 Ying Liu
Ying Liu Yuanman Tang
Yuanman Tang Xiyun Qin2
Xiyun Qin2 Liang Yang
Liang Yang Gaofei Jiang
Gaofei Jiang Wei Ding
Wei Ding- 1Laboratory of Natural Products Pesticides, College of Plant Protection, Southwest University, Chongqing, China
- 2Yunnan Academy of Tobacco Agricultural Research, Yuxi, China
- 3Laboratoire des Interactions Plantes-Microorganismes, Université de Toulouse, INRA, CNRS, Castanet-Tolosan, France
Ralstonia solanacearum, an agent of bacterial wilt, is a highly variable species with a broad host range and wide geographic distribution. As a species complex, it has extensive genetic diversity and its living environment is polymorphic like the lowland and the highland area, so more genomes are needed for studying population evolution and environment adaptation. In this paper, we reported the genome sequencing of R. solanacearum strain CQPS-1 isolated from wilted tobacco in Pengshui, Chongqing, China, a highland area with severely acidified soil and continuous cropping of tobacco more than 20 years. The comparative genomic analysis among different R. solanacearum strains was also performed. The completed genome size of CQPS-1 was 5.89 Mb and contained the chromosome (3.83 Mb) and the megaplasmid (2.06 Mb). A total of 5229 coding sequences were predicted (the chromosome and megaplasmid encoded 3573 and 1656 genes, respectively). A comparative analysis with eight strains from four phylotypes showed that there was some variation among the species, e.g., a large set of specific genes in CQPS-1. Type III secretion system gene cluster (hrp gene cluster) was conserved in CQPS-1 compared with the reference strain GMI1000. In addition, most genes coding core type III effectors were also conserved with GMI1000, but significant gene variation was found in the gene ripAA: the identity compared with strain GMI1000 was 75% and the hrpII box promoter in the upstream had significantly mutated. This study provided a potential resource for further understanding of the relationship between variation of pathogenicity factors and adaptation to the host environment.
Introduction
Plant bacterial wilt disease is caused by a soil-borne pathogen Ralstonia solanacearum, a complex species with extensive diversity (Hayward, 1991; Genin and Denny, 2012). It is widely distributed throughout the world and has a broad host range, including many dicotyledonous and monocotyledonous plants. Previously, R. solanacearum was subdivided into five races (based on the host range) and six biovars (based on their ability to metabolize disaccharides and hexose alcohols) (Buddenhagen et al., 1962; Hayward, 1964; Pegg and Moffett, 1971; He et al., 1983). Recently, it is divided into four phylotypes corresponding to its geographical origin: phylotype I from Asia, phylotype II from the Americas, phylotype III from Africa, and phylotype IV from the Indonesian archipelago (Fegan and Prior, 2005; Prior and Fegan, 2005). Moreover, the species complex has been divided into three species supported by genome analysis (Prior et al., 2016). Because of its highly diverse geographical distribution, host range, and genetic diversity, control of the pathogen is difficult, resulting in large economic losses (Hayward, 1991; Genin and Boucher, 2002; Genin and Denny, 2012).
To better understand the functions of pathogenicity determinants and the traits of aggressiveness under different ecological environments, the whole genome of R. solanacearum was sequenced. Phylotype I strain GMI1000 was the first strain subject to whole genome analysis (Salanoubat et al., 2002). Currently, there are 67 genomes in the National Center for Biotechnology Information (NCBI) database, of which more than ten genomes are complete (Salanoubat et al., 2002; Remenant et al., 2010, 2012; Li et al., 2011, 2016; Xu et al., 2011; Cao et al., 2013; Bocsanczy et al., 2014; Ailloud et al., 2015; She et al., 2015; Guarischi-Sousa et al., 2016). However, because of their high variation, more genome sequences are needed for analyzing the entire species.
Genomes are a very useful resource to understand the mechanism of plant–pathogen interaction and the phylogenetic analyses of the species. Ailloud et al. (2015) compared genomes of different strains to find an explanation for host range adaptation of R. solanacearum strains. In addition, the genome analysis also provided insight into the evolution of virulence, such as hrp gene clusters, and the type III effectors (T3Es) among R. solanacearum strains and other pathogenic bacteria (Genin and Boucher, 2004).
Phylotype I was one of the ongoing diversifying subspecies according to research focused on the evolutionary history of R. solanacearum using multilocus sequence analysis (MLSA) (Wicker et al., 2012). In China, phylotype I R. solanacearum strains infecting tobacco display sequevar diversity and are spreading from the lowlands to the highlands and cold areas (Liu et al., 2017). It is interesting to study the genetic variations of R. solanacearum influenced by highland circumstances and host environment. Here, we report the complete genome sequence of R. solanacearum CQPS-1, a strain isolate from a highland (>1000 m), where soil is severely acidified and tobacco has continuously cropped more than 20 years. Our goal is to explore the molecular traits that the bacterium uses to adapt to its environment and interact with plants. The genome comparison is performed to find dissimilarities between CQPS-1 and other phylotype I genomes as well as genomes of strains belonging to other phylotypes. Furthermore, in order to elucidate pathogenicity variations, the virulence factors of our sequence were compared with strain GMI1000. We found that type III secretion system (T3SS) gene cluster (hrp gene cluster) was conserved, and only some other T3Es had significant gene variations, which may be a result of the strain interacting with its host for a long time.
Materials and Methods
Strains and Genomic DNA Preparation
The R. solanacearum strain CQPS-1, belonging to phylotype I sequevar 17 (Liu et al., 2017), was isolated from a wilting tobacco plant (Nicotiana tabacum). The wilting plant was collected from Pengshui, Chongqing, China, where tobacco has been grown for more than 20 years; the elevation is more than 1000 m and the pH of soil is severely acidic (pH ≈ 5.0). Strains were grown at 30 ± 2°C in B liquid medium (Boucher et al., 1985). Genomic DNA was purified from overnight liquid cultures using the CTAB (hexadecyltrimethylammonium bromide) method (Wilson, 2001).
Sequencing and Assembly
The whole genome was sequenced using the PacBio RS II platform with a 20-kb library. Reads were assembled using HGAP (version 2.3.0, Pacific Biosciences) (Chin et al., 2013). Assembly data for the complete genome have been deposited in GenBank with accession numbers CP016914 and CP016915 (chromosome and megaplasmid, respectively).
Genome Components and Genome Annotation
CDS were predicted using Prodigal (Hyatt et al., 2010). A circular map of the genome was drawn by CIRCOS (Krzywinski et al., 2009). Genomic Islands (GIs) were predicted by using the GI prediction method IslandPath-DIOMB (Dhillon et al., 2015). Clustered regularly interspaced short palindromic repeat sequences (CRISPRs) were found using CRISPRFinder (Grissa et al., 2007b) and PILER-CR (Edgar, 2007). Functional annotation was based on BLASTp searches against the NCBI non-redundant (NR) database and the KEGG, Pfam, Swissprot, and TrEMBL databases. Cluster of Orthologous Group of proteins (COG) analysis was performed to generate functional annotations for coding sequences (reference to orthologous groups1) (Tatusov et al., 2001).
