 Kai Tang
Kai Tang Yingfan Lin
Yingfan Lin- State Key Laboratory for Marine Environmental Science, Institute of Marine Microbes and Ecospheres, Xiamen University, Xiamen, China
Members of phylum Bacteroidetes are distributed across diverse marine niches and Flavobacteria is often the predominant bacterial class decomposing algae-derived polysaccharides. Here, we report the complete genome of Gramella flava JLT2011 (Flavobacteria) isolated from surface water of the southeastern Pacific. A remarkable genomic feature is that the number of glycoside hydrolase (GH) genes in the genome of G. flava JLT2011 is more than 2-fold higher than that of other Gramella species. The functional profiles of the GHs suggest extensive variation in Gramella species. Growth experiments revealed that G. flava JLT2011 has the ability to utilize a wide range of polysaccharides for growth such as xylan and homogalacturonan in pectin. Nearly half of all GH genes were located on the multi-gene polysaccharide utilization loci (PUL) or PUL-like systems in G. flava JLT2011. This species was also found to harbor the two xylan PULs and a pectin PUL, respectively. Gene expression data indicated that more GHs and sugar-specific outer-membrane susC-susD systems were found in the presence of xylan than in the presence of pectin, suggesting a different strategy for heteropolymeric xylan and homoglacturonan utilization. Multi-omics data (transcriptomics, proteomics, and metabolomics) indicated that xylan PULs and pectin PUL are respectively involved in the catabolism of their corresponding polysaccharides. This work presents a comparison of polysaccharide decomposition within a genus and expands current knowledge on the diversity and function of PULs in marine Bacteroidetes, thereby deepening our understanding of their ecological role in polysaccharide remineralization in the marine system.
Introduction
Members of the Bacteroidetes, formerly known as the Cytophaga-Flavobacteria-Bacteroides phylum, are diverse and widely distributed in marine ecosystems (Glöckner et al., 1999; Kirchman, 2002; Alonso et al., 2007). Many have gliding motility as well as the capacity to degrade polymeric substances, possibly enabling them to grow on organic aggregates or algal cells using polymeric substances as carbon and energy sources (Bauer et al., 2006; McBride et al., 2009; Qin et al., 2010; Mann et al., 2013). These bacteria use their wide array of peptidases and carbohydrate-active enzymes (CAZymes), including glycoside hydrolases (GHs), carbohydrate esterases (CEs), polysaccharide lyases (PLs), and glycoside transferases (GTs) to efficiently degrade polymers such as proteins and polysaccharides (Bauer et al., 2006; Fernández-Gómez et al., 2013). Several GH genes in the genomes of Bacteroidetes are organized in distinct polysaccharide utilization loci (PUL), often along with PL and CE genes, and regulatory elements (Martens et al., 2011; Kabisch et al., 2014). These are also likely to be clustered with transporter systems that are involved in the binding and uptake of oligosaccharides, including susD-like oligosaccharide-binding proteins and susC-like TonB-dependent receptors (porins that mediate transport of starch oligosaccharides from the surface of the outer membrane to the periplasm for subsequent degradation; Anderson and Salyers, 1989a,b; Shipman et al., 2000). Despite the fact that PULs can be predicted based on their genomic context (Terrapon et al., 2015), recent studies have focused on the experimental validation of PULs in this group because PULs are integral to the digestion of complex carbohydrates. For example, a xyloglucan PUL, xylan PULs, and a mannan PUL were identified in Bacteroidetes ovatus (Larsbrink et al., 2014; Rogowski et al., 2015) and in B. thetaiotaomicron (Cuskin et al., 2015), respectively.
Phytoplankton contribute approximately 50% of the global primary production (Boyce et al., 2010) and bacteria can consume a substantial fraction of primary production as dissolved organic carbon (DOC) that is released by algal cells (Buchan et al., 2014). Polysaccharides constitute a large percentage of phytoplankton biomass, particulate organic matter, and DOC in the ocean (Benner et al., 1992; Engel et al., 2004). The quantity of CAZymes in phylum Bacteroidetes increases in the presence of phytoplankton blooms (Teeling et al., 2012). Among the members of marine Bacteroidetes that are associated with phytoplankton blooms, a high proportion is represented by class Flavobacteria (Alonso et al., 2007; Teeling et al., 2012). These are the predominant bacteria that utilize phytoplankton-derived transparent exopolymer particles in the surface coastal waters (Taylor et al., 2014). Polysaccharides are constituents of marine algae such as alginate and laminarin in brown algae, xylan in green algae and red algae, and pectin in diatoms (Kröger and Poulsen, 2008; De Jesus Raposo et al., 2014; Based et al., 2015). These algae are significant contributors to primary production in the marine environment (Nelson et al., 1995).
The breakdown of algal polysaccharides by Bacteroidetes is central to marine carbon cycling (Cottrell and Kirchman, 2000); however, the mechanisms underlying the action of complex polysaccharides such as xylan and pectin remain unclear. Gramella forsetii KT0803, which belongs to class Flavobacteria, is the first marine Bacteroidetes that was sequenced and analyzed and was found to contain a substantial number of CAZymes and peptidases, as well as a predicted preference for polymeric carbon sources (Bauer et al., 2006). The presence of a starch PUL, laminarin PUL, and alginate PUL in G. forsetii KT0803 has been further resolved by functional proteomics analysis (Kabisch et al., 2014). Members of genus Gramella occur in a variety of marine habitats, including coastal regions (G. forsetii KT0803) (Bauer et al., 2006), open oceans (G. flava JLT2011) (Liu et al., 2014), marine sediments (G. portivictoriae DSM23547) (Lau et al., 2005), and in phytoplankton- or animal-associated domains (sea urchin-associated G. echinicola DSM19838; Nedashkovskaya et al., 2005). Several more genomes of Gramella have subsequently been sequenced (Panschin et al., 2016), and it is now theoretically possible to compare the CAZyme features of these closely related bacterial species at the genome level.
Here, we present the complete genome of G. flava JLT2011, in which we find more GHs than in G. forsetii, G. portivictoriae, and G. echinicola. This species also contains a xylan PUL and mannan PUL that are absent in the other species. Although G. flava JLT2011 lacks an alginate PUL, it was found to harbor a pectin PUL. These species exhibited differences in the spectrum of polysaccharides that an organism can potentially degrade. Using a combination of transcriptomics, proteomics, and metabolomics, the catabolic pathway models for xylan and pectin in G. flava JLT2011 are proposed. This study expands current knowledge on the diversity and function of PULs in marine Bacteroidetes.
Materials and Methods
Gene Sequencing and Bioinformatics Analysis
Whole genome sequencing of G. flava JLT2011 was performed using a hybrid approach, combining Illumina short read data with PacBio long read data (Koren et al., 2012). The genome sequences were de novo assembled by the HGAP2 program in the SMRT analysis server (v2.3). Illumina pair end reads were mapped to the assembled contigs to improve the accuracy of genome sequences. The final assembled genomes were automatically annotated and analyzed via the IMG/ER platform (http://img.jgi.doe.gov). The comparison and visualization of multiple genomes was conducted using BRIG (Alikhan et al., 2011), and multiple-genome alignment was performed using Mauve (Darling et al., 2009). The amino acid sequences were then submitted to the CAZyme Annotation Toolkit (Park et al., 2010; Lombard et al., 2014; http://mothra.ornl.gov/cgi-bin/cat/cat.cgi) for sequence-based annotation, with an E-value of 1e-40, as well as Pfam-based annotation with an e-value of 0.00001. The results were then further manually checked. Peptidase genes were annotated using the MEROPS peptidases database (Rawlings and Barrett, 1999). The candidates were manually examined in terms of similarity (E-value cutoff 1e-10) to MEROPS proteins and the presence of all catalytic sites. The cluster sequences at the gene cluster level in NCBI GenBank were detected using a MultiGeneBlast (V. 1.1.14) (Medema et al., 2013) architecture search with predicted PULs in Gramella species genomes as queries. The combination of a pairwise based method and phylogeny-based methods to identify orthologous genes. The predicted orthologous genes were firstly identified using OMA (V. 1.0.6) (Roth et al., 2009). The genes were finally identified as orthologs when at least two of their phylogenetic trees were reconciled with the species trees. The phylogenetic trees of the candidate orthologous genes were reconstructed with MEGA (V. 6.0) (Tamura et al., 2013), using maximum likelihood, neighbor joining distance and maximum parsimony algorithms, and implementing a JTT plus a gamma distribution with four categories. The concatenated conserved eight single-copy genes in the phylum Bacteroidetes were used for the reconstruction of the maximum likelihood species tree using the RAxML program (V. 7.4.2) (Stamatakis, 2006), implementing a GTR plus GAMMA model and performing 1000 rapid bootstrap replicates.
