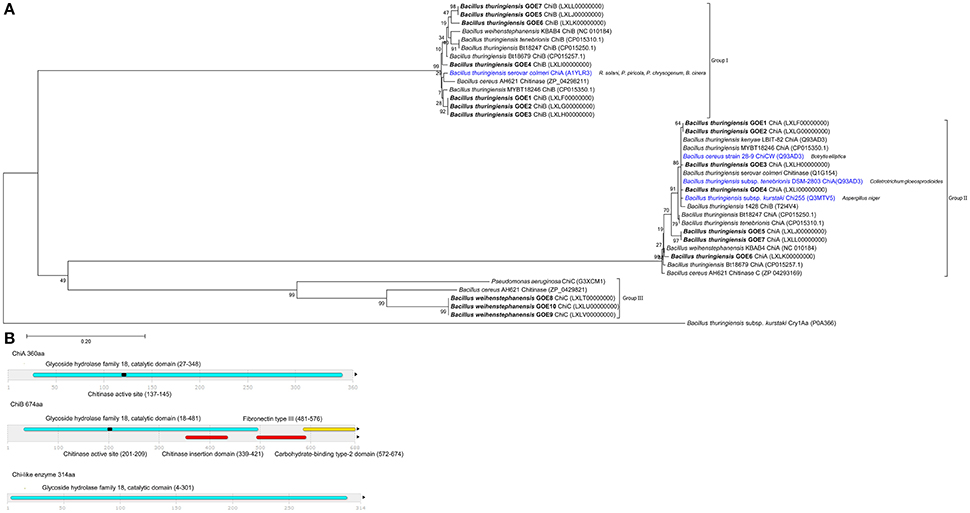
Figure 7. Analysis and comparison of chitinases encoded by the genomes of the isolates. (A) Phylogenetic tree of all identified chitinases (bold black) compared to chitinases of reference strains, with known antifungal activity (blue) target organism is depicted in brackets. The phylogenetic analysis was performed using in MEGA7 (Kumar et al., 2016), details are described in the Materials and Methods Section. (B) Domain structures of three identified chitinase types. InterPro (Mitchell et al., 2015) was used for functional analysis of chitinases, classification into families and for the prediction of domains and active sites. Identified structures are colored: (blue) glycoside hydrolase family 18 with catalytic domain, (red) chitinase insertion domain, fibronectin type III domain, (yellow) carbohydrate-binding type-2 domain, (black) chitinases active site.
Our experiments demonstrate that the classification of ChiA from Bt serovar colmeri should be reconsidered, as the deduced amino acid size of the protein and domain analysis revealed that this chitinase belongs to the ChiB group of chitinases (Group I). The chitinases of the invasive Bw strains were also classified as glycoside hydrolase family 18, containing a catalytic domain. They were most similar to a chitinase of B. cereus AH621 and Pseudomonas aeruginosa (Figure 7). In contrast, the Bw chitinases harbored no chitinase insertion domain, fibronectin type III domain, carbohydrate-binding type-2 domain or a chitinase active site. We suggest that we identified potential mycolytic chitinases in our Bt isolates whereas we can only hypothesize which of the identified groups of chitinases (chiA or chiB) are able to suppress Verticillium. Strain Bt GOE6 exhibited an antifungal effect on LB only but not on PDM. In addition, the genomes of the Bt control strains MYBT18246 and Bt18247 encode two chitinases but showed no effect on the fungal pathogens at all. These results suggest that the growth period and abiotic factors such as pH, temperature, and presence of metal ions, carbon, or nitrogen-sources influence the expression and/or the activity of chitinases. The identified chitinase in the invasive Bw strains exhibited less similarities to known sequences and thus could be a new type of chitinase. To our knowledge, this is the first time that several Bt isolates comprising multiple chitinases, showed the potential to inhibit V. dahliae in vitro. As chitinases are an effective biocontrol substance against a number of phytopathogenic fungi (Bhattacharya et al., 2007), future in planta experiments are needed to elucidate the full potential of chitinases of Bt isolates.