The Virulence Dataset
Virulence factors were predicted based on the virulence factors database (VFDB2). The virulence factors of the R. solanacearum strain CQPS-1 analyzed in this study were selected according to Remenant et al. (2010). T3Es were annotated using the IANT “Ralstonia T3E” database (Peeters et al., 2013). Every gene annotation was then manually validated to ensure homogeneity of the start codon positions and to detect frameshifts and pseudogenization.
Genomic Comparisons
The genome sequences of GMI1000, Y45, YC45, FQY_4, PO82, CFBP2957, CMR15, and PSI07 were downloaded from the NCBI and EMBL databases. Sequences were aligned using Clustal x (Jeanmougin et al., 1998). Phylogenetic analysis was performed using neighbor-joining (NJ) and the algorithm of Jukes and Cantor (1969) with 1,000 bootstrap resamplings in MEGA version 5 (Tamura et al., 2011). Nucleic acid co-linearity was performed using MCScanX according to the alignment results of homology relationships by BLAST (Altschul et al., 1997; Wang et al., 2012). The set of genes unique to strain CQPS-1 was found using OrthoMCL (Chen et al., 2006).
Results
Genome Features
Whole genome sequencing was performed with single molecule real-time sequencing (SMRT) on the PacBio RS II platform (Eid et al., 2009). The completed genome of R. solanacearum strain CQPS-1 was 5.89 Mb (GC%, 66.84%) and contained one circular chromosome (3.83 Mb, Figure 1A) and one megaplasmid (2.06 Mb, Figure 1B). The general features were shown in Table 1. The average GC content of the chromosome was 66.71% and that of the megaplasmid was 67.09%. A total of 5229 CDS were predicted (chromosome and megaplasmid encoded 3573 and 1656 genes, respectively). The CQPS-1 genome contained 12 rRNA and 58 tRNA.
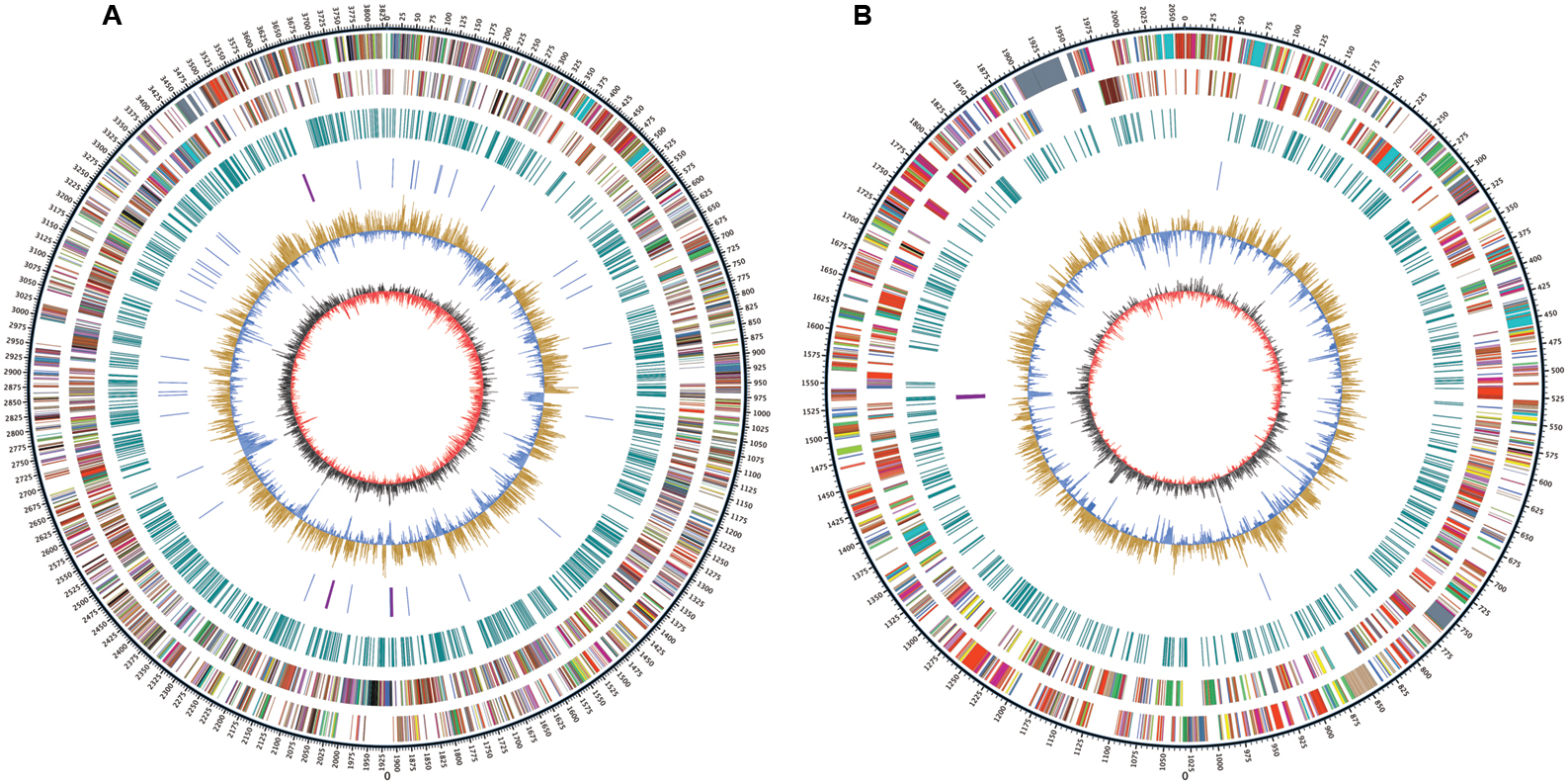
FIGURE 1. Circular map of Ralstonia solanacearum strain CQPS-1 genome. (A) Chromosome; (B) Megaplasmid. The distribution of the circle from outer to inner indicates genome size, forward CDS, reverse CDS, repeat sequences, tRNA (blue) and rRNA (purple), GC ratio (yellow means GC ratio of the region is higher than average GC ratio, blue means GC ratio of the region is lower than average GC ratio), and GC skew (gray represents a region with G content greater than C, red represents a region with C content greater than G).

TABLE 1. General features of the Ralstonia solanacearum strain CQPS-1 genome.
Genomic Islands and CRISPR Prediction
Genomic Islands are evidence of horizontal acquisition (Langille et al., 2010; Remenant et al., 2010). The GIs predicted in CQPS-1 are listed in Supplementary Table S1: a total of 21 GIs were predicted in the chromosome (13 GIs) and megaplasmid (8 GIs). CRISPRs can confer resistance to exogenous genetic elements such as phages and plasmids (Barrangou et al., 2007). To predict the CRISPRs of CQPS-1, the methods PILER-CR and CRISPRFinder were used. From the result predicted by the program PILER-CR, seven CRISPRs were found in the genome of CQPS-1; three were located in the chromosome, and four were in megaplasmid (Supplementary Table S2). Whereas two different questionable CRISPRs were predicted by using CRISPRFinder, one in the chromosome and another in the megaplasmid (Supplementary Table S2). Compared with the previous reports (Li et al., 2016), we knew that the putative CRISPR sequence in the chromosome of CQPS-1 (3,693,731-3,693,841) predicted by CRISPRFinder was completely conserved with the one located in the chromosome of strain GMI1000 (1,445,581-1,445,691).
