Addendum: Comparative Genomic Analysis of the Class Epsilonproteobacteria and Proposed Reclassification to Epsilonbacteraeota (phyl. nov.)
 David W. Waite1
David W. Waite1 Inka Vanwonterghem1
Inka Vanwonterghem1 Christian Rinke1
Christian Rinke1 Donovan H. Parks1
Donovan H. Parks1 Ying Zhang2
Ying Zhang2 Ken Takai3
Ken Takai3 Stefan M. Sievert4
Stefan M. Sievert4 Jörg Simon5
Jörg Simon5 Barbara J. Campbell6
Barbara J. Campbell6 Thomas E. Hanson7
Thomas E. Hanson7 Tanja Woyke8
Tanja Woyke8 Martin G. Klotz9,10
Martin G. Klotz9,10 Philip Hugenholtz1*
Philip Hugenholtz1*- 1Australian Centre for Ecogenomics, School of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, QLD, Australia
- 2Department of Cell and Molecular Biology, College of the Environment and Life Sciences, University of Rhode Island, Kingston, RI, USA
- 3Department of Subsurface Geobiological Analysis and Research, Japan Agency for Marine-Earth Science and Technology, Yokosuka, Japan
- 4Department of Biology, Woods Hole Oceanographic Institution, Woods Hole, MA, USA
- 5Microbial Energy Conversion and Biotechnology, Department of Biology, Technische Universität Darmstadt, Darmstadt, Germany
- 6Department of Biological Sciences, Life Science Facility, Clemson University, Clemson, SC, USA
- 7School of Marine Science and Policy, College of Earth, Ocean, and Environment, Delaware Biotechnology Institute, University of Delaware, Newark, DE, USA
- 8Department of Energy, Joint Genome Institute, Walnut Creek, CA, USA
- 9Department of Biology and School of Earth and Environmental Sciences, Queens College of the City University of New York, New York, NY, USA
- 10State Key Laboratory of Marine Environmental Science, Institute of Marine Microbes and Ecospheres, College of Ocean and Earth Sciences, Xiamen University, Xiamen, China
The Epsilonproteobacteria is the fifth validly described class of the phylum Proteobacteria, known primarily for clinical relevance and for chemolithotrophy in various terrestrial and marine environments, including deep-sea hydrothermal vents. As 16S rRNA gene repositories have expanded and protein marker analysis become more common, the phylogenetic placement of this class has become less certain. A number of recent analyses of the bacterial tree of life using both 16S rRNA and concatenated marker gene analyses have failed to recover the Epsilonproteobacteria as monophyletic with all other classes of Proteobacteria. In order to address this issue, we investigated the phylogenetic placement of this class in the bacterial domain using 16S and 23S rRNA genes, as well as 120 single-copy marker proteins. Single- and concatenated-marker trees were created using a data set of 4,170 bacterial representatives, including 98 Epsilonproteobacteria. Phylogenies were inferred under a variety of tree building methods, with sequential jackknifing of outgroup phyla to ensure robustness of phylogenetic affiliations under differing combinations of bacterial genomes. Based on the assessment of nearly 300 phylogenetic tree topologies, we conclude that the continued inclusion of Epsilonproteobacteria within the Proteobacteria is not warranted, and that this group should be reassigned to a novel phylum for which we propose the name Epsilonbacteraeota (phyl. nov.). We further recommend the reclassification of the order Desulfurellales (Deltaproteobacteria) to a novel class within this phylum and a number of subordinate changes to ensure consistency with the genome-based phylogeny. Phylogenomic analysis of 658 genomes belonging to the newly proposed Epsilonbacteraeota suggests that the ancestor of this phylum was an autotrophic, motile, thermophilic chemolithotroph that likely assimilated nitrogen from ammonium taken up from the environment or generated from environmental nitrate and nitrite by employing a variety of functional redox modules. The emergence of chemoorganoheterotrophic lifestyles in several Epsilonbacteraeota families is the result of multiple independent losses of various ancestral chemolithoautotrophic pathways. Our proposed reclassification of this group resolves an important anomaly in bacterial systematics and ensures that the taxonomy of Proteobacteria remains robust, specifically as genome-based taxonomies become more common.
Introduction
The Epsilonproteobacteria were first described in the early 1990s as the fifth subclass of the Proteobacteria (Tenover et al., 1992) and subsequently assigned class status within this phylum (Garrity et al., 2005). The group is widely known for its pathogenic genera Campylobacter, Helicobacter and, to a lesser extent Arcobacter. However, other members of this class are known to play ecologically important roles across a diverse range of environments in which they thrive as mesophiles or moderate thermophiles (Nakagawa and Takaki, 2009). Epsilonproteobacteria are important chemolithotrophic primary producers in deep-sea hydrothermal vent systems, where they are often the dominant bacterial lineage in vent plumes and deposits (Huber et al., 2010; Flores et al., 2011), and surrounding microbial mats (Moussard et al., 2006; Opatkiewicz et al., 2009; Rassa et al., 2009). On vent chimneys, Epsilonproteobacteria can account for up to 85% of the microbial biomass (Nakagawa et al., 2006). Their metabolic capacity to perform sulfur oxidation coupled to N-oxide reduction while fixing carbon via the reverse TCA cycle (Hügler et al., 2005; Campbell et al., 2009) enables them to be early colonizers of uninhabited vent ecosystems (Alain et al., 2004; Campbell et al., 2006; Gulmann et al., 2015). Non-pathogenic relatives of Campylobacter such as Sulfurospirillum and Thiovulum are often detected in sulfide-rich sediments while others show an affinity for hydrocarbon-rich environments (Hubert et al., 2012). Host-association is also common in this class: Campylobacter, Helicobacter, and some Arcobacter species are known opportunistic pathogens of vertebrates while members of the Caminibacter, Nautilia, and Sulfurospirillum have been reported in association with deep-sea hydrothermal vent fauna. Recent metatranscriptomic data highlighted the role of Sulfurimonas-like bacteria in hydrogenase-driven sulfur oxidation and denitrification on the gills of a deep vent sea snail (Sanders et al., 2013).
While the Epsilonproteobacteria constitute a stable monophyletic group within the bacterial tree of life, a number of studies suggest that they do not reproducibly affiliate with other Proteobacteria, with the exception of the Desulfurellales, which are presently classified as an order of the Deltaproteobacteria (see below). Early conserved marker gene-based studies showing Epsilonproteobacteria branching immediately basal to other Proteobacteria often had inadequate outgroups and/or bootstrap values supporting this placement (Tenover et al., 1992; Trust et al., 1994; Eisen, 1995; Ludwig et al., 1995; Klenk et al., 1999), while others did not resolve this association at all (Gupta, 2000; Reysenbach et al., 2000; Sheridan et al., 2003; Yarza et al., 2014). More recent phylogenomic evidence based on multiple marker proteins and greater outgroup representation have largely failed to recover the Epsilonproteobacteria as reproducibly monophyletic with the rest of the Proteobacteria, further suggesting that taxonomic revision is required at the phylum level (Wu et al., 2009; Di Rienzi et al., 2013; Dodsworth et al., 2013; McLean et al., 2013; Rinke et al., 2013; Zhang and Sievert, 2014; Hug et al., 2016; Yeoh et al., 2016). The class Epsilonproteobacteria currently comprises two orders, Campylobacterales and Nautiliales, encompassing a number of species with ambiguous placement. Particularly problematic are the genera Nitratifractor and Nitratiruptor. The SILVA and LPSN taxonomies (Quast et al., 2013; Parte, 2014; Yilmaz et al., 2014) currently list these organisms as members of the Nautiliaceae, but this classification is not universally accepted (Nakagawa and Takai, 2014) and phylogenetic evidence suggests that they may represent novel families (Nakagawa and Takaki, 2009; Anderson et al., 2011). In a review of the Epsilonproteobacteria, Campbell et al. (2006) used the placeholder family Thiovulgaceae to group the Nitratifractor with Sulfurovum and Sulfurimonas, and the family Nitratiruptoraceae to describe the divergent nature of Nitratiruptor from other families. An identical taxonomy was proposed with recent phylogenomic evidence (Zhang and Sievert, 2014), which also revealed a stable monophyletic clade of Epsilonproteobacteria with the deltaproteobacterial genus Hippea, a member of the order Desulfurellales. Both genera belonging to this order, Desulfurella and Hippea, have low 16S rRNA gene sequence identity to other members of the Deltaproteobacteria and frequently form a clade with members of the Epsilonproteobacteria (Haddad et al., 1995; Moyer et al., 1996; Miroshnichenko et al., 2002; Kersters et al., 2006; Florentino et al., 2016), suggesting that they should be transferred from the Deltaproteobacteria to this group.
The widespread adoption of high-throughput sequencing technologies has resulted in the number of sequenced genomes from bacteria exceeding 70,000 in recent years (Mukherjee et al., 2017)1. Additionally, advances in obtaining high-quality draft genomes from metagenomic data (population genomes; Wrighton et al., 2012; Albertsen et al., 2013) and single cells (Marcy et al., 2007; Rinke et al., 2013) greatly augments genomic coverage of microbial diversity and provides the opportunity to supplant the 16S rRNA gene as the basis for microbial classification. Here, we report a phylogenomic characterization of 624 publicly available Epsilonproteobacteria and Desulfurellales isolate genomes supplemented with 33 Epsilonproteobacteria population genomes. As part of this study, we also sequenced a near-complete genome of Hydrogenimonas thermophila, and analyzed three partial genomes of single cells belonging to the genus Thioreductor. Based on our results, we propose reclassifying the Epsilonproteobacteria and Desulfurellales as a new phylum, the Epsilonbacteraeota (phyl. nov.), together with a number of subordinate changes and additions at the order and family levels.
Materials and Methods
Genome Data
An ingroup comprising 619 Epsilonproteobacteria, four Hippea species and Desulfurella acetivorans were obtained from NCBI RefSeq and GenBank (Supplementary Table S1), and 33 Epsilonproteobacteria population genomes (Supplementary Table S2) were recovered from public metagenomic datasets2. The genome of H. thermophila was sequenced using the Illumina HiSeq 2500 platform (2 × 150 bp chemistry). Raw sequence data (2.4 M reads) were quality filtered using trimmomatic v0.33 (Bolger et al., 2014) in paired end mode, requiring an average quality score of Q ≥ 20 over a sliding window of four bases, and a minimum sequence length of 36 nucleotides. A draft genome was assembled using SPAdes v3.8.1 (Bankevich et al., 2012) with a kmer size range of 35–75 (step size = 4) and automatic coverage cutoff. The genome was then scaffolded using FinishM v0.0.93, and scaffolds assessed for assembly errors using RefineM v0.0.134.
Three partial Thioreductor genomes were obtained by single cell genome sequencing (Supplementary Table S2). Raw sequence data (41 M reads) were quality filtered as per H. thermophila. Quality-filtered sequences were digitally normalized using khmer v2.0 (Crusoe et al., 2015) using the default two-pass approach. Normalized sequences were assembled using SPAdes, and the resulting contigs were scaffolded and refined using RefineM and FinishM as for H. thermophila. The taxonomic identity of each Thioreductor genome was confirmed by screening high-quality reads for 16S rRNA gene sequence fragments using GraftM5. Putative 16S rRNA gene fragments were aligned using the SINA web aligner (Pruesse et al., 2012) and inserted into the SILVA SSU non-redundant database v123.1 using the parsimony insertion tool in ARB.
An outgroup of 4,072 publicly available genomes representing unique species of 24 bacterial phyla were also obtained from NCBI. Completeness and contamination of all genomes was estimated using CheckM v1.0.6 with default settings (Parks et al., 2015).
Phylogenetic Inference
Ingroups for phylogenetic analyses were selected from the 653 Epsilonproteobacteria (including H. thermophila and the 33 population genomes) and five Desulfurellales genomes. The three partial Thioreductor genomes were only included in a reduced concatenated gene analysis due to their low estimated completeness (see below). To resolve the placement of the ingroup in the bacterial domain, 98 ingroup genomes representative at the species-level were selected and combined with the 4,072 outgroup genomes described above. Phylogenetic inference was performed on the 4,170 genomes using a concatenation of 120 conserved protein marker sequences (Ormerod et al., 2016). Protein sequences in each genome were identified and aligned to reference alignments using hmmer v3.1 (Eddy, 1998). Aligned markers were then concatenated and poorly aligned regions removed using Gblocks v0.91b (Castresana, 2000; Talavera and Castresana, 2007).
Maximum likelihood inference of the multiple sequence alignment was performed using the Jones-Taylor-Thornton (JTT), Whelan and Goldman (WAG), and Le and Gascuel (LG) models for amino acid evolution with gamma distributed rate heterogeneity (+Γ) (Jones et al., 1992; Whelan and Goldman, 2001; Le and Gascuel, 2008) implemented in FastTree v2.1.9 (Price et al., 2009). Neighbor joining (NJ) was performed using the Jukes-Cantor and Kimura distance corrections, and with an uncorrected distance matrix implemented in Clearcut v1.0.9 (Sheneman et al., 2006). Under each model/correction, tree building was performed with all sequences included, then once with each phylum or singleton lineage removed, with the exception of Proteobacteria and ingroup genomes (a total of 186 trees). All trees were bootstrap-resampled 100 times to assess the stability of tree topologies. Robustness and reproducibility of the tree topology and association between the Epsilonproteobacteria, Desulfurellales, and Proteobacteria was assessed by manual examination of all tree topologies in ARB (Ludwig et al., 2004).
To resolve the internal structure of Epsilonbacteraeota relationships, a slightly larger data set of 110 ingroup genomes were selected (106 Epsilonproteobacteria, four Desulfurellales) and maximum likelihood inference performed using RAxML v8.1.11 (Stamatakis, 2014). An outgroup of 10 genomes representing six bacterial phyla, including Proteobacteria and Aquificae was used to root this tree. Phylogenetic inference was performed with the WAG+Γ model and 100 bootstrap resamples. To further evaluate the robustness of Epsilonbacteraeota relationships, single gene trees were constructed using FastTree with the WAG+Γ model on the individual protein alignments and tree topologies compared to that of the concatenated alignment using the phytools and ape packages in the R software environment (Paradis et al., 2004; Revell, 2012; R Core Team, 2016).