Conclusion
In the present study, we evaluated the antifungal potential of 20 phenotypically diverse Bacillus isolates toward V. dahliae and V. longisporum. A classification of new isolates based on a combination of morphological, pathogenic, and taxonomic properties revealed 7 B. thuringiensis and 13 B. weihenstephanensis strains. The dual cultivation assays showed a correlation between taxonomy and antagonistic activities. All B. thuringiensis strains exhibited an in vitro antifungal effect against V. dahliae while only limited antagonism was observed against V. longisporum. Additionally, three B. weihenstephanensis isolates showing an invasive growth-type competed with both phytopathogenic fungi. The relation of the rhizoid growth and the mechanism of competition of B. weihenstephanensis strains have not been described previously and thus represent a fascinating new research topic.
The genome analysis of the 20 Bacillus strains revealed that strains with antifungal activity shared genes assigned to bacillibactin production and mycolytic chitinases, which are thus the most promising candidates for encoding the antifungal effect.
The hereby produced genomic and physiological data provide an excellent foundation for the identification, purification, and characterization of the active antifungal substances of Bt. Nonetheless, future in planta experiments are necessary to determine the efficacy of these strains in controlling plant pathogens.
Author Contributions
The study was designed and conceived by HL, RH, RD, and JH. Soil bacterial communities were analyzed by JH and FW. Bacterial isolation, PCR, and bacterial identification were performed by AK, JH, EB, and HL. Genome sequencing, data processing, and genome assembly were done by JH and AP. Genome annotation, data submission, and data interpretation were performed by JH, SDV, and HL. Antagonistic assays were undertaken by AK, RH, and JH. Data Interpretation and manuscript preparation were done by JH, FW, RH, and HL. The whole project was supervised by RH, SB-S, GB, KN, RD, and HL. All authors interpreted the results, read and approved the final manuscript.
Funding
We thank the German Research Foundation (DFG-SPP1399, Grants LI 1690/2-1 and DFG BR1502/15-1) for financial support.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Jan Teer for performing MLST PCR experiments. We would like to thank Nicole Scheiter for technical assistance. We acknowledge support by the German Research Foundation and the Open Access Publication Funds of the Göttingen University.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.02171/full#supplementary-material
Abbreviations
BGSC, Bacillus Genetic Stock Center; NRPS, non-ribosomal polypeptide synthetases; PKS, polyketide synthetases; RiPP, Ribosomally synthesized and post-translationally modified peptides; PGPB, plant growth promoting bacteria; Bcsl, Bacillus cereus sensu lato; Bt, Bacillus thuringiensis; Bw, Bacillus weihenstephanensis; Ba, Bacillus anthracis; Bm, Bacillus mycoides; Bp, Bacillus pseudomycoides; Bc, Bacillus cereus; Bcyt, Bacillus cytotoxicus.
References
Ahemad, M., and Kibret, M. (2014). Mechanisms and applications of plant growth promoting rhizobacteria: current perspective. J. King Saud Univ. Sci. 26, 1–20. doi: 10.1016/j.jksus.2013.05.001
Aleti, G., Sessitsch, A., and Brader, G. (2015). Genome mining: prediction of lipopeptides and polyketides from Bacillus and related Firmicutes. Comput. Struct. Biotechnol. J. 13, 192–203. doi: 10.1016/j.csbj.2015.03.003
Angelopoulou, D. J., Naska, E. J., Paplomatas, E. J., and Tjamos, S. E. (2014). Biological control agents (BCAs) of verticillium wilt: influence of application rates and delivery method on plant protection, triggering of host defence mechanisms and rhizosphere populations of BCAs. Plant Pathol. 63, 1062–1069. doi: 10.1111/ppa.12198
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bateman, A., Martin, M. J., O'Donovan, C., Magrane, M., Apweiler, R., Alpi, E., et al. (2015). UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212. doi: 10.1093/nar/gku989
Bhattacharya, D., Nagpure, A., and Gupta, R. K. (2007). Bacterial Chitinases: properties and potential. Crit. Rev. Biotechnol. 27, 21–28. doi: 10.1080/07388550601168223
