 Vera Thiel
Vera Thiel Michael Hügler
Michael Hügler David M. Ward
David M. Ward Donald A. Bryant
Donald A. Bryant- 1Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA, United States
- 2Department Microbiology and Molecular Biology, DVGW-Technologiezentrum Wasser, Karlsruhe, Germany
- 3Department of Land Resources and Environmental Sciences, Montana State University, Bozeman, MT, United States
- 4Department of Chemistry and Biochemistry, Montana State University, Bozeman, MT, United States
Microbial mat communities in the effluent channels of Octopus and Mushroom Springs within the Lower Geyser Basin of Yellowstone National Park have been extensively characterized. Previous studies have focused on the chlorophototrophic organisms of the phyla Cyanobacteria and Chloroflexi. However, the diversity and metabolic functions of the other portion of the community in the microoxic/anoxic region of the mat are poorly understood. We recently described the diverse but extremely uneven microbial assemblage in the undermat of Mushroom Spring based on 16S rRNA amplicon sequences, which was dominated by Roseiflexus members, filamentous anoxygenic chlorophototrophs. In this study, we analyzed the orange-colored undermat portion of the community of Mushroom Spring mats in a genome-centric approach and discuss the metabolic potentials of the major members. Metagenome binning recovered partial genomes of all abundant community members, ranging in completeness from ~28 to 96%, and allowed affiliation of function with taxonomic identity even for representatives of novel and Candidate phyla. Less complete metagenomic bins correlated with high microdiversity. The undermat portion of the community was found to be a mixture of phototrophic and chemotrophic organisms, which use bicarbonate as well as organic carbon sources derived from different cell components and fermentation products. The presence of rhodopsin genes in many taxa strengthens the hypothesis that light energy is of major importance. Evidence for the usage of all four bacterial carbon fixation pathways was found in the metagenome. Nitrogen fixation appears to be limited to Synechococcus spp. in the upper mat layer and Thermodesulfovibrio sp. in the undermat, and nitrate/nitrite metabolism was limited. A closed sulfur cycle is indicated by biological sulfate reduction combined with the presence of genes for sulfide oxidation mainly in phototrophs. Finally, a variety of undermat microorganisms have genes for hydrogen production and consumption, which leads to the observed diel hydrogen concentration patterns.
Introduction
Microbial mat communities inhabiting the effluent channels of Octopus and Mushroom Springs within the Lower Geyser Basin at Yellowstone National Park (YNP) have been studied for nearly 50 years (Brock, 1967; Ward et al., 2012). In these studies, the chlorophototrophic bacterial populations, i.e., chlorophyll (Chl)-based phototrophs including members of the Cyanobacteria, Chloroflexi, and the recently discovered Chloracidobacterium (Cab.) thermophilum and “Candidatus Thermochlorobacter aerophilum,” have generally been the main focus (Bauld and Brock, 1973; Nold and Ward, 1996; Bryant et al., 2007; van der Meer et al., 2007; Steunou et al., 2008; Becraft et al., 2011; Klatt et al., 2011, 2013b; Liu et al., 2011, 2012; Tank and Bryant, 2015a,b; Tank et al., 2017). Only recently, the diversity of the microbial community in the microoxic/anoxic region of the mat has been explored (Thiel et al., 2016).
The upper green layer of the mats of Mushroom Spring and/or Octopus Spring (Figure 1) have been studied using metagenomic (Klatt et al., 2011), metatranscriptomic (Liu et al., 2011, 2012; Klatt et al., 2013b), metaproteomic (Schaffert et al., 2012), and metametabolomic (Kim et al., 2015) analyses. This layer is dominated by six chlorophototrophic bacterial taxa, in particular two types of cyanobacteria, Synechococcus sp. Types A and B' (Bhaya et al., 2007), chlorophototrophic members of the Chloroflexi (Roseiflexus sp., Chloroflexus sp., and “Ca. Roseilinea gracile”; Klatt et al., 2011, 2013b; Tank et al., 2017) as well as two unusual microaerophilic anoxygenic photoheterotrophs, Chloracidobacterium thermophilum (Bryant et al., 2007; Garcia Costas et al., 2012a,b; Tank and Bryant, 2015a,b) and “Ca. Thermochlorobacter aerophilum” (Liu et al., 2012; Tank et al., 2017). Additionally, two heterotrophic taxa were found in a metagenomic analysis (Klatt et al., 2011). They were later identified as members of the phylum Armatimonadetes and the EM3 group tentatively affiliated with the Thermotogae (Thiel et al., 2016).

Figure 1. Picture of Mushroom Spring in Yellowstone National Park and a microbial mat core, taken at 60°C.
The microbial composition of the orange-colored undermat was assessed in a previous study using metagenomic sequencing and amplicon studies of 16S rRNA genes (iTag) (Thiel et al., 2016). In comparison to the upper green layer, the Mushroom Spring undermat part of the community was shown to be highly diverse but very uneven; it is dominated by members of a single genus, Roseiflexus, that includes filamentous anoxygenic phototrophs, which also inhabit the upper green layer as shown by molecular- and microscopy-based studies (“OS type C”; Nübel et al., 2002; Ward and Cohan, 2005; Klatt et al., 2007, 2013a,b; Thiel et al., 2016; Tank et al., 2017). In addition to the members of the upper green layer of the mat, all of which were also detected in the undermat in lower abundance, further photo- and chemotrophic bacteria were identified. Pseudothermotoga sp. was the second most abundant member. Furthermore, a novel Armatimonadetes, a member of the Aquificae (Thermocrinis sp.) and a novel chlorophototrophic member of the phylum Chloroflexi, “Ca. Roseilinea gracile,” were identified as abundant members of the undermat (Thiel et al., 2016). Less abundant taxa, but still representing ≥1% of the undermat part of the microbial community based on amplicon reads, were a member of the phylum Atribacteria (OP9/JS1); a sulfate-reducing Thermodesulfovibrio sp.; a member of the phylum Planctomycetes; a member of the EM3 group tentatively affiliated with the Thermotogae, as well as a putative member of the Arminicenantes (OP8). According to the iTag analysis, Archaea are rare, and no metagenomic bin representing an archaeon was identified. At present, little is known about the physiological, metabolic, and functional potentials of these heterotrophic members of the undermat layers of the community.
The overall goal of this research is to investigate the complete microbial mat community at Mushroom Spring and to develop a comprehensive understanding of the microbial ecology of the microbial mats of this hot spring. As noted above, the composition and diversity of the undermat was described in a previous report (Thiel et al., 2016). In this report, we focus on a description of the metabolic potentials and hypothesized interactions among mat community members.
Materials and Methods
Sampling, DNA Extraction, Metagenomic Sequencing, and Binning
Sample collection, DNA extraction, and DNA sequencing were performed as described (Thiel et al., 2016). In brief, the samples were collected on August 10th, 2011 from a microbial mat in an effluent channel of the siliceous and slightly alkaline Mushroom Spring in the Lower Geyser Basin of YNP, WY (USA). Samples were collected using a #4 cork borer (⊘ = 9 mm) at a site where the water above the mat was 60°C (Figure 1). The microbial mat is made up of an upper green layer (1–2 mm thick), which mainly consists of different chlorophototrophic bacteria, and an orange-colored undermat layer. Genomic DNA was extracted from the orange-colored undermat layer from a depth of ~3–5 mm; tests showed that DNA from below this level was too degraded to analyze. The metagenomic DNA was sequenced at the DOE Joint Genome Institute (JGI) using HiSeq Illumina technologies. HiSeq DNA sequences were assembled and then clustered into bins by oligonucleotide frequency pattern analyses using the emergent self-organizing map (ESOM) method described by Dick et al. (2009). Initial binning was conducted with sequences ≥5.0 kb in length (Thiel et al., 2016), whereas in this study binning was conducted using sequences ≥2.5 kb. Reference genomes as well as metagenomic bin information from the binning of ≥5.0 kb sequences was used for initial identification of the bin. Metagenomic bins were treated as partial genomes of single taxa, although discussion of ecotype diversity within these taxa is raised where appropriate below; Amphoranet (http://pitgroup.org/amphoranet/; Kerepesi et al., 2014) was used to assess the phylogenetic marker genes present in each bin and to assign taxonomic affiliations. Completeness of the partial genomes was further assessed using CheckM (Parks et al., 2015). The partial genomes and reference genomes of closely related bacteria were annotated using RAST and used to assess and compare the physiological and metabolic potential of mat members (Table 1; Aziz et al., 2008; Overbeek et al., 2014). Detailed descriptions of the methods for DNA extraction, library construction, sequencing, and data analyses are found in the Supplementary Materials in Thiel et al. (2016).
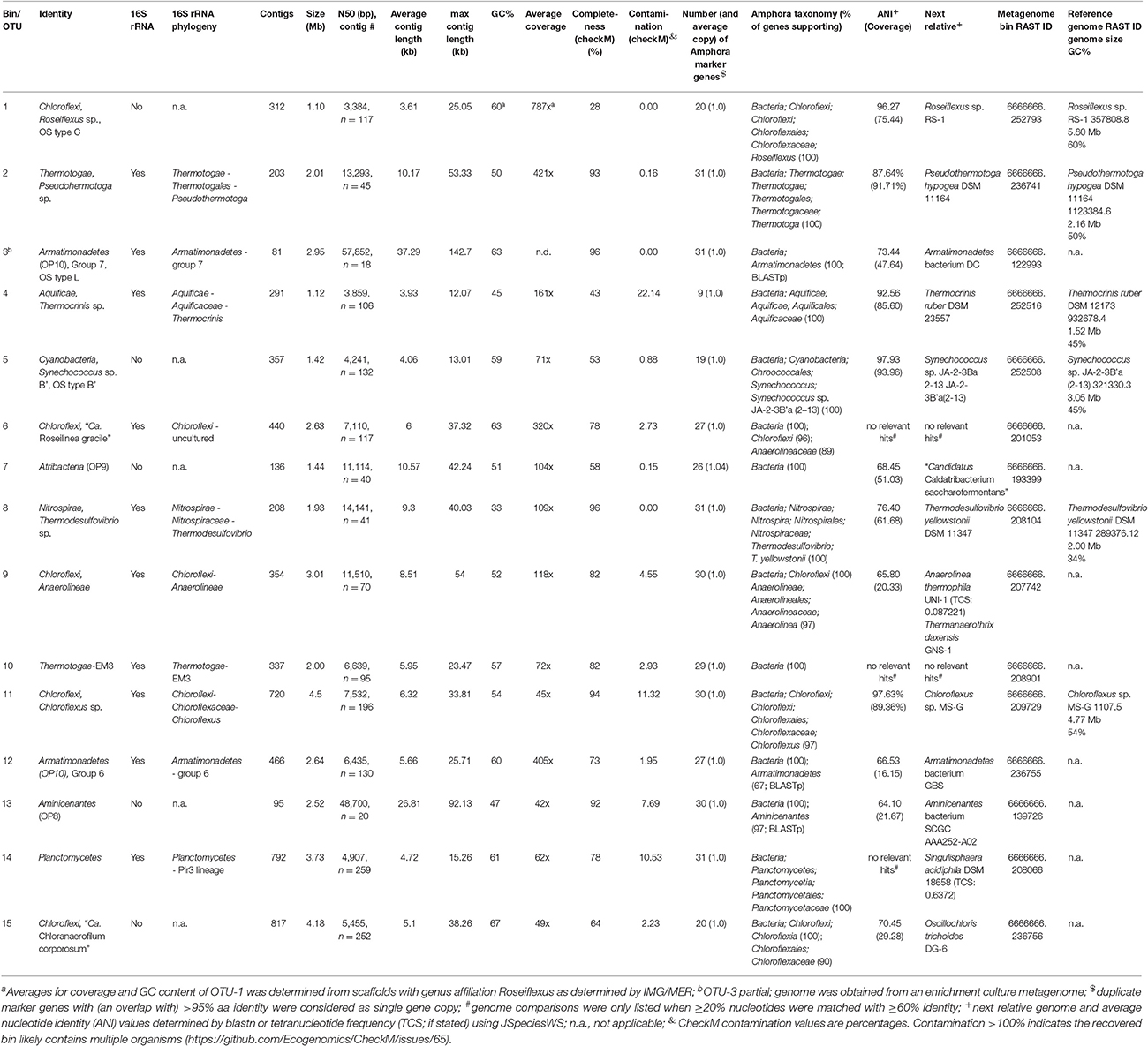
Table 1. List of metagenomic bins and reference genomes used in this study.
Metagenomic Analysis
In addition to the partial genomes, the assembled metagenome was analyzed separately. For functional analyses of the metagenome, the “Gene Search” and “BLAST” functions implemented in the JGI Integrated Microbial Genomes and Microbiome Samples Expert Review (IMG/MER) system (https://img.jgi.doe.gov/cgi-bin/mer/main.cgi) were used. Amino acid sequences of predicted open reading frames were subjected to searches against the NCBI conserved domain database (CDD; http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml; Marchler-Bauer and Bryant, 2004; Marchler-Bauer et al., 2009, 2011, 2015) as well as to BLASTp searches against the “non-redundant sequences” (nr) database (http://blast.ncbi.nlm.nih.gov/Blast.cgi; Altschul et al., 1990, 1997) to verify function and taxonomic affiliation.
Phylogenetic Analyses
Ribosomal RNA genes were extracted and downloaded from the annotated JGI metagenome dataset ID 3300002493. Phylogenetic analyses of ribosomal RNA sequences, as well as functional genes, were conducted using ARB software for sequence analysis (Ludwig et al., 2004). Publicly available databases for 16S rRNA and dsrAB/DsrAB sequences (SILVA SSU Ref NR 123 database released in July 2015: http://www.arb-silva.de/projects/ssu-ref-nr/; and dsrab_dome_v3.arb, http://www.microbial-ecology.net/download; Müller et al., 2015) were used. Databases for additional functional genes were specifically created de novo. Sequences were imported as amino acid or nucleotide sequences in FASTA format and aligned using the ClustalW Protein Aligner (slow and accurate settings) implemented in the ARB software package; the alignments were then inspected and edited manually. Using the PHYML software implemented in the ARB package, phylogenetic trees were constructed with the Maximum Likelihood method; the inferred confidence was based on 100 bootstrap replicates.
Taxonomic Identification
Affiliation to specific mat members was inferred by nucleotide frequency-based binning using ESOM if gene sequences were located on scaffolds with nucleotide lengths of >2.5 kb. Previously observed bins obtained with >5.0 kb length sequences (Thiel et al., 2016), as well as included reference genomes (Table S1) and phylogenetic marker genes determined by Amphoranet (http://pitgroup.org/amphoranet/; Kerepesi et al., 2014), were further used to identify bins. Because Amphoranet uses an outdated NCBI taxonomy file version (last updated 2011; https://www.biostars.org/p/122761/; C. Kerepesi, pers. commun.), additional BLASTp searches were conducted for marker gene sequences that produced ambiguous results and/or were obtained from recently described or candidate phyla. Additional information on the taxonomic identity of scaffolds and genes was obtained from the “phyloDist” method implemented in the JGI IMG/MER software as well as from BLAST search results. All data used in this study is available through public databases. The metagenome is available at IMG/M and IMG/MER under genome ID 330002493. RAST annotation jobs of all (partial) genomes used in this study are publically available using the guest account for the RAST online database (http://rast.nmpdr.org/; username “guest” with password “guest”; for job ID information see Table 1). The JSpecies Web Server (JSpeciesWS; Richter et al., 2015) was used to determine most closely related genomes by tetranucleotide search (TCS) and average nucleotide identity (ANI) values.
