 Horatio H. Morgan
Horatio H. Morgan Maret du Toit
Maret du Toit Mathabatha E. Setati
Mathabatha E. Setati- Department of Viticulture and Oenology, Institute for Wine Biotechnology, Stellenbosch University, Stellenbosch, South Africa
From the time when microbial activity in wine fermentation was first demonstrated, the microbial ecology of the vineyard, grape, and wine has been extensively investigated using culture-based methods. However, the last 2 decades have been characterized by an important change in the approaches used for microbial examination, due to the introduction of DNA-based community fingerprinting methods such as DGGE, SSCP, T-RFLP, and ARISA. These approaches allowed for the exploration of microbial community structures without the need to cultivate, and have been extensively applied to decipher the microbial populations associated with the grapevine as well as the microbial dynamics throughout grape berry ripening and wine fermentation. These techniques are well-established for the rapid more sensitive profiling of microbial communities; however, they often do not provide direct taxonomic information and possess limited ability to detect the presence of rare taxa and taxa with low abundance. Consequently, the past 5 years have seen an upsurge in the application of high-throughput sequencing methods for the in-depth assessment of the grapevine and wine microbiome. Although a relatively new approach in wine sciences, these methods reveal a considerably greater diversity than previously reported, and identified several species that had not yet been reported. The aim of the current review is to highlight the contribution of high-throughput next generation sequencing and metagenomics approaches to vineyard microbial ecology especially unraveling the influence of vineyard management practices on microbial diversity.
Introduction
The conversion of grape juice into wine was first confirmed to be the result of a microbial process by Louis Pasteur in the middle of the nineteenth-century (Barnett, 2003; Jolly et al., 2014; Bokulich et al., 2016b). Since then, the diversity of the vineyard, grape and wine microbiota has been extensively investigated using traditional microbiological methods involving microscopy, cultivation on different agar media and biochemical characteristics. However, the arrival of DNA-based molecular techniques such as polymerase chain reaction (PCR) and the identification of evolutionarily stable molecular marker genes such as ribosomal RNA (rRNA) genes improved our ability to identify microbial species with better resolution and reliability (Justé et al., 2008; Solieri and Giudici, 2008; Cocolin et al., 2013; Sun and Liu, 2014; Wang et al., 2014; Abbasian et al., 2015b). The bacterial small subunit ribosomal RNA gene (16S rRNA) as well as the fungal ITS1-5.8S rRNA-ITS2 gene have been recognized as the gold standard for estimating the phylogenetic diversity in microbial communities (Justé et al., 2008; Cocolin et al., 2013; Sun and Liu, 2014). Consequently, for the past 3 decades, molecular techniques relying on rRNA genes as target molecules, have been employed in conjunction with culture-dependent methodologies to identify microorganisms after isolation and growth in pure cultures (Esteve-Zarzoso, 1999; Alessandria et al., 2013; Cocolin et al., 2013). To date more than 40 yeast species (Jolly et al., 2014), 50 bacterial species (Barata et al., 2012) and ~70 genera of filamentous fungi (Rousseaux et al., 2014) associated with grapevine and wine fermentation processes have been isolated and identified using traditional culture-based methods. These methods are however extremely laborious, time consuming and often inconsistent and biased (Andorrà et al., 2008; Sun and Liu, 2014). In addition, only species that are able to grow on the culture media and under the cultivation conditions used can be isolated and identified, while species that are in low abundance, those species for which the prevailing cultivation conditions are not conducive, as well as viable but non-culturable (VBNC) cells, are often overlooked (Abbasian et al., 2015b). These limitations in culture-based methods as well as the difference between culturable and in situ diversity increased the importance for research into culture-independent molecular approaches (Nocker et al., 2007). Nevertheless, these methods remain important since the microbial species and strains retrieved in such culture-based approaches can be further exploited depending on their biochemical or genetic profiles. Indeed, the wine industry today has access to more than 100 commercial active dry yeast (ADY) strains of Saccharomyces cerevisiae that are used as starter cultures for controlled fermentations (Fernández-Espinar et al., 2001; Guzzon et al., 2014). More recently, strains of non-Saccharomyces yeasts such as Torulaspora delbrueckii, Metschnikowia pulcherrima, Lachancea thermotolerans, and Pichia kluyveri, and several others have been made available as pure starter cultures and in blends with S. cerevisiae (Lu et al., 2016; Padilla et al., 2016).
Introduction of PCR-based methods further created opportunities for the development and improvement of several techniques in molecular ecology. The application of molecular techniques allowed researchers to study microbes not on the basis of their ability to grow on certain media types but rather relied on the presence nucleic acids for detection and identification. Such methods, mostly use DNA extracted directly from the environment as a template for PCR, followed by separation and detection for microbial community profiling. Culture-independent methods are often faster, more sensitive and have a higher accuracy than culture-dependent methods (Justé et al., 2008; Lv et al., 2013). These methods include, single-strand conformational polymorphisms (SSCP), denaturing gradient gel electrophoresis (DGGE), terminal restriction fragment length polymorphisms (T-RFLP), and automated ribosomal intergenic spacer analysis (ARISA; Justé et al., 2008; Kovacs et al., 2010; Slabbert et al., 2010; Balázs et al., 2013; Cocolin et al., 2013; Abbasian et al., 2015b). PCR-DGGE was first applied in wine fermentation by Cocolin et al. (2001) to monitor the diversity and dynamics of yeast populations. Since then, it has remained the most widely used community profiling method in wine fermentation, also including bacteria (Renouf et al., 2006a,b; Cameron et al., 2013). The technique is often employed in combination with culture-dependent methods and has allowed researchers to decipher the complexity and evolution of the microbial population, during berry ripening and throughout the fermentation process (Prakitchaiwattana et al., 2004; Renouf et al., 2005, 2007; Di Maro et al., 2007; Andorrà et al., 2008). Although PCR-DGGE is typically thought to be appropriate for the analysis of less species-rich environments such as grape must, it has low sensitivity (Andorrà et al., 2010) and is unable to detect populations that are present at a relative abundance of <1% of the population (Fasoli et al., 2003; Andorrà et al., 2008). More recently, SSCP (Grube et al., 2011; Schmid et al., 2011; Martins et al., 2014), T-RFLP (Martins et al., 2012; Sun and Liu, 2014), and ARISA (Brežná et al., 2010; Chovanová et al., 2011; Kraková et al., 2012; Pancher et al., 2012; Setati et al., 2012; Ženišová et al., 2014; Ghosh et al., 2015) have been employed to profile the wine microbial diversity. Culture-independent methods also allow researchers to monitor populations that are numerically under-represented as well as those in the VBNC state (Andorrà et al., 2010; Cocolin et al., 2013). It is critical to monitor such populations as they can influence wine quality. For instance, several studies have demonstrated that strains of S. cerevisiae, Zygosaccharomyces baillii, and Brettanomyces bruxellensis when exposed to SO2 can enter into VBNC state and survive for more than a month depending on the pH of the environment (Divol and Lonvaud-Funel, 2005; Salma et al., 2013; Capozzi et al., 2016). During this state a spoilage yeast such as B. bruxellensis can produce volatile phenols that impart off-odors to the final wine thus rendering it unpalatable (Salma et al., 2013; Capozzi et al., 2016). Although the culture-independent methods have allowed researchers to detect and monitor the evolution of microbial communities, and capture species that were previously not detected, or even misrepresented with culture-dependent methods (Peršoh, 2015), they do have several limitations associated with each of the methods (Table 1). Such limitations, e.g., poor band-resolution, co-migration of species, and PCR amplification biases mean that diversity analysis based on these methods still provides a narrow view of the community composition.
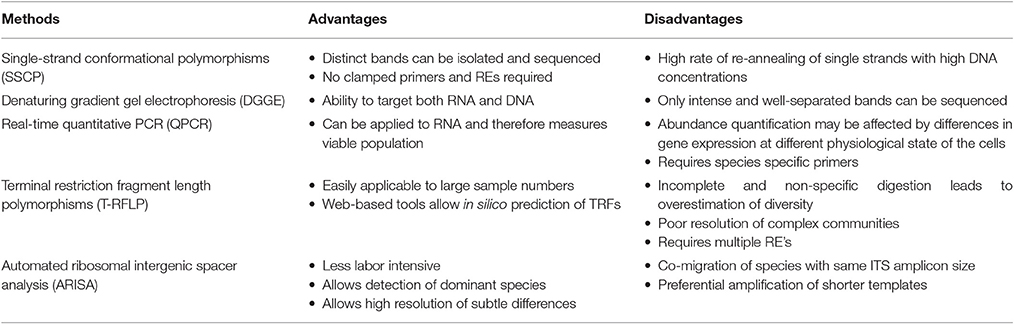
Table 1. A summary of the advantages and disadvantages of PCR-based culture-independent microbial community fingerprinting methods (Arteau et al., 2010; Cocolin et al., 2013).
Improvements in DNA sequencing, expanded the ability of researchers to study the microbial community structure and function with a higher resolution by employing metagenomic approaches. Metagenomics can be defined as the direct genetic analysis of the collective of genomes within an environmental sample (Thomas et al., 2012), this can be achieved either through whole metagenome sequencing or amplicon-based sequencing. Amplicon sequencing, often grouped under the umbrella of metagenomics, is a culture-independent approach for taxonomic, phylogenetic, or functional profiling of microbial communities, accomplished by sequencing specific marker genes amplified directly from environmental DNA without prior enrichment or cultivation of the target population (Franzosa et al., 2015). The innovations in high-throughput, short-amplicon sequencing are revolutionary in a way that they can describe the microbial diversity within and across complex biomes (Bokulich et al., 2013b). Although high-throughput sequencing technologies have been widely used to investigate the microbial ecology of various environments (Ma et al., 2015; Shi et al., 2015; Abbasian et al., 2015a), their application in grapevine and wine fermentation microbial ecology is relatively recent, and their contribution to the field has not been explored. In recent studies it was also shown that grape microbial diversity is driven by cultivar, climatic conditions both macro- and micro-climate, the seasonal environmental conditions, viticultural farming practices as well as wine microbiome by fermentation process applied during the winemaking (Bokulich et al., 2014; David et al., 2014; Gilbert et al., 2014; Setati et al., 2015; Zarraonaindia et al., 2015; Abbasian et al., 2015a; De Filippis et al., 2017). Therefore, with this review, we aim to provide an in-depth overview of the vineyard, grape, and wine microbiome and its functional potential as unraveled through high-throughput sequencing techniques.