Manual inspection of the three partial Thioreductor genomes identified 14 protein families common to all three. Phylogenetic analysis of Thioreductor was performed using the above set of 110 ingroup genomes and associated outgroup, using only these 14 protein markers. Phylogenetic inference was performed using RAxML as described above. To assess the placement of species for which genome data is not available, 16S rRNA gene analysis was performed. Epsilonbacteraeota sequences were obtained from the SILVA Living Tree Project v123 (Yilmaz et al., 2014). As this database does not possess a representative for the genus Thiovulum, a 16S rRNA sequence for this lineage was obtained from NCBI GenBank. Full length 16S rRNA gene sequences of Thiofractor thiocaminus, Candidatus Thioturbo danicus, Cetia pacifica, and Thioreductor species were aligned using the SINA web aligner (Pruesse et al., 2012). An outgroup comprising members of the Proteobacteria, Aquificae, and four other phyla was used to root the tree. The sequence alignment was masked using the LTP 50% SSU conservation filter prior to tree construction. Phylogenetic inference of the masked alignment was performed using RAxML with the general time reversible model with gamma distributed rate heterogeneity and 1,000 bootstrap resamples. Short sequences (<1,000 bp) belonging to Candidatus Thioturbo danicus and Thioreductor sp. Shim25-G were inserted into the resulting topology using the ARB parsimony insert tool. All tree figures were edited for publication in Inkscape v0.48.
Sequence Similarity Comparisons
In order to compare our taxonomic proposals to previously proposed sequence similarity-based thresholds for taxonomic ranking, we performed 16S rRNA gene sequence and amino acid identity (AAI) comparisons between members of reclassified or newly proposed families. 16S rRNA gene sequences belonging to Epsilonproteobacteria and Desulfurellales type strains were extracted from the SILVA Living Tree Project v123 database. Sequences were aligned and hypervariable regions removed using the Lane mask (Lane, 1991). Pairwise sequence distances were calculated for 16S rRNA sequences belonging to the same family but different genera using mothur v1.39.1 (Schloss et al., 2009). AAIs of all best bi-directional diamond (Buchfink et al., 2015) hits between pairwise comparisons of the 110 ingroup genomes were made using CompareM v0.0.216. AAI scores were obtained for genome pairs belonging to the same family, but different genera. Sequence similarity results for each family were visualized using R and compared to previously proposed taxonomic rank boundaries (Konstantinidis and Tiedje, 2005; Yarza et al., 2014).
Functional Profiling of Epsilonbacteraeota
Functional gene predictions for all Epsilonbacteraeota genomes were performed using Prodigal v2.6.3 (Hyatt et al., 2010). Amino acid translations of predicted genes were annotated using diamond v0.8.10.72 (Buchfink et al., 2015) against the Uniref 100 database (downloaded October 2015) and the accessions of target sequences mapped to their KEGG Orthology (KO) group. Annotations were transformed into an abundance matrix using a custom perl script and principal component analysis was performed using the R package vegan v2.3 (Oksanen et al., 2016). Genomes were partitioned into host-associated or ‘environmental’ and indicator analysis was performed using the package indicspecies (De Cáceres and Legendre, 2009; De Cáceres et al., 2011). KO groups that were significantly associated with either the host-associated or environmental lifestyle were grouped into their functional pathway, and fitted to the PCA ordination using the envfit function in vegan. Additional annotation of hydrogenase enzymes was performed using BLAST (Altschul et al., 1990) against a manually curated database (Greening et al., 2016). Homologous sequences were defined as greater than 30% AAI over at least 70% of the target protein length. Annotation of the reference proteins ACM93230, ACM93747, and ACM93557 of the pathway proposed to facilitate nitrite reduction to ammonium in Nautilia profundicola (Campbell et al., 2009; Hanson et al., 2013) was performed with the same BLAST parameters as for hydrogenases.
Phylogenetic analyses of genes involved in carbon fixation, nitrogen and sulfur cycling, and flagella structure and formation were performed using mingle v0.0.187. Protein markers for marker genes (Supplementary Table S3) were downloaded from UniProt and used for initial homolog discovery against the Genome Taxonomy Database (GTDB)8. Putative protein homologs were manually inspected for false positive matches and genes below the identity threshold or with inconsistent annotations were removed. Putative citrate lyase alpha/beta subunits sequences were also removed if a homolog of each protein in the pair was not detected in a given genome to ensure paralogs were not being directly compared. A similar strategy was applied to the Sox thiosulfate oxidation proteins (SoxA and SoxB). For each data set, protein sequences were aligned using MAFFT v7.221 using the L-INS-i algorithm (Katoh et al., 2002; Katoh and Standley, 2013). The alignment was then masked using Gblocks and phylogenetic inference performed with RAxML as described above.
Results and Discussion
Genome Data
A total of 619 Epsilonproteobacteria and five Desulfurellales genomes were obtained from RefSeq version 76 and GenBank version 213 (Supplementary Table S1). Genomes were assessed for completeness and contamination by scoring the presence of conserved single-copy marker genes within each genome using CheckM (Parks et al., 2015). The median estimated genome completeness for this dataset is 99.4% and the minimum is 81.9%. Genomes were estimated to be less than 10% contaminated, with all but eight under 5% (Supplementary Table S1). The taxonomic annotation of the type strain Campylobacter geochelonis (GCA_900063025.1) was manually modified as the NCBI record for this genome incorrectly labels it as C. fetus (Piccirillo et al., 2016). Thirty-three draft population genomes (median completeness 93.8%, contamination 1.1%) belonging to the Epsilonproteobacteria were recovered from publicly available metagenomic data sets as part of a larger study (Parks et al., submitted) and included in our analysis. In addition to the public genomes, we sequenced the type strain of H. thermophila, sole representative of the genus Hydrogenimonas (Takai et al., 2004) and three single cells belonging to the genus Thioreductor (Supplementary Table S2). For H. thermophila, an Illumina-based assembly produced a draft genome of 96 contigs with a predicted completeness of 99.6 and 1.8% contamination. Thioreductor single cells amplifications were assembled into partial genomes with completeness estimates between 27.7 and 36.5%, and with low contamination estimates (0.3–1.2%) (Supplementary Table S2). Owing to their low completeness Thioreductor genomes were excluded from the majority of analyses, resulting in an ingroup comprising 658 quality-filtered genomes (119 complete and 539 draft) for comparative analysis. Outgroup genomes broadly representative of the bacterial domain were selected from a total of 60,258 quality controlled reference genomes available from the Genome Taxonomy Database.
Proposed Genome-Based Taxonomy
Phylogenetic affiliation(s) of the ingroup (Epsilonproteobacteria and Desulfurellales, 98 genomes) to species-level representatives of the outgroup (4,072 genomes) were assessed using two different datasets. The first dataset was a concatenation of 120 single-copy marker proteins (Parks et al., submitted) and the second was a concatenation of the 16S and 23S rRNA gene sequences (Williams et al., 2010; Abby et al., 2012; Kozubal et al., 2013; Guy et al., 2014; Ochoa de Alda et al., 2014; Sen et al., 2014). Note that the 3,144 genomes contributing to the second dataset are a subset of the first as most genome sequences derived from metagenomic data lack complete rRNA gene sequences (Hugenholtz et al., 2016), and is used here primarily to validate the concatenated protein tree. Based on these datasets, phylogenetic trees were inferred using Maximum Likelihood (ML) with the JTT, WAG, and LG models of amino acid substitution (Jones et al., 1992; Whelan and Goldman, 2001; Le and Gascuel, 2008) as well as NJ with Jukes-Cantor and Kimura distance corrections (Jukes and Cantor, 1969; Kimura, 1980). Robustness of tree topologies was analyzed with a combination of bootstrapping and taxon resampling, implemented by removal of one phylum at a time from the outgroup dataset. The consensus of these analyses indicate that the Epsilonproteobacteria and Desulfurellales are robustly monophyletic and not reproducibly affiliated with any other phyla (Figure 1 and Table 1), which is consistent with recent reports also using concatenated protein markers (Zhang and Sievert, 2014; Hug et al., 2016). The phylum-level jackknife analysis suggests a specific association of the ingroup with the Aquificae, which is also supported by bootstrap resampling of this dataset (Figure 1). Tree topologies which suggest a common ancestry between Aquificae and Epsilonproteobacteria have been reported for several marker genes (Gruber and Bryant, 1998; Klenk et al., 1999; Iyer et al., 2004); however, this association is often not statistically robust. Phylogenomic evidence suggests that Aquificae genomes have been shaped by extensive lateral gene transfer from lineages including the Epsilonproteobacteria (Eveleigh et al., 2013), a phenomenon that might have contributed to the observed association. Importantly, removal of the Aquificae in the jackknife analysis did not affect the apparent separation of the Epsilonproteobacteria from the other proteobacterial classes.
FIGURE 1

FIGURE 1. Maximum likelihood phylogenetic inference (WAG + Γ model) of Epsilonproteobacteria and Desulfurellales in the context of 4,170 bacterial genomes and based on 120 concatenated protein sequences. Robustness of the placement of the Epsilonbacteraeota was assessed using bootstrap support and per-phylum jackknifing. Symbols above each branch root (circles) represent bootstrap support of >90% (black) or ≥75% (hollow). Symbols below each root reflect jackknife reproducibility of a node, with black square representing nodes recovered with ≥90% support in ≥90% of tree topologies. Hollow squares indicate ≥75% support in ≥90% of topologies. Under every tree topology explored Epsilonproteobacteria and Desulfurellales were recovered as a strongly supported monophyletic group with no association to the remaining Proteobacteria. Key findings of tree topologies under alternate tree building models are summarized in Table 1.
TABLE 1
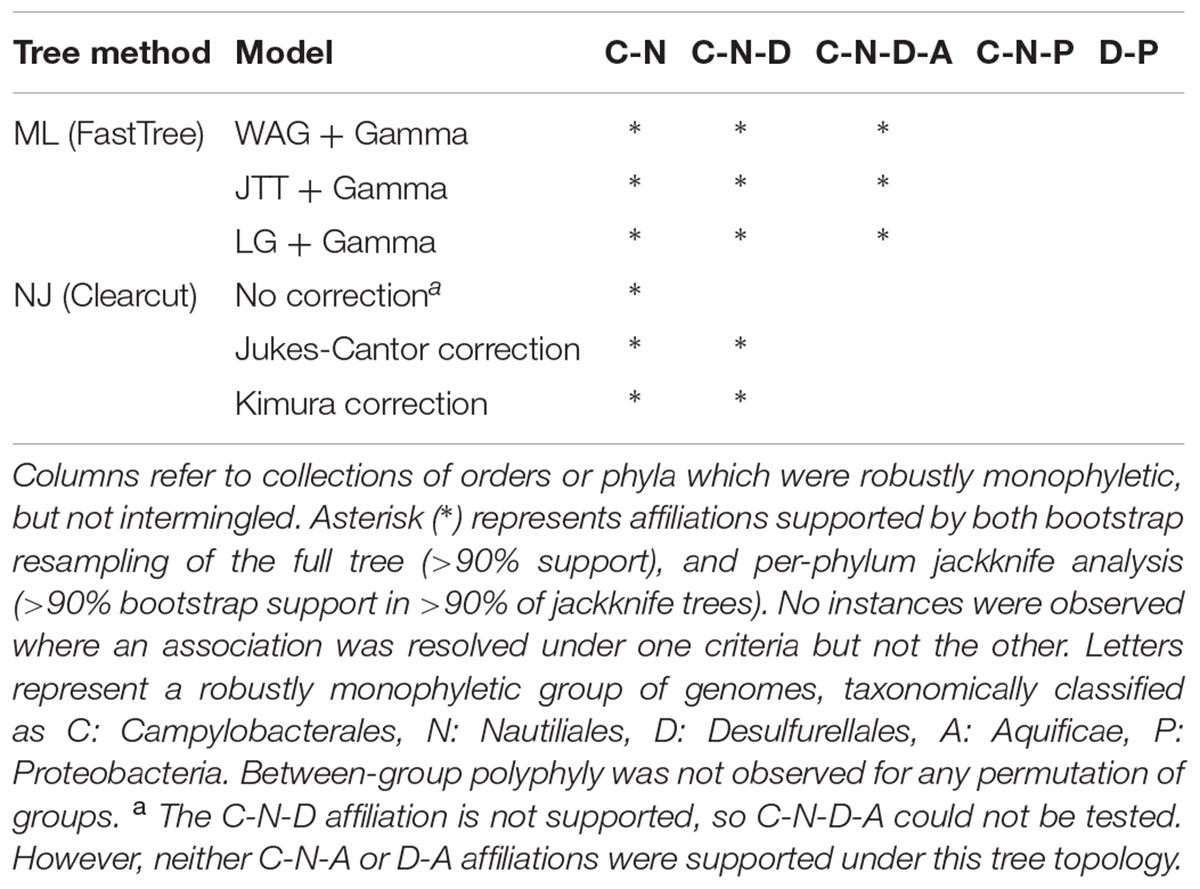
TABLE 1. Support of lineage associations under different tree building methods.
Proposal for a New Phylum
Based on our phylogenetic analyses, we propose to reclassify the Epsilonproteobacteria and Desulfurellales as a single phylum called the Epsilonbacteraeota (phyl. nov.), consistent with a recently proposed naming convention for the phylum rank (Oren et al., 2015). The Epsilonproteobacteria constitute a class-level lineage within this phylum (Figure 2 and Table 2), but we propose to rename it as Campylobacteria (class. nov.) to remove any remaining association with the phylum Proteobacteria. The existing Epsilonproteobacteria orders Campylobacterales and Nautiliales are maintained within this proposed taxonomy as they represent robustly monophyletic groupings and have no direct link to the Proteobacteria in their names (Figure 2 and Supplementary Figure S1 and Table S4). Similarly, the taxon Desulfurellales is retained as the sole order within a new class, the Desulfurellia (class. nov.) (Figure 2 and Supplementary Figure S1). Family and genus level groupings are mostly consistent with existing taxonomic authorities (Table 2), and reclassified or newly proposed families fall within proposed guidelines for 16S rRNA gene similarity (Yarza et al., 2014) and AAI (Konstantinidis and Tiedje, 2005) thresholds (Supplementary Figure S2). Proposed changes to family membership are detailed below.