Genome Annotation
Of the 5229 CDS, 4700 proteins can be assigned to 23 COG families (Supplementary Table S3). Except for the genes predicted to have general (604 genes) or unknown functions (368 genes), the largest group of genes were involved in amino acid transport and metabolism (467 genes, 8.93%). Compared to the distribution of genes in different COG families, the results showed that the megaplasmid had more genes than the chromosome in cell motility (Figure 2), which is consistent with a previous report by Li et al. (2016). In addition, a total of 2539 proteins had KEGG orthologs.
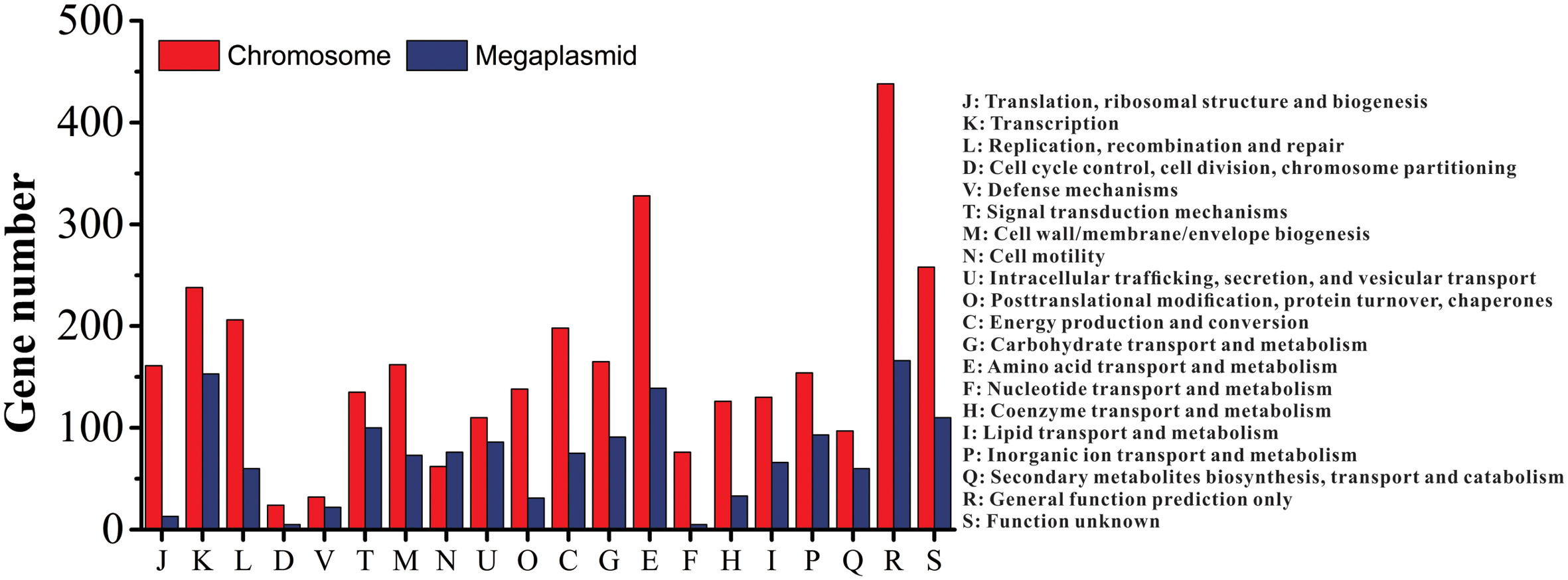
FIGURE 2. Distribution of genes with COG functional categories between the chromosome and the megaplasmid in strain CQPS-1.
Comparative Genome Analysis
Phylogenetic tree was constructed using 16S rRNA. The result showed that CQPS-1 belonged to phylotype I, and was closest to strains YC45, FQY_4, and GMI1000 (Figure 3). When aligning syntenic genes of the CQPS-1 genome with other R. solanacearum genomes, the results demonstrated that the percentages of syntenic genes compared with phylotype I strains were more than other phylotype strains. The number of CDS in synteny with strain GMI1000 was highest (84.97%, Table 2), while the number of CDS in synteny with phylotype IIA strain CFBP2957 was the lowest (70.47%). According to the results of nucleic acid co-linearity, we know that there were a large number of inverse fragments among different R. solanacearum species, and many rearrangements were found in these genomes (Table 2 and Supplementary Figure S1).
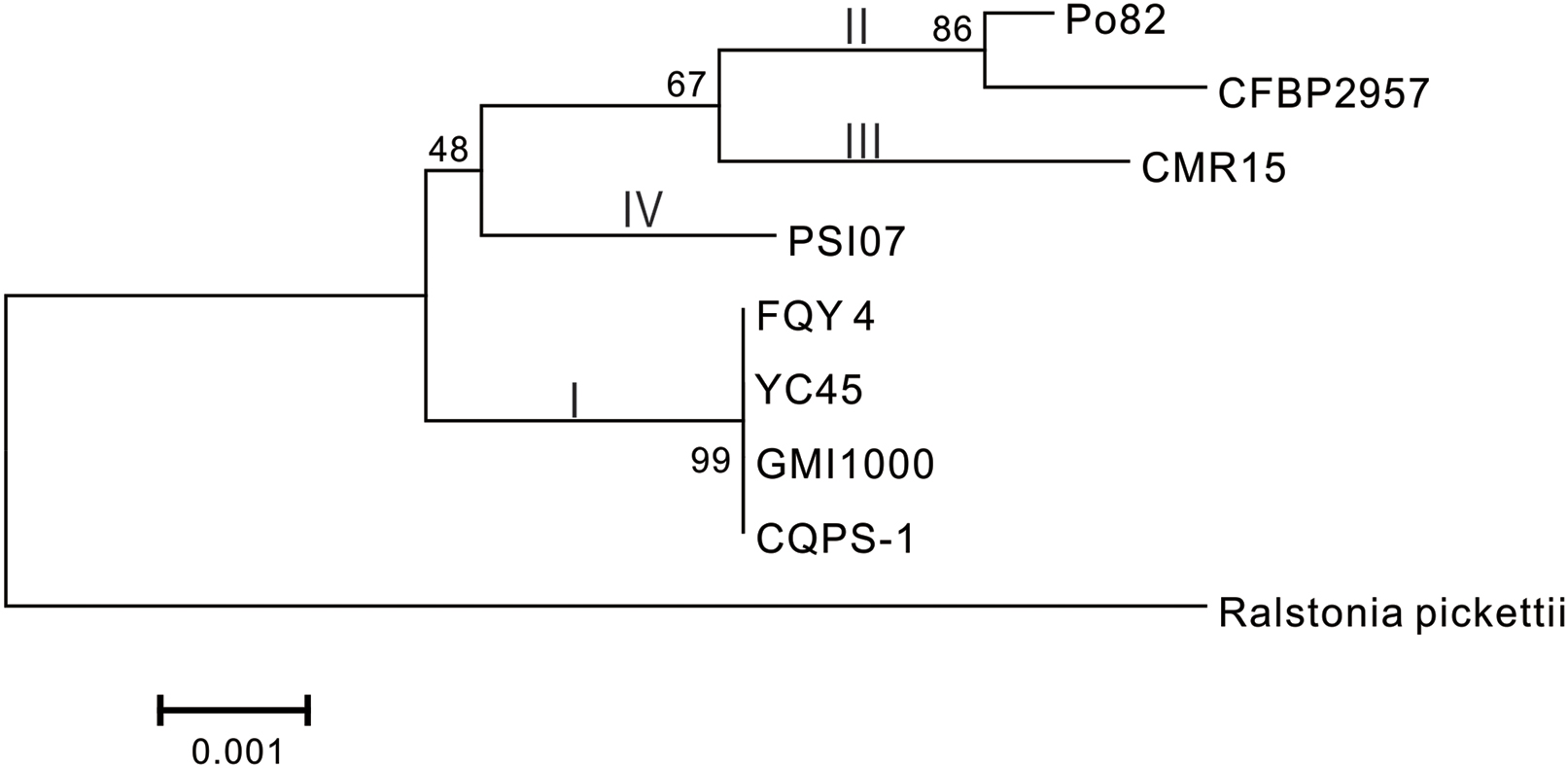
FIGURE 3. Phylogenetic tree of R. solanacearum strain CQPS-1 with other close species based on 16S rRNA. The tree was generated by MEGA-5 software using the neighbor-joining (NJ) and the algorithm of Jukes and Cantor (1969) with 1,000 bootstrap re-samplings. Ralstonia pickettii 12J (NCBI accession NC_010682) was used as an outgroup.
