 Dan-dan Cheng
Dan-dan Cheng Cong He1
Cong He1 Hong-hui Ai
Hong-hui Ai- 1Department of Gastroenterology, The First Affiliated Hospital of Nanchang University, Nanchang, China
- 2Department of Orthopaedics, The Yugan County People's Hospital, Yugan, China
Helicobacter pylori (H. pylori) which colonizes the stomach can cause a wide array of gastric disorders, including chronic gastritis, peptic ulcer, and gastric cancer. Recently, accumulating evidence has implicated H. pylori infection in extragastrointestinal diseases such as cardiovascular diseases, neurological disorders, and metabolic diseases. At the same time, many scholars have noted the relationship between H. pylori infection and non-alcoholic fatty liver disease (NAFLD). Despite the positive association between H. pylori and NAFLD reported in some researches, there are opposite perspectives denying their relationship. Due to high prevalence, unclear etiology and difficult treatment of NAFLD, confirming the pathogenicity of H. pylori infection in NAFLD will undoubtedly provide insights for novel treatment strategies for NAFLD. This paper will review the relationship between H. pylori infection and NAFLD and the possible pathogenic mechanisms.
Introduction
Helicobacter pylori (H. pylori) is a gram-negative microaerophilic bacterium that colonizes the stomach of humans. In 1983, Australian researchers Marshall and Warren (1983) first successfully isolated and cultured H. pylori from the human body. So far, H. pylori is one of the most common human infectious bacteria with a worldwide prevalence of ~50%. In the United States and Europe, the prevalence of H. pylori is estimated to be 20–50%, varying in different socioeconomic, age and ethnic groups, and geography (Ford and Axon, 2010). In developing countries, the prevalence has been reported to be as high as 70% (Mandeville et al., 2009). We all know that H. pylori infection is one of the most important environmental risk factors for the diseases of chronic gastritis, peptic ulcers, gastric mucosa-associated lymphoid tissue (MALT) lymphoma, and gastric cancer. Recently, accumulating evidence implicates H. pylori infection existing in the patients with extragastrointestinal diseases, including idiopathic thrombocytopenic purpura, ischemic heart diseases, obesity, type 2 diabetes mellitus (T2DM) (Wong et al., 2014; Dogan et al., 2015; Nasif et al., 2016). In addition, there is a special focus on the relationship between H. pylori infection and NAFLD.
Unlike alcoholic fatty liver disease, the patients with NAFLD should be an alcohol consumption of < 10 g per day (Neuschwander-Tetri and Caldwell, 2003). NAFLD comprises a spectrum of diseases ranging from simple non-alcoholic fatty liver (NAFL) and non-alcoholic steatohepatitis (NASH) to fibrosis, liver cirrhosis, and ultimately hepatocellular carcinoma (HCC) (Caldwell and Argo, 2010). NAFL is thought to be a relatively benign state, while NASH represents a form of NAFLD that can potentially progress to cirrhosis and HCC. Due to changes in dietary habits and an increase in the number of people engaged in a sedentary lifestyle, the prevalence of NAFLD is increasing worldwide over time (Bhala et al., 2013), which may seriously influence human's health and the quality of life. The worldwide prevalence of NAFLD in the general population is estimated to be 20–30% in Western countries and 5–18% in Asia and is increasing over time (Masarone et al., 2014). Up to 25% of NAFLD patients will evolve into a progressive form of liver diseases named non-alcoholic steatohepatitis (NASH), which is currently the second leading etiology of liver diseases among the adults awaiting liver transplantation in the United States. To date, the exactly pathogenesis mechanisms of NAFLD remain unknown. We know that H. pylori infection has been implicated in the pathogenesis of insulin resistance (IR) (Polyzos et al., 2011), which contributes to the development of NAFLD. Recently, many studies have reported that H. pylori infection is closely related to the development of NAFLD (Polyzos et al., 2013a; Sumida et al., 2015). However, the mechanisms underlying NAFLD remains unclear, and therapeutic options to this disorder are fairly limited nowadays. Thus, investigating the role of H. pylori infection as a risk factor for IR might facilitate understanding its effects on NAFLD. The identification of novel targets for NAFLD therapy is of high priority. Treatment for H. pylori infection is easy and relatively inexpensive, and the interest in exploring its involvement in arising extra-gastric manifestations, is of great interest for public health.
Epidemiology and Clinical Experiments
Specific studies regarding the association between H. pylori infection and NAFLD are increasing. Firstly, H. pylori deoxyribonucleic acid (DNA) has been detected in patients with various etiologies of chronic liver diseases (CLD), including hepatitis, liver fibrosis, and HCC (De Magalhaes Queiroz and Santos, 2001; Castera et al., 2006; Pellicano et al., 2008). In 2008, Cindoruk et al. first found the presence of 16S recombinant RNA of H. pylori in the liver sample of a 44-year-old patient with NASH (Cindoruk et al., 2008). This finding was then been validated by another study, in which the H. pylori DNA was found in 5/11 liver samples of NAFLD patients compared with 2/13 controls (Pirouz et al., 2009). In 2009, it has been demonstrated in an animal model of H. pylori infection that H. pylori inoculated orally, could arrive in the liver and cause hepatitis, further suggested a causative role of H. pylori in CLD (Huang et al., 2009). Surprisingly, H. pylori sequence were found in the liver tissues of chronic hepatitis C patients even though the serology of H. pylori was negative (Castera et al., 2006). The authors speculated two possible mechanisms for the presence of H. pylori in the liver: the bacterium may pass from the stomach to the liver through the duodenum and biliary tract, or may arrive in the liver from the circulation through the hepatic portal vein (Pellicano et al., 2008). Some data indicate the biliary pathway as the most plausible route (Tiwari et al., 2006; Aviles-Jimenez et al., 2016).