FIGURE 2
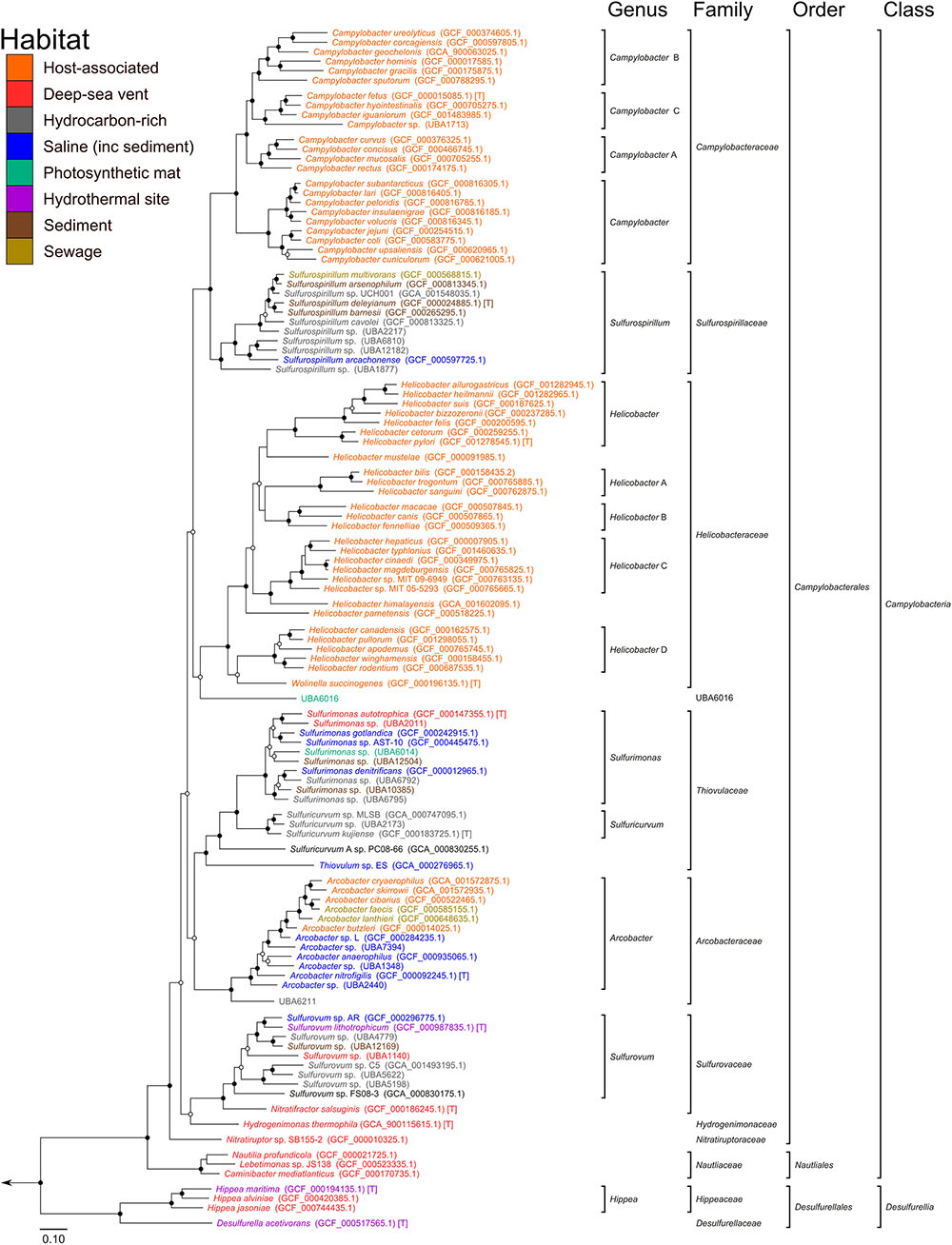
FIGURE 2. Phylogenetic analysis of the Epsilonbacteraeota. Maximum likelihood tree of the Epsilonbacteraeota based on 120 concatenated protein marker sequences using RAxML. Support of internal nodes was calculated using 100 bootstrap iterations, and junctions represent nodes with 100% bootstrap support (solid), and >75% support (hollow). All taxonomic groupings are identical to those in Figure 1 (calculated with FastTree), although the branching positions of Helicobacteraceae and Sulfurovaceae differs between the two topologies. [T]: denotes type species.
TABLE 2
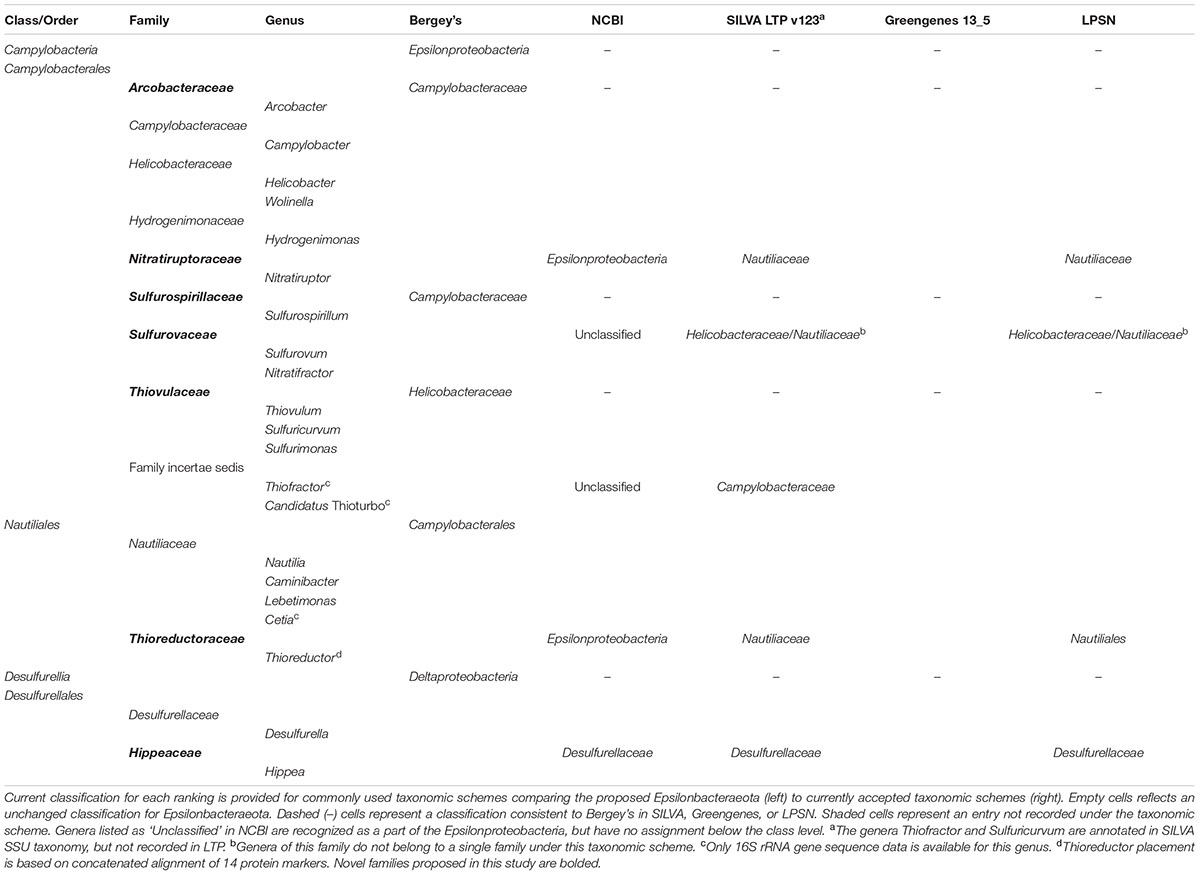
TABLE 2. Taxonomy of the Epsilonbacteraeota compared to existing Epsilonproteobacteria.
Changes to the Campylobacteraceae
The currently defined family Campylobacteraceae, comprised of the genera Arcobacter, Campylobacter, and Sulfurospirillum, is polyphyletic in the concatenated protein tree (Figure 2), although it is monophyletic in the concatenated rRNA gene tree (Supplementary Figure S1). Inspection of individual protein tree topologies revealed that only two of the 120 markers resolve the Campylobacteraceae as monophyletic (Supplementary Figure S3). For this reason, we propose to transfer the Arcobacter and Sulfurospirillum into their own families, the Arcobacteraceae (fam. nov.) and Sulfurospirillaceae (fam. nov.), respectively. Campylobacter is a deeply divergent genus which likely requires reclassification into a number of distinct genera (indicated in Figure 2 by alphabetical suffixing). Note also that Dehalospirillum multivorans and Geospirillum barnesii have been formally transferred to the genus Sulfurospirillum (Finster et al., 1997; Luijten et al., 2003), and Thiomicrospira denitrificans has been reclassified to Sulfurimonas (Takai et al., 2006), although these names persist in some online archives.
Changes to the Helicobacteraceae
The currently defined Helicobacteraceae do not form a monophyletic group to the exclusion of the Campylobacteraceae and Arcobacter; therefore, the genera Thiovulum, Sulfuricurvum, and Sulfurimonas have been removed from the Helicobacteraceae into their own family, the Thiovulaceae (fam. nov.). Monophyly of the Helicobacteraceae is also not robustly supported in the concatenated rRNA gene tree (Supplementary Figure S1). Species in the Sulfurovum and Nitratifractor genera are either currently unclassified at the family level, or classified into the families Helicobacteraceae and Nautiliaceae, respectively (Table 2). Like the Campylobacter, Helicobacter is a deeply divergent genus which likely requires reclassification into a number of distinct genera (indicated in Figure 2 by alphabetical suffixing). Sulfurovum and Nitratifractor are resolved as a robustly monophyletic group, independent of both the Helicobacteraceae and Nautiliaceae, for which we propose the name Sulfurovaceae (fam. nov., Figure 2). However, the concatenated rRNA gene tree does not support the monophyly of the Sulfurovaceae (Supplementary Figure S1). Inspection of individual protein trees reveals that the majority of markers (105/120) supports the association (Supplementary Figure S3), as well as the 16S rRNA gene by itself (Supplementary Figure S4). The revised Helicobacteraceae family comprises only species in the genera Helicobacter and Wolinella (Figure 2). Note that the genus Flexispira (Bryner et al., 1986) is not included in the Helicobacteraceae as it is a defunct basonym of Helicobacter (Dewhirst et al., 2000; Vandamme and On, 2001).
Proposal for New Families
We further propose that the genus Nitratiruptor be placed into its own family Nitratiruptoraceae (fam. nov.) within the order Campylobacterales, a move that, while inconsistent with taxonomic authorities placing this genus in the Nautiliaceae (Table 2), is supported by 16S rRNA gene-based phylogenies (Nakagawa and Takaki, 2009; Anderson et al., 2011; Nakagawa and Takai, 2014). Analysis of the three partial single cell Thioreductor genomes confirms the 16S rRNA-based analysis that this genus is a member of the family Nautiliaceae (Supplementary Figures S4, S5). In addition to the proposed revisions in the class Campylobacteria, we propose that Hippea be transferred to its own family Hippaceae (fam. nov.) within the class Desulfurellia to reflect the depth of its relationship with the genus Desulfurella (Figure 2). A complete summary of proposed changes within the Epsilonbacteraeota are provided in Supplementary Table S3.
Genera Not Yet Represented by Complete Genome Sequences
There are three published genera presently classified as Epsilonproteobacteria for which genomic data are currently unavailable: Thiofractor, Candidatus Thioturbo, and Cetia. These genera are therefore provisionally placed within the Epsilonbacteraeota based on comparative analysis of 16S rRNA gene sequences alone (Supplementary Figure S4); however, their classification may require future revision as genomic information becomes available. Thiofractor and Candidatus Thioturbo appear to be members of the order Campylobacterales consistent with previous findings (Muyzer et al., 2005; Makita et al., 2012), however, neither are clearly resolved into the family level groupings proposed for the Epsilonbacteraeota. Thiofractor may constitute its own family and Candidatus Thioturbo may be incorporated into the Arcobacteraceae, although this latter affiliation is only based on a partial 16S rRNA sequence (Supplementary Figure S4). Cetia pacifica clusters robustly within the family Nautiliaceae, which may or may not require reclassification of the genus Caminibacter (Supplementary Figure S4; Grosche et al., 2015).
Functional Profiling of Epsilonbacteraeota
Overlaying published phenotypic information of cultured Epsilonbacteraeota on the genome-based phylogeny suggests that the ancestor of this phylum was autotrophic and thermophilic (Figure 3). Mesophily arose later in the Campylobacterales, and heterotrophic growth appears to have arisen independently in the Campylobacterales and Desulfurellia (Figure 3). To quantify the extent to which taxonomy reflects functional variation amongst Epsilonbacteraeota, we performed PERMANOVA to quantify the contribution of predicted functional profiles using KO. The largest source of variation was taxonomy (family; R = 0.68, genus; R = 0.70, p = 0.001), followed by habitat (R = 0.28, p = 0.001), indicating that while vertical inheritance is a powerful predictor of functional capacity, a large portion of variation is not captured by this process and likely reflects habitat-specific adaptation. This variation was also reflected in PCA analysis of the functional profiles, where heterotrophic Campylobacter and Helicobacter genomes were clearly separate from other Epsilonbacteraeota (Figure 4). Furthermore, there was pronounced separation within these genera by functional PCA consistent with the suggestion to classify both Campylobacter and Helicobacter into multiple genera (Figure 2). To elucidate the metabolic components driving this separation, indicator analysis (De Cáceres and Legendre, 2009; De Cáceres et al., 2011) was applied to the gene annotation table to highlight key features responsible for functional divergence.