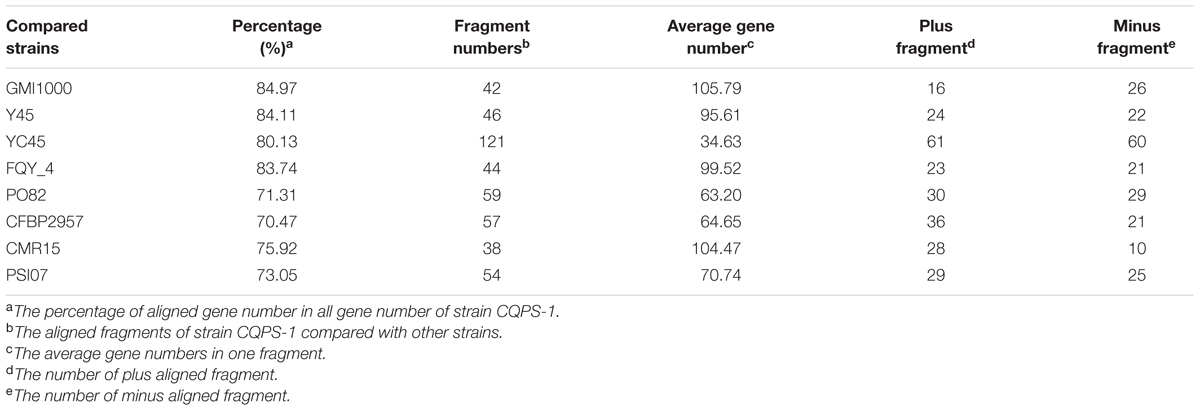
TABLE 2. The co-linearity results of strain CQPS-1compared to that of different R. solanacearum strains.
We also performed a pan-genomic analysis of R. solanacearum strains. First, we compared the genes of strain CQPS-1 to four phylotype I strains: GMI1000, YC45, Y45, and FQY_4. As shown in Figure 4A, 3946 gene families were involved in the core genome, which was shared by all compared strains. In addition, the number of specific gene families in strain CQPS-1 was 16 and contained 442 genes (specific gene numbers were shown in Supplementary Table S4). After annotation, the specific genes encoded a large number of hypothetical proteins and other proteins, such as transposase, LuxR family transcriptional regulator, signal peptide protein, membrane protein, T3E protein, etc. (detailed annotation data was shown in Supplementary Table S5). Then, strain CQPS-1, as a phylotype I strain, was compared with the other four phylotype strains (Po82, CFBP2957, CMR15, and PSI07). The results (shown in Figure 4B) showed that there were 3399 gene families shared by different phylotype strains. The number of CQPS-1-specific gene families was 49, including 625 genes (specific gene numbers were shown in Supplementary Table S6), most of which coded as hypothetical proteins (Supplementary Table S7).
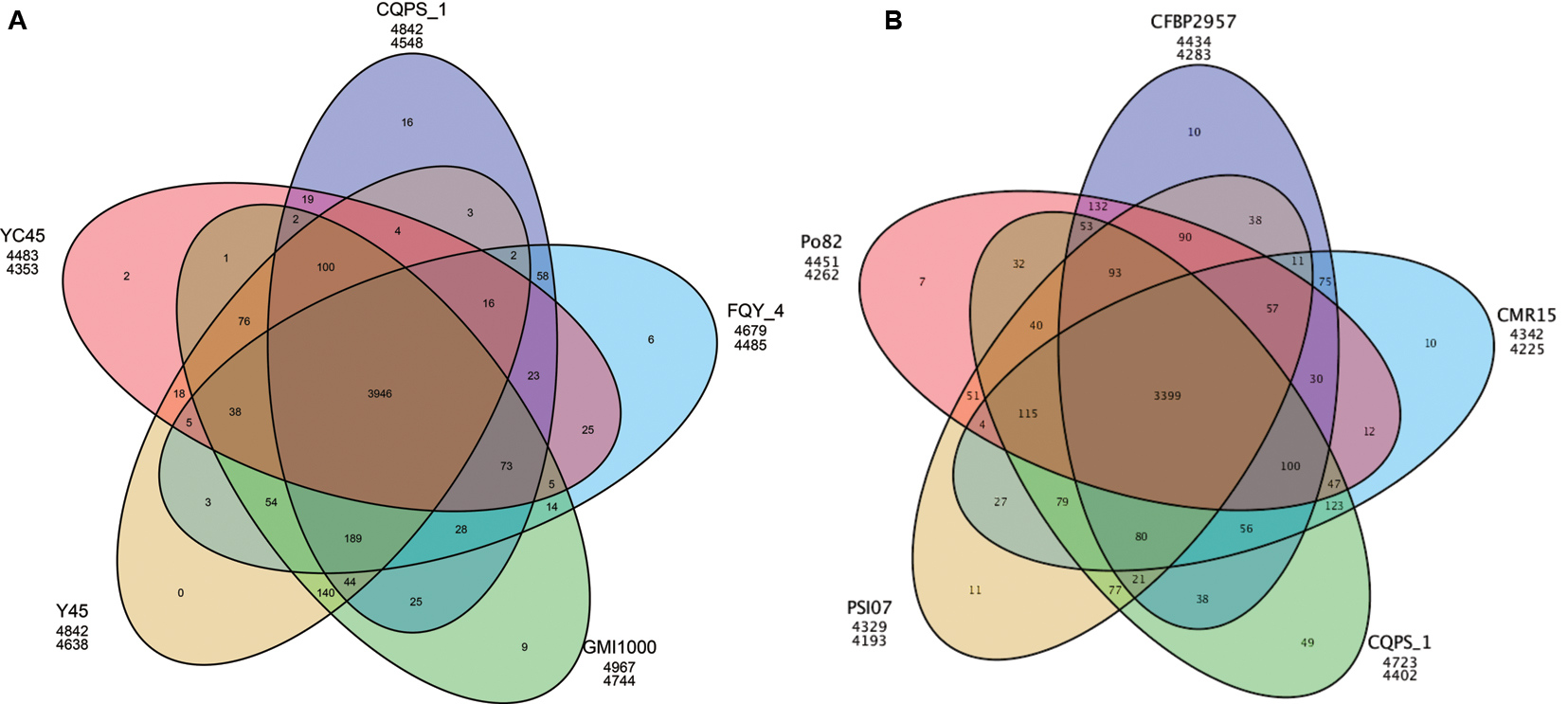
FIGURE 4. Venn diagram showing numbers of specific and shared gene families among five phylotype I species (A), and five different phylotype species (B). The number in the overlapping sections indicate shared numbers of gene families. The first line below each strain name represents the number of genes involved in the clustered gene families, and the second line represents the number of gene families that the strain has.