Next-Generation Sequencing
For many years, microbial community analyses relied on the isolation and identification of individual species, or cloning and sequencing of rRNA genes retrieved by PCR from environmental DNA. These methods mainly relied on first-generation DNA sequencing technology which was developed by Sanger et al. (1977). A few decades later, deep high-throughput, in-parallel sequencing technologies collectively referred to as Next-generation sequencing (NGS) were developed (Bleidorn, 2015). The term NGS therefore specifically refers to non-Sanger-based second and third generation sequencing (TGS) techniques (Türktaş et al., 2015).
After Sanger introduced the chain-terminator DNA sequencing method, commercial second generation sequencing (SGS) platforms were developed. The Genome Sequencer 20 system launched in 2005 by 454 Life Sciences, was the first commercial SGS platform and was soon followed by the Genome Analyzer II launched by Solexa/Illumina in 2006. Both these platforms use a sequencing by synthesis approach. Roughly 2 years later, Lifetechnologies/Applied Biosystems introduced the SOLiD (Sequencing by Oligonucleotide Ligation and Detection) platform which applies fluorophore labeled oligonucleotide panels and ligation chemistry for sequencing. Subsequently, Complete Genomics developed the CGA sequencing technology which employed semi-ordered array of “DNA nanoballs” on a solid surface, while the Ion Torrent, which is regarded as the first of the “post-light sequencing” technologies, was introduced in 2010 (Reuter et al., 2015; Heather and Chain, 2016). The Ion Torrent's semiconductor sequencer is thought to be a technology between second and TGS categories. The technology is capable of sequencing single molecules thus negating the requirement for prior DNA amplification (Heather and Chain, 2016).
The majority of SGS technologies however, still have various limitations, such as errors arising from PCR (Peršoh, 2015), the loss of synchronicity “dephasing” (Schadt et al., 2010; Diaz-Sanchez et al., 2013) and the duration of completion “time to results” (Diaz-Sanchez et al., 2013). To overcome these drawbacks TGS or next-next generation platforms such as Single-molecule real-time (SMRT) sequencing (Schadt et al., 2010; Bleidorn, 2015) and Nanopore DNA Sequencer (Diaz-Sanchez et al., 2013), which open the possibility for single molecule sequencing were developed. These come with several advantages, (i) higher throughput, (ii) faster “time-to-result,” (iii) low cost, (iv) longer read length, (v) increased consensus accuracy enabling rare variant detection and (vi) small starting material (Schadt et al., 2010; Diaz-Sanchez et al., 2013; Bleidorn, 2015). However, these sequencing methodologies are still in development, and/or in the beta stage. Few commercial platforms have been evaluated, however they remain plagued by high error rates, and low output, although the technology is promising (Bleidorn, 2015). As such they cannot yet replace SGS, which remain and continue to be pivotal in microbial ecology surveys.
Next-Generation Sequencing in Microbial Ecology
SGS platforms have revolutionized the landscape of microbial ecology and have been the cornerstone of many phylogenetic surveys. The methods make it possible to compare and analyze the whole microbial community diversity, abundance, and functional genes at far greater sequencing depths. These technologies depend on a parallel process in which each single DNA fragment is sequenced independently and separated in clonal amplicons for downstream analysis between the total sequences generated (Wooley et al., 2010; Diaz-Sanchez et al., 2013). With most SGS methodologies, an uninterrupted operation of a washing and scanning process is used to read tens of thousands of matching strands that are fixed to a specific location (Schadt et al., 2010). The length of the fragments obtained from the analyses differs depending on the sequencing method employed (Wooley et al., 2010; Bokulich et al., 2016b). Until recently, the Illumina and 454 pyrosequencing platforms were the most commonly used platforms for grapevine ecology surveys. At least 48% of the published data on the vineyard, grapevine and wine microbiome is derived from 454 pyrosequencing while the remaining 52% is derived from Illumina sequencing. Both platforms work on a sequencing-by-synthesis approach, however differ in their chemistries.
Illumina
The process of Illumina sequencing, consists of the bridge amplification of adapter-ligated DNA fragments on the surface of a glass (Pettersson et al., 2009). Bases are then determined using a cyclic reversible termination technique, which sequences the template strand, a single nucleotide at a time through progressive rounds of base incorporation, washing, scanning, and cleaning. In this method, labeled dNTPs are used to stop the polymerization reaction, allowing the removal of unincorporated bases. The fluorescent dye is captured to identify the bases added, and then cleaved so that the next nucleotide can be added, this is then repeated (Pettersson et al., 2009; Diaz-Sanchez et al., 2013; Reuter et al., 2015; Heather and Chain, 2016). The earlier Illumina analyser generated at least 1 Gb of sequences with reads averaging 35 bp and the duration of 2–3 days. However, the introduction of HiSeq and MiSeq machines altered the duration time to ~4 days and 24–30 h, and increased the read length to 250–300 bp, respectively with error rates of below 1%, with substitution the most occurring issue (Bleidorn, 2015; Goodwin et al., 2016).
Pyrosequencing
In 454 pyrosequencing an emulsion PCR is used for bridge amplification of adapter-ligated DNA fragments on the surface of a bead. The beads are thereafter distributed and fixed into 44 μm wells, where the sequencing by synthesis occurs. After the nucleotide bases are incorporated an enzymatic luciferase coupled reaction occurs, allowing for the identification of bases, which is measured using a charged couple device (Pettersson et al., 2009; Diaz-Sanchez et al., 2013; Reuter et al., 2015; Heather and Chain, 2016). The Roche 454 FLX platform has the ability to generate 80–120 Mb of sequences averaging in 200–300 bp reads, for a run that averages ~4 h with an error rate of below 1% (Morozova and Marra, 2008), while the FLX titanium is capable of producing read lengths of over 400 bp (Pettersson et al., 2009).
The 454 pyrosequencing technique was reported in 2008, as the most published NGS platform, however, the technology has since been discontinued, and has therefore been surpassed by Illumina which is currently considered to have made the largest contribution to SGS (Huse et al., 2007; Morozova and Marra, 2008; Reuter et al., 2015; Heather and Chain, 2016).
Application of Next-Generation Sequencing in Deciphering the Vineyard Microbiome
The vineyard microbiome broadly describes the collective genomes of microorganisms present in the vineyard ecosystem, including those present in soil, grapevine, cover crops, and the insects associated with the plants. Furthermore, microbial transfer from nearby plants, could be transported aerially or via insects (Gilbert et al., 2014). Consequently, the grape microbiome represents a reservoir of microorganisms comprising filamentous fungi, yeast as well as bacteria. These populations are however variable and are influenced by various external factors, such as grape cultivar, climatic conditions, farming practices, and the vineyard location (Setati et al., 2012; Salvetti et al., 2016). The past decade has seen a significant advancement in the manner in which researchers understand the microbial ecology of the vineyard, due to molecular profiling techniques that have further evolved, to explore microbial ecosystems (Bokulich et al., 2012). Recent studies have employed SGS to decipher the grape and grapevine associated microbiome (David et al., 2014; Pinto et al., 2014), and to determine how viticultural practices could potentially influence these communities (Setati et al., 2015; Kecskeméti et al., 2016; Marzano et al., 2016), their dynamics throughout grape berry development and wine fermentation (Piao et al., 2015; Stefanini et al., 2016) and to unravel their functional potential (Salvetti et al., 2016).
For the comprehensive evaluation of the vineyard and the grape microbiome, two key questions are typically addressed. Firstly, which microorganisms are present within the environment, and secondly the role of the individual species (Ravin et al., 2015). To understand what role the identified species, if any; plays in the grape and wine microbiome requires that standard microbiological methods be applied to isolate the strains and then evaluate them for their potential contribution to grape or wine quality by assessing their phenotypic and genotypic properties and thereafter they will be evaluated in different wine matrices to assess their growth and metabolic profile. To this effect, several species retrieved using culture-dependent methods have been shown to contribute positively in the winemaking process. For instance, some strains of Wickerhamomyces anomalus, Candida pyralidae, T. delbrueckii, and Kluyveromyces wickerhamii were shown to suppress the growth of B. bruxellensis (Comitini et al., 2017), a wine spoilage yeast; M. pulcherrima was highlighted as a desirable co-inoculant for the reduction of ethanol (Morales et al., 2015), while others such as Hanseniaspora vineae, Starmerella bacillaris, L. thermotolerans, P. kluyveri, and T. delbrueckii present various desirable aroma signatures (Jolly et al., 2014; Comitini et al., 2017). In order to explore the untapped diversity revealed by SGS, it would be important to establish enrichment methods that can allow retrieval of those species that have not yet been characterized. Consequently, different sampling strategies are employed depending on what question the researcher seek to address.
Sampling Strategies
The vineyard and grapevine microbiome has been studied from a variety of samples including the soil and different parts of the vines. However, there is currently no standardized sampling strategy or experimental design for vineyard microbiome analysis. For the soil microbiome samples are typically derived from surface soil or from the root zone. Typically, anything from 3 to 5 samples are randomly collected, sifted through a 0–2 mm sieve and then homogenized and composited. Samples are often collected with a spade or with the aid of a 33 inch by 7–8-inch corer, within the alleyways of the vineyard or at a distance of 15–30 cm away from the trunk, at a depth of 0–7 cm (Martins et al., 2014; Burns et al., 2015; Zarraonaindia et al., 2015). In contrast, root soil samples are collected closer to the stem (10–15 cm) although at similar depth to the surface samples (Zarraonaindia et al., 2015). For microbial evaluation of plant material such as roots and branches (Campisano et al., 2014), grapevines of similar age and size are typically chosen, eliminating one source of microbial variability. Only a certain area of the vine is sampled, the material typically peeled or crushed under aseptic conditions for further evaluation. For instance, some studies have used leaves (Leveau and Tech, 2011; Pinto et al., 2014) while others have used the cane, graft union of the trunk as well as the roots (Faist et al., 2016), depending on the aim of the study. In contrast, sampling for analysis of the grape-associated microbiome can vary from a few bunches to kilograms of grapes (David et al., 2014; Taylor et al., 2014; Pinto et al., 2015; Setati et al., 2015; Wang et al., 2015; Salvetti et al., 2016). Careful selection of healthy and undamaged grapes is often critical unless the aim is to investigate botrytized wines (Bokulich et al., 2012) and/or sweet wines (Stefanini et al., 2016). The grapes are subsequently crushed under asceptic conditions and the DNA extracted from the resulting must. In a few cases, samples were collected from commercial wineries as composite grape must (Bokulich et al., 2014, 2016a). In a few studies that monitored population dynamics during fermentation, additional samples are withdrawn at various time points representing the beginning, middle, and end of fermentation (David et al., 2014; Pinto et al., 2015; Wang et al., 2015). In most instances, sample volumes ranging from 5 to 50 mL are then further used for DNA extractions.
