 John C. Alverdy1*
John C. Alverdy1* James N. Luo2
James N. Luo2
- 1Sarah and Harold Lincoln Thompson Professor of Surgery, Pritzker School of Medicine, The University of Chicago, Chicago, IL, USA
- 2Pritzker School of Medicine, The University of Chicago, Chicago, IL, USA
Mammals constantly face stressful situations, be it extended periods of starvation, sleep deprivation from fear of predation, changing environmental conditions, or loss of habitat. Today, mammals are increasingly exposed to xenobiotics such as pesticides, pollutants, and antibiotics. Crowding conditions such as those created for the purposes of meat production from animals or those imposed upon humans living in urban environments or during world travel create new levels of physiologic stress. As such, human progress has led to an unprecedented exposure of both animals and humans to accidental pathogens (i.e., those that have not co-evolved with their hosts). Strikingly missing in models of infection pathogenesis are the various elements of these conditions, in particular host physiologic stress. The compensatory factors released in the gut during host stress have profound and direct effects on the metabolism and virulence of the colonizing microbiota and the emerging pathobiota. Here, we address unanswered questions to highlight the relevance and importance of incorporating host stress to the field of microbial pathogenesis.
Most Models of Infection Pathogenesis do not Incorporate Host Stress
It is well recognized, both in animal experiments and humans, that exposure to an infectious agent alone is insufficient to cause, consistently, clinical manifestations of the disease (Babrowski et al., 2013). Investigators often observe marked heterogeneity of disease manifestation when groups of otherwise similarly appearing and treated hosts are exposed to a given contagion (Connolly et al., 2015). It is for this reason that the “molecular Koch’s postulates” have been proposed to include changes in both host and pathogen phenotypes and their dynamic interaction when studying a single pathogen, or pathogen community, as a cause of an infectious disease (Falkow, 1988). It is well recognized that no two pathogens are alike, even they be from the same origin (Tenaillon et al., 2016). Microbes can also shift their phenotype in vivo in response to a variety of local environmental cues, each of which is particular to a given host, in a given tissue area, and in a given spatial context (Luong et al., 2014). It is for this reason that today, when experimentally modeling infection in small animals, we attempt to control as many variables as possible, such as breeding history, diet, housing conditions, etc. (Stappenbeck and Virgin, 2016). Nonetheless, unaccounted variability still exists frequently.
Traditionally, experiments are designed to detect “between-group” differences while manipulating genes in the infecting agent or host. Yet rarely are “within-group” differences of infection rates or mortality accounted for among the treatments so long as the between group differences are robust and statistically significant. What drives this heterogeneity of response within a highly homogenously treated group in a highly controlled environment? Here, we posit that the degree of physiologic stress of an individual subject plays a key, and regularly dismissed, role in the variability of infection-related outcome. To fully match all animals in a study, hourly measurements of numerous parameters (e.g., sleep, hunger, fear, anxiety, handling, etc.) would be necessary and integrated responses over the course of the experiment would need to be calculated. This is obviously not routinely performed and would be costly, if not impossible to achieve.
Yet in virtually every small animal experiment in which infection or mortality is used as the endpoint, there exists a high degree of variability in outcome that is rarely, if ever, reported or studied (Stough et al., 2016). What is often overlooked may be the emergent properties that develop in the infecting agent and the host as they interact with each other over the entire course of the host–pathogen relationship. Pathogen phenotypes are highly dynamic over the course of this interaction, as is the host physiologic response (hormones, cytokines, metabolome) before, during, and after the infectious inoculum is introduced (Shogan et al., 2014). A complex molecular dialog is developing as these two living organisms interact, exchange signals, and behave as one multi-cellular system (Rhee et al., 2009). Such dynamism will have a profound effect in shaping the social behavior of colonizing microbes.
In order to model more precisely the host pathogen interaction, reductionist experiments with small animal models (i.e., C. elegans) and laboratory pathogens are used (Yuen and Ausubel, 2014). While much is to be gained from these reductionist models, they do not reflect some of the most challenging infections in humans, such as those that occur in modern intensive care units in the developing world (Rasigade et al., 2012). Patients, for example in an ICU are highly traumatized by procedural medicine, cared for under the most physiologically stressful conditions, and confined to the most hostile microbial environment (Zakharkina et al., 2017). Such patients are regularly exposed to healthcare associated pathogens that harbor unique antibiotic resistance patterns and highly virulent phenotypes (Busani et al., 2017). In addition, because of the promiscuous use of antibiotics to care for ICU patients, the protective action of the normal microbiota is essentially eliminated (Arrieta and Finlay, 2012). Hosts are vulnerable on two fronts, loss of the microbiome and the emergence of a virulent and resistant pathobiome (Krezalek et al., 2016). There is also evidence that physiologic or traumatic stress alone causes depletion of the host’s intestinal microbiome by unknown mechanism (Alverdy and Krezalek, 2017). Thus at the same time that compensatory host-derived signaling molecules are released during stress, which shift the phenotype of its colonizing flora, the normal microbiota are collapsing in abundance and function (Hayakawa et al., 2011). Such a scenario begs investigators to understand the role of physiologic and traumatic stress on infection-related outcome beyond their direct effects on the immune system and to apply a more holistic and systems biology approach to model infection, as it likely occurs in vivo (Figure 1).