The complete genome sequence of G. flava JLT2011 has been deposited in GenBank under the accession numbers CP016359 and deposited in the Joint Genome Institute IMG/ER website (http://img.jgi.doe.gov) under the genome ID 2576861820.
Growth Experiments
All chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.), unless otherwise stated. Both G. flava JLT2011 and G. forsetii KT0803 were cultured on minimal media [2.3% (w/v) sea salts, 0.05% (w/v) yeast extract, 0.05% NH4Cl (w/v) and 50 mM Tris-HCl, pH 7.8], with a final concentration of 0.2% of one of the following carbon sources: D-glucose, D-xylose, D-galacturonic acid, L-arabinose, L-rhamnose, D-mannose, D-galactose, N-acetyl-D-glucosamine, D-trehalose, xylan (oat spelts), pectin (apple), pectin (citrus peel), galatcomannan (Ceratonia siliqua), mannan (Saccharomyces cerevisiae), arabinan (sugar beet), arabinogalactan (larch), laminarin (brown algae), alginate (brown algae). and chitin (shrimp shells). Cultures using only minimal media were treated as controls. Bacteria were grown using a mechanical shaker (XMTE-8112, Sukun, China) at 180 rpm and 28°C for 7 days. Cell density was determined by optical density measured at 600 nm (OD600) during cultivation. Cell suspensions at the exponential growth phase were pelleted by centrifugation (14,000 g, 4°C, 20 min) for further analysis. Pectin methylesterase activity was measured as previously described (Tans-kersten et al., 1998).
Microarray Analysis
Total RNA of cells at the exponential growth phase was isolated using the Ambion RiboPure-Bacteria Kit (Ambion Waltham, USA) according to the manufacturer's instructions. Total RNA was polyadenylated using the poly(A) Tailing ATP kit (Thermo Fisher Scientific, U.S.A), and then was converted to cDNA using the T7-oligo (dT) primer and CbcScript enzymes (Capitalbio, Beijing, China) for first strand synthesis of cDNA. Using an RNase H chain, short segments of RNA were cut to obtain single stranded DNA. Using the first cDNA strand as a template, the second strand cDNA was then synthesized and the primer digested using the T7 Enzyme Mix. The double-stranded cDNA was then transcribed into cRNA followed by purification using a Clean-up Kit (MN, Düren, Germany). Then, 5 μg of cRNA along with CbcScript II enzyme (Capitalbio), using Random Primers, was used for reverse transcription. The reverse transcription product was purified by using a PCR NucleoSpinExtract II Kit (MN). This reversely transcribed product was then labeled using the KLENOW enzyme. The labeled PCR product was purified using the NucleoSpinExtract II Kit (MN) and Cy3-dCTP (GE Healthcare, Marlborough, U.S.A.). Labeled DNA was dissolved in a hybridization buffer (2X GEx Hyb Buffer (HI-RPM), 25% formamide), and hybridized overnight at 45°C. Following hybridization, the product was washed in 0.2% SDS and 2 × SSC liquid for approximately 5 min at 42°C. Following this, the product was washed in a 0.2 × SSC for 5 min at room temperature. The 60-mer oligonucleotide probes were designed by CapitalBio with online software Earray (https://earray.chem.agilent.com/earray/), according to the whole genomic sequence of G. flava JLT2011. Custom microarrays (8 × 15 K) with the designed oligonucleotide probes were purchased from Agilent Technologies (Santa Clara, U.S.A.). The gene chips were scanned using an Agilent G2565CA Microarray Scanner (Agilent Technologies). Images from the Agilent arrays were processed using Agilent Feature (Agilent Technologies). Signal values normalized by using the method of percentile shift after raw data were input into the GeneSpring GX software (Agilent Technologies). Differentially expressed gene analysis was carried out using GeneSpring GX software and p-values were calculated. Microarray data details are available at the National Center for Biotechnology Information (GEO series GSE90082).
Proteomics Analysis
Cells at the exponential growth phase were centrifuged (6,600 × g at 4°C for 10 min) and extracellular proteins in the supernatant were precipitated at 4°C using 10% trichloroacetic acid. To extract the intracellular protein fractions, cells were then washed with 10 mM Tris-HCl pH 8.0, and lysed in SDT-lysis buffer (4% SDS, 100 mM DTT, 100 mM Tris-HCl pH 8.0) using a 1:10 sample to buffer ratio for 5 min at 95°C, followed by SDS-PAGE analysis. Extracellular and intracellular protein fractions were then stored at −80°C for further protein analysis. Protein in-solution digestion was performed according to the FASP procedure (Wiśniewski et al., 2009). Proteomics analysis was performed on a Q Exactive mass spectrometer coupled to an Easy nLC (Thermo Fisher Scientific, U.S.A.). The instrument was run with the peptide recognition mode enabled. The MS/MS spectra were identified using the MASCOT engine (Matrix Science, London, UK; V. 2.2) against G. flava JLT2011 genomes. For protein identification, the following options were used: peptide mass tolerance = 20 ppm, MS/MS tolerance = 0.1 Da, enzyme = trypsin, max missed cleavage = 2, fixed modification: carbamidomethyl (C), and variable modification: oxidation (M). All of the reported data were based on 99% confidence for protein identification as determined by false discovery rate (FDR) ≤1%.
Identification and Quantification of Metabolites in G. flava JLT2011 by GC-TOF/MS
Cells at the exponential growth phase were collected by centrifugation (6,600 × g at 4°C for 10 min). Subsequently, re-suspended cells were washed twice with artificial sea water at 4°C. The samples were then quenched with 5 ml 80% methanol, followed by vortex-mixing and centrifugation at 10,000 × g for 20 min at −20°C. Afterwards, 800 μl of each sample and 800 μl of methanol solution were combined in 2 ml EP tubes and mixed. Samples were then centrifuged at 12,000 rpm for 15 min at 4°C to obtain the supernatant. The above steps were repeated three times. Following this, 0.4 ml of methanol:chloroform (V methanol: V chloroform = 3:1) was added, and samples were vortex mixed for 10 s. A ball mill was then used to homogenize the samples for 5 min at 55 Hz followed by sonication for 5 min, and then centrifugation for 10 min at 4°C. Subsequently, the supernatant (approximately 0.4 ml) was transferred to a new 2 ml GC/MS glass vial. We then placed 10 μl of each sample into a new 2 ml GC/MS glass vial to use as a mixed sample for quality control. The extracts were dried in a vacuum concentrator at 30°C for approximately 1.5 h, after which 60 μl of 20 mg/ml methoxymethyl amine salt (dissolved in pyridine) was added to the dried metabolites. The samples were then mixed and sealed, and incubated at 37°C overnight in an oven. Finally, 80 ml BSTFA reagent (containing 1% TMCS) was added into the mixture and reacted at 70°C for an hour.
Metabolites extracted from the quenched G. flava JLT2011 cells were then examined by gas chromatography-mass spectrometry (GC-MS) using an Agilent Technologies 7890 gas chromatograph system coupled to a Pegasus HT time-of-flight (TOF) mass spectrometer (LECO Coporation, U.S.A.). The system utilized a DB-5MS capillary column coated with 5% diphenyl cross-linked with 95% dimethyl polysiloxane (30 m × 250 μm inner diameter, 0.25 μm film thickness; J&W Scientific, Folsom, CA, U.S.A.). A 1 μL aliquot of the analyte was injected in splitless mode. Helium was used as the carrier gas, the front inlet purge flow was 3 mL min−1 and the gas flow rate through the column was 20 mL min−1. The initial temperature was kept at 50°C for 1 min, rose to 330°C at a rate of 20°C min−1, and then maintained for 1 min at 330°C. The injection, transfer line and ion source temperatures were 330, 280, and 250°C, respectively. The energy was −70 eV in electron impact mode. The mass spectrometry data were acquired in full-scan mode with the m/z range of 30–600 at a rate of 20 spectra per second after a solvent delay of 366 s. The Chroma TOF4.3X software (LECO) and LECO-Fiehn Rtx5 database were used for raw peaks extraction, data baselines filtering and calibration, peak alignment, deconvolution analysis, peak identification and integration of the peak area.
Results and Discussion
GHs in the Genome of G. flava JLT2011
The genome of G. flava JLT2011 contains a single circular chromosome of 4,007,868 base pairs (bp) in size and a GC content of 42.1%, which both are larger than that of the three Gramella species from distinct habitats (Table S1). The genome contains 184 genes encoding CAZymes, which is significantly higher than that of other Gramella species (G. forsetii KT0803, 118; G. echinicola DSM19838, 108; G. portivictoriae DSM23547, 119; Table S2). It also harbors 96 genes that encode GHs that represent 34 CAZyme superfamilies (Table S3), compared to G. forsetii KT0803 that has 42 GHs genes that were assigned to 20 superfamilies (Table S2). G. portivictoriae DSM23547 and G. echinicola DSM19838 contained only 42 and 36 GHs genes that were assigned to 19 and 16 superfamilies, respectively (Table S2). G. flava JLT2011 contained genes encoding 18 susCDs.