Target Genes
The target marker genes are universally present in all species evaluated and contain both highly conserved fragments that facilitate the design of PCR primers targeting all members of a community and variable regions that allow for the discrimination of different species within the community (Justé et al., 2008; Cocolin et al., 2013; Sun and Liu, 2014; Wang et al., 2014). In both fungi and bacteria, ribosomal RNA genes are suitable target genes. In bacteria, the 16S rRNA is typically targeted while in fungi the ITS1-5.8S rRNA-ITS2 as well as the 26S rRNA are the target molecules for high throughput amplicon sequencing and microbiome analyses.
The 9 hypervariable regions (V1–V9) of bacteria have all been targeted for the estimation of vineyard bacterial diversity (Leveau and Tech, 2011; Campisano et al., 2014; Perazzolli et al., 2014; Bokulich et al., 2015, 2016a; Burns et al., 2015; Calleja-Cervantes et al., 2015; Piao et al., 2015; Pinto et al., 2015; Zarraonaindia et al., 2015; Holland et al., 2016; Marzano et al., 2016; Portillo et al., 2016). Depending on the region sequenced the data might be similar or differ significantly. For instance, in a study comparing the V4 and V5 region Bokulich et al. (2012), found that the regions resulted in a similar bacterial composition with minor variation in the lower taxa; although the V4 region provided greater taxonomic depth for certain Proteobacteria and lactic acid bacteria (LAB) species. In contrast, Campanaro et al. (2014), targeted the V3–V4 and V5–V6 regions of the 16S rRNA region and evaluated the bacterial community associated with grape marc after crushing and 30 days “post fermentation”/storage. A total of 89 genera were identified, however only 31 of these were common in both target regions evaluated.
The fungal ITS regions are the most commonly targeted region for fungal diversity estimation. The classification of general fungi and arbuscula mycorrhizae (AMF) has been accomplished by targeting the ITS region (Bokulich et al., 2013a, 2015, 2016a; Setati et al., 2015; Bouffaud et al., 2016; Holland et al., 2016; Kecskeméti et al., 2016; Marzano et al., 2016; Stefanini et al., 2016), D1–D2 regions of the 26S rRNA (Holland et al., 2014; Taylor et al., 2014) and the partial 18S rRNA gene (Lumini et al., 2010; David et al., 2014; Holland et al., 2016; Grangeteau et al., 2017; De Filippis et al., 2017). The AMF populations derived from these different targets, were similar in genera and showed compositional differences in samples evaluated, highlighting them all as suitable target genes for AMF evaluation (Lumini et al., 2010; Bouffaud et al., 2016). Furthermore, Pinto et al. (2014, 2015) targeted both the ITS2 region and D2 domain of the 26S rRNA region for fungal community analysis. The results revealed that the taxonomic depth for the two evaluated regions was considerably similar, however of these only a portion of the observed OTU's were shared between the two regions and that overall the ITS region provided a slightly higher coverage. Bokulich and Mills (2013) moreover, evaluated several ITS primers, and they found that targeting the ITS1 region demonstrates higher levels of taxonomic classification accuracy (species and genus), the smallest difference between Ascomycota and Basidiomycota amplicon lengths, as well as a maximized sequence coverage. Therefore, overall the ITS1 locus appears to be the most promising target, for a complete overview of the microbial populations in ecological studies.
Bioinformatics and Analysis
High throughput sequencing techniques generally generate large amounts of sequence data, and the only viable option to handle such information, is via automated approaches. There are currently several open source pipelines accessible for overseeing, almost the complete analysis procedure for NGS data. These include MOTHUR, quantitative insights into microbial ecology (QIIME; Kõljalg et al., 2013), metagenomics rapid annotation using subsystem technology (MG-RAST), server and rapid analysis of multiple metagenomes with clustering and annotation pipeline (RAMMCAP; Wooley et al., 2010). These pipelines provide the tools for basic data analysis steps such as data cleaning, sequence clustering, functional annotation, and taxonomic assignments (Kõljalg et al., 2013).
The current section will provide brief overview in the procedures used to analyze high-throughput sequencing data in targeted amplicon sequencing for the vineyard and wine associated microbiome, followed by a brief overview of whole-metagenomics sequencing.
Target/Amplicon Sequencing
The analysis of amplicon sequencing data typically undergoes three basic steps; (i) Quality trimming and de-noising; (ii) OTU-picking/clustering, and (iii) taxonomic assignment. Quality-trimming is an essential step used to eradicate erroneous reads obtained through PCR, sequencing instruments and the chemistries behind the sequencing reactions (Bokulich et al., 2013a). To minimize the volume of data for annotation, clustering, and OTU-picking is used. During clustering, pairwise comparison of sequencing is performed with a set percentage identity threshold. Subsequently, a single representative of highly similar sequences is chosen and annotated through BLAST or BLAT algorithms. OTUs can be processed through an open-reference or closed-reference OUT-picking approach. Assignment of species or annotation of functional genes is based on percentage similarity to sequences in specific databases such as Greengenes, UNITE, SILVA, NCBI, SWISSPROT etc.
The analysis of data derived from pyrosequencing during quality trimming typically involves; the removal of barcodes, adapters, and primers, followed by denoising which is used to correct problems associated specifically with 454 pyrosequencer. These typically include the removal of sequences, with ≥6 homopolymers, ambiguous bases and those not meeting Phred score of (20–30). Furthermore, sequences of min and max length can be removed, depending on the target region and possible chimeric sequences (Figure 1).
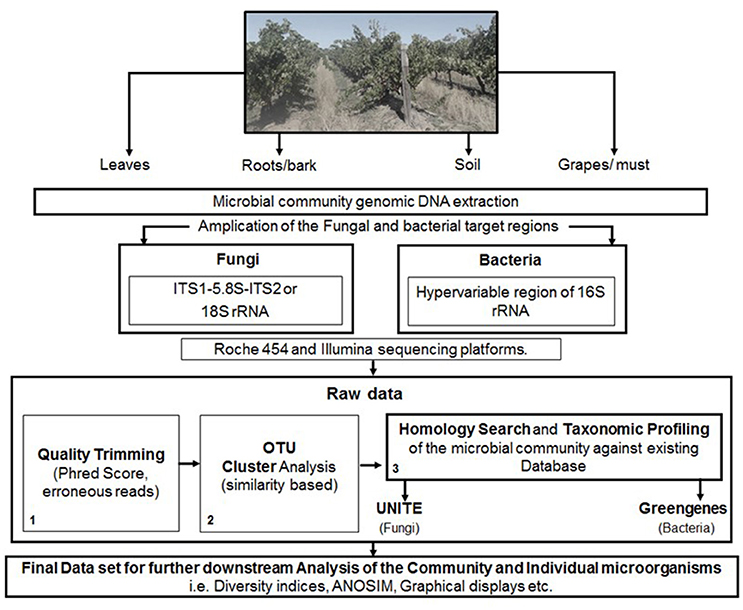
Figure 1. A schematic representation of the steps involved in targeted amplicon sequencing.
The data derived from Illumina sequencing platforms undergoes similar demultiplexing and quality trimming apart from denoising. Reads are typically truncated for ≥3 consecutive bases with a quality <1e−5, and removed when containing ambiguous base calls, primer/barcode errors or a phred score of <20–30. Furthermore, for paired-end sequencing, the reads are typically joined after quality trimming prior to OTU picking, with all sequences retained, even those not overlapping (Figure 1).
Shotgun Metagenomics Sequencing
While the goal in the analysis of the metagenomic data is to reconstruct all the genomes within the environmental sample, the computational intricacy involved makes it unfeasible. Thus, as an alternative two general types of analyses are performed for reconstruction; (i) assembling the reads into contigs, and performing taxonomic classification and functional assignments; (ii) read-based reconstruction of the taxonomic and functional parts of the metagenome. During the assembly of sequences, several problems could arise; for instance, limitation in computational space (Peršoh, 2015), formation of chimeras as a consequence of similarities amongst genomes of related species and variable abundances of genomes within the sample which could potentially result in partial representation (Scholz et al., 2012; Ravin et al., 2015).
Since a mixture of varying amounts of genomic fragments, from different organisms is the result of contig assembly, taxonomic classification can be complicated. Nevertheless, clustering based on the nucleotide composition and coverage carried out by different techniques could sort/bin metagenomic data based on taxonomic status. The clustering efficacy does however rely on various factors. Furthermore, the taxonomic status of the resulting “bins” of contigs is obtained through the identification of phylogenetic marker genes in the bin which was analyzed (Ravin et al., 2015). Additional algorithms have been proposed as an alternative to the cluster based algorithms (Kriseman et al., 2010).
The annotation of the metagenomic contigs can be done using various command-line pipelines and online annotations services, such as MG-RAST, integrated microbial genomes and microbiomes (IMG-M) and community cyberinfrastructure for advanced microbial ecology research and analysis (CAMERA), which in addition to annotation, are able to conduct taxonomic and functional classification as well as pathway reconstruction (Wooley et al., 2010; Desai et al., 2012; Scholz et al., 2012; Ravin et al., 2015). The dependability of the taxonomic assignment and therefore the corresponding information may be decided from scores on sequence similarity and alignment coverage by quality standards or phylogenetic analyses (Peršoh, 2015).
Monitoring complex microbial communities is essential in food fermentations, in which consortia of microbial communities are naturally involved in the processes, such as fermentation and spoilage (Bokulich et al., 2016b). These technological advances, therefore represent an enormous breakthrough for microbial ecology, because metagenomics and NGS allow for in-depth insights into not only the structure, but the function of the most complex microbial communities in their natural environments (Peršoh, 2015). The following section, will therefore focus on metagenomics and how it has been applied to study the vineyard microbial communities.
Vineyard Microbial Communities as Derived from Targeted SGS
SGS technologies have become the tool of choice in deciphering the vineyard and wine microbiome. Most importantly these tools have been employed in microbial surveys that sought to understand how agronomic practices influence microbial community structures and whether there are grapevine organ-specific microbial signatures. Furthermore, it is increasingly becoming important to understand whether there is geographic microbial signatures that contribute to wine typicity.