FIGURE 3
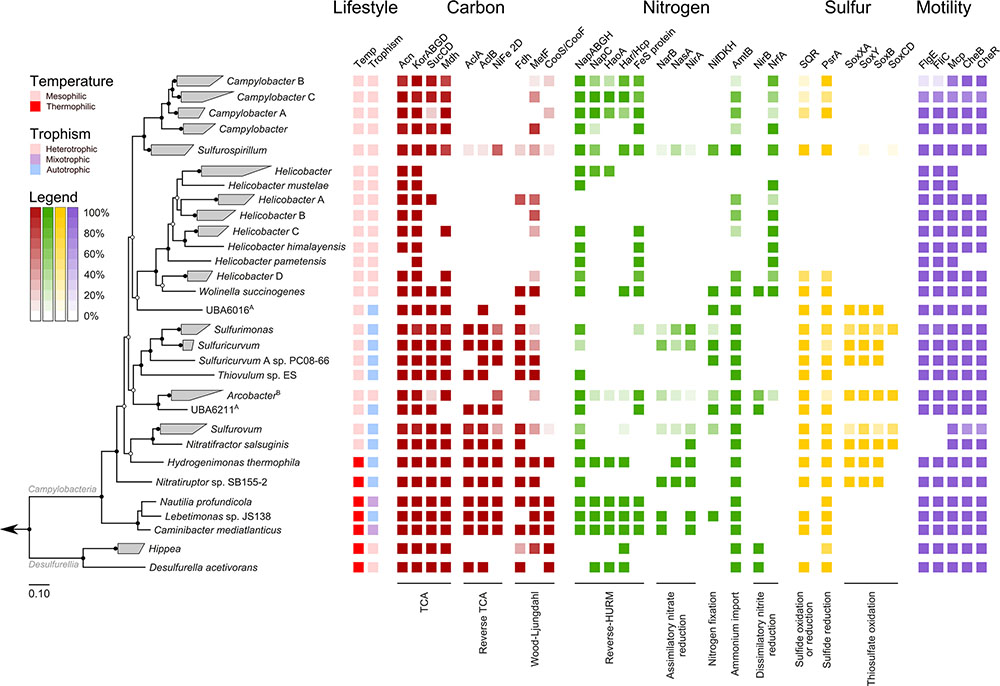
FIGURE 3. Habitat and functional annotation of selected Epsilonbacteraeota pathways. Tree topology is that of Figure 2, collapsed at the genus level. Cell intensity reflects the proportion of genomes within a clade that possessed the column function. A: Trophism is inferred from genomic data. B: Although some members of Arcobacter are capable of autotrophic growth (discussed in text) all genomes in this study were obtained from heterotrophic species. Column descriptions are as follows: Carbon pathways: aconite hydratase (Acn), 2-oxoglutarate oxidoreductase alpha-, beta-, gamma-, and delta- subunits (KorABGD), succinyl-CoA synthetase alpha and beta subunits (SucCD), malate dehydrogenase (Mdh), ATP citrate lyase alpha subunit (AclA) and beta subunit (AclB), [NiFe] hydrogenase family 2D (NiFe 2D), formate dehydrogenase (Fdh), methylenetetrahydrofolate reductase (MetF) and carbon-monoxide dehydrogenase catalytic and iron sulfur subunits (CooS/CooF). Nitrogen pathways: periplasmic nitrate reductase components NapA, NapB, NapG, and NapH (NapABGH), periplasmic nitrate reductase c-type cytochrome (NapC), hydroxylamine oxidoreductase (HaoA), hydroxylamine reductase/hybrid cluster protein (Har/Hcp), predicted reductive Fe-S protein (FeS protein), ferredoxin-nitrate reductase (NarB), assimilatory nitrate reductase (catalytic subunit, NasA), ferredoxin-nitrite reductase (NirA), nitrogenase molybdenum-iron (alpha- and beta-chains) and iron protein (NifDKH), ammonium transporter (AmtB), nitrite reductase (NADH) large subunit (NirB), cytochrome c nitrite reductase (NrfA). Sulfur pathways: sulfide:quinone oxidoreductase (SQR), polysulfide reductase chain A (PsrA), thiosulfate oxidating Sox proteins (SoxXA, SoxY, SoxB, SoxCD). Motility: flagellar hook protein (FlgE), flagellin (FliC), methyl-accepting chemotaxis protein (Mcp), chemotaxis response regulator (CheB), chemotaxis methyltransferase (CheR).
FIGURE 4
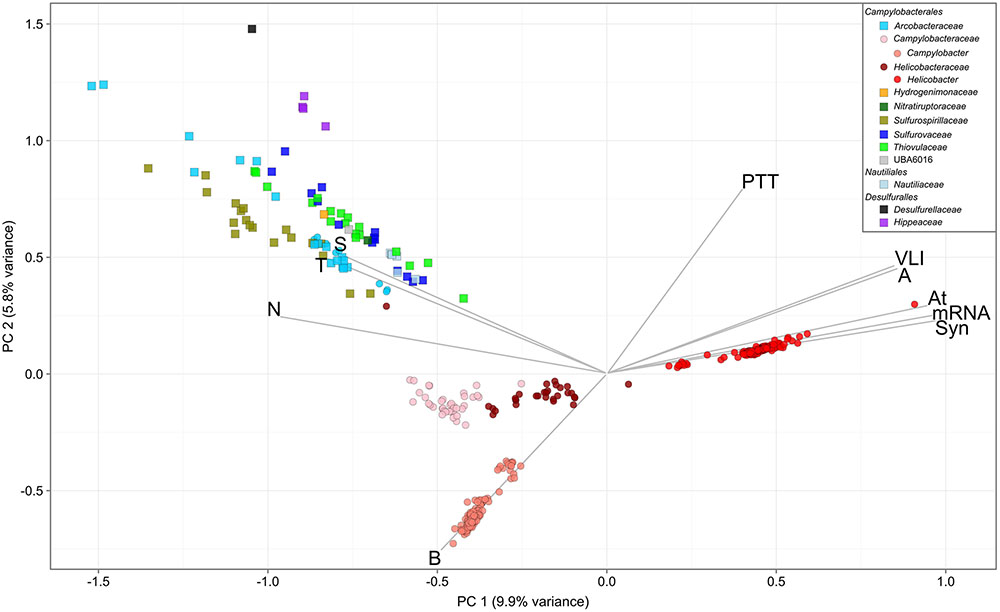
FIGURE 4. Principal component analysis of Epsilonbacteraeota gene annotations. Analysis was performed using the KEGG Orthology (KO) annotation table. Circles denote host-associated individuals, and squares those from other environments. Genomes are colored by family, with exceptions of the Campylobacteraceae and Helicobacteraceae, which are separated based on suffixing described in text. Vectors represent pathway level aggregations of indicator KOs fitted to the ordination using the enfit function in vegan. Only vectors with R > 0.75 are displayed for clarity. Pathways are as follows: A, arginine biosynthesis; At, atrazine degradation; B, biotin metabolism; N, nitrogen metabolism; PTT, phenylalanine, tyrosine, and tryptophan biosynthesis; S, sulfur metabolism; Syn, synthesis and degradation of ketone bodies; T, two-component system; VLI, valine, leucine, and isoleucine degradation.
Genes involved in carbon and nitrogen fixation, assimilatory nitrate and nitrite reduction, thiosulfate oxidation and polysulfide reduction were significantly associated with the environmental ecotype indicating that carbon, nitrogen, and sulfur cycling are primary drivers of functional divergence in environmental Epsilonbacteraeota. These results were visually apparent when aligning functional genes against the concatenated protein phylogeny (Figure 3). Host-associated Epsilonbacteraeota were defined primarily by the absence of these functions, although consistent with their lifestyle the antimicrobial peptide resistance mechanism YejABEF (Wang et al., 2016) was a key signature of host-association. The separation of Helicobacter (containing the type species, H. pylori) from Helicobacter A to D was driven by the presence of genes involved in osmoprotectant and heme transport (Kappes et al., 1999; Létoffé et al., 2006), as well as the CAG pathogenicity island in H. pylori. Similarly, Campylobacter differentiation was in part due to the presence of additional transporters involved in lipopolysaccharide and capsular polysaccharide transport. These findings suggest that the transition toward a host-associated lifestyle in these lineages has occurred in a two-step manner. First, the loss of ‘environmental’ functions associated with the initial transition to host-association in Campylobacter and Helicobacter, followed by gene acquisition to enhance host adaptation. The distinct phylogenetic and functional differences within these genera suggest that future revision of their taxonomy may be warranted.
Carbon Metabolism
Analyzing the carbon metabolism of Epsilonbacteraeota revealed a complete tricarboxylic acid (TCA) cycle in all genomes with the exception of Helicobacter (Figure 3), which functions as a directed pathway producing succinate and α-ketoglutarate in this genus (Pitson et al., 1999). Phylogenetic analysis of the marker protein, 2-oxoglutarate oxidoreductase (KorA), support the TCA cycle being vertically inherited throughout the Epsilonbacteraeota (Supplementary Figure S6). Most genera also encode ATP citrate lyase (AclA and B, Figure 3) indicating the ability to perform autotrophic carbon fixation via reductive TCA (rTCA; Buchanan and Arnon, 1990). The presence of this enzyme coincides with phenotypically established autotrophy (Hügler et al., 2005; Campbell et al., 2006), and genera which have lost the ability to fix carbon have adopted an exclusively heterotrophic lifestyle in carbon rich habitats (Figure 4) with the exception of a number of autotrophic Arcobacter strains (Gevertz et al., 2000; Wirsen et al., 2002) for which genomes are not currently available. Phylogenetic inference of both citrate lyase subunits indicates a monophyletic history of carbon fixation in the Epsilonbacteraeota with independent losses of this capability inferred in the Campylobacter, Helicobacteraceae, and Arcobacter (Figure 3). Citrate lyase was detected in a population genome basal to the Arcobacter genus (UBA6211, Figure 3) which, combined with previous observation of citrate lyase activity and the operation of the rTCA in Arcobacter (Hügler et al., 2005), suggests that carbon fixation was present in the common ancestor of this genus. Vertical inheritance was also observed in Epsilonbacteraeota homologs of the cytosolic [NiFe] hydrogenase group 2D, which is hypothesized to provide reducing power for the rTCA (Brugna-Guiral et al., 2003; Greening et al., 2016).
No other complete carbon fixation pathways were identified in the genomes. Formate dehydrogenase is widely distributed in many autotrophs (Schuchmann and Müller, 2014) and was common in hydrothermal vent-associated lineages (Figures 2, 3), many of which are known to oxidize formate (Macy et al., 1996; Campbell et al., 2006; Grote et al., 2012; Grosche et al., 2015; Florentino et al., 2016). A recent biochemical analysis of hydrothermal vents demonstrated that abiotic processes sequester available carbon dioxide as formate (McDermott et al., 2015), and the presence of formate dehydrogenase may provide the ability to couple respiration with the creation of carbon dioxide to fuel rTCA. The Wood-Ljüngdahl (WL) pathway is more energetically favorable than the rTCA but requires strict anoxic conditions to operate (Berg, 2011), which may be incompatible with the microaerobic growth of many vent-associated Epsilonbacteraeota (Campbell et al., 2006). Monophyletic distribution of the WL-specific marker methylene-tetrahydrofolate reductase (MetF, Figure 3 and Supplementary Figure S7) amongst the Campylobacteria suggests that this pathway was an ancestral trait which has been supplanted by the rTCA, possibly in response to the adoption of (micro)aerobic habitats. Consistent with previous analyses (Hügler et al., 2005, 2011; Takai et al., 2005; Wright et al., 2013), RuBisCO (ribulose-1,5-bisphosphate carboxylase/oxygenase) was not detected in any genome, and the rTCA remains the only known means for carbon fixation amongst Epsilonbacteraeota. The pattern of auto- and heterotrophic lifestyles within the Epsilonbacteraeota suggests that autotrophy is an ancestral trait, punctuated by recent adaptations to heterotrophy mostly in host-associated habitats. Additionally, the Epsilonbacteraeota and Aquificae citrate lyase proteins are robustly monophyletic (Supplementary Figure S8), consistent with the inference that carbon fixation in the Epsilonbacteraeota is descended from an ancestral Aquificae phenotype (Braakman and Smith, 2012).
Nitrogen Metabolism
Anaerobic respiration using nitrate as an electron acceptor is widespread throughout the Epsilonbacteraeota, as evidenced by the presence of soluble periplasmic nitrate reductase (NapA) in most genera (Figure 3), and membrane-bound nitrate reductase (NarG) in Desulfurella (Vetriani et al., 2014). The NAP system of many Campylobacteria lack the NapC protein (Simon et al., 2003; Sievert et al., 2008; Kern and Simon, 2009) and where a homolog is present, it is proposed to act as the quinol-oxidizing protein component of a reversely operating hydroxylamine:quinone redox module. This pathway operates as a quinone-reducing module in ammonia-oxidizing bacteria and Thaumarchaeota, and has recently been proposed in N. profundicola (Klotz and Stein, 2008; Campbell et al., 2009; Hanson et al., 2013; Simon and Klotz, 2013; Kozlowski et al., 2016). As a component of an ammonification pathway, this module produces ammonium by reduction of nitrite either directly (analogous to the NrfHA ammonification module) or via a hydroxylamine intermediate, which is thought to be transported to the cytoplasm and further reduced to ammonium by a putative hydroxylamine reductase (Har/Hcp), powered by an Fe-S ferredoxin (Hanson et al., 2013). While hydroxylamine reduction to ammonium and nitrate-dependent expression of genes encoding the entire pathway has, so far, only been demonstrated in N. profundicola, genes for this pathway are present in Campylobacter concisus, C. curvus, and C. fetus (Hanson et al., 2013). We further identified the complete inventory for this proposed pathway in the remaining Nautiliales genomes, as well as Campylobacter mucosalis, C. geochelonis, C. gracilis, C. hyointestinalis, and C. iguaniorum (Figure 3). Furthermore, H. thermophila and D. acetivorans lack only the predicted Fe-S ferredoxin of this pathway, but as hydroxylamine reductase is known to obtain reductant from other oxidoreductases (Pino et al., 2006), it is possible that these organisms are also capable of ammonification using a hydroxylamine intermediate. The NapC homolog and HaoA proteins are homologous to NrfH and NrfA, respectively (Bergmann et al., 2005; Kern et al., 2011a; Simon and Klotz, 2013) and differ mainly by the infidelity of the HaoA oligomer as an ammonium-producing nitrite reductase. These homologs facilitate incomplete electron transfer and leak hydroxylamine due to looser redox coupling with the NapC homolog compared to that of the NrfA-NrfH module (Simon and Klotz, 2013).