Results and Discussion
Introductory Comments
In this study, we analyzed the 232-Mb assembled metagenome of the orange-colored undermat of the chlorophototrophic microbial mat at Mushroom Spring in Yellowstone National Park. The focus of this analysis was the metabolic lifestyle and genetic potentials of the 15 most abundant mat members identified by 16S rRNA gene amplicon study (Thiel et al., 2016), as well as nutrient cycling within the mat community. Tetranucleotide frequency patterns were used to bin the metagenomic sequences in order to identify partial genomes of uncultured mat members. In an initial approach, binning of 5,362 contigs with length of >5 kb resulted in 37 partial genomes (Thiel et al., 2016). In this study we conducted another binning analysis that included scaffolds with lengths >2.5 kb (13,766 contigs). The resulting metagenomic bins were largely congruent with the previously defined bins (Thiel et al., 2016) and were identified by these congruencies in combination with phylogenetic marker gene analyses and additional reference genomes now included in the binning process (Table S1). The newly obtained metagenomic bins representing the most abundant members of the undermat, as indicated by a 16S rRNA amplicon study (Thiel et al., 2016), are shown in Table 1.
The partial genomes as well as the entire metagenome were independently analyzed to identify genes encoding key enzymes of different metabolic pathways. Results discussed in the following sections are mainly based on genes present in sequence bins. Due to the incomplete character of the genomes, inferences concerning metabolism can only be deduced from the presence but not the absence of genes for diagnostic enzymes. Genes for key enzymes that were present in the assembled but unbinned metagenome were also identified; this analysis was performed to identify specific genes present on short scaffolds that were excluded in the binning approach. When unbinned genes from the metagenome could unambiguously be affiliated with a specific community member, they were included in the metabolic predictions for that organism. In particular, genes encoding key enzymes in phototrophic and chemotrophic energy production, as well as nutrient (carbon, nitrogen, and sulfur) metabolism were analyzed.
The whole mat community was found to be a mixture of photo- and chemo-trophic organisms, using bicarbonate as well as organic carbon sources derived from different cell components and fermentation products. The mat community, which was originally described as a “algal/cyanobacterial mat,” contained a panoply of chlorophototrophs (Doemel and Brock, 1976; Brock, 1995; Bryant et al., 2007; Klatt et al., 2011; Thiel et al., 2016; Tank et al., 2017). Based on 16S rRNA sequence analysis, most of the chlorophototrophs were higher in abundance in the upper green layer, while one abundant as well as two low-abundance chlorophototrophs were detected almost entirely in the undermat (Thiel et al., 2016).
The surprising finding of diverse rhodopsin genes (see below) strengthens the conclusion that light energy is of paramount importance in these mats. Carbon fixation pathways are similarly diverse, and representatives of the Bacteria performing carbon fixation by each of the four known autotrophic pathways in Bacteria were identified in the metagenome. Cyanobacterial nitrogen fixation appears to be the main source for nitrogen, which is made available to the mat community in forms of amino acids and ammonium, while dissimilatory nitrogen metabolism was not detected. A closed sulfur cycle is hypothesized to be maintained by biological sulfate reduction in combination with sulfide:quinone reductase (SQR)-based sulfide oxidation and sulfur oxidation (SOX) system-mediated sulfur, sulfite, and thiosulfate oxidation (Friedrich et al., 2001; Dahl and Friedrich, 2008; Härtig et al., 2014). Sulfur disproportionation is predicted to occur in one low-abundance mat member. Finally, hydrogen is produced and consumed in the mat community by a variety of organisms leading to diel concentration patterns, which have been measured using microsensors (Revsbech et al., 2016).
Light Utilization: Chlorophototrophy and Retinalophototrophy
Chlorophototrophy
Light is the most important energy source for the phototrophic microbial mat at Mushroom Spring. Although it has often been referred to as a “cyanobacterial mat” (Doemel and Brock, 1976; Brock, 1995; Ward et al., 1998; Ramsing et al., 2000; Klatt et al., 2011) highly diverse phototrophic bacteria inhabit the mat community. Sixteen different chlorophototrophic bacteria have been identified by a combination of molecular and isolation- and enrichment-based studies (Table 2; Klatt et al., 2011; Thiel et al., 2016; see Figure 4 in Tank et al., 2017). Genes encoding type-1 photosynthetic reaction center (RCs; psaA/psaB and pscA) and type-2 RCs (pufLM, psbA, and, psbD) of 10 chlorophototrophs were identified in the undermat metagenome at 60°C (Table S2). Taxa containing type-2 RCs are the dominant oxygenic chlorophototrophic Cyanobacteria: Synechococcus spp. Types A and B' (OTUs 22 and 5, respectively); members of the Chloroflexi: Roseiflexus spp. (OTU-1), “Ca. Roseilinea gracile” (OTU-6), Chloroflexus sp. (OTU-11) and “Ca. Chloranaerofilum corporosum” (OTU-15); as well as two less abundant chlorophototrophic Alphaproteobacteria: “Ca. Elioraea thermophila” (OTU-46) and “Ca. Roseovibrio tepidum” (OTU-121; Table S2; Thiel et al., 2016; Tank et al., 2017).
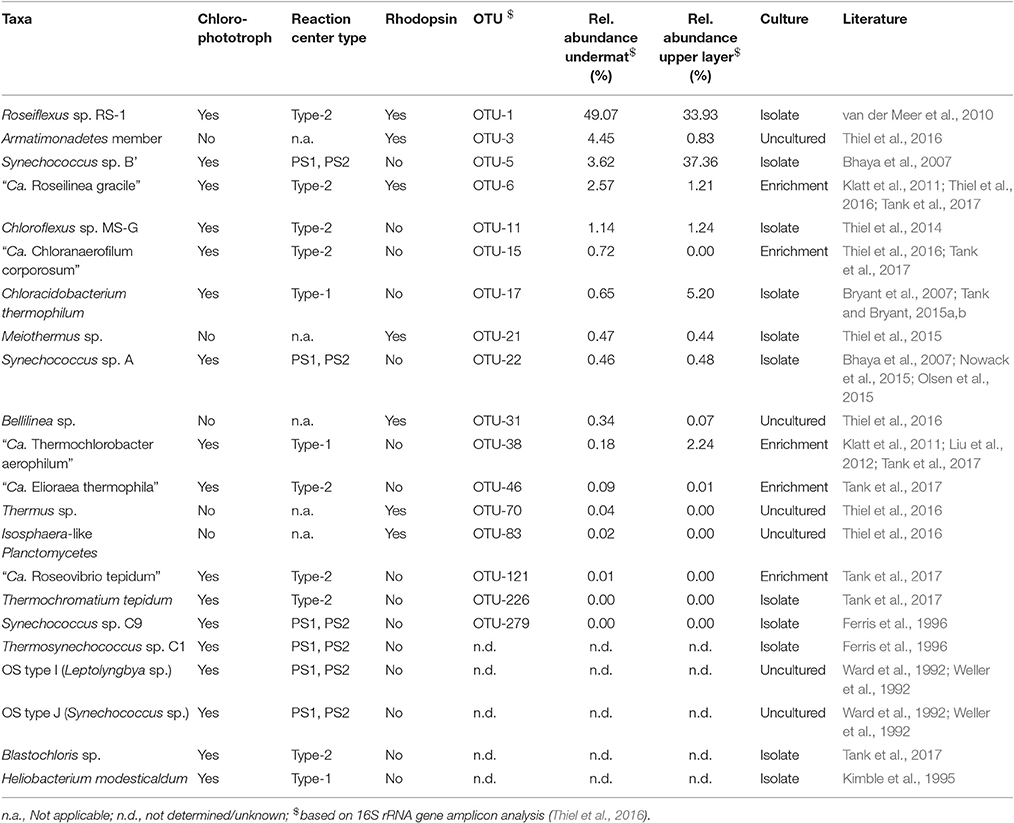
Table 2. Microbial mat members containing genes for chlorophototrophy and/or rhodopsin.
Aside from Synechococcus spp. Types A and B' (OTUs 22 and 5, respectively), taxa containing Type-1 RCs, Chloracidobacterium thermophilum (Bryant et al., 2007; Garcia Costas et al., 2012b; Tank and Bryant, 2015a,b) and “Ca. Thermochlorobacter aerophilum” (Liu et al., 2012), which were first identified in the upper layer of the mat, were also detected in the undermat metagenome (Table S2). Although Heliobacterium modesticaldum was previously isolated from these same mats in a region of lower temperature (Kimble et al., 1995), no pshA sequences, encoding for the P800 photosynthetic reaction center core protein of heliobacteria, were identified nor did the reference genome of this organism recruit any significant hits from the metagenome. This is presumably because H. modesticaldum does not grow at temperatures above ~56°C (Kimble et al., 1995). Similarly, no psaA and psaB genes affiliated with Thermosynechococcus sp. C1, Synechococcus sp. C9, or Leptolyngbya sp. were identified in the metagenome, although these organisms have previously been isolated from these mats (Tank et al., 2017 and references therein). This is probably explained either by their very low abundance and/or optimal growth temperatures lower than that studied here. Similarly, Thermochromatium tepidum and Blastochloris sp. have been obtained through cultivation studies from lower temperature mat samples (~50°C; Tank et al., 2017), but were not detected in the 60°C undermat metagenome.
The highest diversity in genes for type-2 reaction centers was seen for members of OTU-1, Roseiflexus spp. (16 partial pufLM genes with coverage values ranging from 71 to 1227 × Table S2). This indicates that several Roseiflexus spp. ecotypes with differences in their RC proteins are likely present in the mat community. Similarly, cyanobacterial scaffolds containing RC genes showed high diversity, suggesting the presence of different ecotypes adapted to specific light niches as shown previously (Table S2; Becraft et al., 2015; Nowack et al., 2015; Olsen et al., 2015). In both cases the high microdiversity was correlated with assembly difficulties, and only a few long scaffolds were found for these taxa. Physiological information for these two phototrophs was based on reference genomes and isolates (Bhaya et al., 2007; van der Meer et al., 2010; Nowack et al., 2015; Olsen et al., 2015).
Rhodopsins
A surprising finding is that several members of the undermat contain genes belonging to the bacteriorhodopsin superfamily (Table 2). Whether these genes are used to obtain energy from light is not clear, but this is certainly a possibility. In general, blue-green light does not penetrate deeply into the mat (see Figures 4 in Nübel et al., 2002; Becraft et al., 2015), so light wavelengths absorbed by the rhodopsin-like proteins should not be abundant in the undermat. On the other hand, light of around 450–560 nm, which includes the major absorption maxima of xanthorhodopsin (Lanyi and Balashov, 2008), is not well absorbed by Chl a, phycocyanin and carotenoids of cyanobacteria, and thus blue-green light is slightly more abundant in deeper layers than blue or red light (Nübel et al., 2002; Becraft et al., 2015). Seventeen annotated rhodopsin genes in the assembled undermat metagenome were affiliated with seven phylogenetic groups, representing an unexpected diversity of potential retinal-based phototrophy in the mat (Figure 2; Table S3). Both the diversity and the abundance of rhodopsins was considerably lower in the upper layer. Only 10 rhodopsin sequences, representing four community members, Roseiflexus sp. OTU-1, Armatimonadetes member OTU-3 (and possibly other Armatimonadetes members), “Ca. Roseilinea gracile” OTU-6 and Meiothermus sp. OTU-21, were detected in the upper layer metagenome (unpublished data). Identical rhodopsin gene sequences were also detected in the undermat metagenome. It is noteworthy that all of the members showed equal (Meiothermus sp.) or higher relative abundance in the undermat based on 16S rRNA gene amplicon analysis, indicating retinal-based phototrophy to be of unexpected importance in a layer with low irradiance (Table 2; Thiel et al., 2016). Additionally, rhodopsin gene sequences affiliated with Bellilinea sp. OTU-31, Thermus sp. OTU-70, and Isosphaera-like Planctomycetes member OTU-83 were detected exclusively in the undermat metagenome. Most rhodopsin sequences were most closely related to genes annotated as “xanthorhodopsin,” a proton-pumping rhodopsin first described in Salinibacter ruber, which contains a second carotenoid chromophore (salinaxanthin) as a light-harvesting antenna molecule (Balashov et al., 2005). The presence of an antenna chromophore in xanthorhodopsin would certainly be advantageous in niches that receive low irradiance. Additional genes were identified as Na+-pumping rhodopsin (Figure 2).
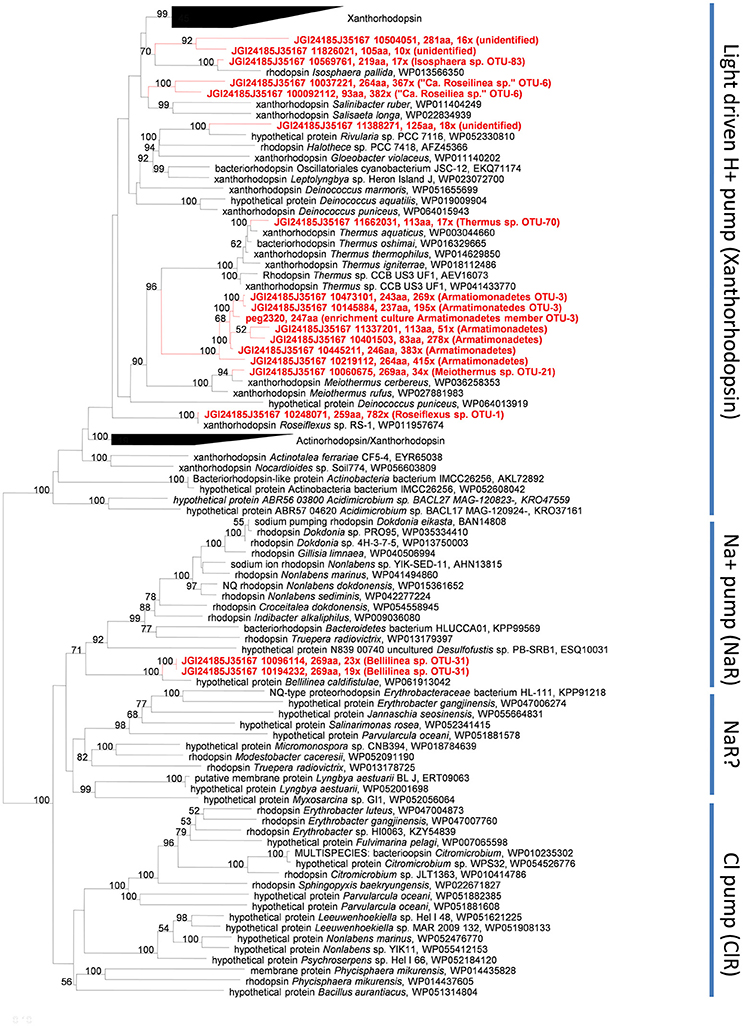
Figure 2. Maximum Likelihood tree based on rhodopsin gene amino acid sequences derived from the Mushroom Spring undermat metagenome (in red) and related sequences.
Xanthorhodopsin genes with phylogenetic affiliation to light-driven, xanthorhodopsin-like H+ pumps were found in the anoxygenic, chlorophototrophic Chloroflexi members Roseiflexus spp. OTU-1 and “Ca. R. gracile” OTU-6. This could indicate the co-existence of xanthorhodopsin and photosynthetic apparatus, as previously suggested for the thylakoid-less cyanobacterium Gloeobacter violaceus, which contains gloeorhodopsin, a rhodopsin closely related to xanthorhodopsin from Salinibacter ruber and Roseiflexus sp. RS-1 (Choi et al., 2014). Rhodopsin genes in cyanobacteria do not seem to be a rare occurrence, as a keyword search in the NCBI database reveals more than 40 entries at the time of writing (March 2017). However, the absence of a carotenoid oxygenase assigned to these organisms in all datasets, the Roseiflexus sp. RS-1 genome, “Ca. Roseilinea gracile” OTU-6 partial genome and the metagenome, suggests that either a currently unidentified protein is responsible for retinal production or retinal is derived from an external source (as discussed below).