Bacterial Communities
Several studies have recently employed high-throughput sequencing to evaluate the bacterial communities associated with the vineyard. The most abundant phyla in vineyard soils and grapevine roots include Proteobacteria, Bacteriodetes, Acidobacteria, Verrucomicrobia, Planctomycetes, Actinobacteria, Chloroflexi, Gemmatimonatedes, and Firmicutes (Burns et al., 2015; Calleja-Cervantes et al., 2015; Zarraonaindia et al., 2015; Faist et al., 2016). Studies suggest that the soil microbial community composition in vineyards closely resembles that of other agricultural ecosystems and is largely structured with respect to soil properties and viticultural area (Burns et al., 2015). Furthermore, soil amendments such as fertilizer and/or compost applications can alter the relative abundances of bacterial groups (Calleja-Cervantes et al., 2015). High-throughput analysis of the grapevine phyllosphere, flowers and grape berry surface, demonstrated that the bacterial communities were predominated by Proteobacteria followed by Firmicutes, Actinobacteria, Acidobacteria, and Bacteroidetes (Perazzolli et al., 2014; Pinto et al., 2014, 2015; Portillo and Mas, 2016; Portillo et al., 2016). The relative abundances of the groups vary depending on the plant tissue or organ. Dominant taxa include members of the genera Pseudomonas, Sphingomonas, Frigoribacterium, Curtobacterium, Bacillus, Enterobacter, Acinetobacter, Erwinia, Citrobacter, Pantoea, and Methylobacterium (Bokulich et al., 2014, 2016a; Perazzolli et al., 2014; Pinto et al., 2015; Zarraonaindia et al., 2015; Kecskeméti et al., 2016; Portillo and Mas, 2016; Portillo et al., 2016). In contrast, the endophytic community in grape berries mainly comprise Ralstonia, Burkholderia, Pseudomonas, Staphylococcus, Mesorhizobium, Propionibacterium, Dyella, and Bacillus species (Campisano et al., 2014). However, it is important to note that the bacterial community structure varies amongst grape cultivars, and is also influenced by agronomic practices (Campisano et al., 2014; Perazzolli et al., 2014; Calleja-Cervantes et al., 2015; Pinto et al., 2015; Kecskeméti et al., 2016). Furthermore, development of diseases can result in establishment of different community structures. For instance, graft unions with crown galls were shown to harbor three bacterial OTUs viz. Agrobacterium vitis, Pseudomonas sp., and Enterobacteriaceae sp., that were most abundant in every season, while the three most abundant OTUs in graft unions without a crown gall differed in every season suggesting that crown galls are colonized by a stable bacterial complex (Faist et al., 2016). In other studies, a higher incidence of acetic acid bacteria (AAB) was shown to develop in positive correlation with the Botryotinia sp. on grapevine leaves and in botrytized wine fermentations (Bokulich et al., 2012; Pinto et al., 2015). However, Portillo and Mas (2016) demonstrated that this group of bacteria, specifically Gluconobacter spp., can persist at high abundance throughout wine fermentation in non-botrytized Grenache fermenting musts, only declining at the end of alcoholic fermentation. Furthermore, the population of Gluconobacter was shown to be highly abundant in organic pied-de-cuve Riesling fermentation compared to the conventional fermentation (Piao et al., 2015). AAB were also shown to dominate in low sulfited, uninoculated wine fermentations, compared to Lactobacillus and Lactobacillaceae that dominated SO2-free uninoculated fermentations (Bokulich et al., 2015). Interestingly a low abundance of LAB is often reported with amplicon sequencing phylogenetic surveys (Bokulich et al., 2012; Pinto et al., 2014, 2015). Most importantly, Oenococcus oeni seems to be rarely encountered in grape must except in one study where it was found to be dominant in fermentations of Grenache and Carignan grapes (Portillo et al., 2016). However, several studies show that the levels of this species increase during malolactic fermentation and that in fact it is in most cases the dominant taxa (Marzano et al., 2016; Portillo et al., 2016). Other LAB often encountered include Lactobacillus, Lactococcus, Leuconostoc, and Pediococcus species (Bokulich et al., 2012, 2014; Piao et al., 2015; Pinto et al., 2015; Portillo et al., 2016).
Overall SGS have made it possible to detect bacterial species often overlooked in culture-based methods and community fingerprinting approaches such as DGGE as it can detect species that represent 0.001–1% of the total population. Furthermore, several novel genera believed to be associated with the wine habitat, including, Candidatus_Liberibacter, Onus, Wolbachia, Komagateaibacter, and Shewanella were detected (Marzano et al., 2016; Portillo and Mas, 2016). In some cases, these rare taxa including Methylobacterium, Sphingomonas, Acinetobacter, Pseudomonas, Wolbachia, and Paracoccus could be detected until the end of alcoholic fermentation (Bokulich et al., 2012; Piao et al., 2015; Portillo and Mas, 2016). A closer look at supplementary data from various publications suggests that over 100 species are newly associated with grapevine or wine. However, since only partial sequences are used, most of the taxanomic assignments are generally reliable to genus level. Nevertheless, Table 2 shows a representation of a few species that have been identified in various studies and have been shown to persist from the vineyard environment and throughout wine fermentation. Some of the species e.g., Methylobacterium populi and Sphingomonas pseudosanguinis, were confirmed to be viable at the end of fermentation (Bokulich et al., 2012) and the populations of these genera were also shown to persist in the winery on non-fermentor surfaces (Bokulich et al., 2013b). Further research into these taxa is, however required to evaluate their possible impact in wine fermentation and/or wine quality.
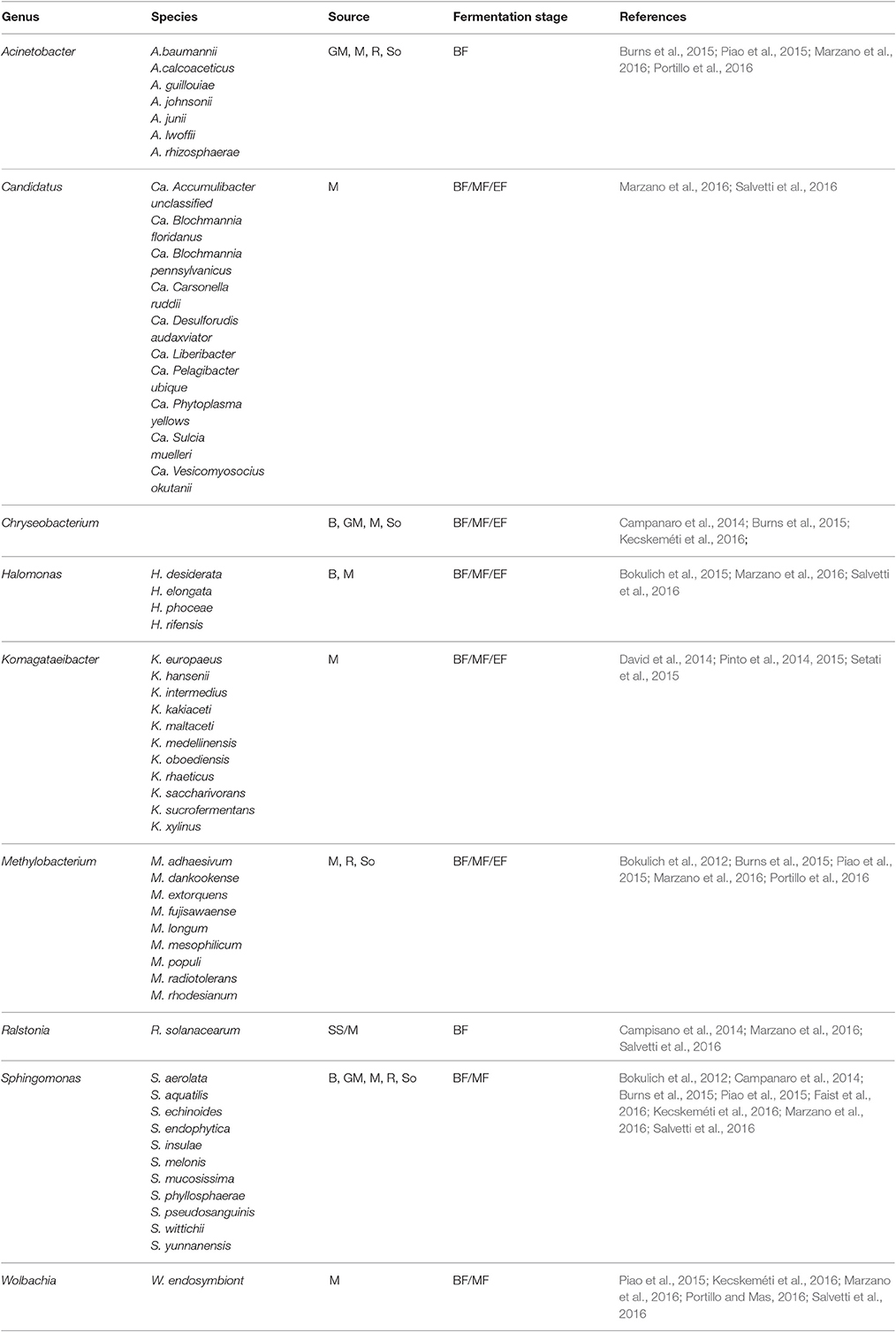
Table 2. A selection of rare bacterial species detected on grapevine leaves (L), Roots (R), Stems, and Shoots (SS), berry surface (B) and in Soil (So), Grape Marc (GM), as well as in must (M) before fermentation (BF), in the middle (MF) and at the end of the alcoholic fermentation (EF).
Fungal Communities
The fungal communities associated with grapevine have mainly been investigated in must after crushing. Overall, the fungal populations at a phylum level are very similar and mainly comprise the Ascomycota and the most abundant phylum followed by the Basidiomycota (Bokulich et al., 2014; David et al., 2014; Taylor et al., 2014; Pinto et al., 2015; Setati et al., 2015; Kecskeméti et al., 2016). Other phyla such as the Zygomycota and Chytridiomycota are only present in low abundance. Frequently encountered genera of filamentous fungi include Aspergillus, Alternaria, Penicillium, Cladosporium, Lewia, Davidiella, Erysiphe, Botrytis and the yeast-like fungus, Aureobasidium pullulans, while the yeast genera include Hanseniaspora, Issatchenkia, Pichia, Candida, Rhodotorula, Lachancea, Metschnikowia, Cryptococcus, Filobasidiella, Sporobolomyces, and Torulaspora (Bokulich et al., 2014; David et al., 2014; Taylor et al., 2014; Pinto et al., 2015; Setati et al., 2015; Wang et al., 2015; Kecskeméti et al., 2016; De Filippis et al., 2017). Generally, the SGS have revealed more filamentous fungal species than yeast species especially those associated with the grape berry surface (Tables 3, 4). These data suggest that most of the yeast genera and species are cultivable but are often missed in culture-based studies due to their presence in minor concentrations. In contrast, for the filamentous fungi, SGS reveals a diversity of possible rot associated taxa such as Botrytis elliptica and Botrytis fabae. Further studies could look into investigating the prevalence of these species and their contribution to rot.