Since then, a train of clinical studies of the relationship between H. pylori infection and NAFLD were successively reported (Table 1; Takuma, 2011; Kountouras et al., 2014; Zhang et al., 2016). At the earliest, a study from Greece recruiting 28 patients with biopsy-confirmed NAFLD (15 with NAFL, and 13 with NASH) and 25 matched healthy controls found higher rates of anti-H. pylori IgG in NAFLD groups compared to control group (p = 0.038) and H. pylori infection could independently predict NAFLD in logistic regression analysis. It is indicated that H. pylori infection may represent one more hit contributing to the pathogenesis of NAFLD (Polyzos et al., 2013a). In 2015, another Japanese clinical study involving 130 patients with biopsy-confirmed NAFLD (43 with NAFL and 87 with NASH) found that the prevalence of NASH was significantly higher in patients with H. pylori IgG seropositivity than in those without (81 and 58%, p = 0.008). Besides, the total NAFLD activity score (NAS) and the grade of hepatocyte ballooning were higher in patients with H. pylori IgG seropositivity than in those without (p = 0.03). This study also found that H. pylori infection could independently predict NASH in logistic regression analysis (p = 0.003). And it further confirmed that H. pylori infection may represent a contributing factor to NAFLD (Sumida et al., 2015). Similarly, another two studies from Turkey and Japan, respectively, also suggested H. pylori infection as one of the independent risk factors for the development of NAFLD (Takuma, 2011; Sumida et al., 2015). Moreover, Abenavoli et al. (2013) described a case report in which the metabolic profile of a 55-year old man, including the homeostatic model assessment of insulin resistance (HOMA-IR), fatty liver index and echographic liver pattern, was improved after H. pylori eradication. It further supports the significance of H. pylori infection in the development of NAFLD. However, there were some studies declared that H. pylori infection is not related to NAFLD (Polyzos et al., 2014; Okushin et al., 2015; Baeg et al., 2016). In recent years, two studies from Korea and Japan involved 3,663 and 13,737 patients, respectively, found that H. pylori infection was not associated with NAFLD and may not be the risk factor for NAFLD (Okushin et al., 2015; Baeg et al., 2016). But, one of the drawbacks of these two studies is the limitation of liver ultrasonography for diagnosis and grading the severity of NAFLD. So, taking into account the differences in diagnosis and the small sample size, clinical trials with larger sample sizes are needed to confirm the exact relationship between H. pylori and NAFLD. If this correlation is verified, the treatment of H. pylori may represent a new specific therapeutic strategy for NAFLD.
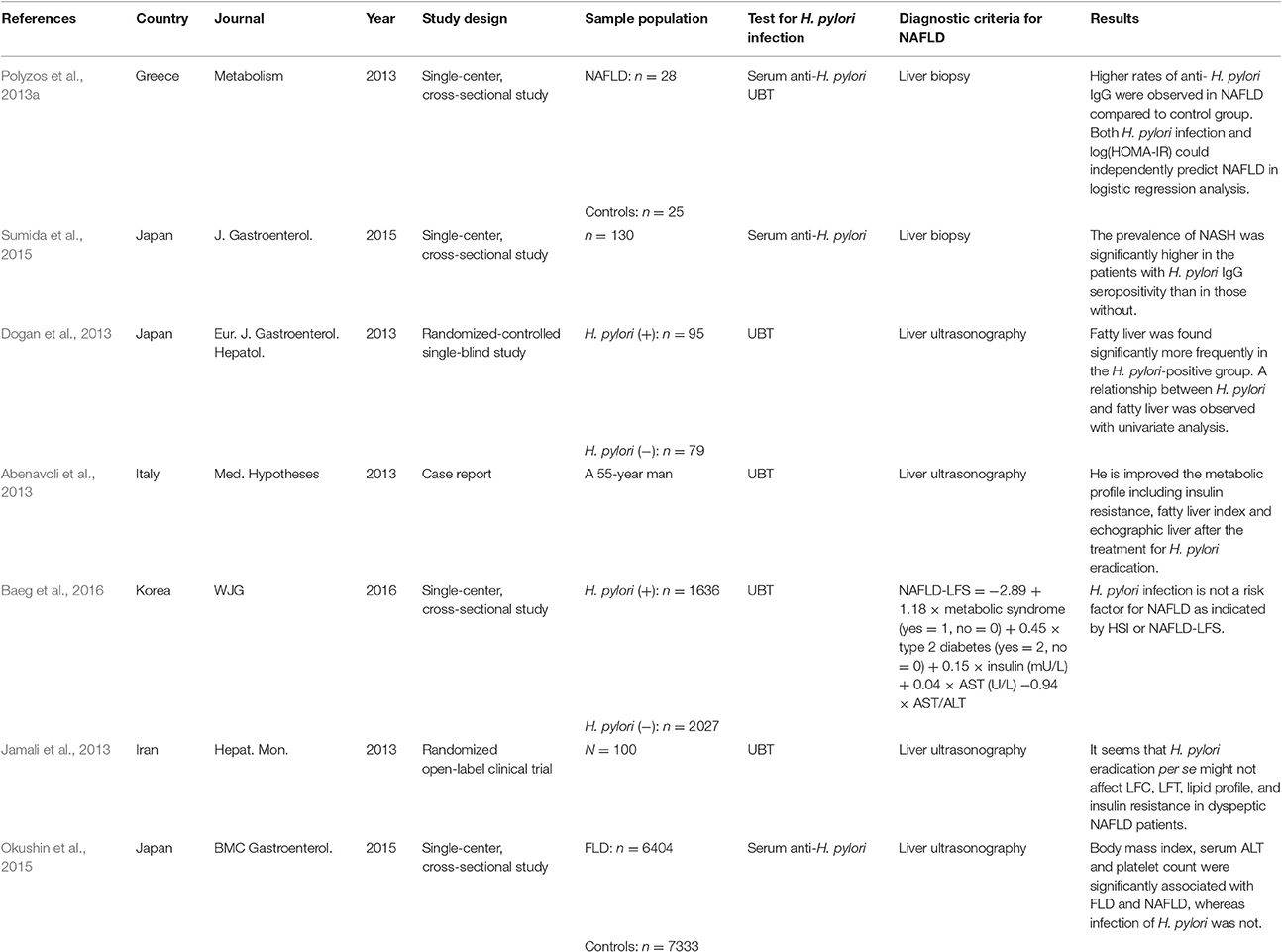
Table 1. Overview of studies regarding the effect of H. pylori infection and NAFLD.
The Possible Mechanisms of the Influence of H. pylori Infection On NAFLD
Insulin Resistance (IR)
IR is closely associated with NAFLD and is also one of the independently risk factors of it (Wang et al., 2013; Birkenfeld and Shulman, 2014). Early in 1998, Day and James (1998) firstly proposed the “two hits” theory for the pathogenesis of NAFLD. They put forward that, the first strike mainly referred to the excessive accumulation of fat in the liver parenchymal cells. This process is associated with IR, which can lead to a dysfunction of intracellular triglyceride synthesis and transport. Recently, some researchers proposed the “multiple hits” hypothesis, which is now widely accepted. They considered multiple insults that interact together on genetically predisposed subjects and induce NAFLD, which provides a more accurate explanation of NAFLD pathogenesis. Such hits include insulin resistance, hormones secreted from the adipose tissue, nutritional factors, gut microbiota, and genetic and epigenetic factors (Takaki et al., 2013; Buzzetti et al., 2016).