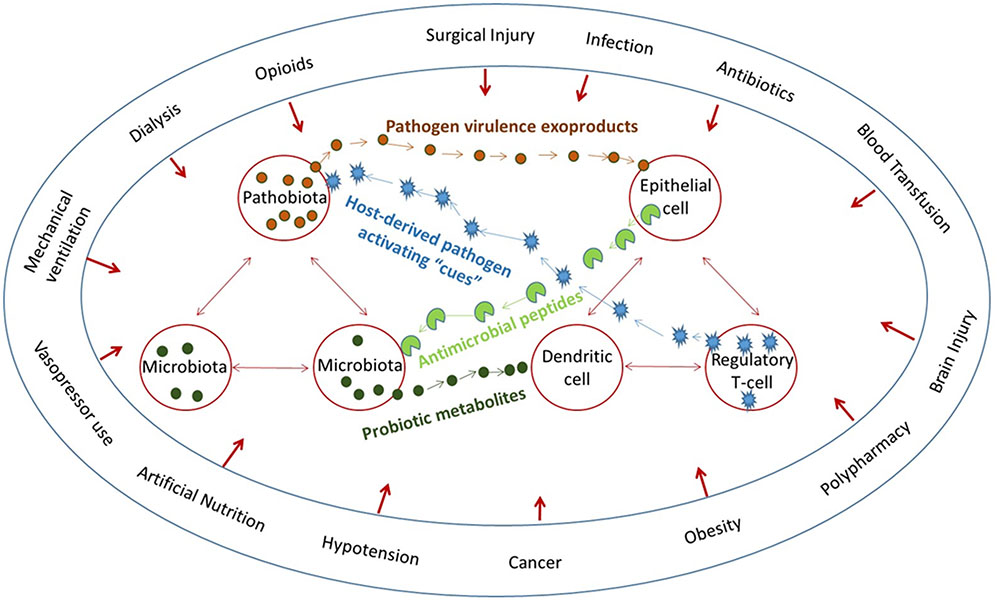
FIGURE 1. The microbiome affects everything and everything affects the microbiome. Multiple converging lines of bidirectional signaling between the host and microbiota and between the microbiota and pathobiota demonstrate that host circumstances directly affect both the microbiota and the immune system.
How Does Acute Host Stress Affect the Abundance and Function of the Microbiota?
It is now well established that following a sudden insult to the host, such as acute trauma, myocardial infarction, or burn injury, the intestinal microbiota decrease in abundance and function by greater than ninety percent (Shimizu et al., 2015). This observation may play an unappreciated role in the general consensus that a stressed host is more vulnerable to infection (Guyton and Alverdy, 2016). The scope and molecular details by which physiologic stress interacts with the intestinal microbiota and causes immunosuppression remains incompletely elucidated. However, ongoing investigations are beginning to shed some light on the mechanisms. In hospitalized patients who are critically ill, we often see a near complete ecological collapse of their endogenous microbiota, which is likely the result of both the patient’s active disease state and the selective pressure imposed upon them by modern intensive care efforts (Modi et al., 2014). Not only does the abundance of the microbiota become reduced in these patients, but low-diversity communities, often difficult to detect, tend to proliferate and are represented by highly resistant and virulent organisms such as Candida albicans, Enterococcus spp., Staphylococcus spp., and Enterobacteriaceae (Zaborin et al., 2014b). In one recent study by our group, Zaborin et al. (2014b), found that 30% of the critically ill patients had “ultra-low-diversity” microbial communities consisting of four or less bacterial taxa.
One of the most obvious and intuitive drivers of this ecologic collapse is the profound selection pressure imposed by the promiscuous use of antimicrobial agents. Extensive work has been reported to understand the effects of antibiotics on the microbiota (Modi et al., 2014). In 2010, more than 70 billion individual doses of antibiotics were consumed world-wide (Blaser, 2016). Broad-spectrum antibiotics can impact up to 30% of the bacteria among the human microbiota, resulting in severe loss of taxonomic and functional diversity (Francino, 2016). This dramatic shift in the microbiota can develop immediately following antibiotic administration, and can sometimes last for years after its cessation (Jakobsson et al., 2010). The perturbation of the endogenous flora has been linked to many disease states including obesity and autoimmunity (Francino, 2016).
While the effects of antibiotics are well studied and appreciated, the microbial collapse associated with critical illness is much more profound and broad when compared to exposure to antimicrobials alone. Many forms of host stress, independent of antimicrobial administration, have been shown to affect the composition and function of the microbiota (Mackenzie et al., 2017). For example, in patients undergoing gastrointestinal surgery, the use of opioid analgesics, withholding of enteral nutrition, and gastric acid suppression have all been shown to have profound effects on the microbiome (Krezalek and Alverdy, 2016; Levesque et al., 2016). Reuland et al. (2016) reported that the use antacids is associated with increased risk of extended-spectrum-β-lactamase producing Enterobacteriaceae carriage. Even surgical procedures themselves, such as colonic resection and reconnection, can be associated with a 500-fold increase in the abundance of Enterococcus faecalis (Shogan et al., 2014). This dynamic reality of microbiome stability further highlights the importance of understanding the complex host–microbiota interaction.