Despite the fact that G. flava JLT2011 and G. forsetii KT0803 share a large amount of homologous regions, the high number of translocations and inversions between chromosomes indicates that extensive genome rearrangements, gene gains, or gene losses have occurred (Figure S1). BLAST analysis showed that 628 genes in the G. flava JLT2011 genome were non-homologous to those in the other Gramella bacteria genomes (>30% amino acid identity; Figure 1A). A total of 1,091 orthologous genes were found among the genomes of Gramella species, in which only 19 GHs were determined to be orthologous (Figure 1B). Both G. flava JLT2011 and G. forsetii KT0803 shared 32 GHs homologs. However, a total of 52 genes belonging to 23 GH superfamilies in G. flava JLT2011 were not detected in the other three Gramella species. The optimal BLASTP hits of approximately two-thirds of the GHs of G. flava JLT2011 were aligned with non-Gramella bacterial species, of which most were homologous to those of bacteria from class Flavobacteria of phylum Bacteroidetes such as Zunongwangia profunda SM-A87 (Table S3, Figure S2). The four GH genes in G. flava JLT2011 that encode alpha-glucosidase, chitinase, and beta-galactosidase shared the highest similarities with those of bacteria from class Bacteroides (Table S3). The top hit for genes encoding endo-1,4-beta-mannosidas was determined to belong to bacteria from class Cytophagia (Table S3). Furthermore, a gene encoding beta-galactosidase in G. flava JLT2011 was best matched to the non-Bacteroidetes bacterium Verrucomicrobiae (Table S3).
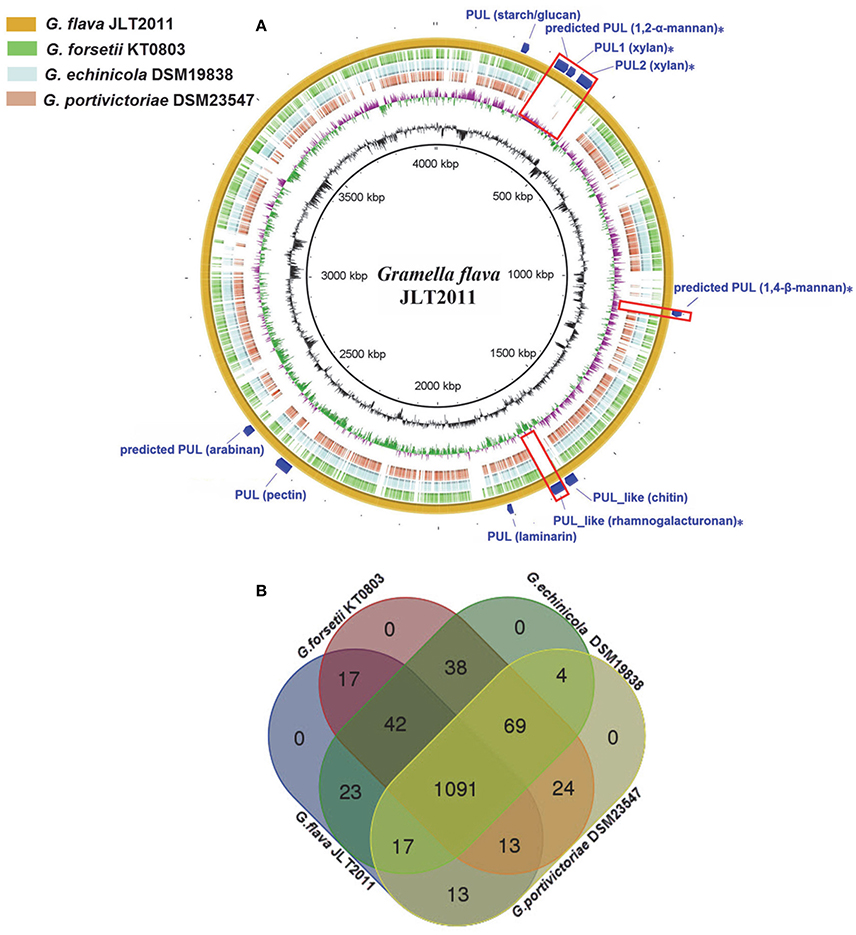
Figure 1. Multiple genome comparison of Gramella species. (A) A composite genome comparison figure was generated using BRIG after performing a BLASTn analysis of G. flava JLT2011 as the reference genome. Each genome mapping to the reference is represented as a colored ring, with a solid color representing >60% sequence identity. The innermost rings show GC content (black) and GC skew (purple/green) in G. flava JLT2011. The remaining rings indicate genome regions of G. echinicola (brown), G. portivictoriae DSM23547 (light blue), G. forsetii KT0803 (green), and G. flava JLT2011 (orange). The predicted PULs and PUL-like system gene clusters in G. flava JLT2011 are labeled on the outermost ring with dark blue arcs. The unique PULs in G. flava JLT2011 are also illustrated as red boxes and labeled with asterisks. The PUL gene cluster information is provided in Figure 3, Figures S3–S5. (B) Venn diagram of the intersections of orthologous genes in four genomes, where each rectangle with round corners represents one of the four genomes.
Comparative Analysis of GHs and Peptidases in Marine Microbes
The GH and peptidase genes were found to be distributed across a variety of divergent bacterial groups, including Proteobacteria, Bacteroidetes, Cyanobacteria, Actinobacteira, Fimicutes, Planctomycetes, and Thermotogae (Tables S2, S5). The numbers of GHs and peptidases in the marine Bacteroidetes ranged from 13 to 135 per genome (median: 46) and from 76 to131 (median: 98), respectively (Tables S2, S4). The median frequency of GHs and peptidases in the marine members of phylum Bacteroidetes was 12 GHs per Mbp and 24 peptidases per Mbp, accounting for 1.4 and 2.6% of the total open reading frames, respectively. This was higher than that of the other taxa that contained more than three genomes (Table S2).
The GH functional profile in phylum Bacteroidetes was distinguishable from most of the other marine bacteria (Figure 2A). The G. flava JLT2011 genome appears to have a high frequency of GHs (>20 genes per Mbp), which is similar to other Bacteroidetes, including Z. profunda SM-A87 from deep-sea sediments (Qin et al., 2010), Echinicola vietnamensis DSM17526 from a lagoon of Nha Trang Bay, South China Sea (Nedashkovskaya et al., 2007), and Leeuwenhoekiella blandensis MED217 from the open ocean (Pinhassi et al., 2006; Figure 2A). Moreover, Echinicola pacifica DSM19836 contained four times as many GHs as that in G. echinicola DSM19838, both of which have been found in sea urchins (Nedashkovskaya et al., 2005, 2006; Table S2). These results indicate that bacteria containing abundant GHs could be found across diverse environments. Several non-Bacteroidetes species also harbored abundant GHs, including Verrucomicrobiae bacterium DG1235 and Saccharophagus degradans 2–40, which are efficient degraders of numerous complex polysaccharides in the marine environment (Figure 2A; Hutcheson et al., 2011; Martinez-Garcia et al., 2012).
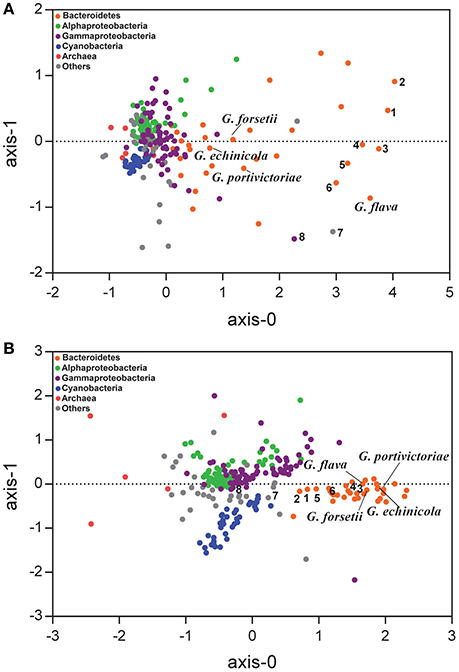
Figure 2. Nonmetric multidimensional scaling analysis based on Bray-Curtis similarity matrix was calculated from the frequency of each GH (A) and peptidase (B) in marine bacteria. A total of 249 marine bacteria genomes were included in the analysis and their GHs and peptidases of each protein superfamily were standardized by the genome size (see details on Tables S2, S4). The bacterial lineages are highlighted in colors. 1. Echinicola vietnamensis DSM17526; 2. Echinicola pacifica DSM19836; 3. Zunongwangia profunda SM-A78; 4. Leeuwenhoekiella blandensis MED217; 5. Pedobacter sp. BAL39; 6. Leeuwenhoekiella sp. Hel_I_48; 7. Verrucomicrobiae bacterium DG1235; 8. Saccharophagus degradans 2-40.