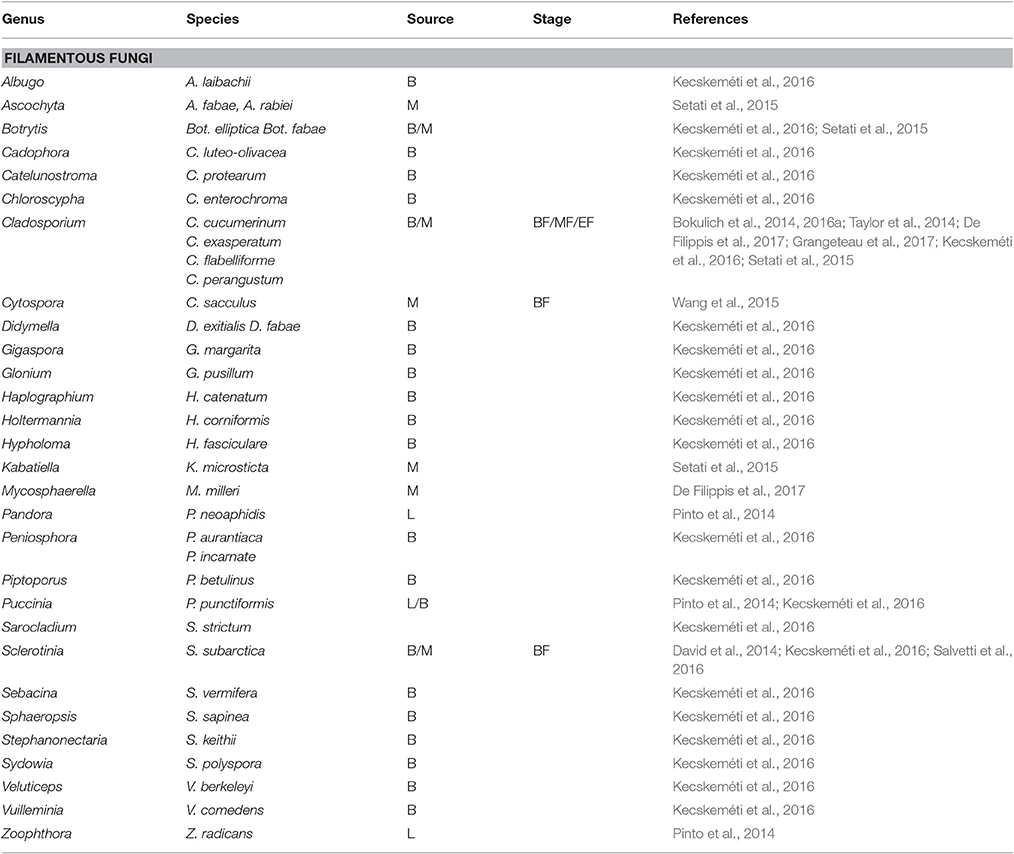
Table 3. Filamentous fungi detected on grapevine leaves (L), berry surface (B) and in must (M) before fermentation (BF), in the middle (MF) and at the end of the alcoholic fermentation (EF).
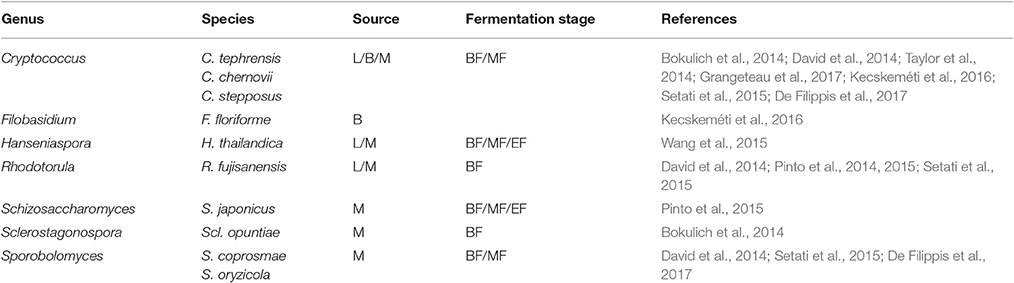
Table 4. Yeasts detected on grapevine leaves (L), berry surface (B) and in must (M) before fermentation (BF), in the middle (MF) and at the end of the alcoholic fermentation (EF).
Several studies have suggested that the microbial community associated with grapevines exhibit regional differentiation (Bokulich et al., 2014, 2016a,b; Taylor et al., 2014; Pinto et al., 2015; Wang et al., 2015). Such regional distinction has been attributed to the dominance of a few species per region. For instance, Bokulich et al. (2014) demonstrated significant association of Aspergillus and Penicillium spp. with the Chardonnay in Napa, while Bacteroides, Actinobacteria, Saccharomycetes, and Erysiphe necator were abundant in Central Coast; and Botryotinia fuckeliana and Proteobacteria in Sonoma. Similarly, Pinto et al. (2015) showed that Lachancea prevailed in the Alentejo appellation, while Rhodotorula and Botrytinia dominated in the Estremadura appellation, Hanseniaspora and Ramularia in Bairrada, Lachancea and Rhodotorula in Dão, Rhodotorula and Erisyphe in Douro, and Rhodotorula and Alternaria in Minho appellation. The fungal diversity associated with grapes is also influenced by agronomic practices. Most importantly, studies have shown that vineyards employing conventional, Integrated Pest management systems, Organic, Biodynamic, and Ecophyto practices harbor different fungal communities (David et al., 2014; Setati et al., 2015; Kecskeméti et al., 2016).
Overall, NGS reveal higher diversity compared to other culture-independent methods such as DGGE and qPCR (David et al., 2014; Wang et al., 2015). Furthermore, these methods have detected minor and rare species that are sometimes overlooked with culture-dependent methods and can detect non-culturable cells at the end of fermentation. For instance, some of the studies show the presence yeast genera such as Kazachstania, Malassezia, Schizosaccharomyces, and Debaryomyces which are typically at low frequency (David et al., 2014; Pinto et al., 2015; Setati et al., 2015; Grangeteau et al., 2017), while cells of Hanseniaspora spp. have been detected at the end of fermentation (Wang et al., 2015). Similar to what has been observed with culture-dependent methods, S. cerevisiae is rarely encountered in grape must even with NGS technologies. However, the fungal community in fermenting musts tends to be less diverse toward the end of fermentation and is dominated by Saccharomyces spp. In some cases, where strong fermentative yeasts such as Lachancea, Starmerella, and Schizosaccharomyces were present at high frequency in the initial population, they persist until the end of fermentation (Pinto et al., 2015; Wang et al., 2015; Bokulich et al., 2016a). Such species have also been shown to contribute toward taxonomic discrimination between growing regions. There is also increasing evidence that there are broad taxonomic trends underlying varietal patterns. For instance, Bokulich et al. (2014) found differences in Chardonnay, Cabernet sauvignon, and Zinfandel, while Wang et al. (2015) demonstrated that Grenache and Carignan grapes harbored certain distinct taxa. Most recently, Aglianico and Greco di Tufo were also found to harbor different yeast communities (De Filippis et al., 2017). Current data show that there is conflicting outcomes regarding the relative abundances of yeast species in must depending on the methods employed. Therefore, although microbial surveys using amplicon sequencing can detect all species that are retrieved by culture-based methods, and other culture-independent methods, the quantity of certain species tends to vary. In addition, there can be variation in community composition depending on the rRNA gene target. For instance, in the study by Pinto et al. (2015) both the D2 region and the ITS-5.8S region were targeted, however, only 13.2% of the taxa were common between the two data sets. This highlights an important gap with regard to the completeness of the databases and accuracy with regard to taxonomic assignment especially at a species level. Furthermore, amplicon sequencing data still comprise significant percentages of “unclassified” or unassigned OTUs which suggests that the diversity is still to some extent under-represented. Studies evaluating fungal diversity in the vineyard remain limited. Orgiazzi et al. (2012) reported that the soil ecosystem is dominated by the genera Penicillium and Cryptococcus, the minor fungal groups are mainly dominated by Glomeromycota or Chytridiomycota. In contrast, the leaf associated microbiome is dominated by early diverging fungal lineages (Zygomycota) such as Rhizopus and Mucor (Pinto et al., 2014), while AMF specific fungi of the soil and grapevine are dominated by Glomeromycota (Lumini et al., 2010; Bouffaud et al., 2016). However, more studies need to be performed in order to confidently elucidate the vineyard and grapevine phyllosphere microbiome.
Whole-Metagenomic Sequencing
Recently, Salvetti et al. (2016) employed whole genome sequencing for the first in-depth evaluation of the microbial consortium associated with Corvina berries post withering performed in two different conditions. A total of 25 bacterial phyla were detected, nine of which were common and consisted of Acidobacteria, Actinobacteria, Cyanobacteria, Firmicutes, and Proteobacteria; the latter was predominant, followed by Firmicutes, Actinobacteria, and Bacteroidetes as reported by Pinto et al. (2014) and Zarraonaindia et al. (2015), who both employed target metagenomics strategies. The class Gammaproteobacteria was dominant, which was further represented by Pseudomonadaceae in high abundances in the traditional withering and Enterobacteriaceae in accelerated withering. Furthermore, both genera Carnobacterium and Enterococcus previously identified as grape associated by Pinto et al. (2015) was detected using the whole genome sequencing approach. Also, evaluating the eukaryotic community, they reported that Ascomycota was the dominant phylum, more specifically the class Eurotiomycetes, specifically genera belonging to Aspergillus and Penicillium, followed by Sordariomycetes and Dothideomycetes. However, common yeast such as Aureobasidium, Cryptococcus, Hanseniaspora, Metschnikowia, and Sporobolomyces which are regularly detected in targeted strategies were not detected.
Beyond providing the inventory of the vineyard, whole metagenomic analysis provides the functional information for the evaluated microbiome. For instance, information regarding defense, amino acid metabolism, transport, transcription and carbohydrate metabolism, potentially allowing a greater comparison to be drawn than the assumed microbial diversity and composition (Campanaro et al., 2014; Salvetti et al., 2016).