Six of the xanthorhodopsin genes in the undermat metagenome are associated with members of the phylum Armatimonadetes (Figure 2). Due to their high sequence similarity and similar coverage values, these sequences are probably derived from different sub-populations of the same OTU, OTU-3, for which a high microdiversity has been observed (Thiel et al., 2016). Alternatively, they could be derived from different members of the Armatimonadetes that are present in the mat community (e.g., OTU-3, OTU-12, and OTU-18). Whether the low-abundance xanthorhodopsin genes that cluster with sequences for Meiothermus cerbereus and Thermus aquaticus in the phylogenetic tree, really represent H+-pumping xanthorhodopsins is uncertain, because the rhodopsin genes in these organisms have not been studied to our knowledge nor has their functionality been verified.
The possibility of light energy usage by members of the microbial mats was further supported by a rhodopsin gene, which is phylogenetically affiliated with a less abundant member of the Chloroflexi, Bellilinea sp. OTU-31. Phylogenetic affiliation placed this gene within a clade of light-driven sodium pumps (Figure 2) and a similar sequence is also present in the genome of the closest relative, Bellilinea caldifistulae, which is a chemotrophic member of the phylum Chloroflexi. Although no specific function has been demonstrated for the rhodopsin gene in the type strain, when the gene obtained from the metagenome was heterologously expressed in Escherichia coli, the resulting strain produced a purple-colored protein and showed sodium-pumping activity (Y. Nakajima, S. Yoshizawa, V. Thiel, D.A. Bryant, unpublished results). The presence of six putative, sodium-dependent transporters in the partial genome of the Bellilinea sp. member in the undermat further strengthens the hypothesis that this rhodopsin has sodium-pumping activity. The evidence suggests that light-driven nutrient uptake/transport using a secondary sodium gradient, similar to the light-driven uptake of vitamin B1 in a marine flavobacterium (Gómez-Consarnau et al., 2016), might be important in this member of the Chloroflexi.
Although it is likely that the xanthorhodopsin genes described above are functional, some uncertainty exists because the partial genomes of organisms with these xanthorhodopsin genes do not generally encode a β-carotene 15,15′-monooxygenase. Xanthorhodopsin in S. ruber was shown to contain retinal and carotenoid salinixanthin (Balashov et al., 2005). Retinal is an oxidative cleavage product of carotenoids, most often β-carotene, which is catalyzed by a β-carotene 15,15′-monooxygenase, sometimes called β-carotene 15,15′-dioxygenase, in eukaryotes (Woggon, 2002; Kim et al., 2009). In bacteria proteins cleaving β-carotene to produce retinal include bacteriopsin-related protein (Brp) and bacteriorhodopsin-related protein-like homolog protein (Blh; (Kim et al., 2009) and references therein). Although they have the same function, mammalian β-carotene 15,15′-dioxygenase (BCD) and prokaryotic Brp/Blh family β-carotene 15,15′-monooxygenases are quite different and share only 20% amino acid sequence similarity (Sabehi et al., 2005; Kim et al., 2009). Only low-abundance sequences in the metagenome, none of which was affiliated with members containing highly abundant rhodopsin genes, showed similarity to β-carotene 15,15′-monooxygenase from Oscillatoria nigroviridis PCC 7112 (Table S4). Only one combination of a gene encoding an enzyme for possible cleavage of β-carotene and a xanthorhodopsin-like gene was identified for an Isosphaera pallida-like organism (OTU-83), a less abundant member of the undermat. This indicates the possibility of a functional xanthorhodopsin and thus light utilization for this organism. However, the failure to detect highly abundant β-carotene 15,15′-monooxygenases affiliated with more abundant rhodopsin-containing complete or partial genomes does not necessarily invalidate the hypothesis that these xanthorhodopsins are used for light energy capture. Firstly, sequences encoding enzymes similar to β-carotene 15,15′-monooxygenases, which could not be identified taxonomically, were present in the unassembled part of the metagenome. Secondly, other oxygenases could substitute for this function in different bacteria, or carotenoids other than retinal could possibly be bound to the rhodopsins. Thirdly, a still unknown family of β-carotene cleaving enzymes/genes with low sequence similarity to known carotenoid oxygenases but exhibiting the same function in other xanthorhodopsin-containing microbes might still be discovered. Finally, β-carotene 15,15′-monooxygenases that produce retinal in some of the major mat organisms (e.g., Synechococcus spp., Cab. thermophilum) might be used as a community resource by other mat members. This last possibility would be of particular ecological interest, as it would emphasize the close relationships and interactions that are connecting the mat members and stabilizing the community. Indications for this kind of co-factor dependency from the environment were recently suggested for the freshwater actinobacterium Rhodoluna lacicola, which expresses a functional rhodopsin gene but exhibits proton pumping activity only when exogenous retinal is provided (Keffer et al., 2015).
Autotrophic Mat Members
In order to assess the autotrophic growth potential of the mat members, we analyzed key genes of different autotrophic carbon fixation pathways. Currently six autotrophic pathways are known (see Fuchs, 2011; Hügler and Sievert, 2011 for recent reviews). Four pathways, namely the Calvin-Benson-Bassham (CBB) cycle, the reverse tricarboxylic acid (rTCA) cycle, the Wood-Ljundahl (WL) pathway, and the 3-hydroxypropionate (3-HP) bi-cycle are known to occur in Bacteria, while the other two pathways are restricted to Archaea (Fuchs, 2011; Hügler and Sievert, 2011; Tang et al., 2011). Suggesting the occurrence of a quite diverse autotrophic community, genes encoding the key enzymes of all four bacterial carbon fixation pathways were detected in the undermat metagenome. This could lead to differences in carbon isotope fractionation (Hayes, 2001), although the CBB and 3-HP pathways are likely to have the most significant impact, due to the greater abundance of organisms containing these pathways.
Calvin-Benson-Bassham Cycle
The key enzyme for the CBB cycle is the CO2-fixing enzyme ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO). Almost all sequences encoding type-I RubisCO that were identified in the metagenome were affiliated with the dominant cyanobacteria, Synechococcus spp. Types A and B'; consistent with their known ability to fix inorganic carbon via the CBB cycle. Only one low-abundance partial sequence appeared to be affiliated with the proteobacterial phylum (JGI24185J35167_11464881; Table S5). Two low-abundance anoxygenic phototrophic Alphaproteobacteria were found in the undermat metagenome, “Ca. Elioraea thermophila” and “Ca. Roseovibrio tepidum” (see above). Because the genome of the closely related Elioraea tepidum is available and does not contain any autotrophic pathway genes, this could be an indication that a CBB cycle occurs in “Ca. Roseovibrio tepidum” represented by OTU-121, which is further supported by the presence of a gene for a type-1 RubisCO in the non-phototrophic but closely related Rhodospirillales/Acetobacteraceae member Roseomonas gilardii subsp. rosea ATCC BAA-691 (Acc. No. NZ_JADY01000025; L882_RS0115610).
3-Hydroxypropionate (3-HP) Bi-Cycle
The 3-HP bi-cycle is a complex autotrophic carbon fixation pathway that has been fully elucidated in Chloroflexus aurantiacus and most likely also operates in other autotrophic members of the Chloroflexaceae (Klatt et al., 2007, 2013b; Zarzycki et al., 2009; van der Meer et al., 2010). The multifunctional enzymes malonyl-CoA reductase, propionyl-CoA synthase, and malyl-CoA/β-methylmalyl-CoA/citramalyl-CoA (MMC) lyase are considered to be the key and diagnostic enzymes of the 3-HP bi-cycle (Fuchs, 2011; Hügler and Sievert, 2011 and references therein). The incorporation of CO2 into biomass is catalyzed by the CO2-fixing enzymes acetyl-CoA and propionyl-CoA carboxylase. Other characteristic enzymes are CoA-transferases and hydratases. The complete pathway is shown in Figure S1. Apart from serving as a carbon fixation pathway for autotrophic growth, the 3-HP bi-cycle may be utilized together with the TCA cycle as mixotrophic pathways for the simultaneous incorporation of organic and inorganic carbon (Klatt et al., 2007, 2013b; Zarzycki and Fuchs, 2011; Bryant et al., 2012). In nature, mixotrophy is most likely the predominant lifestyle for these organisms (also see below).
Sequences for all three of the diagnostic enzymes were detected in the metagenome for Chloroflexi members, the highly abundant Roseiflexus sp. (OTU-1), Chloroflexus sp. MS-G (OTU-11), and “Ca. Chloranaerofilum corporosum”(OTU-15), which indicates that these undermat organisms probably have the capacity to fix inorganic carbon via the 3-HP bi-cycle and grow either mixotrophically or autotrophically (Table S6).
The high microdiversity for malonyl-CoA reductase gene sequences associated with Roseiflexus spp. OTU-1 (Table S7) indicates the presence of putative ecotypes potentially with differences in carbon metabolism—more specifically in enzymes involved in carbon fixation. Several genes encoding important enzymes of the 3-HP bi-cycle, including acetyl-CoA carboxylase, malonyl-CoA reductase and propionyl-CoA synthase, are arranged in an apparent operon in Roseiflexus sp. RS-1 (RoseRS_3199–RoseRS_3203; see Klatt et al., 2007; van der Meer et al., 2010). Transcript levels for the genes in this operon increased during the day in the Roseiflexus spp. population in the upper green layer (Klatt et al., 2013b). A similar increase during day-time hours was observed when RoseRS_3199–3203 were used as mapping targets for RNA-seq data for the undermat (V. Thiel and D. A. Bryant, unpublished results). These data indicate that the 3-HP bi-cycle is probably active during the day when light is available and the mat is highly oxic in the upper layer.
For other members of the Chloroflexi, OTU-6 (“Ca. Roseilinea gracile”) and OTU-9 we could not detect any genes for key enzymes of the 3-HP bi-cycle (Table S6). OTU-9 instead has the genes for the reductive acetyl-CoA pathway (see below), while OTU-6 seems to have a photoheterotrophic lifestyle.
Reductive TCA Cycle
Key enzymes of the reductive or reverse TCA cycle include 2-oxoglutarate synthase (2-oxoglutarate:ferredoxin oxidoreductase), fumarate reductase and especially the citrate-cleaving enzyme. The ATP-dependent cleavage of citrate into acetyl-CoA and oxaloacetate is a complex reaction, that can be accomplished either by the combined action of the enzymes citryl-CoA synthetase (CCS) and citryl-CoA lyase (CCL) or by the heterodimeric enzyme ATP-citrate lyase (ACL; Aoshima, 2007; Hügler et al., 2007; Hügler and Sievert, 2011). Sequences coding for ACL [either the conventional type I, or the novel proposed type II (Hügler and Sievert, 2011; Thiel et al., 2012)] were not found in the metagenome. However, genes encoding the two-enzyme variant of citrate cleavage were present in the metagenome and could be assigned to two mat members. Two copies each of the genes encoding CCS and CCL were affiliated with a highly abundant mat member, Thermocrinis sp. OTU-4, which supports the presence of at least two different populations for this taxon in the mat as previously discussed (Thiel et al., 2016). Chemolithoautotrophic growth has been shown to occur for the closest relative, Thermocrinis ruber with H2, thiosulfate and S0 as electron donors and oxygen as electron acceptor (Huber et al., 1998; Hügler et al., 2007). In addition to CCS and CCL, all other genes necessary for a functional reductive citric acid cycle in Thermocrinis sp. OTU-4 could be identified in the metagenome (see Figure S2). In addition to the two-step citrate cleavage mechanism, Thermocrinis sp. OTU-4 also accomplishes a two-step carboxylation of 2-oxoglutarate to isocitrate using the enzymes 2-oxoglutarate carboxylase and oxalosuccinate reductase instead of the conventional isocitrate dehydrogenase. Both of these two-step reactions, the two-step citrate cleavage, as well as the two-step carboxylation of 2-oxoglutarate, might be relics of an ancient variant of the reductive TCA cycle, that has been shown to occur in other members of the family Aquificaceae, e.g., Hydrogenobacter thermophilus or Aquifex aeolicus (Aoshima, 2007; Hügler et al., 2007).
Another set of genes potentially encoding the citrate-cleaving enzymes CCS and CCL were found on scaffold JGI24185J35167_1000715, which is part of a metagenomic bin identified as OTU-14, a member of the Planctomycetes. The genes were annotated as “succinyl-CoA synthetase alpha and beta subunit” and “citrate synthase” (JGI24185J35167_10007157-9). BLASTp searches against the nr database identified hypothetical proteins from Spirochaetes, Phycisphaerae and Plantomycetes as closest relatives with amino acid sequence identity values of 52–54%. A CDD search identified the genes as “CS_ACL-C_CCL superfamily” citrate synthase as well as “succinyl CoA synthetase alpha and beta subunits.” A phylogenetic analysis using the amino acid sequences of both subunits of ACL and the amino acid sequencing of the concatenated enzymes CCS and CCL is shown in Figure 3. The putative citrate cleaving genes of OTU-14 and related genes from other Plantomycetes cluster between the CCS-CCL of the Aquificae and Leptospirillum and ACL type II sequences. Thus, the genes for citrate-cleaving enzymes of these Planctomycetes bacteria give important insights into the evolution of the reductive TCA cycle and ACL. Phylogenetically, these enzymes are intermediates between the ancient variant that is still present within the Aquificaceae and the type II ACL (Figure 3). Interestingly, only OTU-14 shows separate genes for CCS and CCL, while the closest sequences from the database (e.g., Planctomycetes bacterium DG20) has a heterodimeric ACL; the gene for the second subunit of CCS (ccsB) and ccl are fused to form one gene, aclA (Figure S3). A possible explanation for this difference would be temperature. While OTU-14 is a thermophile (water temperature above the mat was 60°C; Thiel et al., 2016), the closely related Planctomycetes DG20 was detected in a metagenomics study of estuary sediments from North Carolina (Baker et al., 2015), and thus lives in a mesophilic environment. Another surprising feature of the bin assigned to Planctomycetes sp. OTU-14 is, that in addition to the key genes of the reductive TCA cycle, it also harbors genes encoding carbon monoxide dehydrogenase/acetyl CoA synthase and other enzymes of the reductive acetyl-CoA pathway. Thus, this organism has the genomic potential to use two different carbon fixation pathways for autotrophic growth (see below).