In 2005, Aydemir et al. first directly confirmed the association between chronic H. pylori infection and IR, and their results showed that the HOMA-IR of subjects in the H. pylori positive group was significantly higher than that in the H. pylori negative group (p < 0.05; Aydemir et al., 2005). In 2009, a large cross-sectional study including 1,107 subjects, found that the H. pylori seropositivity rate was significantly higher for patients in the IR group (HOMA-IR ≥ 2.5) than in patients in the non-IR group (HOMA-IR < 2.5; 39.4 vs. 28.7%, respectively, p = 0.027). Multiple linear regression analysis showed that H. pylori infection was significantly correlated with the HOMA-IR (95% CI = 0.058–0.246, p = 0.001). The authors therefore proposed that H. pylori infection may be an important independent risk factor for the development of IR (Gunji et al., 2009). Recently, a study of H. pylori infection in NAFLD showed that the rate of infection in NAFLD group is higher than control group, and both H. pylori infection (p = 0.034) and log (HOMA-IR) (p = 0.007) could independently predict NAFLD. Moreover, the study also examined the levels of glucose, insulin, HOMA-IR, ALT, AST, and TNF-α, and found that all of these items were higher in H. pylori-IgG positive group compared to negative group. Therefore, the author put forward that H. pylori infection may contribute to the pathogenesis of NAFLD, mainly by adding to the first hit and this process may be achieved indirectly, or directly, though increasing IR (Polyzos et al., 2013a). Many other studies also demonstrated the causal relationship between H. pylori and IR (Polyzos et al., 2013b; Chen et al., 2015). A prospective study reported that after H. pylori eradication, the fasting plasma insulin level (p < 0.01) and HOMA-IR (p < 0.01) were significantly lower than before treatment, indicating that H. pylori eradication improved IR and may prevent the occurrence of MS, and NAFLD (Abenavoli et al., 2013; Dogan et al., 2015).
The H. pylori-induced IR in NAFLD may be indirectly by causing chronic inflammation or directly by activating certain signaling pathway. Many basic and clinical studies have confirmed that chronic inflammation plays an important role in IR (Hossain et al., 2016). Studies have reported that chronic inflammation due to H. pylori infection can increase the expression of C-reactive protein (CRP), tumor necrosis factor (TNF)-α, and interleukin (IL)-6 (Tsai et al., 2015; Yildirim et al., 2016). These inflammatory cytokines would activate a series of kinases such as IKK/NF-kB and JNK, eventually trigger IR by up-regulating Ser-phosphorylation (Hotamisligil et al., 1996) or inhibition of the autophosphorylation of the tyrosyl of the insulin receptor substrate (IRS)-1 (Dandona et al., 2004). Another study through establishing two kinds of mice models of H. pylori infection and high-fat-diat-fed group, detecting insulin signaling pathway, and relative protein and RNA level in liver tissue, demonstrated that H. pylori infection inhibited miR-203 expression through c-Jun overexpression, and then resulting in the induction of SOCS3, which is a well-known inhibitor of insulin signaling. It manifested H. pylori infection caused hepatic IR by the c-Jun/miR-203/SOCS3 signaling pathway (Zhou et al., 2015).
Inflammation Cytokines or Adipocytokines
The pathogenesis of NASH was originally conceptualized as a disease of consecutive hits: the accumulation of fat in the liver cells (steatosis) that sensitized the liver to a second metabolic insult triggering a cascade of tissue damage (inflammation) resulting in fibrosis. Although, the etiology of NAFLD is maltifactorial and remains largely enigmatic, it is well-accepted that inflammation is a central component of NAFLD pathogenesis (Peverill et al., 2014). A variety of inflammatory cytokines are involved in H. pylori infection, with the closest relationships detected among CRP, TNF-α, IL-6, and interleukin (IL)-1β (Shoelson et al., 2006; Keane et al., 2015). The levels of CRP, TNF-α, and IL-6 in the serum reflect low-grade chronic inflammation in human (Silha et al., 2007). A study regarding H. pylori infection in NAFLD, found TNF-α and IR were significant higher and circulating adiponectin is lower in H. pylori seropositivity compared to H. pylori seronegativity. H. pylori infection may trigger TNF-α, whereas adiponection is secondarily increased to counterbalance the pro-inflammatory cascade. This may be achieved indirectly, through increasing IR which is descripted above, but also directly, given that it could predict NAFLD independently from IR. TNF-α may be a mediator of both direct and indirect effect of H. pylori infection on NAFLD (Polyzos et al., 2013a). CRP is synthesized in liver and adipocytes upon IL-6 and TNF-α stimulation. Hs-CRP levels showed higher in NASH cases vs. non-NASH cases, and it can be used as a non-invasive biomarkers of NAFLD (Maleki et al., 2014). Polyzos's study also detected hs-CRP in H. pylori infection of NAFLD patients, the results showed that the hs-CRP level was higher in H. pylori positive group than in H. pylori negative group (Polyzos et al., 2013a). A prospective, open-label, single-center study involving 159 H. pylori positive patients, who received a 14-day sequential regimen, the results showed that the HOMA-IR and CRP level were significantly higher in patients with H. pylori infection compared to the patients without H. pylori infection (P < 0.05). While, 6 weeks after eradication therapy, the two indicators were significantly decreased from the pretreatment level (P < 0.05; Gen et al., 2010). Thus, we speculate that the H. pylori related-inflammation may play a role in the pathogenesis of NAFLD. The mechanisms of the pathogenesis of H. pylori-related inflammation in NAFLD are directly reducing hepatocyte glycogen levels via a JNK signaling pathway (Li et al., 2010), which can downregulate the expression of key genes of glucose and accelerate lipolysis (Hotamisligil et al., 1996), and indirectly inducing IR through some pathways, which we list above.