Conclusion
The invaluable contribution of metagenomic approaches in deciphering the vineyard microbiome and its application provides great insights in the microbial community composition and structure of both bacteria and fungi. Metagenomic approaches provide an opportunity to study the entire microbial population and not just one group as typically done with culture-based methods. Consequently, it has been possible to assess the population dynamics during fermentation, to evaluate grapevine disease complexes and unravel unique microbial signatures present in grapevine and not in neighboring plants. Furthermore, these approaches have been valuable in understanding the influence of vineyard management practices on the grapevine microbiome. Based on the existing research papers, it appears as though the grapevine microbiome is less complex compared to other ecosystems such as soil and that a large proportion of the yeast species associated with the grape and wine environment are cultivable. This is advantageous as the species can then be evaluated for potential genes, enzymes etc. that can be of importance for winemaking. However, most of the studies show that a significant percentage of the sequence data (OTU's) remained unassigned. This problem highlights existing challenges with sequence databases used for taxonomic assignment that are not complete and for this technology to be furthered in future means that the expansion of the databases are crucial. Nevertheless, based on existing data, sequence-based methods reveal similar fungal species compared to culture-dependent methods, especially regarding the yeasts which are relevant in wine fermentation. The discovery of new species associated with the grape and wine microbiome holds tremendous potential to mine them for novel properties that would improve wine fermentation, aroma and style.
Author Contributions
HM wrote the first draft of the review; MdT and MS proofed the drafts and finalized the review.
Funding
This work was funded by the National Research Foundation (NRF) [grant number 101998] and Winetech SU IWBT 16/02. Opinions expressed and conclusions arrived at are those of the authors and are not necessarily to be attributed to the funding agencies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbasian, F., Lockington, R., Mallavarapu, M., and Naidu, R. (2015a). A pyrosequencing-based analysis of microbial diversity governed by ecological conditions in the Winogradsky column. World J. Microbiol. Biotechnol. 31, 1115–1126. doi: 10.1007/s11274-015-1861-y
Abbasian, F., Lockington, R., Megharaj, M., and Naidu, R. (2015b). The integration of sequencing and bioinformatics in metagenomics. Rev. Environ. Sci. Biotechnol. 14, 357–383. doi: 10.1007/s11157-015-9365-7
Alessandria, V., Giacosa, S., Campolongo, S., Rolle, L., Rantsiou, K., and Cocolin, L. (2013). Yeast population diversity on grapes during on-vine withering and their dynamics in natural and inoculated fermentations in the production of icewines. Food Res. Int. 54, 139–147. doi: 10.1016/j.foodres.2013.06.018
Andorrà, I., Landi, S., Mas, A., Esteve-Zarzoso, B., and Guillamón, J. M. (2010). Effect of fermentation temperature on microbial population evolution using culture-independent and dependent techniques. Food Res. Int. 43, 773–779. doi: 10.1016/j.foodres.2009.11.014
Andorrà, I., Landi, S., Mas, A., Guillamón, J. M., and Esteve-Zarzoso, B. (2008). Effect of oenological practices on microbial populations using culture-independent techniques. Food Microbiol. 25, 849–856. doi: 10.1016/j.fm.2008.05.005
Arteau, M., Labrie, S., and Roy, D. (2010). Terminal-restriction fragment length polymorphism and automated ribosomal intergenic spacer analysis profiling of fungal communities in Camembert cheese. Int. Dairy J. 20, 545–554. doi: 10.1016/j.idairyj.2010.02.006
Balázs, M., Rónavári, A., Németh, A., Bihari, Z., Rutkai, E., Bartos, P., et al. (2013). Effect of DNA polymerases on PCR-DGGE patterns. Int. Biodeterior. Biodegrad. 84, 244–249. doi: 10.1016/j.ibiod.2012.05.011
Barata, A., Malfeito-Ferreira, M., and Loureiro, V. (2012). The microbial ecology of wine grape berries. Int. J. Food Microbiol. 153, 243–259. doi: 10.1016/j.ijfoodmicro.2011.11.025
Barnett, J. A. (2003). Beginnings of microbiology and biochemistry: the contribution of yeast research. Microbiology 149, 557–567. doi: 10.1099/mic.0.26089-0
Bleidorn, C. (2015). Third generation sequencing: technology and its potential impact on evolutionary biodiversity research. Syst Biodivers. 2000, 1–8. doi: 10.1080/14772000.2015.1099575
Bokulich, N. A., Collins, T. S., Masarweh, C., Allen, G., Heymann, H., Ebeler, S. E., et al. (2016a). Associations among wine grape microbiome, metabolome, and fermentation behavior suggest microbial contribution to regional wine characteristics. MBio 7, 1–12. doi: 10.1128/mBio.00631-16
Bokulich, N. A., Joseph, C. M. L., Allen, G., Benson, A. K., and Mills, D. A. (2012). Next-generation sequencing reveals significant bacterial diversity of botrytized wine. PLoS ONE 7:e36357. doi: 10.1371/journal.pone.0036357
Bokulich, N. A., Lewis, Z. T., Boundy-Mills, K., and Mills, D. A. (2016b). A new perspective on microbial landscapes within food production. Curr. Opin. Biotechnol. 37, 182–189. doi: 10.1016/j.copbio.2015.12.008
Bokulich, N. A., and Mills, D. A. (2013). Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-high-throughput profiling of fungal communities. Appl. Environ. Microbiol. 79, 2519–2526.
Bokulich, N. A., Ohta, M., Richardson, P. M., and Mills, D. A. (2013b). Monitoring seasonal changes in winery-resident microbiota. PLoS ONE 8:e66437. doi: 10.1371/journal.pone.0066437
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, I., Knight, R., et al. (2013a). Quality-filtering vastly improves diversity estimates from illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Bokulich, N. A., Swadener, M., Sakamoto, K., Mills, D. A., and Bisson, L. F. (2015). Sulfur dioxide treatment alters wine microbial diversity and fermentation. Progression in a dose-dependent fashion. Am. J. Enol. Vitic. 66, 73–79. doi: 10.5344/ajev.2014.14096
Bokulich, N. A., Thorngate, J. H., Richardson, P. M., and Mills, D. A. (2014). Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. U.S.A. 111, E139–E148. doi: 10.1073/pnas.1317377110
Bouffaud, M. L., Bernaud, E., Colombet, A., Van Tuinen, D., Wipf, D., and Redecker, D. (2016). Regional-scale analysis of arbuscular mycorrhizal fungi: the case of Burgundy vineyards. J. Int. Sci. Vigne Vin. 50, 1–8. doi: 10.20870/oeno-one.2016.50.1.49
Brežná, B., Zenišová, K., Chovanová, K., Chebeňová, V., Kraková, L., Kuchta, T., et al. (2010). Evaluation of fungal and yeast diversity in Slovakian wine-related microbial communities. Antonie Van Leeuwenhoek 98, 519–529. doi: 10.1007/s10482-010-9469-6
Burns, K. N., Kluepfel, D. A., Strauss, S. L., Bokulich, N. A., Cantu, D., and Steenwerth, K. L. (2015). Vineyard soil bacterial diversity and composition revealed by 16S rRNA genes: differentiation by geographic features. Soil Biol. Biochem. 91, 232–247. doi: 10.1016/j.soilbio.2015.09.002
Calleja-Cervantes, M. E., Menéndez, S., Fernández-González, A. J., Irigoyen, I., Cibriáin-Sabalza, J. F., Toro, N., et al. (2015). Changes in soil nutrient content and bacterial community after 12 years of organic amendment application to a vineyard. Eur. J. Soil Sci. 66, 802–812. doi: 10.1111/ejss.12261
Cameron, M., Siebrits, L., du Toit, M., and Witthuhn, R. C. (2013). PCR-based DGGE fingerprinting and identification of the microbial population in South African red grape must and wine. J. Int. Sci. Vigne Vin. 47, 47–54. doi: 10.20870/oeno-one.2013.47.1.1531
Campanaro, S., Treu, L., Vendramin, V., Bovo, B., Giacomini, A., and Corich, V. (2014). Metagenomic analysis of the microbial community in fermented grape marc reveals that Lactobacillus fabifermentans is one of the dominant species: insights into its genome structure. Appl. Microbiol. Biotechnol. 98:60156037. doi: 10.1007/s00253-014-5795-3
Campisano, A., Antonielli, L., Pancher, M., Yousaf, S., Pindo, M., and Pertot, I. (2014). Bacterial endophytic communities in the grapevine depend on pest management. PLoS ONE 9:e112763. doi: 10.1371/journal.pone.0112763
Capozzi, V., Di Toro, M. R., Grieco, F., Michelotti, V., Salma, M., Lamontanara, A., et al. (2016). Viable but not Culturable (VBNC) state of Brettanomyces bruxellensis in wine: new insights on the molecular basis of VBNC behavior using a transcriptomic approach. Food Microbiol. 59, 196–204. doi: 10.1016/j.fm.2016.06.007
Chovanová, K., Kraková, L., Ženišová, K., Turcovská, V., Brežná, B., Kuchta, T., et al. (2011). Selection and identification of autochthonous yeasts in Slovakian wine samples using a rapid and reliable three-step approach. Lett. Appl. Microbiol. 53, 231–237. doi: 10.1111/j.1472-765X.2011.03097.x
Cocolin, L., Heisey, A., and Mills, D. A. (2001). Direct identification of the indigenous yeasts in commercial wine fermentations. Am. J. Enol. Vitic. 52, 49–53.