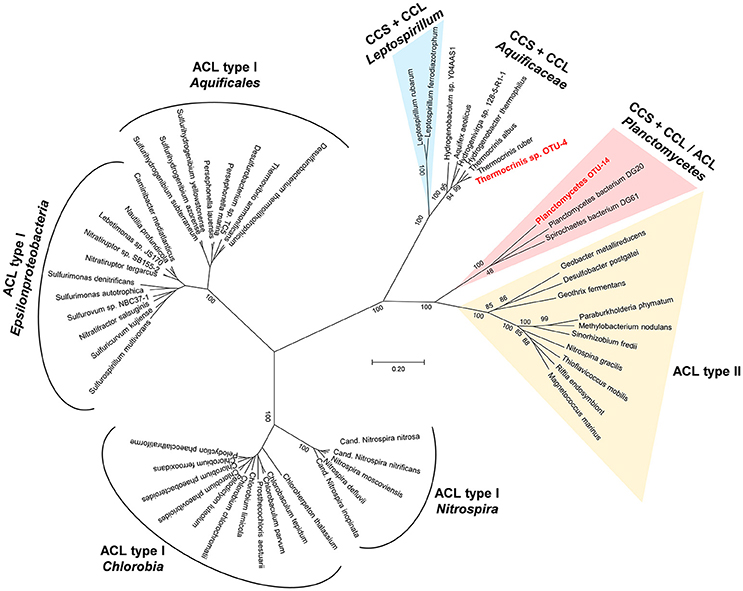
Figure 3. Phylogenetic tree showing the relationship of protein sequences identified in the undermat metagenome (in red) as Types I and II ACL (ATP dependent citrate lyase) as well as citryl-CoA synthetase (CCS) and citryl-CoA lyase (CCL) protein sequences from the public databases.
The reductive carboxylation of either succinyl-CoA or acetyl-CoA to 2-oxoglutarate or pyruvate, respectively, is carried out by the enzymes 2-oxoglutarate or pyruvate:ferredoxin oxidoreductases. Although necessary for autotrophic bacteria using the reductive TCA cycle for carbon fixation, these enzymes can also occur in heterotrophic or mixotrophic microbes. These enzymes perform anaplerotic CO2 fixation reactions that may play an important role within the undermat portion of the community, as genes encoding several versions of these enzymes are found in the metagenome. Thus, several undermat organisms possibly require an ample supply of /CO2 for biosynthetic purposes as has been shown for Cab. thermophilum (Tank and Bryant, 2015a,b) and hypothesized for “Ca. T. aerophilum” (Liu et al., 2012).
Wood-Ljungdahl Pathway
The reductive acetyl-CoA or Wood-Ljungdahl (WL) pathway is the only known carbon fixation pathway used by Bacteria as well as by Archaea, consistent with the hypothesis that it is the most ancient pathway (e.g., Weiss et al., 2016). The key enzyme of the pathway is CO dehydrogenase/acetyl-CoA synthase (CODH/ACS), which catalyzes the reduction of CO2 to CO as well as the synthesis of acetyl-CoA from the methyl- and carbonyl moieties. The reduction of CO2 to the methyl group is accomplished by enzymes that are also characteristic for this pathway (Figure S4; see Ragsdale and Pierce, 2008 for a review).
Four potentially bi-functional CODH/ACS subunit genes were identified in the metagenome (Table S8). The most abundant putative CODH/ACS gene (124 × coverage) belonged to the metagenomic bin for OTU-9, which has been associated with an Anaerolineae-like chemotrophic member of the Chloroflexi (Thiel et al., 2016). The protein shows highest similarity (55%) to CODH/ACS genes from Deltaproteobacteria and Clostridia (e.g., Desulfonatronospira sp., Ammonifex degensii) for which autotrophic growth has been observed, and enzyme activity measurements strongly suggest an active reductive acetyl-CoA pathway (Sorokin et al., 2008). Thus, the metagenomic data suggests that the Anaerolineae-like member of the Chloroflexi OTU-9 has the potential to grow autotrophically via the WL pathway. The next most abundant CODH/ACS sequences had read depths of 68× and 24×; these sequences belonged to two different potential members of the Planctomycetes, one representing OTU-14 (see below), and the other a less abundant member of the undermat layer that was not identified by 16S rRNA (Thiel et al., 2016). The least abundant CODH/ACS gene belonged to a member of the Thermodesulfobacteriaceae (OTU-26), a relative of Caldimicrobium thiodismutans, for which autotrophic growth by hydrogen oxidation or disproportionation of sulfur compounds has been demonstrated (Kojima et al., 2016). Two copies of CODH/ACS sequences were affiliated with Thermodesulfovibrio sp. OTU-8, a member of the phylum Nitrospirae. In addition to that, the genes for all other enzymes of the reductive acetyl-CoA pathway were found in the metagenomic bin representing OTU-8. Similarly, the genes for the reductive acetyl-CoA pathway were also present in the genome of the closely related species Thermodesulfovibrio yellowstonii (Table S8). Many acetotrophic, sulfate-reducing bacteria employ the WL pathway to cleave acetyl-CoA. Yet, the presence of a bifunctional CODH/ACS suggests, that Thermodesulfovibrio spp. are at least facultative autotrophs, although autotrophic growth has not been demonstrated for T. yellowstonii (Henry et al., 1994).
The most intriguing finding is the presence of genes encoding a bifunctional CODH/ACS together with genes for other enzymes of the reductive acetyl-CoA pathway in the Planctomycetes sp. OTU-14, in addition to genes of the reductive TCA cycle. We checked the metagenome of the closely related Planctomycetes sp. DG20 and also found the genes for the bifunctional CODH/ACS. Thus, these uncultured members of the Planctomycetes encode the genes for enzymes of two carbon fixation pathways, the reductive TCA cycle and the reductive acetyl-CoA pathway. So far, the presence of two different carbon fixation pathways in one bacterium has been only been described for the endosymbionts of vestimentiferan tubeworms living near hydrothermal vents and seeps (Markert et al., 2007; Thiel et al., 2012). This endosymbiotic gammaproteobacterium uses the reductive TCA cycle in addition to the CBB cycle for carbon fixation. As mentioned above, the reductive acetyl-CoA is most likely the most ancient autotrophic carbon fixation pathway (Sojo et al., 2016; Weiss et al., 2016). Braakman and Smith (2012) proposed that a hybrid reductive TCA/reductive acetyl-CoA pathway is the basis of all extant carbon fixation pathways. A more detailed study of the genomes of these members of the Planctomycetes might shed more light on the early evolution of carbon fixation.
Heterotrophic Lifestyle
Five of the 15 most abundant microbial undermat members were identified as chlorophototrophic bacteria, but only one group, the dominant cyanobacteria of the upper green layer, are photoautotrophs with regard to carbon fixation. In addition, the capacity for chemolithoautotrophic growth can be assumed for Thermocrinis sp. OTU-4, and putatively for Planctomycetes member OTU-14, Chloroflexi member OTU-9 and Thermodesulfovibrio sp. OTU-8. All other members of the mat probably grow photomixotrophically (e.g., Roseiflexus sp. OTU-1, Chloroflexus sp. OTU-11, and “Ca. Chloranaerofilum corporosum” OTU-15) or heterotropically, and thus they benefit from or depend on organic carbon sources provided by the primary producers (Table 3) by excretion or cell lysis (Figure 4).
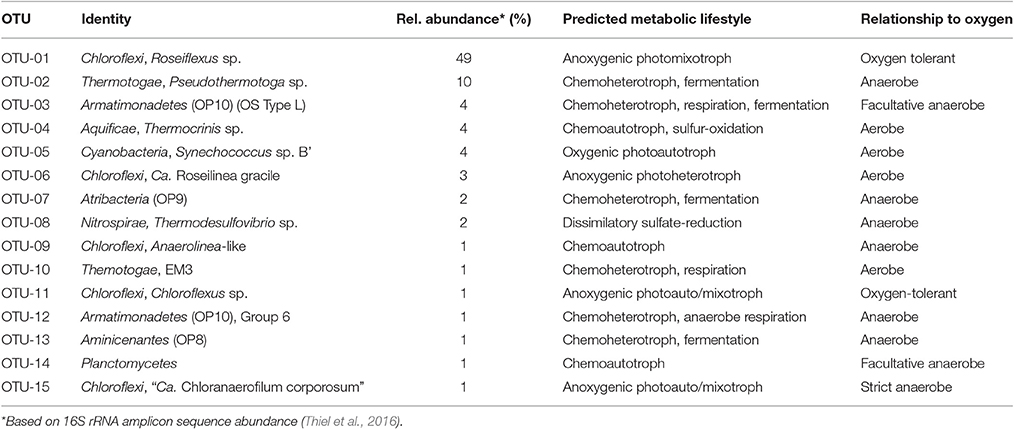
Table 3. List of the 15 most abundant undermat members (based on 16S rRNA gene amplicon sequence abundance) their relative abundance, predicted metabolic life style, and relationship to oxygen.
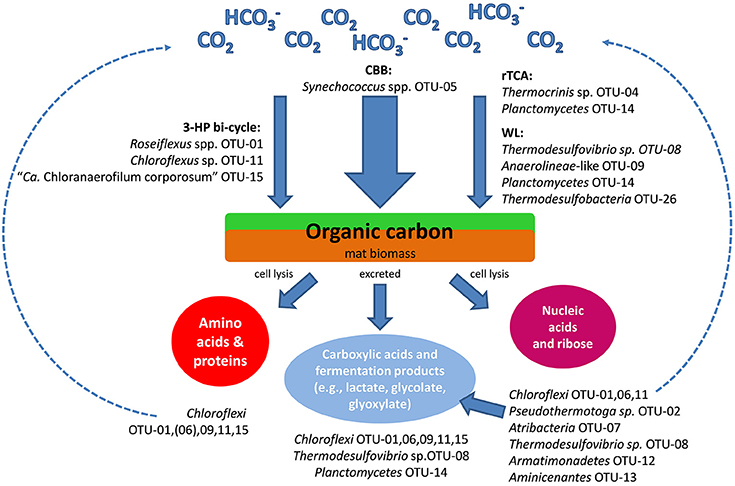
Figure 4. Schematic drawing of the hypothesized carbon cycle and the likely involved mat community members as inferred from analyses of metagenomic gene clusters. CBB, Calvin-Benson-Bassham cycle; rTCA, reverse tricarboxylic acid cycle; WL, Wood-Ljungdahl pathway; 3-HP bi-cycle - 3-hydroxypropionate bi-cycle.
Both aerobic and anaerobic respiration, as well as fermentative, heterotrophic lifestyles are indicated for the different members of the mat community (Table 3). In addition to cellular biopolymers, e.g., nucleic acids and proteins, carboxylic acids, and fermentation products are available to the heterotrophic mat community (Kim et al., 2015). Photoassimilation of fermentation products, such as acetate, glycolate, propionate and lactate, by phototrophic Chloroflexi has been demonstrated both in pure cultures as well as under in situ conditions, and the exchange of glycolate and lactate between Synechococcus spp. and Roseiflexus spp. in the microbial mats has been further supported by metabolomic and metatranscriptomic studies (Anderson et al., 1987; Bateson and Ward, 1988; Liu et al., 2012; Kim et al., 2015). Bulk protein and amino acids have been indicated to play an important role in nutrient cycling in the mat (see below). Amino acids and protein are used by Cab. thermophilum, and presumably also by “Ca. Thermochlorobacter aerophila,” not only as N-sources but also as the principal C-source (Liu et al., 2012; Tank and Bryant, 2015a,b; Tank M, unpublished data). Beyond this, little is presently known about the pathways of nutrient and carbon cycling within the mats. The metagenome and the growing number of isolates should allow one to examine the genetic potential of the heterotrophic mat members and determine more details about carbon and nutrient cycling in the mats.
Lactate Utilization
Lactate is present in the microbial mat in the late afternoon, and it has been suggested that it is exchanged between Synechococcus and Roseiflexus spp. (Kim et al., 2015). Lactate permease genes are present in the genome(s) of Roseiflexus spp.; their relative transcript levels become more abundant during the early evening hours, and lactate levels decline after this occurs. The ability to take up and utilize lactate, as indicated by the presence of genes encoding lactate permease and lactate dehydrogenase genes, is indicated for at least four and up to 7 of the 15 most abundant undermat members (Table S9). Additionally, genes encoding lactate permease are affiliated with some less abundant members, e.g., Thermodesulfobacteria sp. OTU-26, Bellilinea sp. OTU-31, Solibacter-like Acidobacteria OTU-36, and “Ca. E. thermophila” OTU-46.
Due to the high abundance and activity of cyanobacterial cells in the upper green layer of the mat, the measured and available lactate has mostly been attributed to Synechococcus spp. Types A and B' (Kim et al., 2015). Metagenomic data from the undermat layer, however, indicates additional putative producers of lactate as a fermentation end-product: e.g., Armatimonadetes (OP10) member OTU-3, fermentative Atribacteria (OP9) member OTU-7 and Aminicenantes (OP8) member OTU-13 (Table S9).
Saprophytic Life Style
A saprophytic life style, based on cellular components made available by cell lysis of the major primary producer, Synechococcus spp., can be assumed for the majority of the heterotrophic mat community. Excreted proteases, secretion systems, and pathogenesis islands were not observed, so there was little evidence for an active predatory life style for any of the most abundant undermat members, neither in the metagenome nor in the reference genomes employed in this study (data not shown). The presence of viruses in the mat and their probable involvement in cell lysis processes can be assumed (e.g., on the basis of CRISPR systems in genomes), but this has not yet been analyzed and must be shown by future studies.
Protein and Amino Acids
The presence of genes for transporters for oligopeptides and dipeptides, as well as generic amino acid and/or branched chain amino acid (BCAAs) transporters, were identified in the majority of the abundant mat members. This suggests that most organisms in the undermat have the potential to utilize peptides and amino acids as sources of carbon and nitrogen. Nearly complete degradation pathways for BCAAs were found for all four abundant Choroflexi members (Table S10). This indicates the ability to utilize BCAAs as putative carbon sources for these organisms, which is further supported by the ability of the chlorophototrophs to grow in CTM-medium that contains BCAAs (Tank and Bryant, 2015a,b; Tank et al., 2017). However, complete dependency on BCAAs, as demonstrated for Cab. thermophilum (Tank and Bryant, 2015a,b) and postulated for “Ca. Thermochlorobacter aerophilum,” was generally not found except for perhaps Chloroflexi member OTU-9, for which the partial genome so far only indicates biosynthesis of BCAAs from the precursor 2-isopropylmalate. Degradation of BCAAs is also likely to occur for Armatimonadetes member OTU-3 and EM3 member OTU-10 (Table S10).
Nucleic Acids
Utilization of nucleic acids as a nutrient and energy source is suggested to occur in these mats. For example, at least 13 of the 15 most abundant members should be able to utilize ribose, as indicated by the presence of genes for both ribokinase and ribose-5-phosphate isomerase in metagenomic bins and/or reference genomes (Table S11). The presence of ABC-type ribose transporter genes in at least six of undermat members further supports the uptake and utilization of ribose from the environment by these organisms. Furthermore, 23 predicted extracellular nucleases were present in the metagenome, of which 10 were affiliated with 4 of the most abundant members (Table S11). Two highly abundant genes could not be affiliated with a specific taxon (JGI24185J35167_10498681, JGI24185J35167_10496441). Thus, up to eight highly abundant mat members may be able to degrade extracellular nucleic acids and utilization the resulting nucleotides, phosphate, and/or ribose.
Nitrogen Metabolism
Nitrogen metabolism in the mat seems to be purely assimilatory, and no genes suggesting dissimilation of nitrogen compounds were identified in the metagenome (Figure 5; Table S12). Organic nitrogen compounds produced from nitrogen fixation by Synechococcus spp., and probably by Thermodesulfovibrio sp., provide the mat community with reduced nitrogen (e.g., amino acids, nucleic acids, and ammonia). It is possible that some organisms in the mat can use the low levels of ammonium in the spring water (Thiel et al., 2016) as a source of reduced nitrogen. In this respect, nitrate and ammonium do not stimulate the growth of Synechococcus spp. in enrichment cultures (M. Tank, personal communication).