In addition, adipose tissue is not only involved in energy metabolism but also contributes to IR by secreting cytokines such as leptin and adiponectin and participates in the development of NAFLD. Studies suggested that the levels of plasma leptin were elevated in NAFLD group (Friedman, 2008), and it is an independent predictor of hepatic steatosis (Chitturi et al., 2002; Polyzos et al., 2016). A study involving 153 dyspeptic patients, found that H. pylori infection was significantly negatively associated with serum leptin level (P < 0.001). H. pylori infection may influence leptin production (Hemmasi et al., 2013), which can inhibit liver stearoyl-CoA desaturase, thus reducing VLDL-C and fatty deposits in the liver tissue (Ding et al., 2005). Besides, leptin may phosphorylate IRS serine 1,318, thus interfering with insulin signal transduction (Hennige et al., 2006). And it is considered to be a pro-inflammatory cytokine and has structural similarity to other pro-inflammatory cytokines such as IL-6 and IL-12 (La Cava and Matarese, 2004). So we rationally speculate that H. pylori infection may cause NAFLD by influencing fat metabolism and transporting relevant enzymes or by insulin signal transduction. But, other studies revealed that H. pylori infection accelerated the synthesis of leptin (Nishi et al., 2005). Thus, more investigation is needed to determine whether there are relationships between H. pylori and serum leptin levels which could account for the discrepancies.
Lipid Profiles
A standard histological feature of NAFLD is the presence of at least 5% hepatocyte steatosis (Tiniakos, 2010). A common feature of NAFLD is the presence of the hepatic ectopic fat deposition (Liu et al., 2010). A study on the association between hepatic lipids and insulin clearance showed that liver lipid content was significantly linked to insulin clearance (r = 0.43, p < 0.0001) and hepatic insulin sensitivity (r = −0.04, p = 0.0002; Kotronen et al., 2007, 2008). Therefore, IR contributes to liver lipid deposition, while increased hepatic lipid accumulation in turn further aggravates IR, leading to a vicious cycle and promoting the development of liver steatosis and NAFLD. This cycle increases the triglycerides (TG) and free fatty acid (FFA) content in liver cells, producing a serious burden for the metabolism of lipids in hepatocytes. Lipid metabolism further produces large amounts of superoxide anions and reactive oxygen species, leading to liver fat peroxidation and oxidative stress (Vergun and Reynolds, 2005). Thus, this kind of vicious cycle will give a heavy blow to hepatocyte.
In 1999, Laurila et al. found that the serum TG and total cholesterol (T-CHOL) concentrations were significantly higher in the males with positive IgG antibody titers for H. pylori than in the males with no signs of infection (p < 0.001), indicating that chronic H. pylori infection may influence the serum lipid profile (Laurila et al., 1999). Recently, a meta-analysis on the association between H. pylori infection and metabolic syndrome involving 27,544 participants showed that, compared to H. pylori negative groups, H. pylori positive groups were lower in HDL-C (MD = −2.43, 95% CI: −3.75 to −1.12, I2 = 92%) and higher in TG (MD = 8.12, 95% CI: 3.05–13.20, I2 = 71%; Upala et al., 2016). Nam et al. (2015) involving 13,383 participants also found that current infection with H. pylori with 50.5% at baseline increased LDL-C and decreased HDL-C than H. pylori negative group. A study involving in 679 participants, with UPLC-MS analysing the concentrations of molecular lipids, and proton magnetic resonance spectroscopy (1H-MRS) or liver biopsy measuring liver-fat, demonstrated that a serum-lipid signature comprising a lipid triplet (TG[16:0/18:0/18:1], phosphatidylcholine [PC][18:1/22:6], PC[O-24:1/20:4]) could estimate the percentage of liver fat. The significant associations of specific lipid and polar metabolite concentrations with liver-fat content suggested that circulating molecular lipids may be predictive of liver fat (Oresic et al., 2013). Hyperlipidemia is an established risk factor of NAFLD. A prospective, open-label, single-center study which consisted of 159 NAFLD patients, revealed that the HOMA-IR, TC, TG, LDL-C, and CRP levels were significantly higher and HDL-C levels were significantly lower in patients with H. pylori infection (P < 0.05), while HOMA-IR, TC, TG, LDL-C, and CRP levels in patients with successful eradication were significantly decreased compared to the pretreatment levels (P < 0.05). This study showed beneficial effects of H. pylori eradication on insulin resistance, atherogenic lipid abnormalities, and low-grade inflammation. These results suggest that H. pylori eradication may prevent metabolic syndrome including NAFLD (Gen et al., 2010). However, the mechanism by which this occurs is not yet clear.
Intestinal Permeability and Gut Microbiota
In recent years, as the maturity and widely applying of 16s rRNA gene sequencing technique, evidence linking dysbiosis to the pathogenesis of human liver disease has accumulated rapidly, with a primary focus on its role in NAFLD. Animal studies in which the gut microbiota are manipulated, and observational studies in patients with NAFLD, have provided considerable evidence that gut microbiota dysbiosis contributes to the pathogenesis of NAFLD (Turnbaugh et al., 2006; Le Roy et al., 2013; Mouzaki et al., 2013; Jiang et al., 2015; Leung et al., 2016). A retrospective study described a significant imbalance in intestinal flora in patients with NAFLD and intestinal inflammation. NAFLD significantly increased the permeability of the intestinal mucosa, and the author postulated that aside from dysbiosis of the gut microbiota, gut microbiota-mediated inflammation of the intestinal mucosa and the related impairment in mucosal immune function play an important role in the pathogenesis of NAFLD (Jiang et al., 2015). Another retrospective study from France enrolled 57 patients with biopsy-proven NAFLD also revealed that, Bacteroides abundance was significantly increased in NASH and F ≥ 2 patients, whereas Prevotella abundance was decreased, and Ruminococcus abundance was significantly higher in F ≥ 2 patients. By multivariate analysis, Bacteroides abundance was independently associated with NASH and Ruminococcus with F ≥ 2 fibrosis (Boursier et al., 2016). Dysbiosis increases gut permeability to bacterial products and increases hepatic exposure to injurious substances that increase hepatic inflammation and fibrosis.