How Does Host Stress Activate Pathogens to Cause Infection?
Attempts to elucidate the mechanistic details of this microbial shift have aimed mainly at the hypothesis that host stress causes immunosuppression (Vanzant et al., 2014). However, less well explored, is the possibility that host stress diminishes the protective intestinal microbiota, in both abundance and function, and that host stress signals activate colonizing “pathobiota” to express enhanced virulence (Lupp et al., 2007; Alverdy and Krezalek, 2017). It could be postulated that the intestinal microbiota “sense” that the host is under duress and decrease their growth rate and metabolism both anticipating that resources will be limited, and that the host cannot tolerate activation of its immune system by the intestinal microbiota (Babrowski et al., 2013). Alternatively, and in concert with this mechanism, could be the activation of intestinal antimicrobial peptides via IL-22, which is known to be elevated following traumatic and physiologic stress (Bingold et al., 2010; Rendon et al., 2013; Behnsen et al., 2014). In this way, the host keeps its intestinal microbiome “at bay” until which time recovery is established and homeostasis returns. The temporal dynamic of this response, the period of diminution, the refaunation process, and the species and community structure that are involved in this response remain to be clarified. Although some elucidation of this mechanism has been reported with the foodborne pathogen Salmonella; importantly, no host stress was imposed in the experimental model (Behnsen et al., 2014). Although such elegant and insightful models of Salmonella inform the mechanisms of its pathogenesis, they fall short in explaining why most humans exposed to the pathogen never develop an infection (Barak et al., 2009; Spencer et al., 2010). Several key questions remain unanswered. What are the mechanisms by which ingested isolates shift their phenotype to adapt to their new environment so they can express virulence factors that allow them to induce host cytokines (i.e., IL-22) that eliminate the microbiota? Do humans (and mice) who are stressed release host stress-derived compensatory factors that induce Salmonella to express these virulence factors that then determine if and how infection occurs (Krueger and Opp, 2016; Poroyko et al., 2016)? The last is a particularly important question given that we know that host stress depletes the microbiota, activates IL-22 (Bingold et al., 2010) (which further can deplete the microbiota), releases cytokines that directly signal bacteria to activate their quorum sensing circuits (Wu et al., 2005), and diminishes local resources (Long et al., 2008) (i.e., phosphate) in the local milieu (Long et al., 2008; Rendon et al., 2013).
Through a process recently termed “telesensing” (Roux et al., 2009), certain bacteria can not only sense their population density via quorum sensing, but also can detect and respond to host-stress derived signals such as opioids, cytokines, end-products of ischemia, immune cell environments, etc., that are unique to host tissues exposed to stressful conditions (Sansonetti, 2004). This type of interkingdom signaling has traditionally received little attention in the microbial pathogenesis field (Kendall and Sperandio, 2016). While certain physio-chemical cues, such as pH, redox state, phosphate, etc., are well known to influence bacterial virulence activation, an emerging area of interest is how host-stress derived compensatory “cues” drive colonization, invasion, virulence activation, and ultimately, the continuum of infection from symptom development to lethality (Mekalanos, 1992). We and others have described many of these host-stress compensatory elements, the receptors on bacteria to which they bind, and the downstream pathways that become activated leading to a shift in virulence (Seal et al., 2010). For example, the Gram-negative pathogen Pseudomonas aeruginosa can detect host physiologic disturbance by sensing opioids in the host environment, and in response, activate its quorum sensing virulence machinery (Zaborina et al., 2007). This process involves a complex and constant dialog between the pathogen and its host (Alverdy et al., 2000). The host secrete factors in response of microbial presence, the microbe in turn detects these signals and adjusts its virulence accordingly (Patel et al., 2007; Zaborin et al., 2014a). Many commonly encountered bacterial virulence mechanisms are subject to this additional level of host-derived signaling: biofilm formation, swarming, luminescence, toxin production, etc. (Palmer and Blackwell, 2008).
Host–microbe interkingdom signaling and telesensing are not novel developments. Because the microbiota and its human host co-evolved over tens of thousands of years, an elaborate signaling system exists between them (Sansonetti, 2004). It is well known that host catecholamines released during stress can induce bacterial growth, enhance colonization to host tissue, and virulence upregulation (Freestone and Lyte, 2008). In addition, the human “gut-brain axis” is an active area of investigation. We are just beginning to appreciate the level of involvement that the microbiota plays in the development of the human nervous system (Lyte, 2013). So far, we know that this gut-brain axis is a bidirectional dialog involving neural (e.g., GABA), endocrine (e.g., amines), immune, and humoral signals (Carabotti et al., 2015). In addition to host-produced signals, release and sequestration of inorganic compounds, such as phosphate, cooper, iron, have all been implicated in this host–microbe interkingdom signaling (Schaible and Kaufmann, 2004; Zaborin et al., 2014a). These complex mechanisms of communication help to maintain the mutualistic human-microbiome relationship, and is the product of millennia of co-evolution.