Phylogenetic inference of the HaoA and Har/Hcp proteins suggest that this more complex nitrite ammonification pathway is monophyletic and ancestral to the Epsilonbacteraeota, despite widespread partial or complete loss of the pathway in the group, and putative lateral transfers of component genes from Epsilonbacteraeota donors to other bacteria (Supplementary Figure S9). Most Epsilonbacteraeota have multiple potential mechanisms for obtaining fixed nitrogen and the acquisition of chemoorganotrophic catabolic machinery, which provide cells with higher and more reliable reducing power, likely facilitated the functional replacement of the HaoA-type with the NrfA-type nitrite reductase. The majority of extant autotrophic Epsilonbacteraeota possess one or more ammonification pathways including dinitrogen reduction based on nitrogenase and/or a variety of pathways based on the reduction of nitrate (Figure 3). Annotation of the Thiovulum sp. ES genome did not reveal any ammonification inventory; nevertheless and consistent with its original annotation (Marshall et al., 2012), we detected the gene encoding an ammonium transporter (AmtB) in this lineage, shared with most other Epsilonbacteraeota (Figure 3).
Pentaheme cytochrome c nitrite reductase (NrfA) is found almost exclusively in heterotrophic Campylobacteria (Figure 3) and is known to provide protection against nitrosative stress to Campylobacter and Wolinella in their natural environments (Pittman et al., 2007; Kern et al., 2011b). This protein may also serve the same function in the majority of Helicobacter species, although this has not been well studied since the model species, H. pylori, lacks NrfA and has alternative mechanisms for coping with nitrosative stress (Flint et al., 2016). In contrast to the concatenated protein-based monophyly of the heterotrophs (Figure 3), phylogenetic inference of NrfA reveals at least two independent clades, the first comprising Sulfurospirillum, Wolinella and some Arcobacter, and the second Campylobacter and Helicobacter (Supplementary Figure S10). It is likely at least one of these clusters is the result of lateral gene transfer, which we predict to be the Campylobacter and Helicobacter clade as members of these two genera are intermingled indicating multiple lateral transfer events (Supplementary Figure S10), possibly the result of selective pressure exerted by the host immune system (Kern et al., 2011a; Kassem et al., 2012).
Sulfur Metabolism
Anaerobic respiration of Epsilonbacteraeota frequently occurs through the reduction of sulfur compounds, and is a major source of primary production in some environments (Wright et al., 2013). Sulfide-oxidizing sulfide:quinone oxidoreductase (SQR) catalyzes the bidirectional oxidation of hydrogen sulfide to polysulfide, and this gene is inferred to be abundant in environments where Epsilonbacteraeota (Epsilonproteobacteria) are dominant members (Wright et al., 2013; Rossmassler et al., 2016). Consistent with this inference, we observed SQR genes in most Epsilonbacteraeota lineages (Figure 3). In contrast to the monophyletic origins of the carbon and most nitrogen cycling proteins described above, phylogenetic analysis of SQR reveals extensive polyphyly of Epsilonbacteraeota homologs, including a putative lateral transfer from a Bacteroidetes donor to the Nautilaceae ancestor (Supplementary Figure S11). An additional enzyme involved in polysulfide reduction, polysulfide reductase (PsrA) was also common in the Epsilonbacteraeota (Figure 3) and possessed polyphyletic origins (Supplementary Figure S12). These findings suggest that unlike nitrate reduction, sulfur reduction has been acquired in the Epsilonbacteraeota in response to immediate environmental needs. Polysulfide can be formed abiotically under hydrothermal vent conditions (Rushdi and Simoneit, 2005; Boyd and Druschel, 2013) and, similar to formate, presents a resource that early colonizing Epsilonbacteraeota may exploit. It is possible that the acquisition of SQR represents a means of simultaneously detoxifying hydrogen sulfide, and utilizing it as an electron donor, as Epsilonproteobacteria have been seen to bloom in response to hydrogen sulfide releases on the African shelf (Lavik et al., 2009).
Oxidation of sulfur is also a well known trait of Epsilonproteobacteria, although it is only reported in a handful of genera (Campbell et al., 2006). Sulfurimonas-like organisms have been suggested as the primary sulfur-oxidisers in deep-sea vent ecosystems (Akerman et al., 2013) and consistent with this observation, Sulfurimonas was one of the few Epsilonbacteraeota genera to possess the complete sulfur oxidation (sox) pathway (Figure 3). The absence of SoxC, and drastic reduction in electron liberation (Friedrich et al., 2005), in most genera suggests that sulfur oxidation is not the primary means of electron generation amongst Epsilonbacteraeota, with hydrogenases likely a more productive source of reducing power. The evolutionary history of sox in the Epsilonbacteraeota is convoluted. Consistent with previous analysis of sox genes (Meyer et al., 2007; Ghosh et al., 2009), SoxA and SoxB homologs in non-Arcobacter species shared a common ancestor with Aquificae, while Arcobacter acquired a complete sox cassette through lateral gene transfer with Gammaproteobacteria (Supplementary Figure S13). However, homologs of SoxC are monophyletic in the Epsilonbacteraeota, a finding also supported by previously literature (Ghosh et al., 2009). These data suggest that after acquiring the ability to oxidize thiosulfate independently of other Epsilonbacteraeota, an ancestor of Arcobacter introduced soxCD to Sulfurimonas and the Sulfurovaceae.
Motility
The majority of Epsilonbacteraeota possess uni- or bipolar flagella and are highly motile, with Thiovulum majus capable of achieving a speed of over 600 microns per second, or 2 m per hour (Garcia-Pichel, 1989). The flagella of Epsilonbacteraeota differ from that of other bacteria in several respects. For example, the C-ring of model Campylobacter jejuni and Helicobacter hepaticus differs structurally from that of other bacteria (Chen et al., 2011), and recent knock-out experiments identified six novel genes exclusive to flagella activity in Epsilonbacteraeota (Gao et al., 2014). Given the novelty of described Epsilonbacteraeota flagella, we predicted that they should be an ancestral feature of this phylum. Indeed, phylogenetic analysis of the flagellar hook protein (FlgE) and flagellin (FliC, Figure 3) indicate a monophyletic origin of this trait in the Epsilonbacteraeota (Supplementary Figure S14). Flagella are known to be absent in several Epsilonbacteraeota, including Nitratifractor salsuginis, the genus Sulfurovum, and at least two Campylobacter species, C. hominis and C. gracilis (Vandamme et al., 1995; Lawson et al., 2001). The proposed Epsilonbacteraeota taxonomy groups the genera Nitratifractor and Sulfurovum into the novel family Sulfurovaceae, suggesting a single loss of flagella genes in the ancestor of this family. The presence of the chemotactic regulator Mcp and response regulating CheB and CheR within this family (Figure 3) provides additional evidence that the ancestor of the Sulfurovaceae possessed flagella. Campylobacter hominis and C. gracilis are sister species in the Campylobacter B clade (Figure 2) suggesting a single loss of motility occurred in their common ancestor for as yet, unclear reasons given that the rest of the genus has retained motility.
Concluding Remarks
The rise of high-throughput DNA sequencing has fundamentally changed the landscape of microbial ecology and systematics. The 16S rRNA gene has led the way in providing an evolutionary backbone for microbial taxonomy over the past 30 years, but can now be complemented by whole genome methods which use multiple genetic markers to infer evolutionary relationships. More importantly, classifications are not static. As genomically described bacterial diversity expands, historic classifications require revision as new clades are discovered and tree topologies change. The class Epsilonproteobacteria and order Desulfurellales are an excellent case in point. Comparative analysis of these groups with a broad genomic representation of the bacterial domain indicates that they constitute a monophyletic unit not specifically related to the phylum Proteobacteria. We propose the reclassification of this group as the phylum Epsilonbacteraeota, together with a small number of subordinate changes described in this manuscript. Additional changes may be required in future as, e.g., Campylobacter and Helicobacter appear to be phylogenetically and functionally diverse for genera. Metabolic reconstruction of the Epsilonbacteraeota suggests, perhaps unsurprisingly, that the ancestor to this phylum was a chemolithoautotrophic thermophile related to Aquificae, from which several heterotrophic and mesophilic lineages have evolved.
Proposed Taxonomy of Epsilonbacteraeota
Description of Campylobacteria class. nov.
Campylobacteria (Cam.py.lo.bac.te′ri.a. N.L. masc. n. Campylobacter type genus of the type order of the class; suff. -ia, ending to denote a class; N.L. neut. pl. n. Campylobacteria the class of the order Campylobacterales).
Description is the same as for the order Campylobacterales (Garrity et al., 2006b). Type order: Campylobacterales, phylum: Epsilonbacteraeota phyl. nov.
Description of Arcobacteraceae fam. nov.
Arcobacteraceae (Ar.co.bac.ter.a.ce′ae. N.L. masc. n. Arcobacter type genus of the family; suff. -aceae, ending to denote a family; N.L. fem. pl. n. Arcobacteraceae the family of the genus Arcobacter).
Description is identical to that given by Vandamme et al. (1991), with the acknowledgment that some species are capable of autotrophic carbon dioxide fixation via the reverse tricarboxylic acid cycle (Hügler et al., 2005). Type genus: Arcobacter, order: Campylobacterales, class: Campylobacteria, class. nov., phylum: Epsilonbacteraeota phyl. nov.
Emended Description of the Family Campylobacteraceae Vandamme and De Ley (1991)
Description is drawn from that of Vandamme and De Ley (1991), with the removal of the genera Arcobacter and Sulfurospirillum due to a lack of robust monophyly with the type genus. Type genus: Campylobacter, order: Campylobacterales, class: Campylobacteria class. nov., phylum: Epsilonbacteraeota phyl. nov.
Emended Description of the Family Helicobacteraceae Garrity et al. (2006a)
The description is the same as given by Garrity et al. (2006a) for the type genus with the following changes. Currently includes only the genera Helicobacter (type genus) and Wolinella. The genera Sulfurimonas, Sulfuricurvum, Sulfurovum, and Thiovulum are removed owing to a lack of monophyly with the type genus. Type genus: Helicobacter, order: Campylobacterales, class: Campylobacteria class. nov., phylum: Epsilonbacteraeota phyl. nov.
Description of Nitratiruptoraceae fam. nov.
Nitratiruptoraceae (Ni.tra.ti.rup.to.ra.ce′ae. N.L. masc. n. Nitratiruptor type genus of the family; suff. -aceae, ending to denote a family; N.L. fem. pl. n. Nitratiruptoraceae the family of the genus Nitratiruptor).
Described on the basis of divergent nature with all other members of Campylobacterales using both protein marker, as well as 16S and 23S rRNA gene sequence analysis. Description is the same as given by Nakagawa et al. (2005). Type genus: Nitratiruptor, order: Campylobacterales, class: Campylobacteria class. nov., phylum: Epsilonbacteraeota phyl. nov.
Description of Sulfurospirillaceae fam. nov.
Sulfurospirillaceae (Sul.fu.ro.spir.il.la.ce′ae. N.L. neut. n. Sulfurospirillum type genus of the family; suff. -aceae, ending to denote a family; N.L. fem. pl. n. Sulfurospirillaceae the family of the genus Sulfurospirillum).
The description of the family is drawn from the description of the type genus from Schumacher et al. (1992); cells are Gram-negative, motile, microaerobic, and mesophilic. Oxidize hydrogen or formate to reduce a range of electron acceptors. Type genus: Sulfurospirillum, order: Campylobacterales, class: Campylobacteria class. nov., phylum: Epsilonbacteraeota phyl. nov.
Description of Sulfurovaceae fam. nov.
Sulfurovaceae (Sul.fur.o.va.ce′ae. N.L. neut. n. Sulfurovum type genus of the family; suff. -aceae, ending to denote a family; N.L. fem. pl. n. Sulfurovaceae the family of the genus Sulfurovum).
The family is defined based on phylogenetic analysis of protein markers (concatenated and single) and the 16S ribosomal gene sequence. Includes the genera Sulfurovum (type genus) and Nitratifractor. Cells are Gram-negative, non-motile, and mesophilic. Growth occurs in the microaerobic range. Type genus: Sulfurovum, order: Campylobacterales, class: Campylobacteria class. nov., phylum: Epsilonbacteraeota phyl. nov.
Description of Thiovulaceae fam. nov.
Thiovulaceae (Thio.vu.la.ce’ae. N.L. neut. dim. n. Thiovulum type genus of the family; suff. -aceae, ending to denote a family; N.L. fem. pl. n. Thiovulaceae the family of the genus Thiovulum).
The family is defined based on phylogenetic analysis of concatenated protein marker and ribosomal gene sequences. The family currently includes the genera Thiovulum (type genus), Sulfurimonas, and Sulfuricurvum. Cells are Gram-negative, motile, microaerobic, and mesophilic. Growth occurs in the mesophilic range. Type genus: Thiovulum, order: Campylobacterales, class: Campylobacteriaclass. nov., phylum: Epsilonbacteraeota phyl. nov.
Description of Desulfurellia class. nov.
Desulfurellia (De.sul.fu.rel′li.a. N.L. fem. n. Desulfurella type genus of the type order of the class; suff. -ia, ending to denote a class; N.L. neut. pl. n. Desulfurellia the class of the order Desulfurellales).
Description is the same as for the order Desulfurellales (Kuever et al., 2006b). Type order: Desulfurellales, phylum: Epsilonbacteraeota phyl. nov.
Emended Description of the Order Desulfurellales Kuever et al. (2006b)
The order is as previously described (Kuever et al., 2006b) but is transferred from the class Deltaproteobacteria to Desulfurellia class. nov. The order consists of the families Desulfurellaceae and Hippeaceae fam. nov. Cells are Gram-negative, thermophilic, and motile. Type family: Desulfurellaceae, class: Desulfurellia class. nov., phylum: Epsilonbacteraeota phyl. nov.
Emended Description of the Family Desulfurellaceae Kuever et al. (2006a)
Description is as previously (Kuever et al., 2006a), with the exception that the genus Hippea is moved to the family Hippeaceae fam. nov.. Cells are Gram-negative, obligately anaerobic, and motile. Uses short-chain fatty acids including acetate, fumarate, malate, as well as long-chain saturated fatty acids as growth substrates. Type species D. acetivorans is capable of dissimilatory sulfur reduction (Bonch-Osmolovskaya et al., 1990). Type genus: Desulfurella, order: Desulfurellales, class: Desulfurellia class. nov., phylum: Epsilonbacteraeota phyl. nov.