The functional profiles of GHs in G. flava JLT2011 were significantly distinguishable from those of the other three Gramella species (Figure 2A). Furthermore, G. flava contained more GHs that were associated with xylan hydrolysis (GH10, GH30, GH67, and GH115 CAZyme superfamilies) than the other three Gramella species (Table S3). Mannan hydrolyzing GHs (GH26, GH76, GH92, and GH125) were only found in G. flava JLT2011 (Table S3). Pectin hydrolyzing GHs were identified in G. flava JLT2011 and G. echinicola DSM19838 (GH28, GH88, and GH105) (Table S3). While G. forsetii KT0803 contained seven genes that encode PLs, including alginate lyase, only two predicted pectin lyases were present in G. flava JLT2011 (Table S2). Differences in CAZyme distribution among the four Gramella species suggest variations in the genomic potential for polysaccharide degradation. In contrast, their peptidase profiles exhibited little variation (Figure 2B). Notably, G. flava JLT2011 contained 97 peptidases that were distributed across 39 superfamilies, whereas G. forsetii KT0803 comprised 102 peptidases that represented 42 superfamilies (Table S4).
Xylan and Pectin PULs in the Genome of G. flava JLT2011
The PUL and PUL-like systems are variously distributed in the four Gramella strains (Figure 1A). Nearly half of all GHs and all susC-susD systems were distributed in PULs and PUL-like systems in G. flava JLT2011 (Figure 3, Figures S3–S5). G. flava JLT2011 can utilize xylan and its corresponding monosaccharide D-xylose, whereas G. forsetii KT0803 could not (Figure S6). G. flava JLT2011 was found to contain two xylan PULs that were absent in G. forsetii KT0803 (Figure 3). Two xylan PULs in G. flava JLT2011 were distinct from those in Bacteroidetes ovatus (Rogowski et al., 2015). However, the major xylanolytic enzymes were found in the xylan PULs of G. flava JLT2011, including genes that encode xynA-encoding xylanases (GH5 and GH10) and xynB-encoding beta-xylosidase (GH43) (Rogowski et al., 2015; Figure 3). Two xylan PULs both contained genes that encode a susC-susD system. Genes encoding acetyl xylan esterase (xylU), xylulokinase (xylB), xylose isomerase (xylA), and xylose transporters (xylE) were located downstream of a PUL and are probably involved in xylose utilization (Figure 3). Xylans are heteropolymers consisting principally of xylose and a small amount of arabinose, as well as traces of glucuronic acid, galactose, rhamnose, galacturonic acid, and other residues (Bastawde, 1992). The GHs that are potentially involved in the neutral sugars that are linked to the xylan main chain were found in two xylan PULs, which include alpha-N-arabinofurnosidases (abfA-II and arb43, GH43) and alpha-glucuronidases (aguA, GH67) (Rogowski et al., 2015; Figure 3).
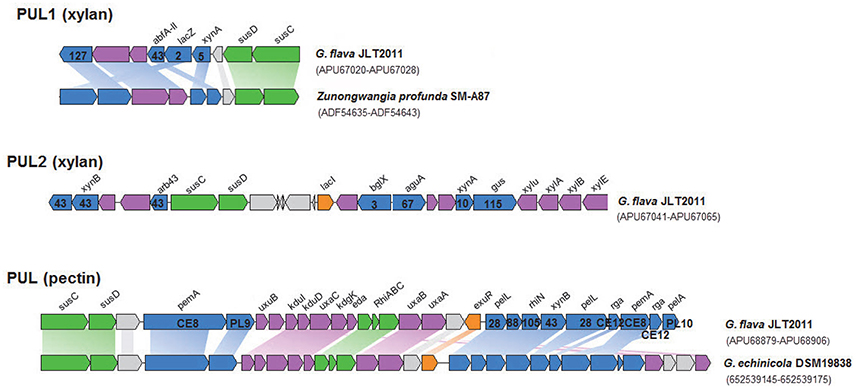
Figure 3. Genetic organization of the two xylanolytic and one pectinolytic operons of G. flava JLT2011. The range of Genbank accession numbers of sequences of the gene cluster in the genome is shown in a bracket. The functions of the proteins are color-coded: blue, CAZymes; green, membrane proteins involved in binding/transport; purple, other enzymes; orange, putative regulation factor; gray, unknown function. Homologous genes are connected by colored bars between two PULs. The CAZymes superfamily classification is listed on a gene. xylan PUL1&2: xynA, Endo-1,4-beta-xylanase; xynB, Beta-xylosidase; abfA-II, Alpha-L-arabinofuranosidase; lacZ, Beta-galactosidase; arb43, Alpha-N-arabinofuranosidase; bglX, Beta-glucosidase; aguA, Alpha-glucuronidase; xylu, Acetyl xylan esterase; xylA, Xylose isomerase; xylB, Xylulose kinase; susC, TonB-dependent receptor; susD, Carbohydrate-binding protein; xylE, Xylose transporter; lacI, LacI family transcriptional regulator; pectin PUL: pemA, Pectinesterase; pelL, Endopolygalacturonase; PL9, Pectate lyase; pelA, Pectate lyase; rhiN, Rhamnogalacturonides degradation protein; uxuB, D-mannonate oxidoreductase; kduI, 4-deoxy-L-threo-5-hexosulose-uronate ketol-isomerase; kduD, 2-deoxy-D-gluconate 3-dehydrogenase; uxaC, Uronate isomerase; kdgK, 2-dehydro-3-deoxygluconate kinase; eda, 2-dehydro-3-deoxyphosphogluconate aldolase; uxaB, Altronate oxidoreductase; uxaA, Altronate hydrolase; rga, Rhamnogalacturonan acetylesterase; susC, TonB-dependent receptor; susD, carbohydrate-binding protein; RhiABC, Predicted rhamnogalacturonide-specific TRAP-type transporter; exuR, Hexuronate utilization operon transcriptional repressor.
Both G. flava JLT2011 and G. forsetii KT0803 can utilize pectin (from apple and citrus, respectively), and their corresponding monosaccharide, D-galacturonic acid (Figure S6, Table S5). G. flava JLT2011 contained a PUL that was predicted to encode for enzymes that are involved in the degradation of pectin, including two pectate lyases (pelL and pelA), two pectin methylesterases (pemA), and two polygalacturonases (pelL) (Figure 3). These enzymes cleave pectin into small oligogalacturonates. The PUL contained other genes that are involved in the catabolism of pectin such as genes encoding rhamnogalacturonides degradation proteins (rhiN, GH88), two rhamnogalacturonan acetylesterases, a susC-susD system, and a predicted transporter system for rhamnogalacturonide (RhiABC), and a set of genes associated with hexuronate metabolism (Figure 3). A similar PUL was identified in G. echinicola DSM19838 (Figure 3). Alishewanella (Jung and Park, 2013) and Alteromonas species within Gammaproteobacteria contained the majority of genes in a pectin PUL found in G. flava JLT2011. On the other hand, G. forsetii KT0803 lacked a pectin PUL, although it harbored a gene that encodes a pectin degradation protein, as well as other genes (including MFS, kdgK, and kdgF) located downstream of the alginate PUL locus that were possibly involved in pectin degradation (Figure S3; Bauer et al., 2006). However, the exact enzymatic mechanism of pectin for G. forsetii KT0803 need further experiments. Pectin methylesterase activity, which is responsible for the removal of methyl-esters from moderately methylated apple pectin, was observed when G. flava cells were grown on apple pectin (Figure S7). In contrast, no pectin methylesterase activity on citrus pectin was detected in G. flava JLT2011 because citrus pectin has a very low capability for methyl esterification. Furthermore, the absence of the pemA gene in G. forsetii KT0803 did not appear to completely prevent it from utilizing apple pectin for growth.