Bhattacharyya, P. N., and Jha, D. K. (2012). Plant growth-promoting rhizobacteria (PGpr): emergence in agriculture. World J. Microbiol. Biotechnol. 28, 1327–1350. doi: 10.1007/s11274-011-0979-9
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bone, E. J., and Ellar, D. J. (1989). Transformation of Bacillus thuringiensis by electroporation. FEMS Microbiol. Lett. 49, 171–177. doi: 10.1111/j.1574-6968.1989.tb03039.x
Bragg, L., Stone, G., Imelfort, M., Hugenholtz, P., and Tyson, G. W. (2012). Fast, accurate error-correction of amplicon pyrosequences using Acacia. Nat. Methods 9, 425–426. doi: 10.1038/nmeth.1990
Bulgarelli, D., Garrido-Oter, R., Münch, P. C., Weiman, A., Dröge, J., Pan, Y., et al. (2015). Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17, 392–403. doi: 10.1016/j.chom.2015.01.011
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
de la Fuente-Salcido, N. M., Casados-Vázquez, L. E., García-Pérez, A. P., Barboza-Pérez, U. E., Bideshi, D. K., Salcedo-Hernández, R., et al. (2016). The endochitinase ChiA Btt of Bacillus tenebrionis subsp. tenebrionis DSM-2803 and its potential use to control the phytopathogen Colletotrichum gloeosporioides. Microbiologyopen 5, 819–829. doi: 10.1002/mbo3.372
de Maagd, R. A., Bravo, A., Berry, C., Crickmore, N., and Schnepf, H. E. (2003). Structure, diversity, and evolution of protein toxins from spore-forming Entomopathogenic Bacteria. Annu. Rev. Genet 37, 409–433. doi: 10.1146/annurev.genet.37.110801.143042
Depotter, J. R., Deketelaere, S., Inderbitzin, P., Tiedemann, A. V., Höfte, M., Subbarao, K. V., et al. (2016). Verticillium longisporum, the invisible threat to oilseed rape and other brassicaceous plant hosts. Mol. Plant Pathol. 17, 1004–1016. doi: 10.1111/mpp.12350
de Serres, J. F., and Hollaender, A. (1982). Chemical Mutagens. Principles and Methods for Their Detection. New York, NY: Plenum Press.
De Vos, P., Garrity, G. M., Jones, D., Krieg, N. R., Ludwig, W., Rainey, F. A., et al. (2009). Bergey's Manual of Systematic Bacteriology Volume 3: The Firmicutes. New York, NY: Springer.
Di Franco, C., Beccari, E., Santini, T., Pisaneschi, G., and Tecce, G. (2002). Colony shape as a genetic trait in the pattern-forming Bacillus mycoides. BMC Microbiol. 2:33. doi: 10.1186/1471-2180-2-33
Dixon, G. R., and Pegg, G. F. (1971). Changes in amino-acid content of tomato xylem sap following infection with strains of Verticillium albo-atrum. Ann. Bot. 36, 147–154.
Driss, F., Kallassy-Awad, M., Zouari, N., and Jaoua, S. (2005). Molecular characterization of a novel chitinase from Bacillus thuringiensis subsp. kurstaki. J. Appl. Microbiol. 99, 945–953. doi: 10.1111/j.1365-2672.2005.02639.x
Eddy, S. (1998). Profile hidden Markov models. Bioinformatics 14, 755–763. doi: 10.1093/bioinformatics/14.9.755
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Enright, A. J., Van Dongen, S., and Ouzounis, C. A. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584. doi: 10.1093/nar/30.7.1575
Enya, J., Koitabashi, M., Shinohara, H., Yoshida, S., Tsukiboshi, T., Negishi, H., et al. (2007). Phylogenetic diversities of dominant culturable Bacillus, Pseudomonas and Pantoea species on tomato leaves and their possibility as biological control agents. J. Phytopathol. 155, 446–453. doi: 10.1111/j.1439-0434.2007.01256.x
Fradin, E. F., and Thomma, B. P. H. J. (2006). Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Mol. Plant Pathol. 7, 71–86. doi: 10.1111/j.1364-3703.2006.00323.x
Fradin, E. F., Zhang, Z., Juarez Ayala, J. C., Castroverde, C. D. M., Nazar, R. N., Robb, J., et al. (2009). Genetic dissection of Verticillium wilt resistance mediated by Tomato Ve1. Plant Physiol. 150, 320–332. doi: 10.1104/pp.109.136762
Frank, M. N. (2003). Development of alternative strategies for management of soilborne pathogens currently controlled with Methylbromide. Annu. Rev. Biochem. 72, 291–336. doi: 10.1146/annurev.phyto.41.052002.095514
Fravel, D. R. (2005). Commercialization and implementation of biocontrol. Annu. Rev. Phytopathol. 43, 337–359. doi: 10.1146/annurev.phyto.43.032904.092924
Gams, W., Hoekstra, E. S., and Aptroot, A. (1998). CBS Course of Mycology. Delft: Centraalbureau voor Schimmelcultures Baarn, Delft.