Virulence Factors
Potential virulence factors in the strain CQPS-1 were identified using the BLAST search in the VFDB database. A total of 622 putative virulence factors were aligned (the chromosome and megaplasmid had 363 and 259 genes, respectively). We also compared the virulence factors reported by Remenant et al. (2010) with strain GMI1000, including exopolysaccharide (EPS) biosynthetic genes, cell wall-degrading enzyme (CWDE) genes, response genes to the host defense and key virulence regulators. The results showed that these genes were highly identical with strain GMI1000 (Supplementary Table S8); the identities were more than 97%, except twitching motility gene pilA, whose identity was 91%.
Comparison Analyses of Type III Secretion Systems and Type III Effectors
Type III secretion systems, which has a syringe-like membrane structure and can inject T3Es into plant cells, causing disease or a hypersensitive response (HR), is important for the pathogenicity of R. solanacearum (Valls et al., 2006; Coll and Valls, 2013). T3SS is coded by hypersensitive response and pathogenicity (hrp) gene cluster, which is in the megaplasmid (Lindgren, 1997; Genin and Boucher, 2004). In strain CQPS-1, the hrp gene cluster contained 30 genes (spanning 29,682 bp, from 1,604,200 to 1,633,881). A comparison showed that the hrp gene cluster of CQPS-1 has a high similarity to that of the strain GMI1000 (the identity was 99%, Figure 5).
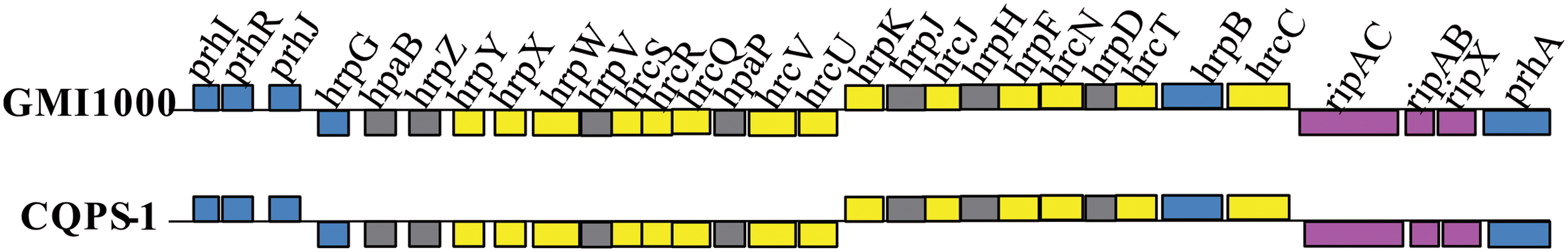
FIGURE 5. Genetic organization and gene comparison of hrp clusters between CQPS-1 and GMI1000.
Type III effectors, presumed to modulate host innate immunity, are important virulence determinants for the pathogen (Poueymiro and Genin, 2009; Peeters et al., 2013). According to Peeters et al. (2013), 32 conserved or core T3Es have been defined. We compared the genes of 32 core T3Es of CQPS-1 to GMI1000. The results showed that 29 core T3Es genes were conserved (the coverage was 100%, and the identity was more than 90%), and some variations were found in others (Table 3). RipB had some base deletion, and the coverage was 93%. The identity of ripG7 was 84%. RipAA (former name avrA), which can encode RipAA, the effector responsible for triggering HR on N. tabacum and N. benthamiana (Carney and Denny, 1990; Poueymiro et al., 2009), had 75% identity with strain GMI1000. In addition, there was a variation in the hrpII box promoter in the upstream of ripAA.
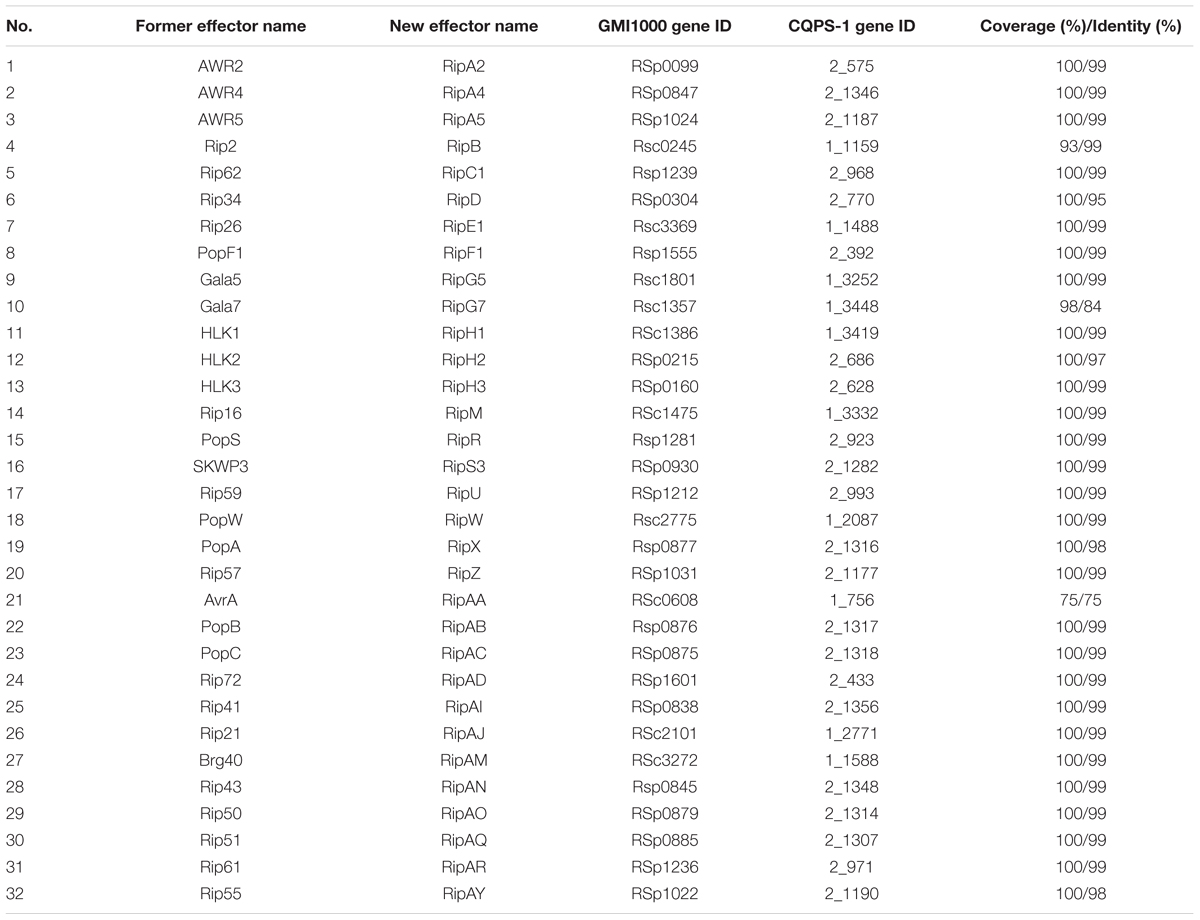
TABLE 3. Comparison of core type III effectors (T3Es) genes of strain CQPS-1 with strain GMI1000.