As such, the occurrence, course, and outcome of infection may be highly influenced by the degree of host stress, not only because stress has a direct effect on immune function, but because physiologic stress has a direct effect on bacterial behavior. In the context of human infection, rarely if ever, is host stress adequately instantiated into experimental models. Host genes are manipulated as are microbial genes, and pathogenicity described. However, a major flaw in this approach is the dismissal of the “within-group” variability in infection occurrence and outcome that may be the most informative of the host–pathogen dialog that must first occur for the process of infection to be initiated. As can be seen in Figure 1, converging lines of host–pathogen interactions make it extremely challenging to organize and study such a dynamic and fluid system in the context of a critically ill patient. It may be for this reason that no new therapies for sepsis in the critically ill have emerged in decades. Yet understanding how the microbiota collapse following host stress, how the pathobiota emerge to achieve a new state of equilibrium with the host, and whether the resilience of the host to achieve recovery depends on the ability of the microbiota to refaunate, remains a challenging but important line of inquiry (Sansonetti, 2004).
Is Host Recovery from Stress Dependent on the Ability of the Microbiota to Refaunate?
While resilience to host injury and recovery from infection is generally attributed to a robust host immune clearance mechanism, emerging knowledge in microbiome science suggests that the intestinal microbiome plays a key role in driving a recovery-directed immune response (Shen et al., 2012). As described above, when a human is injured, both from the injury response itself and from its treatment by modern medicine, the intestinal microbiome can collapse in abundance and function (Shimizu et al., 2006). Yet as injuries are repaired and infections cleared with antibiotics, the ability of the host microbiome to refaunate is often considered to lag behind recovery, rather than to drive it (Jakobsson et al., 2010). However, equally plausible is the possibility that a previously healthy host (no smoking, limited previous antibiotic use, lean diet, regular exercise) may have a capacity to refaunate his/her microbiome to a greater degree than a previously unhealthy patient (Carlisle and Morowitz, 2011; Cosnes, 2016; Munck et al., 2016). The dynamics of refaunation and its correlation to recovery is poorly explored, however, with sequencing and metabolomics becoming more widely available and less costly, this can now be determined. Enhancing the refaunation process with fecal transplantation alongside therapies that are highly catabolic (bone marrow and solid organ transplantation) are underway and may further reinforce the plausibility of this concept (Kazerouni and Wein, 2017). The near disappearance of the intestinal microbiome following severe catabolic stress and injury, while adaptive prior to modern medicine, may be considered maladaptive in the present era, where highly toxic and invasive therapies (chemotherapy, radiation, severe burn injury) are needed to treat life-threatening diseases (Jenq et al., 2012; Pamer, 2016; Taur and Pamer, 2016). Figure 2 depicts our theory involving the uncharted space in the intestinal tract that may play an unappreciated role in recovery from severe host stress.
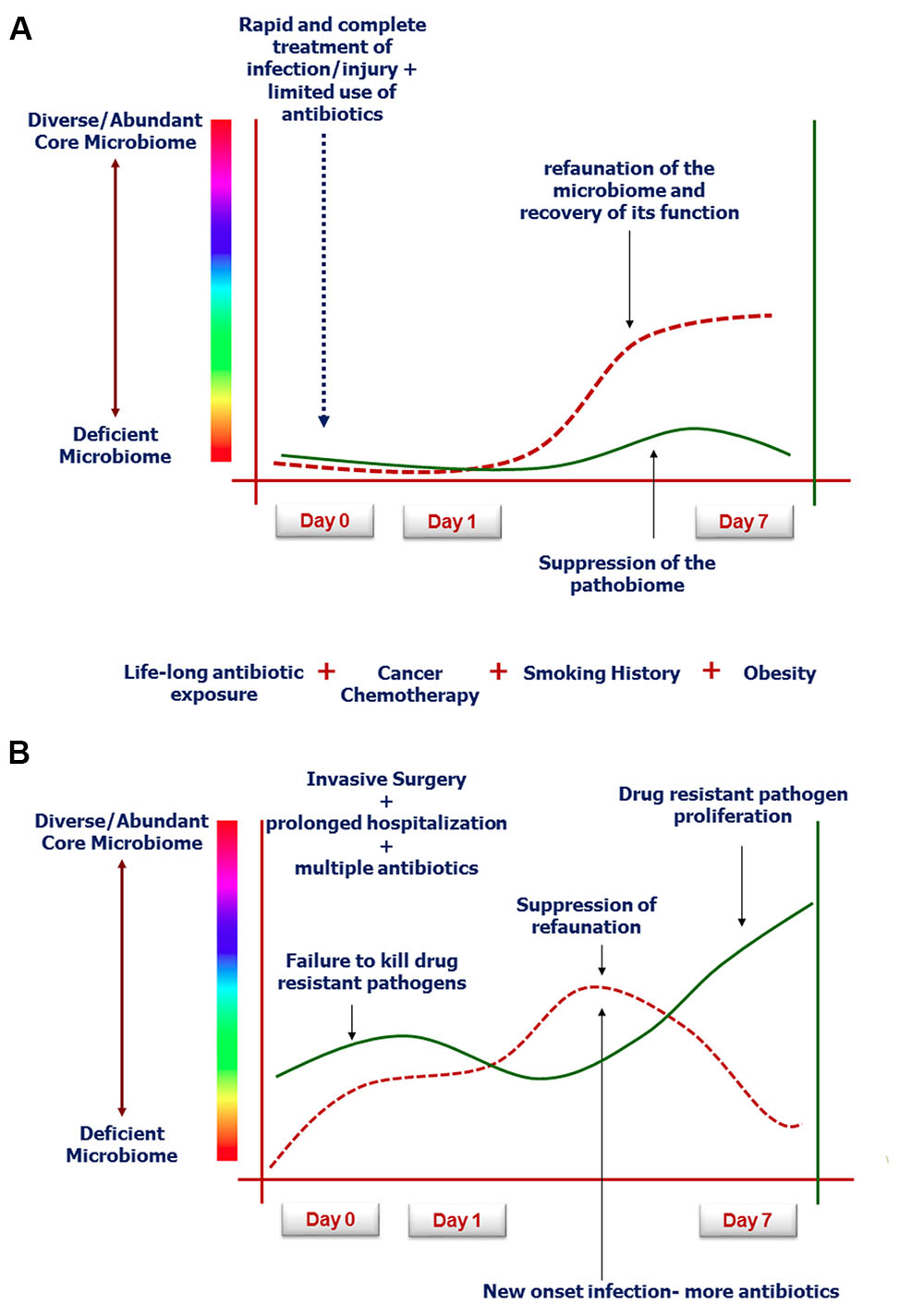