García-Patrone, M. (1985). Bacitracin increases size of parasporal crystals and spores in Bacillus thuringiensis. Mol. Cell. Biochem. 68, 131–137. doi: 10.1007/BF00219377
Glick, B. R. (2012). Plant growth-promoting bacteria: mechanisms and applications. Scientifica 2012, 1–15. doi: 10.6064/2012/963401
González, J. M. Jr., Brown, B. J., and Carlton, B. C. (1982). Transfer of Bacillus thuringiensis plasmids coding for delta-endotoxin among strains of B. thuringiensis and B. cereus. Proc. Natl. Acad. Sci. U.S.A. 79, 6951–6955. doi: 10.1073/pnas.79.22.6951
Haas, D., and Défago, G. (2005). Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat. Rev. Microbiol. 3, 307–319. doi: 10.1038/nrmicro1129
Han, C. S., Xie, G., Challacombe, J. F., Altherr, M. R., Bhotika, S. S., Brown, N., et al. (2006). Pathogenomic sequence analysis of Bacillus cereus and Bacillus thuringiensis isolates closely related to Bacillus anthracis. J. Bacteriol. 188, 3382–3390. doi: 10.1128/JB.188.9.3382-3390.2006
Handelsman, J., Raffel, S., Mester, E. H., Wunderlich, L., and Grau, C. R. (1990). Biological control of damping-off of alfalfa seedlings with Bacillus cereus UW85. Appl. Environ. Microbiol. 56, 713–718.
Helgason, E., Okstad, O. A., Caugant, D. A., Johansen, H. A., Fouet, A., Mock, M., et al. (2000). Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis–one species on the basis of genetic evidence. Appl. Environ. Microbiol. 66, 2627–2630. doi: 10.1128/AEM.66.6.2627-2630.2000
Huang, C. J., Wang, T. K., Chung, S. C., and Chen, C. Y. (2005). Identification of an antifungal chitinase from a potential biocontrol agent, Bacillus cereus 28-9. J. Biochem. Mol. Biol. 38, 82–88. doi: 10.5483/bmbrep.2005.38.1.082
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Inderbitzin, P., Bostock, R. M., Davis, R. M., Usami, T., Platt, H. W., and Subbarao, K. V. (2011). Phylogenetics and taxonomy of the fungal vascular wilt pathogen Verticillium, with the descriptions of five new species. PLoS ONE 6:e28341. doi: 10.1371/journal.pone.0028341
Janiak, A. M., and Milewski, S. (2001). Mechanism of antifungal action of kanosamine. Med. Mycol. 39, 401–408. doi: 10.1080/mmy.39.5.401.408
Jolley, K. A., Chan, M. S., and Maiden, M. C. (2004). mlstdbNet - distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics 5:86. doi: 10.1186/1471-2105-5-86
Jones, J. D., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Katan, J. (1981). Solar heating (Solarization) control of soilborne pests. Ann. Rev. Phytopathol. 19, 211–236. doi: 10.1146/annurev.py.19.090181.001235
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, 1–11. doi: 10.1093/nar/gks808
Kolstø, A. B., Tourasse, N. J., and Økstad, O. A. (2009). What sets Bacillus anthracis apart from other Bacillus species? Annu. Rev. Microbiol. 63, 451–476. doi: 10.1146/annurev.micro.091208.073255
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 7, 1870–1874. doi: 10.1093/molbev/msw054
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lange, B. M., Rujan, T., Martin, W., and Croteau, R. (2000). Isoprenoid biosynthesis: the evolution of two ancient and distinct pathways across genomes. Proc. Natl. Acad. Sci. U.S.A. 97, 13172–13177. doi: 10.1073/pnas.240454797
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Lechner, S., Mayr, R., Francis, K. P., Prüss, B. M., Kaplan, T., Wiessner-Gunkel, E., et al. (1998). Bacillus weihenstephanensis sp. nov. is a new psychrotolerant species of the Bacillus cereus group. Int. J. Syst. Bacteriol. 48(Pt 4), 1373–1382.