Discussion
Ralstonia solanacearum, which causes very large economic losses every year in China, is spreading to high altitudes and cold areas (Liu et al., 2017). This study presented a complete genome of the R. solanacearum strain CQPS-1 collected from a highland area with severely acidified soil and continuous cropping of tobacco. The technology used for sequencing the genome was SMRT (Eid et al., 2009; McCarthy, 2010), which is applied to finished microbial genomes by Pacific Biosciences due to its longer read length. The genome contained a 3.83 Mb chromosome and a 2.06 Mb megaplasmid. A comparative genomics analysis was also performed to identify the differences between strain CQPS-1 and other representative strains. From the results, we found that the genome of strain CQPS-1 showed some degree of variation, which could provide some evidence for a relationship between effectors variance and pathogen adaptation to host and environment.
Phylogenetic analysis was performed based on 16S rRNA. From the result, we knew that strain CQPS-1 was more similar to other phylotype I strains, such as GMI1000, YC45, and FQY_4, than other phylotypes, such as CFBP2957 (phylotype IIA), Po82 (phylotype IIB), CMR15 (phylotype III), and PSI07 (phylotype IV). Co-linearity also supported the result. Genome synteny, which studies the conserved multigene regions, is useful to assess species evolution and predict the gene function (Suyama and Bork, 2001; Bentley and Parkhill, 2004). According to our result of co-linearity analysis, there were different levels of inverse fragments and dissimilarities among these phylotype I strains and different phylotype strains. Remenant et al. (2010) demonstrated that the R. solanacearum genomes were highly syntenic when working on six strains, in addition, intra- and inter-replicon rearrangements occurred in the history of the organisms. In bacteria, rapid evolutionary changes such as chromosomal rearrangements always accompanied by host restriction (Moran and Plague, 2004).
Another important mechanisms in the evolution of pathogens is horizontal gene transfer (HGT) (Bhattacharya et al., 2003; Guidot et al., 2009). Bacteria could get genes from other different species such as archaea, bacteriophage, and eukaryotes (Koonin et al., 2001). R. solanacearum can transfer genes to adapt to novel ecological niches (Guidot et al., 2009). GIs, which known as pathogenicity islands, were thought to be the result of HGT (Langille et al., 2010). There were 21 GIs found in the genome of CQPS-1. The details of these GIs need to be further analyzed. Our complete genome can supply the resource to explore the species evolution interacted with different host plants and study the HGT of R. solanacearum strains occurring in nature.
There were nine CRISPRs predicted in CQPS-1 genome by using two methods, PILER-CR and CRISPRFinder. PILER-CR is a fast and accurate program based on an elegant algorithm to identify the CRISPR properties (Edgar, 2007), and CRISPRFinder is chosen because it can find very small CRISPRs (contained less than three, three or seven spacers) (Grissa et al., 2007a). The results predicted by the two methods showed that there were no intersection, and two small CRISPRs were found by CRISPRFinder.
Pan-genomic analysis of phylotype I strains demonstrated that the numbers of specific genes among compared phylotype I strains were different (ranging from 30 to 478). Phylotype I, of East African/Asian origin, can infect the largest number of host plants (Hayward, 1994). Strain GMI1000 has been isolated from tomatoes (Boucher et al., 1985), strains Y45 and FQY_4 have been known to infect tobacco (Li et al., 2011; Cao et al., 2013), and strain YC45 has been collected from ginger plants (She et al., 2015). The variety of host environments may be one of the reasons that this lineage is highly divergent. The function of specific genes should be further analyzed in depth to understand the relationship between specific genes and the adaptation of strains. For example, several genes encoded T3E proteins were found among specific genes in CQPS-1 when compared with other phylotype I strains (Supplementary Table S5). Whether these genes work is still unknown.
According to our results, the hrp gene cluster of CQPS-1 was conserved compared with GMI1000, which is consistent with the previous report that hrp cluster was highly conserved among phylotype I strains (Li et al., 2016). T3Es, translocated by T3SS, are highly variable and may play a role in shaping or extending the host range of strains according to previous studies (Castaneda et al., 2005; Hajri et al., 2009; Genin, 2010; Baltrus et al., 2011). Furthermore, they could co-evolve with the plant targets, such as the effector RipG7, the essential determinant of R. solanacearum strains for virulence on the legume plant Medicago truncatula (Wang et al., 2016). In this study, we found that the ripAA of strain CQPS-1 was variable compared with that of GMI1000: only 75% was identical with the gene of strain GMI1000, and there was a significant variation in the hrpII box promoter of ripAA in strain CQPS-1. It is known that the RipAA (AvrA) of GMI1000 is the major determinant causing HR on N. tabacum and N. benthamiana (Poueymiro et al., 2009). Strain CQPS-1 was collected from a location where tobacco has been grown for more than 20 years. We speculated that the mutative ripAA may be one of the results of an effector interacting with tobacco for a long time, which could help pathogen to avoid host recognition. This result provided another parameter to analyze effectors co-evolving with hosts.
In summary, this study showed the whole genome of strain CQPS-1 and comparative genomics analyses among different R. solanacearum strains. The genome variability presumably plays an important role when R. solanacearum strains adapt themselves to a host environment, which could provide an essential platform for studying plant–pathogen interactions for a long time.
Author Contributions
Experimental design and authorship: YL and WD; Experiments and data analysis: YL, YT, LY, GJ, and SL; Manuscript revised: GJ, WD, and XQ; All authors read and approved the final manuscript.
Funding
This study was supported by the Key Project from China National Tobacco Corporation (110201502019) and the Key Project from China National Tobacco Corporation Chongqing Branch (NY20130501070005).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00974/full#supplementary-material
FIGURE S1 | Nucleic acid co-linearity of strain CQPS-1 vs. their orthologs, (A) GMI1000, (B) Y45, (C) YC45, (D) FQY_4, (E) PO82, (F) CFBP2957, (G) CMR15, and (H) PSI07, respectively.