FIGURE 2. Rate and degree of microbiota refaunation on recovery from severe catabolic stress such as following human critical illness (Guyton and Alverdy, 2016; Krezalek and Alverdy, 2016; Alverdy and Krezalek, 2017). (A) Demonstrates typical response to successful modern medical care with limited antibiotic exposure and rapid resolution of the infection or injury (Modi et al., 2014). (B) Represents the aging patient population with multiple exposures to western diet, smoking etc, who have fragile microbiomes that cannot recover when invasive surgery and toxic agents are used to treat complex disorders such as cancer (Shogan et al., 2014; Shakhsheer et al., 2016).
Conclusion
Pathogens bring their own unique life histories when they colonize or infected a new host. The complex dynamics of physiologic stress in the host drives these pathogens, and the microbial communities in which they co-exist, into a pathoadaptive process where genes are lost and found, and where new phenotypes emerge. Under such circumstances, emergent phenotypes among the colonizing pathobiota increase in frequency and compete for colonization sites and local resources. As stress becomes a persistent state and antibiotics are added to treat infections, microbial evolution speeds up as the emergent “pathobiome” enters an evolutionarily uncharted environment. As these pathobiomes compete for fixation niches, they become hidden from clinicians in protected sites where they do their dirty work at arms’ length from the immune system. Uncovering the dynamics of this host–pathogen interactome and the sites in which it occurs will lead to novel lines of inquiry and hypotheses to explain more completely the occurrence, course, and outcome of life-threatening infections that develop in the critically ill and all around the world.
Author Contributions
JA: Conducted literature search and review; wrote, revised, and edited the manuscript. JL: Conducted literature search and review; revised and edited the manuscript.
Funding
This work was supported by NIH grant: 2R01GM062344-15 (Principal Investigator: JA).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alverdy, J., Holbrook, C., Rocha, F., Seiden, L., Wu, R. L., Musch, M., et al. (2000). Gut-derived sepsis occurs when the right pathogen with the right virulence genes meets the right host evidence for in vivo virulence expression in Pseudomonas. Ann. Surg. 232, 480–489.
Alverdy, J. C., and Krezalek, M. A. (2017). Collapse of the microbiome, emergence of the pathobiome, and the immunopathology of sepsis. Crit. Care Med. 45, 337–347. doi: 10.1097/CCM.0000000000002172
Arrieta, M. C., and Finlay, B. B. (2012). The commensal microbiota drives immune homeostasis. Front. Immunol. 3:33. doi: 10.3389/fimmu.2012.00033
Babrowski, T., Romanowski, K., Fink, D., Kim, M., Gopalakrishnan, V., Zaborina, O., et al. (2013). The intestinal environment of surgical injury transforms Pseudomonas aeruginosa into a discrete hypervirulent morphotype capable of causing lethal peritonitis. Surgery 153, 36–43. doi: 10.1016/j.surg.2012.06.022
Barak, J. D., Gorski, L., Liang, A. S., and Narm, K. E. (2009). Previously uncharacterized Salmonella enterica genes required for swarming play a role in seedling colonization. Microbiology 155(Pt 11), 3701–3709. doi: 10.1099/mic.0.032029-0
Behnsen, J., Jellbauer, S., Wong, C. P., Edwards, R. A., George, M. D., Ouyang, W., et al. (2014). The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity 40, 262–273. doi: 10.1016/j.immuni.2014.01.003
Bingold, T. M., Ziesche, E., Scheller, B., Sadik, C. D., Franck, K., Just, L., et al. (2010). Interleukin-22 detected in patients with abdominal sepsis. Shock 34, 337–340. doi: 10.1097/SHK.0b013e3181dc07b1
Blaser, M. (2016). Antibiotic use and its consequence for the normal microbiome. Science 352, 544–545. doi: 10.1126/science.aad9358
Busani, S., Serafini, G., Mantovani, E., Venturelli, C., Giannella, M., Viale, P., et al. (2017). Mortality in patients with septic shock by multidrug resistant bacteria: risk factors and impact of sepsis treatments. J. Intensive Care Med. doi: 10.1177/0885066616688165 [Epub ahead of print].