Fukuda et al. (2001) evaluated the effect of H. pylori infection on the permeability of the intestine by oral sucrose tolerance test. The results showed that the existence of H. pylori itself was associated with increased intestinal permeability. Myllyluoma et al. (2007) observed the difference with regard to clostridia and the total number of anaerobic bacteria by detecting the composition of the microbiota between H. pylori-positive and H. pylori-negative individuals. Heimesaat et al. (2014) demonstrated that long-term infection of Mongolian gerbils with an H. pylori WT strain leads to distinct shifts of the microbiota composition in the distal uninflamed intestine. Khosravi et al. (2015) observed that H. pylori infection was related to gut microbiota using germ free (GF) and specific pathogen free (SPF) mice in the presence and absence of H. pylori. The results also indicated that ongoing crosstalk occurs between H. pylori and the normal gut microbiota, which is associated with metabolism and gut inflammation. Thus, some scholars speculate the mechanisms of the development of NAFLD caused by H. pylori related-gut microbiota dysbiosis are follows; H. pylori invasion into intestinal mucosa might increase gut permeability and gut microbiota disorder and then facilitate the passage of bacterial endotoxin (mainly LPS) via the portal vein to the liver, and promote inflammation response (Fukuda et al., 2001; Sumida et al., 2015).
Conclusion and Outlook
NAFLD is a complex disease that is affected by genetic and environmental factors. The incident rate is high, and most of the patients have a good prognosis, however, up to 25% probability develop to NASH, cirrhosis, or liver cancer. Thus, effective treatment regimen for prevention of this progress are extremely urgent. IR is considered to be an crucial part of NAFLD development, and many studies have confirmed that H. pylori infection may be a causal factor for IR. The mechanisms of H. pylori infection that contribute to NAFLD may include the following. H. pylori infection may cause chronic low-grade systemic inflammation, increasing the levels of inflammatory cytokines such as IL-6 and TNF-α, then activating IKK/NF-KB and leading to IR. H. pylori infection may also inhibit white adipose tissue to release leptin, and then promote liver stearoyl-CoA desaturase, thus accelerating VLDL-C and fatty deposits in the liver tissue. Due to the interaction of the stomach and the intestines, H. pylori infection may lead to gastrointestinal flora dysbiota, and increasing serum LPS, stimulating systemic inflammation and causing a decrease in lipoprotein activity followed by dyslipidemia (Figure 1). If we can understanding and confirming the pathogenic role of H. pylori infection in NAFLD, it will provide a new direction for NAFLD treatment strategies.
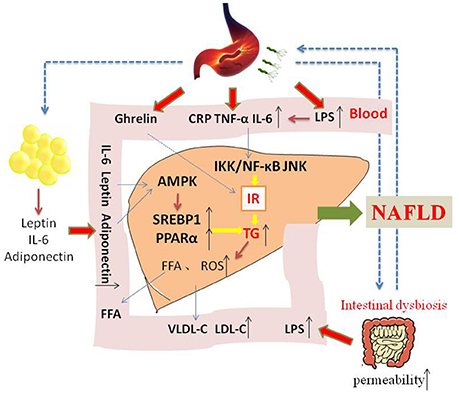
Figure 1. The possible mechanism of how H. pylori infection contribute to NAFLD. Insulin resistance is the central to the development of NAFLD. On the one hand, H. pylori infection may cause chronic low-grade systemic inflammation, increasing the levels of inflammatory cytokines such as IL-6 and TNF-α, which may influence insulin action and its level. On the other hand, H. pylori infection may also stimulate white adipose tissue to release leptin and adiponectin, activating AMPK and then upregulating SREBP1c and PPARα. In addition, other mechanism and mediators may be involved in the possible causative relationship between H. pylori infection and NAFLD.
Author Contributions
NL gives the direction of the paper's conception. DC writes the manuscript. CH helps to combing logic and HA, YH helps in modifying language.
Funding
Supported by The National Natural Science Foundation of China (Nos. 81270479, 81470832 and 81670507).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abenavoli, L., Milic, N., Masarone, M., and Persico, M. (2013). Association between non-alcoholic fatty liver disease, insulin resistance and Helicobacter pylori. Med. Hypotheses 81, 913–915. doi: 10.1016/j.mehy.2013.08.011
Aviles-Jimenez, F., Guitron, A., Segura-Lopez, F., Mendez-Tenorio, A., Iwai, S., Hernandez-Guerrero, A., et al. (2016). Microbiota studies in the bile duct strongly suggest a role for Helicobacter pylori in extrahepatic cholangiocarcinoma. Clin. Microbiol. Infect. 22, 178.e11–178.e22. doi: 10.1016/j.cmi.2015.10.008
Aydemir, S., Bayraktaroglu, T., Sert, M., Sokmen, C., Atmaca, H., Mungan, G., et al. (2005). The effect of Helicobacter pylori on insulin resistance. Dig. Dis. Sci. 50, 2090–2093. doi: 10.1007/s10620-005-3012-z
Baeg, M. K., Yoon, S. K., Ko, S. H., Noh, Y. S., Lee, I. S., and Choi, M. G. (2016). Helicobacter pylori infection is not associated with nonalcoholic fatty liver disease. World J. Gastroenterol. 22, 2592–2600. doi: 10.3748/wjg.v22.i8.2592
Bhala, N., Jouness, R. I., and Bugianesi, E. (2013). Epidemiology and natural history of patients with NAFLD. Curr. Pharm. Des. 19, 5169–5176. doi: 10.2174/13816128113199990336
Birkenfeld, A. L., and Shulman, G. I. (2014). Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 59, 713–723. doi: 10.1002/hep.26672
Boursier, J., Mueller, O., Barret, M., Machado, M., Fizanne, L., Araujo-Perez, F., et al. (2016). The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63, 764–775. doi: 10.1002/hep.28356
Buzzetti, E., Pinzani, M., and Tsochatzis, E. A. (2016). The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65, 1038–1048. doi: 10.1016/j.metabol.2015.12.012