Footnotes
References
Ailloud, F., Lowe, T., Cellier, G., Roche, D., Allen, C., and Prior, P. (2015). Comparative genomic analysis of Ralstonia solanacearum reveals candidate genes for host specificity. BMC Genomics 16:270. doi: 10.1186/S12864-015-1474-8
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J. H., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Baltrus, D. A., Nishimura, M. T., Romanchuk, A., Chang, J. H., Mukhtar, M. S., Cherkis, K., et al. (2011). Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Pathog. 7:e1002132. doi: 10.1371/journal.ppat.1002132
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. doi: 10.1126/science.1138140
Bentley, S. D., and Parkhill, J. (2004). Comparative genomic structure of prokaryotes. Annu. Rev. Genet. 38, 771–792. doi: 10.1146/annurev.genet.38.072902.094318
Bhattacharya, D., Sarma, P. M., Krishnan, S., Mishra, S., and Lal, B. (2003). Evaluation of genetic diversity among Pseudomonas citronellolis strains isolated from oily sludge-contaminated sites. Appl. Environ. Microbiol. 69, 1435–1441. doi: 10.1128/aem.69.3.1435-1441.2003
Bocsanczy, A. M., Huguet-Tapia, J. C., and Norman, D. J. (2014). Whole-genome sequence of Ralstonia solanacearum P673, a strain capable of infecting tomato plants at low temperatures. Genome Announc. 2, e00106-14. doi: 10.1128/genomeA.00106-14
Boucher, C. A., Barberis, P. A., and Demery, D. A. (1985). Transposon mutagenesis of Pseudomonas solanacearum: isolation of Tn5-induced avirulent mutants. Microbiology 131, 2449–2457. doi: 10.1099/00221287-131-9-2449
Buddenhagen, I. W., Sequeira, L., and Kelman, A. (1962). Designation of races in Pseudomonas solanacearum. Phytopathology 52:726.
Cao, Y., Tian, B., Liu, Y., Cai, L., Wang, H., Lu, N., et al. (2013). Genome sequencing of Ralstonia solanacearum FQY_4, isolated from a bacterial wilt nursery used for breeding crop resistance. Genome Announc. 1:e125-13. doi: 10.1128/genomeA.00125-13
Carney, B. F., and Denny, T. P. (1990). A cloned avirulence gene from Pseudomonas solanacearum determines incompatibility on Nicotiana tabacum at the host species level. J. Bacteriol. 172, 4836–4843. doi: 10.1128/jb.172.9.4836-4843.1990
Castaneda, A., Reddy, J. D., El-Yacoubi, B., and Gabriel, D. W. (2005). Mutagenesis of all eight avr genes in Xanthomonas camplestris pv. campestris had no detected effect on pathogenicity, but one avr gene affected race specificity. Mol. Plant Microbe Interact. 18, 1306–1317. doi: 10.1094/MPMI-18-1306
Chen, F., Mackey, A. J., Stoeckert, C. J. Jr., and Roos, D. S. (2006). OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34, 363–368. doi: 10.1093/nar/gkj123
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/Nmeth.2474
Coll, N. S., and Valls, M. (2013). Current knowledge on the Ralstonia solanacearum type III secretion system. Microb. Biotechnol. 6, 614–620. doi: 10.1111/1751-7915.12056
Dhillon, B. K., Laird, M. R., Shay, J. A., Winsor, G. L., Lo, R., Nizam, F., et al. (2015). IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 43, 104–108. doi: 10.1093/nar/gkv401
Edgar, R. C. (2007). PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics 8:18. doi: 10.1186/1471-2105-8-18
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
Fegan, M., and Prior, P. (2005). “How complex is the “Ralstonia solanacearum species complex?”,” in Bacterial wilt Disease and the Ralstonia solanacearum Species Complex, eds C. Allen, P. Prior, and A. C. Hayward (Madison, WI: APS), 449–462.
Genin, S. (2010). Molecular traits controlling host range and adaptation to plants in Ralstonia solanacearum. New Phytol. 187, 920–928. doi: 10.1111/j.1469-8137.2010.03397.x
Genin, S., and Boucher, C. (2002). Ralstonia solanacearum: secrets of a major pathogen unveiled by analysis of its genome. Mol. Plant Pathol. 3, 111–118. doi: 10.1046/j.1364-3703.2002.00102.x
Genin, S., and Boucher, C. (2004). Lessons learned from the genome analysis of Ralstonia solanacearum. Annu. Rev. Phytopathol. 42, 107–134. doi: 10.1146/annurev.phyto.42.011204.104301
Genin, S., and Denny, T. P. (2012). Pathogenomics of the Ralstonia solanacearum species complex. Ann. Rev. Phytopathol. 50, 67–89. doi: 10.1146/annurev-phyto-081211-173000
Grissa, I., Vergnaud, G., and Pourcel, C. (2007a). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172. doi: 10.1186/1471-2105-8-172
Grissa, I., Vergnaud, G., and Pourcel, C. (2007b). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Guarischi-Sousa, R., Puigvert, M., Coll, N. S., Siri, M. I., Pianzzola, M. J., Valls, M., et al. (2016). Complete genome sequence of the potato pathogen Ralstonia solanacearum UY031. Stand. Genomic Sci. 11:7. doi: 10.1186/S40793-016-0131-4
Guidot, A., Coupat, B., Fall, S., Prior, P., and Bertolla, F. (2009). Horizontal gene transfer between Ralstonia solanacearum strains detected by comparative genomic hybridization on microarrays. ISME J. 3, 549–562. doi: 10.1038/ismej.2009.14
Hajri, A., Brin, C., Hunault, G., Lardeux, F., Lemaire, C., Manceau, C., et al. (2009). A <<repertoire for repertoire>> hypothesis: repertoires of type three effectors are candidate determinants of host specificity in Xanthomonas. PLoS ONE 4:e6632. doi: 10.1371/journal.pone.0006632
Hayward, A. C. (1964). Characteristics of Pseudomonas solanacearum. J. Appl. Bacteriol. 27, 265–277. doi: 10.1111/j.1365-2672.1964.tb04912.x
Hayward, A. C. (1991). Biology and epidemiology of bacterial wilt caused by Pseudomonas solanacearum. Ann. Rev. Phytopathol. 29, 65–87. doi: 10.1146/annurev.py.29.090191.000433
Hayward, A. C. (1994). “The hosts of Pseudomonas solanacearum,” in Bacterial Wilt - The Disease and Its Causative Agent, Pseudomonas solanacearum, eds A. C. Hayward and G. L. Hartman (Wallingford: CAB International), 9–24.