Cocolin, L., Alessandria, V., Dolci, P., Gorra, R., and Rantsiou, K. (2013). Culture independent methods to assess the diversity and dynamics of microbiota during food fermentation. Int. J. Food Microbiol. 167, 29–43. doi: 10.1016/j.ijfoodmicro.2013.05.008
Comitini, F., Capece, A., Ciani, M., and Romano, P. (2017). New insights on the use of wine yeasts. Curr. Opin. Food Sci. 13, 44–49. doi: 10.1016/j.cofs.2017.02.005
David, V., Terrat, S., Herzine, K., Claisse, O., Rousseaux, S., Tourdot-Maréchal, R., et al. (2014). High-throughput sequencing of amplicons for monitoring yeast biodiversity in must and during alcoholic fermentation. J. Ind. Microbiol. Biotechnol. 41, 811–821. doi: 10.1007/s10295-014-1427-2
De Filippis, F., La Storia, A., and Blaiotta, G. (2017). Monitoring the mycobiota during Greco di Tufo and Anglianico wine fermentation by 18S rRNA gene sequencing. Food Microbiol. 63, 117–122. doi: 10.1016/j.fm.2016.11.010
Desai, N., Antonopoulos, D., Gilbert, J. A., Glass, E. M., and Meyer, F. (2012). From genomics to metagenomics. Curr. Opin. Biotechnol. 23, 72–76. doi: 10.1016/j.copbio.2011.12.017
Diaz-Sanchez, S., Hanning, I., Pendleton, S., and D'Souza, D. (2013). Next-generation sequencing: the future of molecular genetics in poultry production and food safety. Poult. Sci. 92, 562–572. doi: 10.3382/ps.2012-02741
Di Maro, E., Ercolini, D., and Coppola, S. (2007). Yeast dynamics during spontaneous wine fermentation of the Catalanesca grape. Int. J. Food Microbiol. 117, 201–210. doi: 10.1016/j.ijfoodmicro.2007.04.007
Divol, B., and Lonvaud-Funel, A. (2005). Evidence for viable but nonculturable yeasts in Botrytis-affected wine. J. Appl. Microbiol. 99, 85–93. doi: 10.1111/j.1365-2672.2005.02578.x
Esteve-Zarzoso, B. (1999). Identification of yeasts by RFLP analysis of the 5.8 S rRNA gene and the two ribosomal internal transcribed spacers. Int. J. Syst. Bacteriol. 49, 329–337. doi: 10.1099/00207713-49-1-329
Faist, H., Keller, A., Hentschel, U., and Deeken, R. (2016). Grapevine (Vitis vinifera) crown galls host distinct microbiota. Appl. Environ. Microbiol. 82, 5542–5552. doi: 10.1128/AEM.01131-16
Fasoli, S., Marzotto, M., Rizzotti, L., Rossi, F., Dellaglio, F., and Torriani, S. (2003). Bacterial composition of commercial probiotic products as evaluated by PCR-DGGE analysis. Int. J. Food Microbiol. 82, 59–70. doi: 10.1016/S0168-1605(02)00259-3
Fernández-Espinar, M. T., López, V., Ramón, D., Bartra, E., and Querol, A. (2001). Study of the authenticity of commercial wine yeast strains by molecular techniques. Int. J. Food Microbiol. 70, 1–10. doi: 10.1016/S0168-1605(01)00502-5
Franzosa, E. A., Hsu, T., Sirota-Madi, A., Shafquat, A., Abu-Ali, G., Morgan, X. C., et al. (2015). Sequencing and beyond: integrating molecular “omics” for microbial community profiling. Nat. Rev. Microbiol. 13, 360–372. doi: 10.1038/nrmicro3451
Ghosh, S., Bagheri, B., Morgan, H. H., Divol, B., and Setati, M. E. (2015). Assessment of wine microbial diversity using ARISA and cultivation-based methods. Ann. Microbiol. 65, 1833–1840. doi: 10.1007/s13213-014-1021-x
Gilbert, J. A., van der Lelie, D., and Zarraonaindia, I. (2014). Microbial terroir for wine grapes. Proc. Natl. Acad. Sci. U.S.A. 111, 5–6. doi: 10.1073/pnas.1320471110
Goodwin, S., McPherson, J. D., and McCombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351. doi: 10.1038/nrg.2016.49
Grangeteau, C., Roullier-Gall, C., Rousseaux, S., Guogeon, R. D., Schmitt-Kopplin, P., Alexanddre, H., et al. (2017). Wine microbiology is driven by vineyard and winery anthropogenic factors. Microb. Biotechnol. 10, 354–370. doi: 10.1111/1751-7915.12428
Grube, M., Schmid, F., and Berg, G. (2011). Black fungi and associated bacterial communities in the phyllosphere of grapevine. Fungal Biol. 115, 978–986. doi: 10.1016/j.funbio.2011.04.004
Guzzon, R., Nicolini, G., Nardin, T., Malacarne, M., and Larcher, R. (2014). Survey about the microbiological features, the oenological performance and the influence on the character of wine of active dry yeast employed as starters of wine fermentation. Int. J. Food Sci. Technol. 49, 2142–2148. doi: 10.1111/ijfs.12610
Heather, J. M., and Chain, B. (2016). The sequence of sequencers: the history of sequencing DNA. Genomics 107, 1–8. doi: 10.1016/j.ygeno.2015.11.003
Holland, T. C., Bowen, P. A., Bogdanoff, C. P., Lowery, T. D., Shaposhnikova, O., Smith, S., et al. (2016). Evaluating the diversity of soil microbial communities in vineyards relative to adjacent native ecosystems. Agric. Ecosyst. Environ. Appl. Soil Ecol. 100, 91–103. doi: 10.1016/j.apsoil.2015.12.001
Holland, T. C., Bowen, P., Bogdanoff, C., and Hart, M. (2014). Arbuscular mycorrhizal fungal communities associated with Vitis vinifera vines under different frequencies of irrigation. Am. J. Enol. Vitic. 65, 222–229. doi: 10.5344/ajev.2014.13101
Huse, S. M., Huber, J. A., Morrison, H. G., Sogin, M. L., and Welch, D. M. (2007). Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 8:R143. doi: 10.1186/gb-2007-8-7-r143
Jolly, N. P., Varela, C., and Pretorius, I. S. (2014). Not your ordinary yeast: non-Saccharomyces yeasts in wine production uncovered. FEMS Yeast Res. 14, 215–237. doi: 10.1111/1567-1364.12111
Justé, A., Thomma, B. P. H. J., and Lievens, B. (2008). Recent advances in molecular techniques to study microbial communities in food-associated matrices and processes. Food Microbiol. 25, 745–761. doi: 10.1016/j.fm.2008.04.009
Kecskeméti, E., Berkelmann-Löhnertz, B., and Reineke, A. (2016). Are epiphytic microbial communities in the carposphere of ripening grape clusters (Vitis vinifera L.) different between conventional, organic, and biodynamic grapes? PLoS ONE 11:e0160852. doi: 10.1371/journal.pone.0160852
Kõljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F. S., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Kovacs, A., Yacoby, K., and Gophna, U. (2010). A systematic assessment of automated ribosomal intergenic spacer analysis (ARISA) as a tool for estimating bacterial richness. Res. Microbiol. 161, 192–197. doi: 10.1016/j.resmic.2010.01.006
Kraková, L., Chovanová, K., Ženišová, K., Turcovská, V., Brežná, B., Kuchta, T., et al. (2012). Yeast diversity investigation of wine-related samples from two different Slovakian wine-producing areas through a multistep procedure. LWT Food Sci Technol. 46, 406–411. doi: 10.1016/j.lwt.2011.12.010
Kriseman, J., Busick, C., Szelinger, S., and Dinu, V. (2010). BING: biomedical informatics pipeline for next generation sequencing. J. Biomed. Inform. 43, 428–434. doi: 10.1016/j.jbi.2009.11.003
Leveau, J. H. J., and Tech, J. J. (2011). Grapevine microbiomics: bacterial diversity on grape leaves and berries revealed by high-throughput sequence analysis of 16S rRNA amplicons. Acta Hortic. 905, 31–42. doi: 10.17660/ActaHortic.2011.905.2
Lu, Y., Huang, D., Lee, P. R., and Liu, S. Q. (2016). Assessment of volatile and non-volatile compounds in durian wines fermented with four commercial non-Saccharomyces yeasts. J. Sci. Food Agric. 96, 1511–1521. doi: 10.1002/jsfa.7253
Lumini, E., Orgiazzi, A., Borriello, R., Bonfante, P., and Bianciotto, V. (2010). Disclosing arbuscular mycorrhizal fungal biodiversity in soil through a land-use gradient using a pyrosequencing approach. Environ. Microbiol. 12, 2165–2179. doi: 10.1111/j.1462-2920.2009.02099.x
Lv, X. C., Huang, R. L., Chen, F., Zhang, W., Rao, P. F., and Ni, L. (2013). Bacterial community dynamics during the traditional brewing of Wuyi Hong Qu glutinous rice wine as determined by culture-independent methods. Food Control. 34, 300–306. doi: 10.1016/j.foodcont.2013.05.003
Ma, Q., Qu, Y., Shen, W., Zhang, Z., Wang, J., Liu, Z., et al. (2015). Bacterial community compositions of coking wastewater treatment plants in steel industry revealed by Illumina high-throughput sequencing. Bioresour. Technol. 179, 436–443. doi: 10.1016/j.biortech.2014.12.041
Martins, G., Miot-Sertier, C., Lauga, B., Claisse, O., Lonvaud-Funel, A., Soulas, G., et al. (2012). Grape berry bacterial microbiota: impact of the ripening process and the farming system. Int. J. Food Microbiol. 158, 93–100. doi: 10.1016/j.ijfoodmicro.2012.06.013
Martins, G., Vallance, J., Mercier, A., Albertin, W., Stamatopoulos, P., Rey, P., et al. (2014). Influence of the farming system on the epiphytic yeasts and yeast-like fungi colonizing grape berries during the ripening process. Int. J. Food Microbiol. 177, 21–28. doi: 10.1016/j.ijfoodmicro.2014.02.002
Marzano, M., Fosso, B., Manzari, C., Grieco, F., Intranuovo, M., Cozzi, G., et al. (2016). Complexity and dynamics of the winemaking bacterial communities in berries, musts, and wines from apulian grape cultivars through time and space. PLoS ONE 11:e0157383. doi: 10.1371/journal.pone.0157383
Morales, P., Rojas, V., Quirós, M., and Gonzalez, R. (2015). The impact of oxygen on the final alcohol content of wine fermented by a mixed starter culture. Appl. Microbiol. Biotechnol. 99, 3993–4003.