Lee, J. Y., Passalacqua, K. D., Hanna, P. C., and Sherman, D. H. (2011). Regulation of Petrobactin and Bacillibactin biosynthesis in Bacillus anthracis under iron and oxygen variation. PLoS ONE 6:e20777. doi: 10.1371/journal.pone.0020777
Li, B., Li, Q., Xu, Z., Zhang, N., Shen, Q., Zhang, R., et al. (2014). Responses of beneficial Bacillus amyloliquefaciens SQR9 to different soilborne fungal pathogens through the alteration of antifungal compounds production. Front. Microbiol. 5:636. doi: 10.3389/fmicb.2014.00636
Li, J. G., Di Ren, G., Jia, Z. J., and Dong, Y. H. (2014). Composition and activity of rhizosphere microbial communities associated with healthy and diseased greenhouse tomatoes. Plant Soil 380, 337–347. doi: 10.1007/s11104-014-2097-6
Liu, D., Cai, J., Xie, C. C., Liu, C., and Chen, Y.-H. (2010). Purification and partial characterization of a 36-kDa chitinase from Bacillus thuringiensis subsp. colmeri, and its biocontrol potential. Enzyme Microb. Technol. 46, 252–256. doi: 10.1016/j.enzmictec.2009.10.007
Luo, X., Xie, C., Dong, J., Yang, X., and Sui, A. (2014). Interactions between Verticillium dahliae and its host: vegetative growth, pathogenicity, plant immunity. Appl. Microbiol. Biotechnol. 98, 6921–6932. doi: 10.1007/s00253-014-5863-8
Markowitz, V. M., Mavromatis, K., Ivanova, N. N., Chen, I. M., Chu, K., and Kyrpides, N. C. (2009). IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 25, 2271–2278. doi: 10.1093/bioinformatics/btp393
Martin, D. M., Gershenzon, J., and Bohlmann, J. (2003). Induction of volatile terpene biosynthesis and diurnal emission by methyl jasmonate in foliage of Norway spruce. Plant Physiol. 132, 1586–1599. doi: 10.1104/pp.103.021196
Martin, M. (2011). Cutadapt removes adapter sequcences from high-throughput sequencing reads. EMBnet. J. 17, 5–7. doi: 10.14806/ej.17.1.200
Masri, L., Branca, A., Sheppard, A. E., Papkou, A., Laehnemann, D., Guenther, P. S., et al. (2015). Host–pathogen coevolution: the selective advantage of Bacillus thuringiensis virulence and its cry toxin genes. PLos Biol. 13:e1002169. doi: 10.1371/journal.pbio.1002169
Mathur, H., Rea, M. C., Cotter, P. D., Hill, C., and Ross, R. P. (2015). The sactibiotic subclass of bacteriocins: an update. Curr. Protein Pept. Sci. 16, 549–558. doi: 10.2174/1389203716666150515124831
Milutinovic, B., Fritzlar, S., and Kurtz, J. (2014). Increased survival in the red flour beetle after oral priming with bacteria-conditioned media. J. Innate Immun. 6, 306–314. doi: 10.1159/000355211
Milutinovic, B., Stolpe, C., Peu, R., Armitage, S. A. O., Kurtz, J., and Milutinovic, B. (2013). The red flour beetle as a model for bacterial oral infections. PLoS ONE 8:e64638. doi: 10.1371/journal.pone.0064638
Mitchell, A., Chang, H. Y., Daugherty, L., Fraser, M., Hunter, S., Lopez, R., et al. (2015). The interpro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221. doi: 10.1093/nar/gku1243
Müller, S., Garcia-Gonzalez, E., Mainz, A., Hertlein, G., Heid, N. C., Mösker, E., et al. (2014). Paenilamicin: structure and biosynthesis of a hybrid nonribosomal peptide/polyketide antibiotic from the bee pathogen Paenibacillus larvae. Angew Chem. Int. Ed. Engl. 53, 10821–10825. doi: 10.1002/anie.201404572
Nübel, U., Engelen, B., Felske, A., Snaidr, J., Wieshuber, A., Amann, R. I., et al. (1996). Sequence heterogeneities of genes encoding 16S rRNA in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. Appl. Environ. Microbiol. 178, 5636–5643.