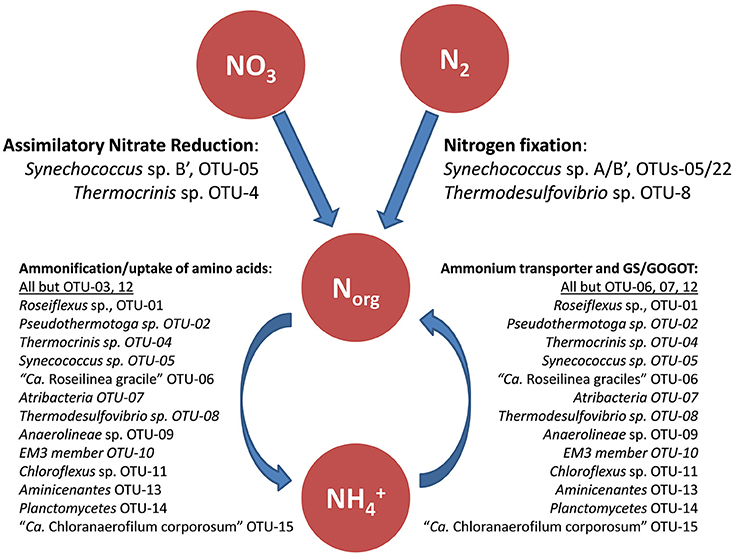
Figure 5. Schematic drawing of the hypothesized nitrogen metabolism in the mat including likely involved mat community members. GS, Glutamine synthetase; GOGAT, glutamate synthase.
Nitrogen Fixation
Genes encoding putative nitrogenases (nifHDK) were found in four members of the mat: Synechococcus spp., Roseiflexus spp., “Ca. Chloranaerofilum corporosum” and Thermodesulfovibrio sp. (Table S12). However, active nitrogen fixation has only been shown for one of these organisms, Synechococcus spp. (Steunou et al., 2006, 2008), which are believed to provide the mat community with bio-available fixed nitrogen. It is not clear what the function of the nifHDK-like genes in Roseiflexus spp. might be, because most other accessory proteins required for maturation of nitrogenase are not present in any available Roseiflexus spp. genome (only nifB is present), and e.g., Roseiflexus castenholzii is unable to grow on dinitrogen as sole nitrogen source (D.A. Bryant, unpublished results). “Ca. Chloranaerofilum corporosum” contains the same set of genes as Roseiflexus spp. and is similarly not expected to grow on dinitrogen as sole nitrogen source. Frank et al. (2016) have reported that three Thermodesulfovibrio spp., T. yellowstonii, T. aggregans, and T. islandicus, genomes have the complete set of genes for nitrogenase, strongly suggesting that these organsims use dinitrogen as a nitrogen source; however, diazotrophic growth has not been reported for any of these isolates. The presence of additional nif genes (nifENXB) as well as a transcriptional repressor of the nif operon, suggests that this Thermodesulfovibrio sp. is also likely to be a nitrogen-fixing organism. This suggestion is strengthened by the observation that transcription of the nif genes in Thermodesulfovibrio sp. occurs and varies as a function of the diel cycle (unpublished observations). The presence of similar organisms in other hot spring microbial mats, especially in those dominated by non-nitrogen fixing cyanobacteria (Everroad et al., 2012), supports the possibility that they have the ability to fix nitrogen in situ and thus contribute to nitrogen input to the microbial mat community.
Assimilatory Use of Ammonium
Despite the low concentration of ammonium in the hot spring water (values ranging from <3 to 35 μM have been measured over the years; Doemel and Brock, 1976; Papke et al., 2003; Ball et al., 2004), ammonium seems to be the preferred nitrogen source, along with organic nitrogen compounds such as amino acids (see above). Ammonium probably becomes available through ammonification and through the degradation of organic compounds, specifically of cyanobacterial proteins and nucleic acids. In accordance with this, numerous annotated genes for ammonium permease (amtB) are found in the undermat metagenome (95 partial genes in total). Putative ammonium permease genes were identified in 10 of the 15 most abundant mat members (Table S12). Genes for the key enzymes utilized in both ammonification (amino acid catabolism) and use of ammonium in (amino acid) anabolism, glutamine synthetase (GS), and glutamate:2-oxoglutarate amidotransferase (GOGAT; glutamate synthase) are present in high numbers in the metagenome.
Nitrate Reduction
Based on the metagenomic data, nitrate reduction appears to be uncommon in the microbial undermat layer. Although genes encoding nitrite reductase (nirB) and nitrate reductase (narB) in combination with genes for a nitrate ABC transporter (nrtABC), are present in 2 of the 15 most abundant mat members (Table S12), assimilatory nitrate reduction is only assumed to occur in Thermocrinis sp. OTU-4.
Based on the presence of nirA and narB genes in the genomes of isolates as well as the metagenome, assimilatory nitrate reduction has long been assumed to occur for the dominant Synechococcus spp. Moreover, nitrate has traditionally been the only nitrogen source used in cultivation experiments of uni-cyanobacterial mixed cultures of Synechococcus spp. Types A and B' obtained from the mats (Nowack et al., 2015; Olsen et al., 2015). However, in experiments with Synechococcus spp. isolates on agar plates aiming to produce axenic cultures, nitrate did not support growth under constant light and oxygen conditions nor did it enhance growth under conditions that favor nitrogen fixation. Thus, nitrate may not be assimilated by Synechococcus spp. under laboratory conditions (M. Tank, N. Soulier, D.A. Bryant, unpublished). These observations might be explained by micro-niche conditions favoring nitrogen fixation or a nitrate-reducing “helper” bacterium present in these mixed cultures. In contrast, assimilatory nitrate reduction can be assumed for Thermocrinis sp. OTU-4, for which nirA and narB genes showing closest similarity to Thermocrinis ruber were identified in the metagenome. Assimilatory nitrate reduction for this organism is further supported by chemolithoautotrophic growth of its closest relative, Thermocrinis ruber, in media containing nitrate as sole nitrogen source (Härtig et al., 2014). Similarly, the absence of narGHI genes in the genome of the type strain, as well as the metagenome sequences, correlate with the observation that nitrate is not an electron acceptor that can support the growth of Thermocrinis ruber strain OC1/4 (Huber et al., 1998).
Nitrification and Denitrification
Nitrification does not seem to occur in any of the abundant microbial undermat members. Ammonia monooxygenase (amoA), the key enzyme responsible for the ammonium oxidation step in nitrification, was not identified in the metagenome. Similarly, dentrification genes were not detected in the undermat metagenome. Therefore, denitrification does not appear to occur in any abundant mat organism. This is consistent with the low concentration of nitrate in the spring water (<0.1 mg/L; ~ <1.6 μM; see Thiel et al., 2016).
Sulfur Metabolism
Despite the low sulfate concentration in the spring water (<200 μM; USGS report 2001–2002 Ball et al., 2004; Dillon et al., 2007), an active sulfur cycle is likely maintained in the mats (Figure 6). Sulfate reduction has previously been shown in the mats (Dillon et al., 2007), while the oxidation of sulfide and sulfur has not been studied in detail. Metagenomic analysis of the undermat supports the hypothesis of a closed sulfur cycle and identified the key organisms involved.
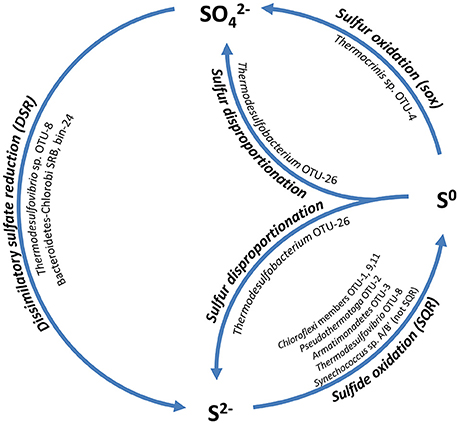
Figure 6. Schematic drawing of the hypothesized sulfur cycle and the involved mat community members. SQR, Sulfide:quinone oxidoreductase; sox, genes for sulfur oxidation pathway; DSR, dissimilatory sulfate reductase.
Sulfate Reduction
Dissimilatory sulfate reduction is likely to occur in at least three members of the mat community. Previous studies revealed four distinct clusters of dsrAB sequences by a PCR-based cloning survey (Dillon et al., 2007). Two complete sets of sulfate reduction genes (sat, aprBA, dsrAB, dsrTMKJOP, qmoABC) were present in the metagenomic dataset. The more abundant set of genes (~127 × coverage for the dsrAB genes) belonged to Thermodesulfovibrio sp. OTU-8, which was associated with measured sulfate reduction activity in previous studies (“clade-1,” Dillon et al., 2007). A second set of less abundant dsrAB genes (63 × coverage) were the same as unidentified drsAB clone sequences that were previously retrieved from the mats (“clade-2,” Dillon et al., 2007). These sequences were affiliated with a metagenomic bin (bin-24, Thiel et al., 2016), with an uncertain phylogenetic affiliation within the Bacteroidetes-Chlorobi group. Due to the lack of a 16S rRNA gene sequence in the metagenome bin, phylogenetic affiliation was based on phylogenetic marker genes, most of which were affiliated with either the Bacteroidetes or the Chlorobi group. Phylogenetic analyses of 17 concatenated phylogenetic marker genes placed the unidentified novel uncultured organism in a cluster with Bacteroidetes and both heterotrophic as well as phototrophic Chlorobi representatives (Thiel et al., in preparation). Furthermore, the presence of a 1023-bp partial 23S rRNA gene sequence with closest relationship to Bacteroidetes-Chlorobi group members strengthens the affiliation. “Clade-3” sequences were also found in the metagenome at very low abundance (6–8×) (Table S13). A previous phylogenetic analysis by Müller et al. (2015), as well as this study, places these sequences into an unclassified cluster within a group of Firmicutes-like dsrAB sequences (dsrAB ARB database available at http://www.microbial-ecology.net/download). “Clade-4” sequences reported by Dillon et al. (2007) were not found in the undermat metagenome.
In addition to the previously known dsrAB phylotypes, sequences encoding dissimilatory sulfite reductase from a Thermodesulfobacteria member, putatively OTU-26, were recovered from the metagenome. Three low abundance partial dsrAB sequences (16–26 × coverage) showed closest similarity to Caldimicrobium thiodismutans with 87–93% amino acid identity and 96–98% similarity (Table S13). C. thiodismutans has recently been isolated from a hot spring in Japan and displays sulfur disproportionating activity (Kojima et al., 2016). A similar sulfur-disproportionating metabolism is hypothesized for the Mushroom Spring mat member (Figure 6).
Assimilatory sulfate reduction does not seem to be prevalent in the undermat metagenome. This indicates that the majority of mat members rely on reduced sulfur sources, as has been shown for Chloracidobacterium thermophilum (Tank and Bryant, 2015a,b). Genes for ferredoxin-dependent sulfite reductase (sir) were only identified for 3 of the 15 most abundant undermat members (OTUs-5, 11, and 14; Table S14). Additional low-coverage gene sequences were affiliated with less abundant members, e.g., Armatimonadetes member OTU-18, Acidobacterium member OTU-36 (bin-20, Thiel et al., 2016) as well as two unidentified Planctomycetes members (bins-23 and 29; Thiel et al., 2016), while some remained unidentified. Roseiflexus spp. strains do not contain complete reduction pathways and presumably cannot perform assimilatory sulfate reduction (Bryant et al., 2012). Among members of the phylum Chloroflexi, genes for sulfite reductase were only identified in Chloroflexus sp. OTU-11.
Sulfur Oxidation
Although the concentration of sulfide present in the hot spring source water is at the limit of detection (0.003 mg/L = ~0.1 μM, Ball et al., 2004), sulfide accumulates in the mats as a result of biological sulfate reduction (Dillon et al., 2007). Little is known about the oxidative part of the sulfur cycle in the mats. Sulfide can be used as an electron donor by some anoxygenic phototrophic bacteria, but it is also toxic to many organisms, and detoxification mechanisms have evolved. Chloroflexi are known to contain sulfide:quinone oxidoreductase (sqr) of the type II family (Bryant et al., 2012) and have the ability to oxidize sulfide to polysulfides (Madigan and Brock, 1975), which sometimes even support autotrophic growth (Madigan et al., 1974; Madigan and Brock, 1977; Keppen et al., 2000; Gich et al., 2003; Klappenbach and Pierson, 2004; Thiel et al., 2014). SQR genes were also found in the metagenome and were affiliated with chlorophototrophic and chemotrophic members of the Chloroflexi, Thermotogae, Armatimonadetes, and Nitrospirae (Table S14).
When sulfide is oxidized by Chloroflexus and Oscillochloris spp., globules of elemental sulfur are deposited outside the cells (Madigan and Brock, 1975; Keppen et al., 2000). Elemental sulfur was shown to be a sulfur source for the growth of Chloracidobacterium thermophilum (Tank and Bryant, 2015a,b). It might also be a sulfur source for other mat members that lack the genes for assimilatory sulfate reduction (e.g., OTU-7, see Table S14). Furthermore, elemental sulfur is known to be an electron donor for Thermocrinis ruber (Huber et al., 1998), which is the closest type-strain relative to the microbial mat member Thermocrinis sp. OTU-4 (Thiel et al., 2016), for which a complete set of sox genes was identified in the metagenome. It is thus expected that this mat member can oxidize elemental sulfur to sulfate, which would close the sulfur cycle and provide sulfate that can act as electron acceptor for biological sulfate-reduction by Thermodesulfovibrio sp. OTU-8 and any other putative sulfate-reducers in the mat (Figure 6).
There is evidence that Synechococcus spp. in the mats can also oxidize sulfide. In situ studies showed that sulfide addition strongly stimulated 13C-bicarbonate incorporation into Synechococcus spp. lipid biomarkers (van der Meer et al., 2005). Some cyanobacteria are known to possess sulfide quinone reductase, but this gene is not present in the genomes of Synechococcus spp. that occur in the mats. Cyanobacteria that do not produce polysulfides from sulfide oxidation produce thiosulfate as the product (De Wit and van Gemerden, 1987; Rabenstein et al., 1995); however, the mechanism of thiosulfate production by such organisms is currently unknown.
Hydrogen Metabolism
Revsbech et al. (2016) have recently studied the diel H2 dynamics in the Mushroom Spring mat. Light-stimulated H2 production as a by-product of cyanobacterial N2 fixation was shown to occur in the morning, while H2 accumulated in the evening and slowly decreased until just before sunrise. Metagenomic analyses indicate fermentative sources for the production of H2 in the evening and night by Pseudothermotoga sp., Thermodesulfovibrio sp. and members of the Atribacteria and Aminicenantes (Figure 7). H2 levels during the day are low (Kim et al., 2015; Revsbech et al., 2016), which presumably is due to its oxidation by chlorophototrophic members of the Chloroflexi as well as Thermocrinis sp., Thermodesulfovibrio sp., Planctomycetes, and Thermodesulfobacteria (Table S15, Figure 7).
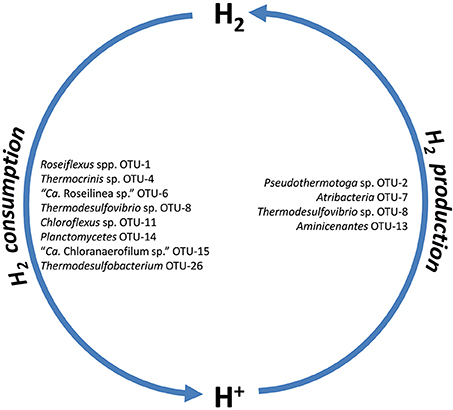
Figure 7. Putative H2 producers and consumers of the microbial mat community as inferred from metagenome analysis.