He, L. Y., Sequeira, L., and Kelman, A. (1983). Characteristics of strains of Pseudomonas solanacearum from China. Plant Dis 67, 1357–1361. doi: 10.1094/PD-67-1357
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jeanmougin, F., Thompson, J. D., Gouy, M., Higgins, D. G., and Gibson, T. J. (1998). Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 23, 403–405. doi: 10.1016/S0968-0004(98)01285-7
Jukes, T. H., and Cantor, C. R. (1969). “Evolution of protein molecules,” in Mammalian Protein Metabolism, ed. H. N. Munro (New York, NY: Academic Press), 121–132. doi: 10.1016/b978-1-4832-3211-9.50009-7
Koonin, E. V., Makarova, K. S., and Aravind, L. (2001). Horizontal gene transfer in prokaryotes: quantification and classification. Annu. Rev. Microbiol. 55, 709–742. doi: 10.1146/annurev.micro.55.1.709
Krzywinski, M., Schein, J., Birol,İ., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Langille, M. G. I., Hsiao, W. W. L., and Brinkman, F. S. L. (2010). Detecting genomic islands using bioinformatics approaches. Nat. Rev. Microbiol. 8, 372–382. doi: 10.1038/nrmicro2350
Li, P., Wang, D. C., Yan, J. L., Zhou, J. A., Deng, Y. Y., Jiang, Z. D., et al. (2016). Genomic analysis of phylotype I strain EP1 reveals substantial divergence from other strains in the Ralstonia solanacearum species complex. Front. Microbiol. 7:1719. doi: 10.3389/Fmicb.2016.01719
Li, Z., Wu, S., Bai, X., Liu, Y., Lu, J., Liu, Y., et al. (2011). Genome sequence of the tobacco bacterial wilt pathogen Ralstonia solanacearum. J. Bacteriol. 193, 6088–6089. doi: 10.1128/JB.06009-11
Lindgren, P. B. (1997). The role of hrp genes during plant-bacterial interactions. Ann. Rev. Phytopathol. 35, 129–152. doi: 10.1146/annurev.phyto.35.1.129
Liu, Y., Wu, D., Liu, Q., Zhang, S., Tang, Y., Jiang, G., et al. (2017). The sequevar distribution of Ralstonia solanacearum in tobacco-growing zones of China is structured by elevation. Eur. J. Plant Pathol. 147, 541–551. doi: 10.1007/s10658-016-1023-6
McCarthy, A. (2010). Third generation DNA sequencing: pacific biosciences’ single molecule real time technology. Chem. Biol. 17, 675–676. doi: 10.1016/j.chembiol.2010.07.004
Moran, N. A., and Plague, G. R. (2004). Genomic changes following host restriction in bacteria. Curr. Opin. Genet Dev. 14, 627–633. doi: 10.1016/j.gde.2004.09.003
Peeters, N., Carrere, S., Anisimova, M., Plener, L., Cazale, A. C., and Genin, S. (2013). Repertoire, unified nomenclature and evolution of the Type III effector gene set in the Ralstonia solanacearum species complex. BMC Genomics 14:859. doi: 10.1186/1471-2164-14-859
Pegg, K., and Moffett, M. L. (1971). Host range of the ginger strain of Pseudomonas solanacearum in Queensland. Aust. J. Exp. Agric. 11, 696–698. doi: 10.1071/EA9710696
Poueymiro, M., Cunnac, S., Barberis, P., Deslandes, L., Peeters, N., Cazale-Noel, A. C., et al. (2009). Two type III secretion system effectors from Ralstonia solanacearum GMI1000 determine host-range specificity on tobacco. Mol. Plant Microbe Interact. 22, 538–550. doi: 10.1094/Mpmi-22-5-0538
Poueymiro, M., and Genin, S. (2009). Secreted proteins from Ralstonia solanacearum: a hundred tricks to kill a plant. Curr. Opin. Microbiol. 12, 44–52. doi: 10.1016/j.mib.2008.11.008
Prior, P., Ailloud, F., Dalsing, B. L., Remenant, B., Sanchez, B., and Allen, C. (2016). Genomic and proteomic evidence supporting the division of the plant pathogen Ralstonia solanacearum into three species. BMC Genomics 17:90. doi: 10.1186/s12864-016-2413-z
Prior, P., and Fegan, M. (2005). Recent developments in the phylogeny and classification of Ralstonia solanacearum. Acta Hortic. 695, 127–136. doi: 10.17660/ActaHortic.2005.695.14
Remenant, B., Babujee, L., Lajus, A., Médigue, C., Prior, P., and Allen, C. (2012). Sequencing of K60, type strain of the major plant pathogen Ralstonia solanacearum. J. Bacteriol. 194, 2742–2743. doi: 10.1128/JB.00249-12
Remenant, B., Coupat-Goutaland, B., Guidot, A., Cellier, G., Wicker, E., Allen, C., et al. (2010). Genomes of three tomato pathogens within the Ralstonia solanacearum species complex reveal significant evolutionary divergence. BMC Genomics 11:379. doi: 10.1186/1471-2164-11-379
Salanoubat, M., Genin, S., Artiguenave, F., Gouzy, J., Mangenot, S., Arlat, M., et al. (2002). Genome sequence of the plant pathogen Ralstonia solanacearum. Nature 415, 497–502. doi: 10.1038/415497a
She, X., Tang, Y., He, Z., and Lan, G. (2015). Genome sequencing of Ralstonia solanacearum race 4, biovar 4, and phylotype I, strain YC45, isolated from Rhizoma kaempferiae in southern China. Genome Announc. 3, e01110-15. doi: 10.1128/genomeA.01110-15
Suyama, M., and Bork, P. (2001). Evolution of prokaryotic gene order: genome rearrangements in closely related species. Trends Genet. 17, 10–13. doi: 10.1016/S0168-9525(00)02159-4
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tatusov, R. L., Natale, D. A., Garkavtsev, I. V., Tatusova, T. A., Shankavaram, U. T., Rao, B. S., et al. (2001). The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29, 22–28. doi: 10.1093/Nar/29.1.22
Valls, M., Genin, S., and Boucher, C. (2006). Integrated regulation of the type III secretion system and other virulence determinants in Ralstonia solanacearum. PLoS Pathog. 2:e82. doi: 10.1371/journal.ppat.0020082
Wang, K. K., Remigi, P., Anisimova, M., Lonjon, F., Kars, I., Kajava, A., et al. (2016). Functional assignment to positively selected sites in the core type III effector RipG7 from Ralstonia solanacearum. Mol. Plant Pathol. 17, 553–564. doi: 10.1111/mpp.12302
Wang, Y. P., Tang, H. B., DeBarry, J. D., Tan, X., Li, J. P., Wang, X. Y., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi: 10.1093/nar/gkr1293
Wicker, E., Lefeuvre, P., de Cambiaire, J. C., Lemaire, C., Poussier, S., and Prior, P. (2012). Contrasting recombination patterns and demographic histories of the plant pathogen Ralstonia solanacearum inferred from MLSA. ISME J. 6, 961–974. doi: 10.1038/ismej.2011.160
Wilson, K. (2001). Preparation of genomic DNA from bacteria. Curr. Protoc. Mol. Biol Chap. 2, Unit2.4. doi: 10.1002/0471142727.mb0204s56
Keywords: genome sequencing, Ralstonia solanacearum, virulence factors, type III effectors, comparative genomic analysis
Citation: Liu Y, Tang Y, Qin X, Yang L, Jiang G, Li S and Ding W (2017) Genome Sequencing of Ralstonia solanacearum CQPS-1, a Phylotype I Strain Collected from a Highland Area with Continuous Cropping of Tobacco. Front. Microbiol. 8:974. doi: 10.3389/fmicb.2017.00974
Received: 30 March 2017; Accepted: 15 May 2017;
Published: 31 May 2017.
Edited by:
Philippe Prior, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Madhaiyan Munusamy, Temasek Life Sciences Laboratory, SingaporeNiklas Schandry, University of Tübingen, Germany
Copyright © 2017 Liu, Tang, Qin, Yang, Jiang, Li and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Ding, dingw@swu.edu.cn