Okinaka, R. T., and Keim, P. (2016). The phylogeny of Bacillus cereus sensu lato. Microbiol. Spectr. 4, 1–12. doi: 10.1128/microbiolspec.tbs-0012-2012
Ongena, M., and Jacques, P. (2008). Bacillus lipopeptides: versatile weapons for plant disease biocontrol. Trends Microbiol. 16, 115–125. doi: 10.1016/j.tim.2007.12.009
Palma, L., Muñoz, D., Berry, C., Murillo, J., and Caballero, P. (2014). Bacillus thuringiensis toxins: an overview of their biocidal activity. Toxins 6, 3296–3325. doi: 10.3390/toxins6123296
Patel, K. D., Bhanshali, F. C., and Ingle, S. S. (2011). Diversity and characterization of Bacillus thuringiensis isolates from alluvial soil of mahi river basin, India. J. Adv. Dev. Res. 2, 14–20.
Pegg, G. F., and Brady, B. L. (2002). Verticillium Wilts. Wallingford, CT; Oxfordshire: CABI Publishing.
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Pii, Y., Borruso, L., Brusetti, L., Crecchio, C., Cesco, S., and Mimmo, T. (2016). The interaction between iron nutrition, plant species and soil type shapes the rhizosphere microbiome. Plant Physiol. Biochem. 99, 39–48. doi: 10.1016/j.plaphy.2015.12.002
Priest, F. G., Barker, M., Baillie, L. W., Holmes, E. C., and Maiden, M. C. (2004). Population structure and evolution of the Bacillus cereus Group. Society 186, 7959–7970. doi: 10.1128/jb.186.23.7959-7970.2004
Priest, F. G., Kaji, D. A., Rosato, Y. B., and Canhos, V. P. (1994). Characterization of Bacillus thuringiensis and related bacteria by ribosomal RNA gene restriction fragment length polymorphisms. Microbiology 140, 1015–1022. doi: 10.1099/13500872-140-5-1015
R Core Team (2014). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://www.R-project.org/
Rocha, L. O., Tralamazza, S. M., Reis, G. M., Rabinovitch, L., Barbosa, C. B., and Corrêa, B. (2014). Multi-method approach for characterizing the interaction between Fusarium verticillioides and Bacillus thuringiensis subsp. kurstaki. PLoS ONE 9:e92189. doi: 10.1371/journal.pone.0092189
Romero, F. M., Marina, M., and Pieckenstain, F. L. (2014). The communities of tomato (Solanum lycopersicum L.) leaf endophytic bacteria, analyzed by 16S-ribosomal RNA gene pyrosequencing. FEMS Microbiol. Lett. 351, 187–194. doi: 10.1111/1574-6968.12377
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Sambrook, J., and Russell, D. W. (2001). Molecular Cloning: A Laboratory Manua. 3rd Edn., New York, NY: Cold Spring Harbor Laboratory Press.
Scherling, C., Ulrich, K., Ewald, D., and Weckwerth, W. (2009). A metabolic signature of the beneficial interaction of the endophyte Paenibacillus sp. isolate and in vitro-grown poplar plants revealed by metabolomics. Mol. Plant. Microbe. Interact. 22, 1032–1037. doi: 10.1094/MPMI-22-8-1032
Schnathorst, W. C. (1981). “Life cycle and epidemiology of Verticillium,” in Fungal Wilt Diseases of Plants, eds M. E. Mace, A. A. Bell, and C. H. Beckman (San Francisco, CA: Academic Press), 81–111.
Schnepf, E., Crickmore, N., Van Rie, J., Lereclus, D., Baum, J., Feitelson, J., et al. (1998). Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol. Mol. Biol. Rev. 62, 775–806.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shange, R. S., Ankumah, R. O., Ibekwe, A. M., Zabawa, R., and Dowd, S. E. (2012). Distinct soil bacterial communities revealed under a diversely managed agroecosystem. PLoS ONE 7:e40338. doi: 10.1371/journal.pone.0040338
Sheppard, A. E., Nakad, R., Saebelfeld, M., Masche, A. C., Dierking, K., and Schulenburg, H. (2016). High instability of a nematicidal cry toxin plasmid in Bacillus thuringiensis. J. Invertebr. Pathol. 133, 34–40. doi: 10.1016/j.jip.2015.11.009
Sheppard, A. E., Poehlein, A., Rosenstiel, P., Liesegang, H., and Schulenburg, H. (2013). Complete genome sequence of Bacillus thuringiensis strain 407 cry. Genome Announc. 1, 158–112. doi: 10.1128/genomeA.00158-12
Silo-Suh, L. A., Lethbridge, B. J., Raffel, S. J., He, H., Clardy, J., and Handelsman, J. (1994). Biological activities of two fungistatic antibiotics produced by Bacillus cereus UW85. Appl. Environ. Microbiol. 60, 2023–2030.