Morozova, O., and Marra, M. A. (2008). Applications of next-generation sequencing technologies in functional genomics. Genomics 92, 255–264. doi: 10.1016/j.ygeno.2008.07.001
Nocker, A., Burr, M., and Camper, A. K. (2007). Genotypic microbial community profiling: a critical technical review. Microb. Ecol. 54, 276–289. doi: 10.1007/s00248-006-9199-5
Orgiazzi, A., Lumini, E., Nilsson, R. H., Girlanda, M., Vizzini, A., Bonfante, P., et al. (2012). Unravelling soil fungal communities from different Mediterranean land-use backgrounds. PLoS ONE 7:e34847. doi: 10.1371/journal.pone.0034847
Padilla, B., Gil, J. V., and Manzanares, P. (2016). Past and future of non-saccharomyces yeast: from spoilage microorganisms to biotechnological tools for improving wine aroma complexity. Front. Microbiol. 7:411. doi: 10.3389/fmicb.2016.00411
Pancher, M., Ceol, M., Corneo, P. E., Longa, C. M. O., Yousaf, S., Pertot, I., et al. (2012). Fungal endophytic communities in grapevines (Vitis vinifera L.) respond to crop management. Appl. Environ. Microbiol. 78, 4308–4317. doi: 10.1128/AEM.07655-11
Perazzolli, M., Antonielli, L., Storari, M., Puopolo, G., Pancher, M., Giovannini, O., et al. (2014). Resilience of the natural phyllosphere microbiota of the grapevine to chemical and biological pesticides. Appl. Environ. Microbiol. 80, 3585–3596. doi: 10.1128/AEM.00415-14
Peršoh, D. (2015). Plant-associated fungal communities in the light of meta'omics. Fungal Divers. 75, 1–25. doi: 10.1007/s13225-015-0334-9
Pettersson, E., Lundeberg, J., and Ahmadian, A. (2009). Generations of sequencing technologies. Genomics 93, 105–111. doi: 10.1016/j.ygeno.2008.10.003
Piao, H., Hawley, E., Kopf, S., DeScenzo, R., Sealock, S., Henick-Kling, T., et al. (2015). Insights into the bacterial community and its temporal succession during the fermentation of wine grapes. Front. Microbiol. 6:809. doi: 10.3389/fmicb.2015.00809
Pinto, C., Pinho, D., Cardoso, R., Custódio, V., Fernandes, J., Sousa, S., et al. (2015). Wine fermentation microbiome: a landscape from different Portuguese wine appellations. Front. Microbiol. 6:905. doi: 10.3389/fmicb.2015.00905
Pinto, C., Pinho, D., Sousa, S., Pinheiro, M., Egas, C., and Gomes, A. C. (2014). Unravelling the diversity of grapevine microbiome. PLoS ONE 9:e85622. doi: 10.1371/journal.pone.0085622
Portillo, M. C., Franquès, J., Araque, I., Reguant, C., and Bordons, A. (2016). Bacterial diversity of Grenache and Carignan grape surface from different vineyards at Priorat wine region (Catalonia, Spain). Int. J. Food Microbiol. 219, 56–63. doi: 10.1016/j.ijfoodmicro.2015.12.002
Portillo, M. C., and Mas, A. (2016). Analysis of microbial diversity and dynamics during wine fermentation of Grenache grape variety by high-throughput barcoding. LWT Food Sci. Technol. 72, 317–321. doi: 10.1016/j.lwt.2016.05.009
Prakitchaiwattana, C. J., Fleet, G. H., and Heard, G. M. (2004). Application and evaluation of denaturing gradient gel electrophoresis to analyse the yeast ecology of wine grapes. FEMS Yeast Res. 4, 865–877. doi: 10.1016/j.femsyr.2004.05.004
Ravin, N. V., Mardanov, A. V., and Skryabin, K. G. (2015). Metagenomics as a tool for the investigation of uncultured microorganisms. Russ. J. Genet. 51, 431–439. doi: 10.1134/S1022795415050063
Renouf, V., Claisse, O., and Lonvaud-Funel, A. (2005). Understanding the microbial ecosystem on the grape berry surface through numeration and identification of yeast and bacteria. Aust. J. Grape Wine Res. 11, 316–327. doi: 10.1111/j.1755-0238.2005.tb00031.x
Renouf, V., Claisse, O., and Lonvaud-Funel, A. (2006a). rpoB gene: a target for identification of LAB cocci by PCR-DGGE and melting curves analyses in real time PCR. J. Microbiol. Methods 67, 162–170. doi: 10.1016/j.mimet.2006.03.008
Renouf, V., Claisse, O., and Lonvaud-Funel, A. (2007). Inventory and monitoring of wine microbial consortia. Appl. Microbiol. Biotechnol. 75, 149–164. doi: 10.1007/s00253-006-0798-3
Renouf, V., Claisse, O., Miot-Sertier, C., and Lonvaud-Funel, A. (2006b). Lactic acid bacteria evolution during winemaking: use of rpoB gene as a target for PCR-DGGE analysis. Food Microbiol. 23, 136–145. doi: 10.1016/j.fm.2005.01.019
Reuter, J. A., Spacek, D. V., and Snyder, M. P. (2015). High-throughput sequencing technologies. Mol. Cell 58, 586–597. doi: 10.1016/j.molcel.2015.05.004
Rousseaux, S., Diguta, C. F., Radoï-Matei, F., Alexandre, H., and Guilloux-Bénatier, M. (2014). Non-Botrytis grape-rotting fungi responsible for earthy and moldy off-flavors and mycotoxins. Food Microbiol. 38, 104–121. doi: 10.1016/j.fm.2013.08.013
Salma, M., Rousseaux, S., Sequeira-Le Grand, A., Divol, B., and Alexandre, H. (2013). Characterization of the Viable but Nonculturable (VBNC) state in Saccharomyces cerevisiae. PLoS ONE 8:e77600. doi: 10.1371/journal.pone.0077600
Salvetti, E., Campanaro, S., Campedelli, I., Fracchetti, F., Gobbi, A., Tornielli, G. B., et al. (2016). Whole-metagenome-sequencing-based community profiles of Vitis vinifera L. cv. Corvina berries withered in two post-harvest conditions. Front. Microbiol. 7:937. doi: 10.3389/fmicb.2016.00937
Sanger, F., Nicklen, S., and Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A. 74, 5463–5467. doi: 10.1073/pnas.74.12.5463
Schadt, E. E., Turner, S., and Kasarskis, A. (2010). A window into third-generation sequencing. Hum. Mol. Genet. 19, 227–240. doi: 10.1093/hmg/ddq416
Schmid, F., Moser, G., Müller, H., and Berg, G. (2011). Functional and structural microbial diversity in organic and conventional viticulture: organic farming benefits natural biocontrol agents. Appl. Environ. Microbiol. 77, 2188–2191. doi: 10.1128/AEM.02187-10
Scholz, M. B., Lo, C. C., and Chain, P. S. G. (2012). Next generation sequencing and bioinformatic bottlenecks: the current state of metagenomic data analysis. Curr. Opin. Biotechnol. 23, 9–15. doi: 10.1016/j.copbio.2011.11.013
Setati, M. E., Jacobson, D., Andong, U. C., and Bauer, F. (2012). The vineyard yeast microbiome, a mixed model microbial map. PLoS ONE 7:e52609. doi: 10.1371/journal.pone.0052609
Setati, M. E., Jacobson, D., and Bauer, F. F. (2015). Sequence-based analysis of the Vitis vinifera L. cv Cabernet Sauvignon grape must mycobiome in three South African vineyards employing distinct agronomic systems. Front. Microbiol. 6:1358. doi: 10.3389/fmicb.2015.01358
Shi, Y., Lou, K., Li, C., Wang, L., Zhao, Z., Zhao, S., et al. (2015). Illumina-based analysis of bacterial diversity related to halophytes Salicornia europaea and Sueada aralocaspica. J. Microbiol. 53, 678–685. doi: 10.1007/s12275-015-5080-x
Slabbert, E., Kongor, R. Y., Esler, K. J., and Jacobs, K. (2010). Microbial diversity and community structure in Fynbos soil. Mol. Ecol. 19, 1031–1041. doi: 10.1111/j.1365-294X.2009.04517.x
Solieri, L., and Giudici, P. (2008). Yeasts associated to traditional balsamic vinegar: ecological and technological features. Int. J. Food Microbiol. 125, 36–45. doi: 10.1016/j.ijfoodmicro.2007.06.022
Stefanini, I., Albanese, D., Cavazza, A., Franciosi, E., De Filippo, C., Donati, C., et al. (2016). Dynamic changes in microbiota and mycobiota during spontaneous “Vino Santo Trentino” fermentation. Microb. Biotechnol. 9, 195–208. doi: 10.1111/1751-7915.12337
Sun, Y., and Liu, Y. (2014). Investigating of yeast species in wine fermentation using terminal restriction fragment length polymorphism method. Food Microbiol. 38, 201–207. doi: 10.1016/j.fm.2013.09.001
Taylor, M. W., Tsai, P., Anfang, N., Ross, H. A., and Goddard, M. R. (2014). Pyrosequencing reveals regional differences in fruit-associated fungal communities. Environ. Microbiol. 16, 2848–2858. doi: 10.1111/1462-2920.12456
Thomas, T., Gilbert, J., and Meyer, F. (2012). Metagenomics - a guide from sampling to data analysis. Microb. Inform. Exp. 2:3. doi: 10.1186/2042-5783-2-3
Türktaş, M., Kurtoglu, K. Y., Dorado, G., Zhang, B., Hernandez, P., and Ünver, T. (2015). Sequencing of plant genomes - a review. Turk. J. Agric. Forest. 39, 361–376. doi: 10.3906/tar-1409-93
Wang, C., Garcia-Fernández, D., Mas, A., and Esteve-Zarzoso, B. (2015). Fungal diversity in grape must and wine fermentation assessed by massive sequencing, quantitative PCR and DGGE. Front. Microbiol. 6:1156. doi: 10.3389/fmicb.2015.01156
Wang, Z. K., Yang, Y. S., Stefka, A. T., Sun, G., and Peng, L. H. (2014). Review article: fungal microbiota and digestive diseases. Aliment. Pharmacol. Ther. 39, 751–766. doi: 10.1111/apt.12665
Wooley, J. C., Godzik, A., and Friedberg, I. (2010). A primer on metagenomics. PLoS Comput. Biol. 6:e1000667. doi: 10.1371/journal.pcbi.1000667
Zarraonaindia, I., Owens, S. M. S., Weisenhorn, P., West, K., Hampton-Marcell, J., Lax, S., et al. (2015). The soil microbiome influences grapevine-associated microbiota. Mbio 6, e02527–e02514. doi: 10.1128/mBio.02527-14
Ženišová, K., Chovanová, K., Chebeňová-Turcovská, V., Godálová, Z., Kraková, L., Kuchta, T., et al. (2014). Mapping of wine yeast and fungal diversity in the Small Carpathian wine-producing region (Slovakia): evaluation of phenotypic, genotypic and culture-independent approaches. Ann. Microbiol. 64, 1819–1828. doi: 10.1007/s13213-014-0827-x
Keywords: amplicon sequencing, vineyard microbiome, microbial diversity, wine fermentation, non-Saccharomyces yeasts
Citation: Morgan HH, du Toit M and Setati ME (2017) The Grapevine and Wine Microbiome: Insights from High-Throughput Amplicon Sequencing. Front. Microbiol. 8:820. doi: 10.3389/fmicb.2017.00820
Received: 18 December 2016; Accepted: 21 April 2017;
Published: 11 May 2017.
Edited by:
Sandra Torriani, University of Verona, ItalyReviewed by:
Vittorio Capozzi, University of Foggia, ItalyCarmen Portillo, Universidad Rovira i Virgili, Spain
Copyright © 2017 Morgan, du Toit and Setati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mathabatha E. Setati, setati@sun.ac.za