Carabotti, M., Scirocco, A., Maselli, M. A., and Severi, C. (2015). The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 28, 203–209.
Carlisle, E. M., and Morowitz, M. J. (2011). Pediatric surgery and the human microbiome. J. Pediatr. Surg. 46, 577–584. doi: 10.1016/j.jpedsurg.2010.12.018
Connolly, J. P., Finlay, B. B., and Roe, A. J. (2015). From ingestion to colonization: the influence of the host environment on regulation of the LEE encoded type III secretion system in enterohaemorrhagic Escherichia coli. Front. Microbiol. 6:568. doi: 10.3389/fmicb.2015.00568
Cosnes, J. (2016). Smoking and diet: impact on disease course? Dig. Dis. 34, 72–77. doi: 10.1159/000442930
Falkow, S. (1988). Molecular Koch’s postulates applied to microbial pathogenicity. Rev. Infect. Dis. 10(Suppl. 2), S274–S276.
Francino, M. P. (2016). Antibiotics and the human gut microbiome: dysbioses and accumulation of resistances increased susceptibility to infections. Front. Microbiol. 6:1543. doi: 10.3389/fmicb.2015.01543
Freestone, P. P., and Lyte, M. (2008). Microbial endocrinology: experimental design issues in the study of interkingdom signalling in infectious disease. Adv. Appl. Microbiol. 64, 75–105. doi: 10.1016/S0065-2164(08)00402-4
Guyton, K., and Alverdy, J. C. (2016). The gut microbiota and gastrointestinal surgery. Nat. Rev. Gastroenterol. Hepatol. 14, 43–54. doi: 10.1038/nrgastro.2016.139
Hayakawa, M., Asahara, T., Henzan, N., Murakami, H., Yamamoto, H., Mukai, N., et al. (2011). Dramatic changes of the gut flora immediately after severe and sudden insults. Dig. Dis. Sci. 56, 2361–2365. doi: 10.1007/s10620-011-1649-3
Jakobsson, H. E., Jernberg, C., Andersson, A. F., Sjolund-Karlsson, M., Jansson, J., and Engstrand, L. (2010). Short-term antibiotic treatment has differing long- term impacts on the human throat and gut microbiome. PLoS ONE 5:e9836. doi: 10.1371/journal.pone.0009836
Jenq, R. R., Ubeda, C., Taur, Y., Menezes, C. C., Khanin, R., Dudakov, J. A., et al. (2012). Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J. Exp. Med. 209, 903–911. doi: 10.1084/jem.20112408
Kazerouni, A., and Wein, L. M. (2017). Exploring the efficacy of pooled stools in fecal microbiota transplantation for microbiota-associated chronic diseases. PLoS ONE 12:e0163956. doi: 10.1371/journal.pone.0163956
Kendall, M. M., and Sperandio, V. (2016). What a dinner party! mechanisms and functions of interkingdom signaling in host-pathogen associations. MBio 7:e01748. doi: 10.1128/mBio.01748-15
Krezalek, M. A., and Alverdy, J. C. (2016). The role of the microbiota in surgical recovery. Curr. Opin. Clin. Nutr. Metab. Care 19, 347–352. doi: 10.1097/MCO.0000000000000299
Krezalek, M. A., DeFazio, J., Zaborina, O., Zaborin, A., and Alverdy, J. C. (2016). The shift of an intestinal “microbiome” to a “pathobiome” governs the course and outcome of sepsis following surgical injury. Shock 45, 475–482. doi: 10.1097/SHK.0000000000000534
Krueger, J. M., and Opp, M. R. (2016). Sleep and microbes. Int. Rev. Neurobiol. 131, 207–225. doi: 10.1016/bs.irn.2016.07.003
Levesque, C. L., Turner, J., Li, J., Wizzard, P., St Pierre, B., and Lim, D. (2016). In a neonatal piglet model of intestinal failure, administration of antibiotics and lack of enteral nutrition have a greater impact on intestinal microflora than surgical resection alone. J. Parenter. Enteral Nutr. 20, 1–9. doi: 10.1177/0148607115626903
Long, J., Zaborina, O., Holbrook, C., Zaborin, A., and Alverdy, J. (2008). Depletion of intestinal phosphate after operative injury activates the virulence of P. aeruginosa causing lethal gut-derived sepsis. Surgery 144, 189–197. doi: 10.1016/j.surg.2008.03.045
Luong, P. M., Shogan, B. D., Zaborin, A., Belogortseva, N., Shrout, J. D., Zaborina, O., et al. (2014). Emergence of the P2 Phenotype in Pseudomonas aeruginosa PAO1 strains involves various mutations in mexT or mexF. J. Bacteriol. 196, 504–513. doi: 10.1128/JB.01050-13
Lupp, C., Robertson, M. L., Wickham, M. E., Sekirov, I., Champion, O. L., Gaynor, E. C., et al. (2007). Article host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2, 119–129. doi: 10.1016/j.chom.2007.06.010
Lyte, M. (2013). Microbial endocrinology in the microbiome-gut-brain axis: how bacterial production and utilization of neurochemicals influence behavior. PLoS Pathog. 9:e1003726. doi: 10.1371/journal.ppat.1003726
Mackenzie, B. W., Waite, D. W., Hoggard, M., Douglas, R. G., Taylor, M. W., and Biswas, K. (2017). Bacterial community collapse: a meta-analysis of the sinonasal microbiota in chronic rhinosinusitis. Environ. Microbiol. 19, 381–392. doi: 10.1111/1462-2920.13632
Mekalanos, J. J. (1992). Environmental signals controlling expression of virulence determinants in bacteria. J. Bacteriol. 174, 1–7.