Caldwell, S., and Argo, C. (2010). The natural history of non-alcoholic fatty liver disease. Dig. Dis. 28, 162–168. doi: 10.1159/000282081
Castera, L., Pedeboscq, A., Rocha, M., Le Bail, B., Asencio, C., De Ledinghen, V., et al. (2006). Relationship between the severity of hepatitis C virus-related liver disease and the presence of Helicobacter species in the liver: a prospective study. World J. Gastroenterol 12, 7278–7284. doi: 10.3748/wjg.v12.i45.7278
Chen, L. W., Chien, C. Y., Yang, K. J., Kuo, S. F., Chen, C. H., and Chien, R. N. (2015). Helicobacter pylori infection increases insulin resistance and metabolic syndrome in residents younger than 50 years old: a community-based study. PLoS ONE 10:e0128671. doi: 10.1371/journal.pone.0128671
Chitturi, S., Farrell, G., Frost, L., Kriketos, A., Lin, R., Fung, C., et al. (2002). Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: a manifestation of lipotoxicity? Hepatology 36, 403–409. doi: 10.1053/jhep.2002.34738
Cindoruk, M., Cirak, M. Y., Unal, S., Karakan, T., Erkan, G., Engin, D., et al. (2008). Identification of Helicobacter species by 16S rDNA PCR and sequence analysis in human liver samples from patients with various etiologies of benign liver diseases. Eur. J. Gastroenterol. Hepatol. 20, 33–36. doi: 10.1097/MEG.0b013e3282efa4f2
Dandona, P., Aljada, A., and Bandyopadhyay, A. (2004). Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. 25, 4–7. doi: 10.1016/j.it.2003.10.013
Day, C. P., and James, O. F. (1998). Steatohepatitis: a tale of two “hits”? Gastroenterology 114, 842–845. doi: 10.1016/S0016-5085(98)70599-2
De Magalhaes Queiroz, D. M., and Santos, A. (2001). Isolation of a Helicobacter strain from the human liver. Gastroenterology 121, 1023–1024. doi: 10.1053/gast.2001.28574
Ding, X., Saxena, N. K., Lin, S., Xu, A., Srinivasan, S., and Anania, F. A. (2005). The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am. J. Pathol. 166, 1655–1669. doi: 10.1016/S0002-9440(10)62476-5
Dogan, Z., Filik, L., Ergul, B., Sarikaya, M., and Akbal, E. (2013). Association between Helicobacter pylori and liver-to-spleen ratio: a randomized-controlled single-blind study. Eur. J. Gastroenterol. Hepatol. 25, 107–110. doi: 10.1097/MEG.0b013e3283590c10
Dogan, Z., Sarikaya, M., Ergul, B., and Filik, L. (2015). The effect of Helicobacter pylori eradication on insulin resistance and HbA1c level in people with normal glucose levels: a prospective study. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 159, 242–245. doi: 10.5507/bp.2014.036
Ford, A. C., and Axon, A. T. (2010). Epidemiology of Helicobacter pylori infection and public health implications. Helicobacter 15(Suppl. 1), 1–6. doi: 10.1111/j.1523-5378.2010.00779.x
Friedman, S. L. (2008). Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 88, 125–172. doi: 10.1152/physrev.00013.2007
Fukuda, Y., Bamba, H., Okui, M., Tamura, K., Tanida, N., Satomi, M., et al. (2001). Helicobacter pylori infection increases mucosal permeability of the stomach and intestine. Digestion 63(Suppl. 1), 93–96. doi: 10.1159/000051918
Gen, R., Demir, M., and Ataseven, H. (2010). Effect of Helicobacter pylori eradication on insulin resistance, serum lipids and low-grade inflammation. South Med. J. 103, 190–196. doi: 10.1097/SMJ.0b013e3181cf373f
Gunji, T., Matsuhashi, N., Sato, H., Fujibayashi, K., Okumura, M., Sasabe, N., et al. (2009). Helicobacter pylori infection significantly increases insulin resistance in the asymptomatic Japanese population. Helicobacter 14, 144–150. doi: 10.1111/j.1523-5378.2009.00705.x
Heimesaat, M. M., Fischer, A., Plickert, R., Wiedemann, T., Loddenkemper, C., Gobel, U. B., et al. (2014). Helicobacter pylori induced gastric immunopathology is associated with distinct microbiota changes in the large intestines of long-term infected Mongolian gerbils. PLoS ONE 9:e100362. doi: 10.1371/journal.pone.0100362
Hemmasi, G., Zamani, F., Khonsari, M., Sohrabi, M., Abdollahi, N., and Ajdarkosh, H. (2013). Association between Helicobacter pylori and serum leptin in iranian dyspeptic patients. Middle East J. Dig. Dis. 5, 158–162.
Hennige, A. M., Stefan, N., Kapp, K., Lehmann, R., Weigert, C., Beck, A., et al. (2006). Leptin down-regulates insulin action through phosphorylation of serine-318 in insulin receptor substrate 1. FASEB J. 20, 1206–1208. doi: 10.1096/fj.05-4635fje
Hossain, I. A., Akter, S., Bhuiyan, F. R., Shah, M. R., Rahman, M. K., and Ali, L. (2016). Subclinical inflammation in relation to insulin resistance in prediabetic subjects with nonalcoholic fatty liver disease. BMC Res. Notes 9:266. doi: 10.1186/s13104-016-2071-x
Hotamisligil, G. S., Peraldi, P., Budavari, A., Ellis, R., White, M. F., and Spiegelman, B. M. (1996). IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271, 665–668. doi: 10.1126/science.271.5249.665
Huang, Y., Tian, X. F., Fan, X. G., Fu, C. Y., and Zhu, C. (2009). The pathological effect of Helicobacter pylori infection on liver tissues in mice. Clin. Microbiol. Infect. 15, 843–849. doi: 10.1111/j.1469-0691.2009.02719.x
Jamali, R., Mofid, A., Vahedi, H., Farzaneh, R., and Dowlatshahi, S. (2013). The effect of Helicobacter pylori eradication on liver fat content in subjects with non-alcoholic Fatty liver disease: a randomized open-label clinical trial. Hepat. Mon. 13:e14679. doi: 10.5812/hepatmon.14679
Jiang, W., Wu, N., Wang, X., Chi, Y., Zhang, Y., Qiu, X., et al. (2015). Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 5:8096. doi: 10.1038/srep08096