Apple and citrus pectins are mainly composed of D-galacturonic acid-forming homogalacturonans. The backbone of apple pectin also contains a small amount of α-1,2-rhamnose that alternates with α-1,4-D-galacturonate (ramnogalacturonan; Wu and Mort, 2014). Other sugars, including galactose, rhamnose, glucose, arabinose, xylose, and mannose, were also detected in apple pectin (Wu and Mort, 2014). G. flava JLT2011 is capable of utilizing L-rhamnose as a carbon source for growth (Figure S6) due to the presence of complete L-rhamnose utilization genes (rha) in the genome (Figure S5). These genes were clustered with pectin lyase and GHs to form a predicted PUL-like system for rhamnogalacturonan utilization (Figure S5). Polygalacturonases act on the homoglacturonan chains of the smooth region of pectin, whereas rhamnogalacturonases digest the rhamnogalacturonan chain of the hair region of pectin (Ochiai et al., 2007). Phylogenetic analysis indicated that four genes encoding polygalacturonases in G. flava JL2011 functionally belonged to the endopoylygalacturonase or exo-polygalacturonase family, but were distinct from rhamnogalacturonases (Figure 4). The alternative enzymatic route for rhamnogalacturonan degradation includes rhamnogalacturonan lyase (PL4 or PL11) and rhamnogalacturonan hydrolase (GH88 and GH105) (Ochiai et al., 2007). G. flava JL2011 contained five rhamnogalacturonan hydrolases but lacked rhamnogalacturonan lyase. These results suggest that a PUL-like system in G. flava JLT2011 does not appear to function in rhamnogalacturonan hydrolysis. Both G. flava JLT2011 and G. forsetii KT0803 could only utilize the homogalacturonan in apple pectin due to the lack of rhamnogalacturonan hydrolyase or rhamnogalacturonan lyase.
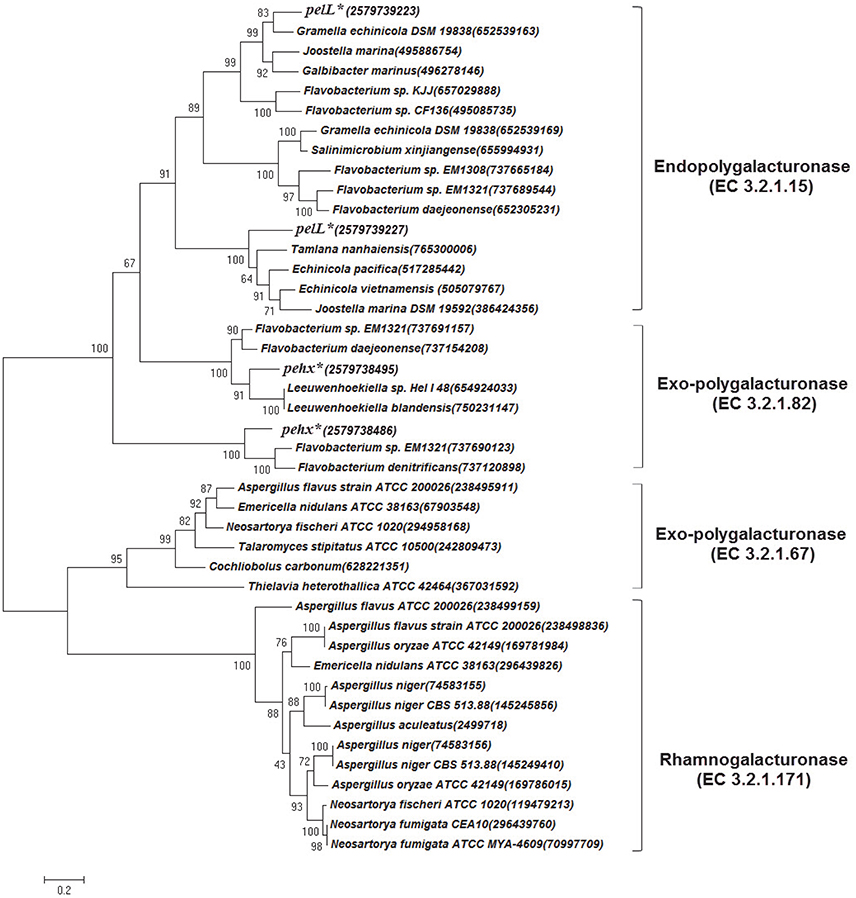
Figure 4. Phylogenetic tree based on polygalacturonase amino acid sequence comparisons constructed using the maximum-likelihood method in MEGA6. The pehX and pelL genes in G. flava JLT2011 are marked by asterisks and IMG/ER accession numbers are given in the bracket. The classes of polygalacturonase are listed on the right. The Genebank accession number for each reference sequence is given in the bracket. Bootstrap percentages are shown on each branch (1,000 replications). The scale bar indicates 20% sequence divergence.
Both G. flava JLT2011 and G. forsetii KT0803 harbored other PULs (Figures S3–S5); therefore, these are capable of utilizing trehalose, laminarin, starch, arabinan, arabinogalactan, galactomannan, and their corresponding monosaccharides (D-glucose, D-galactose, D-galacturonic acid, and L-arabinose; Figure S6, Table S5). These polysaccharides and corresponding monosaccharides have frequently been found in various marine algae that function in storage, or act as a cell wall constituent or as extracellular polysaccharides (Kröger and Poulsen, 2008; De Jesus Raposo et al., 2014; Based et al., 2015). Potential PUL identification and carbon utilization experiments revealed that Gramella species can degrade a wide range of algal polysaccharides, thus indicating a strong specialization toward an “opportunitrophic” lifestyle in the marine environment.
Reconstruction of Xylan and Pectin Catabolic Pathways Based on Transcriptomics, Proteomics, and Metabolomics Data
Global gene expression patterns for G. flava JLT2011 at the exponential growth phase in the presence of glucose, xylan, and pectin were assayed using microarrays (Figure 5). Overall, compared to the glucose treatment condition, a total of 1,234 and 951 genes were differentially expressed under xylan and pectin treatment conditions, respectively (fold change >2 or < 0.5; p < 0.05) (Tables S6, S7). In the presence of xylan, some genes in two PULs for xylan showed increased transcript levels compared to glucose, and the induction levels varied between 2.3 and 89.6 when relative to that in glucose, including the xylan main chain degradation genes (xynA and xynB), whereas the side chain sugar degradation genes (arb43, abfA-II, and aguA), and xylose metabolism genes (xylU, xylA, xylB, and xylE) were also up-regulated (Figure 5A). Two susC-susD systems in xylan PULs were up-regulated, in which induction levels were up to 35.5 and 89.6 for susC and susD (Table S6), respectively, thereby suggesting that these are involved in the uptake of oligomeric xylan. In the presence of pectin, the transcription of endopolygalacturonase (pelL) and hexuronate metabolism (kdgK, uxaA, uxaB, uxaC, and eda) in the pectin PUL were significantly enhanced compared to glucose (Figure 5B). However, a transcription of susC-susD system in the pectin PUL was not significantly up-regulated, and the transcripts of RhiABC genes were also significantly down-regulated compared to glucose (Table S7), suggesting that these are not involved in pectin degradation. The transcripts of genes in a rhamnogalacturonan PUL-like system did not significantly change in the presence of pectin, thus supporting the notion that G. flava JLT2011 does not utilize heteropolymeric rhamnogalacturonans in apple pectin.
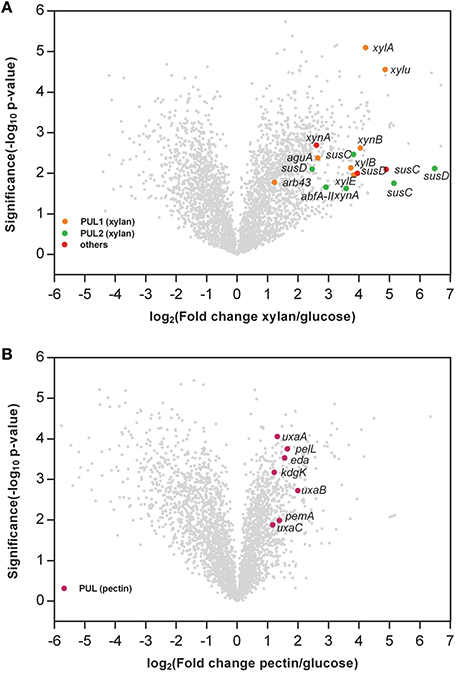
Figure 5. Volcano plot comparison of the gene expression between xylan and glucose (A) and pectin and glucose (B). The x axis indicates the differential expression profiles, plotting the fold-induction ratios in a log-2 scale. The y axis indicates the statistical significance of the difference in expression (p-value from a t-test) in a log10 scale. Genes within the xylan and pectin PULs are shown in colors (fold change > 2, or < 0.5; p < 0.05). (A) xynA, Endo-1,4-beta-xylanase; xynB, Beta-xylosidase; abfA-II, Alpha-L-arabinofuranosidase; arb43, Alpha-N-arabinofuranosidase; aguA, Alpha-glucuronidase; xylu, Acetyl xylan esterase; xylA, Xylose isomerase; xylB, Xylulose kinase; susC, TonB-dependent receptor; susD, carbohydrate-binding protein; xylE, Xylose transporter. (B) pemA, Pectinesterase; pelL, Endopolygalacturonase; uxaC, Uronate isomerase; kdgK, 2-dehydro-3-deoxygluconate kinase; eda, 2-dehydro-3-deoxyphosphogluconate aldolase; uxaB, Altronate oxidoreductase; uxaA, Altronate hydrolase; susC, TonB-dependent receptor; susD, carbohydrate-binding protein.