Modi, S. R., Collins, J. J., and Relman, D. A. (2014). Antibiotics and the gut microbiota. J. Clin. Investig. 124, 4212–4218. doi: 10.1172/JCI72333
Munck, C., Helby, J., Westergaard, C. G., Porsbjerg, C., Backer, V., and Hansen, L. H. (2016). Smoking cessation and the microbiome in induced sputum samples from cigarette smoking asthma patients. PLoS ONE 11:e0158622. doi: 10.1371/journal.pone.0158622
Palmer, A. G., and Blackwell, H. E. (2008). Deciphering a protolanguage for bacteria – host communication. Nat. Chem. Biol. 4, 452–454. doi: 10.1038/nchembio0808-452
Pamer, E. G. (2016). Resurrecting the intestinal microbiota to combat antibiotic-resistant pathogens. Science 352, 535–538. doi: 10.1126/science.aad9382
Patel, N. J., Zaborina, O., Wu, L., Wang, Y., Wolfgeher, J. D., Valuckaite, V., et al. (2007). Recognition of intestinal epithelial HIF-1α activation by Pseudomonas aeruginosa. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G134–G142. doi: 10.1152/ajpgi.00276.2006
Poroyko, V. A., Carreras, A., Khalyfa, A., Khalyfa, A. A., Leone, V., Peris, E., et al. (2016). Chronic sleep disruption alters gut microbiota, induces systemic and adipose tissue inflammation and insulin resistance in mice. Sci. Rep. 6:35405. doi: 10.1038/srep35405
Rasigade, J. P., Raulin, O., Picaud, J. C., Tellini, C., Bes, M., Grando, J., et al. (2012). Methicillin-resistant Staphylococcus capitis with reduced vancomycin susceptibility causes late-onset sepsis in intensive care neonates. PLoS ONE 7:e31548. doi: 10.1371/journal.pone.0031548
Rendon, J. L., Li, X., Akhtar, S., and Choudhry, M. A. (2013). Interleukin-22 modulates gut epithelial and immune barrier functions following acute alcohol exposure and burn injury. Shock 39, 11–18. doi: 10.1097/SHK.0b013e3182749f96
Reuland, E. A., Al Naiemi, N., Kaiser, A. M., Heck, M., Kluytmans, J. A., Savelkoul, P. H., et al. (2016). Prevalence and risk factors for carriage of ESBL-producing Enterobacteriaceae in Amsterdam. J. Antimicrob. Chemother. 71, 1076–1082. doi: 10.1093/jac/dkv441
Rhee, S. H., Pothoulakis, C., and Mayer, E. A. (2009). Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 6, 306–314. doi: 10.1038/nrgastro.2009.35
Roux, A., Payne, S. M., and Gilmore, M. S. (2009). Microbial telesensing: probing the environment for friends, foes, and food. Cell Host Microbe 6, 115–124. doi: 10.1016/j.chom.2009.07.004
Sansonetti, P. J. (2004). War and peace at mucosal surfaces. Nat. Rev. Immunol. 4, 953–964. doi: 10.1038/nri1499
Schaible, U. E., and Kaufmann, S. H. (2004). Iron and microbial infection. Nat. Rev. Microbiol. 2, 946–953. doi: 10.1038/nrmicro1046
Seal, J. B., Morowitz, M., Zaborina, O., An, G., and Alverdy, J. C. (2010). The molecular Koch’s postulates and surgical infection: a view forward. Surgery 147, 757–765.