H2 Production
Microbes can produce molecular hydrogen (H2) via fermentation and dinitrogen fixation (Nielsen et al., 2015; Revsbech et al., 2016). Hydrogen accumulation has been detected in these mats following a diel pattern. In the evening, H2 accumulated rapidly after the onset of darkness, reaching peak values of up to 30 μmol H2 L−1 at about 1-mm depth below the mat surface, and slowly decreasing to about 11 μmol H2 L−1 just before sunrise (Revsbech et al., 2016). Another pulse of H2 production, reaching a peak concentration of 46 μmol H2 L−1, was found in the early morning under dim light conditions that were too low to cause the accumulation of O2 in the mat, indicating that the nitrogenase activity of the dominant cyanobacteria Synechococcus spp. Types A and B' is of greatest importance for this morning peak. During the night, fermentation has been proposed to contribute to the formation of H2 (Revsbech et al., 2016).
In this study, we identified several microbial community members that could possibly contribute to fermentative H2 evolution. The anaerobic fermenter, Pseudothermotoga sp. OTU-2, is probably the most abundant hydrogen-producing mat member. The partial genome of this bacterium contains genes encoding a [FeFe]-hydrogenase (H2ase; Table 3, Table S15) enabling fermentative H2 production, as has been shown in the next closest relative (Pseudo)thermotoga hypogea strain NBRC 106472 (Fardeau et al., 1997; Yang et al., 2015).
The microbial mat also contains other fermentative bacteria: e.g., an uncultured member of the Atribacteria (Candidate phylum OP9), OTU-7 and a member (OTU-13) of the phylum Aminicenantes (Candidate phylum OP8; Table 3, Table S15). Analysis of the OTU-7 metagenomic bin indicates an anaerobic, fermentative lifestyle for this mat member, similar to that deduced from single-cell genomics previously obtained for members of the phylum Atribacteria that occur in hot springs in California and Nevada, USA (Dodsworth et al., 2013). The presence of a gene for a monomeric, periplasmic [FeFe]-H2ase in the partial genome further suggests the ability of this organism to produce H2 fermentatively. For OTU-13 a H2-evolving group 4 H2ase (formate hydrogenlyase complex) and a bidirectional [NiFe]-H2ase III could be responsible for fermentative H2 evolution, as shown in Thiocapsa roseopersicina (Rákhely et al., 2007). Thermodesulfovibrio sp. OTU-8 may grow on fumarate and acetate, as observed for Thermodesulfovibrio yellowstonii (Henry et al., 1994), and may subsequently produce H2 via a type-IV hydrogenase (fumarate hydrogenlyase complex; Table S15).
H2 Consumption
Several members of the Chloroflexi (OTUs-1, 11, 15), Thermocrinis sp. OTU-4, but also the sulfate reducing Thermodesulfovibrio sp. OTU-8, presumably the Planctomycetes member OTU-14 as well as a less abundant member of the Thermodesulfobacteria, OTU-26, were identified as putative H2 consumers due to the presence of genes encoding [NiFe]-H2ases (Table S15). Because it is the most abundant member of the undermat, Roseiflexus sp. OTU-1 is expected to be the major consumer of H2. The genome of Roseiflexus sp. strain RS-1 contains a type-I uptake [NiFe]-H2ase, encoded by genes hydAB (hyaAB), as well as a complete suite of hyp genes potentially involved in the biosynthesis and maturation of this H2ase. Accordingly, this organism has been suggested to utilize H2 (van der Meer et al., 2010). All of these genes were also present in the undermat metagenome. A high diversity (between 1 and 14 sequences) of different (partial) H2ases with 66–100% amino acid similarity to the Roseiflexus sp. RS-1 reflects the high microdiversity for this taxon (Ferris and Ward, 1997; Thiel et al., 2016) and might indicate the presence of putative ecotypes with different H2ases gene sequences. [NiFe]-H2ase genes (hydABCD, RoseRS_2319–RoseRS_2322) were shown to be expressed, and the relative transcript abundances for these genes exhibited a nocturnal expression pattern during the evening and night hours in the upper green layer of the microbial mat. This pattern led to the suggestion that H2 is an important source of electrons for hypothesized light-driven CO2 fixation by filamentous anoxygenic phototrophs (FAPs) in the morning, when light is available but while the mat remains anoxic (van der Meer et al., 2005; Klatt et al., 2013b).
Thermocrinis sp. OTU-4 is likely to be the second most abundant H2 consumer in the mat. Although no H2ase genes affiliated with this organism were detected in the assembled metagenome, their presence was confirmed in the unbinned portion of the sequences (Table S15). The closest relative of the organism(s) represented in this bin, Thermocrinis ruber, which was isolated from Octopus Spring from temperatures above 80°C, grows on H2, elemental sulfur, and thiosulfate (Huber et al., 1998; Jahnke et al., 2001; Härtig et al., 2014) and a similar metabolism can be assumed for the undermat member at 60°C based on the corresponding metagenomic bin (Table 3). The presence of affiliated H2ase genes only in the unassembled part of the metagenome, as well as the low number of scaffolds in the metagenome bin are indicative of assembly difficulties probably arising from the presence of closely related populations with highly similar but non-identical genome sequences. Another mat member, Thermodesulfovibrio sp. OTU-8, is assumed to grow on H2 plus acetate, as shown for its closest relative, T. yellowstonii (Henry et al., 1994). This suggestion is supported by the presence of genes encoding a [FeFe]-H2ase in the genome of the type strain and the metagenomic bin. However, genes for additional group-4 H2ases (formate hydrogen lyase) are present in both the isolate genome (Lim et al., 2010; Bhatnagar et al., 2015) and metagenomic bin-8 (Table S15). These genes indicate an ability to produce H2, e.g., when growing on formate and acetate as described by Henry et al. (1994). A less abundant putatively H2-utilizing member of the mat is the Thermodesulfobacteria member OTU-26. Genes encoding a [NiFe]-H2ase and an [NiFe]-H2ase maturation protease, closely related to sequences from Caldimicrobium thiodismutans have been identified in the undermat metagenome (scaffold JGI24185J35167_1006546). Additionally, H2ase maturation genes indicating the presence of [NiFe]-H2ases with unknown function and identity were identified in the novel chlorophototrophic FAP “Ca. R. gracile” OTU-6 and the chemotrophic Chloroflexi member OTU-9, as well as the unidentified Planctomycetes member OTU-14 (Table S15).
Conclusion
In this study, we used deep metagenomic sequencing to further our understanding of the microbial mat community ecology at Mushroom Spring in Yellowstone National Park, and added metabolic information about the understudied orange-colored undermat layer. Genomic analyses for the 15 most abundant members of the undermat were conducted based on reference genomes and partial genomes obtained by binning a 232-Mb assembled metagenome from a full lane HiSeq sequencing run. The “cyanobacterial” mat harbors a panoply of phototrophs, some of which were initially detected in the undermat for the first time. We detected 10 of the 16 chlorophototrophs previously obtained from these mats in the undermat, and 5 of them were among the 15 most abundant members (Thiel et al., 2016; Tank et al., 2017). Although, most chlorophototrophic members were more abundant in the upper green layer, some chlorophototrophs were almost exclusively detected in the undermat (Thiel et al., 2016). The undermat portion of the community was more uneven than the upper layer. The most dominant member of the undermat is Roseiflexus sp., a, filamentous anoxygenic phototrophic bacterium (Thiel et al., 2016). The detection of rhodopsin genes, mostly xanthorhodopsins, in many taxa, specifically in the undermat, was surprising and strengthens the conclusion that light energy is of major importance in this community, even under very low light conditions.
The mixture of phototrophic and chemotrophic organisms in the undermat use bicarbonate as well as organic carbon sources derived from different cell components and fermentation products. The overall mat community has been suggested to ultimately depend upon the primary productivity of cyanobacteria in the upper green layer (Tank et al., 2017), which also provide the majority of nutrient and carbon sources utilized for heterotrophic and mixotrophic growth in the undermat. Further, this study indicates autotrophic growth for several undermat members. The four known bacterial carbon fixation pathways are apparently used for inorganic carbon fixation in the undermat metagenome. Additionally, inorganic carbon is probably used in anaplerotic CO2 fixation reactions by several undermat members of the community using 2-oxoglutarate or pyruvate:ferredoxin oxidoreductases in the reductive TCA branch as has been shown for Cab. thermophilum (Tank and Bryant, 2015a,b) and as hypothesized for “Ca. Thermochlorobacter aerophilum” (Liu et al., 2012). Nitrogen metabolism seems to be limited to assimilatory reactions. Many community members are capable of using organic nitrogen in form of proteins and amino acids. Nitrogen input into the system is likely to be driven by a low diversity of nitrogen fixing bacteria; the dominant cyanobacteria, Synechococcus spp. Types A and B' in the upper green layer (Steunou et al., 2006, 2008) and the anaerobe Thermodesulfovibrio sp. in the undermat. A closed sulfur cycle is indicated by biological sulfate reduction combined with evidence of genes for sulfide oxidation mainly in chlorophototrophs. Finally, hydrogen is produced and consumed in the mat by a variety of microorganisms, which leads to the observed diel hydrogen concentration patterns. Nitrogenase activity of cyanobacterial members of the upper layer in combination with fermentative bacteria in the undermat are the main sources of hydrogen, which can be consumed by phototrophic and chemotrophic members of the microbial mat community in both layers.
In this metagenomic analysis of the orange-colored undermat, we describe metabolic potentials and putative interactions among mat community members, leading to an initial overview of the metabolic potential of the entire mat community. Metabolic co-dependencies among various community members of both layers are indicated by (almost) closed nutrient cycles as well as nutrient, e.g., vitamin, requirements in different mat members (e.g., Tank and Bryant, 2015a,b; Tank et al., 2017). Oxygen production by the dominant upper layer cyanobacteria in concert with heterotrophic oxygen respiration in the undermat leads to a variety of oxygen microenvironments over the diel cycle affecting the microbial community and vertical distribution of different members within the mat. The analysis further disclosed the metabolic potentials of previously unknown and unidentified microbes, such as a sulfate-reducing member of the deep-branching Bacteroidetes-Chlorobi group, a member of the Planctomycetes with two types of CO2 fixation capabilities, and a member of the Chloroflexi using the Wood-Ljungdahl pathway. Although microbial studies at Mushroom Spring extend over 50 years, these microbial mats still harbor the potential for new discoveries and for gaining a deeper understanding of hot spring microbial mat ecology and physiology.
Author Contributions
VT conducted sequence analysis after assembly for metagenome sequences, including binning, phylogenetic analysis, annotation and phylogenetic marker genes analysis of metagenome bins, as well as reference targeted mapping studies, and generated tables and figures. MH analyzed carbon fixation pathway affiliated genes in the metagenome and the (partial) genomes, designed the corresponding figures, and wrote the corresponding sections of the manuscript. Sequencing, quality check, assembly, and annotation of the metagenome was conducted by JGI staff. DW and DB planned the experiments, acquired funding, organized and led field excursions, and provided scientific infrastructure. VT and DB wrote the manuscript with contributions from all other authors.
Funding
This study was partly funded by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the Department of Energy through Grant DE-FG02-94ER20137. DB and DW additionally acknowledge support from the NASA Exobiology program (NX09AM87G and NNX16AJ62G). This work was also partly supported by the U. S. Department of Energy (DOE), Office of Biological and Environmental Research (BER), as part of BER's Genomic Science Program395 (GSP). This contribution originates from the GSP Foundational Scientific Focus Area (FSFA) at the Pacific Northwest National Laboratory (PNNL) under a subcontract to DB. The nucleotide sequencing was performed as part of a Community Sequencing Program (Project CSP-411) and was performed by the U.S. Department of Energy Joint Genome Institute, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank all of the JGI staff members who contributed to obtaining the sequence data. The materials used in this study were collected under permit #YELL-SCI-0129 held by DW and administered under the authority of Yellowstone National Park. The authors especially thank Christie Hendrix and Stacey Gunther for their advice and assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00943/full#supplementary-material
Figure S1. Schematic map of metabolic reactions of the 3-hydroxypropionate bi-cycle and enzymes involved.
Figure S2. Genes for rTCA cycle enzymes identified in the metagenome and affiliated with Thermocrinis sp. OTU-04.
Figure S3. Schematic presentation of the different versions of genes encoding an ATP dependent citrate lyase (CCS, CCL, and ACL) of the reductive TCA cycle. Representative organisms for each gene variant are given in blue.
Figure S4. Schematic presentation of the two branches (methyl branch and carbonyl branch) of the reductive acetyl-CoA pathway. The two key-enzymes CO dehyodrogenase (CODH) and acetyl-CoA synthase (ACS) are given in red.
Table S1. Reference genomes used in emergent self-organizing map binning.
Table S2. List of photosynthetic reaction center genes detected in the undermat metagenome, and their taxonomic affiliation.
Table S3. List of rhodopsin encoding genes identified in the undermat metagenome, and their taxonomic affiliation.
Table S4. List of bacteriorhodopsin-related protein (Brp) and bacteriorhodopsin-related protein-like homolog protein (Blh) encoding genes identified in the undermat metagenome, and their taxonomic affiliation.
Table S5. List of RubisCO genes detected in the undermat metagenome, length, and sequence coverage of scaffolds as well as the BLASTp results and taxonomic affiliation.
Table S6. List of 3-hydroxypropionate bi-cycle key-enzyme genes in metagenome bins and reference genomes of microbial mat Chloroflexi.
Table S7. List of malonyl-CoA reductase encoding genes identified in the undermat metagenome, and their taxonomic affiliation.
Table S8. List of Wood-Ljungdahl/reductive acetyl-CoA pathway enzyme genes in metagenome bins and reference genomes of microbial mat members.
Table S9. List of genes affiliated with lactate utilization and production found in the (partial) genomes and metagenomic dataset.
Table S10. List of genes involved in putative utliziation of proteins and amino acids by the 15 most abundant mat members based on (partial) genome/metagenomic sequence information.
Table S11. List of genes affiliated with ribose utilization in (partial) genomes of the 15 most abundant mat members.
Table S12. List of genes assigned to nitrogen metabolism pathways in the most abundant mat member partial genomes/reference genomes and/or metagenome dataset.
Table S13. List of dissimilatory sulfite-reductase encoding genes (dsrAB) identified in the undermat metagenome, and their taxonomic affiliation.
Table S14. List of genes assigned to sulfur metabolism pathways in the most abundant mat member partial genomes/reference genomes and/or metagenome dataset.
Table S15. List of hydrogenase genes identified in (partial) genomes, reference genomes, and metagenome affiliated with the most abundant mat members.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl. Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Anderson, K. L., Tayne, T. A., and Ward, D. M. (1987). Formation and fate of fermentation products in hot spring cyanobacterial mats. Appl. Environ. Microbiol. 53, 2343–2352.
Aoshima, M. (2007). Novel enzyme reactions related to the tricarboxylic acid cycle: phylogenetic/functional implications and biotechnological applications. Appl. Microbiol. Biotechnol. 75, 249–255. doi: 10.1007/s00253-007-0893-0
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Baker, B. J., Lazar, C. S., Teske, A. P., and Dick, G. J. (2015). Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3, 14. doi: 10.1186/s40168-015-0077-6
Balashov, S. P., Imasheva, E. S., Boichenko, V. A., Antón, J., Wang, J. M., and Lanyi, J. K. (2005). Xanthorhodopsin: a proton pump with a light-harvesting carotenoid antenna. Science 309, 2061–2064. doi: 10.1126/science.1118046
Ball, J. W., McCleskey, R. B., Nordstrom, D. K., Holloway, J., and Verplanck, P. L. (2004). Water-Chemistry Data for Selected Springs, Geysers and Streams in Yellowstone National Park, Wyoming, 2001-2002. U. S. Geological Survey Open-File Report 2004–1316.