Description of Hippeaceae fam. nov.
Hippeaceae (Hippe.a.ce′ae. N.L. fem. n. Hippea type genus of the family; suff. -aceae, ending to denote a family; N.L. fem. pl. n. Hippeaceae the family of the genus Hippea).
The family is defined based on phylogenetic analysis of concatenated marker proteins, and is separated from the Desulfurellaceae due to deep branching distance. Cells are Gram-negative, obligately anaerobic, thermophilic, and motile. Capable of heterotrophic growth using acetate and various other organic substrates by species. Type genus: Hippea, order: Desulfurellales, class: Desulfurellia class. nov., phylum: Epsilonbacteraeota phyl. nov.
Description of Epsilonbacteraeota phyl. nov.
Epsilonbacteraeota (Ep.si.lon.bac.ter.ae.o′ta. Gr. n. epsilon, name of the fifth letter of Greek alphabet; Gr. n. baktêria, staff, cane; suff. -aeota, proposed ending to denote a phylum; N.L. neut. pl. n. Epsilonbacteraeota the phylum of bacteria sharing a common ancestry and possessing ribosomal gene sequence and protein marker similarity to those of the members of the classes Campylobacteria class. nov. and Desulfurellia class. nov)
The phylum is described based upon concatenated protein marker and ribosomal gene sequence phylogeny. Owing to the diverse ecological range of members of this phylum there is little unifying physiology. All members are Gram-negative, and motility as well as the reduction of nitrate and polysulfide are ancestral and common traits. The phylum Epsilonbacteraeota currently comprises two classes: Campylobacteria class. nov. and Desulfurellia class. nov.; and three orders: Campylobacterales, Nautiliales, and Desulfurellales. The type class of the phylum is Campylobacteria class. nov.
Author Contributions
DW, DP, and PH performed the bioinformatic analysis, interpretation, and proposed taxonomic changes. KT, MK, SS, and TW sequenced additional genomes for analysis. BC, IV, JS, and MK proposed avenues for functional analysis and assisted with interpretation of data. DW and PH wrote the draft manuscript and all authors contributed to, and approved, the final manuscript.
Funding
The study was supported by a Discovery Outstanding Researcher Award (DP120103498) and an Australian Laureate Fellowship (FL150100038) from the Australian Research Council.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Maria Chuvochina, Aharon Oren, and Brian Tindall for advice on etymology. The genomes of Hydrogenimonas thermophila and Thioreductor were sequenced by the US Department of Energy Joint Genome Institute (http://www.jgi.doe.gov/) in collaboration with the user community.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00682/full#supplementary-material
Footnotes
- ^https://gold.jgi.doe.gov/
- ^http://gtdb.ecogenomic.org/downloads
- ^https://github.com/wwood/finishm
- ^https://github.com/dparks1134/RefineM
- ^https://github.com/geronimp/graftM
- ^https://github.com/dparks1134/CompareM
- ^https://github.com/Ecogenomics/mingle
- ^gtdb.ecogenomic.org
References
Abby, S. S., Tannier, E., Gouy, M., and Daubin, V. (2012). Lateral gene transfer as a support for the tree of life. Proc. Natl. Acad. Sci. U.S.A. 109, 4962–4967. doi: 10.1073/pnas.1116871109
Akerman, N. H., Butterfield, D. A., and Huber, J. A. (2013). Phylogenetic diversity and functional gene patterns of sulfur-oxidizing subseafloor Epsilonproteobacteria in diffuse hydrothermal vent fluids. Front. Microbiol. 4:185. doi: 10.3389/fmicb.2013.00185
Alain, K., Zbinden, M., Le Bris, N., Lesongeur, F., Quérellou, J., Gaill, F., et al. (2004). Early steps in microbial colonization processes at deep-sea hydrothermal vents. Environ. Microbiol. 6, 227–241. doi: 10.1111/j.1462-2920.2003.00557.x
Albertsen, M., Hugenholtz, P., Skarshewski, A., Nielsen, K. L., Tyson, G. W., and Nielsen, P. H. (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31, 533–538. doi: 10.1038/nbt.2579
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anderson, I., Sikorski, J., Zeytun, A., Nolan, M., Lapidus, A., Lucas, S., et al. (2011). Complete genome sequence of Nitratifractor salsuginis type strain (E9I37-1T). Stand. Genomic Sci. 4, 322–330. doi: 10.4056/sigs.1844518
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Berg, I. A. (2011). Ecological aspect of the distribution of different autotrophic CO2 fixation pathways. Appl. Environ. Microbiol. 77, 1925–1936. doi: 10.1128/AEM.02473-10
Bergmann, D. J., Hooper, A. B., and Klotz, M. G. (2005). Structure and sequence conservation of hao cluster genes of autotrophic ammonia-oxidizing bacteria: evidence for their evolutionary history. Appl. Environ. Microbiol. 71, 5371–5382. doi: 10.1128/AEM.71.9.5371-5382.2005
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bonch-Osmolovskaya, E. A., Sokolova, T. G., Kostrikina, N. A., and Zavarzin, G. A. (1990). Desulfurella acetivorans gen. nov. and sp. nov. A new thermophilic sulfur-reducing eubacterium. Arch. Microbiol. 153, 151–155. doi: 10.1007/BF00247813
Boyd, E. S., and Druschel, G. K. (2013). Involvement of intermediate sulfur species in biological reduction of elemental sulfur under acidic, hydrothermal conditions. Appl. Environ. Microbiol. 79, 2061–2068. doi: 10.1128/AEM.03160-12
Braakman, R., and Smith, E. (2012). The emergence and early evolution of biological carbon-fixation. PLoS Comput. Biol. 8:e1002455. doi: 10.1371/journal.pcbi.1002455.
Brugna-Guiral, M., Tron, P., Nitschke, W., Stetter, K. O., Burlat, B., Guigliarelli, B., et al. (2003). [NiFe] hydrogenases from the hyperthermophilic bacterium Aquifex aeolicus: properties, function, and phylogenetics. Extremophiles 7, 145–157. doi: 10.1007/s00792-002-0306-3
Bryner, J. H., Littleton, J., Gates, C., Kirkbridge, C. A., and Richie, A. E. (1986). “Flexispira rappini gen. nov., sp. nov., a Gram-negative rod from mammalian fetus and feces,” in Proceedings of the XIV International Congress of Microbiology, Manchester.
Buchanan, B. B., and Arnon, D. I. (1990). A reverse KREBS cycle in photosynthesis: consensus at last. Photosynth. Res. 24, 47–53. doi: 10.1007/BF00032643
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Campbell, B. J., Engel, A. S., Porter, M. L., and Takai, K. (2006). The versatile 𝜖-proteobacteria: key players in sulphidic habitats. Nat. Rev. Microbiol. 4, 458–468. doi: 10.1038/nrmicro1414
Campbell, B. J., Smith, J. L., Hanson, T. E., Klotz, M. G., Stein, L. Y., Lee, C. K., et al. (2009). Adaptations to submarine hydrothermal environments exemplified by the genome of Nautilia profundicola. PLoS Genet. 5:e1000362. doi: 10.1371/journal.pgen.1000362
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chen, S., Beeby, M., Murphy, G. E., Leadbetter, J. R., Hendrixson, D. R., Briegel, A., et al. (2011). Structural diversity of bacterial flagellar motors. EMBO J. 30, 2972–2981. doi: 10.1038/emboj.2011.186
Crusoe, M. R., Alameldin, H. F., Awad, S., Boucher, E., Caldwell, A., Cartwright, R., et al. (2015). The khmer software package: enabling efficient nucleotide sequence analysis. F1000Research 4, 900. doi: 10.12688/f1000research.6924.1
De Cáceres, M., and Legendre, P. (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. doi: 10.1890/08-1823.1
De Cáceres, M., Sol, D., Lapiedra, O., and Legendre, P. (2011). A framework for estimating niche metrics using the resemblance between qualitative resources. Oikos 120, 1341–1350. doi: 10.1111/j.1600-0706.2011.19679.x
Dewhirst, F. E., Fox, J. G., Mendes, E. N., Paster, B. J., Gates, C. E., Kirkbride, C. A., et al. (2000). “Flexispira rappini” strains represent at least 10 Helicobacter taxa. Int. J. Syst. Evol. Microbiol. 50, 1781–1787.
Di Rienzi, S. C., Sharon, I., Wrighton, K. C., Koren, O., Hug, L. A., Thomas, B. C., et al. (2013). The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. Elife 2:e01102. doi: 10.7554/eLife.01102
Dodsworth, J. A., Blainey, P. C., Murugapiran, S. K., Swingley, W. D., Ross, C. A., Tringe, S. G., et al. (2013). Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for members of the OP9 lineage. Nat. Commun. 4, 1854. doi: 10.1038/ncomms2884
Eddy, S. R. (1998). Profile hidden Markov models. Bioinformatics 14, 755–763. doi: 10.1093/bioinformatics/14.9.755
Eisen, J. A. (1995). The RecA protein as a model molecule for molecular systematic studies of bacteria: comparison of trees of RecAs and 16S rRNAs from the same species. J. Mol. Evol. 41, 1105–1123. doi: 10.1007/BF00173192
Eveleigh, R. J. M., Meehan, C. J., Archibald, J. M., and Beiko, R. G. (2013). Being Aquifex aeolicus: untangling a hyperthermophile’s checkered past. Genome Biol. Evol. 5, 2478–2497. doi: 10.1093/gbe/evt195
Finster, K., Liesack, W., and Tindall, B. J. (1997). Sulfurospirillum arcachonense sp. nov., a new-microaerophilic sulfur-reducing bacterium. Int. J. Syst. Bacteriol. 47, 1212–1217.
Flint, A., Stintzi, A., and Saraiva, L. M. (2016). Oxidative and nitrosative stress defences of Helicobacter and Campylobacter species that counteract mammalian immunity. FEMS Microbiol. Rev. 40, 938–960.
Florentino, A. P., Brienza, C., Stams, A. J. M., and Sánchez-Andrea, I. (2016). Desulfurella amilsii sp. nov., a novel acidotolerant sulfur-respiring bacterium isolated from acidic river sediments. Int. J. Syst. Evol. Microbiol. 66, 1249–1253. doi: 10.1099/ijsem.0.000866
Flores, G. E., Campbell, J. H., Kirshtein, J. D., Meneghin, J., Podar, M., Steinberg, J. I., et al. (2011). Microbial community structure of hydrothermal deposits from geochemically different vent fields along the Mid-Atlantic Ridge. Environ. Microbiol. 13, 2158–2171. doi: 10.1111/j.1462-2920.2011.02463.x
Friedrich, C. G., Bardischewsky, F., Rother, D., Quentmeier, A., and Fischer, J. (2005). Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 8, 253–259. doi: 10.1016/j.mib.2005.04.005
Gao, B., Lara-Tejero, M., Lefebre, M., Goodman, A. L., and Galán, J. E. (2014). Novel components of the flagellar system in Epsilonproteobacteria. MBio 5:e01349-14. doi: 10.1128/mBio.01349-14
Garcia-Pichel, F. (1989). Rapid bacterial swimming measured in swarming cells of Thiovulum majus. J. Bacteriol. 171, 3560–3563.
Garrity, G. M., Bell, J. A., and Lilburn, T. (2005). “Bergey’s manual of systematic bacteriology,” in The Proteobacteria, Vol. 2, eds D. J. Brenner, N. R. Krieg, J. T. Staley, and G. M. Garrity (New York, NY: Springer).
Garrity, G. M., Bell, J. A., and Lilburn, T. (2006a). “Family II. Helicobacteraceae fam. nov,” in Bergey’s Manual of Systematic Bacteriology, (The Proteobacteria), Part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria), Vol. 2, 2nd Edn. eds D. J. Brenner, N. R. Krieg, J. T. Staley, and G. M. Garrity (New York, NY: Springer), 1168.
Garrity, G. M., Bell, J. A., and Lilburn, T. (2006b). “Order I. Campylobacterales ord. nov,” in Bergey’s Manual of Systematic Bacteriology, (The Proteobacteria), Part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria), Vol. 2, 2nd Edn. eds D. J. Brenner, N. R. Krieg, J. T. Staley, and G. M. Garrity (New York, NY: Springer), 1145.
Gevertz, D., Telang, A. J., Voordouw, G., and Jenneman, G. E. (2000). Isolation and characterization of strains CVO and FWKO B, two novel nitrate-reducing, sulfide-oxidizing bacteria isolated from oil field brine. Appl. Environ. Microbiol. 66, 2491–2501. doi: 10.1128/AEM.66.6.2491-2501.2000
Ghosh, W., Mallick, S., and DasGupta, S. K. (2009). Origin of the Sox multienzyme complex system in ancient thermophilic bacteria and coevolution of its constituent proteins. Res. Microbiol. 160, 409–420. doi: 10.1016/j.resmic.2009.07.003
Greening, C., Biswas, A., Carere, C. R., Jackson, C. J., Taylor, M. C., Stott, M. B., et al. (2016). Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J. 10, 761–777. doi: 10.1038/ismej.2015.153
Grosche, A., Sekaran, H., Pérez-Rodríguez, I., Starovoytov, V., and Vetriani, C. (2015). Cetia pacifica gen. nov., sp. nov., a chemolithoautotrophic, thermophilic, nitrate-ammonifying bacterium from a deep-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 65, 1144–1150. doi: 10.1099/ijs.0.000070
Grote, J., Schott, T., Bruckner, C. G., Glöckner, F. O., Jost, G., Teeling, H., et al. (2012). Genome and physiology of a model Epsilonproteobacterium responsible for sulfide detoxification in marine oxygen depletion zones. Proc. Natl. Acad. Sci. U.S.A. 109, 506–510. doi: 10.1073/pnas.1111262109
Gruber, T. M., and Bryant, D. A. (1998). Characterization of the group 1 and group 2 sigma factors of the green sulfur bacterium Chlorobium tepidum and the green non-sulfur bacterium Chloroflexus aurantiacus. Arch. Microbiol. 170, 285–296. doi: 10.1007/s002030050644
Gulmann, L. K., Beaulieu, S. E., Shank, T. M., Ding, K., Seyfried, W. E., and Sievert, S. M. (2015). Bacterial diversity and successional patterns during biofilm formation on freshly exposed basalt surfaces at diffuse-flow deep-sea vents. Front. Microbiol. 6:901. doi: 10.3389/fmicb.2015.00901
Gupta, R. S. (2000). The phylogeny of proteobacteria: relationships to other eubacterial phyla and eukaryotes. FEMS Microbiol. Rev. 24, 367–402. doi: 10.1016/S0168-6445(00)00031-0
Guy, L., Saw, J. H., and Ettema, T. J. G. (2014). The archaeal legacy of eukaryotes: a phylogenomic perspective. Cold Spring Harb. Perspect. Biol. 6:a016022. doi: 10.1101/cshperspect.a016022
Haddad, A., Camacho, F., Durand, P., and Cary, S. C. (1995). Phylogenetic characterization of the epibiotic bacteria associated with the hydrothermal vent polychaete Alvinella pompejana. Appl. Environ. Microbiol. 61, 1679–1687.