LC-MS/MS analysis identified a total of 1,778, 1,638, and 1,429 proteins at the exponential growth phase in the presence of xylan, pectin, and glucose, respectively (Table S8). A total of 1,190 proteins were detected among all samples, representing 33.7% of the protein-coding genes in G. flava. JLT2011. A higher number of proteins were associated with carbohydrate metabolism in cells cultured with xylan or pectin compared to those cultured with glucose (Figure 6A). The xylan degradation enzymes (XynA and XynB) and the endopolygalacturonases (PelL) were detected in the presence of xylan and pectin, respectively, which coincided with the results of microarray analysis. However, the SusC-susD system and RhiABC transporter in the pectin PUL were not detected, indicating that these were not involved in the transport of pectin degradation products. However, the GntP transporter was detected (Table S8) and is reported to have a high specificity for importing D-galacturonate (Suvorova et al., 2011). All Gramella species have the complete glycolysis and uncompleted Entner–Duodoroff and pentose phosphate pathways due to the presence of a gene that encodes glucose-6-phosphaste (P)-1-dehydrogenase. However, transketolase and transaldolase were detected in the proteomic samples (Table S8), which could convert xylose-5P into fructose-6P and glyceraldehyde-3P, which subsequently enter the glycolysis cycle. Gliding motility-related proteins, which possibly play a role in the digestion of biopolymers, were detected in the presence of xylan and pectin (Table S8).
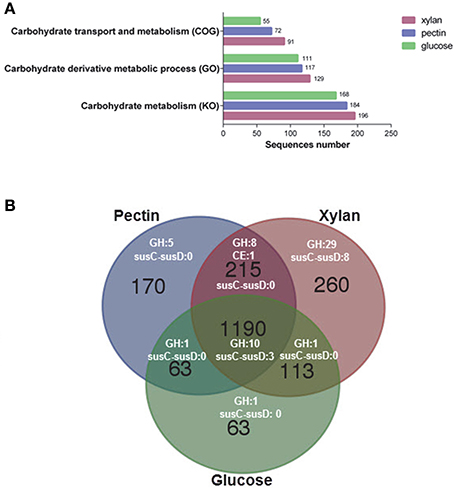
Figure 6. Functional analysis of the proteome of G. flava JLT2011 grown on xylan, pectin, and glucose. (A) Hits of identified proteins associated with carbohydrate metabolism classified by cluster of orthologous groups (COG), gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KO), respectively. (B) Venn diagram of counts of the detected CAZymes and susC-susD systems under different treatments with xylan, pectin and glucose. GH, glycoside hydrolase; CE, carbohydrate esterase.
Comparative analysis of the transcriptomics and proteomics data shows a good consistency for the PUL genes at the transcription level. All significantly up-regulated gene transcripts of xylan PULs and pectin PULs were detected at the protein level of the bacterial exponential growth phase (Table S8). Compared to glucose, a total of 25 GH and seven susC-susD system transcripts were up-regulated in the presence of xylan (Table S6), whereas only six GHs and a susC-susD system were up-regulated in the presence of pectin (Table S7). A total of 48, 24, and 13 GHs at the protein level were detected in the presence of xylan, pectin, and glucose, respectively (Figure 6B; Table S8). In addition, a total of 29 GH and eight SusC-SusD system proteins were only detected in the presence of xylan (Figure 6B). In contrast, only three SusC-SusD systems were detected in the presence of pectin and glucose (Figure 6B). Compared to glucose, the up-regulated transcript of xynA and its adjacent susC-susD system, xynB, were found in non-xylan PUL (Figure 5A), which possibly accelerates the degradation of the main chain of xylan. Moreover, other up-regulated transcripts of GHs, including beta-galactosidase and alpha-glucosidase, allows sufficient hydrolysis of various sugar residues of xylan (Table S6). These up-regulated transcripts in the presence of xylan are also detected at the protein level (Table S8). These results suggest that G. flava JLT2011 exhibited contrasting gene expression patterns on GHs and susC-susD systems in response to heteropolymeric xylan and homopolymeric pectin utilization, respectively. It was noted that some GH genes were shown differentially expressed revealed by transcriptomics in barely, while their corresponding proteins were not detected in the proteomic data possibly due to reasons such as gene expression and the limitation of techniques.
GC-MS analysis of bacterial metabolites showed that the highest concentration of xylose was observed in xylan-grown cells, whereas the highest concentration of D-galacturonic acid was detected in pectin-grown cells (Table 1). These are degradation products from xylan and pectin, respectively. The metabolites of pectin-grown cells that are involved in glycolysis (glucose-6-phosphate and fructose-6-phosphate) and the citric acid cycle cycle (citric acid, succinic acid, and fumaric acid) were down-regulated compared to that in the glucose-grown and xylan-grown cells (Table 1), indicating an decrease in the rate of glycolysis and the citric acid cycle.
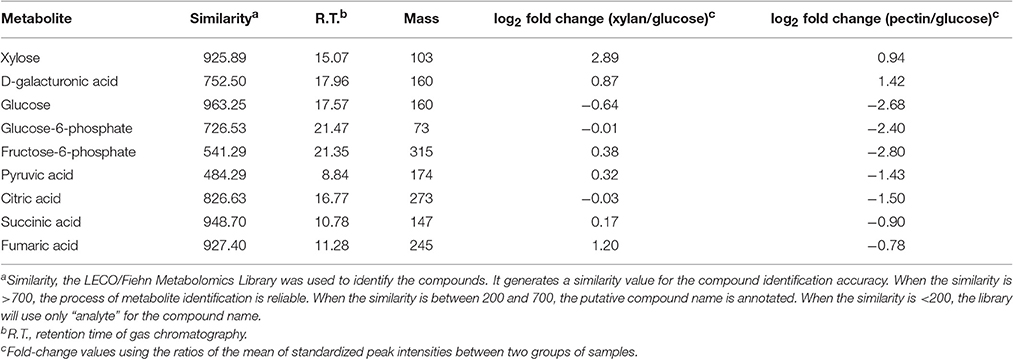
Table 1. Relative fold-change in selected metabolites in G. flava JLT2011 grown in different conditions (t-test, p < 0.05).
The integrated multi-omics data of this study reveal that PULs play a vital role in catabolic xylan and pectin pathways and we hereby propose a xylan and pectin utilization pathway as follows. The enzyme XynA hydrolyzes the main chain of xylan into oligomeric polymers that are subsequently transported into cells by the SusC-SusD system of G. flava JLT2011 (Figure 7). The enzymes XynB and Xylu convert xylan oligosaccharides into xylose, which then enter the cell and move into the cytoplasm through the mediation of XylE (Figure 7). The enzymes XylA and XylB yield the final product, xylose-5P, which is then directed into the pentose phosphate pathway (Figure 7). The enzymes Arb43, AbfA-II, and AguA might be involved in hydrolysis of the side chain of xylan (Figure 7). The homogalacturonan in pectin is degraded into D-galacturonic acid via the demethylating enzyme PemA, and PelL is used in pectin hydrolysis in G. flava JLT2011 (Figure 7). D-galacturonic acid is further utilized by the consequent actions of five hexuronate metabolism enzymes (UxaC, UxaB, UxaA, KdgK, and Eda), converting D-galacturonic acid into pyruvate and D-glyceraldehyde-3-phosphate, which subsequently enter the glycolytic pathway (Figure 7). Further biochemical analysis of the proteins involved in xylan and pectin utilization is necessary to fully understand the systems and validate the proposed pathway.