Bateson, M. M., and Ward, D. M. (1988). Photoexcretion and fate of glycolate in a hot spring cyanobacterial mat. Appl. Environ. Microbiol. 54, 1738–1743.
Bauld, J., and Brock, T. D. (1973). Ecological studies of Chloroflexus, a gliding photosynthetic bacterium. Arch. Fuer Mikrobiol. 92, 267–284. doi: 10.1007/BF00409281
Becraft, E. D., Cohan, F. M., Kuehl, M., Jensen, S. I., and Ward, D. M. (2011). Fine-scale distribution patterns of Synechococcus ecological diversity in the microbial mat of Mushroom Spring, Yellowstone National Park. Appl. Env. Microbiol. 77, 7689–7697. doi: 10.1128/AEM.05927-11
Becraft, E. D., Wood, J. M., Rusch, D. B., Kühl, M., Jensen, S. I., Bryant, D. A., et al. (2015). The molecular dimension of microbial species: 1. Ecological distinctions among, and homogeneity within, putative ecotypes of Synechococcus inhabiting the cyanobacterial mat of Mushroom Spring, Yellowstone National Park. Front. Microbiol. 6:590. doi: 10.3389/fmicb.2015.00590
Bhatnagar, S., Badger, J. H., Madupu, R., Khouri, H. M., Connor, E. M. O., Robb, F. T., et al. (2015). Genome sequence of the sulfate-reducing thermophilic bacterium Thermodesulfovibrio yellowstonii strain DSM 11347(T) (Phylum Nitrospirae). Genome Announc. 3:e01489–14. doi: 10.1128/genomeA.01489-14
Bhaya, D., Grossman, A. R., Steunou, A.-S., Khuri, N., Cohan, F. M., Hamamura, N., et al. (2007). Population level functional diversity in a microbial community revealed by comparative genomic and metagenomic analyses. ISME J. 1, 703–713. doi: 10.1038/ismej.2007.46
Braakman, R., and Smith, E. (2012). The emergence and early evolution of biological carbon-fixation. PLoS Comput. Biol. 8:e1002455. doi: 10.1371/journal.pcbi.1002455
Brock, T. D. (1995). The road to Yellowstone - and beyond. Annu. Rev. Microbiol. 49, 1–28. doi: 10.1146/annurev.mi.49.100195.000245
Brock, T. D. (1967). Micro-organisms adapted to high temperatures. Nature 214, 882–885. doi: 10.1038/214882a0
Bryant, D. A., Costas, A. M. G., Maresca, J. A., Gomez Maqueo Chew, A., Klatt, C. G., Bateson, M. M., et al. (2007). Candidatus Chloracidobacterium thermophilum: an aerobic phototrophic Acidobacterium. Science 317, 523–526. doi: 10.1126/science.1143236
Bryant, D. A., Liu, Z., Li, T., Zhao, F., Garcia Costas, A. M., Klatt, C. G., et al. (2012). “Comparative and functional genomics of anoxygenic green bacteria from the taxa Chlorobi, Chloroflexi, and Acidobacteria,” in Advances in Photosynthesis and Respiration, Vol. 35, Functional Genomics and Evolution of Photosynthetic Systems, eds R. L. Burnap and W. Vermaas (Dordrecht: Springer), 47–102.
Choi, A. R., Shi, L., Brown, L. S., and Jung, K.-H. (2014). Cyanobacterial light-driven proton pump, Gloeobacter rhodopsin: complementarity between rhodopsin-based energy production and photosynthesis. PLoS ONE 9:e110643. doi: 10.1371/journal.pone.0110643
Dahl, C., and Friedrich, C. G. (2008). Microbial Sulfur Metabolism, 1st Edn. Berlin, New York, NY: Springer. doi: 10.1007/978-3-540-72682-1
De Wit, R., and van Gemerden, H. (1987). Oxidation of sulfide to thiosulfate by Microcoleus chtonoplastes. FEMS Microbiol. Lett. 45, 7–13.
Dick, G. J., Andersson, A. F., Baker, B. J., Simmons, S. L., Thomas, B. C., Yelton, A. P., et al. (2009). Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10:R85. doi: 10.1186/gb-2009-10-8-r85
Dillon, J. G., Fishbain, S., Miller, S. R., Bebout, B. M., Habicht, K. S., Webb, S. M., et al. (2007). High rates of sulfate reduction in a low-sulfate hot spring microbial mat are driven by a low level of diversity of sulfate-respiring microorganisms. Appl. Environ. Microbiol. 73, 5218–5226. doi: 10.1128/AEM.00357-07
Dodsworth, J. A., Dohnalkova, A., Blainey, P. C., Murugapiran, S. K., Wesley, D., Ross, C. A., et al. (2013). Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for member of the OP9 lineage. Nat Commun. 4, 1854. doi: 10.1038/ncomms2884
Doemel, W. N., and Brock, T. D. (1976). Vertical distribution of sulfur species in benthic algal mats. Limnol. Oceanogr. 21, 237–244. doi: 10.4319/lo.1976.21.2.0237
Everroad, R. C., Otaki, H., Matsuura, K., and Haruta, S. (2012). Diversification of bacterial community composition along a temperature gradient at a thermal spring. Microbes Environ. 27, 374–381. doi: 10.1264/jsme2.ME11350
Fardeau, M.-L., Ollivier, B., Patel, B. K. C., Magot, M., Thomas, P., and Rimbault, A. (1997). Thermotoga hypogea sp. nov., a xylanolytic, thermophilic bacterium from an oil-producing well. Int. J. Syst. Bacteriol. 47, 1013–1019. doi: 10.1099/00207713-47-4-1013
Ferris, M. J., and Ward, D. M. (1997). Seasonal distributions of dominant 16S rRNA-defined populations in a hot spring microbial mat examined by denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 63, 1375–1381.
Ferris, M. J., Kopczynski, E. D., Bateson, M. M., and Ward, D. M. (1996). Enrichment culture and microscopy conceal diverse thermophilic Synechococcus populations in a single hot spring mat habitat. Microbiology 62, 1045–1050.
Frank, Y. A., Kadnikov, V. V., Lukina, A. P., Banks, D., Beletsky, A. V., Mardanov, A. V., et al. (2016). Characterization and genome analysis of the first facultatively alkaliphilic Thermodesulfovibrio isolated from the deep terrestrial subsurface. Front. Microbiol. 7:2000. doi: 10.3389/fmicb.2016.02000
Friedrich, C. G., Rother, D., Bardischewsky, F., Ouentmeier, A., and Fischer, J. (2001). Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl. Environ. Microbiol. 67, 2873–2882. doi: 10.1128/AEM.67.7.2873-2882.2001
Fuchs, G. (2011). Alternative pathways of carbon dioxide fixation: insights into the early evolution of life? Annu. Rev. Microbiol. 65, 631–658. doi: 10.1146/annurev-micro-090110-102801
Garcia Costas, A. M., Liu, Z., Tomsho, L. P., Schuster, S. C., Ward, D. M., and Bryant, D. A. (2012a). Complete genome of “Candidatus Chloracidobacterium thermophilum,” a chlorophyll-based photoheterotroph belonging to the phylum Acidobacteria. Environ. Microbiol. 14, 177–190. doi: 10.1111/j.1462-2920.2011.02592.x
Garcia Costas, A. M., Tsukatani, Y., Rijpstra, W. I. C., Schouten, S., Welander, P. V., Summons, R. E., et al. (2012b). Identification of the bacteriochlorophylls, carotenoids, quinones, lipids, and hopanoids of “Candidatus Chloracidobacterium thermophilum.” J. Bacteriol. 194, 1158–1168. doi: 10.1128/JB.06421-11
Gich, F., Airs, R. L., Danielsen, M., Keely, B. J., Abella, C. A., Garcia-Gil, J., et al. (2003). Characterization of the chlorosome antenna of the filamentous anoxygenic phototrophic bacterium Chloronema sp. strain UdG9001. Arch. Microbiol. 180, 417–426. doi: 10.1007/s00203-003-0608-6
Gómez-Consarnau, L., González, J. M., Riedel, T., Jaenicke, S., Wagner-Döbler, I., Sa-udo-Wilhelmy, S. A., et al. (2016). Proteorhodopsin light-enhanced growth linked to vitamin-B1 acquisition in marine flavobacteria. ISME J. 10, 1101–1112. doi: 10.1038/ismej.2015.196
Härtig, C., Lohmayer, R., Kolb, S., Horn, M. A., Inskeep, W. P., and Planer-Friedrich, B. (2014). Chemolithotrophic growth of the aerobic hyperthermophilic bacterium Thermocrinis ruber OC 14/7/2 on monothioarsenate and arsenite. FEMS Microbiol. Ecol. 90, 747–760. doi: 10.1111/1574-6941.12431
Hayes, J. M. (2001). Fractionation of carbon and hydrogen isotopes in biosynthetic processes. Rev. Mineral. Geochemistry 43, 225–277. doi: 10.2138/gsrmg.43.1.225
Henry, E. A., Devereux, R., Maki, J. S., Gilmour, C. C., Woese, C. R., Mandelco, L., et al. (1994). Characterization of a new thermophilic sulfate-reducing bacterium Thermodesulfovibrio yellowstonii, gen. nov. and sp. nov.: its phylogenetic relationship to Thermodesulfobacterium commune and their origins deep within the bacterial domain. Arch. Microbiol. 161, 62–69. doi: 10.1007/BF00248894
Huber, R., Eder, W., Heldwein, S., Huber, H., Rachel, R., Karl, O., et al. (1998). Thermocrinis ruber gen. nov., sp. nov., a pink-filament-forming hyperthermophilic bacterium isolated from Yellowstone National Park. Appl. Environ. Microbiol. 64, 3576–3583.
Hügler, M., and Sievert, S. M. (2011). Beyond the Calvin cycle: autotrophic carbon fixation in the ocean. Ann. Rev. Mar. Sci. 3, 261–289. doi: 10.1146/annurev-marine-120709-142712
Hügler, M., Huber, H., Molyneaux, S. J., Vetriani, C., and Sievert, S. M. (2007). Autotrophic CO2 fixation via the reductive tricarboxylic acid cycle in different lineages within the phylum Aquificae: evidence for two ways of citrate cleavage. Environ. Microbiol. 9, 81–92. doi: 10.1111/j.1462-2920.2006.01118.x
Jahnke, L. L., Eder, W., Huber, R., Hope, J. M., Hinrichs, K. U., Hayes, J. M., et al. (2001). Signature lipids and stable carbon isotope analyses of Octopus Spring hyperthermophilic communities compared with those of Aquificales representatives. Appl. Environ. Microbiol. 67, 5179–5189. doi: 10.1128/AEM.67.11.5179-5189.2001
Keffer, J. L., Hahn, M. W., and Maresca, J. A. (2015). Characterization of an unconventional rhodopsin from the freshwater actinobacterium Rhodoluna lacicola. J. Bacteriol. 197, 2704–2712. doi: 10.1128/JB.00386-15
Keppen, O. I., Tourova, T. P., Kuznetsov, B. B., Ivanovsky, R. N., and Gorlenko, V. M. (2000). Proposal of Oscillochloridaceae fam. nov. on the basis of a phylogenetic analysis of the filamentous anoxygenic phototrophic bacteria, and emended description of Oscillochloris and Oscillochloris trichoides in comparison with further new isolates. Int. J. Syst. Evol. Microbiol. 50, 1529–1537. doi: 10.1099/00207713-50-4-1529
Kerepesi, C., Bánky, D., and Grolmusz, V. (2014). AmphoraNet: the webserver implementation of the AMPHORA2 metagenomic workflow suite. Gene 533, 538–540. doi: 10.1016/j.gene.2013.10.015
Kim, J., Smith, J. J., Tian, L., and Dellapenna, D. (2009). The evolution and function of carotenoid hydroxylases in Arabidopsis. Plant Cell Physiol. 50, 463–479. doi: 10.1093/pcp/pcp005
Kim, Y.-M., Nowack, S. P., Olsen, M. T., Becraft, E. D., Wood, J. M., Thiel, V., et al. (2015). Diel metabolomics analysis of a hot spring chlorophototrophic microbial mat leads to new hypotheses of community member metabolisms. Front. Microbiol. 6:209. doi: 10.3389/fmicb.2015.00209
Kimble, L. K., Mandelco, L., Woese, C. R., and Madigan, M. T. (1995). Heliobacterium modesticaldum, sp. nov., a thermophilic heliobacterium of hot springs and volcanic soils. Arch. Microbiol. 163, 259–267. doi: 10.1007/BF00393378
Klappenbach, J. A., and Pierson, B. K. (2004). Phylogenetic and physiological characterization of a filamentous anoxygenic photoautotrophic bacterium “Candidatus Chlorothrix halophila,” gen. nov., sp. nov., recovered from hypersaline microbial mats. Arch. Microbiol. 181, 17–25. doi: 10.1007/s00203-003-0615-7
Klatt, C. G., Bryant, D. A., and Ward, D. M. (2007). Comparative genomics provides evidence for the 3-hydroxypropionate autotrophic pathway in filamentous anoxygenic phototrophic bacteria and in hot spring microbial mats. Environ. Microbiol. 9, 2067–2078. doi: 10.1111/j.1462-2920.2007.01323.x
Klatt, C. G., Inskeep, W. P., Herrgard, M. J., Jay, Z. J., Rusch, D. B., Tringe, S. G., et al. (2013a). Community structure and function of high-temperature chlorophototrophic microbial mats inhabiting diverse geothermal environments. Front. Microbiol. 4:106. doi: 10.3389/fmicb.2013.00106
Klatt, C. G., Liu, Z., Ludwig, M., Kühl, M., Jensen, S. I., Bryant, D. A., et al. (2013b). Temporal metatranscriptomic patterning in phototrophic Chloroflexi inhabiting a microbial mat in a geothermal spring. ISME J. 7, 1775–1789. doi: 10.1038/ismej.2013.52
Klatt, C. G., Wood, J. M., Rusch, D. B., Bateson, M. M., Hamamura, N., Heidelberg, J. F., et al. (2011). Community ecology of hot spring cyanobacterial mats: predominant populations and their functional potential. ISME J. 5, 1262–1278. doi: 10.1038/ismej.2011.73
Kojima, H., Umezawa, K., and Fukui, M. (2016). Caldimicrobium thiodismutans sp. nov., a sulfur-disproportionating bacterium isolated from a hot spring, and emended description of the genus Caldimicrobium. Int. J. Syst. Evol. Microbiol. 66, 1828–1831. doi: 10.1099/ijsem.0.000947
Lanyi, J. K., and Balashov, S. P. (2008). Xanthorhodopsin: a bacteriorhodopsin-like proton pump with a carotenoid antenna. Biochim. Biophys. Acta Bioenerg. 1777, 684–688. doi: 10.1016/j.bbabio.2008.05.005
Lim, J. K., Kang, S. G., Lebedinsky, A. V., Lee, J. H., and Lee, H. S. (2010). Identification of a novel class of membrane-bound [NiFe]-Hydrogenases in Thermococcus onnurineus NA1 by in silico analysis. Appl. Environ. Microbiol. 76, 6286–6289. doi: 10.1128/AEM.00123-10
Liu, Z., Klatt, C. G., Ludwig, M., Rusch, D. B., Jensen, S. I., Kühl, M., et al. (2012). “Candidatus Thermochlorobacter aerophilum:” an aerobic chlorophotoheterotrophic member of the phylum Chlorobi defined by metagenomics and metatranscriptomics. ISME J. 6, 1869–1882. doi: 10.1038/ismej.2012.24
Liu, Z., Klatt, C. G., Wood, J. M., Rusch, D. B., Ludwig, M., Wittekindt, N., et al. (2011). Metatranscriptomic analyses of chlorophototrophs of a hot-spring microbial mat. ISME J. 5, 1279–1290. doi: 10.1038/ismej.2011.37
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, et al. (2004). ARB: a software environment for sequence data. Nucl. Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Madigan, M. T., and Brock, T. D. (1975). Photosynthetic sulfide oxidation by Chloroflexus aurantiacus, a filamentous, photosynthetic, gliding bacterium. J. Bacteriol.122, 782–784.