Hanson, T. E., Campbell, B. J., Kalis, K. M., Campbell, M. A., and Klotz, M. G. (2013). Nitrate ammonification by Nautilia profundicola AmH: experimental evidence consistent with a free hydroxylamine intermediate. Front. Microbiol. 4:180. doi: 10.3389/fmicb.2013.00180
Huber, J. A., Cantin, H. V., Huse, S. M., Mark Welch, D. B., Sogin, M. L., and Butterfield, D. A. (2010). Isolated communities of Epsilonproteobacteria in hydrothermal vent fluids of the Mariana Arc seamounts. FEMS Microbiol. Ecol. 73, 538–549. doi: 10.1111/j.1574-6941.2010.00910.x
Hubert, C. R. J., Oldenburg, T. B. P., Fustic, M., Gray, N. D., Larter, S. R., Penn, K., et al. (2012). Massive dominance of Epsilonproteobacteria in formation waters from a Canadian oil sands reservoir containing severely biodegraded oil. Environ. Microbiol. 14, 387–404. doi: 10.1111/j.1462-2920.2011.02521.x
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J., et al. (2016). A new view of the tree of life. Nat. Microbiol. 1, 16048. doi: 10.1038/nmicrobiol.2016.48
Hugenholtz, P., Skarshewski, A., and Parks, D. H. (2016). Genome-based microbial taxonomy coming of age. Cold Spring Harb. Perspect. Biol. 8:a018085. doi: 10.1101/cshperspect.a018085
Hügler, M., Petersen, J. M., Dubilier, N., Imhoff, J. F., and Sievert, S. M. (2011). Pathways of carbon and energy metabolism of the epibiotic community associated with the deep-sea hydrothermal vent shrimp Rimicaris exoculata. PLoS ONE 6:e16018. doi: 10.1371/journal.pone.0016018
Hügler, M., Wirsen, C. O., Fuchs, G., Taylor, C. D., and Sievert, S. M. (2005). Evidence for autotrophic CO2 fixation via the reductive tricarboxylic acid cycle by members of the ε subdivision of proteobacteria. J. Bacteriol. 187, 3020–3027. doi: 10.1128/JB.187.9.3020-3027.2005
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Iyer, L. M., Koonin, E. V., and Aravind, L. (2004). Evolution of bacterial RNA polymerase: implications for large-scale bacterial phylogeny, domain accretion, and horizontal gene transfer. Gene 335, 73–88. doi: 10.1016/j.gene.2004.03.017
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Bioinformatics 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Jukes, T. H., and Cantor, C. R. (1969). “Evolution of protein molecules,” in Mammalian Protein Metabolism, Vol. 3, ed. H. N. Munro (New York, NY: Academic Press), 21–132.
Kappes, R. M., Kempf, B., Kneip, S., Boch, J., Gade, J., Meier-Wagner, J., et al. (1999). Two evolutionarily closely related ABC transporters mediate the uptake of choline for synthesis of the osmoprotectant glycine betaine in Bacillus subtilis. Mol. Microbiol. 32, 203–216. doi: 10.1046/j.1365-2958.1999.01354.x
Kassem, I. I., Khatri, M., Esseili, M. A., Sanad, Y. M., Saif, Y. M., Olson, J. W., et al. (2012). Respiratory proteins contribute differentially to Campylobacter jejuni’s survival and in vitro interaction with hosts’ intestinal cells. BMC Microbiol. 12:258. doi: 10.1186/1471-2180-12-258
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kern, M., Klotz, M. G., and Simon, J. (2011a). The Wolinella succinogenes mcc gene cluster encodes an unconventional respiratory sulphite reduction system. Mol. Microbiol. 82, 1515–1530. doi: 10.1111/j.1365-2958.2011.07906.x
Kern, M., Volz, J., and Simon, J. (2011b). The oxidative and nitrosative stress defence network of Wolinella succinogenes: Cytochrome c nitrite reductase mediates the stress response to nitrite, nitric oxide, hydroxylamine and hydrogen peroxide. Environ. Microbiol. 13, 2478–2494. doi: 10.1111/j.1462-2920.2011.02520.x
Kern, M., and Simon, J. (2009). Electron transport chains and bioenergetics of respiratory nitrogen metabolism in Wolinella succinogenes and other Epsilonproteobacteria. Biochim. Biophys. Acta 1787, 646–656. doi: 10.1016/j.bbabio.2008.12.010
Kersters, K., De Vos, P., Gillis, M., Swings, J., Vandamme, P., and Stakebrandt, E. (2006). Introduction to the Proteobacteria. New York, NY: Springer. doi: 10.1007/0-387-30745-1-1
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Klenk, H. P., Meier, T. D., Durovic, P., Schwass, V., Lottspeich, F., Dennis, P. P., et al. (1999). RNA polymerase of Aquifex pyrophilus: implications for the evolution of the bacterial rpoBC operon and extremely thermophilic bacteria. J. Mol. Evol. 48, 528–541. doi: 10.1007/PL00006496
Klotz, M. G., and Stein, L. Y. (2008). Nitrifier genomics and evolution of the nitrogen cycle. FEMS Microbiol. Lett. 278, 146–156. doi: 10.1111/j.1574-6968.2007.00970.x
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Kozlowski, J. A., Stieglmeier, M., Schleper, C., Klotz, M. G., and Stein, L. Y. (2016). Pathways and key intermediates required for obligate aerobic ammonia-dependent chemolithotrophy in bacteria and Thaumarchaeota. ISME J. 10, 1–10. doi: 10.1038/ismej.2016.2
Kozubal, M. A., Romine, M., Jennings, R. D., Jay, Z. J., Tringe, S. G., Rusch, D. B., et al. (2013). Geoarchaeota: a new candidate phylum in the Archaea from high-temperature acidic iron mats in Yellowstone National Park. ISME J. 7, 622–634. doi: 10.1038/ismej.2012.132
Kuever, J., Rainey, F. A., and Widdel, F. (2006a). “Family I. Desulfurellaceae fam. nov,” in Bergey’s Manual of Systematic Bacteriology, (The Proteobacteria), Part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria), Vol. 2, 2nd Edn. eds D. J. Brenner, N. R. Krieg, J. T. Staley, and G. M. Garrity (New York, NY: Springer), 923.
Kuever, J., Rainey, F. A., and Widdel, F. (2006b). “Order I. Desulfurellales ord. nov,” in Bergey’s Manual of Systematic Bacteriology, (The Proteobacteria), part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria)2, Vol. 2, 2nd Edn. eds D. J. Brenner, N. R. Krieg, J. T. Staley, and G. M. Garrity (New York, NY: Springer), 922.
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (New York, NY: Wiley), 115–175.
Lavik, G., Stuhrmann, T., Bruchert, V., Van der Plas, A., Mohrholz, V., and Lam, P. (2009). Detoxification of sulphidic African shelf waters by blooming chemolithotrophs. Nature 457, 581. doi: 10.1017/CBO9781107415324.004
Lawson, A. J., On, S. L. W., Logan, J. M. J., and Stanley, J. (2001). Campylobacter hominis sp. nov., from the human gastrointestinal tract. Int. J. Syst. Evol. Microbiol. 51, 651–660. doi: 10.1099/00207713-51-2-651
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Létoffé, S., Delepelaire, P., and Wandersman, C. (2006). The housekeeping dipeptide permease is the Escherichia coli heme transporter and functions with two optional peptide binding proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 12891–12896. doi: 10.1073/pnas.0605440103
Ludwig, W., Rossellö-Mora, R., Aznar, R., Klugbauer, S., Spring, S., Reetz, K., et al. (1995). Comparative sequence analysis of 23S rRNA from Proteobacteria. Syst. Appl. Microbiol. 18, 164–188. doi: 10.1016/S0723-2020(11)80388-7
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, A., et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Luijten, M. L. G. C., de Weert, J., Smidt, H., Boschker, H. T. S., de Vos, W. M., Schraa, G., et al. (2003). Description of Sulfurospirillum halorespirans sp. nov., an anaerobic, tetrachloroethene-respiring bacterium, and transfer of Dehalospirillum multivorans to the genus Sulfurospirillum a Sulfurospirillum multivorans comb. nov. Int. J. Syst. Evol. Microbiol. 53, 787–793. doi: 10.1099/ijs.0.02417-0
Macy, J. M., Schröder, I., Thauer, R. K., and Kröger, A. (1996). Growth the Wolinella succinogenes on H2S plus fumarate and on formate plus sulfur as energy sources. Arch. Microbiol. 144, 147–150. doi: 10.1007/BF00414725
Makita, H., Nakagawa, S., Miyazaki, M., Nakamura, K. I., Inagaki, F., and Takai, K. (2012). Thiofractor thiocaminus gen. nov., sp. nov., a novel hydrogen-oxidizing, sulfur-reducing epsilonproteobacterium isolated from a deep-sea hydrothermal vent chimney in the Nikko seamount weld of the Northern Mariana Arc. Arch. Microbiol. 194, 785–794. doi: 10.1007/s00203-012-0814-1
Marcy, Y., Ouverney, C., Bik, E. M., Losekann, T., Ivanova, N., Martin, H. G., et al. (2007). Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc. Natl. Acad. Sci. U.S.A. 104, 11889–11894. doi: 10.1073/pnas.0704662104
Marshall, I. P. G., Blainey, P. C., Spormann, A. M., and Quake, S. R. (2012). A single-cell genome for Thiovulum sp. Appl. Environ. Microbiol. 78, 8555–8563. doi: 10.1128/AEM.02314-12
McDermott, J. M., Seewald, J. S., German, C. R., and Sylva, S. P. (2015). Pathways for abiotic organic synthesis at submarine hydrothermal fields. Proc. Natl. Acad. Sci. U.S.A. 112, 7668-7672. doi: 10.1073/pnas.1506295112
McLean, J. S., Lombardo, M. J., Badger, J. H., Edlund, A., Novotny, M., Yee-Greenbaum, J., et al. (2013). Candidate phylum TM6 genome recovered from a hospital sink biofilm provides genomic insights into this uncultivated phylum. Proc. Natl. Acad. Sci. U.S.A. 110, E2390–E2399. doi: 10.1073/pnas.1219809110
Meyer, B., Imhoff, J. F., and Kuever, J. (2007). Molecular analysis of the distribution and phylogeny of the soxB gene among sulfur-oxidizing bacteria–Evolution of the Sox sulfur oxidation enzyme system. Environ. Microbiol. 9, 2957–2977. doi: 10.1111/j.1462-2920.2007.01407.x
Miroshnichenko, M. L., Kostrikina, N. A., L’Haridon, S., Jeanthon, C., Hippe, H., Stackebrandt, E., et al. (2002). Nautilia lithotrophica gen. nov., sp. nov., a thermophilic sulfur-reducing epsilon-proteobacterium isolated from a deep-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 52, 1299–1304. doi: 10.1099/ijs.0.02139-0
Moussard, H., Corre, E., Cambon-Bonavita, M. A., Fouquet, Y., and Jeanthon, C. (2006). Novel uncultured Epsilonproteobacteria dominate a filamentous sulphur mat from the 13°N hydrothermal vent field, East Pacific Rise. FEMS Microbiol. Ecol. 58, 449–463. doi: 10.1111/j.1574-6941.2006.00192.x
Moyer, C. L., Tiedje, J. M., Dobbs, F. C., and Karl, D. M. (1996). A computer-simulated restriction fragment length polymorphism analysis of bacterial small-subunit rRNA genes: efficacy of selected tetrameric restriction enzymes for studies of microbial diversity in nature. Appl. Environ. Microbiol. 62,2501–2507.