Keane, K. N., Cruzat, V. F., Carlessi, R., De Bittencourt, P. I. Jr., and Newsholme, P. (2015). Molecular events linking oxidative stress and inflammation to insulin resistance and beta-cell dysfunction. Oxid. Med. Cell. Longev. 2015:181643. doi: 10.1155/2015/181643
Khosravi, Y., Seow, S. W., Amoyo, A. A., Chiow, K. H., Tan, T. L., Wong, W. Y., et al. (2015). Helicobacter pylori infection can affect energy modulating hormones and body weight in germ free mice. Sci. Rep. 5:8731. doi: 10.1038/srep08731
Kotronen, A., Juurinen, L., Tiikkainen, M., Vehkavaara, S., and Yki-Jarvinen, H. (2008). Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 135, 122–130. doi: 10.1053/j.gastro.2008.03.021
Kotronen, A., Vehkavaara, S., Seppala-Lindroos, A., Bergholm, R., and Yki-Jarvinen, H. (2007). Effect of liver fat on insulin clearance. Am. J. Physiol. Endocrinol. Metab. 293, E1709–E1715. doi: 10.1152/ajpendo.00444.2007
Kountouras, J., Polyzos, S. A., Zavos, C., Deretzi, G., Kountouras, C., Vardaka, E., et al. (2014). Helicobacter pylori might contribute to nonalcoholic fatty liver disease-related cardiovascular events by releasing prothrombotic and proinflammatory factors. Hepatology 60, 1450–1451. doi: 10.1002/hep.27168
La Cava, A., and Matarese, G. (2004). The weight of leptin in immunity. Nat. Rev. Immunol. 4, 371–379. doi: 10.1038/nri1350
Laurila, A., Bloigu, A., Nayha, S., Hassi, J., Leinonen, M., and Saikku, P. (1999). Association of Helicobacter pylori infection with elevated serum lipids. Atherosclerosis 142, 207–210. doi: 10.1016/S0021-9150(98)00194-4
Le Roy, T., Llopis, M., Lepage, P., Bruneau, A., Rabot, S., Bevilacqua, C., et al. (2013). Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 62, 1787–1794. doi: 10.1136/gutjnl-2012-303816
Leung, C., Rivera, L., Furness, J. B., and Angus, P. W. (2016). The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 13, 412–425. doi: 10.1038/nrgastro.2016.85
Li, L., He, Q., Huang, X., Man, Y., Zhou, Y., Wang, S., et al. (2010). NOX3-derived reactive oxygen species promote TNF-alpha-induced reductions in hepatocyte glycogen levels via a JNK pathway. FEBS Lett. 584, 995–1000. doi: 10.1016/j.febslet.2010.01.044
Liu, Q., Bengmark, S., and Qu, S. (2010). The role of hepatic fat accumulation in pathogenesis of non-alcoholic fatty liver disease (NAFLD). Lipids Health Dis. 9:42. doi: 10.1186/1476-511X-9-42
Maleki, I., Rastgar, A., Hosseini, V., Taghvaei, T., Rafiei, A., Barzin, M., et al. (2014). High sensitive CRP and pentraxine 3 as noninvasive biomarkers of nonalcoholic fatty liver disease. Eur. Rev. Med. Pharmacol. Sci. 18, 1583–1590.
Mandeville, K. L., Krabshuis, J., Ladep, N. G., Mulder, C. J., Quigley, E. M., and Khan, S. A. (2009). Gastroenterology in developing countries: issues and advances. World J. Gastroenterol. 15, 2839–2854. doi: 10.3748/wjg.15.2839
Marshall, B., and Warren, J. (1983). Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1, 1273–1275.
Masarone, M., Federico, A., Abenavoli, L., Loguercio, C., and Persico, M. (2014). Non alcoholic fatty liver: epidemiology and natural history. Rev. Recent Clin. Trials 9, 126–133. doi: 10.2174/1574887109666141216111143
Mouzaki, M., Comelli, E. M., Arendt, B. M., Bonengel, J., Fung, S. K., Fischer, S. E., et al. (2013). Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 58, 120–127. doi: 10.1002/hep.26319
Myllyluoma, E., Ahlroos, T., Veijola, L., Rautelin, H., Tynkkynen, S., and Korpela, R. (2007). Effects of anti-Helicobacter pylori treatment and probiotic supplementation on intestinal microbiota. Int. J. Antimicrob. Agents 29, 66–72. doi: 10.1016/j.ijantimicag.2006.08.034
Nam, S. Y., Ryu, K. H., Park, B. J., and Park, S. (2015). Effects of Helicobacter pylori infection and its eradication on lipid profiles and cardiovascular diseases. Helicobacter 20, 125–132. doi: 10.1111/hel.12182
Nasif, W. A., Mukhtar, M. H., Nour Eldein, M. M., and Ashgar, S. S. (2016). Oxidative DNA damage and oxidized low density lipoprotein in Type II diabetes mellitus among patients with Helicobacter pylori infection. Diabetol. Metab. Syndr. 8:34. doi: 10.1186/s13098-016-0149-1
Neuschwander-Tetri, B. A., and Caldwell, S. H. (2003). Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology 37, 1202–1219. doi: 10.1053/jhep.2003.50193
Nishi, Y., Isomoto, H., Uotani, S., Wen, C. Y., Shikuwa, S., Ohnita, K., et al. (2005). Enhanced production of leptin in gastric fundic mucosa with Helicobacter pylori infection. World J. Gastroenterol. 11, 695–699. doi: 10.3748/wjg.v11.i5.695
Okushin, K., Takahashi, Y., Yamamichi, N., Shimamoto, T., Enooku, K., Fujinaga, H., et al. (2015). Helicobacter pylori infection is not associated with fatty liver disease including non-alcoholic fatty liver disease: a large-scale cross-sectional study in Japan. BMC Gastroenterol. 15:25. doi: 10.1186/s12876-015-0247-9
Oresic, M., Hyotylainen, T., Kotronen, A., Gopalacharyulu, P., Nygren, H., Arola, J., et al. (2013). Prediction of non-alcoholic fatty-liver disease and liver fat content by serum molecular lipids. Diabetologia 56, 2266–2274. doi: 10.1007/s00125-013-2981-2
Pellicano, R., Menard, A., Rizzetto, M., and Megraud, F. (2008). Helicobacter species and liver diseases: association or causation? Lancet Infect. Dis. 8, 254–260. doi: 10.1016/S1473-3099(08)70066-5
Peverill, W., Powell, L. W., and Skoien, R. (2014). Evolving concepts in the pathogenesis of NASH: beyond steatosis and inflammation. Int. J. Mol. Sci. 15, 8591–8638. doi: 10.3390/ijms15058591