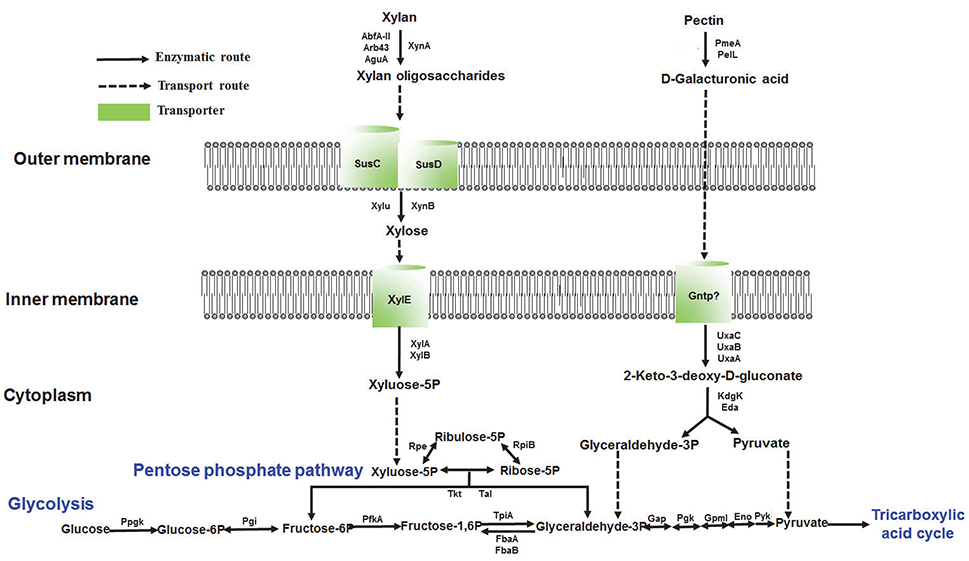
Figure 7. Reconstructed pathways of xylan and pectin utilization in G. flava JLT2011 based on multi-omics data. All labeled genes were expressed at the proteomic level. Xylan utilization pathway: XynA, Endo-1,4-beta-xylanase; XynB, Beta-xylosidase; AbfA-II, Alpha-L-arabinofuranosidase; Arb43, Alpha-N-arabinofuranosidase; AguA, Alpha-glucuronidase; SusC, TonB-dependent receptor; SusD, Carbohydrate-binding protein; Xylu, Acetyl xylan esterase; XylE, Xylose transporter; XylA, Xylose isomerase; XylB, Xylulose kinase. Pectin utilization pathway: PemA, Pectinesterase; PelL, Pectate lyase; PehX, Exo-poly-alpha-D-galacturonosidase; UxaC, Uronate isomerase; UxaB, Altronate oxidoreductase; UxaA, Altronate hydrolase; GntP, Gluconate transporter; KdgK, 2-dehydro-3-deoxygluconate kinase; Eda, 2-dehydro-3-deoxyphosphogluconate aldolase. Pentose phosphate pathway: Rpe, Ribulose-phosphate 3-epimerase; RpiB, Ribose 5-phosphate isomerase B; Tkt, Transketolase; Tal, Transaldolase. Glycolysis: PpgK, Polyphosphate glucokinase; Pgi, Glucose-6-phosphate isomerase; PfkA, 6-phosphofructokinase; TtpiA, Triosephosphate isomerase; FbaA, Fructose-bisphosphate aldolase class II; FbaB, Fructose-bisphosphate aldolase class I; Gap, NAD-dependent glyceraldehyde-3-phosphate dehydrogenase; Pgk, Phosphoglycerate kinase; GpmI, 2,3-bisphosphoglycerate-independent phosphoglycerate mutase; Eno, Enolase; Pyk, Pyruvate kinase.
In summary, this study has shown that the members of Gramella spp. have different potentials in hydrolyzing polysaccharides based on their differences in GHs and PUL genes. Genes encoding GHs and PULs in G. flava JLT2011 could function in the degradation of polysaccharides; however, these have distinct gene expression patterns in response to xylan and pectin, respectively. Our multi-omics studies have allowed us to functionally characterize GHs and PULs in a bacterial model that belongs phylum Bacteroidetes, thereby facilitating in better understanding their ecological role in algal polysaccharide decomposition in relation to marine carbon cycling.
Author Contributions
KT and NJ conceived and designed the experiments; KT, YL, and YH carried out the experiments and analyzed the data. All of the authors assisted in writing the manuscript, discussed the results and commented on the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China project (41276131), the National Key Research and Development Program of China (2016YFA0601100 & 2013CB955700), the National Program on Global Change and Air-Sea Interaction (GASI-03-01-02-05), the Natural Science Foundation of Fujian Province (2014J01164).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00220/full#supplementary-material
References
Alikhan, N.-F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Alonso, C., Warnecke, F., Amann, R., and Pernthaler, J. (2007). High local and global diversity of Flavobacteria in marine plankton. Environ. Microbiol. 9, 1253–1266. doi: 10.1111/j.1462-2920.2007.01244.x
Anderson, K. L., and Salyers, A. A. (1989a). Biochemical evidence that starch breakdown by Bacteroides thetaiotaomicron involves outer membrane starch-binding sites and periplasmic starch-degrading enzymes. J. Bacteriol. 171, 3192–3198.
Anderson, K. L., and Salyers, A. A. (1989b). Genetic evidence that outer membrane binding of starch is required for starch utilization by Bacteroides thetaiotaomicron. J. Bacteriol. 171, 3199–3204.
Based, F., Synytsya, A., Kim, W. J., and Park, Y. (2015). “Cell wall polysaccharides of marine algae,” in Springer Handbook of Marine Biotechnology, ed S. K. Kim (New York, NY: Springer Dordrecht Heidelberg), 543–590.
Bastawde, K. B. (1992). Xylan structure, microbial xylanases, and their mode of action. World J. Microbiol. Biotechnol. 8, 353–368. doi: 10.1007/BF01198746
Bauer, M., Kube, M., Teeling, H., Richter, M., Lombardot, T., Allers, E., et al. (2006). Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 8, 2201–2213. doi: 10.1111/j.1462-2920.2006.01152.x
Benner, R., Pakulski, J., McCarthy, M., Hedges, J., and Hatcher, P. (1992). Bulk chemical characteristics of dissolved organic matter in the ocean. Science 255, 1561–1564.
Boyce, D. G., Lewis, M. R., and Worm, B. (2010). Global phytoplankton decline over the past century. Nature 466, 591–596. doi: 10.1038/nature09268
Buchan, A., Lecleir, G. R., Gulvik, C. A., and González, J. M. (2014). Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat. Rev. Mircobiol. 12, 686–698. doi: 10.1038/nrmicro3326
Cottrell, M. T., and Kirchman, D. L. (2000). Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl. Environ. Microbiol. 66, 1692–1697. doi: 10.1128/AEM.66.4.1692-1697.2000
Cuskin, F., Lowe, E. C., Temple, M. J., Zhu, Y., Cameron, E. A., Pudlo, N. A., et al. (2015). Human gut Bacteroidetes can utilize yeast mannan through a selfish mechanism. Nature 517, 165–169. doi: 10.1038/nature13995
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2009). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
De Jesus Raposo, M. F., de Morais, A. M. M. B., and de Morais, R. M. S. C. (2014). “Bioactivity and Applications of polysaccharides from marine microalgae,” in Polysaccharides, eds J. M. Merillon and K. G. Ramawat (Porto: Springer), 1683–1727.
Engel, A., Thoms, S., Riebesell, U., Rochelle-Newall, E., and Zondervan, I. (2004). Polysaccharide aggregation as a potential sink of marine dissolved organic carbon. Nature 428, 929–932. doi: 10.1038/nature02453
Fernández-Gómez, B., Richter, M., Schüler, M., Pinhassi, J., Acinas, S. G., González, J. M., et al. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7, 1026–1037. doi: 10.1038/ismej.2012.169
Glöckner, F. O., Fuchs, B. M., Fuchs, B. M., Glo, F. O., and Amann, R. (1999). Bacterioplankton compositions of lakes and oceans : a first comparison based on fluorescence in situ hybridization. Appl. Environ. Microbiol. 65, 3721–3726.