Mukherjee, S., Stamatis, D., Bertsch, J., Ovchinnikova, G., Verezemska, O., Isbandi, M., et al. (2017). Genomes OnLine Database (GOLD) v.6: data updates and feature enhancements. Nucleic Acids Res. 45, D446–D456. doi: 10.1093/nar/gkw992
Muyzer, G., Yildirim, E., Van Dongen, U., Kühl, M., and Thar, R. (2005). Identification of “Candidatus Thioturbo danicus,” a microaerophilic bacterium that builds conspicuous veils on sulfidic sediments. Appl. Environ. Microbiol. 71, 8929–8933. doi: 10.1128/AEM.71.12.8929-8933.2005
Nakagawa, S., and Takai, K. (2014). “The family Nautiliaceae: the genera Caminibacter, Lebetimonas, and Nautilia,” in The Prokaryotes: Deltaproteobacteria and Epsilonproteobacteria, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt and F. Thompson (Berlin: Springer), 393–399. doi: 10.1007/978-3-642-39044-9-276
Nakagawa, S., Takai, K., Inagaki, F., Horikoshi, K., and Sako, Y. (2005). Nitratiruptor tergarcus gen. nov., sp. nov. and Nitratifractor salsuginis gen. nov., sp. nov., nitrate-reducing chemolithoautotrophs of the E-Proteobacteria isolated from a deep-sea hydrothermal system in the Mid-Okinawa Trough. Int. J. Syst. Evol. Microbiol. 55, 925–933. doi: 10.1099/ijs.0.63480-0
Nakagawa, S., and Takaki, Y. (2009). “Nonpathogenic Epsilonproteobacteria,” in Encyclopedia of Life Sciences (ELS) (Chichester: John Wiley & Sons Ltd), 1–11. doi: 10.1002/9780470015902.a0021895
Nakagawa, T., Takai, K., Suzuki, Y., Hirayama, H., Konno, U., Tsunogai, U., et al. (2006). Geomicrobiological exploration and characterization of a novel deep-sea hydrothermal system at the TOTO caldera in the Mariana Volcanic Arc. Environ. Microbiol. 8, 37–49. doi: 10.1111/j.1462-2920.2005.00884.x
Ochoa de Alda, J. A G., Esteban, R., Diago, M. L., and Houmard, J. (2014). The plastid ancestor originated among one of the major cyanobacterial lineages. Nat. Commun. 5, 4937. doi: 10.1038/ncomms5937
Oksanen, J., Blanchet, F., Kindt, R., Legendre, P., and O’Hara, R. (2016). Vegan: Community ecology package. R Packag. 2.3-3. Available at: https://CRAN.R-project.org/package=vegan. doi: 10.4135/9781412971874.n145
Opatkiewicz, A. D., Butterfield, D. A., and Baross, J. A. (2009). Individual hydrothermal vents at Axial Seamount harbor distinct subseafloor microbial communities. FEMS Microbiol. Ecol. 70, 413–424. doi: 10.1111/j.1574-6941.2009.00747.x
Oren, A., Da Costa, M. S., Garrity, G. M., Rainey, F. A., Rosselló-Móra, R., Schink, B., et al. (2015). Proposal to include the rank of phylum in the international code of nomenclature of prokaryotes. Int. J. Syst. Evol. Microbiol. 65, 4284–4287. doi: 10.1099/ijsem.0.000664
Ormerod, K. L., Wood, D. L. A., Lachner, N., Gellatly, S. L., Daly, J. N., Parsons, J. D., et al. (2016). Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome 4, 36. doi: 10.1186/s40168-016-0181-2
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parte, A. C. (2014). LPSN - List of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 42, D613–D616. doi: 10.1093/nar/gkt1111
Piccirillo, A., Niero, G., Calleros, L., Pérez, R., Naya, H., and Iraola, G. (2016). Campylobacter geochelonis sp. nov. isolated from the western Hermann’s tortoise (Testudo hermanni hermanni). Int. J. Syst. Evol. Microbiol. 66, 3468–3476. doi: 10.1099/ijsem.0.001219
Pino, C., Olmo-Mira, F., Cabello, P., Martínez-Luque, M., Castillo, F., Roldán, M. D., et al. (2006). The assimilatory nitrate reduction system of the phototrophic bacterium Rhodobacter capsulatus E1F1. Biochem. Soc. Trans. 34, 127–129. doi: 10.1042/BST0340127
Pitson, S. M., Mendz, G. L., Srinivasan, S., and Hazell, S. L. (1999). The tricarboxylic acid cycle of Helicobacter pylori. Eur. J. Biochem. 260, 258–267. doi: 10.1046/j.1432-1327.1999.00153.x
Pittman, M. S., Elvers, K. T., Lee, L., Jones, M. A., Poole, R. K., Park, S. F., et al. (2007). Growth of Campylobacter jejuni on nitrate and nitrite: electron transport to NapA and NrfA via NrfH and distinct roles for NrfA and the globin Cgb in protection against nitrosative stress. Mol. Microbiol. 63, 575–590. doi: 10.1111/j.1365-2958.2006.05532.x
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2016). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. doi: 10.1038/sj.hdy.6800737.
Rassa, A. C., McAllister, S. M., Safran, S. A., and Moyer, C. L. (2009). Zeta-Proteobacteria dominate the colonization and formation of microbial mats in low-temperature hydrothermal vents at Loihi Seamount, Hawaii. Geomicrobiol. J. 26, 623–638. doi: 10.1080/01490450903263350
Revell, L. J. (2012). phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Reysenbach, A. L., Longnecker, K., and Kirshtein, J. (2000). Novel bacterial and archaeal lineages from an in situ growth chamber deployed at a mid-atlantic ridge hydrothermal vent. Appl. Environ. Microbiol. 66, 3798–3806. doi: 10.1128/AEM.66.9.3798-3806.2000
Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N. N., Anderson, I. J., Cheng, J. -F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437. doi: 10.1038/nature12352
Rossmassler, K., Hanson, T. E., and Campbell, B. J. (2016). Diverse sulfur metabolisms from two subterranean sulfidic spring systems. FEMS Microbiol. Lett. 363:fnw162. doi: 10.1093/femsle/fnw162
Rushdi, A. I., and Simoneit, B. R. T. (2005). Abiotic synthesis of organic compounds from carbon disulfide under hydrothermal conditions. Astrobiology 5, 749–769. doi: 10.1089/ast.2005.5.749
Sanders, J. G., Beinart, R. A., Stewart, F. J., Delong, E. F., and Girguis, P. R. (2013). Metatranscriptomics reveal differences in in situ energy and nitrogen metabolism among hydrothermal vent snail symbionts. ISME J. 7, 1556–1567. doi: 10.1038/ismej.2013.45
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schuchmann, K., and Müller, V. (2014). Autotrophy at the thermodynamic limit of life: a model for energy conservation in acetogenic bacteria. Nat. Rev. Microbiol. 12, 809–821. doi: 10.1038/nrmicro3365
Schumacher, W., Kroneck, P. M. H., and Pfennig, N. (1992). Comparative systematic study on “Spirillum” 5175, Campylobacter and Wolinella species–description of “Spirillum” 5175 as Sulfurospirillum deleyianum gen. nov., spec. nov. Arch. Microbiol. 158, 287–293. doi: 10.1007/BF00245247
Sen, A., Daubin, V., Abrouk, D., Gifford, I., Berry, A. M., Normand, P., et al. (2014). Phylogeny of the class Actinobacteria revisited in the light of complete genomes. Int. J. Syst. Evol. Microbiol. 64, 3821–3832. doi: 10.1099/ijs.0.063966-0
Sheneman, L., Evans, J., and Foster, J. A. (2006). Clearcut: a fast implementation of relaxed neighbor joining. Bioinformatics 22, 2823–2824. doi: 10.1093/bioinformatics/btl478
Sheridan, P. P., Freeman, K. H., and Brenchley, J. E. (2003). Estimated minimal divergence times of the major bacterial and archaeal phyla estimated minimal divergence times of the major bacterial and archaeal phyla. Geomicrobiol. J. 20, 1–14. doi: 10.1080/01490450390144330
Sievert, S. M., Scott, K. M., Klotz, M. G., Chain, P. S. G., Hauser, L. J., Hemp, J., et al. (2008). Genome of the epsilonproteobacterial chemolithoautotroph Sulfurimonas denitrificans. Appl. Environ. Microbiol. 74, 1145–1156. doi: 10.1128/AEM.01844-07
Simon, J., and Klotz, M. G. (2013). Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations. Biochim. Biophys. Acta 1827, 114–135. doi: 10.1016/j.bbabio.2012.07.005
Simon, J., Sänger, M., Schuster, S. C., and Gross, R. (2003). Electron transport to periplasmic nitrate reductase (NapA) of Wolinella succinogenes is independent of a NapC protein. Mol. Microbiol. 49, 69–79. doi: 10.1046/j.1365-2958.2003.03544.x
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Takai, K., Campbell, B. J., Cary, S. C., Suzuki, M., Oida, H., Nunoura, T., et al. (2005). Enzymatic and genetic characterization of carbon and energy metabolisms by deep-sea hydrothermal chemolithoautotrophic isolates of Epsilonproteobacteria. Appl. Environ. Microbiol. 71, 7310–7320. doi: 10.1128/AEM.71.11.7310-7320.2005
Takai, K., Nealson, K. H., and Horikoshi, K. (2004). Hydrogenimonas thermophila gen. nov., sp. nov., a novel thermophilic, hydrogen-oxidizing chemolithoautotroph within the E-Proteobacteria, isolated from a black smoker in a Central Indian Ridge hydrothermal field. Int. J. Syst. Evol. Microbiol. 54, 25–32. doi: 10.1099/ijs.0.02787-0
Takai, K., Suzuki, M., Nakagawa, S., Miyazaki, M., Suzuki, Y., Inagaki, F., et al. (2006). Sulfurimonas paralvinellae sp. nov., a novel mesophilic, hydrogen- and sulfur-oxidizing chemolithoautotroph within the Epsilonproteobacteria isolated from a deep-sea hydrothermal vent polychaete nest, reclassification of Thiomicrospira denitrificans as Sulfurimonas denitrificans comb. nov. and emended description of the genus Sulfurimonas. Int. J. Syst. Evol. Microbiol. 56, 1725–1733. doi: 10.1099/ijs.0.64255-0
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tenover, F. C., Fennel, C. L., Tanner, A., and Paster, B. J. (1992). “Subdivision C5: Epsilon subclass,” in The Prokaryotes, 2nd Edn, eds A. Balows, H. G. Troper, M. Dworkin, and W. Harder (Berlin: Springer-Verlag), 3499–3512.
Trust, T. J., Logan, S. M., Gustafson, C. E., Romaniuk, P. J., Kim, N. W., Chan, V. L., et al. (1994). Phylogenetic and molecular characterization of a 23S rRNA gene positions the genus Campylobacter in the epsilon subdivision of the Proteobacteria and shows that the presence of transcribed spacers is common in Campylobacter spp. J. Bacteriol. 176, 4597–4609.
Vandamme, P., Daneshvar, M. I., Dewhirst, F. E., Paster, B. J., Kersters, K., Goossens, H., et al. (1995). Chemotaxonomic analyses of Bacteroides gracilis and Bacteroides ureolyticus and reclassification of B. gracilis as Campylobacter gracilis comb. nov. Int. J. Syst. Bacteriol. 45, 145–152.
Vandamme, P., and De Ley, J. (1991). Proposal for a new family, Campylobacteraceae. Int. J. Syst. Bacteriol. 41, 451–455.
Vandamme, P., Falsen, E., Rossau, R., Hoste, B., Segers, P., Tytgat, R., et al. (1991). Revision of Campylobacter, Helicobacter, and Wolinella taxonomy: emendation of generic descriptions and proposal of Arcobacter gen. nov. Int. J. Syst. Bacteriol. 41, 88–103. doi: 10.1099/00207713-41-1-88
Vandamme, P., and On, S. L. W. (2001). Recommendations of the subcommittee on the taxonomy of Campylobacter and related bacteria. Int. J. Syst. Evol. Microbiol. 51, 719–721. doi: 10.1099/ijs.0.02333-0
Vetriani, C., Voordeckers, J. W., Crespo-Medina, M., O’Brien, C. E., Giovannelli, D., and Lutz, R. A. (2014). Deep-sea hydrothermal vent Epsilonproteobacteria encode a conserved and widespread nitrate reduction pathway (Nap). ISME J. 8, 1510–1521. doi: 10.1038/ismej.2013.246
Wang, Z., Bie, P., Cheng, J., Lu, L., Cui, B., and Wu, Q. (2016). The ABC transporter YejABEF is required for resistance to antimicrobial peptides and the virulence of Brucella melitensis. Sci. Rep. 6:31876. doi: 10.1038/srep31876
Whelan, S., and Goldman, N. (2001). A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699. doi: 10.1093/oxfordjournals.molbev.a003851
Williams, K. P., Gillespie, J. J., Sobral, B. W. S., Nordberg, E. K., Snyder, E. E., Shallom, J. M., et al. (2010). Phylogeny of Gammaproteobacteria. J. Bacteriol. 192, 2305–2314. doi: 10.1128/JB.01480-09
Wirsen, C. O., Sievert, S. M., Cavanaugh, C. M., Molyneaux, S. J., Ahmad, A., Taylor, L. T., et al. (2002). Characterization of an autotrophic sulfide-oxidizing marine Arcobacter sp. that produces filamentous sulfur. Appl. Environ. Microbiol. 68, 316–325. doi: 10.1128/AEM.68.1.316-325.2002
Wright, K. E., Williamson, C., Grasby, S. E., Spear, J. R., and Templeton, A. S. (2013). Metagenomic evidence for sulfur lithotrophy by Epsilonproteobacteria as the major energy source for primary productivity in a sub-aerial arctic glacial deposit, Borup Fiord Pass. Front. Microbiol. 4:63. doi: 10.3389/fmicb.2013.00063
Wrighton, K. C., Thomas, B. C., Sharon, I., Miller, C. S., Castelle, C. J., VerBerkmoes, N. C., et al. (2012). Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337, 1661–1665. doi: 10.1126/science.1224041
Wu, D., Hugenholtz, P., Mavromatis, K., Pukall, R., Dalin, E., Ivanova, N. N., et al. (2009). A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462, 1056–1060. doi: 10.1038/nature08656
Yarza, P., Yilmaz, P., Pruesse, E., Glöckner, F. O., Ludwig, W., Schleifer, K. -H., et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645. doi: 10.1038/nrmicro3330
Yeoh, Y. K., Sekiguchi, Y., Parks, D. H., and Hugenholtz, P. (2016). Comparative genomics of candidate phylum TM6 suggests that parasitism is widespread and ancestral in this lineage. Mol. Biol. Evol. 33, 915–927. doi: 10.1093/molbev/msv281
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Keywords: Epsilonproteobacteria, taxonomy, classification, genome, phylogenomics, Epsilonbacteraeota, evolution
Citation: Waite DW, Vanwonterghem I, Rinke C, Parks DH, Zhang Y, Takai K, Sievert SM, Simon J, Campbell BJ, Hanson TE, Woyke T, Klotz MG and Hugenholtz P (2017) Comparative Genomic Analysis of the Class Epsilonproteobacteria and Proposed Reclassification to Epsilonbacteraeota (phyl. nov.). Front. Microbiol. 8:682. doi: 10.3389/fmicb.2017.00682
Received: 03 March 2017; Accepted: 04 April 2017;
Published: 24 April 2017.
Edited by:
Svetlana N. Dedysh, Winogradsky Institute of Microbiology (RAS), RussiaReviewed by:
Barny Whitman, University of Georgia, USACraig Lee Moyer, Western Washington University, USA
Copyright © 2017 Waite, Vanwonterghem, Rinke, Parks, Zhang, Takai, Sievert, Simon, Campbell, Hanson, Woyke, Klotz and Hugenholtz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philip Hugenholtz, p.hugenholtz@uq.edu.au