Shakhsheer, B. A., Versten, L. A., Luo, J. N., Defazio, J. R., Klabbers, R., Christley, S., et al. (2016). Morphine promotes colonization of anastomotic tissues with collagenase - producing Enterococcus faecalis and causes leak. J. Gastrointest. Surg. 20, 1744–1751. doi: 10.1007/s11605-016-3237-5
Shen, Y., Giardino Torchia, M. L., Lawson, G. W., Karp, C. L., Ashwell, J. D., and Mazmanian, S. K. (2012). Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 12, 509–520. doi: 10.1016/j.chom.2012.08.004
Shimizu, K., Ogura, H., Asahara, T., Nomoto, K., Matsushima, A., Hayakawa, K., et al. (2015). Gut microbiota and environment in patients with major burns - a preliminary report. Burns 41, e28–e33. doi: 10.1016/j.burns.2014.10.019
Shimizu, K., Ogura, H., Goto, M., Asahara, T., Nomoto, K., Morotomi, M., et al. (2006). Altered gut flora and environment in patients with severe SIRS. J. Trauma 60, 126–133.
Shogan, B. D., Smith, D. P., Christley, S., Gilbert, J. A., Zaborina, O., and Alverdy, J. C. (2014). Intestinal anastomotic injury alters spatially defined microbiome composition and function. Microbiome 2, 1–10. doi: 10.1186/2049-2618-2-35
Spencer, H., Karavolos, M. H., Bulmer, D. M., Aldridge, P., Chhabra, S. R., Winzer, K., et al. (2010). Genome-wide transposon mutagenesis identifies a role for host neuroendocrine stress hormones in regulating the expression of virulence genes in Salmonella. J. Bacteriol. 192, 714–724. doi: 10.1128/JB.01329-09
Stappenbeck, T. S., and Virgin, H. W. (2016). Accounting for reciprocal host-microbiome interactions in experimental science. Nature 534, 191–199. doi: 10.1038/nature18285
Stough, J. M., Dearth, S. P., Denny, J. E., LeCleir, G. R., Schmidt, N. W., Campagna, S. R., et al. (2016). Functional characteristics of the gut microbiome in C57BL/6 mice differentially susceptible to Plasmodium yoelii. Front. Microbiol. 7:1520. doi: 10.3389/fmicb.2016.01520
Taur, Y., and Pamer, E. G. (2016). Microbiome mediation of infections in the cancer setting. Genome Med. 8, 40. doi: 10.1186/s13073-016-0306-z
Tenaillon, O., Barrick, J. E., Ribeck, N., Deatherage, D. E., Blanchard, J. L., Dasgupta, A., et al. (2016). Tempo and mode of genome evolution in a 50,000-generation experiment. Nature 536, 165–170.
Vanzant, E. L., Lopez, C. M., Ozrazgat-Baslanti, T., Ungaro, R., Davis, R., Cuenca, A. G., et al. (2014). Persistent inflammation, immunosuppression, and catabolism syndrome after severe blunt trauma. J Trauma Acute Care Surg. 76, 21–29. doi: 10.1097/TA.0b013e3182ab1ab5
Wu, L., Estrada, O., Zaborina, O., Bains, M., Shen, L., Kohler, J. E., et al. (2005). Recognition of host immune activation by Pseudomonas aeruginosa. Science 309, 774–777.
Yuen, G. J., and Ausubel, F. M. (2014). Enterococcus infection biology: lessons from invertebrate host models. J. Microbiol. 52, 200–210. doi: 10.1007/s12275-014-4011-6
Zaborin, A., Defazio, J. R., Kade, M., Kaiser, B. L., Belogortseva, N., Camp, D. G., et al. (2014a). Phosphate-containing polyethylene glycol polymers prevent lethal sepsis by multidrug-resistant pathogens. Antimicrob. Agents Chemother. 58, 966–977. doi: 10.1128/AAC.02183-13
Zaborin, A., Smith, D., Garfield, K., Quensen, J., Shakhsheer, B., Kade, M., et al. (2014b). Membership and behavior of ultra-low-diversity pathogen critical illness. MBio 5, e01361-14. doi: 10.1128/mBio.01361-14.Invited
Zaborina, O., Lepine, F., Xiao, G., Valuckaite, V., Chen, Y., Li, T., et al. (2007). Dynorphin activates quorum sensing quinolone signaling in Pseudomonas aeruginosa. PLoS Pathog. 3:e35. doi: 10.1371/journal.ppat.0030035
Zakharkina, T., Martin-loeches, I., Matamoros, S., Povoa, P., Torres, A., Kastelijn, J. B., et al. (2017). The dynamics of the pulmonary microbiome during mechanical ventilation in the intensive care unit and the association with occurrence of pneumonia. Thorax doi: 10.1136/thoraxjnl-2016-209158 [Epub ahead of print].
Keywords: microbiome, injury, surgery, pathogens, sepsis, interkingdom interactions, interkingdom signaling
Citation: Alverdy JC and Luo JN (2017) The Influence of Host Stress on the Mechanism of Infection: Lost Microbiomes, Emergent Pathobiomes, and the Role of Interkingdom Signaling. Front. Microbiol. 8:322. doi: 10.3389/fmicb.2017.00322
Received: 01 November 2016; Accepted: 15 February 2017;
Published: 02 March 2017.
Edited by:
José Eduardo González-Pastor, Centro de Astrobiología (CSIC-INTA), SpainReviewed by:
Åsa Sjöling, Karolinska Institutet, SwedenWilliam Cheadle, University of Louisville, USA
Copyright © 2017 Alverdy and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John C. Alverdy, jalverdy@surgery.bsd.uchicago.edu