Pirouz, T., Zounubi, L., Keivani, H., Rakhshani, N., and Hormazdi, M. (2009). Detection of Helicobacter pylori in paraffin-embedded specimens from patients with chronic liver diseases, using the amplification method. Dig. Dis. Sci. 54, 1456–1459. doi: 10.1007/s10620-008-0522-5
Polyzos, S. A., Aronis, K. N., Kountouras, J., Raptis, D. D., Vasiloglou, M. F., and Mantzoros, C. S. (2016). Circulating leptin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Diabetologia 59, 30–43. doi: 10.1007/s00125-015-3769-3
Polyzos, S. A., Kountouras, J., Papatheodorou, A., Patsiaoura, K., Katsiki, E., Zafeiriadou, E., et al. (2013a). Helicobacter pylori infection in patients with nonalcoholic fatty liver disease. Metabolism 62, 121–126. doi: 10.1016/j.metabol.2012.06.007
Polyzos, S. A., Kountouras, J., Zavos, C., and Deretzi, G. (2011). The association between Helicobacter pylori infection and insulin resistance: a systematic review. Helicobacter 16, 79–88. doi: 10.1111/j.1523-5378.2011.00822.x
Polyzos, S. A., Kountouras, J., Zavos, C., and Deretzi, G. (2013b). Helicobacter pylori Infection and insulin resistance. Helicobacter 18, 165–166. doi: 10.1111/hel.12019
Polyzos, S. A., Nikolopoulos, P., Stogianni, A., Romiopoulos, I., Katsinelos, P., and Kountouras, J. (2014). Effect of Helicobacter pylori eradication on hepatic steatosis, NAFLD fibrosis score and HSENSI in patients with nonalcoholic steatohepatitis: a MR imaging-based pilot open-label study. Arq. Gastroenterol. 51, 261–268. doi: 10.1590/S0004-28032014000300017
Shoelson, S. E., Lee, J., and Goldfine, A. B. (2006). Inflammation and insulin resistance. J. Clin. Invest. 116, 1793–1801. doi: 10.1172/JCI29069
Silha, J. V., Nyomba, B. L., Leslie, W. D., and Murphy, L. J. (2007). Ethnicity, insulin resistance, and inflammatory adipokines in women at high and low risk for vascular disease. Diabetes Care 30, 286–291. doi: 10.2337/dc06-1073
Sumida, Y., Kanemasa, K., Imai, S., Mori, K., Tanaka, S., Shimokobe, H., et al. (2015). Helicobacter pylori infection might have a potential role in hepatocyte ballooning in nonalcoholic fatty liver disease. J. Gastroenterol. 50, 996–1004. doi: 10.1007/s00535-015-1039-2
Takaki, A., Kawai, D., and Yamamoto, K. (2013). Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 14, 20704–20728. doi: 10.3390/ijms141020704
Takuma, Y. (2011). Helicobacter pylori infection and liver diseases. Gan To Kagaku Ryoho 38, 362–364.
Tiniakos, D. G. (2010). Nonalcoholic fatty liver disease/nonalcoholic steatohepatitis: histological diagnostic criteria and scoring systems. Eur. J. Gastroenterol. Hepatol. 22, 643–650. doi: 10.1097/MEG.0b013e32832ca0cb
Tiwari, S. K., Khan, A. A., Ibrahim, M., Habeeb, M. A., and Habibullah, C. M. (2006). Helicobacter pylori and other Helicobacter species DNA in human bile samples from patients with various hepato-biliary diseases. World J. Gastroenterol. 12, 2181–2186. doi: 10.3748/wjg.v12.i14.2181
Tsai, C. C., Kuo, T. Y., Hong, Z. W., Yeh, Y. C., Shih, K. S., Du, S. Y., et al. (2015). Helicobacter pylori neutrophil-activating protein induces release of histamine and interleukin-6 through G protein-mediated MAPKs and PI3K/Akt pathways in HMC-1 cells. Virulence 6, 755–765. doi: 10.1080/21505594.2015.1043505
Turnbaugh, P. J., Ley, R. E., Mahowald, M. A., Magrini, V., Mardis, E. R., and Gordon, J. I. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031. doi: 10.1038/nature05414
Upala, S., Jaruvongvanich, V., Riangwiwat, T., Jaruvongvanich, S., and Sanguankeo, A. (2016). Association between Helicobacter pylori infection and metabolic syndrome: a systematic review and meta-analysis. J. Dig. Dis. 17, 433–440. doi: 10.1111/1751-2980.12367
Vergun, O., and Reynolds, I. J. (2005). Distinct characteristics of Ca(2+)-induced depolarization of isolated brain and liver mitochondria. Biochim. Biophys. Acta 1709, 127–137. doi: 10.1016/j.bbabio.2005.07.006
Wang, Y., Li, Y. Y., Nie, Y. Q., Zhou, Y. J., Cao, C. Y., and Xu, L. (2013). Association between metabolic syndrome and the development of non-alcoholic fatty liver disease. Exp. Ther. Med. 6, 77–84. doi: 10.3892/etm.2013.1090
Wong, F., Rayner-Hartley, E., and Byrne, M. F. (2014). Extraintestinal manifestations of Helicobacter pylori: a concise review. World J. Gastroenterol. 20, 11950–11961. doi: 10.3748/wjg.v20.i34.11950
Yildirim, Z., Bozkurt, B., Ozol, D., Armutcu, F., Akgedik, R., Karamanli, H., et al. (2016). Increased exhaled 8-isoprostane and interleukin-6 in patients with Helicobacter pylori infection. Helicobacter. 21, 389–394. doi: 10.1111/hel.12302
Zhang, C., Guo, L., Qin, Y., and Li, G. (2016). Correlation between Helicobacter pylori infection and polymorphism of adiponectin gene promoter-11391G/A, superoxide dismutase gene innonalcoholic fatty liver disease. Zhong Nan Da Xue Xue Bao Yi Xue Ban 41, 359–366. doi: 10.11817/j.issn.1672-7347.2016.04.004
Keywords: H. pylori, NAFLD, insulin resistance, lipid profile, gut microbiota
Citation: Cheng D-d, He C, Ai H-h, Huang Y and Lu N-h (2017) The Possible Role of Helicobacter pylori Infection in Non-alcoholic Fatty Liver Disease. Front. Microbiol. 8:743. doi: 10.3389/fmicb.2017.00743
Received: 03 September 2016; Accepted: 10 April 2017;
Published: 10 May 2017.
Edited by:
Awdhesh Kalia, University of Texas MD Anderson Cancer Center, USAReviewed by:
Haider Abdul-Lateef Mousa, University of Basrah, IraqXinhua Qu, Shanghai Ninth People's Hospital, China
Copyright © 2017 Cheng, He, Ai, Huang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nong-hua Lu, lunonghua@ncu.edu.cn