Singh, S., Braus-Stromeyer, S. A., Timpner, C., Tran, V. T., Lohaus, G., Reusche, M., et al. (2010). Silencing of Vlaro2 for chorismate synthase revealed that the phytopathogen Verticillium longisporum induces the cross-pathway control in the xylem. Appl. Microbiol. Biotechnol. 85, 1961–1976. doi: 10.1007/s00253-009-2269-0
Soufiane, B., and Côté, J. C. (2009). Discrimination among Bacillus thuringiensis H serotypes, serovars and strains based on 16S rRNA, gyrB and aroE gene sequence analyses. Antonie Van Leeuwenhoek 95, 33–45. doi: 10.1007/s10482-008-9285-4
Swiontek Brzezinska, M., Jankiewicz, U., Burkowska, A., and Walczak, M. (2014). Chitinolytic microorganisms and their possible application in environmental protection. Curr. Microbiol. 68, 71–81. doi: 10.1007/s00284-013-0440-4
Tamura, K., Nei, M., and Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U.S.A. 101, 11030–11035. doi: 10.1073/pnas.0404206101
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Tran, V. T., Braus-Stromeyer, S. A., Timpner, C., and Braus, G. H. (2013). Molecular diagnosis to discriminate pathogen and apathogen species of the hybrid Verticillium longisporum on the oilseed crop Brassica napus. Appl. Microbiol. Biotechnol. 97, 4467–4483. doi: 10.1007/s00253-012-4530-1
van Dongen, S. (2007). Graph Clustering by Flow Simulation. Dissertation, Amsterdam: Centre for Mathematics and Copmuter Science (CWI).
Wang, J., Zhang, L., Teng, K., Sun, S., Sun, Z., and Zhong, J. (2014). Cerecidins, novel lantibiotics from Bacillus cereus with potent antimicrobial activity. Appl. Environ. Microbiol. 80, p2633–p2643. doi: 10.1128/AEM.03751-13
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., et al. (2015). antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. doi: 10.1093/nar/gkv437
Wemheuer, B., Güllert, S., Billerbeck, S., Giebel, H. A., Voget, S., Simon, M., et al. (2014). Impact of a phytoplankton bloom on the diversity of the active bacterial community in the southern North Sea as revealed by metatranscriptomic approaches. FEMS Microbiol. Ecol. 87, 378–389. doi: 10.1111/1574-6941.12230
Wemheuer, B., Wemheuer, F., and Daniel, R. (2012). RNA-Based assessment of diversity and composition of active archaeal communities in the German Bight. Archaea 2012, 1–8. doi: 10.1155/2012/695826
Keywords: Bacillus thuringiensis, Bacillus weihenstephanensis, Verticillium, bacterial-fungal interaction, antifungal activity, biopesticide, plant pathogen, chitinase
Citation: Hollensteiner J, Wemheuer F, Harting R, Kolarzyk AM, Diaz Valerio SM, Poehlein A, Brzuszkiewicz EB, Nesemann K, Braus-Stromeyer SA, Braus GH, Daniel R and Liesegang H (2017) Bacillus thuringiensis and Bacillus weihenstephanensis Inhibit the Growth of Phytopathogenic Verticillium Species. Front. Microbiol. 7:2171. doi: 10.3389/fmicb.2016.02171
Received: 06 October 2016; Accepted: 23 December 2016;
Published: 18 January 2017.
Edited by:
Yunrong Chai, Northeastern University, USAReviewed by:
Gao Xuewen, Nanjing Agricultural University, ChinaDennis Halterman, Agricultural Research Service (USDA), USA
Copyright © 2017 Hollensteiner, Wemheuer, Harting, Kolarzyk, Diaz Valerio, Poehlein, Brzuszkiewicz, Nesemann, Braus-Stromeyer, Braus, Daniel and Liesegang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heiko Liesegang, hlieseg@gwdg.de
†Present Address: Anna M. Kolarzyk, Department of Bacterial Genetics, Institute of Microbiology, University of Warsaw, Warsaw, Poland