Hutcheson, S. W., Zhang, H., and Suvorov, M. (2011). Carbohydrase systems of Saccharophagus degradans degrading marine complex polysaccharides. Mar. Drugs 9, 645–665. doi: 10.3390/md9040645
Jung, J., and Park, W. (2013). Comparative genomic and transcriptomic analyses reveal habitat differentiation and different transcriptional responses during pectin metabolism in Alishewanella species. Appl. Environ. Microbiol. 79, 6351–6361. doi: 10.1128/AEM.02350-13
Kabisch, A., Otto, A., König, S., Becher, D., Albrecht, D., Schüler, M., et al. (2014). Functional characterization of polysaccharide utilization loci in the marine Bacteroidetes ‘Gramella forsetii’ KT0803. ISME J. 8, 1492–1502. doi: 10.1038/ismej.2014.4
Kirchman, D. L. (2002). The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39, 91–100. doi: 10.1016/S0168-6496(01)00206-9
Koren, S., Schatz, M. C., Walenz, B. P., Martin, J., Howard, J. T., Ganapathy, G., et al. (2012). Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 30, 693–700. doi: 10.1038/nbt.2280
Kröger, N., and Poulsen, N. (2008). Diatoms-from cell wall biogenesis to nanotechnology. Annu. Rev. Genet. 42, 83–107. doi: 10.1146/annurev.genet.41.110306.130109
Larsbrink, J., Rogers, T. E., Hemsworth, G. R., McKee, L. S., Tauzin, A. S., Spadiut, O., et al. (2014). A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature 506, 498–502. doi: 10.1038/nature12907
Lau, S. C. K., Tsoi, M. M. Y., Li, X., Plakhotnikova, I., Dobretsov, S., Wong, P.-K., et al. (2005). Gramella portivictoriae sp. nov., a novel member of the family Flavobacteriaceae isolated from marine sediment. Int. J. Syst. Evol. Microbiol. 55, 2497–2500. doi: 10.1099/ijs.0.63824-0
Liu, K., Li, S., Jiao, N., and Tang, K. (2014). Gramella flava sp. nov., a member of the family Flavobacteriaceae isolated from seawater. Int. J. Syst. Evol. Microbiol. 64, 165–168. doi: 10.1099/ijs.0.051987-0
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, 490–495. doi: 10.1093/nar/gkt1178
Mann, A. J., Hahnke, R. L., Huang, S., Werner, J., Xing, P., Barbeyron, T., et al. (2013). The genome of the alga-associated marine flavobacterium Formosa agariphila KMM 3901T reveals a broad potential for degradation of algal polysaccharides. Appl. Environ. Microbiol. 79, 6813–6822. doi: 10.1128/AEM.01937-13
Martens, E. C., Lowe, E. C., Chiang, H., Pudlo, N. A., Wu, M., McNulty, N. P., et al. (2011). Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 9:e1001221. doi: 10.1371/journal.pbio.1001221
Martinez-Garcia, M., Brazel, D. M., Swan, B. K., Arnosti, C., Chain, P. S. G., Reitenga, K. G., et al. (2012). Capturing single cell genomes of active polysaccharide degraders: an unexpected contribution of Verrucomicrobia. PLoS ONE 7:e35314. doi: 10.1371/journal.pone.0035314
McBride, M. J., Xie, G., Martens, E. C., Lapidus, A., Henrissat, B., Rhodes, R. G., et al. (2009). Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl. Environ. Microbiol. 75, 6864–6875. doi: 10.1128/AEM.01495-09
Medema, M. H., Takano, E., and Breitling, R. (2013). Detecting sequence homology at the gene cluster level with multigeneblast. Mol. Biol. Evol. 30, 1218–1223. doi: 10.1093/molbev/mst025
Nedashkovskaya, O. I., Kim, S. B., Hoste, B., Shin, D. S., Beleneva, I. A., Vancanneyt, M., et al. (2007). Echinicola vietnamensis sp. nov., a member of the phylum Bacteroidetes isolated from seawater. Int. J. Syst. Evol. Microbiol. 57, 761–763. doi: 10.1099/ijs.0.64546-0
Nedashkovskaya, O. I., Kim, S. B., Lysenko, A. M., Frolova, G. M., Mikhailov, V. V., Bae, K. S., et al. (2005). Gramella echinicola gen. nov., sp. nov., a novel halophilic bacterium of the family Flavobacteriaceae isolated from the sea urchin Strongylocentrotus intermedius. Int. J. Syst. Evol. Microbiol. 55, 391–394. doi: 10.1099/ijs.0.63314-0
Nedashkovskaya, O. I., Kim, S. B., Vancanneyt, M., Lysenko, A. M., Shin, D. S., Park, M. S., et al. (2006). Echinicola pacifica gen. nov., sp. nov., a novel flexibacterium isolated from the sea urchin Strongylocentrotus intermedius. Int. J. Syst. Evol. Microbiol. 56, 953–958. doi: 10.1099/ijs.0.64156-0
Nelson, D. M., Treguer, P., Brzezinski, M. A., Leynaert, A., and Queguiner, B. (1995). Production and dissolution of biogenic silica in the ocean: revised global estimates, comparision with regional data and relationship to biogenic sedimentation. Global Biogeochem. Cycles 9, 359–372.
Ochiai, A., Itoh, T., Kawamata, A., Hashimoto, W., and Murata, K. (2007). Plant cell wall degradation by saprophytic Bacillus subtilis strains: gene clusters responsible for rhamnogalacturonan depolymerization. Appl. Environ. Microbiol. 73, 3803–3813. doi: 10.1128/AEM.00147-07
Panschin, I., Huang, S., Meier-Kolthoff, J. P., Tindall, B. J., Rohde, M., Verbarg, S., et al. (2016). Comparing polysaccharide decomposition between the type strains Gramella echinicola KMM 6050T (DSM 19838T) and Gramella portivictoriae UST040801-001T (DSM 23547T), and emended description of Gramella echinicola Nedashkovskaya et al. 2005 emend. Shahina et. Stand. Genomic Sci. 11:37. doi: 10.1186/s40793-016-0163-9
Park, B. H., Karpinets, T. V., Syed, M. H., Leuze, M. R., and Uberbacher, E. C. (2010). CAZymes Analysis Toolkit (cat): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 20, 1574–1584. doi: 10.1093/glycob/cwq106
Pinhassi, J., Bowman, J. P., Nedashkovskaya, O. I., Lekunberri, I., Gomez-Consarnau, L., and Pedrós-Alió, C. (2006). Leeuwenhoekiella blandensis sp. nov., a genome-sequenced marine member of the family Flavobacteriaceae. Int. J. Syst. Evol. Microbiol. 56, 1489–1493. doi: 10.1099/ijs.0.64232-0
Qin, Q. L., Zhang, X. Y., Wang, X. M., Liu, G. M., Chen, X. L., Xie, B. B., et al. (2010). The complete genome of Zunongwangia profunda SM-A87 reveals its adaptation to the deep-sea environment and ecological role in sedimentary organic nitrogen degradation. BMC Genomics 11:247. doi: 10.1186/1471-2164-11-247
Rawlings, N. D., and Barrett, A. J. (1999). MEROPS: the peptidase database. Nucleic Acids Res. 27, 325–331. doi: 10.1093/nar/27.1.325
Rogowski, A., Briggs, J. A., Mortimer, J. C., Tryfona, T., Terrapon, N., Lowe, E. C., et al. (2015). Glycan complexity dictates microbial resource allocation in the large intestine. Nat. Commun. 6:7481. doi: 10.1038/ncomms8481
Roth, A. C., Gonnet, G. H., and Dessimoz, C. (2009). Algorithm of OMA for large-scale orthology inference. BMC Bioinformatics 10:220. doi: 10.1186/1471-2105-10-220
Shipman, J. A., Berleman, J. E., and Salyers, A. A. (2000). Characterization of four outer membrane proteins involved in binding starch to the cell surface of Bacteroides thetaiotaomicron. J. Bacteriol. 182, 5365–5372. doi: 10.1128/JB.182.19.5365-5372.2000
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Suvorova, I. A., Tutukina, M. N., Ravcheev, D. A., Rodionov, D. A., Ozoline, O. N., and Gelfand, M. S. (2011). Comparative genomic analysis of the hexuronate metabolism genes and their regulation in gammaproteobacteria. J. Bacteriol. 193, 3956–3963. doi: 10.1128/JB.00277-11
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tans-kersten, J., Guan, Y., and Allen, C. (1998). Ralstonia solanacearum pectin methylesterase is required for growth on methylated pectin but not for bacterial wilt virulence. Appl. Environ. Microbiol. 64, 4918–4923.
Taylor, J. D., Cottingham, S. D., Billinge, J., and Cunliffe, M. (2014). Seasonal microbial community dynamics correlate with phytoplankton-derived polysaccharides in surface coastal waters. ISME J. 8, 245–248. doi: 10.1038/ismej.2013.178
Teeling, H., Fuchs, B. M., Becher, D., Klockow, C., Gardebrecht, A., Bennke, C. M., et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611. doi: 10.1126/science.1218344
Terrapon, N., Lombard, V., Gilbert, H. J., and Henrissat, B. (2015). Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics 31, 647–655. doi: 10.1093/bioinformatics/btu716
Wiśniewski, J. R., Zougman, A., Nagaraj, N., and Mann, M. (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. doi: 10.1038/nmeth.1322
Keywords: glycoside hydrolases, polysaccharide utilization loci, Bacteroidetes, genome, microarray, proteomics, marine bacteria
Citation: Tang K, Lin Y, Han Y and Jiao N (2017) Characterization of Potential Polysaccharide Utilization Systems in the Marine Bacteroidetes Gramella Flava JLT2011 Using a Multi-Omics Approach. Front. Microbiol. 8:220. doi: 10.3389/fmicb.2017.00220
Received: 19 August 2016; Accepted: 31 January 2017;
Published: 14 February 2017.
Edited by:
Rex Malmstrom, DOE Joint Genome Institute, USAReviewed by:
Zongze Shao, State Oceanic Administration, ChinaThomas Schweder, University of Greifswald, Germany
Fiona Cuskin, Newcastle University, UK
Copyright © 2017 Tang, Lin, Han and Jiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Tang, tangkai@xmu.edu.cn
Nianzhi Jiao, jiao@xmu.edu.cn