Madigan, M. T., and Brock, T. D. (1977). Adaptation by hot spring phototrophs to reduced light intensities. Arch. Microbiol. 113, 111–120. doi: 10.1007/BF00428590
Madigan, M. T., Petersen, S. R., and Brock, T. D. (1974). Nutritional studies on Chloroflexus, a filamentous photosynthetic, gliding bacterium. Arch. Microbiol. 100, 97–103. doi: 10.1007/BF00446309
Marchler-Bauer, A., and Bryant, S. H. (2004). CD-search: protein domain annotations on the fly. Nucl. Acids Res. 32, 327–331. doi: 10.1093/nar/gkh454
Marchler-Bauer, A., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., Deweese-Scott, C., Fong, J. H., et al. (2009). CDD: specific functional annotation with the Conserved Domain Database. Nucl. Acids Res. 37, 205–210. doi: 10.1093/nar/gkn845
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI's conserved domain database. Nucl. Acids Res. 43, 222–226. doi: 10.1093/nar/gku1221
Marchler-Bauer, A., Lu, S., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., DeWeese-Scott, C., et al. (2011). CDD: a conserved domain database for the functional annotation of proteins. Nucl. Acids Res. 39, 225–229. doi: 10.1093/nar/gkq1189
Markert, S., Arndt, C., Felbeck, H., Becher, D., Sievert, S. M., Hügler, M., et al. (2007). Physiological proteomics of the uncultured endosymbiont of Riftia pachyptila. Science 315, 247–250. doi: 10.1126/science.1132913
Müller, A. L., Kjeldsen, K. U., Rattei, T., Pester, M., and Loy, A. (2015). Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 9, 1152–1165. doi: 10.1038/ismej.2014.208
Nielsen, M., Revsbech, N. P., and Kühl, M. (2015). Microsensor measurements of hydrogen gas dynamics in cyanobacterial microbial mats. Front. Microbiol. 6:726. doi: 10.3389/fmicb.2015.00726
Nold, S. C., and Ward, D. M. (1996). Photosynthate partitioning and fermentation in hot spring microbial mat communities. Appl. Environ. Microbiol. 62, 4598–4607.
Nowack, S. P., Olsen, M. T., Schaible, G. A., Becraft, E. D., Shen, G., Klapper, I., et al. (2015). The molecular dimension of microbial species: 2. Synechococcus strains representative of putative ecotypes inhabiting different depths in the Mushroom Spring microbial mat exhibit different adaptive and acclimative responses to light. Front. Microbiol. 6:626. doi: 10.3389/fmicb.2015.00626
Nübel, U., Bateson, M. M., Vandieken, V., Kühl, M., and Ward, D. M. (2002). Microscopic examination of distribution and phenotypic properties of phylogenetically diverse Chloroflexaceae-related bacteria in hot spring microbial mats. Appl. Environ. Microbiol. 68, 4593–4603. doi: 10.1128/AEM.68.9.4593-4603.2002
Olsen, M. T., Nowack, S. P., Wood, J. M., Becraft, E. D., LaButti, K., Lipzen, A., et al. (2015). The molecular dimension of microbial species: 3. Comparative genomics of Synechococcus strains with different light responses and in situ diel transcription patterns of associated putative ecotypes in the Mushroom Spring microbial mat. Front. Microbiol. 6:604. doi: 10.3389/fmicb.2015.00604
Overbeek, R. A., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucl. Acids Res. 42, 1–9. doi: 10.1093/nar/gkt1226
Papke, R. T., Ramsing, N. B., Bateson, M. M., and Ward, D. M. (2003). Geographical isolation in hot spring cyanobacteria. Environ. Microbiol. 5, 650–659. doi: 10.1046/j.1462-2920.2003.00460.x
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Rabenstein, A., Rethmeier, J., and Fischer, U. (1995). Sulphite as intermediate sulfur compound in anaerobic sulphide oxidation to thiosulphate by marine cyanobacteria. Z. Naturforsch. 50c, 769–774.
Ragsdale, S. W., and Pierce, E. (2008). Acetogenesis and the Wood-Ljungdahl pathway for CO2 fixation. Biochem. Biophys. Acta 1784, 1873–1898. doi: 10.1016/j.bbapap.2008.08.012
Rákhely, G., Laurinavichene, T. V., Tsygankov, A. A., and Kovács, K. L. (2007). The role of Hox hydrogenase in the H2 metabolism of Thiocapsa roseopersicina. Biochim. Biophys. Acta 1767, 671–676. doi: 10.1016/j.bbabio.2007.02.004
Ramsing, N. B., Ferris, M. J., and Ward, D. M. (2000). Highly ordered vertical structure of Synechococcus populations within the one-millimeter-thick photic zone of a hot spring cyanobacterial mat. Appl. Environ. Microbiol. 66, 1038–1049. doi: 10.1128/AEM.66.3.1038-1049.2000
Revsbech, N. P., Trampe, E., Lichtenberg, M., Ward, D. M., and Kühl, M. (2016). In situ hydrogen dynamics in a hot spring microbial mat during a diel cycle. Appl. Environ. Microbiol. 82, 4209–4217. doi: 10.1128/AEM.00710-16
Richter, M., Rosselo-Mora, R., Oliver Glöckner, F., and Peplies, J. (2015). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Sabehi, G., Loy, A., Jung, K.-H., Partha, R., Spudich, J. L., Isaacson, T., et al. (2005). New insights into metabolic properties of marine bacteria encoding proteorhodopsins. PLoS Biol. 3:e273. doi: 10.1371/journal.pbio.0030273
Schaffert, C. S., Klatt, C. G., Ward, D. M., Pauley, M. A., and Steinke, L. (2012). Identification and distribution of high abundance proteins in an Octopus Spring microbial mat community. Appl. Environ. Microbiol. 78, 8481–8484. doi: 10.1128/aem.01695-12
Sojo, V., Herschy, B., Whicher, A., Camprubi, E., and Lane, N. (2016). The origin of life in alkaline hydrothermal vents. Astrobiology 16, 181–197. doi: 10.1089/ast.2015.1406
Sorokin, D. Y., Tourova, T. P., Henstra, A. M., Stams, A. J. M., Galinski, E. A., and Muyzer, G. (2008). Sulfidogenesis under extremely haloalkaline conditions by Desulfonatronospira thiodismutans gen. nov., sp. nov., and Desulfonatronospira delicat sp. nov. - A novel lineage of Deltaproteobacteria from hypersaline soda lakes. Microbiology 154, 1444–1453. doi: 10.1099/mic.0.2007/015628-0
Steunou, A.-S., Bhaya, D., Bateson, M. M., Melendrez, M. C., Ward, D. M., Brecht, E., et al. (2006). In situ analysis of nitrogen fixation and metabolic switching in unicellular thermophilic cyanobacteria inhabiting hot spring microbial mats. Proc. Natl. Acad. Sci. U.S.A. 103, 2398–2403. doi: 10.1073/pnas.0507513103
Steunou, A.-S., Jensen, S. I., Brecht, E., Becraft, E. D., Bateson, M. M., Kilian, O., et al. (2008). Regulation of nif gene expression and the energetics of N2 fixation over the diel cycle in a hot spring microbial mat. ISME J. 2, 364–378. doi: 10.1038/ismej.2007.117
Tang, K.-H., Tang, Y. J., and Blankenship, R. E. (2011). Carbon metabolism pathways in phototrophic bacteria and their broader evolutionary implications. Front. Microbiol. 2:165. doi: 10.3389/fmicb.2011.00165
Tank, M., and Bryant, D. A. (2015a). Nutrient requirements and growth physiology of the photoheterotrophic acidobacterium, Chloracidobacterium thermophilum. Front. Microbiol. 6:226. doi: 10.3389/fmicb.2015.00226
Tank, M., and Bryant, D. A. (2015b). Chloracidobacterium thermophilum gen. nov., sp. nov.: an anoxygenic microaerophilic chlorophotoheterotrophic acidobacterium. Int. J. Syst. Evol. Microbiol. 65, 1426–1430. doi: 10.1099/ijs.0.000113
Tank, M., Thiel, V., Ward, D. M., and Bryant, D. A. (2017). “A panoply of phototrophs: a photomicrographic overview of the thermophilic chlorophototrophs of the microbial mats of alkaline siliceous hot springs in Yellowstone National Park, WY, USA,” in Modern Topics in the Phototrophic Prokaryotes: Environmental and Applied Aspects, ed P. C. Hallenbeck (Cham: Springer International Publishing), 87–137. doi: 10.1007/978-3-319-46261-5_3
Thiel, V., Hamilton, T. L., Tomsho, L. P., Burhans, R., Gay, S. E., Schuster, S. C., et al. (2014). Draft genome sequence of a sulfide-oxidizing, autotrophic filamentous anoxygenic phototrophic bacterium, Chloroflexus sp. strain MS-G (Chloroflexi). Genome Announc. 2, e00872–e00814. doi: 10.1128/genomeA.00872-14
Thiel, V., Tomsho, L. P., Burhans, R., Gay, S. E., Schuster, S. C., Ward, D. M., et al. (2015). Draft genome sequence of the Deinococcus-Thermus bacterium Meiothermus ruber strain A. Genome Announc. 3:e00202–15. doi: 10.1128/genomeA.00202-15
Thiel, V., Hügler, M., Blümel, M., Baumann, H. I., Gärtner, A., Schmaljohann, R., et al. (2012). Widespread occurrence of two carbon fixation pathways in tubeworm endosymbionts: lessons from hydrothermal vent associated tubeworms from the Mediterranean Sea. Front. Microbiol. 3:423. doi: 10.3389/fmicb.2012.00423
Thiel, V., Wood, J. M., Olsen, M. T., Tank, M., Klatt, C. G., Ward, D. M., et al. (2016). The dark side of the Mushroom Spring microbial mat : life in the shadow of chlorophototrophs. I. Microbial diversity based on 16S rRNA amplicon and metagenome sequencing. Front. Microbiol. 7:919. doi: 10.3389/fmicb.2016.00919
van der Meer, M. T. J., Klatt, C. G., Wood, J., Bryant, D. A., Bateson, M. M., Lammerts, L., et al. (2010). Cultivation and genomic, nutritional, and lipid biomarker characterization of Roseiflexus strains closely related to predominant in situ populations inhabiting Yellowstone hot spring microbial mats. J. Bacteriol. 192, 3033–3042. doi: 10.1128/JB.01610-09
van der Meer, M. T. J., Schouten, S., Bateson, M. M., Nübel, U., Wieland, A., Kühl, M., et al. (2005). Diel variations in carbon metabolism by green nonsulfur-like bacteria in alkaline siliceous hot spring microbial mats from Yellowstone National Park. Appl. Environ. Microbiol. 71, 3978–3986. doi: 10.1128/AEM.71.7.3978-3986.2005
van der Meer, M. T. J., Schouten, S., Damsté, J. S. S., and Ward, D. M. (2007). Impact of carbon metabolism on 13C signatures of cyanobacteria and green non-sulfur-like bacteria inhabiting a microbial mat from an alkaline siliceous hot spring in Yellowstone National Park (USA). Environ. Microbiol. 9, 482–491. doi: 10.1111/j.1462-2920.2006.01165.x
Ward, D. M., and Cohan, F. M. (2005). “Microbial diversity in hot spring cyanobacterial mats: pattern and prediction,” in Geothermal Biology and Geochemistry in Yellowstone National Park, eds W. P. Inskeep and T. R. McDermott (Bozeman: Montana State University Publications), 185–202.
Ward, D. M., Bateson, M. M., Weller, R., and Ruff-Roberts, A. L. (1992). “Ribosomal RNA analysis of microorganisms as they occur in nature,” in Advances in Microbial Ecology, ed K. C. Marshall (Boston, MA: Springer US), 219–286.
Ward, D. M., Castenholz, R. W., and Miller, S. R. (2012). “Ecology of cyanobacteria II: Cyanobacteria in geothermal habitats,” in Ecology of cyanobacteria II, ed B. A. Whitton (Dordrecht: Springer Netherlands), 39–63. doi: 10.1007/978-94-007-3855-3_3
Ward, D. M., Ferris, M. J., Nold, S. C., and Bateson, M. M. (1998). A natural view of microbial biodiversity within hot spring cyanobacterial mat communities. Microbiol. Mol. Biol. Rev. 62, 1353–1370.
Weiss, M. C., Sousa, F. L., Mrnjavac, N., Neukirchen, S., Roettger, M., Nelson-Sathi, S., et al. (2016). The physiology and habitat of the last common ancestor. Nat. Microbiol. 1, 16116. doi: 10.1038/nmicrobiol.2016.116
Weller, R., Bateson, M. M., Heimbuch, B. K., Kopczynski, E. D., and Ward, D. M. (1992). Uncultivated cyanobacteria, Chloroflexus-like inhabitants, and spirochete-like inhabitants of a hot spring microbial mat. Appl. Environ. Microbiol. 58, 3964–3969.
Woggon, W.-D. (2002). Oxidative cleavage of carotenoids catalyzed by enzyme models and beta-carotene 15,15′-monooxygenase. Pure Appl. Chem. 74, 1397–1408. doi: 10.1351/pac200274081397
Yang, X., Hao, L., Zhu, H., and Ma, K. (2015). Purification and molecular characterization of an [FeFe]-hydrogenase from Thermotoga hypogea. Curr. Biotechnol. 4, 118–127. doi: 10.2174/2211550104666150414185950
Zarzycki, J., and Fuchs, G. (2011). Coassimilation of organic substrates via the autotrophic 3-hydroxypropionate bi-cycle in Chloroflexus aurantiacus. Appl. Environ. Microbiol. 77, 6181–6188. doi: 10.1128/AEM.00705-11
Keywords: hot spring, microbial community, microbial diversity, extreme environments, chlorophototrophic bacteria, metagenomics
Citation: Thiel V, Hügler M, Ward DM and Bryant DA (2017) The Dark Side of the Mushroom Spring Microbial Mat: Life in the Shadow of Chlorophototrophs. II. Metabolic Functions of Abundant Community Members Predicted from Metagenomic Analyses. Front. Microbiol. 8:943. doi: 10.3389/fmicb.2017.00943
Received: 27 March 2017; Accepted: 10 May 2017;
Published: 06 June 2017.
Edited by:
Robert Duran, University of Pau and Pays de l'Adour, FranceReviewed by:
Richard Allen White III, Pacific Northwest National Laboratory (DOE), United StatesJeremy Dodsworth, California State University, San Bernardino, United States
Copyright © 2017 Thiel, Hügler, Ward and Bryant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vera Thiel, vthiel@tmu.ac.jp
Donald A. Bryant, dab14@psu.edu
†Present Address: Vera Thiel, Department of Biological Sciences, Tokyo Metropolitan University, Hachioji, Tokyo, Japan